

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Phenylalanyl-tRNA synthetase (PheRS) is shown to be among the most complex of the aminoacyl-tRNA synthetases (aaRSs) with the tetrameric subunit organization of cytoplasmic PheRS markedly conserved during evolution. The structure of Thermus thermophilus PheRS complexed with tRNAPhe has explained the functional necessity for PheRS to be (αβ)2 heterodimeric. The divalent cation detected at the interface of the α- and β-subunits is important for the enzyme activity and αβ heterodimer stability. The heterodimeric structure is not a prerequisite of the phenylalanylation activity: monomeric mitochondrial PheRSs are also active. Structurally, the enzyme belongs to class II, as its catalytic domain is built around an antiparallel β sheet, but functionally it resembles class I, aminoacylating the 2' OH group of the tRNA terminal ribose. The major determinants of tRNAPhe specificity are three nucleotides of the anticodon, directly recognized by the RNP (the subclass IIc specific) domain of the β-subunit. Multiple minor elements scattered over tRNAPhe contribute to the recognition of its general shape, mostly through backbone interactions with the N-terminal coiled-coil domain of the α-subunit, and to mutual steric adaptation of the tRNA and PheRS. The proper positioning of the tRNAPhe acceptor end corresponding to the structure of the productive complex is promoted only in the presence of phenylalanyl-adenylate; the base-specific contacts of the terminal adenosine dictate the conformational rearrangement of the tRNA in aminoacylation reaction. Specific recognition of the phenylalanine substrate is achieved by interactions of the phenyl ring with two neighbouring phenylalanine residues in the protein. No metal ions have been observed within the active site of PheRS complexed with phenylalanyl-adenylate. The peculiarity of the aminoacylation site in this system is governed by stepwise adjustment of all the reactants and intermediates in the active site. The structural fragments of PheRS reveal multiple similarities to those of different DNA/RNA binding proteins, suggesting a puzzling evolution of the enzyme and existence of a range of functions additional to the main activity.
Quaternary Organization
Phenylalanyl-tRNA synthetase (PheRS) is known to be among the most complex and large enzymes of the aaRS family (for a review see refs. 1 and 2). The subunit organization of cytoplasmic PheRS is markedly conserved during the evolution from prokaryotes to eukaryotes and forms tetramers in all known species (Table 1). The structural studies6 provide the basis for revision of the subunit organization formula of PheRS from the previously used term α2β2, which does not reflect the true molecular topology, to (αβ)2, that is a dimer built of two heterodimers. In this notation bacterial PheRS from T. thermophilus is an (αβ)2 enzyme of 350 residues per α- and 785 residues per β-subunit. Neither the α or β monomers, nor the α2 or β2 dimers manifest catalytic activity in tRNA aminoacylation and aminoacyl-adenylate formation.7-9 Both subunits are involved in the binding of three functional substrates as shown by affinity labeling.10-12
Table 1
Primary and quaternary structure of phenylalanyl-tRNA synthetases.
The identification of PheRS as a member of class II is based on the discovery6 of the characteristic active site topology13,14 (seven-stranded β-sheet flanked by four α-helices) and three signature motifs (1, 2 and 3) in the small catalytic α-subunit. The clustering of aaRSs into two classes surprisingly correlates with differences in their enzymatic mechanism: class I aaRSs attach amino acids to the 2'OH of the ribose of the tRNA terminal adenosine, and class II to the 3'OH.15 There is one exception to this rule: for PheRS the site of amino acid attachment is the 2'OH group.16,17 It is notable that bisphenylalanyl-tRNA (tRNA bearing two phenylalanyl residues) formation was observed in the reaction of tRNA aminoacylation catalyzed by T. thermophilus PheRS in vitro, however the second phenylalanyl residue is attached to tRNA approximately 50 times more slowly than the first one.18
The structure of the PheRS-tRNAPhe complex19 offers a clearer view of the functional necessity for PheRS to be (αβ)2 heterodimer (Fig. 1). The CCA-end and the acceptor stem of the tRNAPhe molecule interact with the active site located in the α-subunit and with the N-terminal domain of the β-subunit from the same heterodimer, while the anticodon loop of the tRNAPhe is specifically recognized by the C-terminal domain (B8 or RNP) of the β*-subunit (where * indicates the second heterodimer). The N-terminus of the α-subunit forms a helical arm built up from a two long antiparallel α-helices. The helical arm of the α*-subunit approaches the tRNA from the variable loop side. Thus, one tRNAPhe molecule interacts with all four subunits of the enzyme and the structures of native PheRS and those complexed with tRNAPhe account for the enzyme to be a functional (αβ)2 dimer. At the same time the β-subunit is not directly involved in the catalysis of aminoacylation and apparently its major function is in recognition and binding of tRNAPhe.11,12,19,20 In accordance with existing classification, the availability of the RNP domain involved in tRNA anticodon recognition and subunit composition places bacterial PheRS into subclass 2c.

The (αβ)2 subunit organization is not canonical for mitochondrial PheRSs (mitPheRS). MitPheRSs from yeast21 and human4 can effectively charge tRNAPhe as monomers. Multiple sequence alignment (Fig. 2) indicates that the N-terminal fragments of mitPheRSs bear close similarity to the catalytic α-subunit of the cytoplasmic enzymes, while C-terminal regions display sequence homology to the C-terminus (B8 domain) of the β-subunits. Monomeric enzymes in the aminoacylation reaction demonstrate turnover rates comparable to the heterodimeric aaRSs.21 This in turn suggests that availability of the “α-subunit” and “RNP-domain” at the C-terminus is the minimal structural set to construct an enzyme with phenylalanylation activity. Furthermore, the existence of insertions (from 35 to 60-70 residues) into the amino acid sequence of mitochondrial enzymes immediately after motif 2, appear to be a general characteristic of mitPheRSs. These insertions form structural domains that may participate in tRNA binding.22

Primary structure analyses of PheRSs reveal that the lengths of the polypeptide chains of eukaryotic enzymes are significantly higher than those of the homologous prokaryotic counterparts (see Fig. 2). The elongation of the chains mostly occurs at the Nor C-terminal extremities of the subunits, rather than by insertion into the catalytic domain region. For example the human and yeast catalytic α-subunits obey this rule of thumb, whereas the β-subunits turn out to be ˜200 residues shorter than their respective prokaryotic analogs. Multiple sequence alignment, greatly facilitated for the β-subunit by taking advantage of atomic models of PheRS6 and its complexes with functional ligands,19,23 shows the absence of the residues immediately after motif 3 in human and yeast β-subunits. Thus the RNP (or B8 in the nomenclature of the T. thermophilus PheRS structure) domain that performs anticodon-binding functions in bacteria is missing from eukaryotic PheRSs. The direct consequence of this observation is that binding and recognition modes of tRNAPhe will differ in prokaryotes and eukaryotes. Thus, cytoplasmic PheRSs from prokaryotes and eukaryotes differ substantially in the structure of the anticodon binding domains and should be partitioned in different subclasses, with respect to the current classification of aaRSs.
Three-Dimensional Structure
The (αβ)2 molecule of T. thermophilus PheRS has the shape of a leatherback turtle with large flippers formed by the N-terminal parts of the β-subunits (see Fig. 1). The span of the flippers is about 65 Å.6 The crystal structure of native PheRS highlighted 10 structural domains which clustered into four structural modules: catalytic—CAM (A1-A2, α-subunit), N-terminal—NTM (B1-B5, β-subunit), “catalytic-like”—CLM (B6-B7, β-subunit) and C-terminal—CTM (B8, β-subunit). In general, they do not follow each other successively along the polypeptide chain. Insertions in the subunits of A1, B1, B3 and B6 create the individual domains A2, B2, B4 and B7, respectively (Fig. 3). The three modules of β-subunits, which are covalently connected by the extended polypeptide segments, are separated in space by large cavities. Despite the fact that no signature motifs 1, 2 and 3 have been detected in the β-subunit, it turns out that B6 together with B7 possesses an overall fold very similar to that of CAM, and the catalytic domains of class II aaRS. Motifs 2 and 3 of β-subunit do not contain the invariant amino acids that are directly involved in the aminoacylation process that was the rationale behind calling the whole module “catalytic-like”. Thus, T. thermophilus PheRS possesses four characteristic, class II antiparallel folds of which only two are catalytically active.24 A remarkable feature of the (αβ)2 subunit communication is that heterodimerization mode between CAM and CLM (i.e., between α and β) is essentially the same as in the known homodimers of class II aaRSs; that is, the modules are related by a pseudo two-fold axis perpendicular to motif 1 interface helices. At the same time the dimerization mode produced by the true two-fold axis (between the αβ heterodimers) differs from other aaRSs in class II, as the helices are nearly parallel to this axis. The core of the (αβ)2 interface looks quasi-tetrahedral and comprises a four-helix bundle with an interhelical separation of 14 Å. It is of interest that positively and negatively charged residues exposed to this interface by all four subunits fully compensate each other and total charge within the area equals zero.

