By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2000, Geoffrey M Cooper.
Bookshelf ID: NBK9900

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000.

DNA, like any other molecule, can undergo a variety of chemical reactions. Because DNA uniquely serves as a permanent copy of the cell genome, however, changes in its structure are of much greater consequence than are alterations in other cell components, such as RNAs or proteins. Mutations can result from the incorporation of incorrect bases during DNA replication. In addition, various chemical changes occur in DNA either spontaneously (Figure 5.19) or as a result of exposure to chemicals or radiation (Figure 5.20). Such damage to DNA can block replication or transcription, and can result in a high frequency of mutations—consequences that are unacceptable from the standpoint of cell reproduction. To maintain the integrity of their genomes, cells have therefore had to evolve mechanisms to repair damaged DNA. These mechanisms of DNA repair can be divided into two general classes: (1) direct reversal of the chemical reaction responsible for DNA damage, and (2) removal of the damaged bases followed by their replacement with newly synthesized DNA. Where DNA repair fails, additional mechanisms have evolved to enable cells to cope with the damage.
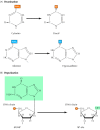
Spontaneous damage to DNA. There are two major forms of spontaneous DNA damage: (A) deamination of adenine, cytosine, and guanine, and (B) depurination (loss of purine bases) resulting from cleavage of the bond between the purine bases and deoxyribose, (more...)
Examples of DNA damage induced by radiation and chemicals. (A) UV light induces the formation of pyrimidine dimers, in which two adjacent pyrimidines (e.g., thymines) are joined by a cyclobutane ring structure. (B) Alkylation is the addition of methyl (more...)
Most damage to DNA is repaired by removal of the damaged bases followed by resynthesis of the excised region. Some lesions in DNA, however, can be repaired by direct reversal of the damage, which may be a more efficient way of dealing with specific types of DNA damage that occur frequently. Only a few types of DNA damage are repaired in this way, particularly pyrimidine dimers resulting from exposure to ultraviolet (UV) light and alkylated guanine residues that have been modified by the addition of methyl or ethyl groups at the O6 position of the purine ring.
UV light is one of the major sources of damage to DNA and is also the most thoroughly studied form of DNA damage in terms of repair mechanisms. Its importance is illustrated by the fact that exposure to solar UV irradiation is the cause of almost all skin cancer in humans. The major type of damage induced by UV light is the formation of pyrimidine dimers, in which adjacent pyrimidines on the same strand of DNA are joined by the formation of a cyclobutane ring resulting from saturation of the double bonds between carbons 5 and 6 (see Figure 5.20A). The formation of such dimers distorts the structure of the DNA chain and blocks transcription or replication past the site of damage, so their repair is closely correlated with the ability of cells to survive UV irradiation. One mechanism of repairing UV-induced pyrimidine dimers is direct reversal of the dimerization reaction. The process is called photoreactivation because energy derived from visible light is utilized to break the cyclobutane ring structure (Figure 5.21). The original pyrimidine bases remain in DNA, now restored to their normal state. As might be expected from the fact that solar UV irradiation is a major source of DNA damage for diverse cell types, the repair of pyrimidine dimers by photoreactivation is common to a variety of prokaryotic and eukaryotic cells, including E. coli, yeasts, and some species of plants and animals. Curiously, however, photoreactivation is not universal; many species (including humans) lack this mechanism of DNA repair.
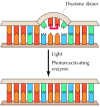
Direct repair of thymine dimers. UV-induced thymine dimers can be repaired by photoreactivation, in which energy from visible light is used to split the bonds forming the cyclobutane ring.
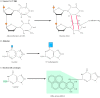
Another form of direct repair deals with damage resulting from the reaction between alkylating agents and DNA. Alkylating agents are reactive compounds that can transfer methyl or ethyl groups to a DNA base, thereby chemically modifying the base (see Figure 5.20B). A particularly important type of damage is methylation of the O6 position of guanine, because the product, O6-methylguanine, forms complementary base pairs with thymine instead of cytosine. This lesion can be repaired by an enzyme (called O6-methylguanine methyltransferase) that transfers the methyl group from O6-methylguanine to a cysteine residue in its active site (Figure 5.22). The potentially mutagenic chemical modification is thus removed, and the original guanine is restored. Enzymes that catalyze this direct repair reaction are widespread in both prokaryotes and eukaryotes, including humans.
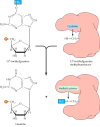
Repair of O6-methylguanine. O6-methylguanine methyltransferase transfers the methyl group from O6-methylguanine to a cysteine residue in the enzyme's active site.
