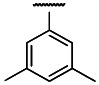
Heterotrimeric G-protein coupled receptors (GPCRs), the largest family of membrane-bound receptors, are major targets for therapeutic applications due to their broad tissue distribution, structural diversity, varied modes of action, and disease-associated mutations. The recently de-orphanized GPCR, GPR7, is distributed predominantly in the central nervous system. Neuropeptides W (NPW) and B (NPB) have been identified as endogenous ligands of GPR7. GPR7 represents a new and expanding target for drug development as it has been demonstrated to modulate the release of pituitary-derived hormones, and implicated in feeding behavior, the development of obesity, and mediating the inflammatory pain response. There is a significant medical need for novel compounds for treating chronic pain that are effective, long lasting and safe. To date, no small molecules that bind to the GPR7 receptor have been reported in the literature. The Scripps Research Institute Molecular Screening Center (SRIMSC), part of the Molecular Libraries Probe Production Centers Network (MLPCN), identified a potent and selective GPR7 antagonist probe, ML181, by high-throughput screening using a cell-based fluorescence assay. ML181 inhibited human GPR7 expression in the presence of a GPR7 agonist NPW with an IC50 of 272 nM. In a counterscreen for melanin-concentrating hormone receptor 1 (MCH1) antagonism in the presence of an agonist, ML181 has an IC50 of 3.9 μM, for a 14-fold selectivity for GPR7. ML181 is nontoxic to human colon adenocarcinoma (HT29) cells, with a CC50 of > 20 μM. As the first reported small molecule to modulate GPR7 activity, ML181 represents a significant milestone that will allow experiments aimed at elucidating the diverse roles of this receptor in physiological and pathological processes. ML181 also represents a potential therapeutic option in treating chronic pain.
Assigned Assay Grant #: 1-R03-DA026557-01
Screening Center Name & PI: The Scripps Research Institute Molecular Screening Center (SRIMSC), H Rosen
Chemistry Center Name & PI: SRIMSC, H Rosen
Assay Submitter & Institution: Olivier Civelli, University of California Irvine
PubChem Summary Bioassay Identifier (AID): 1880

Recommendations for scientific use of the probe
Heterotrimeric G-protein coupled receptors (GPCRs), the largest family of membrane-bound receptors, are major targets for therapeutic applications due to their broad tissue distribution, structural diversity, varied modes of action, and disease-associated mutations (1–4). The recently de-orphanized GPCR, GPR7 (5), is distributed predominantly in the central nervous system (5, 6). Neuropeptides W (NPW) and B (NPB) have been identified as endogenous ligands of GPR7 (5, 8). GPR7 represents a new and expanding target for drug development as it has been demonstrated to modulate the release of pituitary-derived hormones, and implicated in feeding behavior, the development of obesity, and mediating the inflammatory pain response (9–11). There is a significant medical need for novel compounds for treating chronic pain that are effective, long-lasting and safe (12).
To date, no small molecules that bind to the GPR7 receptor have been reported in the literature. The probe compound ML181 reported here inhibits NPW activation of GPR7 receptor with an IC50 in the low nanomolar range, and displays high selectivity versus MCH1. As the first reported small molecule to modulate GPR7 activity, this probe represents a significant milestone that will allow experiments aimed at elucidating the diverse roles of this receptor in physiological and pathological processes. This probe also represents a potential therapeutic option in treating chronic pain.
1. Introduction
Heterotrimeric G-protein coupled receptors (GPCRs) are the largest family of membrane-bound receptors and major targets for therapeutic applications due in part to their broad tissue distribution, structural diversity, varied modes of action, and disease-associated mutations (1–4). In particular, GPCRs are widely distributed in the peripheral and central nervous systems, where they are located on the plasma membrane of neurons along the nociceptive pathways (1, 7). They have been shown to play an important role in the modulation of pain and are one of the most important therapeutic targets in the area of pain management. (1,13). Chronic pain is a debilitating condition that exerts a high social cost in terms of productivity, economic impact, and quality of life. Currently available therapies yield limited success in treating such pain, suggesting the need for new insight into underlying mechanisms (12).
The recently de-orphanized GPCR, GPR7 (5), represents a new and expanding target for drug development, as it has been demonstrated to modulate the release of pituitary-derived hormones, regulate feeding behavior, and manipulate pain pathways (14). The genes for GPR7 and the closely related GPR8 receptor share 70% sequence identity with each other, and significant similarities with the transmembrane regions of the opioid and somatostatin receptors (15). Several studies have shown that GPR7 is distributed predominantly in the central nervous system, with the highest expression found in the hippocampus and amygdale (6, 7). Studies identifying the energy-regulating Neuropeptide W (NPW) and Neuropeptide B (NPB) as endogenous ligands of GPR7 (9, 10), and the development of abnormally increased appetite and obesity in male GPR7 knockout mice (7, 8), implicate GPR7 in feeding behavior.
Most notably, while under normal conditions GPR7 is expressed at low levels in the spinal cord, its expression is dramatically increased in patients suffering from inflammatory/immune-mediated neuropathies (11). Similar results have been found in animal models using immune-inflammatory and ligation-induced nerve injuries (9, 16). NPB-deficient mice demonstrate a super sensitivity in response to inflammatory pain, directly linking the NPB/GPR7 system to pain sensation (9). Therefore, understanding the role of GPR7 and its cognate ligands NPB and NPW may provide new physiological insights and may offer novel targets for therapeutic drug development (14).
The aim of this project is to identify potent and selective small molecule GPR7 antagonists to be used as valuable basic research tools that will greatly facilitate understanding of the involvement of GPR7 in chronic pain, its role in the mechanism of pain transmission and its interaction with other pain-related pathways. In particular, small compounds will be useful to study GPR7 with regard to its role in inflammatory neuropathies where established animal models are available.