An intriguing question is related to the understanding of the structural and functional role of CLM. This module is likely to carry out dual functions, shared between domains B6 and B7. Firstly, both participate in nucleation of the αβ interface, generating a vast area of hydrophobic contacts with the convex side of CAM β-sheet. Secondly, insertion domain B7 topologically resembles insertion domain A2 with minor exceptions. As it appears, such a similarity is related to the additional structural stability that B7 and A2 contribute to heterodimer formation: they create one, common to both subunits, four-stranded antiparallel β-sheet. Together with the symmetry mate (molecular two-fold axis) two β-sheets protect the interface region and the cavities of the active sites.
A variety of familiar “nucleic acid binding” domains are found in the β-subunit: B1 and B5 domains form a characteristic dimer with architecture similar to that of the catabolite gene activator domain responsible for DNA binding by means of the helix-turn-helix motif;25 B2 or “EMAP II-like” β-barrel domain with the Greek-key topology (staphylococcal nuclease type) similar to the anticodon binding domain of AspRS and LysRS, class IIb aaRSs,22 but almost free from the contacts with tRNAPhe; B8 or “RNP” domain performs anticodon recognition of tRNAPhe in prokaryotic Phe-system (see below). B4 or “SH3-like” domain (associated with signal transduction in a number of eukaryotic proteins) is the insertion domain between the two βαβ motifs of a larger structural fragment B3.26 The topology of CTM or B8 domain is very similar to that of the RNA-binding domain (RBD) of the U1A spliceosomal protein.27 Moreover, the B8 sequence contains two short motifs highly conserved in RNA-binding proteins,27,28 RNP1 and RNP2.
The N-terminal fragment of the α-subunit is disordered and was not detected in the electron density maps of the native enzyme6 or complexed with phenylalanine23 and phenylalanyl-adenylate.29 Upon formation of PheRS-tRNAPhe complex, both the enzyme and the substrate undergo conformational changes that conceivably allow better complementarity of interacting surfaces.19 The most pronounced difference between the native and tRNA-complexed states is the ordering of the N-terminal region of the α-subunit, which appeared to comprise a coiled-coil structure (helical arm). The helices form the 11th structural domain of the αβ-heterodimer and stretch out into the solvent by 65 Å (see Fig. 1).
A tightly bound divalent cation was first noted for a magnesium ion in an apoenzyme structure.6 This ion is coordinated by six electrostatic interactions with side chains of Aspβ-452, Aspβ-458, Gluβ-461, Gluβ-462 and Asnβ-163, emerging from the β-subunit, and Gluα-262 from the α-subunit. The latter residue belongs to the amino acid binding loop of the protein active site. The position of the metal ion at the α/β-subunit interface, near the active site, obviously emphasizes its crucial role in PheRS structure and function: it is required for strengthening the heterodimer association, coordinating intersubunit electrostatic interactions, and anchors the amino acid binding loop 255-263 containing the functionally important Pheα-258 and Pheα-260.
Interaction of PheRS with Its Cognate tRNAPhe: Binding and Recognition
One of the remarkable features revealed by the structure of the PheRS-tRNAPhe complex is that specific recognition of the tRNAPhe anticodon (GAA) is achieved by its interaction with domain B8 (Fig. 4). In contrast to conformation of the anticodon loops of the various aaRSs-bound tRNA molecules, T. thermophilus tRNAPhe keeps the conformation of the anticodon loop relatively similar to that of free yeast tRNAPhe. The main reason for this difference is that other aaRSs approach the anticodon loop from the major-groove side, and the anticodon bases have to protrude out to form base-specific contacts, whereas in PheRS such contacts exist on the minor groove side of the anticodon loop, maintaining its almost undistorted conformation. It is intriguing that the structure of the 30S ribosomal subunit complexed with mRNA and anticodon stem-loop (ASL) of tRNAPhe reveals conformation of the ASL fragment very similar to that observed in the T. thermophilus PheRS-tRNAPhe complex.30

Each tRNA binds across all four subunits of the enzyme yielding ˜2700 Å2 to the contact area of the tRNA substrate. Extensive regions of the tRNA interactions with the enzyme suggest the involvement of large numbers of nucleotides in this process. The tRNAPhe nucleotides required for recognition by PheRS of evolutionary diverged species have been analyzed by in vitro studies using mutant transcripts.31-38 They are scattered over different tRNAPhe regions (Table 2). The anticodon nucleotides are the major determinants of tRNAPhe specificity in all systems studied including plants. Among them, G34 provides maximum contribution to the recognition, with A35 and A36 playing a smaller role. As it follows from X-ray data,19 recognition of G34 is accomplished by means of stacking interaction between G34 and Tyrβ*-731, as well as by two base-specific contacts (see Fig. 4). The first is a hydrogen bond between O6 of G34 and Serβ*-742, which belongs to the group of amino acids identified as an RNP1 motif.28 The second base-specific interaction is between N2 of G34 and Aspβ*-729. In addition, the hydrogen bond between N7 of G34 and Argβ*-780 may favor purine bases in this position. All four aforementioned amino-acid residues are strictly conserved in the tetrameric PheRSs. A van der Waals contact of Alaβ*-698 with base of A35 has an important role because any longer side chain (instead of the conserved Ala) would interfere with the anticodon base. Recognition of A36 can be achieved through van der Waals contact between the Cα atom of Leuβ*-697 and C2 atom of the adenine. A hydrogen bond between the sugar O2' and the Oδ atom of Aspβ*-696 probably stabilizes the conformation of tRNA in this area. Formally, residues Aspβ*-696, Leuβ*-697 and Alaβ*-698 interacting with bases of A35 and A36 belong to the characteristic RNP2 motif. It is interesting that the binding modes of RNA loops with the spliceosomal protein U1A and with B8 of PheRS appear to be different.19 The structure of the PheRS-tRNAPhe complex reveals nucleotide G34 to make the largest number of contacts, confirming its vital importance for recognition. The role of domain B8 in tRNAPhe binding was additionally supported by mutation experiments: the removal of B8 resulted in dramatic reduction of the association constant of the PheRS-tRNAPhe complex.20
Table 2
Recognition elements in tRNAPhe.
Nucleotide 73 may also be assigned to the common recognition nucleotides but its contribution to aminoacylation efficiency varies greatly: the strongest effects of mutation are seen in human while in other species such as T. thermophilus the strength is minor or moderate (see Table 2). The conformation of the acceptor (CCA) end of the complexed T. thermophilus tRNAPhe is determined by base-specific interaction of the 3'-terminal adenosine with strictly conserved residues Serα-180, Gluα-220 and Pheα-258 and a network of contacts between the protein and the tRNA sugar-phosphate backbone.19 Thus, in addition to the anticodon, the 3' terminal nucleotide makes base-specific interactions in the PheRS-tRNAPhe complex. In the partially unwound conformation of the tRNAPhe acceptor backbone, bases of C72 and A73 are not strictly stacked on each other, as occurs in free yeast tRNAPhe. The base of A73 points away from the CAM and the B1 domain, not participating in the contacts with the protein and in agreement with its minor contribution to recognition.32 Binding of two adjacent nucleotides of the CCA end by the two PheRS subunits at functional conditions is evidenced by affinity labeling experiments.39,40 This mode of acceptor end binding seems universal for prokaryotic and eukaryotic heterodimeric PheRSs.40a
The position of the terminal adenosine in PheRS-tRNAPhe complex19 partially interferes with the position of substrate amino acid as it follows from the comparison with the complexes of PheRS with phenylalanine, aminoacyl-adenylate or its synthetic analog.23,29 It is common for aaRSs, with a few exceptions (GlnRS, GluRS and ArgRS), that the formation of aminoacyl-adenylate, the first intermediate of the aminoacylation reaction, does not require the presence of tRNA.41 Moreover, adenylate formation is a prerequisite for effective tRNA binding, as seen for some other class II synthetases—SerRS42 and AspRS.43 The proper positioning of the tRNAPhe terminal nucleotide corresponding to the structure of the productive complex is promoted only in the presence of phenylalanyl-adenylate (see below) as evidenced also from biochemical studies.40
The role of other nucleotides from the anticodon arm and acceptor stem has not been extensively studied (see Table 2). Substitution of base pairs 27-43 and 28-42 had only minor effects on aminoacylation in vitro with two prokaryotic enzymes but they are included in the identity set for E. coli tRNAPhe.44 It seems more likely that these nonconserved tRNAPhes45 nucleotides serve as anti-determinants to hinder interaction of tRNAPhe with noncognate synthetases. The base pairs A31-U(pseudo-U)39 and G30-C40 which are strictly conserved among tRNAPhes contribute to the catalytic efficiency of tRNAPhe aminoacylation in all species studied with the strength of determinants being phylum-dependent. Phylogenetically conserved in prokaryotic and eukaryotic tRNAPhe, nucleotide 20 is also a common recognition element. The strongest effect of its mutation is seen in human while in other species the strength is minor or moderate. Some of the differences between PheRSs stem from nonidentical experimental conditions. The relative strength of recognition elements depends on Mg2+ concentration.32,46 Losses of the aminoacylation efficiency determined for T. thermophilus PheRS at the optimal ion concentration (9 mM) are stronger than at the higher concentration (15 mM).32 The recognition set for some species was analyzed in a heterologous tRNAPhe background; however, quantitative differences are seen when the same mutation is compared in the two backgrounds. It is worth noting that observed diversity in mutational effects may arise also from the differences in the tRNA binding and recognition modes for prokaryotic and eukaryotic PheRSs.