Although direct repair is an efficient way of dealing with particular types of DNA damage, excision repair is a more general means of repairing a wide variety of chemical alterations to DNA. Consequently, the various types of excision repair are the most important DNA repair mechanisms in both prokaryotic and eukaryotic cells. In excision repair, the damaged DNA is recognized and removed, either as free bases or as nucleotides. The resulting gap is then filled in by synthesis of a new DNA strand, using the undamaged complementary strand as a template. Three types of excision repair—base-excision repair, nucleotide-excision repair, and mismatch repair—enable cells to cope with a variety of different kinds of DNA damage.
The repair of uracil-containing DNA is a good example of base-excision repair, in which single damaged bases are recognized and removed from the DNA molecule (Figure 5.23). Uracil can arise in DNA by two mechanisms: (1) Uracil (as dUTP [deoxyuridine triphosphate]) is occasionally incorporated in place of thymine during DNA synthesis, and (2) uracil can be formed in DNA by the deamination of cytosine (see Figure 5.19A). The second mechanism is of much greater biological significance because it alters the normal pattern of complementary base pairing and thus represents a mutagenic event. The excision of uracil in DNA is catalyzed by DNA glycosylase, an enzyme that cleaves the bond linking the base (uracil) to the deoxyribose of the DNA backbone. This reaction yields free uracil and an apyrimidinic site—a sugar with no base attached. DNA glycosylases also recognize and remove other abnormal bases, including hypoxanthine formed by the deamination of adenine, pyrimidine dimers, alkylated purines other than O6-alkylguanine, and bases damaged by oxidation or ionizing radiation.

Base-excision repair. In this example, uracil (U) has been formed by deamination of cytosine (C) and is therefore opposite a guanine (G) in the complementary strand of DNA. The bond between uracil and the deoxyribose is cleaved by a DNA glycosylase, leaving (more...)
The result of DNA glycosylase action is the formation of an apyridiminic or apurinic site (generally called an AP site) in DNA. Similar AP sites are formed as the result of the spontaneous loss of purine bases (see Figure 5.19B), which occurs at a significant rate under normal cellular conditions. For example, each cell in the human body is estimated to lose several thousand purine bases daily. These sites are repaired by AP endonuclease, which cleaves adjacent to the AP site (see Figure 5.23). The remaining deoxyribose moiety is then removed, and the resulting single-base gap is filled by DNA polymerase and ligase.
Whereas DNA glycosylases recognize only specific forms of damaged bases, other excision repair systems recognize a wide variety of damaged bases that distort the DNA molecule, including UV-induced pyrimidine dimers and bulky groups added to DNA bases as a result of the reaction of many carcinogens with DNA (see Figure 5.20C). This widespread form of DNA repair is known as nucleotide-excision repair, because the damaged bases (e.g., a thymine dimer) are removed as part of an oligonucleotide containing the lesion (Figure 5.24).
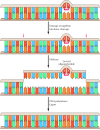
Nucleotide-excision repair of thymine dimers. Damaged DNA is recognized and then cleaved on both sides of a thymine dimer by 3′ and 5′ nucleases. Unwinding by a helicase results in excision of an oligonucleotide containing the damaged (more...)
In E. coli, nucleotide-excision repair is catalyzed by the products of three genes (uvrA, B, and C) that were identified because mutations at these loci result in extreme sensitivity to UV light. The protein UvrA recognizes damaged DNA and recruits UvrB and UvrC to the site of the lesion. UvrB and UvrC then cleave on the 3′ and 5′ sides of the damaged site, respectively, thus excising an oligonucleotide consisting of 12 or 13 bases. The UvrABC complex is frequently called an excinuclease, a name that reflects its ability to directly excise an oligonucleotide. The action of a helicase is then required to remove the damage-containing oligonucleotide from the double-stranded DNA molecule, and the resulting gap is filled by DNA polymerase I and sealed by ligase.
