

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Checkpoints are mechanisms that establish dependence relationships between biochemically unrelated cellular processes. The temporal order of many critical cell cycle events must be strictly maintained to ensure cell survival and integrity. A simple example is that of genome duplication which must be completed before cell division. The relationship between these processes is controlled by the S-phase checkpoint. After S-phase, the topoisomerase II-dependent checkpoint ensures that the topology of the newly replicated DNA has been correctly organized before cells begin mitosis. During mitosis itself, distinct checkpoints monitor mitotic spindle assembly, preventing the onset of chromosome segregation until all the chromosomes are correctly aligned on the mitotic spindle, and prevent exit from mitosis until anaphase chromosome segregation has been completed. In this chapter, we discuss these checkpoint control systems (Fig. 1).

An elegant way to define checkpoint pathways has been by analysis of loss-of-function mutants in the genetically manipulatable yeast systems. For example, the S-phase checkpoint was described in budding yeast by the isolation of mutants that initiate mitosis despite a replication block enforced by the ribonucleotide reductase inhibitor hydroxyurea (HU). Other checkpoint systems were originally described in mammalian cells. Indeed, the existence of checkpoint controls had been inferred from mammalian cell-fusion studies earlier than the genetic analyses performed in yeast (Fig. 2).1 More recently, mammalian checkpoints have been investigated by demonstrating that some checkpoint arrests can be overridden by drugs such as caffeine.2-9 Caffeine inhibits the kinase ATM, a key component of eukaryotic checkpoint pathways.10-13 Although mammalian cells are less amenable to genetic studies than are yeast, they have proven to be important for the study of checkpoint biology.

Our descriptions of checkpoint controls use a commonly adopted format which divides the pathways into (1) the sensor, (2) the transducer and (3) the target. Checkpoints do not necessarily follow simple linear pathways, however. Many of the sensor and transducer components are likely to be assembled into large complexes. Still, this nomenclature allows for a framework to be drawn up, on which the details can be built. The sensor components are those that monitor completion of the relevant process, for example DNA replication. The transducer transmits the signal from the sensor to the target of the checkpoint. It is the activity of the target that controls cell cycle progression.
Budding Yeast Versus Higher Eukaryotes
Since checkpoint pathways in both budding yeast and higher eukaryotes will be discussed in this chapter, it is important to describe a fundamental difference in budding yeast cell cycle organization that sets it apart from other species. In budding yeast, checkpoints promote the activity of anaphase inhibitors, whereas in most eukaryotes, checkpoints inhibit the activity of the mitotic kinase (cyclin/Cdk), required for passage through the G2/M transition. A need for distinct modes of control is related to differences in the spindle assembly pathway. In many eukaryotes, including mammals, the mitotic spindle does not assemble until mitosis. However, budding yeast spindles assembled during S-phase; checkpoints must inhibit spindle elongation even while DNA is being replicated. In addition, sister chromatid cohesion, established during DNA replication, must be maintained until the onset of anaphase. In budding yeast, an inhibitor of anaphase, Pds1, can perform both of these tasks.14-18 Before anaphase, Pds1 binds to protease Esp1 and thereby inhibits the anaphase-promoting activity of Esp1.18 During an unperturbed cell cycle, Pds1 becomes poly-ubiquitinated at the metaphase to anaphase transition by a multi-subunit enzyme complex known as the APC (Anaphase Promoting Complex); the modified forms are recognized and degraded by 26S proteasomes.16 Once released from Pds1, Esp1 induces cleavage of Scc1, a cohesin required to maintain cohesion between sister chromatids.18-21 Concurrently with loss of sister cohesion, Esp1 induces spindle elongation.22 Not surprisingly, Pds1 is a major target of checkpoints controlling anaphase onset. Vertebrate proteins named Securins, that are at least partial functional homologues of Pds1, have been identified,23 making the study of checkpoints in budding yeast highly relevant. Moreover, budding yeast and higher eukaryotes employ a common strategy for controlling exit from mitosis. In this case, regulation of cyclin/Cdk activity appears to be a universally adopted mode of control.
