Abstract
Many drugs are inhibitors of enzymes involved in mediating the disease processes. Understanding the mechanism of action (MOA) of the target enzyme is critical in early discovery and development of drug candidates through extensive Structure-Activity Relationship (SAR) studies. This chapter contains a primer on the MOA of enzymes and its significance in drug discovery, types of inhibition, development and validation of MOA assays, data analysis and guidelines for performing these assays. New and experienced investigators will find this chapter useful when starting new projects involving enzyme targets.
Overview of MOA in Drug Discovery
The purpose of a mechanism of action (MOA) study is to characterize the interaction of a compound with its target to understand how the compound interacts with the target and how natural substrates at physiologic concentrations will modulate this activity. These compounds are often inhibitors of enzymes but only rarely become drugs due to the requirements for a drug to not only inhibit the target but to have acceptable solubility, permeability, protein binding, and selectivity, metabolism and toxicity profiles. This potential for the compound to become a drug is slowly revealed through the analysis and tracking of these characteristics, as chemistry elaborates the structure-activity relationship (SAR). As described in the body of this document, certain types of biochemical behavior are associated with good drug-like properties both in vitro and in vivo.
Most biochemical screens are designed to provide a chemical starting point based upon the most robust, simple and inexpensive modality for screening. This is due to the required reproducibility in the screening process and the potentially large number of molecules to be run through the screen. Most enzymatic screens are designed to identify inhibitors regardless of their mode of action. Thus, screens are usually run at or below the Km for the substrate(s). In the case of an enzyme with two substrates, the screen is often designed to run under pseudo-first order kinetics by running the assay under conditions where one substrate is at saturation, well above its Km, and the second is at or below its Km for the enzyme. One can therefore identify inhibitors that have competitive, noncompetitive and uncompetitive behavior with regard to the substrate at or below Km and noncompetitive or uncompetitive behavior with regard to the other substrate at well above its Km for the enzyme.
In the drug discovery process, the screening phase casts a wide net and the ability to further analyze compounds in more detail is limited, therefore the number of actives isolated from a screen for follow-up are determined by the overall hit rate, the repeat rate upon retesting and determination of the IC50 in a concentration response curve (CRC) test. In general, activities range from mid-micromolar to sub-micromolar for enzyme inhibitors right out of the screen. It is this piece of information (the IC50), along with an analysis of the structural classes of active molecules by a medicinal chemist, which defines the initial SAR, if there is one in the data. It is after this initial analysis that MOA studies can prove valuable by further defining the nature of the inhibitor from a biochemical point of view. MOA studies at this point in the drug discovery process define the nature of the SAR by elucidating the type of inhibition by which the discovered molecules operate. Thus, one can define if the discovered inhibitor is competitive with substrate, for example, and as described below, potentially suffers from certain liabilities associated with this mechanism.
Cell based assays of biochemical actives are usually utilized to identify promising molecules in a second round of low to medium throughput screening. If a molecule shows significant activity in a cell based assay, then it continues through the flow scheme. The lack of cell based activity of biochemically potent actives is usually attributed to lack of cellular permeability, with a wave of the hand; however, an understanding the MOA of a compound at this stage can add depth to the interpretation of cellular activity or its absence. Knowing a compound is competitive with a substrate helps establish the binding pocket and, in combination with structural and SAR information, provides an immediate direction for further chemical synthesis. However, these competitive compounds with promising structure and potent biochemical activity might compete with a cellular substrate present at high intracellular concentration thus show no significant cell based activity. Alternatively, more potent cell based activity than is biochemically predicted from IC50 curves might correlate with unusual kinetic behaviors such as slow binding behavior and/or slow off rates (tight binding). As there is no single, unique answer, biochemical MOA studies help in interpretation of cell based activities and provides further support for molecules with desirable characteristics to move forward in the flow scheme. Traditionally, as MOA studies were slow, laborious efforts, only a few selected molecules could be readily analyzed. With the advent of laboratory automation and enhanced data processing, it is now possible to assess a larger number of compounds rapidly. Therefore, it is feasible (and desirable) to examine the results of a screening campaign, in addition to standard cell based assays in the second tier, by an analysis of MOA.
Types of Inhibition
There are three main types of inhibition (competitive, noncompetitive, and uncompetitive) that are most commonly used to describe the binding of an inhibitor to a target enzyme (). However, a complete analysis of the mechanism of action requires the scientist to also evaluate other potential inhibition events, including allosteric, partial, tight-binding, and time-dependent inhibition. A review of these types of inhibition is provided in the sections that follow.
