In contrast to the previously described EE, a large proportion of patients with Dravet syndrome have mutations in known genes, leading to their systematic testing if the clinical context is suggestive.
De novo mutations in SCN1A are the main cause of Dravet syndrome
A common genetic predisposition between SMEI and febrile seizures was first suggested by Benlounis et al. in 2001. 32 Furthermore, Singh et al., (2001) considered SMEI to be a severe phenotype of the “generalised epilepsy and febrile seizures plus” (GEFS+) familial context, particularly because of the presence of patients with SMEI in families with GEFS+ (MIM 604233). 33 GEFS+ is a variable autosomal dominant epileptic condition that also associates febrile and afebrile seizures. Affected family members present with phenotypes ranging from isolated febrile seizures to various idiopathic generalized epilepsy subtypes (epilepsy with grand mal seizures, childhood absence or juvenile myoclonic epilepsy) or can remain asymptomatic. The outcome is usually benign and patients are sensitive to classical antiepileptic treatments. 34 However, family members can occasionally experience focal seizures, be severely affected and/or pharmacoresistant, and even present with DS. 33 This common genetic background was confirmed by Claes and coworkers (2001) who identified seven de novo mutations in SCN1A, which had previously been incriminated in GEFS+,35 in 7 sporadic cases of SMEI. 36 The vast majority (6/7) of these mutations led to premature termination codon. 36 More recent studies have confirmed the high frequency of mutations of SCN1A in sporadic SMEI. 37–42
Different types of mutation in SCN1A cause Dravet syndrome
All types of mutations have been identified in the coding sequence of the SCN1A gene in patients with Dravet syndrome: missense, nonsense, and splice-site mutations, small deletions and insertions. The mutations are located all along the SCN1A gene. Missense mutations are the most common mutation type identified (about 40%). 42 The main remaining mutation types (nonsense, splice-site mutations, frameshift) introduce PTCs into the mRNA, which are probably recognized and degraded via the NMD system. This hypothesis is compatible with the effects of the other mutation types detected, including alteration of the initiation codon and the microdeletions that delete essential regions of the proteins, introduce PTC, or delete the whole gene at the heterozygous state. Indeed, during the past few years, many techniques have been developed in order to detect genomic rearrangements such as deletions or duplications: quantitative PCR (QPCR), 43 multiplex ligation-dependent probe amplification (MLPA) 44 and microarrays. 45 The analysis of series of patients without point mutations with these methods has identified several heterozygous rearrangements in SCN1A, variable in length and breakpoints and encompassing from a single exon to the whole gene. 42, 46, 47 Deletion of the whole gene was the most common rearrangement found, supporting haploinsufficiency as the main molecular mechanism responsible for Dravet syndrome. Analysis of the parents showed that most deletions occurred de novo. The nearby genes comprised in the deletion, including other genes encoding voltage-gated sodium channels (SCN7A and SCN9A), were different between patients, without evident variability of the phenotype. 42 However, patients with very large deletions could have consistent dysmorphic features including ear abnormalities, microcephaly, micrognathia and brachysyndactyly, likely related to the size of deletion and deletions of other genes than SCN1A. 48
In spite of many functional studies, it remains unclear how missense mutations can cause a clinical phenotype indistinguishable from that of PTC mutations or whole-gene deletion and why some missense mutations are associated with a severe phenotype, such as Dravet syndrome, and others with curable epilepsies as in GEFS+ families. Kanai et al. (2004) suggested a preferential location of the missense mutations leading to Dravet syndrome in the S5 and S6 segments and the S5–S6 intracellular boucle, which together form the “pore” of the Nav1.1 channel.21. 49 Depienne and colleagues (2009) reported that most missense .mutations causing Dravet syndrome affected highly conserved amino acids located in ion-transport sequences and resulted in chemically dissimilar changes in amino acid classes. However, these mutations were not preferentially located in S5–S6 segments, in contrast to the previous report (). 42
Schematic representation of the missense mutations and in-frame deletions in the Nav1.1 protein. Each star represents a missense mutation. Red stars: mutations identified in a single patient; green stars: recurrent mutations; blue triangles: inframe deletions (more...)