Nucleotide-excision repair systems have also been studied extensively in eukaryotes, particularly in yeasts and in humans. In yeasts, as in E. coli, several genes involved in DNA repair (called RAD genes for radiation sensitivity) have been identified by the isolation of mutants with increased sensitivity to UV light. In humans, DNA repair genes have been identified largely by studies of individuals suffering from inherited diseases resulting from deficiencies in the ability to repair DNA damage. The most extensively studied of these diseases is xeroderma pigmentosum (XP), a rare genetic disorder that affects approximately one in 250,000 people. Individuals with this disease are extremely sensitive to UV light and develop multiple skin cancers on the regions of their bodies that are exposed to sunlight. In 1968 James Cleaver made the key discovery that cultured cells from XP patients were deficient in the ability to carry out nucleotide-excision repair. This observation not only provided the first link between DNA repair and cancer, but also suggested the use of XP cells as an experimental system to identify human DNA repair genes. The identification of human DNA repair genes has been accomplished by studies not only of XP cells, but also of two other human diseases resulting from DNA repair defects (Cockayne's syndrome and trichothiodystrophy) and of UV-sensitive mutants of rodent cell lines. The availability of mammalian cells with defects in DNA repair has allowed the cloning of repair genes based on the ability of wild-type alleles to restore normal UV sensitivity to mutant cells in gene transfer assays, thereby opening the door to experimental analysis of nucleotide-excision repair in mammalian cells.
Molecular cloning has now identified seven different repair genes (designated XPA through XPG) that are mutated in cases of xeroderma pigmentosum, as well as in some cases of Cockayne's syndrome, trichothiodystrophy, and UV-sensitive mutants of rodent cells. Table 5.1 lists the enzymes encoded by these genes. Some UV-sensitive rodent cells have mutations in yet another repair gene, called ERCC1 (for excision repair cross complementing), which has not been found to be mutated in known human diseases. It is notable that the proteins encoded by these human DNA repair genes are closely related to proteins encoded by yeast RAD genes, indicating that nucleotide-excision repair is highly conserved throughout eukaryotes.
Enzymes Involved in Nucleotide-Excision Repair.
With cloned yeast and human repair genes available, it has been possible to purify their encoded proteins and develop in vitro systems to study the repair process. Although some steps remain to be fully elucidated, these studies have led to the development of a basic model for nucleotide-excision repair in eukaryotic cells. In mammalian cells, the XPA protein (and possibly also XPC) initiates repair by recognizing damaged DNA and forming complexes with other proteins involved in the repair process. These include the XPB and XPD proteins, which act as helicases that unwind the damaged DNA. In addition, the binding of XPA to damaged DNA leads to the recruitment of XPF (as a heterodimer with ERCC1) and XPG to the repair complex. XPF/ERCC1 and XPG are endonucleases, which cleave DNA on the 5′ and 3′ sides of the damaged site, respectively. This cleavage excises an oligonucleotide consisting of approximately 30 bases. The resulting gap then appears to be filled in by DNA polymerase δ or ε (in association with RFC and PCNA) and sealed by ligase.
An intriguing feature of nucleotide-excision repair is its relationship to transcription. A connection between transcription and repair was first suggested by experiments showing that transcribed strands of DNA are repaired more rapidly than nontranscribed strands in both E. coli and mammalian cells. Since DNA damage blocks transcription, this transcription-repair coupling is thought to be advantageous by allowing the cell to preferentially repair damage to actively expressed genes. In E. coli, the mechanism of transcription-repair coupling involves recognition of RNA polymerase stalled at a lesion in the DNA strand being transcribed. The stalled RNA polymerase is recognized by a protein called transcription-repair coupling factor, which displaces RNA polymerase and recruits the UvrABC excinuclease to the site of damage.
Although the molecular mechanism of transcription-repair coupling in mammalian cells is not yet known, it is noteworthy that the XPB and XPD helicases are components of a multisubunit transcription factor (called TFIIH) that is required to initiate the transcription of eukaryotic genes (see Chapter 6). Thus, these helicases appear to be required for the unwinding of DNA during both transcription and nucleotide-excision repair, providing a direct biochemical link between these two processes. Patients suffering from Cockayne's syndrome are also characterized from a failure to preferentially repair transcribed DNA strands, suggesting that the proteins encoded by the two genes known to be responsible for this disease (CSA and CSB) function in transcription-coupled repair. In addition, one of the genes responsible for inherited breast cancer in humans (BRCA1) appears to encode a protein specifically involved in transcription-coupled repair of oxidative DNA damage, suggesting that defects in this type of DNA repair can lead to the development of one of the most common cancers in women.
A third excision repair system recognizes mismatched bases that are incorporated during DNA replication. Many such mismatched bases are removed by the proofreading activity of DNA polymerase. The ones that are missed are subject to later correction by the mismatch repair system, which scans newly replicated DNA. If a mismatch is found, the enzymes of this repair system are able to identify and excise the mismatched base specifically from the newly replicated DNA strand, allowing the error to be corrected and the original sequence restored.