Presently there are no small molecules available which could be used to modulate the GPR7 system. A search of patents posted on the US Patent and Trademark Office website on September 29, 2010, returned no results for GPR7-specific inhibitors or antagonists.
2. Materials and Methods
The following cell lines were provided by the Assay Provider: hGPR7 HEK293T/Gqi3, hMCHR1 HEK293T/Gqi3, and HT29. The following reagents were obtained from Invitrogen: DMEM (11965), Hank’s Balanced Salt Solution (14025-092), Geneticin (10131-027), Hygromycin-B (10687-010), Trypsin-EDTA solution (25200-056), 100X Penicillin-Streptomycin-Neomycin mix (15640-055), Zeocin (46-0509), and Fetal Bovine Serum (16140-071). Fluo-8 No Wash Calcium Assay Kit (36316) and Trypan red plus (2456) were obtained from ABD Bioquest. Fetal Bovine Serum (FB-02) was obtained from Omega Scientific. T-175 tissue culture flasks (431080), 150 mm tissue culture dishes (430599), and 384-well plates (3570) were obtained from Corning. 1536-well plates (19326) were obtained from Aurora. Neuropeptide W-23 NPW-23) (Human) (61653) was obtained from AnaSpec. Melanin-Concentrating Hormone (MCH) (Human, Mouse, Rat) (070-47), Neuropeptide W-23 (NPW-23)/L8 (Human) (005-60), and Neuropeptide B-23 (NPB-23)/L8 (Human) (005-53) were obtained from Phoenix Pharmaceuticals. T-75 tissue culture flasks (178905) were obtained from Nunc. 384-well plates (781964) were obtained from Greiner. Rotenone (R8875) and DMSO (472301) were obtained from Sigma. CellTiter Glo (S-G7570) was obtained from Promega. 96-well plates (T-3073-46) were obtained from ISC BioExpress. Reagents for the Ricerca HitProfilingScreen + CYP450 assay were provided by Ricerca Biosciences, LLC.
2.1. Assays
LC-MS/MS
All analytical methods are in MRM mode where the parent ion is selected in Q1 of the mass spectrometer. The parent ion is fragmented and a characteristic fragment ion is monitored in Q3. MRM mass spectroscopy methods are particularly sensitive because additional time is spent monitoring the desired ions and not sweeping a large mass range. Methods will be rapidly set up using Automaton® (Applied Biosystems), where the compounds are listed with their name and mass in an Excel datasheet. Compounds are submitted in a 96-well plate to the HPLC autosampler and are slowly injected without a column present. A narrow range centered on the indicated mass is scanned to detect the parent ion. The software then evaluates a few preselected parameters to determine conditions that maximize the signal for the parent ion. The molecule is then fragmented in the collision cell of the mass spectrometer and fragments with m/z larger than 70 but smaller than the parent mass are determined. Three separate collision energies are evaluated to fragment the parent ion and the largest three ions are selected. Each of these three fragment ions is further optimized and the best fragment is chosen. The software then inserts the optimized masses and parameters into a template method and saves it with a unique name that indicates the individual compound being optimized. Spectra for the parent ion and the fragmentation pattern are saved and can be reviewed later.
Solubility
The solubility of compounds was tested in phosphate buffered saline, pH 7.4. Compounds were inverted for 24 hours in test tubes containing 1–2 mg of compound with 1 mL of PBS. The samples were centrifuged and analyzed by HPLC (Agilent 1100 with diode-array detector). Peak area was compared to a standard of known concentration. In cases when the concentration was too low for UV analysis or when the compound did not possess a good chromophore, LC-MS/MS analysis was used.
Stability
Demonstration of stability in PBS was conducted under conditions likely to be experienced in a laboratory setting. The compound was dissolved in 1 mL of PBS at a concentration of 10 μM, unless its maximum solubility was insufficient to achieve this concentration. Low solubility compounds were tested between ten and fifty percent of their solubility limit. The solution was immediately aliquoted into seven standard polypropylene microcentrifuge tubes which were stored at ambient temperature in a block microcentrifuge tube holder. Individual tubes were frozen at −80°C at 0, 1, 2, 4, 8, 24, and 48 hours. The frozen samples were thawed in a room temperature and an equal volume of acetonitrile was added prior to determination of concentration by LC-MS/MS.
Primary uHTS assay to identify GPR7 antagonists (AIDs 1861, 1952, and 2251)
Assay Overview: The purpose of this assay was to identify compounds that inhibit GPR7 activity. Although GPR7 is naturally coupled to Gαi, which decreases cAMP levels upon activation, this assay employs a chimeric cell line that forces the receptor to use Gqi3, and therefore the assay readout is calcium release. In this assay, HEK cells stably co-transfected with the human GPR7 receptor and Gαqi3 (hGPR7 HEK293T/Gqi3 cell line) were treated with test compounds, followed by measurement of intracellular calcium as monitored by the FLUO-8 fluorescent, cell permeable calcium indicator dye. As designed, compounds that act as GPR7 antagonists will decrease calcium mobilization, resulting in decreased relative fluorescence of the indicator dye, and thus decreased well fluorescence. Test compounds were assayed in singlicate at a final nominal concentration of 4.4 μM (AID 1861), in triplicate at a final nominal concentration of 4.4 μM (AID 1952), or in triplicate in a 10-point 1:3 dilution series starting at a nominal test concentration of 44 μM (AID 2251).