Side by side with the subtle differences in binding and recognition mode of cognate tRNA by aaRSs from the same subclass, there are strict and possibly ancient characteristics of tRNA-aaRS complex formation, common for different subclasses. A typical illustration of this point is the presence of the long helical arm in SerRS and PheRS forming a vast area of nonspecific contacts with cognate tRNA molecules within subclasses IIa and IIc, respectively. Judging from the structure of the Ser-tRNASer complex47 and keeping in mind that the anticodon is not recognized by SerRS, the coiled-coil domain is a major tRNA-recognition element. The existence of the long variable loop in tRNASer was believed to be a prerequisite for the helical arm to perform its function. The Phe-system shows that, along with specific recognition of the anticodon, the coiled-coil domain is also characteristic of the PheRS structure. This domain plays an important role in PheRS even in the absence of the long variable loop in tRNAPhe. Analysis of the contacts between the coiled-coil and tRNA in both systems reveals that the general patterns of interaction have much in common. The two separated areas of contacts are the middle region of the helices, which interacts with a row of paired bases running in a perpendicular direction (along the variable loop in SerRS and the anticodon stem and short variable loop in PheRS), and the end of the helical arm, where a saddle is formed for base pair 19-56. These findings are indicative of the considerable importance of the coiled-coil domain for recognition processes in the Phe-system: a) the role of the coiled-coil domain consists of recognizing a certain structural pattern characteristic for tRNA (a stem-like structure and an exposed base pair about 15 Å away); b) nonspecific coiled-coil/tRNAPhe interactions localized at widely spaced regions of the tRNA and involving tertiary nucleotides are important for recognition of the general shape of the tRNAPhe. This correlates well with the fact that precise conformation of the tRNAPhe is a critical requirement for efficient aminoacylation.31,32,34,36,37,48 The strongest effects of mutations in the tertiary nucleotides were observed when tRNAPhe folding was disrupted.
Phosphorothioate footprinting49 and s4U-induced crosslinking12 experiments basically agree with the X-ray data. However, the biochemical studies have revealed dynamic interactions of PheRS with tRNA, supplementing the crystal structure. The regions of cleavage enhancement are located at the sites where bending of tRNA is likely to occur. Most of the protected nucleotides are found in those fragments of tRNA that are in close contact with the enzyme. Nucleotides of major crosslinks are located in regions where conformational changes of the bound tRNA were supposed to occur or their bases are unstacked. Both the flexibility of the protein domains and deformability of the tRNA structure allows favourable contacts and facilitate the crosslinking reactions. Notably, nucleotides 8, 20, 39 and 45, involved in the crosslinks and in the tRNAPhe recognition set (see Table 2) show no close contacts with the enzyme in the crystal structure. Conformational flexibility suggested from the crosslinking data allow one to propose a mechanism for their indirect recognition: the structure of tRNAPhe at the flexible positions can be deformed by PheRS in the process of mutual adaptation. Such a mechanism has been described recently for the recognition of the G3-U70 wobble pair in tRNAAla.50 The role of these determinants of specificity is to adjust the local tRNA conformation or its stability. Interestingly, the mutational effect of nucleotide 20 on aminoacylation by yeast PheRS is mediated by the rate of pyrophosphate dissociation.51 In the presence of inorganic pyrophosphatase the effect of this mutation was significantly reduced, while aminoacylation of tRNAs substituted at the anticodon or the discriminator base was unchanged. Along with the mutant at position 20, pyrophosphatase relieved the effects of tRNAPhe structural variants, thereby suggesting that G20 exerts some action on the conformation of tRNA in the process of recognition by eukaryotic PheRS.
Selection of the Phenylalanine Substrate
A distinctive feature of the topology of the PheRS active site is the presence of a deep phenylalanine-binding pocket.23,29 The bottom surface of the pocket is parallel to the phenyl ring of the substrate and is covered by the invariant glycines, thus providing the space required for the Phe and ATP moieties. One of the walls and the top surface of the pocket are covered by hydrophobic residues. Another wall of the pocket is built up entirely of residues, which may participate in electrostatic interactions and in hydrogen bonding. Such an anisotropy in the distribution of hydrophobic and hydrophilic residues within the pocket unambiguously orients the amino and carbonyl groups of the amino acid moiety of aminoacyl-adenylate (Phe-AMP). Remarkably, only minor conformational changes have been observed in the active site of the PheRS complexed with phenylalanine as compared with the structure of native enzyme. The r.m.s. deviation between PheRS-Phe complex and PheRS is 1.08 Å for all nonhydrogen atoms. Totally, the substrate Phe forms 52 van der Waals contacts with the PheRS. The conformation and position of phenylalanine in this complex is very similar to these observed in the complexes with Phe-AMP and its synthetic analogue (PheOH-AMP).
The specific recognition of phenylalanine is achieved by interactions, wherein the substrate phenyl ring and two neighboring phenyl rings of Pheα-258 and Pheα-260 make a “network” of interactions and each aromatic pair is arranged with “edge-to-face” contacts (Fig. 5). The appearance of the Phe substrate in such an environment makes the attractive potential energy of interaction twice as large as a single “edge-to-face” aromatic-aromatic interaction52 and thus makes the Phe-PheRS recognition highly specific and favorable energetically. It is interesting to note that the active site of PheRS itself contains a stretched “network” of aromatic-aromatic interactions, that successively include Pheα-258 => Pheα-260 => Trpα-149 => Pheα-134 and Trpα-153.

Figure 5
Stereo view of three aromatic rings that form triple “edge-to-face” aromatic interactions at the PheRS active site: the phenyl ring of phenylalanyl-adenylate interacts with Pheα-258 and Pheα-260.
The correct orientation of the Phe substrate, in agreement with an in-line mechanism for phenylalanine activation, is achieved by anchoring of its α-amino and COO groups. In the binary PheRS-Phe complex, the amino group of the substrate forms direct hydrogen bonds with Glnα-218 and Gluα-220. In the PheRS-Phe-AMP complex this group interacts with Glnα-218 and Gluα-220 via well-ordered water molecule S9 and forms additional hydrogen bonds with Serα-180 and Hisα-178 (Fig. 6).
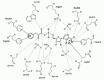
Figure 6
Schematic representation of principal interactions between PheRS and phenylalanyl-adenylate at the active site. Hydrogen bonds are shown by dashed lines and the van der Waals interactions by solid arrows.
Among 20 amino acids there are only three candidates that might be considered specific for binding by PheRS, since the recognition process is essentially driven by a “network” of aromatic-aromatic interactions (see above). In fact, the scheme of van der Waals contacts in the phenylalanine-binding pocket shows why tryptophan cannot be correctly positioned at this site to react with ATP. A predetermined net of interactions with the α-amino group and carbonyl oxygen of the amino acid gives no way to insert the bulky side chain of Trp without serious rearrangement of the active site. Thus, the only amino acid in question is tyrosine, which differs from phenylalanine by an additional OH group attached to the phenyl ring. However, both steric hindrance and the hydrophobic nature of the back wall of the amino acid binding pocket (Vala-261 and Alaa-314) would not favor Tyr binding in this place. Thus, hydrophobic interactions and van der Waals contacts should discriminate between the three amino acids, achieving highly specific recognition of phenylalanine.
