

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Gruber A, Durham AM, Huynh C, et al., editors. Bioinformatics in Tropical Disease Research: A Practical and Case-Study Approach [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2008.

Bioinformatics in Tropical Disease Research: A Practical and Case-Study Approach [Internet].
Show detailsThis chapter refers to diseases that represent major public health problems, such as tuberculosis, leprosy, and Buruli ulcer, and emerging diseases, caused by other mycobacterial species. A brief account of bacteria responsible for each disease and the respective global situation is followed by a description of finished and ongoing genome projects and the impact of genome projects on mycobacteria research. Mycobacterium is one of the most sequenced bacterial genera. This wealth of comparative genome sequence information provides unique opportunities for new insights into the biology of these globally important pathogens to address the scientific imperatives of better drugs, vaccines, and diagnostics for mycobacterial diseases.
The Global Burden of Tuberculosis, Leprosy, and Other Mycobacterioses
Diseases Caused by Mycobacterium tuberculosis Complex
Mycobacterium tuberculosis complex (MTBC) includes closely related bacteria showing higher than 99% similarity at the DNA level. The group comprises several main members, such as M. tuberculosis, responsible for most human tuberculosis cases, Mycobacterium bovis, the agent of bovine tuberculosis, which can also infect other animals as well as humans, Mycobacterium africanum, a prevalent cause of human tuberculosis on the African continent, and the vole bacillus Mycobacterium microti (1) (Genus Mycobacterium). It also contains the vaccine strain Bacille Calmette-Guérin (BCG). Other recently proposed members of the MTBC are Mycobacterium caprae, primarily infecting goats in Spain (2, 3), and Mycobacterium pinnipedii, infecting seals in Australia and Argentina (4). A variant human isolate of M. tuberculosis, the canetti strain, was also described recently and has been associated with the “ancient” tuberculosis lineages (5). Despite this close relationship, they show a large variability in their phenotypic properties, epidemiology, and incidence in human tuberculosis, and for this and other historical reasons, they are still considered different species (6). However, the extent of MTBC speciation is not yet resolved (7).
According to WHO reports, one-third of the world’s population is currently infected with the tuberculosis bacillus, and each year, about two million people die of tuberculosis (WHO Tuberculosis). Lack of control of tuberculosis is largely attributable to delayed or under diagnosis, non-efficient protection by BCG vaccination, the use of multiple drugs and prolonged treatment regimens, which leads to the existence and spread of multidrug-resistant strains, and association with HIV infection. Other social factors also have strong influence, such as increase in migration and poverty. The evolution of human tuberculosis incidence since 1990 is depicted in the maps in Figure 1 and Figure 2.

Figure 1
TB incidence, all forms (per 100,000 population per year), By Country, Total, 1990. From: WHO|Global TB database (http://www.who.int/tb/country/global_tb_database/en/index1.html).
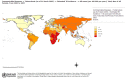
Figure 2
TB incidence, all forms (per 100,000 population per year), By Country, Total, 2004. From: WHO|Global TB database (http://www.who.int/tb/country/global_tb_database/en/index1.html).
Bovine tuberculosis is a chronic zoonotic disease whose etiological agent is M. bovis. It constitutes a serious animal health problem, causing economic losses due to decreased meat and milk production and to low exportation of cattle products (TB in cattle). Although the main host of M. bovis is cattle, other animals, including humans, may be affected. A study performed in the main milk production region of Argentina showed that in the period 1984 to 1989, M. bovis was responsible for 2.4 to 6.2% of tuberculosis cases in human beings, of which 64% were rural and meat workers (8). The AIDS epidemic has also increased the risk of transmission of M. bovis to humans. Nosocomial transmission of bovine tuberculosis produced by multidrug-resistant M. bovis strains among HIV-positive individuals was described recently in Spain (9).
Leprosy: A Public Health Problem
Leprosy remains a public health problem, mainly in developing countries, because in spite of the actions promoted by WHO, prevalence has been reduced, but the number of new cases has remained constant, with more than 690,000 cases reported (WHO; New Case Detection). The discovery of the leprosy bacillus by Hansen in 1873 constituted the first demonstration of a clear association of a microorganism to human disease. Mycobacterium leprae, the etiological agent of leprosy, is still unable to be grown as axenic culture and has an extremely slow doubling time in tissues (approximately 14 days). Large quantities of bacilli could be isolated for biochemical and genetic studies using the nine-banded armadillo as a surrogate host. The complete genome sequence of the armadillo-derived Indian isolate, the TN strain, was performed (10).
Leprosy is considered a low transmissible disease, and the means of transmission are uncertain. However, M. leprae infection is thought to spread by the respiratory route, because bacilli could be isolated from nasal swabs of patients. Leprosy develops very slowly in the infected host (2 to 10 years); there are two polar, stable forms based on skin smears (named multibacillary (MB) and paucibacillary (PB) leprosy, respectively) and a complex spectrum of unstable forms (Classification).
Using comparative genomics, seven strains of the leprosy bacilli from separate geographic origins were recently analyzed, showing a surprisingly stable genome. Authors concluded that cases of leprosy around the world could be attributable to a single clone, providing a general evolutionary scheme for M. leprae on the basis of single nucleotide polymorphism (SNP) data (11) (Figure 3). More recently, the computer analysis of substitutions in the pseudogenes, and its comparison with the functional orthologs of closely related genomes, has allowed the reconstruction of the gene content of the common ancestor of M. leprae and mycobacteria (12).
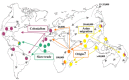
Figure 3
From Leprae Monot, Science 308:1040-42, 2005.
Other Mycobacterioses: Neglected Diseases
Buruli Ulcer
Mycobacterium ulcerans is an emerging human pathogen responsible for Buruli ulcer, a necrotizing skin disease found most commonly in West Africa (13), but outbreaks have also been reported in the Americas, Australia, and Asia (WHO Buruli Ulcer). The first detailed clinical description of ulcers caused by M. ulcerans has been attributed to Dr. Albert Cook, a missionary physician who worked in Uganda in the late 1800s (Buruli ulcer). Since the 1980s, Buruli ulcer has emerged as a serious public health problem in an increasing number of countries. In terms of numbers of cases, it is probably the third most common mycobacterial disease in humans, after tuberculosis and leprosy (Figure 4).

Figure 4
Buruli ulcer: global situation.
Knowledge of M. ulcerans at the molecular level has come only in the last 5 years, primarily from research focused on developing tools for either rapid diagnosis or molecular epidemiological investigation. M. ulcerans has an unexpectedly close genetic relationship with Mycobacterium marinum. Analysis of the 16S rRNA gene of both species revealed greater than 99.8% sequence identity. Biochemically, M. ulcerans has also been found to resemble M. marinum. However, there are also substantial phenotypic differences and dramatic differences in the pathologies and treatment of the diseases caused by each of these organisms. Also at a genetic level, multi-copy insertion sequences, IS2404 and IS2606, are present in the genome of M. ulcerans and absent from M. marinum (14).
This close relationship has been exploited in comparative genetic analysis by multi-locus sequencing (15). The results of this analysis suggest that M. ulcerans has recently diverged from M. marinum by the acquisition and concomitant loss of DNA driven mainly by the activity of mobile DNA elements. M. ulcerans, but not M. marinum, expresses a plasmid-encoded immunomodulatory macrolide toxin, mycolactone, which plays an important role in virulence and pathology (16). Recently, it was suggested that the acquisition of this plasmid and the subsequent ability to produce mycolactone appears to be a key issue in the evolution of M. ulcerans from a common M. marinum progenitor (17)
Johne's Disease
Johne’s disease or paratuberculosis is a chronic disease produced by Mycobacterium avium subsp. paratuberculosis (MAP) that causes chronic enteritis in bovine, ovine, and other small ruminants, thus leading to production losses because of deficient food conversion. The disease has also been diagnosed in primates, deer, rabbits, foxes, and camels, as well as in a wide variety of wild animals (Johne´s disease, paratuberculosis). It is currently associated with Crohn’s disease in humans; therefore, it is considered a zoonotic disease (18).