Protocol Summary: The hGPR7 HEK293T/Gqi3 cell line was routinely cultured in T-175 sq cm flasks at 37 degrees C and 95% relative humidity (RH). The growth media consisted of Dulbecco’s Modified Eagle’s Media (DMEM) supplemented with 10% v/v heat-inactivated qualified fetal bovine serum, 25 mM HEPES, 200 μg/mL Hygromycin-B, 200 μg/mL Geneticin, 0.625 μg/mL Puromycin, and 1X antibiotic mix (penicillin, streptomycin, and neomycin). The day before the assay 1500 cells in 3 μL of growth media were seeded into each well of 1536-well microtiter plates and allowed to incubate at 37 degrees C, 5% CO2, and 95 % RH for 23 hours. Next, 2 μL of the fluorogenic Fluo-8 intracellular calcium indicator mixture with 1 mM trypan red plus (prepared according to the manufacturer’s protocol) was added to each well. After incubation for 1 hour at 37 degrees C, 5% CO2, and 95 % RH, 22 nL of test compound in DMSO, or DMSO alone, were dispensed to the appropriate wells. The assay was started after a 30-minute incubation at room temperature by performing a basal read of plate fluorescence (470–495 nm excitation and 515–575 nm emission) for 5 seconds on the FLIPR Tetra (Molecular Devices). Next, 15 nL of GPR7 agonist NPW (2 nM final concentration) in DMSO, or DMSO alone, were dispensed to the appropriate wells. Then a real-time fluorescence measurement was immediately performed for the remaining 180 seconds of the assay. Assay Cutoff: 1) Plate-based cutoff (AID 1861); 2) Compounds that inhibited GPR7 greater than 23.76% were considered active (AID 1952); 3) Compounds with an IC50 < 10 μM were considered active.
Counterscreen uHTS assay to identify MCH1 antagonists (AIDs 2148 and 2257)
Assay Overview: The purpose of this assay was to identify compounds that inhibit the melanin-concentrating hormone receptor 1 (MCH1) (GPR24), the receptor for MCH, a cyclic hypothalamic neuropeptide that promotes food intake (17), increases leptin and insulin release (18), and modulates energy metabolism (19). This assay also serves as a counterscreen for compounds identified as active in a previous set of experiments entitled, “Fluorescence-based primary cell-based high throughput screening assay to identify antagonists of the G-protein coupled receptor 7 (GPR7)” (AID 1861), and that confirmed activity in a previous set of experiments entitled, “Fluorescence-based confirmation cell-based high throughput screening assay to identify antagonists of the G-protein coupled receptor 7 (GPR7)” (AID 1952). This assay employs HEK cells stably co-transfected with human MCH1 and a chimeric Gαqi3. Cells are treated with test compounds followed by measurement of intracellular calcium as monitored by the FLUO-8 fluorescent, cell permeable calcium indicator dye. As designed, compounds that act as human MCH1 antagonists will decrease calcium mobilization, resulting in decreased relative fluorescence of the indicator dye, and thus decreased well fluorescence. Test compounds were assayed in triplicate at a final nominal concentration of 4.4 μM (AID 2148) or in triplicate in a 10-point 1:3 dilution series starting at a nominal test concentration of 44 μM (AID 2257).
Protocol Summary: The hMCHR1 HEK293T/Gqi3 cell line was routinely cultured in T-175 sq cm flasks at 37 degrees C and 95% RH. The growth media consisted of DMEM supplemented with 10% v/v heat-inactivated qualified fetal bovine serum, 25 mM HEPES, 200 μg/mL Hygromycin-B, 100 μg/mL Zeocin, 200 μg/mL Geneticin and 1X antibiotic mix (penicillin, streptomycin, and neomycin). The day before the assay, 1500 cells in 3 μL of growth media were seeded into each well of 1536-well microtiter plates and allowed to incubate at 37 degrees C, 5% CO2, and 95 % RH for 23 hours. Next, 2 μL of the fluorogenic Fluo-8 intracellular calcium indicator mixture with 1 mM trypan red plus (prepared according to the manufacturer’s protocol) was added to each well. After incubation for 1 hour at 37 degrees C, 5% CO2, and 95 % RH, 22 nL of test compound in DMSO, or DMSO alone, were dispensed to the appropriate wells. The assay was started after an additional 30 minute incubation at room temperature by performing a basal read of plate fluorescence (470–495 nm excitation and 515–575 nm emission) for 5 seconds on the FLIPR Tetra (Molecular Devices). Next, 15 nL of MCH peptide agonist (0.2 nM final concentration) in DMSO, or DMSO alone, were dispensed to the appropriate wells. Then a real-time fluorescence measurement was immediately performed for the remaining 180 seconds of the assay. Assay Cutoff: 1) Compounds that inhibited MCH1 greater than 21.36% were considered active (AID 2148); 2) Compounds with an IC50 ≤ 10 μM were considered active.
Late-stage assay to identify GPR7 antagonists (AID 463251)
Assay Overview: The purpose of this assay was to determine dose response curves for synthesized compounds in a 384-well plate format for antagonism of GPR7. The assay was as described above for the uHTS assay (AID 1861, AID 1952, and AID 2251). Test compounds were assayed in triplicate in an 8-point 1:3 dilution series starting at a nominal test concentration of 20 μM.
