At the end of July 2010, there were 10,168 references under “kainic acid” and 5,149 references under “kainic acid AND neurons” in PubMed. The first reference to kainate 1 reported the structure of kainate and allokainate. In the next 20 years, attention to kainate was focused on its strong excitatory actions on crayfish muscles and other systems. The first references by the groups of Curtis and Watkins described the excitatory actions of this new agent that had just been purified from a sea weed known to exert an anti-ascaris action 2;3. Kainate –a glutamate analogue- is a stronger excitant than other amino acids, and pharmacological observations pointed to its unique structure/actions features that suggested the presence of a specific receptor, an observation that was rapidly confirmed. The identification of kainate-specific receptors stimulated a flurry of studies on the excitatory actions of kainate that were soon to be viewed as “excitotoxic” when Olney, Nadler and colleagues discovered that kainate in fact selectively destroys various neuronal populations 4–7. Their finding was the starting point for two parallel lines of research focused on the following questions: Why does kainate kill neurons selectively? How can this action be used as a tool to reproduce animal models of neurological disorders?
During the late seventies and early eighties, extensive investigation showed that kainate excites a large number of neuronal populations where receptors are present; this excitation is followed by a selective neuronal loss at the site of injection, while the axons en passant are spared. Given this finding, McGeers, Schwartz and Coyle and other investigators produced several animal models of neurological disorders, relying on the “axon sparing” excitotoxic actions of kainate to model Parkinson’s disease and Huntington’s disease 8–12. The technique was also used to selectively destroy cell bodies (i.e., spare axons), thus providing a method to trace neuronal connections in the brain and to determine the selective sequelae produced by “neuron-specific” lesions of a brain structure without altering en passant fibres. Thus, the first phase of kainate research exploited the lesioning capabilities of kainate to delete specific neuronal populations and determine the consequences of this loss on brain operation.
In 1978, during an investigation aimed at using the axon sparing effects of kainate in the amygdala, Lagowska and Ben-Ari discovered the epileptogenic actions of amygdaloid injections of kainate 13. Injections of low concentrations of kainic acid in the amygdala generated a status epilepticus that persisted for several hours, often until the animal died. Injections of benzodiazepine were used to interrupt the seizures, providing an animal model of limbic status epilepticus in which the mechanisms underlying the generation of a status and its effects on brain operation can be investigated 14. This publication, in turn, led to a flurry of studies; in three decades, over 2,495 references to “kainate AND epilepsies” appeared on PubMed, (July 2010). Extensive investigations used injections of kainate parenterally and intra-cerebrally to test the properties of epileptogenic neurons and networks. These studies have provided a wide range of advances in our understanding of the mechanisms of seizure generation, propagation, and their sequelae, with the emergence of concepts extending from apoptotic cell death to reactive plasticity and sprouting of mossy fibres in TLE to the selective loss of GABAergic neurons (see ) 15. These concepts were subsequently confirmed with other animal models of TLE, notably pilocarpine 16 and electrical brain stimulation 17. Parallel studies led to the discovery of kainate high-affinity receptors in the brain and having a specific distribution—notably in regions that are known to play a central role in seizures, particularly in the hippocampus 18–20. With the development of molecular biology and genetic tools, kainate receptors were cloned, and shown to belong to the glutamate receptor family, with several subunits conferring unique properties; these characterizations provided clear examples of the intricate links between biologically active natural molecules and recognition signals in the brain for these molecules 21. This insight was further expanded by the development of relatively specific antagonists that block AMPA but not kainate receptor-mediated currents (see below). Investigators were then able to identify centrally active kainatergic synapses, that when activated by glutamate generate synaptic currents. Compelling evidence that kainate plays a role in central transmission was obtained in studies showing spontaneous kainatergic EPSCs and miniature kainatergic PSCs with electrical properties that differ from the predominant fast acting AMPA receptor mediated PSCs generated by glutamate. Glutamate activates a series of receptors, with kainate receptors playing a specific role different from NMDA and AMPA receptors. This finding, in turn, raised formidable questions: Why do certain neuronal populations use these long-lasting PSCs mediated through kainatergic receptors? Why are these receptors enriched in regions known to be highly vulnerable to seizures and to be destroyed in patients suffering from TLE? The evolution of an “en passant” discovery of the destructive properties of a biologically active molecule to an endogenous selective set of synapses employing these receptors to carry out physiological functions stands at the core of present kainate research. This discovery has provided important insights into brain operation and will undoubtedly also lead to novel therapeutic strategies for TLE.