The importance of Alaα-294 in the E. coli PheRS (Alaα-314 in the T. thermophilus PheRS) as a determinant of amino acid specificity was demonstrated by biochemical studies.53 Replacement of Ala-294 by Gly or Ser, thereby increasing or decreasing the size of the binding pocket, respectively, reduced affinity for phenylalanine. The Ala294Gly mutant shows a relaxed specificity toward a number of para-halogenated analogs of phenylalanine, the apparent dissociation constant KM increasing in direct relation to an increase of the van der Waals radius of the para group, thus confirming the role of position 294 in determining amino acid binding pocket size. Tyrosine was activated by mutant Gly294PheRS, but not by either wild-type or Ser294PheRS. The resulting enzyme-Tyr-adenylate complex was rapidly hydrolyzed, indicating the existence of a proofreading mechanism. This proofreading is specific for tyrosine since the complex with activated p-Cl-Phe was of comparable stability to that of phenylalanine. Gly294PheRS can attach p-Cl-Phe and p-Br-Phe to tRNAPhe and incorporate them into full-length luciferase in vitro.54 In vivo incorporation of p-Cl-Phe into cellular protein by the mutant PheRS was also shown.53,54 The existence of a proofreading mechanism in E. coli PheRS53 was supported by comparison with S. cerevisiae PheRS for which hydrolysis of the noncognate complexes has been reported55 and the fact that the wild-type yeast PheRS contains a glycine at the position analogous to the referenced Ala-294 in E. coli (and other bacterial enzymes).56 The presence of Gly may be the reason for the low specificity of yeast PheRS. A given specificity is similar to those observed for class I ValRS, for which the lowest specificity was observed.56 For discrimination between phenylalanine and 18 other naturally occurring noncognate amino acids by the yeast PheRS, discrimination factors of 190-6300 were determined and pretransfer proofreading was shown to be the main correction step. Whether proofreading is carried out at a site distinct from the amino-acid-binding site of PheRS remains to be determined.
However, based on analysis of the PheRS active site we are inclined to believe that Tyr-AMP formation by PheRS is a characteristic property of the engineered Gly294PheRS only, but not of the wild-type PheRS. Comparison of the sequences of S. cerevisiae and human enzymes (see Fig. 2) reveals Ala in human PheRS analogously to bacterial enzymes. Bearing in mind the close similarity of the active sites topology of these two eukaryotic aaRSs, we may conclude, that substitution of Ala for Gly is not the only reason triggering the misactivation of tyrosine by both prokaryotic and eukaryotic PheRSs.
Structural Aspects of Phenylalanyl-Adenylate Formation
Crystal structures of T. thermophilus PheRS complexed with aminoalkyl-adenylate (PheOH-AMP)23 and with true intermediate (Phe-AMP)29 elucidated the class- and system-specific net of interactions of the amino acid, adenine and sugar moieties of the Phe-AMP and its analogue. The structures show that the peculiarity of the aminoacylation site in the Phe-system is governed by stepwise adjustment of all the reactants and intermediates in the active site. The analysis of a few class II aaRSs complexed with ATP, aminoacyl-adenylate or aminoacyl-adenylate analogues, revealed the presence of magnesium ions in the active sites. The availability of the Mg2+ or other divalent cations presumably should stabilize the bent conformation of ATP and was supposed to be necessary for transition state formation in the majority of class II systems.43,57-59
Attempts at PheRS-Phe-AMP complex formation, by soaking PheRS crystals grown in the presence of saturated ammonium sulfate in solution containing Phe, ATP, and Mg2+ have not been successful.23 When Mg2+ ions in the soaking solution have been replaced by the Mn2+ ions, the Fourier difference map displayed extra positive density that could be referred to the entire Phe-AMP molecule in the active site,29 thus showing PheRS in the crystal to be active for the formation of Phe-AMP in the presence of manganese.
Conformational changes of PheRS observed upon its complex formation with Phe-AMP or PheOH-AMP have the effect of a general shift towards the area where the Phe-AMP molecule is bound. PheRS complexed with Phe-AMP reveals less dramatic conformational changes in motif 2 relative to the apoenzyme, compared with T. thermophilus SerRS57 and yeast AspRS.43 In fact, a whole region of electron density adjacent to the Phe-AMP, including the side chain of Argα-204 is well ordered in both the native and complex structures. The fully extended conformation of Argα-204 makes possible simultaneous contacts of its side chain with the phosphate group and carbonyl oxygen of Phe-AMP (see Fig. 6). When compared to native PheRS the side chain has been pulled out towards the adenylate from its original conformation. It is of interest that the lack of the carbonyl group in the synthetic analogue of Phe-AMP disrupts the balance of the hydrogen bonds, and Trpa-149 appears to be disordered.23 Superposition of these two complexes (PheRS-Phe-AMP and PheRS-PheOH-AMP) shows the largest conformational shift of ˜1.6 Å for the polypeptide segment 138-151 comprising Trpα-149. In the PheRS-Phe-AMP structure this segment is displaced towards the adenylate, thus providing hydrogen bonding between nitrogen of the indole ring and the carbonyl oxygen.
The binding mode of the adenosine portion of phenylalanyl-adenylate has much in common with those observed for other class II aaRSs complexes. The adenine is located between the phenyl ring of Pheα-216 and the aliphatic portion of the Argα-321 side chain (see Fig. 6). These residues are strictly conserved within class II aaRSs and belong to motif 2 and 3, respectively. Upon the formation of the PheRS-Phe-AMP complex the side chain of Argα-321 undergoes large conformational changes. It is switched to interact with the adenine ring and the observed movement is larger than 4.5 Å for the CZ atom. However, the reoriented guanidinium group is directed outward from the adenine ring. In the PheRS-PheOH-AMP complex, the guanidinium group of Argα-321 is positioned in the vicinity of the adenine ring being almost coplanar to it. The adenine moiety in the PheRS-Phe-AMP complex forms hydrogen bonds between N6 and N1 of the adenine ring and the side chain of Gluα-206 as well as with the main chain atoms of Gluα-213. It is notable that the position occupied by loop 204-213 and the conformation of the Gluα-213 side chain provide the Phe-system with a strengthened network of hydrogen bonding between N6 and two oxygen atoms of Gluα-206, as compared with other class II aaRSs. The ribose moiety is held in position by Glnα-218 and a water molecule bonded to the 2'OH and the amide group of Argα-321. Another water molecule bridges the 3'OH of the ribose and the side chain of the invariant Gluα-279. Here we emphasize that there is no direct interaction between Gluα-279 and the 3'OH group: the carboxylic group of the Gluα-279 side chain swings aside 90° from the ribose moiety and participates in tetracoordination of water molecule S195 together with Argα-252, Glnα-254 and Glnα-266. Among class II aaRSs, PheRS is not exceptional in this respect: in E. coli HisRS the Glu residue in this position is replaced by Ala, which lacks the ability for hydrogen bonding and thus the 3'OH of histidyl-adenylate is free of direct interactions with the enzyme.60 A different situation emerges for AspRS, SerRS and LysRS, where highly conserved Glu residues interact directly with 3'OH of the ribose. Moreover, a repulsive electronegative environment created by topologically equivalent triads of negatively charged residues in each of the above-mentioned synthetases (for AspRS: Asp-475, Asp-282, and Glu-482; for SerRS: Asp-332, Glu-345, and Glu-334; for LysRS: Glu-414, Glu-421, and Glu-380) prevents reorientation of the referred glutamic acid residues away from the ribose in the active site to the protein surface as happens in PheRS. Thus, conformational constraints of the adenylate ribose appear to be dictated exclusively by a local, system specific net of hydrogen bonding, and are not necessarily retained in class II or in their associated subclasses.
The anchoring of the amino group of the Phe moiety is achieved by its interactions with Serα-180, Hisα-178 and water-mediated interactions through the S9 molecule with Gluα-220, Glnα-218 and Thrα-179 (see Fig. 6). It is interesting to note the concerted conformational switch of Gluα-220 and Glnα-218, compared to the native structure. Under this realignment, the S9 water molecule occupies a highly coordinated position in the vicinity of Phe-AMP and new position of Glnα-218 favors its hydrogen bonding with the ribose in addition to the carbonyl oxygen of the amino acid moiety. Multiple sequence alignment between PheRSs from different sources (see Fig. 2) shows that residues Glnα-218 and Gluα-220 are strictly conserved for all referenced sequences. For two residues, Thrα-179 and Serα-180, conservation should be assigned to the hydroxyl group, which occurs in these residues. Amino acids Ser and Thr substitute for one another at positions 179 and 180. Hisα-178 in some cases is replaced by Gln, which is uncharged but has a polar amide group with extensive hydrogen-bonding capacity.