Competitive Inhibition
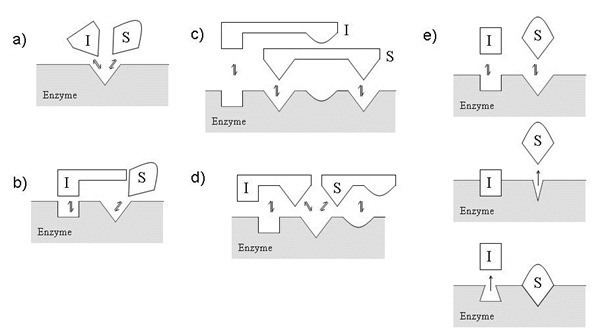
A competitive inhibitor binds only to free enzyme. Often this binding event occurs on the active site of the target, precisely where substrate also binds. Although this is the case for a majority of competitive inhibitors, it is a misleading oversimplification. It is more appropriate to state that the binding of a competitive inhibitor and the binding of substrate are mutually exclusive events. provides illustrations of some possible mutually exclusive binding events.
Despite the differences in binding to the free enzyme illustrated in , all competitive inhibitors have the same effects on substrate binding and catalysis. A competitive inhibitor will raise the apparent Km value for its substrate with no change in the apparent Vmax value. As a result, it is often stated that competitive inhibition can be overcome, observed by an increase in the apparent Ki value, at higher concentrations of substrate. This characteristic will have physiological consequences on the observed efficacy of drugs. As an enzyme’s reaction is inhibited by a competitive inhibitor, there is an increase in the local concentration of substrate. Without a mechanism to clear the substrate, a competitive inhibitor will lose potency. This is not the case for a noncompetitive inhibitor.
Noncompetitive Inhibition
A noncompetitive inhibitor binds equally well to both free enzyme and the enzyme-substrate complex. These binding events occur exclusively at a site distinct from the precise active site occupied by substrate. provides some illustrations of the more common noncompetitive binding events.
In contrast to a competitive inhibitor, a noncompetitive inhibitor will lower the apparent Vmax value, yet there is no effect on the apparent Km value for its substrate. Essentially, the Ki of the inhibitor does not change as a function of the substrate concentration.
In some circumstances, a compound may have unequal affinity for both free enzyme and the enzyme-substrate complex. This mixture of competitive and noncompetitive phenotypes is called mixed inhibition.
Uncompetitive Inhibition
An uncompetitive inhibitor binds exclusively to the enzyme-substrate complex yielding an inactive enzyme-substrate-inhibitor complex (). When encountered, the apparent Vmax value and the apparent Km value should both decrease. Despite their rarity in drug discovery programs, uncompetitive inhibitors could have dramatic physiological consequences. As the inhibitor decreases the enzyme activity, there is an increase in the local concentration of substrate. Without a mechanism to clear the buildup of substrate, the potency of the uncompetitive inhibitor will increase.
Allosteric Inhibition
An allosteric inhibitor decreases activity by binding to an allosteric site other than or in addition to the active site on the target. This interaction is characterized by a conformational change in the target enzyme that is required for inhibition. These conformational changes can affect the formation of the usual enzyme-substrate active site complex, stabilization of the transition state, or reduce the ability to lower the activation energy of catalysis. and are classical examples of allosteric inhibition. As such, an allosteric inhibitor may display a competitive, noncompetitive, or uncompetitive phenotype with respect to substrate binding.
Partial Inhibition
Partial inhibition results from the formation of an enzyme-substrate-inhibitor complex that can generate product with less facility than the enzyme-substrate complex. This can be illustrated in . When “I” is a partial inhibitor bound in the enzyme-substrate-inhibitor complex, the catalytic center may retain some ability to align near the substrate and facilitate catalysis. As a consequence of these structural changes, partial inhibitors can also be allosteric inhibitors of enzyme activity. In direct contrast, full inhibition results in an enzyme-substrate-inhibitor complex where the catalytic center is not capable of aligning near the substrate for catalysis.
Tight-Binding Inhibition
In this type of inhibition, the population of free, soluble inhibitor is significantly depleted by the formation of the enzyme-inhibitor or enzyme-substrate-inhibitor complex. While tight-binding inhibitors can bind to the target enzyme in a competitive, noncompetitive, or uncompetitive manner with respect to substrate binding, they can display noncompetitive phenotypes. However, a tight-binding inhibitor typically binds with an apparent affinity (Ki) near the concentration of enzyme (active sites) present in the biochemical assay.