MAP belongs to the M. avium complex (MAC), which comprises a group of opportunistic pathogenic mycobacteria able to infect humans and other animals and is also widely distributed in the environment. The group includes four species: M. avium, Mycobacterium intracellulare, and the recently described Mycobacterium chimaera (19) and Mycobacterium colombiense (20). M. avium has now been divided into four subspecies: M. avium subsp. paratuberculosis, M. avium subsp. silvaticum, M. avium subsp. avium, and M. avium subsp. hominisuis (21). The subspecies paratuberculosis presents higher than 95% sequence similarity with M. avium subsp. avium but is unique in the fact that the majority of its isolates require the iron-chelating agent mycobactin for growth.
Diseases Caused by Rapidly Growing Mycobacteria
Infections caused by rapidly growing mycobacteria (RGM) are not reportable diseases, and consequently their incidence and prevalence are largely unknown. Nevertheless, an increasing number of outbreaks and clusters of infections caused by RGM have been reported worldwide in the last years (22-27). RGM species are widely distributed in the environment and have been isolated from potable water, wastewater, soil, and inanimate surfaces (28).
Mycobacterial taxonomy is in constant evolution, and genotypic studies led to recent splitting and redefinition of species defined previously by phenotype-based methods (29). The rationale of genotypic taxonomy consists in the identification of highly conserved genomic regions harboring hypervariable sequences that show species-specific deletions, insertions, or replacements of single nucleotides. The 16S rRNA gene was, for many years, the primary target of molecular taxonomic studies, combined with analysis of unique mycobacterial cell wall lipids, the mycolic acids, and led to classification of RGM in three main groups: the Mycobacterium chelonae-abscessus group, the M. fortuitum group, and the M. smegmatis group.
RGM species are closely related to each other, and the indication that bacterial strains belong to the same species if they have fewer than 5- to 15-base differences in their 16S rRNA gene sequences is not applicable to RGM (30). Therefore, genes other than 16S rRNA have recently been included to assess phylogenetic relationships and to help clarify the taxonomy of RGM (31-34). Phylogenetic trees based upon individual sequences of five genes—16S rRNA, recA, rpoB, sodA, and hsp65—of 19 RGM species were compared with trees based on the combined dataset of the five genes and combined datasets of recA and rpoB. Six phylogenetic groups were recognized: the M. chelonae-abscessus group, M. mucogenicum group, M. fortuitum group, M. mageritense group, M. wolinsky group, and M. smegmatis group (35).
Information derived from the ribosomal RNA has been applied for further recognition of species within RGM. Mycobacteria have a minimum number of ribosomal operons, one or two copies per genome. Typical RGM have two copies. It was demonstrated that the chromosomal location of the two rrn operons in RGM is conserved within each species, as indicated by the Restriction Fragment Length Polymorphism (16S-RFLP)-derived patterns (36). Since the first description in 1994 (37), this approach has been demonstrated to complement the DNA-DNA hybridization data in the delimitation of closely related RGM species (38, 39).
The near completion of the genomes of M. smegmatis, M. chelonae, and M. abscessus will certainly further improve taxonomy studies and our understanding of these emerging pathogens.
Mycobacteria and AIDS
Soon after the beginning of AIDS pandemy, it was evident that mycobacterial infections, not only tuberculosis but also opportunistic infections, were a major cause of morbidity and mortality among patients living with HIV/AIDS worldwide. MAC members are responsible for most opportunistic bacterial infections in late stages of the disease, and other non-tuberculous mycobacteria can also be involved. The panorama dramatically changed in countries where highly active antiretroviral treatment (HAART) became widely available, with the beneficial effect of restoration of pathogen-specific immune responses. Thus, the incidence of mycobacterial infections, including tuberculosis, decreased in treated cohorts living in high- and low-income countries (40).
MAC bacilli are acquired from the environment (person-to-person transmission had not been demonstrated), enter the host by the gastrointestinal mucosa, and cause disseminated disease with fatal consequences in patients with AIDS. Epidemiological evidence indicates that most AIDS patients with disseminated MAC infections have recently acquired these organisms, suggesting that infection was not due to previously viable MAC bacilli that could reactivate; therefore, MAC bacteria seem not to be able to establish latent infection, compared with that of M. tuberculosis (41).
Association of HIV infection and tuberculosis is the major adult infectious killer in the developing world, and approximately 10% of all global tuberculosis cases are attributable to HIV. About 13 million people are infected with both causative organisms. The biggest impact has been described in the sub-Saharan Africa: tuberculosis notifications have tripled since the mid-1980s, and death rates are fourth times higher compared with countries with good tuberculosis-control programs. Probably because of its association with HIV, Africa is the only continent where tuberculosis incidence is rising, causing a global increase of about 1% per year (42) (Figure 5).
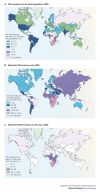
Figure 5
From Nunn et al. (42).
Molecular epidemiology studies on MTBC strains isolated from AIDS patients showed that both re-infection and new infection occur in these patients. It has been speculated that HIV/AIDS patients could be an ecological niche for M. tuberculosis low virulent clones without the selective pressure provided by the immunocompetent patient (43). As a consequence of the increase in the number of tuberculosis cases due to HIV, the performance of tuberculosis-control programs has deteriorated. The lack of human resources is the major bottleneck for implementation of services in developing countries. Global development efforts have been established until 2015 within the Millenium development goals by the United Nations with prominent features addressed against Tuberculosis and HIV/AIDS. The achievements will depend on progress in Africa.
Mycobacterial Genomes
Several mycobacterial genomes have been completed or are near completion. The updated list of mycobacterial genome projects can be viewed at Mycobacterial genome-sequencing projects and GOLD: Genomes OnLine Database Homepage
Mycobacterium tuberculosis and M. canetti
The The Wellcome Trust Sanger Institute sequenced M. tuberculosis strain H37Rv using cosmids and BAC clones supplied by researchers at the Institut Pasteur. This strain, isolated in 1905, retained full virulence in animal models and is widely used in tuberculosis research. The sequence of 4,411,529 base pairs (bp) has a G + C content of 65.6%. The genome is rich in repetitive DNA, particularly insertion sequences, new multigene families, and duplicated housekeeping genes. The annotated genome published in 1998 (44) identified 3,974 genes. Annotation was reviewed 4 years later (45) with identification of 82 additional genes. The annotated sequence is deposited in public databases with accession number AL123456. Information about genes and predicted proteins can be obtained at TubercuList Web Server.
A second M. tuberculosis strain was sequenced at The Institute for Genomic Research (TIGR). This virulent isolate, CDC1551, was involved in a cluster of tuberculosis cases in the USA and was highly transmissible to humans. Comparison of the two genome sequences revealed large-sequence and single-nucleotide polymorphisms (46).
The M. tuberculosis strain 210, from the Beijing family, widely distributed in the USA (47), is now complete at TIGR. http://www.tigr.org/tdb/mdb/mdbinprogress.htmlThe Broad Institute is engaged in sequencing additional M. tuberculosis strains: A1, C, Ekat-4, F11, Haarlen, KZN 605 (XDR), KZN 1435 (MDR), KZN 4207 (DS), Peruvian 1, Peruvian 2, W-148, and M. tuberculosis spp. Recent achievements of these genome projects are: F11: Finished as a single supercontig (scaffold) with high quality; C: The current assembly contains 160 sequence contigs in 4 supercontigs (scaffolds); and Haarlem: The current assembly contains 65 sequence contigs in 8 supercontig (scaffold). The entire genomes can be downloaded
M. canetti is a member of the tubercle bacilli, with untypical smooth colony morphology, that has been isolated from tuberculosis patients in East Africa. Shotgun sequencing of strain CIP 140010059 will begin soon at the Sanger Institute
Mycobacterium bovis, BCG, M. africanum, and M. microti
The complete sequence of M. bovis strain AF2122/97, a fully virulent UK isolate from a cow showing caseous lesions in lung and bronchomediastinal lymph nodes, was obtained by the The Wellcome Trust Sanger Institute in collaboration with Institut Pasteur and is available at the BoviList Web Server. Annotation was managed using ARTEMIS with single-nucleotide polymorphism (SNP) identification performed by using the EMBOSS package. The sequence and annotation have been deposited in the EMBL database under accession number BX248333. The sequence and analysis have now been published (48).