A. Seizure and brain damage produced by kainate in the adult brain
1. The kainate model of Temporal Lobe Epilepsy (TLE)
From the late 1970s, studies revealed that intra-amygdaloid, intrahippocampal, intracerebroventricular, or systemic injections of kainate generates a syndrome of seizures and brain damage that mimics human TLE (reviewed in 15;22–28. Injections of kainic acid either peripherally, or centrally in various brain structures generate seizures and brain damage syndromes in which limbic structures play key roles. Clinically, after a variable delay, animals display wet dog shakes, facial motor signs and chewing, paw tremor associated with rearing and falling, and other complex motor manifestations. EEG and depth recordings reveal recurrent tonic-clonic severe ictal discharges and a long-lasting status epilepticus that are reminiscent of those observed in TLE. EEG recordings established that the seizures are initiated in regions that are highly excitable and central to TLE, notably the hippocampal CA3 region and the amygdala. The description of the propagation of seizures discharges has been repeatedly shown to include triggering zones –notably within the hippocampus the CA3 region and various amygdaloid and adjacent pyriform and entorhinal cortex regions - that appear to have among the lowest thresholds for seizure generation in the rat brain. The anatomical pathways that enable seizures to express motor manifestations are consistent with classical axonal pathways that interconnect limbic structures and their projection targets. Thus, the facial signs are activated when the seizure activity in amygdala propagates to its brain stem targets, as defined by extensive human and animal investigation. Studies using 2-deoxyglucose metabolic methods as well as a wide range of other imaging and recording techniques, have confirmed the general pattern of structures involved and the crucial role of limbic structures. In addition, following kainate-induced status epilepticus, there is a silent period followed by ongoing, chronic seizure activity; although the initial status is not often observed in humans, this pattern of seizure development in animal models has been useful for dissecting out the mechanisms underlying cell death and chronic epilepsies. The kainate and pilocarpine models of TLE have been extensively used for such investigations. Other closely related models include kindling, in which daily electrical stimuli (usually of the amygdale) transforms a naïve structure to one that seizes upon stimulation 29;29;30. The pros and cons of these models have been reviewed and discussed extensively. The goal here is to stress that they provide, collectively, useful contexts in which to study mechanisms of epileptogenesis in limbic structures 31–33. The clinical and pathological effects of kainate are reminiscent of those observed in partial temporal lobe epilepsies 15;26;30;32.
2. Seizure-specific and non-specific cell loss produced by kainate
Administration of kainate produces a seizure and brain damage syndrome. Following recurrent seizures, there is a pattern of cell loss primarily –but not solely – in limbic structures including the hippocampus, amygdala and pyriform cortex 15;34. Within the hippocampus, there is a gradient of vulnerability, with CA3 pyramidal neurons being particularly vulnerable, as they can be produced with low doses (that may be truly homeopathic concentrations) of kainate applied locally or at distant sites 15;23. These observations raised considerable interest in determining the mechanisms underlying cell loss. One question was whether the damage is due to the direct actions of kainate (i.e., directly exciting the cell, increasing calcium influx, and initiating a cascade of events that have been extensively investigated in relation to “neurotoxicity” 4;7. Two types of cell loss have been identified on the basis of morphological and chemical studies: apoptotic –or programmed - cell loss and necrosis (i.e., swelling followed by cell death 35;36). It is most likely that the mechanisms underlying cell death in the kainate model (as in other models such as ischemic cell loss 35) depend on the severity of seizures and the concentrations of kainate injected; small doses appear to produce preferentially apoptosis and higher doses produce necrotic cell damage. In keeping with this scheme, the regional susceptibility of hippocampal neurons in seizure models parallels the pattern of vulnerability to ischemic insults.
These observations raise yet another important question: Does kainate induce cell loss because of its direct actions or because of the seizures it generates? The following observations suggest that seizures generated by kainate can induce selective secondary damage due to seizures per se: i) following intra-amygdaloid injections of kainate, injections of valium bock seizures and prevents “distal” hippocampal damage but not local cell loss in the amygdala14;37;ii) quantitative measures of the severity of intra-hippocampal electrographic seizures revealed an excellent relation between the severity of paroxysmal events (including the duration of ictal events and post ictal depression) and subsequent damage38; iii) the damage in the hippocampus is prevented by lesions of mossy fibre synapses on CA3 neurons, confirming the essential role of propagated activity in these synapses in the high degree of vulnerability of target neurons to seizures 39–42.; iv) other types of limbic seizures, produced by pilocarpine or by long-lasting recurrent electrical stimulation of the fascia dentate, also trigger cell death with a similar pattern of vulnerability 16;43; v) direct local determination of the blood flow, oxygen consumption and PCO2 showed that during recurrent seizures, the increase of blood flow over-compensates for the enhanced oxygen consumption, suggesting that the damage is not due to a metabolic failure44. Therefore, recurrent seizures can produce directly cell loss in vulnerable neurons.
3. Loss of GABAergic interneurons and failure of inhibition
Because GABA-mediated inhibition controls the excitability of principal neurons and networks, a failure of inhibition has long been favoured to explain seizure generation and propagation. In keeping with this hypothesis, GABA receptor antagonists generate seizures and agents thought to specifically reinforce the GABAergic drive often reduce seizures and are used extensively as antiepileptic agents. However, there is a large variety of GABAergic populations, each of which innervate specific targets controlling highly specific cellular and network features 45–47. By means of their cell- or domain-specific targets, activation of GABAergic synapses will differently alter neuronal and network excitability 45–47. For example, some GABAergic neurons innervate the axon hillock of projection cells where spikes are generated; the release of GABA at these synapses alters the generation of action potentials by a large population of pyramidal neurons 48–50. Other GABAergic neurons innervate the dendrites of pyramidal neurons; Oriens Lacunosum Moleculare interneurons (OLM) release GABA that would be expected to reduces the excitatory drive that impinges on pyramidal neurons (see ) 48. The former synapses will entrain large ensembles of principal neurons in coordinated patterns, whereas the latter will more locally alter the responses of these neurones to inputs. GABAergic neurons also exert their inhibitory actions by a plethora of different mechanisms, including a shunting action and an intracellular accumulation of chloride that would tend to shift GABA reversal potential (EGABA) which results in an excitatory action of GABA (see below).
Somatic projecting GABAergic interneurons are more active in the hippocampus of epileptic animals. Recurrent seizures were generated by pilocarpine injections and weeks later once the reactive reaction has taken place, slices were recorded from the sclerotic (more...)