Protocol Summary: The hGPR7 HEK293T/Gqi3 cell line was routinely cultured in T-75 sq cm flasks at 37 degrees C and 95% RH. The growth media consisted of DMEM supplemented with 10% v/v heat-inactivated qualified fetal bovine serum, 25 mM HEPES, 200 μg/mL Hygromycin-B, 200 μg/mL Geneticin, 0.625 μg/mL Puromycin, and 1X antibiotic mix (penicillin, streptomycin, and neomycin). The day before the assay 10,000 cells in 25 μL of growth media were seeded into each well of 384-well microtiter plates and allowed to incubate at 37 degrees C, 5% CO2, and 95 % RH for 23 hours. Next, 25 μL of the fluorogenic Fluo-8 intracellular calcium indicator mixture with 1 mM trypan red plus (prepared according to the manufacturer’s protocol) was added to each well. After incubation for 50 minutes at 37 degrees C, 5% CO2, and 95 % RH, 100 nL of test compound in DMSO, or DMSO alone, were dispensed to the appropriate wells. The assay was started after an additional 15-minute incubation at room temperature by performing a basal read of plate fluorescence (470–495 nm excitation and 515–575 nm emission) for 5 seconds on the FLIPR Tetra (Molecular Devices). Next, 5.5 nL of GPR7 agonist (20 nM final concentration) in FLIPR buffer (HBSS/20 mM Hepes/0.1% BSA) was dispensed to the appropriate wells. Then a real time fluorescence measurement was immediately performed for the remaining 180 seconds of the assay. Assay Cutoff: Compounds with an IC50 ≤ 10 μM were considered active.
Late-stage counterscreen assay to identify MCH1 antagonists (AID 463250)
Assay Overview: The purpose of this assay was to determine dose response for a set of synthesized compounds in a 384-well format counterscreen assay for antagonism of MCH1. The assay was as described above for the uHTS assay (AID 2148 and AID 2257). Test compounds were assayed in triplicate in an 8-point 1:3 dilution series starting at a nominal test concentration of 20 μM.
Protocol Summary: The hMCHR1 HEK293T/Gqi3 cell line was routinely cultured in T-75 sq cm flasks at 37 degrees C and 95% RH. The growth media consisted of DMEM supplemented with 10% v/v heat-inactivated qualified fetal bovine serum, 25 mM HEPES, 200 μg/mL Hygromycin-B, 100 μg/mL Zeocin, 200 μg/mL Geneticin and 1X antibiotic mix (penicillin, streptomycin, and neomycin). The day before the assay, 10,000 cells in 3 μL of growth media were seeded into each well of 384-well microtiter plates and allowed to incubate at 37 degrees C, 5% CO2, and 95 % RH for 23 hours. Next, 25 μL of the fluorogenic Fluo-8 intracellular calcium indicator mixture with 1 mM trypan red plus (prepared according to the manufacturer’s protocol) was added to each well. After incubation for 50 minutes at 37 degrees C, 5% CO2, and 95 % RH, 100 nL of test compound in DMSO, or DMSO alone, were dispensed to the appropriate wells. The assay was started after an additional 15-minute incubation at room temperature, by performing a basal read of plate fluorescence (470–495 nm excitation and 515–575 nm emission) for 5 seconds on the FLIPR Tetra (Molecular Devices). Next, 5.5 nL of MCH peptide agonist (30 nM final concentration) in FLIPR buffer (HBSS/20 mM Hepes/0.1% BSA) were dispensed to the appropriate wells. Then a real time fluorescence measurement was immediately performed for the remaining 180 seconds of the assay. Assay Cutoff: Compounds with an IC50 ≤ 10 μM were considered active.
Cytotoxicity assay (AID 463253)
Assay Overview: The purpose of this assay was to determine cytotoxicity of a powder sample of a compound identified as active in the late-stage assay to identify GPR7 antagonists (AID 463251). In this assay, HT29 cells were incubated with test compound, followed by determination of cell viability. The assay utilizes the CellTiter-Glo luminescent reagent to measure intracellular ATP in viable cells. Luciferase present in the reagent catalyzes the oxidation of beetle luciferin to oxyluciferin and light in the presence of cellular ATP. Well luminescence is directly proportional to ATP levels and cell viability. As designed, compounds that reduce cell viability will reduce ATP levels, luciferin oxidation and light production, resulting in decreased well luminescence. Compounds were tested in triplicate in a 7-point 1:3 dilution series starting at a nominal test concentration of 20 mM.
Protocol Summary: HT29 cells were grown in DMEM supplemented with 10% v/v fetal bovine serum, 2 mM L-Glutamine, and 100U/mL penicillin and streptomycin. Prior to the start of the assay, 200,000 HT29 cells in 20 μL HBSS were dispensed into wells of a 384-well tissue culture-treated microtiter plate. Test compound was diluted 1:100 in growth medium (100 μM final concentration) and then serially diluted 1:3 in growth medium. The assay was started immediately by dispensing 5 μL of test compound, media alone, or rotenone as a positive control (150 μM final concentration) to the appropriate wells. The plates were then incubated for 2 hours at 37 degrees C. The plate was then equilibrated at room temperature for 30 minutes. The reaction was stopped by dispensing 25 μL of CellTiter-Glo reagent to each well, followed by incubation in the dark at room temperature for 10 minutes. Well luminescence was measured on the ViewLux plate reader. Activity Cutoff: Compounds with a CC50 value of ≤ 10 μM were considered active (cytotoxic).
Late-stage intracellular calcium release assay (AID 485339)
Assay Overview: The purpose of this assay was to identify compounds that inhibit GPR7 activity. Although GPR7 is naturally coupled to Gαi, which decreases cAMP levels upon activation, this assay employs a chimeric cell line that forces the receptor to use Gqi3, and therefore the assay readout is calcium release. In this assay HEK cells stably co-transfected with the human GPR7 receptor and Gαqi3 (hGPR7 HEK293T/Gqi3 cell line) are treated with test compounds, followed by measurement of intracellular calcium as monitored by the FLUO-8 fluorescent, cell permeable calcium indicator dye. As designed, compounds that act as GPR7 antagonists will decrease calcium mobilization, resulting in decreased relative fluorescence of the indicator dye, and thus decreased well fluorescence. Test compound was assayed in triplicate in an 8-point 1:3 dilution series starting at a nominal test concentration of 25 μM.