The genome sequence of M. bovis AF2122/97 is >99.95% identical to that of M. tuberculosis, but deletion of genetic information has led to a reduced genome size (4,345,492 bp), which is about 66 kb smaller than the one from M. tuberculosis H37Rv. Cell wall components and secreted proteins show the greatest variation.
As an extension to the M. bovis genome project, the The Wellcome Trust Sanger Institute, in collaboration with the Institut Pasteur, determined the genome sequence of M. bovis BCG-Pasteur 1173P2 using clones from the ordered BAC library and the physical map as tools. A 4×-coverage shotgun sequence of Mycobacterium bovis BCG Pasteur was obtained. Comparison of the BCG genome sequence with those of virulent tubercle bacilli highlighted the genetic rearrangements that led to attenuation of this live vaccine strain (49). A second strain of M. bovis BCG, Moreau RDJ, the Brazilian vaccine strain of Mycobacterium bovis that is very immunogenic with few side effects, is being sequenced using random shotgun by researchers from FIOCRUZ and Fundação Ataulpho de Paiva, in Brazil. This strain will be used for comparative genomics.
The Sanger Institute is sequencing the genome of M. africanum strain GM041182. Shotgun sequencing is complete. A database of reads is available for searching at the Blast Server, or for download from the FTP site.
The Sanger Institute is sequencing the genome of M. microti strain OV254 (OV stands for Orkney Vole), in collaboration with the Pasteur Institute (Unité de Génétique Moléculaire Bactérienne). A whole genome screen based on BAC maps was also undertaken (50).
Mycobacterium leprae
The sequencing project of the complete genome of a strain of M. leprae, originally isolated in Tamil Nadu (TN), is finished (Mycobacterium leprae genome project). The complete sequence is 3,268,203 bp in length with a G+C content of 57.8% and was generated from a combination of cosmid and whole-genome shotgun sequencing. There are 1,604 protein-coding genes and 1,116 pseudogenes (10). Both the sequence and annotation have been deposited in the public databases with the accession number AL450380. The project was undertaken as a collaboration between the Sanger Institute and the Pasteur Institute (Leproma Web Server).
Analysis of the leprosy genome indicates that it had suffered important genomic downsizing, which appears to have resulted from extensive recombination events between dispersed repetitive sequences. As a consequence, it had lost essential metabolic routes, including most of the microaerophilic and anaerobic respiratory chains, providing explanation for its intracellular parasitic lifestyle (10). In this respect, the M. leprae genome is very different from the other mycobacterial genomes analyzed thus far.
In a recent paper, the use of bioinformatics and comparative genomics methods allowed the identification of putative leprosy-specific antigenic proteins suitable for use for diagnostic purposes (51).
Mycobacterium marinum and M. ulcerans
The genome project of M. marinum, M strain, is now finished. It is 6,636,827 bp in length, with an average G+C content of 65.73%. The project has been accomplished by the Sanger Institute in collaboration with research centers from the USA, France, and Australia (Mycobacterium marinum genome project).
Institut Pasteur is sequencing the 4.4-Mb genome of an epidemic strain of M. ulcerans (Unité de Génétique Moléculaire Bactérienne). The BuruList Web Server is currently operating only as a BLAST server because the genome is still in a preliminary stage of assembly. In parallel with the genome project, pulsed field gel electrophoresis experiments and sequence analysis have led to the identification of a large plasmid that is required for the production of mycolactone (52).
The Clemson University Genomics Institute has also started a sequencing project of M. ulcerans.
Mycobacterium avium subsp. avium and M. avium subsp. paratuberculosis
The genome sequence of M. avium subsp. avium (strain 104) is in progress at TIGR. The genome has 4.70 million base pairs (Mb). The sequence is complete, and all gaps are closed. This strain is a common representative of the species complex, isolated from an AIDS patient.
The sequence of MAP (strain K-10) has been recently completed at Minnesota University and consists of a circular chromosome of 4.83 Mb and contains about 4,350 open reading frames (53). This strain is a virulent bovine clinical isolate, isolated in Wisconsin in the mid-1970s. A random shotgun approach was adopted to sequence the genome of MAP K-10.
Rapidly Growing Mycobacteria
The sequence of M. smegmatis strain mc2155 is in progress at TIGR. New sequences generated for gap closure have been assembled and added to the data release. This strain is a highly efficient plasmid transformation mutant, widely used in mycobacterial research (54).
Genoscope is undertaking sequencing of type strains M. abscessus ATCC 19977, isolated from a knee abscess in 1953, and M. chelonae ATCC 35752, isolated from turtle in 1921, which were already compared by subtractive hybridization by the same group.
Mycobacterial strains involved in degradation of environmentally hazardous chemicals (55, 56) are being sequenced by DOE Joint Genome Institute.
Impact of Genome Projects on Mycobacterial Research
The recent determination of genomic nucleotide sequences of M. tuberculosis H37Rv and CDC 1551, M. bovis, M. leprae, and MAP makes Mycobacterium one of the most sequenced bacterial genera (57). This wealth of comparative genome sequence information provides unique opportunities for new insights into the biology of these globally important pathogens.
Microarrays
Concurrent emergence of new functional genomic technologies such as microarrays, in addition to recently developed genetic recombination tools for mycobacteria, creates new opportunities to exploit genome information to address the scientific imperatives of better drugs, vaccines, and diagnostics for mycobacterial diseases. Tuberculosis microarrays have mainly been used (31) to compare genomes within the M. tuberculosis complex (35), simultaneously monitor the relative expression of every gene in the genome by comparing mRNA levels, and (16) for analysis of SNPs for genotyping and drug resistance detection using high-density oligonucleotides array. Whole genome microarrays for M. tuberculosis are available through the Pathogen Functional Genomics Resource Center at TIGR and St. George's Hospital Medical School.
Tuberculosis is a complex disease influenced by the genotypes of both host and pathogen as well as the immunological status of the host; then microarrays containing genes from the host can also be used to study host–M. tuberculosis interactions. Experimental mouse infection with M. tuberculosis (in vivo experiments) (58) or mouse macrophage infection models (in vitro experiments) (59) represent an approach to study M. tuberculosis pathogenesis. Affymetrix supplies a gene chip with murine genes for these experiments.
The microarray methodology has been extensively applied to analyze one of the most intriguing features that characterize the infection caused by the tubercle bacilli: the dormant stage. Transcriptomic analyses have been performed in both in vivo and in vitro models of dormancy, and a dormancy regulon was identified in the conditions tested. This regulon is under the control of the two-component system dosR/S and encompasses up to 48 different genes; unfortunately, nearly one-third of the genes putatively related to dormancy in M. tuberculosis are of unknown function, indicating the scarce information available concerning this infective situation (60-63).
A collaborative research between VLA of Weybridge, UK and the Argentinean laboratory was done using a DNA microarray containing non-redundant CDS from the two sequenced M. tuberculosis strains, CDC1551 and H37Rv, and from M. bovis AF2122/97 to evaluate genetic variability between M. tuberculosis complex strains (64, 65). In M. microti, a new deletion was identified that removes five genes that code for ESAT-6 family antigens and PE-PPE proteins. This region, called MiD4, was also found to be deleted from M. pinnipedii, supporting the previous idea of a common evolutionary lineage for both species (65). To extend the repertoire of these deletion markers, a whole genome microarray analysis of the recently defined M. pinnipedii species was done (64). Two deletions that are exclusive to M. pinnipedii were found: PiD1 that removes the orthologs of the M. tuberculosis genes Rv3530c and Rv3531c, and PiD2 that encompasses genes Rv1977 and Rv1978 (64).