Experiments using kainate and/or pilocarpine illustrate the multiple facets of the suggested failure of inhibition and its implications in seizures and epilepsies. A decrease in the number of GAD mRNA-containing neuron has been observed following pilocarpine seizures primarily in interneurons labelled for somatostatin only (O-LM and bistratified cells) 17;51. Electron microscopic observations suggest that the loss of somatostatin-containing neurons corresponds preferentially to the degeneration of interneurons with an axon projecting to stratum lacunosum-moleculare (O-LM cells) 51. In contrast, stratum oriens interneurons labelled for parvalbumin were preserved, suggesting that somatic inhibition is maintained whereas dendritic inhibition is not. In keeping with this, direct recording form the somata and dendrites of principal neurons using the same model 52 revealed a marked reduction of GABAergic inhibition in the dendrites whereas somatic inhibition was preserved 52;53. Also, the activity of basket cells that have not been destroyed by the seizures 52–54 is enhanced thereby compensating for the loss of other interneurons. These observations collectively suggest that the glutamatergic inputs to the dendrites of principal cells will be less inhibited in epileptic neurons whereas somatic inhibition will not. Other mechanisms affecting the strength of inhibition have been suggested including notably: i) a reduction of miniature GABAergic Post Synaptic Currents (mIPSCs), suggesting a decrease release of GABA from presynaptic elements 53, and ii) post synaptic reduction of the efficacy of zinc 55–57, NMDA receptors and Ca2+ signals 58, and the density of GABAergic synapse subunit composition 59;60.
The suggestion that inhibition is not reduced per se, but rather there is a loss of the excitatory drive onto basket cells following a seizure (an induced denervation of interneurons –the so called “dormant basket cell” 61 - has been confirmed by pair recordings from interneurons and their target pyramidal neurons in TLE hippocampi. These studies showed that GABAergic inhibition is “operative” 54. It bears stressing that alterations of GABAergic inhibition are area and cell specific (but also see 62;63. Therefore, some (but not all) facets of GABAergic actions are reduced in TLE, most likely explaining why seizures are not continuously generated by the sclerotic hippocampus.
4. Physiological actions of kainate on hippocampal neurons
One important question is whether kainate exerts it actions through specific kainate receptors. Direct recordings from limbic neurons have revealed that the hippocampus and particularly CA3 neurons are strongly excited by kainate. Submicromolar applications of kainate excite CA3 pyramidal neurons 64–66, generating large currents and trigger action potentials. Kainate-induced currents are also observed in the presence of TTX (ibid and 67;68. At higher concentrations, kainate also activates AMPA receptors 25 [How is that distinguished from the activation seen at lower concentrations, as described above?]. In the presence of specific AMPA and NMDA receptor blockers, kainate depolarises rat and mouse hippocampal interneurons 69–71.
How is this excitatory drive generated? A large repertoire of actions underlies the excitatory actions of kainate. Kainate strongly reduces evoked GABAergic PSCs 71–74, an effect that would augment excitability. However, small sub-micromolar concentrations of kainate augment spontaneous IPSCs in interneurons 75 as well as GABAergic mIPSPs in CAI interneurons (also see 74 suggesting a presynaptic regulation by kainate receptors of GABA release that increases the efficacy of GABAergic transmission between interneurons. In keeping with this, in paired recordings, low concentrations of kainate (300nM) reduced the failures and increase the occurrence of post synaptic responses 76 but higher concentrations (5μM) depressed IPSCs confirming, the dose dependence of the dual actions of kainate.
Kainate also acts on voltage gated currents. It reduces the slow hyperpolarising K+ current 77;78, effects that are mediated by intracellular second messengers 79–81. These effects can be induced with nanomolar concentrations of kainate, suggesting that they are mediated by the high affinity receptors observed on these neurons (see below). This strong excitatory action mediated by high affinity kainate receptors plays an important role in the epileptogenic actions of kainate (also see82).
5. The Yin and the Yan of kainate receptors: molecular considerations
The advent of novel genetic tools has enabled investigators to decipher between different subtypes of glutamate receptors, including kainate receptors. Kainate receptors are composed of various combinations of five subunits: GluR5, GluR6, GluR7, KA1 and KA2 21;83–85. In studies of hyper-excitability and epilepsy, the GluR6 and GluR5 subtypes of kainate receptors have been of particular interest. The former has been thought to be related to limbic epilepsies because of its distribution, especially in the vulnerable CA3 pyramidal neurons86–88. In addition, GluR6 knockouts have a greatly reduced vulnerability to seizures and to kainite-induced injury 68
39;89. In contrast, GluR5 subunits, are mostly expressed by certain types of interneurons, notably somatostatin interneurons that are enriched within stratum oriens; in these cells the activation of kainate receptors leads to a dramatic increase of cell activity, and of the consequent inhibitory drive onto target pyramidal cells (see ) 69;70;90. Given these data, it appears that kainate may exert a dual action: It may have pro-convulsive effects in principal neurons due to the activation of GluR6 receptor subtypes, and anticonvulsive effects due to the activation of GluR5 subunits in interneurons. The strong excitation by GluR5 receptor agonists could explain the vulnerability of interneurons to seizures, as this action will dramatically excite these neurons. Additional subunit-specific actions have been reported and reviewed extensively elsewhere 25;91;92.
Dual actions of kainate: The GluR5 agonist ATPA excites interneurons thereby inhibiting the pyramidal neurons. A pyramidal neuron and an interneuron were recorded simultaneously and the GluR 5 selective agonist applied. Note the dramatic increase activity (more...)