Protocol Summary: The hGPR7 HEK293T/Gqi3 cell line was routinely cultured in T-75 sq cm flasks at 37 degrees C and 95% RH. The growth media consisted of Dulbecco’s Modified Eagle’s Media (DMEM) supplemented with 10% v/v heat-inactivated qualified fetal bovine serum, 25 mM HEPES, 200 μg/mL Hygromycin-B, 200 μg/mL G418, 0.625 μg/mL Puromycin, and 1X antibiotic mix (penicillin and streptomycin). The day before the assay 70,000 cells in 100 μL of growth media were seeded into each well of 96-well microtiter plates and allowed to incubate at 37 degrees C, 5% CO2, and 95 % RH for 23 hours. Next, 100 μL of the fluorogenic Fluo-8 intracellular calcium indicator mixture with 1 mM trypan red plus (prepared according to the manufacturer’s protocol) was added to each well. After incubation for 50 minutes at 37 degrees C, 5% CO2, and 95 % RH, a basal read of plate fluorescence (470–495 nm excitation and 515–575 nm emission) for 1 second on the FLIPR1 (Molecular Devices) was taken. 50 μL of test compound in FLIPR buffer, or buffer alone, were dispensed to the appropriate wells. After an additional 10 minute incubation at 37 degrees C, 50 μL of GPR7 agonist NPB (10 nM final concentration) in FLIPR buffer (HBSS/20 mM Hepes/0.1% BSA) was dispensed to the appropriate wells. Then real time fluorescence was continuously monitored for the remaining 180 seconds of the assay. Assay Cutoff: Compounds with an IC50 ≤ 10 μM were considered active.
Ricerca HitProfilingScreen + CYP450 panel assay (AID 504401)
Assay Overview: The purpose of this panel of binding assays performed by Ricerca Biosciences, LLC, is to identify a subset of potential receptors, transporters, ion channels, etc. for which the GPR7 antagonist compound CID 46172919 displays affinity.
Protocol Summary: Assays for CYP450, 1A2; CYP450, 2C19; CYP450, 2C9; CYP450, 2D6; and CYP450, 3A4 were enzyme assays using human recombinant insect Sf9 cells with 5 μM 3-cyano-7-ethoxycoumarin as substrate (except for CYP450, 3A4, which used 50 μM 7-benzyloxy-4-(trifluoromethyl)-coumarin as substrate). Detection was based on spectrofluorimetric quantitation of the enzymatic product produced. Assays for the other targets were radioligand binding assays. Assay Cutoff: A response of ≥ 50% inhibition was considered active.
2.2. Probe Chemical Characterization
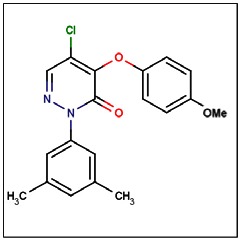
CID 46172919
SID 96021160
ML181
The probe structure was verified by NMR and high resolution MS. Purity was assessed to be greater than 95% by LC-MS (Figure 1).
Figure 1
LC-MS results for probe ML181.
MS (EI) m/z 358, 359 (M+). 1H NMR (300 MHz, CDCl3): δ 7.94 (s, 1H), 7.14 (bs, 2H), 7.00 (s, 1H), 6.98 (d, J = 8.9 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 3,77 (s, 3H), 2.33 (s, 6H).
The solubility of probe compound ML181 in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic, pH 7.4) at 23 degrees C was determined to be 2 μM, and the probe has a half-life of > 48 hours in PBS at 23 degrees C when tested at 1.4 μM (100% compound remaining at 48 hours) (Figure 2).

Figure 2
Stability of Probe ML181 in PBS.
The following compounds (Table 1) have been submitted to the SMR collection. Compound numbers refer to the SAR Table.
Table 1
Compounds Submitted to the MLSMR.
2.3. Probe Preparation
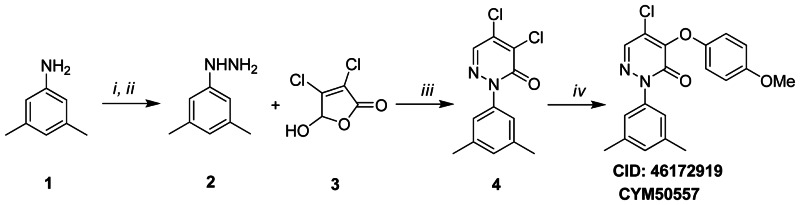
Figure 3.1Synthesis scheme for ML181
Reagents and conditions (i) (a) NaNO2, 6N HCL, −5 °C, 0.5 h; (b) SnCl2 · 2H2O, conc. HCl; (ii) 15% NaOH; (iii) (a) EtOH, rt, 3 h; (b) AcOH, reflux, 3 h; (iv) NaH, p-MeOPhOH, 1,4-dioxane, 0 °C→ rt, overnight
To 3,5-dimethylaniline 1 (8.0 mM) in 6N HCl (8.5 mL) was added dropwise at −5 degrees C a solution of NaNO2 (8.0 mM) in water (2.5 mL). The resulting mixture was stirred at −5 degrees C for 0.5 hour, then SnCl2·2H2O solution (4.5 g in 4 mL concentrated HCl) was added while keeping the temperature at −5 degrees C. The solid precipitated out and was collected and washed with cold ether. The crude product was dispersed in a 15% NaOH solution (20 mL) and extracted with ethyl acetate to afford (3,5-dimethylphenyl)hydrazine 2 (3.0 mM). A solution of 2 (3.0 mM) and mucochloric acid 3 (3.0 mM) in ethanol (8 mL) was stirred for 3 hours at room temperature. After removing the solvent by vacuum, AcOH (8 mL) was added and the obtained mixture was refluxed for 3 hours. The reaction mixture was neutralized with saturated Na2CO3 and extracted with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and evaporated under reduced pressure. Subsequent purification by flash chromatography (10% EtOAc/Hexane) afforded pyridazinone (1.5 mM) 4. MS (EI) m/z 270, 271 (M+). To a solution of para-methoxyphenol (0.2 mM) in anhydrous 1,4-dioxane (1.0 mL) was added NaH (60% mineral oil dispersion) (0.2 mM) at 0 degrees C. The mixture was stirred for 10 minutes at room temperature, then 4 (0.2 mM) was added at 0 degrees C. The resulting mixture was stirred at room temperature overnight. The reaction mixture was dispersed in saturated NaHCO3 and extracted with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography (10% EtOAc/Hexane) to afford the title compound in high purity (>95%).