New Typing Methods
The classical molecular epidemiology that was applied in the analysis of the mycobacterial diseases was based on Restriction Fragment Polymorphism Analysis (RFLP) patterns attributable to the chromosomal distribution of specific insertion sequences. Comparison of the patterns derived from the IS6110–RFLP analysis has been an essential tool to track the transmission and distribution of M. tuberculosis isolates worldwide, helping in the control of the disease. Data derived from completion of genome sequencing allowed the development of new, quicker, and also highly discriminative PCR-based typing methods, such as MIRU-VNTR and Spoligotyping, the latter only applicable to MTBC thus far.
MIRU-VNTR
Special tandem repeat sequences such as the Variable Number Tandem Repeat (VNTR) loci, called Mycobacteria Interspersed Repetitive Units (MIRU), have been described in M. tuberculosis (MIRU Inventory), M. bovis (Mycobacterium bovis molecular typing database), M. leprae and M. avium complex (66-75). The properties of MIRU that distinguish them from other tandem repeat sequences are that they have regulatory elements, such as initiation and stop codons and consensus to bacterial ribosome-binding site 5'-TGA GGA GGA GC-3'. Most MIRU overlap the termination and initiation codons of their flanking genes (74). Polymorphism at these tandem repeat loci can occur as a result of either nucleotide sequence changes among individual repeat units or variation in the number of repeat units.
Spoligotyping
In human tuberculosis, molecular typing has advanced by the use of the insertion sequence IS6110 (76), which is repeated many times in the M. tuberculosis genome, producing a genotypic heterogeneity of isolates. In M. bovis, IS6110 is less useful because the genome of most strains contains only one or very few IS6110 copies (77). The insertion element IS6110 is frequently found in a unique locus of the M. tuberculosis complex genome called the direct repeat (DR) region. A more recent technique, called spoligotyping (78), is highly efficient for typing MTBC, including M. bovis. With this method, DNA polymorphisms in the genomic DR locus of MTBC isolates are visualized. This locus contains multiple, well-conserved 36-bp DRs interspersed with nonrepetitive 34- to 41-bp DNA spacer sequences. Spoligotyping involves the amplification of the whole DR region, followed by hybridization of the amplified DNA to a set of spacer oligonucleotides, covalently linked to a membrane (Mycobacterium bovis spoligotype database).
Considering that spoligotyping is a rapid and easy-to-apply technique, it is important to improve its capacity for differentiation. New spacers of the DR region were analyzed in Argentinian isolates of M. bovis. These isolates were discriminated more accurately by the novel probe spoligotyping than by traditional spoligotyping (79).
Two databases, SPOLDB4 and MIRU-VNTRplus, containing MIRU-VNTR and spoligotype patterns, are now available for online comparison and analysis of patterns obtained in the laboratory.
Comparative Genomics
Comparison of mycobacterial genomes can be performed by a set of different approaches. One of them, the array technology, easily identify deletion events but cannot readily detect insertions or duplications. Other powerful method includes whole-genome sequence comparison that covers from SNP to gene rearrangements (80) and allows detection of large sequence polymorphisms (LSP), considered the major contributor to genetic diversity within members of the genus. Several of these methodologies have been used to compare members of MTBC, and more rarely were applied to other members of the genus.
Comparative Genomics between Members of MTBC
Whole genome comparisons of M. tuberculosis H37Rv and M. bovis BCG Pasteur were undertaken using two different approaches. Using BAC-arrays and direct comparison of canonical BACs from ordered libraries, several polymorphic genomic regions were uncovered (48). By performing comparative hybridization experiments on a DNA microarray comprising 4896 spots, representing 3902 ORFs of M. tuberculosis H37Rv, 16 regions of difference (RD1-16) were shown to be absent from some M. bovis BCG relative to M. tuberculosis H37Rv (81). In parallel, five regions deleted in H37Rv were described (RvD)1-5 (82) compared with genomes of other members of the complex. The combined findings of these studies can be found at UnitéGénétique Moléculaire Bactérienne.
{kind=link}
Recently, a M. tuberculosis-specific deletion (TbD1) was identified. This deletion is characterized by the absence of a 2,153-bp fragment truncating genes mmpS6 and mmpL6 in “modern” strains of M. tuberculosis; however, this region is present in all other members of the M. tuberculosis complex (6).
In another study (83), M. tuberculosis H37Rv and M. bovis AF2122/97 genomic sequences were compared using blastn and MSPcrunch (84) software and visualized with the Artemis Comparison Tool (ACT) version 4.0 for Apple. The criteria used to select loci for further studies were that they were not IS6110 insertions, PE-PPE family genes, single-nucleotide polymorphisms, or previously published regions of difference. Four polymorphisms were found: 1) a deletion of Rv3479 specific to M. bovis; 2) that the rpfA gene is shortened to various extents in M. bovis; 3) an insertion in Rv0648 in M. bovis,; and 4) a duplication of lppA/B in M. bovis. Interestingly, lppA is also duplicated in M. tuberculosis CDC 1551.
Comparative genomics has given also some clues to understand the evolution of members of the complex. Thus, the analysis of the distribution of variable regions indicates that, contrary to the initial proposed scheme, M. tuberculosis did not descend from M. bovis; instead, both bacteria descended from a single common ancestor named “ancient” TB, which appears to be closely related to M. canetti, a member of the complex isolated from humans described recently. Both M. bovis and “modern” M. tuberculosis evolved from that common ancestor, as well as all other components (82) (Figure 6).

Figure 6
From Brosch et al. (82).
Identification of the different members of MTBC in the laboratory is difficult using standard methodologies; however, their correct differentiation has important influence in the treatment to be chosen and the consequent management of the patients. The development of comparative genomics has allowed the description of PCR-based schemes for such purposes. On the basis of variable distribution of insertion and deletions in the genomes of the several members, some flow charts were described for M. tuberculosis complex differentiation (80, 85).
Comparative Genomics between Members of MAC
Some few studies have been performed mainly focused on the detection of specific targets for identification of MAP, by comparison of its genome with that of other related members of MAC. The annotated sequence of M. avium subsp. avium strain 104 (provided by TIGR) was used initially to assemble a whole-genome DNA microarray representing the predicted coding sequences. Seventy-base-pair-long oligonucleotide probes were designed and synthesized for 4,158 of 4,480 predicted open reading frames (ORFs). Each probe was printed in duplicate onto microarray slides to permit genomic DNA comparison of M. avium subsp. avium strain 104 and the following strains: 1) M. avium subsp. paratuberculosis K10 (cow strain); 2) M. avium subsp. paratuberculosis LN20 (sheep strain); and 3) M. avium subsp. silvaticum 49884 (ATCC strain). Microarray comparison revealed 14 LSP regions that distinguish a single strain of M. avium subsp. avium (MAA) from other strains of MAP (86). These LSP regions encompass 572 genes, more than 700 Kb, that represents 13.5% of the MAA genome. Remarkably higher diversity was demonstrated in this complex compared with the genomic variability described among M. tuberculosis complex isolates (87). This could be in agreement with consideration of MAP and MAA as different subspecies within the species M. avium.
As part of this ongoing work to characterize M. avium complex organisms, sequenced genome of M. avium subsp. avium strain 104 (TIGR) and M. avium subsp. paratuberculosis strain K10 (GenBank accession number NC_002944) were compared using Artemis software for visualization of the M. avium subsp. paratuberculosis strain K10 genome and annotation of M. avium subsp. avium strain 104. Genome sequence comparison, via visual inspection in Artemis, revealed two types of LSPs: those present in the former but missing in the latter (LSPAs), and those only present in the latter (LSPPs). Three LSPAs and 17 LSPPs were found. The distribution of these LSPs were examined across a panel of M. avium complex isolates, revealing 1 LSPA absent in all MAP isolates tested; then this absence was described as 100% specific of the MAP genome, and 7 LPSPs genomic regions present only in the analyzed MAP strains, and described as highly specific of MAP isolates; other 10 LPSP regions were not specific of the MAP genome (86).