B. Seizures but no brain damage produced by kainate in pups
1. maturation of the kainate system
The syndrome of seizures and brain damage produced by kainate is age dependent. Injections of kainate into immature pups generate seizures, but these seizures are not followed by cell loss 93–97. Conspicuous damage is first seen at the end of the 2nd post natal week; before then, even high doses of systemic kainate (that generate seizures) fail to produce cell damage 93–97. The clinical reflection of kainite-induced seizures is quite restricted initially –little or no facial movements or other manifestations that implicate the activation of the amygdaloid complex and its projections to the brainstem - suggesting that the critical network is not yet mature. This assumption was confirmed by direct recordings and by 2-deoxyglucose studies showing that the kainate-induced seizures are initially local, with no sign of propagation to the entire limbic system {Tremblay, 1984 15796/id;Albala, 1984 13047/id}. This time-frame correlates with the progressive and protracted maturation of the fascia dentate-mossy fibre system that extends to the 3rd week of age at least in rodents 98. Consistent with this finding, lesions of the mossy fibres (by irradiation or other means) also protect CA3 pyramidal neurons from kainate-induced damage (see below). In addition to reduced hippocampal cell loss following status epilepticus, there is also less mossy fibres sprouting in young animals than adult animals suggesting that the neuronal damage and subsequent reactive plasticity are correlated 99;100. Interestingly, cell loss is also less readily produced in neonatal neurons by other experimental models of TLE suggesting that the full maturation of networks and connections is indeed required for the cell loss to be produced 101.
We do not fully comprehend why immature neurons are more resistant than adults to insults and epilepsies. This is likely a general phenomenon as suggested by the observation that immature neurons are also much more resistant to anoxic insults 102
103 and have reduced pro-inflammatory cytokines associated with seizures 104. The reduced sensitivity to glutamate 105–108 and reduced oxidative stress compared to adult seizures 109 may be pertinent in this context. Also, GABA synthesis is better preserved during status epilepticus in neonatal neurons than adults 110. Therefore, although this reduced vulnerability is not restricted to the maturation of the kainate system, it illustrates its general pertinence to epilepsies of the developing brain.
2. Recurrent seizures in the immature brain produce long term effects
Although seizures in pups produce a less severe syndrome and no cell loss until the end of the 2nd week, they are not harmless. Extensive human and experimental data suggest that seizures early in life can lead to life-long severe intractable neurological disorders, a result of particular concern since. infants and children are at a high risk for seizures compared with adults 111;112. Although most seizures in children are benign and result in no long-term consequences, experimental animal data strongly suggests that frequent or prolonged seizures in the developing brain result in long-lasting sequel 113–115. Behavioural effects of acute seizures or status epilepticus are also associated with the age of the animal, adult animals having substantial deficits in learning, memory and behaviour113–115.
Recurrent seizures in children and infants can be harmful, causing long lasting sequel 116;117. Similarly, recurrent seizures in pups can cause long term behavioural and physiological alterations. Neonatal seizures induced by the inhalant fluothyl produces impairment of visual memory without any discernible cell loss 118. This study also shows that after recurrent seizures, the number of newly formed granule cells is reduced in neonates but increased in adults. Since recurrent seizures do not readily cause cell loss and extensive damage, the long-term deficits must be due to other mechanisms.
What do these observations suggest and how can they be reconciled and unified on a coherent theory? Can these seemingly diverse observations be related to a similar feature of developing neurons? If immature neurons are more resistant to insults in terms of cell loss BUT are still affected by early insults as reflected by the long term consequences these produce, then it is reasonable to suggest that the insults are “programmatic rather than lesional “and due to alterations of developmental programs. I have recently suggested the “neuroarcheology” concept of presymptomatic electrical or morphological signatures of the developing brain that has experienced seizures or other insults 119. The suggestion is that recurrent seizures –or other insults- produce early malformations that produce an ensemble of misplaced and/or misconnected neurons but also alter the properties of adjacent neurons that have already reached their assigned region and developed as programmed their synaptic connections. The former remain with immature features in the adult brain including a high input resistance and high tendency to oscillate and generate intrinsic activities that can entrain other neurons to seizures. The latter notably in the neocortex are also affected and show a re-expression of immature features –notably excitatory actions of GABA 120–127. This “post insult recapitulation of ontogenesis” 119;128 has important implications, since the determination of the properties of neurons in the sclerotic hippocampus will help guide the development of suitable therapeutic agents. Although this issue deserves a large description and debate, it is important to keep in mind the major differences between immature and adult mechanisms that connect an insult and the subsequent expression of epilepsies.
C. Role of Kainatergic Synapses in Seizure-induced epileptogenesis
One of the most salient and specific properties of neurons and synapses is their plasticity. In fact, almost all the procedures and mechanisms that take place during the operation of brain networks are altered by incoming information, and depend on the magnitude of neuronal activity that impinges on its synapses. Activity-dependent alterations of synapse operation are classically observed with recurrent stimuli such as in Long Term Potentiation (LTP), considered as a cellular model of memory processes (at least with respect to cellular mechanisms and signalling pathways). Activity-dependent alterations of almost all the steps of brain maturation have been reported, including neuronal proliferation and migration, synapse formation, network construction and sequential development of voltage and synapse mediated currents.