Time-Dependent Inhibition
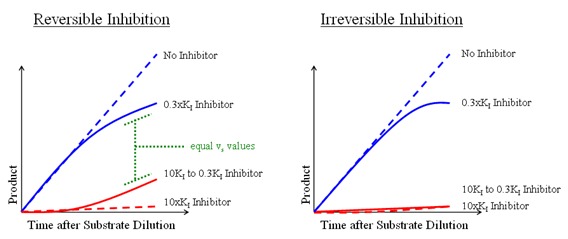
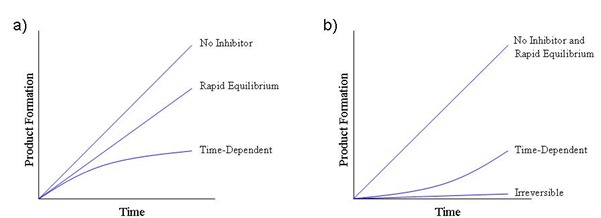
Time-dependent inhibitors bind slowly to the enzyme on the time scale of enzymatic turnover, thus displaying a change in initial velocity with time. This has the effect of slowing the observed onset of inhibition. Time-dependent inhibitors also impede the observed recovery of enzyme activity following inhibition, resulting in slow koff values. As illustrated in , these inhibitors typically yield nonlinear initial velocities and nonlinear recoveries of enzyme activity.
Interestingly, many successful therapeutic drugs are time-dependent inhibitors. For these inhibitors with slow koff values, the rate of release of inhibitor from the enzyme-inhibitor complex (recovery of enzyme activity) proceeds independent of the substrate concentration and the physiological mechanism to remove inhibitor. This makes time-dependent inhibition a very attractive and proven strategy for the discovery and development of drugs.
Some time-dependent inhibitors covalently attach to the target enzyme. For those inhibitors, the koff value is zero and the inhibition is said to be irreversible. These are typically less attractive molecules, unless the formation of the covalent species is specific to the reaction mechanism of the enzyme. Some inhibitors are for all practical purposes irreversible, with very low koff values, despite their inability to covalently attach to the enzyme. This stands in direct contrast to rapid equilibrium, reversible inhibitors that bind to and release from the enzyme at rates that are rapid in comparison to the rate of enzyme turnover.
References
Literature Cited
- 1.
Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, second edition. 2000, Wiley, New York.
- 2.
Segal IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. 1975, Wiley, New York.
- 3.
Additional References
Copeland RA. Mechanistic Considerations in High-Throughput Screening.
Analytical Biochemistry 2003, v320, pgs 1-12. [
PubMed: 12895464]
Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. 2005, Wiley, New York, [
PubMed: 16350889]
Copeland RA, Pompliano DL, Meek TD. Drug-Target Residence Time and Its Implications for Lead Optimization.
Nature Reviews Drug Discovery 2006, v5, pgs 730-739. [
PubMed: 16888652]
Fehrst A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. 1999, W.H. Freeman, New York.
Kenakin TP. A Pharmacology Primer: Theory, Application, and Methods. 2004, Elsevier, San Diego.
Robertson JG. Mechanistic Basis of Enzyme-Targeted Drugs.
Biochemistry 2006, v44, pgs 5561-5571. [
PubMed: 15823014]
Swinney DC. Biochemical Mechanisms of Drug Action: What Does It Take For Success?
Nature Reviews Drug Discovery 2004, v3, pgs 801-808. [
PubMed: 15340390]
Swinney DC. Biochemical Mechanisms of New Molecular Entities (NMEs) Approved by United States FDA During 2001-2004: Mechanisms Leading to Optimal Efficacy and Safety. Current Topics in Medicinal Chemistry 2006, v6, pgs 461-478. [
PubMed: 16719803]
Glossary of MOA Terms
The definitions for these terms were gathered from references
- Active site
the specific and precise location on the target responsible for substrate binding and catalysis.
- Allosteric activators
an allosteric effector that operates to enhance active site substrate affinity and/or catalysis. (Copeland, Enzymes, pg368)
- Allosteric effector
small molecule that can bind to sites other than the enzyme active site and, as a result of binding, induce a conformational change in the enzyme that regulates the affinity and/or catalysis of the active site for its substrate (or other ligands). (Copeland, Enzymes, pg368)
- Allosteric repressors
an allosteric effector that operates to diminish active site substrate affinity and/or catalysis. (Copeland, Enzymes, pg368)
- Allosteric site
a site on the target, distinct from the active site, where binding events produce an effect on activity through a protein conformational change. (Kenakin, A Pharmacology Primer, p195).