According to the literature, nucleophilic addition at 4,5-dihalo pyridazin-3(2H)-one can occur selectively at C-4 by using non polar solvents such as anhydrous 1,4-dioxane (20). Unsubstituted derivative (x = H, Center internal number CYM50544, Scheme 1) was synthesized for both SAR analysis and structure assignment purposes. CYM50544 was obtained by dechlorination of the re-synthesized hit compound (CID 1479652, CYM50434) using Pd/C (10%) under hydrogen atmosphere. The 1H-NMR spectroscopy of the obtained CYM50544 supported the regiochemistry of the hit. The pyridazin-3(2H)-one protons originated two doublets at δ 6.22 ppm and 7.65 ppm with a coupling constant (J) of 4.6 Hz consistent with H5-H6 J of pyridazin-3(2H)-one ring (Fig. 3.2).

Figure 3.2
Analog Synthesis Scheme 1.
MS (EI) m/z 309 (M+). 1H NMR (400 MHz, CDCl3): 7.65 (d, J = 4.6 Hz, 1H), 7.48 (d, J = 7.5 Hz, 2H), 7.26 (d, J = 7.5 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.22 (d, J = 4.6 Hz, 1H), 3.81 (s, 3H), 2.39 (s, 3H).
3. Results
Compound 1 satisfies the goals for the probe characteristics identified in the CPDP: 1) probes should exhibit potent (≤ 1 mM) antagonistic activity against human and/or rat GPR7 in stably transfected HEK cells and 2) probes should have ≥ 10-fold selectivity for GPR7. Probe ML181 inhibited human GPR7 expression in the presence of a GPR7 agonist NPW with an IC50 of 272 nM (AID 463251). In a late-stage screen, probe ML181 inhibited human GPR7 expression in the presence of a GPR7 agonist NPB with an IC50 of 281 nM (AID 485339). In a counterscreen for MCH1 antagonism in the presence of an agonist, it has an IC50 of 3.9 μM (AID 463250), for a 14-fold selectivity for GPR7. ML181 is nontoxic to HT29 cells, with a CC50 of > 20 μM (AID 463253).
3.1. Summary of Screening Results
In the primary cell-based uHTS assay (AID 1861), 290K compounds were screened using a fluorogenic intracellular calcium indicator. A total of 3157 compounds (1.1%) were active. 2380 out of these 3157 compounds were available. For the uHTS confirmation assay (AID 1952), these 2380 active compounds were retested in triplicate, and 1246 compounds (52.4%) were confirmed as active. These 1246 confirmed active compounds were tested in triplicate in a uHTS counterscreen assay against MCH1 (AID 2148), and 130 compounds were found not to be active against MCH1. 126 out of these 130 compounds were available, and were tested in a dose response assay against GPR7 (AID 2251). 19 out of the 126 compounds had an IC50 < 10 μM. These 19 compounds were tested in a dose response counterscreen assay against MCH1, and 18 were found to be inactive, with an IC50 > 10 μM.

Figure 4GPR7 antagonist HTS overview
With the aim of discovering a novel class of small molecule GPR7 antagonists, a hit-to-lead research program was performed by exploring the structure-activity-relationships (SAR) of the identified hit compound 7 (see SAR Table). Various analogs modifying either the para-substituent (x) of the pendant aryloxy ring, the N-aromatic group (Ar), or the substituent (y) at the 5-position of the central core were synthesized. The SAR Table shows representative examples of compounds used for probe optimization. A subset of these compounds was submitted to the Molecular Libraries Small Molecule Repository (MLSMR) (Table 1). Generally, replacement of x with various substituents resulted in comparable or slightly increased GPR7 activity with respect to the original hit, whereas more variability was observed in terms of selectivity, thus indicating a possible role of x as binding site in the MCH1 rather than GPR7 interaction. Indeed, as some polar groups such as hydroxymethylene (compound 16) were tolerated, this site was found to be an important molecular feature to achieve enhanced physico-chemical properties.
Modifications of Ar significantly modified both potency and selectivity, thus suggesting a possible role of this portion as a pharmacophore site in the binding interaction with both GPR7 and MCH1. Submicromolar potency but increased MCH1 activity were observed in the presence of longer, linear or branched, alkylic groups such as ethyl, tert- and iso-butyl (compounds 4, 19, and 20), as well as for the meta-methyl regioisomer (compound 21).
Notably, higher inhibitory activity in the low nanomolar range was observed in the presence of iso-propyl at either the para- or meta-position (compounds 24 and 23), and for the meta-methyl-para-isopropyl derivative (compound 5), although with 5.5- to 10-fold higher activity for MCH1.
Moreover, the 1-naphthalyl analog (compound 22) showed high activity in combination with higher selectivity with respect to compound 7 (>3-fold). In particular, an attractive potency/selectivity profile with low nanomolar activity and satisfactory selectivity against MCH1 (14.4-fold), in combination with better drug-like properties was found for the 3,5-dimethyl derivative (compound 1), which was therefore selected as probe molecule for the target receptor.