This plasticity is not restricted to development and integration of activity in the adult brain. Following a wide range of insults -- including traumatic brain injury, sensory deprivation or brain lesions and cell loss -- there is a considerable synaptic reorganization, with formation of novel synapses and changes in the properties of the involved cortical regions. This plasticity has been demonstrated after visual impairments, limb de-afferentation, and central insults; cortical regions are invaded by elements from adjacent structures, with neuronal sprouting and formation of novel aberrant synaptic connections in sites where they are not present normally. There is little doubt that recurrent severe seizures trigger a similar cascade of events and the history and recurrence of seizures cannot be obliterated as if the environment is not affected. In keeping with this, seizures –notably TLE – are associated with cell loss and degeneration of a substantial part of the hippocampus and other limbic structures that are known to engage in various forms of reactive plasticity129;130. For over two decades, there has been direct evidence that at least in TLE in humans and animal models, there is a considerable sprouting of fibres and establishment of novel synapses that may in turn lead to enhanced excitability, and thus contribute to the generation of further seizures. In this domain, studies using kainate have provided major breakthroughs in understanding the alterations produced by insults.
1. The mossy fibres: an ideal location to look for changes
The story starts with the mossy fibres, which are a helpful target in studies to determine long term changes produced by seizures. The mossy fibres constitute the main and sole output of the granule neurons of the fascia dentate –a major gate to the hippocampus through which an inflow of information that originates in the entorhinal cortex reaches the Ammon’s horn and modulates its operation (see ) 98;131;132. Mossy fibres innervate a wide range of interneurons, but their most visible and investigated targets are the giant CA3 pyramidal neurons. Mossy fibres establish their synapses with the proximal apical dendrites of pyramidal neurons – within the stratum lucidum that in most rodents is immediately above the pyramidal layer. In primates and humans, the region innervated is interspersed within the pyramidal layer, in part because the pyramidal neurons are not aligned as tightly as in rodent. Yet, in all species, the mossy fibres terminals and innervations zones are readily visible because mossy fibre terminals are very large and enriched in zinc; the latter property enables investigators to visualize the terminals with simple histological techniques133;134. Mossy fibres accumulate and release zinc in very large amounts, although the role of zinc not fully understood 135.
2. Mossy fibres have an intimate relation with kainate signalling
In the early times of receptor identification, one could use autoradiographic investigations to determine the distribution of subtypes of receptors. Several observations have shown that KAR-mediated synaptic transmission in hippocampus is strongly linked to the presence of mossy fiber terminals: (1) the stratum lucidum (the target zone of mossy fibres on CA3 pyramidal neurons) contains among the highest density of KARs in the brain 18; (2) the stimulation of mossy fibres selectively generate EPSCKA in CA3 pyramidal cells 67;68;136;137; (3) lesions of the mossy fibres both reduce the density of KARs 19;137) and suppress KAR-mediated synaptic transmission in CA3 pyramidal cells 137; (4) the expression of EPSCKA in CA3 pyramidal cells is correlated with the postnatal development of mossy fibre synapses 138; (5) the severity of kainate-induced seizures and brain damage is reduced in GluR6 KOs39;68. The EPSCKA evoked by mossy fibre stimulation in CA3 pyramidal neurons 67;69;136;139 has several interesting features. Stimulation of a single mossy fibre generates 3 types of EPSCS: 1) fast EPSCs that are mediated by AMPA receptors –the traditional ionotropic receptor mediated currents; 2) slow EPSCs that are selectively mediated by kainate receptors; and 3) mixed EPSCs composed of both AMPA and kainate EPSCS137. Blocking ongoing activity with TTX revealed the presence of three comparable types of miniature EPSCs, indicating that kainate receptors are located at the core of synaptic terminals and act as a conventional transmitter-mediated signalling device (i.e., are not on extrasynaptic sites as initially thought) 137. EPSCKA are selectively restricted to some neuronal populations (i.e., are not found on every neuron, in contrast to AMPA receptors that mediate the general fast glutamatergic ionotropic synaptic current. In particular, in the hippocampus, EPSCKA are enriched on CA3 pyramidal neurons and certain interneurons but not on CA1 pyramidal neurons or on granule cells. Neurons enriched with “kainatergic synapses” are also the ones that degenerate most readily in human and animal TLE –stratum oriens somatostatin containing interneurons, CA3 pyramidal neurons etc- suggesting that the presence of a high density of kainatergic synapses triggers a cascade of events that is deleterious to neurons. In addition, EPSCKA have long lasting slow kinetics with important implications.
A multitude of effects of kainate on synaptic transmission have been reported. Kainate inhibits mossy fibres synaptic inputs 140. Low and high concentrations of kainite, respectively, facilitate and inhibit synaptic transmission 89;141;142. Presynaptic KARs (with GluR 7 subunits) are localized in the presynaptic active zone, close to release sites of mossy fibres, where they facilitate the release of glutamate143;144. Therefore, at both pre and post synaptic sites, kainate modulates the effects of mossy fibre synapses in a dose dependent manner.
3. Mossy fibres sprout after seizures: anatomical observations
That mossy fibres sprout after kainate injections (and consequent seizure generation) was reported over 2 decades ago 19;145;146. These studies identified a novel band of mossy fibre terminals below the CA3 pyramidal neuron layer after recurrent seizures caused by the convulsive agent. This sprouting was also associated with the formation of a band of mossy fibres immediately above the granule layer, suggesting that an additional sprouting of fibres - and formation of synapses - had also occurred within the granule cells. Investigators speculated that in epileptic tissue, sprouting mossy fibres may innervate other granule cells (in contrast to the case in seizure-naïve granule cells layer) (see ) 19;41;147–150. Interestingly, this new band of mossy fibre terminals was characterised by both a high concentration of zinc (Timm stain) and high affinity kainate receptors, suggesting that the innervations of neurons by mossy fibres entrain the post synaptic expression of the features that normally characterise these synapses 19;145;147;151;152. Direct demonstration that the newly formed mossy fibre terminals have indeed all the features and constituents of conventional mossy fibres was then demonstrated using electron microscopy and kainate binding 19;150;151. As stressed above, mossy fibres terminals have unique features (very large diameter with multiple invaginations, large numbers of vesicles). After recurrent seizures, typical mossy fibres terminals were observed in the aberrant regions above the granule layers and the infra-pyramidal (CA3) zone, where they are not present in naive animals. Whether mossy fibres also sprout and increase their innervation of GABAergic interneurons has not been firmly established.