A Scenario of the Phenylalanylation Process
Analysis of three-dimensional structures of PheRS complexed with cognate tRNA, amino acid and aminoacyl-adenylate allows us to propose structural guidelines for phenylalanine activation and conformational rearrangement of the 3'-terminal portion of cognate tRNAPhe in the presence of adenylate. The location of the Phe moiety observed in the PheRS-Phe-AMP complex,29 and in the PheRS structure with phenylalanine alone23 shows that the free amino acid binds and holds a position close to that in Phe-AMP. Furthermore, the class II conserved mode of ATP binding, and the hydrogen-bonded distance of Argα-204 from the α-phosphate of ATP and the carbonyl oxygen of phenylalanine are good evidence that the reactants are located in the active site of PheRS in accordance with an in-line mechanism for the activation. In the mechanism of amino acid activation described for class II aaRSs,41 the geometry and charge of a pentacovalent transition state should be stabilized by positively charged amino acids and a divalent cation (Mg2+ or Mn2+). However, in the His-system,60 the divalent cation is replaced by HisRS-specific Arg-259, which directly interacts with the adenylate phosphate on the side opposite to the guanidinium group of the class II conserved Arg-113. These two Arg residues increase the electrophilicity of the α-phosphate group of ATP and lead to nucleophilic attack by the carboxylic group of the amino acid on the α-phosphate of ATP, and then expel pyrophosphate.
In the Phe-system (see Fig. 6), the negative charge of the carbonyl oxygen in the transition state is stabilized by three positively charged polar residues located within hydrogen-bonding distance: Argα-204 (invariant for all class II aaRSs), Hisα-178 (substituted by Gln in some PheRSs), Glnα-218 (strictly conserved in all PheRSs), as well as by Trpα-149 (substituted by His or Gln in other PheRSs). Currently available experimental evidence suggests two possibilities, (a) the formation of the stable intermediate Phe-AMP in the Phe-system may occur without divalent cation(s) or (b) these ions are released after Phe-AMP formation. The detection of Mn2+ ion in the interface area ensures that the absence of the anomalous signal in the active site is not accidental but is an intrinsic characteristic of the Phe-system. We propose that role of Mg2+ (acting as an electrophilic catalyst) in the system is shared by two partners: the polar NH group of Trpα-149 and the water molecule S290. Trpα-149 is not conserved in the other PheRS sequences (see Fig. 2), but His and Gln at this position in other PheRSs (both in prokaryotes and eukaryotes) are capable of participating in such an interaction. The circumstantial evidence of this contact is the lack of electron density on Trpα-149 in the absence of the carbonyl group of aminoacyl-adenylate. On the other hand, it is unlikely that the electron density identified as the water molecule S290 occupies the position of a “principal magnesium ion”57 as there are no obvious candidates among the amino acids with polar side chains, capable of participating in octahedral coordination of a Mg2+ ion, as happens in SerRS, AspRS, GlyRS and AsnRS. In fact, the carbonyl oxygen of Glyα-282 (in place of Ser in SerRS), Gluα-279 and Glnα-266 (involved in solvent-mediated interactions with Argα-252; see above) occur in PheRS at positions suitable for electrostatic interaction in other class II aaRSs with a “principal ion” (bridging the α- and β-phosphates of ATP) Mg2+ (or Mn2+). We hypothesize that if ATP coupled with the metal ions triggers conformational changes disrupting the cluster of polar residues (Argα-252, Glnα-254, Glnα-266 and Gluα-279) linked to the water molecule S195, then the side chains of Gluα-279 and Glnα-266, being reoriented towards the substrate, will come into play in the coordination of the divalent ion. Additional structural information on the PheRS-ATP+Mg2+(or Mn2+) complex would be required to shed light on this issue.
The necessity for the tRNAPhe terminal adenosine rearrangement in the presence of adenylate follows both from structural (see above) and biochemical studies. The data on affinity crosslinking of T. thermophilus PheRS with reactive tRNAPhe derivatives give a clear indication of the acceptor end rearrangement in the presence of other substrates: the interaction of the enzyme with Phe and ATP and synthesis of adenylate influence the orientation of the tRNA 3' terminus.40,40b The kinetic data on substrate activity of modified tRNAs40,61 suggest a functional importance of base-specific contacts of the terminal adenosine for the productive interaction of tRNAPhe with the PheRS: their disruption is reflected in a reduction in the rate of catalytic transformation.
The position of the terminal adenosine in the active site, as shown in the PheRS-tRNAPhe complex19 is stabilized by three hydrogen bonds. N6 of A76 makes contacts with Serα-180 and Gluα-220; and the indole ring of Trpα-149 (which is approximately perpendicular to the base of A76) makes a hydrogen bond between Nε1 and N7 of the adenosine. As indicated above, in the PheRS-Phe-AMP complex, the side chain of Gluα-220 is swung away from its position in the PheRS-tRNA complex, making only indirect contact with the amino group of the Phe moiety. Trpα-149 is shifted out from its position towards the hydrogen bonding position with the carbonyl oxygen of the intermediate. Both phenyl rings of Pheα-258 and Pheα-260 also change their orientation. In this new environment only Serα-180 keeps its orientation shown in the PheRS-tRNAPhe complex.
The proposed mode of simultaneous positioning of Phe-AMP and the 3' end of tRNAPhe in the active site of the protein is shown schematically (Fig. 7). Compared with its location in the PheRS-tRNA complex, A76 may occupy a new position approaching adenylate from the 2'OH group of its ribose moiety. This conformational rearrangement may be considered as an “aromatic-aromatic exchange”. Penetrating to the amino acid binding pocket in the PheRS-tRNAPhe complex, A76 forms an aromatic “network” with Pheα-258 and Phea-260, whereas in the presence of Phe-AMP it participates in formation of a “network” with Pheα-258 and Trpα-149. Thus, phenylalanine from Phe-AMP and its neighboring Pheα-258 and Pheα-260 create one aromatic triad, while the adenine ring of A76 and Pheα-258 together with Trpα-149 create another triad. These two adjacent aromatic-aromatic triads in turn may create a six-member “network” that may stabilize the ternary PheRS-Phe-AMP-tRNAPhe complex within the active site and orient the reactants in a proper way, ready for the aminoacylation to proceed.

Noncanonical Functions
Along with the key activity of aminoacylation of specific tRNAs, aaRSs are known to perform various other biological functions (for a review see refs. 62, 63). The molecular basis of these alternative functions of aaRSs lies in their modular composition64 and T. thermophilus PheRS is a particularly significant example. This enzyme consists of 22 structural domains and only four of them (A1 and A2 from the α-subunit and their symmetry mates) are directly involved in the catalytic reaction. The noncatalytic β-subunit comprising a “catalytic-like” module (B6 and B7), OB-, RNP-, SH3-, and DNA-binding-like domains suggests a puzzling evolution of the enzyme that resulted in functions in addition to the main activity. A striking similarity between the catalytic domain structures of biotin synthetase/repressor protein (BirA) and class II aaRSs have been observed65 and T. thermophilus PheRS additionally reveals significant similarities outside the catalytic domains. Apart from the close similarity of the CLM (B6 and B7) with the catalytic domain of BirA, the β-subunit also shares two noncatalytic domains with BirA: a DNA-binding-like domain (B5) containing a helix-turn-helix (HTH) motif and a Src-homology 3 (SH3-like domain). This similarity provides an interesting example in which all domains of one multidomain protein (BirA) appear to be constituents of another multidomain protein (PheRS) and supports the concept of a common ancestor for the two different synthetase families.26 Structural relationships between the DNA-binding domain of BirA and the analogous B5 domain of the T. thermophilus PheRS β-subunit made it possible to assume that PheRS participates in cellular processes via DNA binding.
Based on the structural predictions, the DNA-binding properties of T. thermophilus PheRS were demonstrated, reaching the conclusion that the recognition of double-stranded genomic DNA is modulated by its sequence.66 The subsequent detailed investigation revealed that PheRS recognizes a certain DNA structural motif rather than a particular consensus sequence.67 The “wings” of the HTH motifs belonging to B5 and B5* domains, symmetry related by the intersubunit 2-fold axis, are the relevant structural DNA-binding fragments. It was suggested that two parts of the same DNA molecule are bound simultaneously in the saddle-like cavity formed by the “winged” B5 domains of the heterodimer. These fragments should be aligned in parallel, while the DNA joining them forms a loop structure. Interaction of T. thermophilus PheRS with single-stranded DNA was also demonstrated and this binding was not competitive to the interaction of PheRS with tRNAPhe.68 While the function of PheRS related to DNA binding is still unknown, localization of PheRS in the nucleus69 may indicate involvement of the protein in DNA replication/transcription processes. In view of the discovery of transcription-coupled protein biosynthesis in the nucleus,70 PheRS could provide its specific functions here, rather than contributing to other systems.