The proportion of patients with SCN1A mutations is highly variable in reported studies, ranging from 33% 39, 50 to 80–100%.40, 51, 52 These discrepancies may be due to the sizes of the series, the methods of screening, and use of different clinical criteria to define Dravet syndrome. Supporting this hypothesis, the proportion of positive patients is higher in SMEI using strict criteria than in epileptic syndromes closely related to SMEI including ICEGTC (intractable childhood epilepsy with generalised tonic–clonic seizures) or SMEB (borderline SMEI). 51, 53 Considering only the patients with typical Dravet syndrome, approximately 20% of them do not have mutations in SCN1A, even when microrearrangements have been excluded. 41, 42 In addition, recent studies have established that rare pathogenic mutations in SCN1A, some of which are de novo, can also be identified in other infantile EEs such as cryptogenic generalized epilepsies, cryptogenic focal epilepsies or infantile spasms.50, 53
54 This extension of the clinical spectrum related to SCN1A has called into question the initial concept of SMEI, because neither clinical nor genetic criteria are sufficient to delimit accurately the various syndromes.
Dravet syndrome in a familial context of epilepsy
Few patients with DS have a parent or relative with a milder epileptic phenotype. Depienne and colleagues (2010) showed that mutations inherited from an asymptomatic or mildly affected parent were identified in 10% of the DS patients. Mosaicism (the mutation is present only in a fraction of germinal [germinal mosaicism] or non-germinal [somatic mosaicism] cells) was the main event associated with inherited SCN1A mutations in DS patients (about 70%). Parental mosaicism in DS was not a rare situation since it was found in at least 7% of families with an SCN1A mutation. When the level of somatic mosaicism was high, the parent could present with seizures, although he/she was less severely affected than his/her child who carried the mutation in all cells. The clinical status of the mosaic parent appeared to be somehow correlated with the amount of the mutation in his/her blood cells although this correlation was not strict. 55
Nevertheless, mosaicism is not the only situation accounting for inherited mutations as DS can also be encountered in the context of GEFS+ families. In that case, SCN1A missense mutations segregating in the family are associated with a wide phenotypic variability, with DS at the severe end of the spectrum. 33 Distinguishing mosaicism from de novo constitutional mutations or other situations is of particular concern in disorders that are frequently sporadic, such as DS, in order to give appropriate genetic counseling. 56 The risk of recurrence is, therefore, not null when the mutation is apparently de novo, which should be taken into account for genetic counseling.
A question that remains is whether SCN1A neomutations causing DS act through the same pathophysiological mechanisms as mutations found in GEFS+. Basically, mutations associated with clear loss-of-function are always associated with DS, except in one family recently described by Suls et al. (2010), in which a microdeletion of SCN1A segregated with a very variable phenotype in a 4-generation family. 57 In mosaic patients, the mutations with a loss-of-function effect can be associated with milder phenotypes reminiscent of those seen in GEFS+ if present in only some neurons. Conversely, missense mutations found in GEFS+ are generally associated with mild epileptic phenotypes but can occasionally cause DS. From a mechanistic point of view, it is likely that the pathophysiological pathways are different: de novo mutations would be sufficient to cause DS whereas missense mutations associated with GEFS+ would not, and additional genetic or non-genetic factors would be necessary to cause DS in the latter case.
Dravet-like syndrome in females associated with mutations in PCDH19 gene
At least 20% of patients with Dravet syndrome are negative for mutations or rearrangements in SCN1A. In addition, rare mutations in GABRG2 have been described in cases of SMEI belonging to GEFS+ families 58 but were not demonstrated in 29 sporadic cases. 38 These findings suggest that Dravet syndrome could be genetically heterogeneous (i.e. other genes are involved in DS).