S-Phase Checkpoint
The S-phase checkpoint ensures that the onset of mitosis is dependent on the completion of DNA replication.24-26 Since little is known about S-phase checkpoint control in mammals, the components of this pathway in budding yeast will be described (Fig. 3). As mentioned above, budding yeast cells initiate DNA replication and mitotic spindle formation at a common cell cycle point, early in S-phase. To prevent the generation of aneuploid daughter cells that are inviable, it is essential that the mitotic spindle does not elongate before DNA replication has been completed. The order of these two processes is normally maintained by a timing mechanism rather than a checkpoint control: DNA replication takes only 20–30 minutes and spindle formation takes around 60 minutes; thus, spindle assembly is not completed before genome replication. This example illustrates how the temporal order of two events can be maintained independently of checkpoint controls, i.e. if the processes have a common starting point and each require a differing minimum amount of time for their completion.

A checkpoint pathway does exist, however, to ensure that the dependence between spindle elongation and DNA replication is always maintained. If DNA replication is inhibited with HU,27 the cells arrest with fully assembled short G2 spindles. After removal of the HU, spindle elongation is delayed until replication is complete. The S-phase checkpoint does not only control the mitotic spindle, however. All eukaryotes establish sister cohesion during DNA replication, and it must be maintained until the onset of anaphase.20,21 Maintenance of sister chromatid cohesion is of great importance to mammals and yeast alike and is a function of the S-phase checkpoint. At least in yeast, cohesion is established at some loci very early in S-phase and must therefore be maintained for the remainder of the S-phase period as well as during G2 and until the moment of anaphase onset. The homologs of yeast S-phase checkpoint components are therefore likely to be important regulators of mammalian sister chromatid cohesion.
To define the budding yeast S-phase checkpoint, loss-of-function mutations causing sensitivity to HU were identified. The proteins encoded by these genes were determined to have S-phase checkpoint functions by showing that the loss-of-function mutations allowed entry into mitosis when DNA replication was blocked with HU. Thus the S-phase checkpoint is defined as that which restrains entry into mitosis when replication is blocked. However, kinetic analyses of various checkpoint mutants, grown in the presence of a concentration of HU that allows replication to proceed, but more slowly than in an unperturbed cell cycle, have revealed genetically distinct S-phase checkpoint systems (see below).
To monitor ongoing DNA replication, cells seem to have replication sensors that reside at replication forks. In budding yeast, the putative sensor components include Pol2, Rfc5, Dpb11, Drc1 and Sgs1.28-32 POL2 encodes the replicative DNA polymerase, Pole, and Rfc5 is a replication factor C subunit involved in recruiting polymerases to replication forks. Dpb11 is also required for DNA replication; it can bind to Pole and is thought to help recruit Pola-primase complexes to ARS sequences at replication origins.33 Dpb11 also binds to Drc1, which is itself essential for DNA replication.31 Together with Srs2, Sgs1 has a redundant but essential role in DNA replication.34
Evidently, the sensor proteins also play important roles in DNA replication itself, and with hindsight it might seem elementary that components of the replication fork machinery are involved in generating the checkpoint signal. For each of these components, it was important to know that their checkpoint functions could be distinguished from their roles in DNA replication. This is the case because the S-phase checkpoint cannot be activated until DNA replication has begun,35 a fact illustrated by the phenotype of cells carrying a heat-inducible cdc45 degron mutant.36 Cdc45 binds to replication origins before S-phase in budding yeast and is required for origin firing. The cdc45 degron mutant is rapidly degraded at the restrictive temperature. When degradation was induced in G1 of the cell cycle, replication origins could not fire, and the cells entered mitosis without replicating any DNA. When the temperature shift was performed within S-phase, DNA replication was immediately inhibited because Cdc45 is also needed for elongation of replication forks, but in this case mitotic progression was inhibited. Thus, the S-phase checkpoint signal requires the presence of replication forks that have fired, and Cdc45 is not a component of the checkpoint response.