While modifications of the putative binding site (Ar) were carried out, the central core was initially modified by derivatizing the 5-position (y) with various groups. The original hit compound 7 and compound 24 were selected as reference compounds. Exchanging of chlorine with different groups was generally deleterious, with the bromine (compound 18) being the only exception with slightly higher potency (2.8-fold). Unsubstituted analog (x = H) as well as derivatives with electron-donating groups were devoid of activity (structures not shown). Slight loss of potency (2.8-fold) was found for the cyclopropyl derivative (compound 12), while more significant decrements were observed in the presence of furan, thiophene, or methyl ester (compounds 9, 6, and 16) (6.5- to 10-fold), and particularly for methyl, phenyl, and various electron- withdrawing groups such as aldehyde and cyano (compounds 11, 8, 13, and 14) (15.5- to 27.8-fold). Complete loss of activity was observed with the introduction of a carboxylic group (structure not reported), which may be dissociated at physiological pH, thus suggesting that negative charges are not tolerated in the binding site. The activity over MCH1 was comparable to the reference compounds; the most significant differences were found for the cyclopropyl (compound 12) and methyl (compound 11) derivatives, which showed 3-fold increased and 3-fold decreased MCH1 activity, respectively. Noteworthy, based on the acquired information, further modifications at this site may be advantageous for drug-like properties by allowing removal of the halogen atom which may negatively affect the pharmacokinetic profile.
Additional modifications are currently in progress in order to acquire more detailed SAR information with the aim of further improving the potency/selectivity profile and the physico-chemical properties of the probe molecule.
3.2. Dose Response Curves for Probe

Figure 5Dose response curve for probe ML181
3.3. Scaffold/Moiety Chemical Liabilities
No reactive functional groups are observed in the probe molecule, which is very stable with a half life of > 48 hours.
3.4. SAR Table
Table
SAR Table.
3.5. Cellular Activity
The primary GPR7 inhibition assay is a cell-based assay (AID 1861, AID 1952, and AID 2251). We also employed a MCH1 cell-based counterscreen assay (AID 2148 and AID 2257). We tested the probe compound 1 in a cytotoxicity assay (AID 463253), where no cytotoxicity was observed in HT29 cells at 20 μM, the highest concentration tested. Thus the probe compound has been tested in three cellular assays.
3.6. Profiling Assays
To date, the lead hit compound 7 (CID 1479652) has been tested in 350 other bioassays deposited in PubChem, and has shown activity in only six of those assays, three of which are for the GPR7 antagonist project. The other three assays give a hit rate of 0.9%, indicating that this series is not generally active across a broad range of cell-based and non-cell based assays.
The selectivity profile of the probe compound ML181 was further investigated against the Ricerca panel of 35 off-target proteins, including therapeutically relevant GPCRs, enzymes and ion channels. The compound was found to be highly selective against the tested targets (stimulation/inhibition < 50% at 10 μM). A moderate but significant response of 53% inhibition was found only for CYP450 3A4 at 10 μM concentration.
4. Discussion
The lead hit compound was a weak hit with relatively little selectivity against the off target receptor MCH1. Probe ML181 inhibits human GPR7 expression in the presence of a GPR7 agonist with an IC50 of 272 nM (AID 463251). In a counterscreen for MCH1 antagonism in the presence of agonist NPW, it has an IC50 of 3.9 μM (AID 463250), for a 14-fold selectivity for GPR7. ML181 is nontoxic to HT29 cells, with a CC50 of > 20 μM (AID 463253). Its good antagonist activity has been confirmed by Dr Olivier Civelli at UC Irvine, the submitting PI (AID 485339).
4.1. Comparison to existing art and how the new probe is an improvement
To date, no small molecules that bind to the GPR7 receptor have been reported in the literature. The probe compound ML181 reported here inhibits NPW activation of GPR7 receptor with an IC50 below 300 nM, and displays selectivity versus MCH1 receptor. As the first reported small molecule to modulate GPR7 activity, this probe represents a significant milestone that will allow experiments aimed to elucidate the diverse roles of this receptor in GPR7 function in physiological and pathological processes. This probe also represents potential therapeutic option in treating chronic pain.
4.2. Mechanism of Action Studies
An intracellular calcium release assay was performed. Although GPR7 is naturally coupled to Gαi, which decreases cAMP levels upon activation, this assay employs a chimeric cell line that forces the receptor to use Gqi3, and therefore the assay readout is calcium release. The probe compound was effective at inhibiting calcium release in this assay, indicating that the probe interacts with GPR7 to inhibit downstream signaling events.
4.3. Planned Future Studies
The probe herein reported represents a promising starting point for a lead-optimization program aimed at developing innovative GPR7 antagonists with an improved potency and selectivity profile, and desirable pharmacokinetic and toxicokinetic properties suitable for clinical development. In the extended probe development period, in addition to simple analogue SAR, we propose to perform some in-house scaffold hopping using available software. We hope this will allow is to identify a second or third generation chemotype. The most promising compounds will be tested against the Ricerca panel of GPCRs to highlight selectivity, and will also be tested for selectivity against the panel of off-target proteins including GPCRs and ion channels. The pharmacokinetic (PK) properties of the lead compounds will also be optimized using a series of in vitro and in vivo (mouse) PK studies to identify a compound that is suitable for use in animal studies.
Dr. Civelli will perform experiments using animal models of neuropathic and inflammatory pain, including analgesic effects observed in the paw flick test and formalin test in rats, the behavioral response observed after acute thermal stimuli or chemically induced pain (4, 9), and a ligation-induced nerve injury model (11, 16).
5. References
- 1.
- Pan HL, Wu ZZ, Zhou HY, Chen SR, Zhang HM, Li DP. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol Ther. 2008;117(1):141–161. [PMC free article: PMC2965406] [PubMed: 17959251]
- 2.