Epileptic hippocampus exhibits an aberrant mossy fibre terminal zone that is not present in naïve animals. Similar observations have been repeatedly made in a variety of animal models of TLE, including kindling and pilocarpine, providing a potential substrate for seizure-induced epileptogenesis (see below)41;148;149;153–166. Sprouting has also been observed in human epileptic patients 148;154;167–171. Parallel studies performed in post mortem hippocampi, first in non TLE types of infantile seizures and then on many different types of epilepsies, suggest that this plasticity is not restricted to rodents 148;149. If functional, this sprouting could lead to profound rearrangements in the operation of the hippocampal circuit, with an increased excitation of CA3 pyramidal neurons and granule cells.
4. Recurrent mossy fibre synapses in TLE include kainatergic synapses that are not observed in naïve neurons
In spite of overwhelming anatomical evidence that mossy fibres sprout, it remained to be shown that they also are functional in their aberrant localization, and that sprouting therefore contributes to enhance the excitability of their targets. Experimental evidence suggests that changes in voltage gated conductances - in addition to mossy fibre sprouting- could promote the generation of epileptiform activity 172–177. Since mossy fibre synapses are closely associated with kainate signalling, one can hypothesise that the formation of aberrant mossy fibre synapses onto Dentate Granule Cells (DCG) would trigger the formation of functional KAR-operated synapses in chronic epileptic rats. Indeed, recordings from granule cells, in an animal model of TLE – selected because they have no mossy fibre terminals in naïve conditions - revealed major differences from control cells. In control granule cells, stimulation of the perforant pathway generated exclusively fast AMPA receptor mediated EPSCs (EPSCAMPA) 137. In contrast, in TLE DCGs, a similar stimulation paradigm generated long-lasting EPSCKA, originating from recurrent mossy fibre synapses 178 (see and ). Therefore, epileptic DG neurons operate by means of aberrant glutamatergic synaptic currents that are not observed in naïve neurons.
A Kainate receptor antagonist blocks the epileptiform events generated by an electrical stimulation of the perforant pathway. In a naïve slice, this generated an all or none field EPSP, in an epileptic slices –prepared weeks after in vivo (more...)
How do aberrant KAR-operated synapses, with their slow kinetics, impact the temporal precision of EPSP-spike coupling in DGCs of epileptic rats and generate seizures? There are indications that the loss of time locked EPSCs evoked action potentials, and a large degree of jitter, facilitate the generation of paroxysmal synchronised activities 179. Many studies have shown that the shape of excitatory synaptic event, and its modulation by voltage-gated conductance, are important determinants of the temporal precision of hippocampal and neocortical cell operation 180–185. In naïve DCGs, the generation of an EPSPAMPA leads to a time locked spike with a fixed latency and very little jitter. This precision is instrumental in the operation of the entorhinal cortex/perforant pathway/Ammon’s horn and most likely underlies the behaviourally relevant patterns that this pathway entrains. In contrast, in TLE DCGs, there is a dramatic decrease in the spike timing precision 186. ”Jittery” spikes are selectively evoked by EPSPKA (not by EPSPAMPA that only generate highly time-locked spikes in both control and TLE conditions) 178;186. A direct proof of the link between kainatergic synapses and reduced time locked responses was shown in an experiment in which a simulated electrical pulse, with kainate-like kinetics, was injected intracellularly in control DCGs - which are endowed only with fast kinetic EPSPAMPA. The injection of a kainate-like EPSC converted the time-locked spikes to jittery responses. In other words, it is not the amplitude of the EPSC but its kinetics that is here determinant: EPSPKA are endowed with a long lasting decay time constant that is ideal for the loss of these time locked responses; Indeed, the activation by EPSPs of voltage-gated conductances near threshold is an important parameter in the modulation of EPSP time course and of EPSP-spike coupling temporal precision 182;187–190; We showed that EPSPKA but not by EPSPAMPA activate voltage gated currents and specifically the persistent Na+ current (INaP) which is activated below firing threshold and amplifies EPSPs in hippocampal and neocortical neurons 188;191;192. Using two blockers of INaP, phenytoin 180;188;193;194 and a low dose of TTX 195;196, we found that blockade of INaP restore the temporal precision of EPSP-spike coupling in DGCs of epileptic rats. Therefore, DG epileptic but not naive neurons generate an EPSPKA that trigger selectively the activation of INaP that in turn facilitates the generation of bursts of action potentials in a dispersed ”jittery” pattern instead of the time locked EPSPAMPA time locked response. These observations suggest that a selective interplay between an aberrant EPSPKA and INaP alters the temporal precision of EPSP-spike coupling in epileptic but not naïve DCGs. This action is not due to an enhancement of INaP but to the unique long lasting kinetics of kainatergic EPSCs that are needed to activate INaP. Interestingly, INaP is enhanced in neurons in both animal models and patients with temporal lobe epilepsy {Agrawal, 2003 17799/id;Vreugdenhil, 2004 17800/id}. These aberrant KAR-operated synapses will exert an strong influence on the operation of hippocampal circuitry, given the high frequency of ongoing excitatory synaptic events in DGCs from epileptic rats 197 It is likely that these mechanisms underlie the alterations of place cell and phase/precession pattern and temporal organization of firing among pairs of neurons in TLE 198;199.