- Alpha
typically noted as the ratio, KI’/KI. It reflects the effect of an inhibitor on the affinity of the enzyme for its substrate, and likewise the effect of the substrate on the affinity of the enzyme for the inhibitor. (Copeland, Enzyme, pg268)
- Biochemical assay
the in vitro based mechanism used to measure the activity of a biological macromolecule (enzyme).
- Cofactor
nonprotein chemical groups required for an enzyme reaction.
- Enzyme
protein that acts as a catalyst for specific biochemical reaction, converting specific substrates into chemically distinct products.
- Multivariate fitting
Fitting a more than 2 variable model (Example: Response, [Inhibitor], [Substrate]) to all of the data from a MoA experiment using nonlinear regression.
- Inhibitor
any compound that reduces the velocity of an enzyme-catalyzed reaction measured in a biochemical assay, as represented by percent inhibition or IC50.
- Initial velocity
the initial linear portion of the enzyme reaction when less than 10% of the substrate has been depleted or 10% of the product has formed. (QB Manual, Section IV, pg5)
- In vitro
(to be defined later)
- Ligand
a molecule that binds to the target. (Kenakin, A Pharmacology Primer, pg 198)
- Linearity
A relationship between two variables that is best described by a straight line. In MoA experiments, the amount of product formed should be linear with respect to time.
- Substrate
a molecule that binds to the active site of an enzyme target and is chemically modified by the enzyme target to produce a new chemical molecule (product).
- Target
a macromolecule or macromolecular complex in a biochemical pathway that is responsible for the disease pathology. (QB manual, Section XII, pg3)
- kcat
turnover number representing the maximum number of substrate molecules converted to products per active site per unit time. (Fehrst, Str Mech Prot Sci, pg109)
- KI
the affinity of the inhibitor for free enzyme.
- KI’
the affinity of the inhibitor for the enzyme-substrate complex.
- KM
the concentration of substrate at ½ Vmax, according to the Henri-Michaelis-Menten kinetic model (QB manual, Section IV, pg9)
- koff
the off-rate associated with the release of inhibitor from an enzyme-inhibitor complex.
- kon
the on-rate associated with the formation of an enzyme-inhibitor complex.
-
*
Edited by James McGee and Jeffrey Weidner
Tables
Table 1:
Summary of competitive, non-competitive and uncompetitive inhibition models.
View in own window
| Inhibition | Description | Ki | Ki’ | Ki’/Ki |
|---|
| Competitive | The inhibitor binds only to free enzyme. This binding most often occurs in the active site at the precise location where substrate or cofactor (being evaluated in the MOA study) also binds. | finite | Infinite | infinite |
| Mixed | These inhibitors display properties of both competitive and noncompetitive inhibition. | finite | Finite | > 1 |
| Noncompetitive | The inhibitor binds equally well to both free enzyme and the enzyme-substrate complex. Consequently, these binding events occur outside the active site. | finite | Finite | = 1 |
| Uncompetitive | The inhibitor binds only to the enzyme-substrate complex at a location outside the active site. | infinite | Finite | = 0 |




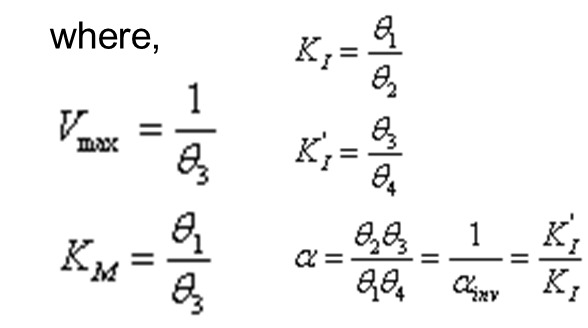











![Figure 9 . – Plotting the IC50 vs [Enzyme] will reveal whether a compound is tight binding.](/books/NBK92001/bin/enzymemoa-Image019.jpg)
![Figure 10 . – A plot of the IC50 vs [substrate] will reveal the binding modality for a tight binding inhibitor.](/books/NBK92001/bin/enzymemoa-Image020.jpg)

![Figure 12 . – A plot of the kobs vs [inhibitor] will allow for the determination of the appKi value for a time dependent inhibitor.](/books/NBK92001/bin/enzymemoa-Image022.jpg)
![Figure 13 . – A plot of the appKi (and appKi*) vs [substrate] will allow for the determination of the true binding potency and modality.](/books/NBK92001/bin/enzymemoa-Image023.jpg)
![Figure 14 . – A plot of the kobs vs [substrate] will reveal the binding modality for a time dependent inhibitor.](/books/NBK92001/bin/enzymemoa-Image024.jpg)