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–357. [PubMed: 18382464]
- 3.
- Thompson MD, Cole DE, Jose PA. Pharmacogenomics of G protein-coupled receptor signaling: insights from health and disease. Methods Mol Biol. 2008;448:77–107. [PubMed: 18370232]
- 4.
- Bosier B, Hermans E. Versatility of GPCR recognition by drugs: from biological implications to therapeutic relevance. Trends Pharmacol Sci. 2007;28(8):438–446. [PubMed: 17629964]
- 5.
- Tanaka H, Yoshida T, Miyamoto N, Motoike T, Kurosu H, Shibata K, Yamanaka A, Williams SC, Richardson JA, Tsujino N, Garry MG, Lerner MR, King DS, O’Dowd BF, Sakurai T, Yanagisawa M. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc Natl Acad Sci U S A. 2003;100(10):6251–6256. [PMC free article: PMC156358] [PubMed: 12719537]
- 6.
- Brezillon S, Lannoy V, Franssen JD, Le Poul E, Dupriez V, Lucchetti J, Detheux M, Parmentier M. Identification of natural ligands for the orphan G protein-coupled receptors GPR7 and GPR8. J Biol Chem. 2003;278(2):776–783. [PubMed: 12401809]
- 7.
- Lee DK, Nguyen T, Porter CA, Cheng R, George SR, O’Dowd BF. Two related G protein-coupled receptors: the distribution of GPR7 in rat brain and the absence of GPR8 in rodents. Brain Res Mol Brain Res. 1999;71(1):96–103. [PubMed: 10407191]
- 8.
- Fujii R, Yoshida H, Fukusumi S, Habata Y, Hosoya M, Kawamata Y, Yano T, Hinuma S, Kitada C, Asami T, Mori M, Fujisawa Y, Fujino M. Identification of a neuropeptide modified with bromine as an endogenous ligand for GPR7. J Biol Chem. 2002;277(37):34010–34016. [PubMed: 12118011]
- 9.
- Kelly MA, Beuckmann CT, Williams SC, Sinton CM, Motoike T, Richardson JA, Hammer RE, Garry MG, Yanagisawa M. Neuropeptide B-deficient mice demonstrate hyperalgesia in response to inflammatory pain. Proc Natl Acad Sci U S A. 2005;102(28):9942–9947. [PMC free article: PMC1174999] [PubMed: 15983370]
- 10.
- Ishii M, Fei H, Friedman JM. Targeted disruption of GPR7, the endogenous receptor for neuropeptides B and W, leads to metabolic defects and adult-onset obesity. Proc Natl Acad Sci U S A. 2003;100(18):10540–10545. [PMC free article: PMC193597] [PubMed: 12925742]
- 11.
- Zaratin PF, Quattrini A, Previtali SC, Comi G, Hervieu G, Scheideler MA. Schwann cell overexpression of the GPR7 receptor in inflammatory and painful neuropathies. Mol Cell Neurosci. 2005;28(1):55–63. [PubMed: 15607941]
- 12.
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25(6):319–325. [PubMed: 12086751]
- 13.
- Ahmad S, Dray A. Novel G protein-coupled receptors as pain targets. Curr Opin Investig Drugs. 2004;5(1):67–70. [PubMed: 14983976]
- 14.
- Singh G, Davenport AP. Neuropeptide B and W: neurotransmitters in an emerging G-protein-coupled receptor system. Br J Pharmacol. 2006;148(8):1033–1041. [PMC free article: PMC1752024] [PubMed: 16847439]
- 15.
- O’Dowd BF, Scheideler MA, Nguyen T, Cheng R, Rasmussen JS, Marchese A, Zastawny R, Heng HH, Tsui LC, Shi X, Asa A, Puy L, George SR. The cloning and chromosomal mapping of two novel human opioid-somatostatin-like receptor genes GPR7 and GPR8, expressed in discrete areas of the brain. Genomics. 1995;1;28(1):84–91. [PubMed: 7590751]
- 16.
- Yamamoto T, Saito O, Shono K, Tanabe S. Effects of intrathecal and i.c.v. administration of neuropeptide W-23 and neuropeptide B on the mechanical allodynia induced by partial sciatic nerve ligation in rats. Neuroscience. 2006;137(1):265–273. [PubMed: 16289588]
- 17.
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380(6571):243–247. [PubMed: 8637571]
- 18.
- Bradley RL, Mansfield JP, Maratos-Flier E, Cheatham B. Melanin-concentrating hormone activates signaling pathways in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2002;283(3):E584–592. [PubMed: 12169453]
- 19.
- Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E. Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest. 2001;107(3):379–386. [PMC free article: PMC199192] [PubMed: 11160162]
- 20.
- Suree N, Yi SW, Thieu W, Marohn M, Damoiseaux R, Chan A, Jung ME, Clubb RT. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg Med Chem. 2009;17(20):7174–7185. [PMC free article: PMC2888031] [PubMed: 19781950]
Publication Details
Author Information and Affiliations
Authors
Miguel Guerrero,* Mariangela Urbano,* Zhiwei Wang,† Jian Zhao,* Subash Velaparthi,* Marie-Therese Schaeffer,‡ Steven J Brown,‡ S Adrian Saldanha,§ Peter Chase,§ Jill Ferguson,‡ Olivier Civelli,† Edward Roberts,* Peter Hodder,§ and Hugh Rosen‡.
Affiliations
Publication History
Received: October 13, 2010; Last Update: November 21, 2011.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Guerrero M, Urbano M, Wang Z, et al. Optimization and Characterization of an Antagonist for G-Protein Coupled Receptor 7 (GPR7) 2010 Oct 13 [Updated 2011 Nov 21]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.