Mec1 and Rad53 kinases are traditionally described as components of the signal transduction element of the S-phase checkpoint.24,25,37 When replication is perturbed, these checkpoint kinases are activated in a manner dependent on the sensor components. Exactly how Rad53 and Mec1 activation occurs is not known, but several physical interactions have been identified that may represent key steps. Sgs1 was found to co-localize with Rad53 in discrete nuclear foci during S-phase. Intriguingly, Sgs1 is reported to interact with the FHA domain of Rad53,38 the domain required for the formation of the Rad53-Rad9 complex, required for DNA damage checkpoint signaling.39 Rad53-Sgs1 association may have revealed an Sgs1-dependent loading of Rad53 onto specific chromatin regions that might be involved in monitoring replication. That Sgs1 is involved in S-phase checkpoint control,32 adds weight to this model. But, the S-phase checkpoint defect of sgs1 mutants is rather weak, not nearly as substantial as other S-phase checkpoint mutants. This suggests that Sgs1 has a redundant checkpoint function, perhaps with another helicase such as Srs2.
Another activator of signal transduction may be Ddc2 (also known as Lcd1), a component that physically associates with Mec1 and is phosphorylated by Mec1.40,41 Phosphorylated Ddc2 is present in unperturbed S-phase cells and in HU-treated cells, and Ddc2 is required for cell cycle arrest in the presence of HU. Phosphorylation and activation of Rad53 in response to replication arrest is Ddc2-dependent. Therefore, Ddc2 appears to mediate between Mec1 and Rad53 in response to ongoing DNA replication and when fork progression is blocked. It remains to be tested whether the association of Ddc2 with Mec1, or the phosphorylation of Ddc2 by Mec1, depends on the sensor components, and how these events are regulated.
In response to replication inhibition, Mec1 and Rad53 enforce cell cycle arrest by blocking Pds1 degradation. In addition, these kinases induce transcription of genes involved in DNA repair and that help deal with the perturbed replication process.24 This safety system most likely protects stalled replication forks, allowing them to be re-initiated when conditions have improved. The transcription pathway depends on Rad53-dependent phosphorylation of the kinase Dun1 (damage uninducible).24,42 Activation of Dun1, in response to DNA damage or DNA replication blocks, induces transcription of genes that promote efficient DNA repair.24 This transcription response is partially initiated by Crt1 hyperphosphorylation.42 Crt1 represses transcription of DNA damage-inducible genes by binding to their promoter regions; binding is prevented by hyperphosphorylation. Activation of a transcription program clearly has the potential to enforce a wide range of Mec1- and Rad53-dependent functions, and a growing literature has made clear that Mec1 and Rad53 are involved in numerous cellular processes. For example, Mec1 and Rad53 inhibit the firing of late replication origins during early S-phase. Eukaryotic cells replicate their genomes by initiating DNA synthesis from multiple replication origins. Some fire early in S-phase, others are initiated part way through S-phase. When cells are arrested in early S-phase with HU, late firing origins are kept dormant by a dominant process that requires the Rad53-Mec1 pathway.43 Mec1 and Rad53 are also involved in the regulation of telomere length and in transcriptional silencing at telomeres.44-46
It is not clear, however, whether the Mec1/Rad53/Dun1-dependent transcriptional response contributes to cell cycle arrest in the presence of HU, since dun1 null mutants are not S-phase checkpoint defective. In agreement with this, the cell cycle checkpoint defects of mec1 and rad53 mutants are somewhat different. Both mutants elongate their mitotic spindles when DNA replication is blocked with HU, so it seems that no checkpoint response remains in these cells. However, rad53 mutants delay in anaphase, while mec1 mutants exit mitosis. Thus some aspects of mitotic progression are inhibited in rad53 mutants. Light was shed on the basis of this difference by analysis of pds1 mutants, revealing that there are several S-phase checkpoint targets. Pds1 is not an essential target in early S-phase