B2 is another interesting domain of the T. thermophilus PheRS. It is similar to the anticodon-binding domain of AspRS and LysRS and, on the other hand, has 36% identity to one of six domains of the p43 component of the mammalian multisynthetase complex.71 A dual function of p43 has been suggested. In normal cells, p43 may be required for tRNA channeling and, after proteolytic processing in tumor cells, it would acquire inflammatory properties presumably related to apoptosis. The similarity between a fragment of p43 and B2 domains of the large PheRS subunits seems very interesting in the quest to discover noncanonical RNA-dependent functions of PheRSs.
Mammalian PheRSs were believed to be involved in human autoimmune diseases such as polymyositis, dermatomyositis and juvenile rheumatoid arthritis (for a review see ref. 3). A connection between PheRS and tumorigenic events was supported by the enhanced expression of the human PheRS catalytic subunit during the development of myeloid leukemia.72 The authors suggested that overproduction of the α-subunit would cause the β-subunit to be sequestered in the cytoplasm by its association with the α-subunit and thus abstracted from its putative alternative function in the nucleus. This is the first example of tumor-selective and cell cycle stage- and differentiation-dependent expression of aaRSs. Northern blot hybridization analysis of malignant and normal human tissues enabled an assessment of the relative expression levels of the α- and β-subunits independently.3 Similar levels of mRNA corresponding to the α- and β-subunits were detected in all cell types and tissues examined and indicate the implication of the entire (αβ)2 heterodimer in tumorigenic events. Cloning of the full-length cDNA for the murine PheRS regulatory β-subunit-like protein was reported.73 The level of PheRS β-subunit-like mRNA is regulated during differentiation but not during cell cycle progression. It is extremely attractive to exploit the PheRS β-subunit-like clone for the specific identification of the nature of mutations associated with diseases.
PheRSs from different species were shown to produce bis(5'-nucleosidyl) polyphosphates such as Ap4A and Ap3A.74-76 By using E. coli PheRS, an aminoacyl-adenylate was established as intermediate in Ap4A and Ap3A synthesis.75 The synthesis of Ap4A catalyzed by the E. coli and yeast PheRSs was stimulated by Zn2+ or Cd2+ ions, while the tRNA aminoacylation was inhibited.74 The significance of this observation for the regulation of the cellular level of diadenosine oligophosphates is still unclear. Competitive inhibitors of amino acid activation showed weak or no action on the Ap4A/Ap3A synthesis catalyzed by PheRS in the presence of Zn2+ ions.77 These data are indicative of differences between tRNA aminoacylation and Ap4A/Ap3A synthesis at the intermediate steps of the enzyme-catalyzed reaction under certain conditions. Despite the fact that the functions of bis(5'-nucleosidyl) polyphosphates are still debated (for a review see ref. 2), a diverse range of their actions on cellular processes has been demonstrated (for a review see refs. 63, 78 and 79). In this way, PheRSs could contribute to the regulation of the multiple processes in living cells.
AaRSs have emerged as a leading target for the development of new antibiotics.80 The advance in structural studies of the bacterial PheRS6,23,29 and the availability of the human counterpart for comparative analysis80a allow us to regard them as prospective systems for the design of drug molecules. Based on the differences in the primary sequences, one can speculate that it may be feasible to create therapeutics directed against bacterial PheRSs that will not affect the human enzyme activity.
Acknowledgements
We thank our colleagues Reshetnikova L, Ankilova V, Mosyak L, Goldgur Y, Fishman R, Khodyreva S, Vasil'eva I, Stepanov V, Bobkova E who contributed to the results presented in this chapter. We are also grateful to Tvorowski D for technical assistance in preparing the manuscript, Wolfson A for comments and advice and Horton J for reading the manuscript. This work was supported by Kimmelman Center for Biomolecular Structure and Assemblies and INTAS grant (No. 97-2110). M.N. was partly supported by the Russian Fund for Basic Research (Grants No. 99-04-49856, 03-04-48384). M.S. was aided by a grant from Israel Science Foundation (No. 1034/03-16.1).
References
- 1.
- Mirande M. Aminoacyl-tRNA synthetase family from prokaryotes and eukaryotes: structural domains and their implications. Prog Nucleic Acid Res Mol Biol. 1991;40:95–142. [PubMed: 2031086]
- 2.
- Meinnel T, Mechulam Y, Blanquet S. Aminoacyl-tRNA synthetases: occurrence, structure, and function. In: Sö ll D, RajBhandary U, eds. RNA: structure, biosynthesis and function. Washington, DC 20005: American Society for Microbiology. 1995:251–292.
- 3.
- Rodova M, Ankilova V, Safro MG. Human phenylalanyl-tRNA synthetase: cloning, characterization of the deduced amino acid sequences in terms of the structural domains and coordinately regulated expression of the α and β subunits in chronic myeloid leukemia cells. Biochem Biophys Res Commun. 1999;255:765–773. [PubMed: 10049785]
- 4.
- Bullard JM, Cai YC, Demeler B. et al. Expression and characterization of a human mitohondrial phenylalanyl-tRNA synthetase. J Mol Biol. 1999;288:567–577. [PubMed: 10329163]
- 5.
- Das R, Vothknecht UC. Phenylalanyl-tRNA synthetase from the archaeon Methanobacterium thermoautotrophicum is an (αβ)2 heterotetrameric protein. Biochimie. 1999;81:1037–1039. [PubMed: 10575359]
- 6.
- Mosyak L, Reshetnikova L, Goldgur Y. et al. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat Stuct Biol. 1995;2:537–547. [PubMed: 7664121]
- 7.
- Ducruix A, Hounwanou N, Reinbolt J. et al. Purification and reversible subunit dissociation of overproduced Escherichia coli phenylalanyl-tRNA synthetase. Biochim Biophys Acta. 1983;741:244–250. [PubMed: 6360212]
- 8.
- Hanke T, Bartman P, Holler E. Quaternary structure and catalytic functioning of L-phenylalanyne:tRNA ligase of Escherichia coli K10. Eur J Biochem. 1975;56:605–615.
- 9.
- Bobkova EV, Mashanov-Golikov AV, Wolfson A. et al. Comparative study of subunits of phenylalanyl-tRNA synthetase from Escherichia coli and Thermus thermophilus. FEBS Lett. 1991;290:95–98. [PubMed: 1915899]
- 10.
- Baltzinger M, Fasiolo F, Remy P. Yeast phenylalanyl-tRNA synthetase. Affinity and photoaffinity labeling of stereospecific binding sites. Eur J Biochem. 1979;97:481–494. [PubMed: 380996]
- 11.
- Khodyreva SN, Moor NA, Ankilova VN. et al. Phenylalanyl-tRNA synthetase from E. coli MRE600: analysis of the active site distribution on the enzyme subunits by affinity labelling. Biochim Biophys Acta. 1985;830:206–212. [PubMed: 3893548]
- 12.
- Moor NA, Ankilova VN, Lavrik OI. et al. Determination of tRNAPhe nucleotides contacting the subunits of Thermus thermophilus phenylalanyl-tRNA synthetase by photoaffinity crosslinking. Biochim Biophys Acta. 2001;1518:226–236. [PubMed: 11311934]
- 13.
- Eriani G, Delarue M, Poch O. et al. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. [PubMed: 2203971]
- 14.
- Cusack S, Berthet-Colominas C, Hartlein M. et al. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-trasfer tRNA synthetase at 2.5 Å Nature. 1990;347:249–255. [PubMed: 2205803]
- 15.
- Moras D. Structural and functional relationships between aminoacyl-tRNA synthetases. Trends Biochem Sci. 1992;17:159–164. [PubMed: 1585461]
- 16.
- Fraser TH, Rich A. Amino acids are not all initially attached to the same position on transfer RNA molecules. Proc Natl Acad Sci USA. 1975;72:3044–3048. [PMC free article: PMC432915] [PubMed: 1103136]
- 17.
- Sprinzl M, Cramer F. Site of aminoacylation of tRNAs from E. coli with respect to the 2'- or 3'-hydroxyl group of the terminal adenosine. Proc Natl Acad Sci USA. 1975;72:3049–3053. [PMC free article: PMC432916] [PubMed: 1103137]
- 18.