The analysis of a larger number of MAP isolates showed that these isolates have a high degree of genetic conservation, with no differences with the reference strain K10. Deletions detected by comparison with other members of the MAC are, as expected, associated with the presence of mobile genetic elements (88).
Comparative Genomics as a Tool for the Analysis of Mycobacterial Speciation
The pool of different mycobacterial genome projects currently in progress opens new possibilities for comparison between members of different species in the genus, a task still to be undertaken. LSPs, based on insertion/deletions and rearrangements of the genomes, are being considered the main mechanism that occurs during the evolution of the bacterial genomes. These changes are considered important in the separation of isolates in different genomic species and could putatively be studied by determining a gene’s location and its organization in the chromosome.
Recently, a new website (Microbesonline) has been described and organized for quick comparison of bacterial genomes (89). Only five mycobacterial genomes are included in that site thus far, three of them members of MTBC, plus MAP and M. leprae.
Using the facilities offered at that website (MicrobesOnline Comparative Genome Browser), a multispecies comparison of the location and gene organization of the available mycobacterial genomes was performed. An expected result was found when the M. leprae genome was analyzed; this genome showed major differences from all other mycobacterial genomes, differences that were scattered along the chromosome. As an exception, only short regions showed conserved gene organization in the M. leprae genome when compared with the equivalent region in other mycobacterial genomes. For example, the region corresponding to recN (ML1360 and Rv1696) shows gene conservation in about a 10-Kb length among all five compared mycobacterial genomes; and the region corresponding to dnaA (ML0001 and Rv0001) is also conserved along 12 Kb in all five genomes.
The genome of M. tuberculosis H37Rv was considered as reference for organization of the contained ORFs (from Rv0001 to Rv3924) and was theoretically divided into eight regions (containing approximately 500 ORFs each), taking the following ORFs as references: Rv0001 (dnaA); Rv0500 (proC); Rv1001 (arcA); Rv1522 (mmpL12); Rv2031 (acr/hspX); Rv2500 (fadE19); Rv3065 (emr); and Rv3504 (fadE26). Approximately 50 Kb were checked upstream and downstream from each of the previous genes. A genome multispecies browser was applied to each of the selected regions, and as a result, the eight genomic regions could be divided into three groups.
Group I regions were conserved in members of the MTBC as well as in MAP. This includes Rv0001, where genes related to chromosome replication and partitioning are located. Group II regions were conserved in members of the MTBC but were different than the corresponding region in MAP. This includes Rv0500 (this region encloses the icl gene, which has been related to dormancy in M. tuberculosis) and Rv3065 (this region includes components of the cluster nrd involved in DNA replication). Group III variable regions were conserved in all four mycobacteria, including members of the MTBC. This includes four of the remaining regions, such as Rv1522 (the mmpL12 gene thought to be involved in the fatty acid transport), Rv2031 (gene coding for the stress protein α-crystallin), Rv2500 (belonging to a family of genes associated with fatty acid degradation and whose components have more or less scattered distribution along the chromosome), and Rv3504 (several genes coding for members of the PE-PGRS family of proteins, which have been related to the pathogenesis of the genus, are located within this region). A curious result was that the region defined by the gene Rv1001 divided two genomic zones: one belongs to Group II, located in the corresponding downstream region, and the other to Group III, located in the corresponding upstream region.
The previous data are deeply biased because of the small number of species compared (actually only three clearly different species); however, as more mycobacterial genomes were included in this site, a similar approach can be used to help in the detection of genomic regions that putatively would participate in mycobacterial speciation.
References
- 1.
- Euzéby JP. 2004, posting date. List of Bacterial names with Standing in Nomenclature. Societé de Bactériologie Systématique et Vétérinaire [online.]
- 2.
- Aranaz A , Cousins D , Mateos A , Dominguez L . Elevation of Mycobacterium tuberculosis subsp. caprae Aranaz et al. 1999 to species rank as Mycobacterium caprae comb. nov., sp. nov. Int J Syst Evol Microbiol. 2003;53:1785–1789. [PubMed: 14657105]
- 3.
- Niemann S , Richter E , Rusch-Gerdes S . Biochemical and genetic evidence for the transfer of Mycobacterium tuberculosis subsp. caprae Aranaz et al. 1999 to the species Mycobacterium bovis Karlson and Lessel 1970 (approved lists 1980) as Mycobacterium bovis subsp. caprae comb. nov. Int J Syst Evol Microbiol. 2002;52:433–436. [PubMed: 11931153]
- 4.
- Cousins DV , Bastida R , Cataldi A , Quse V , Redrobe S , Dow S , Duignan P , Murray A , Dupont C , Ahmed N , Collins DM , Butler WR , Dawson D , Rodriguez D , Loureiro J , Romano MI , Alito A , Zumarraga M , Bernardelli A . Tuberculosis in seals caused by a novel member of the Mycobacterium tuberculosis complex: Mycobacterium pinnipedii sp. nov. Int J Syst Evol Microbiol. 2003;53:1305–1314. [PubMed: 13130011]
- 5.
- Gutierrez MC , Brisse S , Brosch R , Fabre M , Omais B , Marmiesse M , Supply P , Vincent V . Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005;1:e5. [PMC free article: PMC1238740] [PubMed: 16201017]
- 6.
- Brosch R, Behr MA. Comparative Genomics and evolution of Mycobacterium bovis BCG. In: Cole ST, et al., editors. Tuberculosis and Tubercle bacilli. ASM press; 2005. pp. 155–164.
- 7.
- Mostowy S , Behr MA . The origin and evolution of Mycobacterium tuberculosis. Clin Chest Med. 2005;26:207–216. [PubMed: 15837106]
- 8.
- Latini MS , Latini OA , Lopez ML , Cecconi JO . Tuberculosis bovina en seres humanos. Rev Argent Torax. 1990;51:13–16.
- 9.
- Samper S , Martin C , Pinedo A , Rivero A , Blazquez J , Baquero F , van Soolingen D , van Embden J . Transmission between HIV-infected patients of multidrug-resistant tuberculosis caused by Mycobacterium bovis. AIDS. 1997;11:1237–1242. [PubMed: 9256941]
- 10.
- Cole ST , Eiglmeier K , Parkhill J , James KD , Thomson NR , Wheeler PR , Honore N , Garnier T , Churcher C , Harris D , Mungall K , Basham D , Brown D , Chillingworth T , Connor R , Davies RM , Devlin K , Duthoy S , Feltwell T , Fraser A , Hamlin N , Holroyd S , Hornsby T , Jagels K , Lacroix C , Maclean J , Moule S , Murphy L , Oliver K , Quail MA , Rajandream MA , Rutherford KM , Rutter S , Seeger K , Simon S , Simmonds M , Skelton J , Squares R , Squares S , Stevens K , Taylor K , Whitehead S , Woodward JR , Barrell BG . Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. [PubMed: 11234002]
- 11.
- Monot M , Honore N , Garnier T , Araoz R , Coppee JY , Lacroix C , Sow S , Spencer JS , Truman RW , Williams DL , Gelber R , Virmond M , Flageul B , Cho SN , Ji B , Paniz-Mondolfi A , Convit J , Young S , Fine PE , Rasolofo V , Brennan PJ , Cole ST . On the origin of leprosy. Science. 2005;308:1040–1042. [PubMed: 15894530]
- 12.