The importance of sprouting and neosynapse formation in chronic epileptogenesis has been challenged by Mello and colleagues on the basis of experiments using the blocker of protein synthesis cycloheximide. In their experiments, pre-treatment with cycloheximide following pilocarpine-induced status epilepticus allowed epileptogenesis but prevented aberrant mossy fiber sprouting as assessed by Timm staining. This result suggested that mossy fibre sprouting is not required for seizures to beget seizures in this model 200;201. However, Dudek and colleagues 197;202 showed that pre-treatment with cycloheximide neither altered the spontaneous motor seizure rate post-treatment (compared to untreated TLE animals) nor changed the pattern of Timm stain. Cycloheximide also did not prevent hilar, CA1, or CA3 neuronal loss compared to the untreated TLE rats. Direct evidence for a functional aberrant mossy fibre synapses was obtained, suggesting that pre-treatment with cycloheximide does not affect aberrant mossy fibre sprouting in epileptic rats and does not prevent the formation of recurrent excitatory circuits (also see168).
5. Implications of Kainatergic Pathways on the development of efficient antiepileptic agents
Several lessons can be drawn from the use of kainate –and other animal models of TLE. First, to understand and eventually cure TLE, reactive plasticity must be incorporated and taken into account in the animal models used for basic and applied research. This conclusion may be valid in general for other insults and neurological disorders, since although mossy fibre synapses offer a unique opportunity to demonstrate post-lesional plasticity, it is by no means restricted to mossy fibres. Axonal sprouting and innervation of aberrant targets have been observed in CA1 pyramidal neurons that are deprived of an important input by seizures 203;204, and a wide range of lesions and insults produce fibre sprouting and formation of novel connections that impact the operation of brain networks. 205–208. The implications of these observations are that animal models used to mimic human neurological disorders must be chronic and must incorporate alterations. After seizures, sprouting and functional aberrant synapse operation require several weeks to take place and to express the aberrant features (including the novel aberrant kainatergic synapses); then and only then –weeks after the inaugurating status- can one investigate genuine epileptic networks and develop suitable antiepileptic drugs. In keeping with this important point, in epileptic animals, activation of granule cell synaptic inputs generate all-or-none epileptiform bursts (instead of the normal field population spike evoked in naive neurons) that was blocked by SYM2081, a relatively specific kainate receptor antagonist 178. This observation suggests that this aberrant synapse plays an important role in the generation of seizures by epileptic neurons, and that suitable kainate receptor antagonists could provide novel therapeutic avenues. This conclusion could not have been reached had the experimental procedures only relied upon naïve acutely treated animals.
D. Seizures beget seizures in vitro in the developing hippocampus
The observations that seizures induced by kainate (or other convulsive agents or procedures) do not produce cell loss and mossy fibre sprouting in immature tissue reflect an important difference between adult and infantile brain. Yet, recurrent seizures do produce long term, often severe, neurological and behavioural sequel. To address this puzzle, we have developed an in vitro preparation that has allowed several important observations regarding how recurrent seizures generated by kainate could directly lead to long lasting alterations of neuronal activity.
1. An experimental protocol to generate an epileptogenic mirror focus
In spite of their essential contributions, in vivo studies do not readily provide access to the full mechanisms involved in seizure generation. Acute slice preparations also have some limitations, including the difficulties in generating ictal events similar to those observed in vivo. To circumvent these limitations, Khalilov and colleagues developed a technique for studying an intact ex-vivo hippocampus preparation; the neonatal hippocampus is dissected and the entire repertoire of recording techniques applied in vitro can be utilised 209. This intact preparation can be used for long-lasting recordings, with excellent preservation of essential variables and physiological parameters. It is, however limited to neonatal preparations, and requires suitable relatively fast perfusion rates. Recordings from this preparation revealed larger and better network driven signals than observed in slices, allowing the generation and propagation of Giant Depolarising Potentials (GDPs) – the dominant synaptic pattern of developing cortical networks 210;211 along the entire hippocampal axis – to be described. Combined whole-cell and extracellular field recordings from the CA3 hippocampal region and the septum indicated that spontaneous GDPs are most often initiated in the septal poles of hippocampus and propagate to medial septum and temporal poles of both hippocampi simultaneously 212.
This preparation was subsequently extended to a triple chamber that accommodates the two interconnected hippocampi and their connecting commissures; each hippocampus is placed in an independent chamber, so that a convulsive agent can be applied exclusively to one chamber (whereas the naïve contralateral hippocampus and/or the associative/commissural connections are perfused with a different solution 213. This approach provides a unique opportunity to separate the network submitted to a convulsive agent from the “naïve” network that experiences only recurrent seizures that propagate from the other side. Indeed, one can allow the propagation of a predetermined number of seizures before interrupting the flow of activity. Further, putative AEDs can be applied on the treated or on the naïve network.