In E. coli, the ability of the mismatch repair system to distinguish between parental DNA and newly synthesized DNA is based on the fact that DNA of this bacterium is modified by the methylation of adenine residues within the sequence GATC to form 6-methyladenine (Figure 5.25). Since methylation occurs after replication, newly synthesized DNA strands are not methylated and thus can be specifically recognized by the mismatch repair enzymes. Mismatch repair is initiated by the protein MutS, which recognizes the mismatch and forms a complex with two other proteins called MutL and MutH. The MutH endonuclease then cleaves the unmethylated DNA strand at a GATC sequence. MutL and MutS then act together with an exonuclease and a helicase to excise the DNA between the strand break and the mismatch, with the resulting gap being filled by DNA polymerase and ligase.

Mismatch repair in E. coli. The mismatch repair system detects and excises mismatched bases in newly replicated DNA, which is distinguished from the parental strand because it has not yet been methylated. MutS binds to the mismatched base, followed by (more...)
Eukaryotes have a similar mismatch repair system, although the mechanism by which eukaryotic cells identify newly replicated DNA differs from that used by E. coli. In mammalian cells, it appears that the strand-specificity of mismatch repair is determined by the presence of single-strand breaks (which would be present in newly replicated DNA) in the strand to be repaired (Figure 5.26). The eukaryotic homologs of MutS and MutL then bind to the mismatched base and direct excision of the DNA between the strand break and the mismatch, as in E. coli. The importance of this repair system is dramatically illustrated by the fact that mutations in the human homologs of MutS and MutL are responsible for a common type of inherited colon cancer (hereditary nonpolyposis colorectal cancer, or HNPCC). HNPCC is one of the most common inherited diseases; it affects as many as one in 200 people and is responsible for about 15% of all colorectal cancers in this country. The relationship between HNPCC and defects in mismatch repair was discovered in 1993, when two groups of researchers cloned the human homolog of MutS and found that mutations in this gene were responsible for about half of all HNPCC cases. Subsequent studies have shown that most of the remaining cases of HNPCC are caused by mutations in one of three human genes that are homologs of MutL.
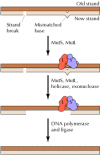
Mismatch repair in mammalian cells. Mismatch repair in mammalian cells is similar to E. coli, except that the newly replicated strand is distinguished from the parental strand because it contains strand breaks. MutS and MutL bind to the mismatched base (more...)
The direct reversal and excision repair systems act to correct DNA damage before replication, so that replicative DNA synthesis can proceed using an undamaged DNA strand as a template. Should these systems fail, however, the cell has alternative mechanisms for dealing with damaged DNA at the replication fork. Pyrimidine dimers and many other types of lesions cannot be copied by the normal action of DNA polymerases, so replication is blocked at the sites of such damage. Downstream of the damaged site, however, replication can be initiated again by the synthesis of an Okazaki fragment and can proceed along the damaged template strand (Figure 5.27). The result is a daughter strand that has a gap opposite the site of damage to the parental strand. One of two types of mechanisms may be used to repair such gaps in newly synthesized DNA: recombinational repair or error-prone repair.
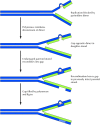
Postreplication repair. The presence of a thymine dimer blocks replication, but DNA polymerase can bypass the lesion and reinitiate replication at a new site downstream of the dimer. The result is a gap opposite the dimer in the newly synthesized DNA (more...)
Recombinational repair depends on the fact that one strand of the parental DNA was undamaged and therefore was copied during replication to yield a normal daughter molecule (see Figure 5.27). The undamaged parental strand can be used to fill the gap opposite the site of damage in the other daughter molecule by recombination between homologous DNA sequences (see the next section). Because the resulting gap in the previously intact parental strand is opposite an undamaged strand, it can be filled in by DNA polymerase. Although the other parent molecule still retains the original damage (e.g., a pyrimidine dimer), the damage now lies opposite a normal strand and can be dealt with later by excision repair. By a similar mechanism, recombination with an intact DNA molecule can be used to repair double strand breaks, which are frequently introduced into DNA by radiation and other damaging agents.
In error-prone repair, a gap opposite a site of DNA damage is filled by newly synthesized DNA. Since the new DNA is synthesized from a damaged template strand, this form of DNA synthesis is very inaccurate and leads to frequent mutations. It is used only in bacteria that have been subjected to potentially lethal conditions, such as extensive UV irradiation. Such treatments induce the SOS response, which may be viewed as a mechanism for dealing with extreme environmental stress. The SOS response includes inhibition of cell division and induction of repair systems to cope with a high level of DNA damage. Under these conditions, error-prone repair mechanisms are used, presumably as a way of dealing with damage so extensive that cell death is the only alternative.
Molecular Medicine : Colon Cancer and DNA Repair.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...