- Gomez-Valero L , Rocha ECP , Latorre A , Silva FJ . Reconstructing the ancestor of Mycobacterium leprae: the dynamics of gene loss and genome reduction. Genome Res. 2007;17:1178–1185. [PMC free article: PMC1933519] [PubMed: 17623808]
- 13.
- Debacker M , Aguiar J , Steunou C , Zinsou C , Meyers WM , Guedenon A , Scott JT , Dramaix M , Portaels F . Mycobacterium ulcerans disease (Buruli ulcer) in rural hospital, Southern Benin, 1997-2001. Emerg Infect Dis. 2004;10:1391–1398. [PMC free article: PMC3320395] [PubMed: 15496239]
- 14.
- Stinear T , Ross BC , Davies JK , Marino L , Robins-Browne RM , Oppedisano F , Sievers A , Johnson PD . Identification and characterization of IS2404 and IS2606: two distinct repeated sequences for detection of Mycobacterium ulcerans by PCR. J Clin Microbiol. 1999;37:1018–1023. [PMC free article: PMC88643] [PubMed: 10074520]
- 15.
- Stinear TP , Jenkin GA , Johnson PD , Davies JK . Comparative genetic analysis of Mycobacterium ulcerans and Mycobacterium marinum reveals evidence of recent divergence. J Bacteriol. 2000;182:6322–6330. [PMC free article: PMC94777] [PubMed: 11053375]
- 16.
- Adusumilli S , Mve-Obiang A , Sparer T , Meyers W , Hayman J , Small PL . Mycobacterium ulcerans toxic macrolide, mycolactone modulates the host immune response and cellular location of M. ulcerans in vitro and in vivo. Cell Microbiol. 2005;7:1295–1304. [PubMed: 16098217]
- 17.
- Yip MJ , Porter JL , Fyfe JAM , Lavender CJ , Portaels F , Rhodes M , Kator H , Colorni A , Jenkin GA , Stinear T . Evolution of Mycobacterium ulcerans and other mycolactone-producing mycobacteria from a common Mycobacterium marinum progenitor. J Bacteriol. 2007;189:2021–2029. [PMC free article: PMC1855710] [PubMed: 17172337]
- 18.
- Hermon-Taylor J , Bull T . Crohn's disease caused by Mycobacterium avium subspecies paratuberculosis: a public health tragedy whose resolution is long overdue. J Med Microbiol. 2002;51:3–6. [PubMed: 11800469]
- 19.
- Tortoli E , Rindi L , Garcia MJ , Chiaradonna P , Dei R , Garzelli C , Kroppenstedt RM , Lari N , Mattei R , Mariottini A , Mazzarelli G , Murcia MI , Nanetti A , Piccoli P , Scarparo C . Proposal to elevate the genetic variant MAC-A, included in the Mycobacterium avium complex, to species rank as Mycobacterium chimaera sp. nov. Int J Syst Evol Microbiol. 2004;54:1277–1285. [PubMed: 15280303]
- 20.
- Murcia MI , Tortoli E , Menendez MC , Palenque E , Garcia MJ . Mycobacterium colombiense sp. nov., a novel member of the Mycobacterium avium complex and description of MAC-X as a new ITS genetic variant. Int J Syst Evol Microbiol. 2006;56:2049–2054. [PubMed: 16957098]
- 21.
- Mijs W , de Haas P , Rossau R , Van der Laan T , Rigouts L , Portaels F , van Soolingen D . Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and 'M. avium subsp. hominissuis' for the human/porcine type of M. avium. Int J Syst Evol Microbiol. 2002;52:1505–1518. [PubMed: 12361252]
- 22.
- Freitas D , Alvarenga L , Sampaio J , Mannis M , Sato E , Sousa L , Vieira L , Yu MC , Martins MC , Hoffling-Lima A , Belfort R . An outbreak of Mycobacterium chelonae infection after LASIK. Ophthalmology. 2003;110:276–285. [PubMed: 12578767]
- 23.
- Sampaio JLM , Chimara E , Ferrazoli L , Telles MAS , Del Guercio VMF , Jericó ZVN , Miyashiro K , Fortaleza CMCB , Padoveze MC , Leao SC . Use of four molecular methods for typing of Mycobacterium fortuitum group strains causing post augmentation mammaplasty infections. Clin Microbiol Infect. 2006;12:142–149. [PubMed: 16441452]
- 24.
- Tiwari TS , Ray B , Jost KC , Rathod MK , Zhang Y , Brown-Elliott BA , Hendricks K , Wallace RJ . Forty years of disinfectant failure: outbreak of postinjection Mycobacterium abscessus infection caused by contamination of benzalkonium chloride. Clin Infect Dis. 2003;36:954–962. [PubMed: 12684906]
- 25.
- Wallace RJ , Brown BA , Griffith DE . Nosocomial outbreaks/pseudo-outbreaks caused by nontuberculous mycobacteria. Annu Rev Microbiol. 1998;52:453–490. [PubMed: 9891805]
- 26.
- Winthrop KL , Abrams M , Yakrus M , Schwartz I , Ely J , Gillies D , Vugia DJ . An outbreak of mycobacterial furunculosis associated with footbaths at a nail salon. N Engl J Med. 2002;346:1366–1371. [PubMed: 11986410]
- 27.
- Campos-Herrero MI , Garcia D , Figuerola A , Suarez P , Campo C , Garcia MJ . Bacteremia caused by the novel species Mycobacterium canariasense. Eur J Clin Microbiol Infect Dis. 2006;25:58–60. [PubMed: 16391913]
- 28.
- Primm TP , Lucero CA , Falkinham JO . Health impacts of environmental mycobacteria. Clin Microbiol Rev. 2004;17:98–106. [PMC free article: PMC321467] [PubMed: 14726457]
- 29.
- Tortoli E . Impact of genotypic studies on mycobacterial taxonomy: the new mycobacteria of the 1990s. Clin Microbiol Rev. 2003;16:319–354. [PMC free article: PMC153139] [PubMed: 12692101]
- 30.
- Fox GE , Wisotzkey JD , Jurtshuk P . How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166–170. [PubMed: 1371061]
- 31.
- Adekambi T , Colson P , Drancourt M . rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol. 2003;41:5699–5708. [PMC free article: PMC308974] [PubMed: 14662964]
- 32.
- Blackwood KS , He C , Gunton J , Turenne CY , Wolfe J , Kabani AM . Evaluation of recA sequences for identification of Mycobacterium species. J Clin Microbiol. 2000;38:2846–2852. [PMC free article: PMC87126] [PubMed: 10921937]
- 33.
- Domenech P , Jimenez MS , Menendez MC , Bull TJ , Samper S , Manrique A , Garcia MJ . Mycobacterium mageritense sp. nov. Int J Syst Bacteriol. 1997;47:535–540. [PubMed: 9103645]
- 34.
- McNabb A , Eisler D , Adie K , Amos M , Rodrigues M , Stephens G , Black WA , Isaac-Renton J . Assessment of partial sequencing of the 65-kilodalton heat shock protein gene (hsp65) for routine identification of Mycobacterium species isolated from clinical sources. J Clin Microbiol. 2004;42:3000–3011. [PMC free article: PMC446301] [PubMed: 15243051]
- 35.
- Adekambi T , Drancourt M . Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. Int J Syst Evol Microbiol. 2004;54:2095–2105. [PubMed: 15545441]
- 36.
- Menendez MC , Garcia MJ , Navarro MC , Gonzalez-y-Merchand JA , Rivera-Gutierrez S , Garcia-Sanchez L , Cox RA . Characterization of an rRNA operon (rrnB) of Mycobacterium fortuitum and other mycobacterial species: implications for the classification of mycobacteria. J Bacteriol. 2002;184:1078–1088. [PMC free article: PMC134815] [PubMed: 11807068]
- 37.
- Domenech P , Menendez MC , Garcia MJ . Restriction fragment length polymorphisms of 16S rRNA genes in the differentiation of fast-growing mycobacterial species. FEMS Microbiol Lett. 1994;116:19–24. [PubMed: 7907567]
- 38.