- Stepanov VG, Moor NA, Ankilova VN. et al. A peculiarity of the reaction of tRNA aminoacylation catalyzed by phenylalanyl-tRNA synthetase from the extreme thermophile Thermus thermophilus. Biochim Biophys Acta. 1998;1386:1–15. [PubMed: 9675230]
- 19.
- Goldgur Y, Mosyak L, Reshetnikova L. et al. The crystal structure of phenylalanyl-tRNA synthetase from T. thermophilus complexed with cognate tRNAPhe. Structure. 1997;5:59–68. [PubMed: 9016717]
- 20.
- Lechler A, Kreutzer R. Domains of phenylalanyl-tRNA synthetase from Thermus thermophilus required for aminoacylation. FEBS Lett. 1997;420:139–142. [PubMed: 9459297]
- 21.
- Sanni A, Walter P, Boulanger Y. et al. Evolution of aminoacyl-tRNA synthetase quaternary structure and activity: Saccharomyces cerevesiae mitohondrial phenylalanyl-tRNA synthetase. Proc Natl Acad Sci USA. 1991;88:8387–8391. [PMC free article: PMC52513] [PubMed: 1924298]
- 22.
- Cusack S. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1997;7:881–889. [PubMed: 9434910]
- 23.
- Reshetnikova L, Moor N, Lavrik O. et al. Crystal structures of phenylalanyl-tRNA synthetase complexed with phenylalanine and a phenylalanyl-adenylate analogue. J Mol Biol. 1999;287:555–568. [PubMed: 10092459]
- 24.
- Mosyak L, Safro M. Phenylalanyl-tRNA synthetase from Thermus thermophilus has four antiparallel folds of which only two are catalytically functional. Biochimie. 1993;75:1091–1098. [PubMed: 8199244]
- 25.
- Schultz S, Shields G, Steitz T. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. [PubMed: 1653449]
- 26.
- Safro M, Mosyak L. Structural similarities in noncatalytic domain of phenylalanyl-tRNA and biotin synthetases. Protein Sci. 1995;4:2429–2432. [PMC free article: PMC2143022] [PubMed: 8563641]
- 27.
- Nagai K, Oubridge C, Jessen TH. et al. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature. 1990;348:515–520. [PubMed: 2147232]
- 28.
- Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. [PubMed: 8036511]
- 29.
- Fishman R, Ankilova V, Moor N. et al. Structure at 2.6 Å resolution of phenylalanyl-tRNA synthetase complexed with phenylalanyladenylate in the presence of manganese. Acta Crystallogr. 2001;D57:1534–1544. [PubMed: 11679717]
- 30.
- Ogle JM, Brodersen DE, Clemons WM. et al. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. [PubMed: 11340196]
- 31.
- Tinkle Peterson E, Uhlenbeck O. Determination of recognition nucleotides for Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry. 1992;31:10380–10389. [PubMed: 1420156]
- 32.
- Moor NA, Ankilova VN, Lavrik OI. Recognition of tRNAPhe by phenylalanyl-tRNA synthetase of Thermus thermophilus. Eur J Biochem. 1995;234:897–902. [PubMed: 8575450]
- 33.
- Moor N, Nazarenko I, Ankilova V. et al. Determination of tRNAPhe recognition nucleotides for phenylalanyl-tRNA synthetase from Thermus thermophilus. Biochimie. 1992;74:353–356. [PubMed: 1379078]
- 34.
- Aphasizhev R, Senger B, Rengers JU. et al. Conservation in evolution for a small monomeric phenylalanyl-tRNA synthetase of the tRNAPhe recognition nucleotides and initial aminoacylation site. Biochemistry. 1996;35:117–123. [PubMed: 8555164]
- 35.
- Sampson JR, Behlen LS, DiRenzo AB. et al. Recognition of yeast tRNAPhe by its cognate yeast phenylalanyl-tRNA synthetase: an analysis of specificity. Biochemistry. 1992;31:4161–4167. [PubMed: 1567862]
- 36.
- Sampson JR, DiRenzo AB, Behlen LS. et al. Role of the tertiary nucleotides in the interaction of yeast phenylalanine tRNA with its cognate synthetase. Biochemistry. 1990;29:2523–2532. [PubMed: 2334680]
- 37.
- Nazarenko IA, Tinkle Peterson E, Zakharova OD. et al. Recognition nucleotides for human phenylalanyl-tRNA synthetase. Nucleic Acids Res. 1992;20:475–478. [PMC free article: PMC310410] [PubMed: 1741281]
- 38.
- Carneiro VT, Dietrich A, Maréchal-Drouard L. et al. Characterization of some major identity elements in plant alanine and phenylalanine transfer RNAs. Plant Mol Biol. 1994;26:1843–1853. [PubMed: 7532029]
- 39.
- Moor NA, Ankilova VN, Favre A. et al. Localization of the binding site for the 3'-terminal sequence of tRNAPhe in subunits of phenylalanyl-tRNA synthetase from Thermus thermophilus. Biochemistry (Moscow). 1998;63:1051–1056. [PubMed: 9795274]
- 40.
- Vasil'eva IA, Ankilova VN, Lavrik OI. Interaction of T. thermophilus phenylalanyl-tRNA synthetase with the 3'-terminal nucleotide of tRNAPhe. Biochemistry (Moscow). 2000. pp. 1157–1166. [PubMed: 11092959]
- 40a.
- Moor N, Lavrik O, Favre A. et al. Prokaryotic and eukaryotic tetrameric phenylalanyl-tRNA synthetases display conservation of the binding mode of the tRNAPhe CCA end. Biochemistry. 2003;42:10697–10708. [PubMed: 12962494]
- 40b.
- Vasil'eva I, Bogachev V, Favre A. et al. Role of low-molecular-weight substrates in functional binding of the tRNAPhe acceptor end by phenylalanyl-tRNA synthetase. Biochemistry (Moscow). 2004;69:143–153. [PubMed: 15000680]
- 41.
- Arnez JG, Moras D. Structural and functional considerations of the aminoacylation reaction. Trends Biochem Sci. 1997;22:211–216. [PubMed: 9204708]
- 42.
- Belrhali H, Yaremchuk A, Tukalo M. et al. Crystal structures at 2.5 angstrom resolution of seryl-tRNA synthetase complexed with two analogs of seryl-adenylate. Science. 1994;263:1432–1436. [PubMed: 8128224]
- 43.
- Cavarelli J, Eriani G, Rees B. et al. The active site of yeast aspartyl-tRNA synthetase: structural and functional aspects of the aminoacylation reaction. EMBO J. 1994;13:327–337. [PMC free article: PMC394812] [PubMed: 8313877]
- 44.
- McClain WH, Foss K. Nucleotides that contribute to the identity of Escherichia coli tRNAPhe. J Mol Biol. 1988;202:697–709. [PubMed: 2459397]
- 45.
- Sprinzl M, Horn C, Brown M. et al. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. [PMC free article: PMC147216] [PubMed: 9399820]
- 46.
- Kholod NS, Pan'kova NV, Mayorov SG. et al. Transfer RNAPhe isoacceptors possess nonidentical set of identity elements at high and low Mg2+ concentration. FEBS Lett. 1997;411:123–127. [PubMed: 9247156]
- 47.
- Biou V, Yaremchuk A, Tukalo M. et al. The 2.9 Å crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNASer. Science. 1994;263:1404–1410. [PubMed: 8128220]
- 48.
- Perret V, Florentz C, Puglisi JD. et al. Effect of conformational features on the aminoacylation of tRNAs and consequences on the permutation of tRNA specificities. J Mol Biol. 1992;226:323–333. [PubMed: 1640453]
- 49.
- Kreutzer R, Kern D, Giege R. et al. Footprinting of tRNAPhe transcripts from Thermus thermophilus HB8 with the homologous phenylalanyl-tRNA synthetase reveals a novel mode of interaction. Nucleic Acids Res. 1995;23:4598–4602. [PMC free article: PMC307431] [PubMed: 8524648]
- 50.
- Chang KY, Varani G, Bhattacharya S. et al. Correlation of deformability at a tRNA recognition site and aminoacylation specificity. Proc Natl Acad Sci USA. 1999;96:11764–11769. [PMC free article: PMC18360] [PubMed: 10518524]
- 51.
- Khvorova A, Motorin Y, Wolfson AD. Pyrophosphate mediates the effect of certain tRNA mutations on aminoacylation of yeast tRNAPhe. Nucleic Acids Res. 1999;27:4451–4456. [PMC free article: PMC148729] [PubMed: 10536155]
- 52.
- Burley SK, Petsko GA. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. [PubMed: 3892686]
- 53.