Mutations in PCDH19 mainly affect females with a Dravet-like syndrome
In order to identify new genes responsible for the disorder in SCN1A mutation-negative patients, Depienne and colleagues screened 41 patients with DS for micro-rearrangements with high-density SNP microarrays. Interestingly, a hemizygous deletion on chromosome Xq22.1, encompassing the PCDH19 gene, was identified in one male patient. To confirm that PCDH19 was responsible for a Dravet-like syndrome, its coding region was sequenced in 73 additional SCN1A mutation-negative patients. Different point mutations (four missense and five truncating mutations) were identified in 11 unrelated female patients (15%). The spectrum of mutations includes non-sense mutations, small deletions/insertions introducing a frameshift as well as missense mutations affecting highly conserved amino acids in the protein, predominantly in the extracellular domain, which is presumably involved in cell-cell interaction. These mutations are therefore predicted to result in a loss-of-function of the mutated allele. 59
Protocadherin 19 is a 1148 amino acid transmembrane protein belonging to the protocadherin delta2 subclass of the cadherin superfamily, which is highly expressed in neural tissues and at different developmental stages. 60–63 The precise functions of the protein remain so far unknown. However, Delta protocadherins were reported to mediate cell-cell adhesion in vitro and cell sorting in vivo, and could regulate the establishment of neuronal connections during brain development. 64, 65
Patients with PCDH19 mutations could present with clinical features similar to patients with SCN1A mutations, including the association of early febrile and afebrile seizures, seizures occurring in clusters, developmental and language delays, behavioral disturbances, and cognitive regression. There were, however, slight but constant differences in the evolution of epilepsy, including fewer polymorphic seizures (in particular rare myoclonic jerks and atypical absences) in patients with PCDH19 mutations. These results show that PCDH19 plays a major role in epileptic encephalopathies, with a clinical spectrum overlapping that of DS. 59
PCDH19-linked epilepsies: an unusual mode of inheritance
Mutations in PCDH19 were first reported to cause EFMR (epilepsy and mental retardation limited to females). The clinical features of EFMR, unlike those of DS, are highly variable, even in members of the same family: onset of seizures is between 6 and 36 months and affected females present with a combination of febrile and afebrile seizures of various types and a variable degree of psychomotor delay and cognitive impairment, ranging from mild to severe mental retardation. 66 Dibbens et al. reported PCDH19 mutations in six large families and one small family with two affected sib pairs. 60 All the patients were familial cases that were, for the most part, already adults at the time of examination, and appeared socially integrated in that most of them were married and had children.
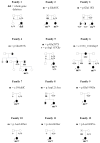
PCDH19-related epileptic encephalopathy therefore mainly affects females. In a large series of PCDH19 mutation-positive index cases in whom inheritance could be assessed, half of the mutations occurred de novo and half were inherited from fathers who were healthy, had no cognitive impairment and had never had febrile seizures or epilepsy. 59 (Depienne at al., personal communication) This heredity is very different from the X-linked mode of inheritance encountered for ARX or CDKL5 mutations since in this case only the heterozygous females are affected whereas the hemizygous males are asymptomatic and spared ().
Segregation analysis of the PCDH19 deletion and point mutations in 12 families. del/+, m/+ and v/+ denote individuals heterozygous for the deletion, mutation or variant, respectively; +/+ denotes individuals carrying homozygous wild-type alleles. Squares (more...)
Several mechanisms have been suggested to account for the unusual mode of inheritance observed in PCDH19-linked epilepsy, one of which is cellular interference, a mechanism reminiscent of metabolic interference. 60
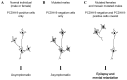
67, 68 This concept postulates that random inactivation of one X chromosome in mutated females generates tissue mosaicism (i.e., coexistence of PCDH19-positive an PCDH19-negative cells), which would be pathogenic by altering cell-cell interactions; normal individuals and mutated males, who are homogeneous for PCDH19-positive or PCDH19-negative cells, respectively, would not develop the disease (). The identification of an affected male who was mosaic for the PCDH19 deletion in his fibroblasts, and therefore had PCDH19-positive and PCDH19-negative cells in this tissue, strongly supports the hypothesis of cellular interference as the main pathogenic mechanism associated with PCDH19 mutations. 59
Schematic illustration of the cellular interference mechanism associated with PCDH19 mutations. A) In normal individuals characterized by a homogeneous population of PCDH19-positive cells, neurons are able to form normal neuronal networks. B) In male (more...)