- Brown BA , Springer B , Steingrube VA , Wilson RW , Pfyffer GE , Garcia MJ , Menendez MC , Rodriguez-Salgado B , Jost KC , Chiu SH , Onyi GO , Bottger EC , Wallace RJ . Mycobacterium wolinskyi sp. nov. and Mycobacterium goodii sp. nov., two new rapidly growing species related to Mycobacterium smegmatis and associated with human wound infections: a cooperative study from the International Working Group on Mycobacterial Taxonomy. Int J Syst Bacteriol. 1999;49(Pt 4):1493–1511. [PubMed: 10555330]
- 39.
- Jimenez MS , Campos-Herrero MI , Garcia D , Luquin M , Herrera L , Garcia MJ . Mycobacterium canariasense sp. nov. Int J Syst Evol Microbiol. 2004;54:1729–1734. [PubMed: 15388736]
- 40.
- Lawn SD , Bekker LG , Miller RF . Immune reconstitution disease associated with mycobacterial infections in HIV-infected individuals receiving antiretrovirals. Lancet Infect Dis. 2005;5:361–373. [PubMed: 15919622]
- 41.
- Horsburgh CR . The pathophysiology of disseminated Mycobacterium avium complex disease in AIDS J Infect Dis 1999179Suppl 3S461–S465. [PubMed: 10099120]
- 42.
- Nunn P , Williams B , Floyd K , Dye C , Elzinga G , Raviglione M . Tuberculosis control in the era of HIV. Nat Rev Immunol. 2005;5:819–826. [PubMed: 16200083]
- 43.
- Ahmed N , Hasnain SE . Genomics of Mycobacterium tuberculosis: old threats & new trends. Indian J Med Res. 2004;120:207–212. [PubMed: 15520478]
- 44.
- Cole ST , Brosch R , Parkhill J , Garnier T , Churcher C , Harris D , Gordon SV , Eiglmeier K , Gas S , Barry CE , Tekaia F , Badcock K , Basham D , Brown D , Chillingworth T , Connor R , Davies R , Devlin K , Feltwell T , Gentles S , Hamlin N , Holroyd S , Hornsby T , Jagels K , Krogh A , McLean J , Moule S , Murphy L , Oliver K , Osborne J , Quail MA , Rajandream MA , Rogers J , Rutter S , Seeger K , Skelton J , Squares R , Squares S , Sulston JE , Taylor K , Whitehead S , Barrell BG . Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. [PubMed: 9634230]
- 45.
- Camus JC , Pryor MJ , Medigue C , Cole ST . Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148:2967–2973. [PubMed: 12368430]
- 46.
- Fleischmann RD , Alland D , Eisen JA , Carpenter L , White O , Peterson J , DeBoy R , Dodson R , Gwinn M , Haft D , Hickey E , Kolonay JF , Nelson WC , Umayam LA , Ermolaeva M , Salzberg SL , Delcher A , Utterback T , Weidman J , Khouri H , Gill J , Mikula A , Bishai W , Jacobs Jr WR , Venter JC , Fraser CM . Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–5490. [PMC free article: PMC135346] [PubMed: 12218036]
- 47.
- Beggs ML , Eisenach KD , Cave MD . Mapping of IS6110 insertion sites in two epidemic strains of Mycobacterium tuberculosis. J Clin Microbiol. 2000;38:2923–2928. [PMC free article: PMC87149] [PubMed: 10921952]
- 48.
- Garnier T , Eiglmeier K , Camus JC , Medina N , Mansoor H , Pryor M , Duthoy S , Grondin S , Lacroix C , Monsempe C , Simon S , Harris B , Atkin R , Doggett J , Mayes R , Keating L , Wheeler PR , Parkhill J , Barrell BG , Cole ST , Gordon SV , Hewinson RG . The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci U S A. 2003;100:7877–7882. [PMC free article: PMC164681] [PubMed: 12788972]
- 49.
- Gordon SV , Brosch R , Billault A , Garnier T , Eiglmeier K , Cole ST . Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol. 1999;32:643–655. [PubMed: 10320585]
- 50.
- Brodin P , Eiglmeier K , Marmiesse M , Billault A , Garnier T , Niemann S , Cole ST , Brosch R . Bacterial artificial chromosome-based comparative genomic analysis identifies Mycobacterium microti as a natural ESAT-6 deletion mutant. Infect Immun. 2002;70:5568–5578. [PMC free article: PMC128332] [PubMed: 12228284]
- 51.
- Araoz R , Honore N , Cho S , Kim JP , Cho SN , Monot M , Demangel C , Brennan PJ , Cole ST . Antigen discovery: a postgenomic approach to leprosy diagnosis. Infect Immun. 2006;74:175–182. [PMC free article: PMC1346601] [PubMed: 16368971]
- 52.
- Stinear TP , Mve-Obiang A , Small PL , Frigui W , Pryor MJ , Brosch R , Jenkin GA , Johnson PD , Davies JK , Lee RE , Adusumilli S , Garnier T , Haydock SF , Leadlay PF , Cole ST . Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci U S A. 2004;101:1345–1349. [PMC free article: PMC337055] [PubMed: 14736915]
- 53.
- Li L , Bannantine JP , Zhang Q , Amonsin A , May BJ , Alt D , Banerji N , Kanjilal S , Kapur V . The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci U S A. 2005;102:12344–12349. [PMC free article: PMC1194940] [PubMed: 16116077]
- 54.
- Snapper SB , Melton RE , Mustafa S , Kieser T , Jacobs WR . Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. [PubMed: 2082148]
- 55.
- Khan AA , Kim SJ , Paine DD , Cerniglia CE . Classification of a polycyclic aromatic hydrocarbon-metabolizing bacterium, Mycobacterium sp. strain PYR-1, as Mycobacterium vanbaalenii sp. nov. Int J Syst Evol Microbiol. 2002;52:1997–2002. [PubMed: 12508859]
- 56.
- Miller CD , Hall K , Liang YN , Nieman K , Sorensen D , Issa B , Anderson AJ , Sims RC . Isolation and characterization of polycyclic aromatic hydrocarbon-degrading Mycobacterium isolates from soil. Microb Ecol. 2004;48:230–238. [PubMed: 15107954]
- 57.
- Brosch R , Pym AS , Gordon SV , Cole ST . The evolution of mycobacterial pathogenicity: clues from comparative genomics. Trends Microbiol. 2001;9:452–458. [PubMed: 11553458]
- 58.
- Lopez B , Aguilar D , Orozco H , Burger M , Espitia C , Ritacco V , Barrera L , Kremer K , Hernandez-Pando R , Huygen K , van Soolingen D . A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol. 2003;133:30–37. [PMC free article: PMC1808750] [PubMed: 12823275]
- 59.
- Schnappinger D , Ehrt S , Voskuil MI , Liu Y , Mangan JA , Monahan IM , Dolganov G , Efron B , Butcher PD , Nathan C , Schoolnik GK . Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. [PMC free article: PMC2194186] [PubMed: 12953091]
- 60.
- Bacon J , James BW , Wernisch L , Williams A , Morley KA , Hatch GJ , Mangan JA , Hinds J , Stoker NG , Butcher PD , Marsh PD . The influence of reduced oxygen availability on pathogenicity and gene expression in Mycobacterium tuberculosis. Tuberculosis (Edinb). 2004;84:205–217. [PubMed: 15207490]
- 61.
- Kendall SL , Rison SC , Movahedzadeh F , Frita R , Stoker NG . What do microarrays really tell us about M. tuberculosis? Trends Microbiol. 2004;12:537–544. [PubMed: 15539113]
- 62.
- Muttucumaru DG , Roberts G , Hinds J , Stabler RA , Parish T . Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis (Edinb). 2004;84:239–246. [PubMed: 15207493]
- 63.
- Voskuil MI , Visconti KC , Schoolnik GK . Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb). 2004;84:218–227. [PubMed: 15207491]
- 64.