- Ibba M, Kast P, Hennecke H. Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry. 1994;33:7107–7112. [PubMed: 8003476]
- 54.
- Ibba M, Hennecke H. Relaxing the substrate specificity of an aminoacyl-tRNA synthetase allows in vitro and in vivo synthesis of proteins containing unnatural amino acids. FEBS Lett. 1995;364:272–275. [PubMed: 7758582]
- 55.
- Lin SX, Baltzinger M, Remy P. Fast kinetic study of yeast phenylalanyl-tRNA synthetase: an efficient discrimination between tyrosine and phenylalanine at the level of the aminoacyladenylateenzyme complex. Biochemistry. 1983;22:681–689. [PubMed: 6340722]
- 56.
- Freist W, Sternbach H, Cramer F. Phenylalanyl-tRNA synthetase from yeast and its discrimination of 19 amino acids in aminoacylation of tRNAPhe-C-C-A and tRNAPhe-C-C-A(3'NH2). Eur J Biochem. 1996;240:526–531. [PubMed: 8856050]
- 57.
- Belrhali H, Yaremchuk A, Tukalo M. et al. The structural basis for seryl-adenylate and Ap4A synthesis by seryl-tRNA synthetase. Structure. 1995;3:341–352. [PubMed: 7613865]
- 58.
- Arnez JG, Dock-Bregeon AC, Moras D. Glycyl-tRNA synthetase uses a negatively charged pit for specific recognition and activation of glycine. J Mol Biol. 1999;286:1449–1459. [PubMed: 10064708]
- 59.
- Berthet-Colominas C, Seignovert L, Hartlein M. et al. The crystal structure of asparaginyl-tRNA synthetase from Thermus thermophilus and its complexes with ATP and asparaginyl-adenylate: the mechanism of discrimination between asparagine and aspartic acid. EMBO J. 1998;17:2947–2960. [PMC free article: PMC1170635] [PubMed: 9582288]
- 60.
- Arnez JG, Harris DC, Mitschler A. et al. Crystal structure of histidyl-tRNA synthetase from Escherichia coli complexed with histidyl-adenylate. EMBO J. 1995;14:4143–4155. [PMC free article: PMC394497] [PubMed: 7556055]
- 61.
- Moor NA, Repkova MN, Yamkovoy VI. et al. Alterations at the 3'-CCA end of Escherichia coli and Thermus thermophilus tRNAPhe do not abolish their acceptor activity. FEBS Lett. 1994;351:241–242. [PubMed: 8082771]
- 62.
- Martinis SA, Plateau P, Cavarelli J. et al. Aminoacyl-tRNA synthetases: a family of expanding functions. EMBO J. 1999;18:4591–4596. [PMC free article: PMC1171533] [PubMed: 10469639]
- 63.
- Ivanov KA, Moor NA, Lavrik OI. Noncanonical functions of aminoacyl-tRNA synthetases. Biochemistry (Moscow). 2000;65:888–897. [PubMed: 11002181]
- 64.
- Jasin M, Regan L, Schimmel P. Modular arrangement of functional domains along the sequence of an aminoacyl tRNA synthetase. Nature. 1983;306:441–447. [PubMed: 6358898]
- 65.
- Artymiuk PJ, Rice DW, Poirrette AR. et al. A tale of two synthetases. Nat Struct Biol. 1994;1:758–760. [PubMed: 7634083]
- 66.
- Lechler A, Kreutzer R. The phenylalanyl-tRNA synthetase specifically binds DNA. J Mol Biol. 1998;278:897–901. [PubMed: 9600851]
- 67.
- Dou X, Limmer S, Kreutzer R. DNA-binding of phenylalanyl-tRNA synthetase is accompanied by loop formation of the double-stranded DNA. J Mol Biol. 2001;305:451–458. [PubMed: 11152603]
- 68.
- Ivanov KA, Moor NA, Ankilova VN. et al. Phenylalanyl-tRNA synthetase interacts with DNA: studies on activity using deoxyribonucleotides. Biochemistry (Moscow). 2000;65:436–441. [PubMed: 10810180]
- 69.
- Arts GL, Kuersten S, Romby P. et al. The role of exportin-t in selective nuclear export of mature tRNAs. EMBO J. 1998;17:7430–7441. [PMC free article: PMC1171087] [PubMed: 9857198]
- 70.
- Iborra FJ, Jackson DA, Cook PR. Coupled transcription and translation within nuclei of mammalian cells. Science. 2001;29:1139–1142. [PubMed: 11423616]
- 71.
- Quevillon S, Agou F, Robinson JC. et al. The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J Biol Chem. 1997;272:32573–32579. [PubMed: 9405472]
- 72.
- Sen S, Zhou H, Ripmaster T. et al. Expression of a gene encoding a tRNA synthetase-like protein is enhanced in tumorigenic human myeloid leukemia cells and is cell cycle stage- and differentiation-dependent. Proc Natl Acad Sci USA. 1997;94:6164–6169. [PMC free article: PMC21020] [PubMed: 9177188]
- 73.
- Zhou X, Richon VM, Ngo L. et al. Cloning of the cDNA encoding phenylalanyl-tRNA synthetase regulatory alpha-subunit-like protein whose expression is down-regulated during differentiation. Gene. 1999;233:13–19. [PubMed: 10375616]
- 74.
- Plateau P, Mayaux JF, Blanquet S. Zinc (II)-dependent synthesis of diadenosine 5',5"'-P1,P4-tetraphosphate by Escherechia coli and yeast phenylalanyl transfer ribonucleic acid synthetases. Biochemistry. 1981;20:4654–4662. [PubMed: 7028092]
- 75.
- Goerlich O, Foeckler R, Holler E. Mechanism of synthesis of adenosine(5')tetraphospho(5') adenosine (AppppA) by aminoacyl-tRNA synthetases. Eur J Biochem. 1982;126:135–142. [PubMed: 7128581]
- 76.
- Biryukov AI, Ankilova VN, Lavrik OI. Diadenosine oligophosphates: peculiarities of synthesis by phenylalanyl-tRNA synthetases from E. coli MRE600 and Thermus thermophilus HB8. Nucleic Acids Symp Ser. 1991;24:19–20. [PubMed: 1841280]
- 77.
- Biryukov AI, Zhukov YuN, Lavrik OI. et al. Influence of the aminoacyl-tRNA synthetase inhibitors and the diadenosine-5'- tetraphosphate phosphonate analogues on the catalysis of diadenosyl oligophosphates formation. FEBS Lett. 1990;273:208–210. [PubMed: 2226855]
- 78.
- Kisselev LL. Aminoacyl-tRNA synthetases (codases) and their noncanonical functions. Mol Biol (Moscow). 1990;24:1445–1473. [PubMed: 2094804]
- 79.
- Kisselev LL, Justesen J, Wolfson AD. et al. Diadenosine oligophosphates (Ap4A), a novel class of signalling molecules? FEBS Lett. 1998;427:157–163. [PubMed: 9607303]
- 80.
- Schimmel P, Tao J, Hill J. Aminoacyl-tRNA synthetases as targets for new anti-infectives. FASEB J. 1998;12:1599–1609. [PubMed: 9837850]
- 80a.
- Moor N, Linshiz G, Safro M. Cloning and expression of human phenylalanyl-tRNA synthetase in E coli comparative study of purified recombinant enzymes. Protein Expression Purif. 2002;24:260–267. [PubMed: 11858721]
- 81.
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950.
- 82.
- Szymanski M, Barciszewski J. Aminoacyl-tRNA synthetase base Y2K. Nucleic Acids Res. 2000;28:326–328. [PMC free article: PMC102446] [PubMed: 10592262]
- 83.
- Combet C, Blanchet C, Geourjon C. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25:147–150. [PubMed: 10694887]
- 84.
- Merrit E, Murphy M. RASTER3D version 2.0: a program for photorealistic molecular graphics. Acta Crystallogr. 1994;D50:869–873. [PubMed: 15299354]
- Quaternary Organization
- Three-Dimensional Structure
- Interaction of PheRS with Its Cognate tRNAPhe: Binding and Recognition
- Selection of the Phenylalanine Substrate
- Structural Aspects of Phenylalanyl-Adenylate Formation
- A Scenario of the Phenylalanylation Process
- Noncanonical Functions
- Acknowledgements
- References
- Phenylalanyl-tRNA Synthetases - Madame Curie Bioscience DatabasePhenylalanyl-tRNA Synthetases - Madame Curie Bioscience Database
- -3 eggc.vipS8P (14281)BioProject
Your browsing activity is empty.
Activity recording is turned off.
See more...