Using this preparation several important observations have been made as to the mechanisms underlying the effects of kainate-induced seizures. First, gradual developmentally-dependent actions of kainate were observed. Kainate did not generate ictal seizures at Post Natal day 2 (P2) tissue, but triggered ictal seizures at P 7. The propagation of seizures is also developmentally regulated, with interictal seizures propagating to the contralateral hippocampus and septum at P2 but to the entorhinal cortex; the latter pattern is seen only starting from P4, confirming the crucial role of the hippocampus at an early age to act as the pacemaker of kainate induced seizures 214. Applications of kainate (300nM) to one hippocampus generated a tonic-clonic seizure pattern with ictal High Frequency Oscillations (HFOs->40Hz-) similar to those observed in vivo in experimental animals and in epileptic patients215–219. The seizures propagate to the other hippocampus, leading to the generation of a similar electrographic event. If the connections between the two hippocampi are interrupted after one seizure, the contralateral hippocampus does not generate seizures spontaneously; i.e., it does not become epileptic. In contrast, after 10–15 kainate applications to the stimulated hippocampus, the contralateral hippocampus – that has never been perfused with kainate - becomes epileptic; after disconnection from the stimulated side, it generates ongoing seizures. The networks are “chronically” epileptic; even after 2 days in vitro, ongoing seizures are generated by slices prepared from the mirror foci and artificially maintained. This preparation therefore enables to directly assess the consequences of seizures on a naïve network and how seizures beget seizures.
2. Conditions required for recurrent seizures to generate a ‘mirror’ focus
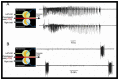
Using this preparation, we first determined the conditions required for seizures to beget seizures and to form a mirror focus. Recurrent seizures must include High Frequency Oscillations (HFOs, >40Hz) to transform a naïve network to an epileptic one. Seizures without HFOs do not generate a mirror focus (see ). To test this, we applied repetitively kainate to one hippocampus –referred to as the treated hippocampus- and NMDA or GABA receptor antagonists were applied to the other hippocampus –referred to as the naïve hippocampus as it did not receive kainate. The GABA or NMDA receptor antagonists did not block the propagated seizures but eliminated only their HFO components and completely prevented the formation by seizures of a mirror focus 122;123;220. Clearly GABA and/NMDA receptors are necessary and sufficient for both the generation of HFOs AND the formation by seizures of a mirror focus. An interesting implication of these observations is that seizures generated by GABA receptor blockers are NOT epileptogenic as they do not lead to long term consequences. This was directly demonstrated by applications of GABA receptor antagonists to both hippocampi that produced “seizures “but not epileptogenic mirror foci, as the hippocampi did not generate ongoing seizures when the drugs were washed out. Therefore, at least in the neonatal hippocampus, GABA and NMDA receptors must be operative for seizures to include HFOs, and this determines whether seizures will beget seizures. Interestingly, observations in human and animal TLE also reflect the importance of HFOs 216–218.
Recurrent seizures generated in one intact hippocampus by kainate propagate to the other hippocampus and form an epileptogenic mirror focus. Triple chamber with the two interconnected hippocampi and their connecting commissures. After 15 kainate applications (more...)
This preparation was then used to determine the persistent alterations occurring in an epileptogenic mirror focus formed by the propagation or recurrent seizures from the other hippocampus. Several alterations have been reported. GABA strongly depolarises and excites epileptic neurons, because of a permanent shift of E GABA; indeed single channel recordings of GABA channels showed a highly significant alteration of [Cl−]I and the Driving Force of GABA (DF GABA). GABA excites neurons in the epileptic tissue, generating action potentials (122. The accumulation of chloride is most likely mediated by a loss of the chloride exporter KCC2, perturbing the capacity of neurons to remove chloride that accumulates during recurrent seizures 221(but also see 124. This excitatory to inhibitory shift of GABA actions (I to E) has been reported in several other preparations including human TLE neurons 121 and animal models -- although other underlying mechanisms (notably, an increased efficacy of the chloride importer NKCC1) have been suggested 222;222. This preparation also has been quite useful in testing the actions of known and novel AEDs. For example, the NKCC1 chloride co transporter antagonist, bumetanide (that has been used for decades as a diuretic agent) also reduces seizures 124;222223;224. In the triple chamber, bumetanide applied to the naïve hippocampus while kainate was repeatedly applied to the other hippocampus, failed to prevent the formation by seizures of a mirror focus but efficiently reduced ongoing seizures generated by the mirror focus 223. These actions are mediated by its potent reduction of intracellular chloride that counteracts the depolarising and excitatory actions of GABA in the mirror focus, and thereby reinforces the inhibitory actions of GABA. This effect of modulating GABA effects is also illustrated in a recent study where the Dynamic Regulation of Chloride (DCR) was determined using a perforated patch clamp recording with focal applications of GABA and V rest selected so as to have a nil DF GABA –i.e., no net current at that voltage 224. A large depolarising step led to a chloride influx, and then the time required for chloride to return to control values was determined. In mirror foci neurons, this time course was significantly augmented and this was mimicked by specific antagonists of NKCC1/KCC2. As seizures also beget seizures in NKCC1 KO mice and lead to a permanent rise of DF GABA, it appears that NKCC1 is neither necessary nor sufficient (see contrary from 225. We also showed that in this preparation, KCC2 is internalised – and thus not operational - in epileptic neurons. Therefore, recurrent seizures reduce DCR, leading to more excitatory actions of GABA, at least in part because of a down regulation of KCC2 (now known to be heavily controlled by tyrosine phosphorylation and dimerization 226. An important consequence of these effects is that phenobarbital will efficiently block seizure inauguration but aggravate established seizures 225. Therefore, the history of seizures prior to phenobarbital administration is instrumental in determining its effects.