- Bigi F , Garcia-Pelayo MC , Nunez-Garcia J , Peralta A , Caimi KC , Golby P , Hinds J , Cataldi A , Gordon SV , Romano MI . Identification of genetic markers for Mycobacterium pinnipedii through genome analysis. FEMS Microbiol Lett. 2005;248:147–152. [PubMed: 15979818]
- 65.
- Garcia-Pelayo MC , Caimi KC , Inwald JK , Hinds J , Bigi F , Romano MI , van Soolingen D , Hewinson RG , Cataldi A , Gordon SV . Microarray analysis of Mycobacterium microti reveals deletion of genes encoding PE-PPE proteins and ESAT-6 family antigens. Tuberculosis (Edinb). 2004;84:159–166. [PubMed: 15207485]
- 66.
- Bull TJ , Sidi-Boumedine K , McMinn EJ , Stevenson K , Pickup R , Hermon-Taylor J . Mycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complex. Mol Cell Probes. 2003;17:157–164. [PubMed: 12944117]
- 67.
- Chanchaem W , Palittapongarnpim P . A variable number of tandem repeats result in polymorphic alpha -isopropylmalate synthase in Mycobacterium tuberculosis. Tuberculosis (Edinb). 2002;82:1–6. [PubMed: 11914056]
- 68.
- Frothingham R , Meeker-O'Connell WA . Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144(Pt 5):1189–1196. [PubMed: 9611793]
- 69.
- Mazars E , Lesjean S , Banuls AL , Gilbert M , Vincent V , Gicquel B , Tibayrenc M , Locht C , Supply P . High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A. 2001;98:1901–1906. [PMC free article: PMC29354] [PubMed: 11172048]
- 70.
- Romano MI , Amadio A , Bigi F , Klepp L , Etchechoury I , Llana MN , Morsella C , Paolicchi F , Pavlik I , Bartos M , Leao SC , Cataldi A . Further analysis of VNTR and MIRU in the genome of Mycobacterium avium complex, and application to molecular epidemiology of isolates from South America. Vet Microbiol. 2005;110:221–237. [PubMed: 16171956]
- 71.
- Roring S , Scott A , Brittain D , Walker I , Hewinson G , Neill S , Skuce R . Development of variable-number tandem repeat typing of Mycobacterium bovis: comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J Clin Microbiol. 2002;40:2126–2133. [PMC free article: PMC130792] [PubMed: 12037076]
- 72.
- Savine E , Warren RM , van der Spuy GD , Beyers N , van Helden PD , Locht C , Supply P . Stability of variable-number tandem repeats of mycobacterial interspersed repetitive units from 12 loci in serial isolates of Mycobacterium tuberculosis. J Clin Microbiol. 2002;40:4561–4566. [PMC free article: PMC154626] [PubMed: 12454152]
- 73.
- Sola C , Filliol I , Legrand E , Lesjean S , Locht C , Supply P , Rastogi N . Genotyping of the Mycobacterium tuberculosis complex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics. Infect Genet Evol. 2003;3:125–133. [PubMed: 12809807]
- 74.
- Supply P , Magdalena J , Himpens S , Locht C . Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997;26:991–1003. [PubMed: 9426136]
- 75.
- Supply P , Mazars E , Lesjean S , Vincent V , Gicquel B , Locht C . Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–771. [PubMed: 10844663]
- 76.
- van Embden JD , Cave MD , Crawford JT , Dale JW , Eisenach KD , Gicquel B , Hermans P , Martin C , McAdam R , Shinnick TM . Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31:406–409. [PMC free article: PMC262774] [PubMed: 8381814]
- 77.
- Romano MI , Alito A , Fisanotti JC , Bigi F , Kantor I , Cicuta ME , Cataldi A . Comparison of different genetic markers for molecular epidemiology of bovine tuberculosis. Vet Microbiol. 1996;50:59–71. [PubMed: 8810008]
- 78.
- Kamerbeek J , Schouls L , Kolk A , van Agterveld M , van Soolingen D , Kuijper S , Bunschoten A , Molhuizen H , Shaw R , Goyal M , van Embden J . Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–914. [PMC free article: PMC229700] [PubMed: 9157152]
- 79.
- Caimi K , Romano MI , Alito A , Zumarraga M , Bigi F , Cataldi A . Sequence analysis of the direct repeat region in Mycobacterium bovis. J Clin Microbiol. 2001;39:1067–1072. [PMC free article: PMC87874] [PubMed: 11230428]
- 80.
- Cole ST . Comparative and functional genomics of the Mycobacterium tuberculosis complex. Microbiology. 2002;148:2919–2928. [PubMed: 12368425]
- 81.
- Behr MA , Wilson MA , Gill WP , Salamon H , Schoolnik GK , Rane S , Small PM . Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science. 1999;284:1520–1523. [PubMed: 10348738]
- 82.
- Brosch R , Gordon SV , Marmiesse M , Brodin P , Buchrieser C , Eiglmeier K , Garnier T , Gutierrez C , Hewinson G , Kremer K , Parsons LM , Pym AS , Samper S , van Soolingen D , Cole ST . A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–3689. [PMC free article: PMC122584] [PubMed: 11891304]
- 83.
- Amadio A , Romano MI , Bigi F , Etchechoury I , Kubica T , Niemann S , Cataldi A , Caimi K . Identification and characterization of genomic variations between Mycobacterium bovis and M. tuberculosis H37Rv. J Clin Microbiol. 2005;43:2481–2484. [PMC free article: PMC1153726] [PubMed: 15872289]
- 84.
- Sonnhammer EL , Durbin R . A workbench for large-scale sequence homology analysis. Comput Appl Biosci. 1994;10:301–307. [PubMed: 7922687]
- 85.
- Parsons LM , Brosch R , Cole ST , Somoskovi A , Loder A , Bretzel G , Van Soolingen D , Hale YM , Salfinger M . Rapid and simple approach for identification of Mycobacterium tuberculosis complex isolates by PCR-based genomic deletion analysis. J Clin Microbiol. 2002;40:2339–2345. [PMC free article: PMC120548] [PubMed: 12089245]
- 86.
- Semret M , Alexander DC , Turenne CY , de Haas P , Overduin P , van Soolingen D , Cousins D , Behr MA . Genomic polymorphisms for Mycobacterium avium subsp. paratuberculosis diagnostics. J Clin Microbiol. 2005;43:3704–3712. [PMC free article: PMC1234005] [PubMed: 16081899]
- 87.
- Semret M , Zhai G , Mostowy S , Cleto C , Alexander D , Cangelosi G , Cousins D , Collins DM , van Soolingen D , Behr MA . Extensive genomic polymorphism within Mycobacterium avium. J Bacteriol. 2004;186:6332–6334. [PMC free article: PMC515132] [PubMed: 15342607]
- 88.
- Paustian ML , Kapur V , Bannantine JP . Comparative genomic hybridizations reveal genetic regions within the Mycobacterium avium complex that are divergent from Mycobacterium avium subsp. paratuberculosis isolates. J Bacteriol. 2005;187:2406–2415. [PMC free article: PMC1065244] [PubMed: 15774884]
- 89.
- Alm EJ , Huang KH , Price MN , Koche RP , Keller K , Dubchak IL , Arkin AP . The MicrobesOnline Web site for comparative genomics. Genome Res. 2005;15:1015–1022. [PMC free article: PMC1172046] [PubMed: 15998914]
- Tuberculosis, Leprosy, and Other Mycobacterioses - Bioinformatics in Tropical Di...Tuberculosis, Leprosy, and Other Mycobacterioses - Bioinformatics in Tropical Disease Research
- LOC9629518 [Selaginella moellendorffii]LOC9629518 [Selaginella moellendorffii]Gene ID:9629518Gene
- LOC9629432 [Selaginella moellendorffii]LOC9629432 [Selaginella moellendorffii]Gene ID:9629432Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...