Molecular Pathogenesis
The von Willebrand facto (VWF) protein is comprised of a 22-amino acid signal peptide, a 741-amino acid propeptide, and a 2,050-amino acid mature protein (see ). VWF has two key functions:
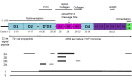
VWF protein structure and location of VWD-causing VWF variants by VWD type. Bold horizontal lines indicate the approximate position of exons where VWD-causing variants are most prevalent; thinner lines indicate exons with variants of lower frequency. (more...)
Binding to the subendothelium at sites of vascular damage and recruiting platelets to sites of clotting; and
Protecting factor VIII (FVIII) from proteolytic degradation in circulation and transporting it to sites of clot generation.
VWF has two sites of synthesis: endothelial cells and megakaryocytes, the precursors of platelets. During synthesis, tail-to-tail disulfide-linked dimers are formed through the C-terminal cystine knot (CK) domains, followed by head-to-head disulfide-linked VWF oligomers of up to 40 dimers in length.
During intracellular processing, the VWF propeptide (VWFpp) is cleaved by furin in the Golgi, plays a critical role in VWF multimerization, remains non-covalently associated with VWF through intracellular trafficking and storage, and is secreted into the plasma along with VWF [Haberichter 2015]. The ratio of VWFpp to mature VWF (von Willebrand factor antigen or VWF:Ag) can be used to estimate relative half-life of mature VWF [Haberichter et al 2008] and provide information about the mechanism of pathogenicity [Eikenboom et al 2013]. The VWFpp-to-VWF:Ag ratio correlates poorly with rapid VWF clearance in individuals with VWF levels of 30-50 IU/dL [Doherty et al 2023].
In circulation, high-molecular-wight VWF is cleaved to smaller VWF multimer forms by ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif) between amino acids 1605 and 1606 following secretion. The pattern of multimer proteolysis products by agarose-SDS gel analysis can help inform the underlying mechanism [Saadalla et al 2023].
Type 1 von Willebrand disease (VWD). Partial quantitative deficiency in type 1 VWD is mostly associated with missense variants [Seidizadeh et al 2024].
Type 1C VWD. VWD-causing variants associated with type 1C VWD are predominantly missense [Haberichter et al 2008, Millar et al 2008, Eikenboom et al 2013] or other structural variants [Sadler et al 2023] located in the D3 and A1 domains and reduce the residence time of VWF in plasma. The "Vicenza" variant, p.Arg1205His, is the most common of these VWD-causing variants.
Type 2 VWD. Qualitative deficiency (type 2 VWD) almost always results from VWD-causing missense variants in functionally important areas of VWF. Most VWD-causing variants seen in types 2A, 2B, and 2M VWD are located in exon 28.
Type 3 VWD. Both alleles are affected by VWD-causing variants (null or missense) that result in lack of VWF secretion from the cell. Most individuals with type 3 VWD have two null alleles and therefore produce no significant quantity of VWF. Approximately 25% of alleles have missense variants (see ) [Seidizadeh et al 2024]. Some may impair VWF multimerization, resulting in intracellular retention and lack of secretion into plasma.
Mechanism of disease causation. Loss of function for types 1, 2A, 2M, 2N, and 3 VWD; gain of function for type 2B VWD
VWF-specific laboratory technical considerations.
VWF has a partial pseudogene, VWFP1 (exons 23-34), which complicates analysis of these exons [Laffan et al 2021]. Domain structure and exons encoding each VWF domain are shown in . Gene conversion events with VWFP1 may impact exons 23-34 [Goodeve 2010, Laffan et al 2021]. There can be sufficient differences between VWF and VWFP1 to recognize a conversion event, usually through two or more sequential sequence variants from the pseudogene sequence (VWFP1) [Ahmad et al 2019]. Conversions of 6 bp to 335 bp are most commonly seen and have been reported in types 1, 2B, 2M, and 3 VWD.
Table 9.
VWF Pathogenic Variants Referenced in This GeneReview
View in own window
| Reference Sequences | DNA Nucleotide Change | Predicted Protein Change | Comment [Reference] |
|---|
NM_000552.5
NP_000543.3
| c.3614G>A | p.Arg1205His | Assoc w/type 1C VWD (See Molecular Pathogenesis.) |
| c.3931C>T | p.Gln1311Ter | Founder variant in Romani population in Spain; homozygotes present w/type 3 VWD [Casaña et al 2000] |
| c.4120C>T | p.Arg1374Cys | Founder variant in Amish population from Indiana & Ohio, US [Gupta et al 2016] |
| c.4414G>C | p.Asp1472His | Common benign variant that causes artificially ↓ results on VWF:GPIbR & VWF:RCo assays |
VWD = von Willebrand disease; VWF = von Willebrand factor; VWF:GPIbR = ristocetin-induced binding of VWF to glycoprotein Ib (GPIb); VWF:RCo = ristocetin cofactor activity testing ability of VWF to agglutinate platelets
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.



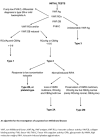
![Figure 1. . An approach to the diagnosis of von Willebrand disease (VWD) [James et al 2021].](/books/NBK7014/bin/von-willebrand-Image001.gif)