because pds1 mutants can inhibit spindle elongation when replication is blocked with HU in early S. However, kinetic analyses determined that, part way through S-phase, a critical point is reached where Pds1 becomes essential: pds1 mutants elongate spindles and lose sister chromatid cohesion when roughly 2/3 of the genome has been replicated.47 These experiments were performed in the presence of a concentration of HU that does not fully block replication, but instead, slows the rate of DNA replication. In these experiments, mec1 or rad53 mutant cells began anaphase when very little DNA had been replicated (as is the case when replication is blocked). Therefore, a Pds1-independent system restrains spindle elongation in early S-phase, but later in S-phase, Pds1 is required. A reasonable prediction is that Pds1 and Rad53 function downstream of Mec1 in the context of S-phase checkpoint control, and that these pathways run in parallel, and are temporally regulated; one necessary in early S-phase, the other part way through S-phase. Presumably, Mec1 controls Pds1 stability in late S-phase. Several details remain unresolved, however. For example, the fact that pds1 null mutants are able to restrain spindle elongation and prevent premature loss of sister chromatid cohesion in early S-phase necessitates a novel Mec1/Rad53 target at that point in the cell cycle.
An explanation for the duality of S-phase checkpoint control in budding yeast is the linkage of spindle elongation with regulation of sister cohesion. Sister cohesion is established during DNA replication and must be maintained until the onset of anaphase.20,21 Once cohesion is established, part way through S-phase,48 checkpoint control of anaphase must coordinate release of cohesion with spindle elongation. Early in S-phase, prior to replication of critical cohesion regions and concomitant establishment of cohesion, spindle elongation might be regulated independently of cohesion. Therefore, the switch in the mode of checkpoint control from the Mec1-Rad53 pathway to the Mec1-Pds1 pathway may be controlled by the establishment of sister chromatid cohesion.
It remains to be determined how Pds1 levels are controlled in late S-phase when DNA replication is perturbed. Recent evidence has linked two yeast genes to regulation of Pds1 in this context.49 Rad23 or Ddi1 overproduction was found to rescue the sensitivity of pds1 mutant cells to HU. Rad23 is a nucleotide excision repair protein, but recent studies suggest a novel role of Rad23 in ubiquitin-dependent proteolysis.50 Rad23 binds to mono- or di-ubiquitinated proteins but cannot bind when the ubiquitin chains have been elongated. Crucially, Rad23 blocks extension of the ubiquitin chains. For most ubiquitinated proteins that are targeted for degradation, ubiquitin chain elongation is critical for efficient recognition by the 26S proteasome. Therefore, Rad23 might have an important function in preventing or delaying the degradation of proteasome targets. Although this mechanism has not been tested directly in the context of S-phase checkpoint control, overexpression of Rad23 is able to stabilize a mutant pds1 protein, suggesting that Rad23 might play a role in S-phase checkpoint signaling.49 A role of Rad23 in checkpoint signaling may be utilized by virally expressed proteins. The HIV-1 encoded protein Vpr has been shown to bind to the C-terminal UBA (Ubiquitin associated domain) of human Rad23 (HHR23A).51-53 This interaction is needed for one of the cellular functions of Vpr, the ability of Vpr to induce G2 cell cycle arrest, which allows time for viral replication. It seems plausible that Vpr mimics an endogenous cellular checkpoint response that involves binding of the Rad23 UBA to an unknown protein, inducing G2 arrest.
Mammalian cells also need an S-phase checkpoint. The initiation of mitosis must be prevented during S-phase, and chromatid cohesion must be maintained. Mammalian Sgs1 homologs are clearly important for S-phase regulation.38 Sgs1 is a budding yeast member of the Escherichia coli recQ helicase family, and sgs1 mutants are genomically unstable.54 Mammalian recQ helicase family members include WRN (mutated in Werner's syndrome patients)55 and BLM (mutated in Bloom's syndrome patients).56 Bloom's syndrome is characterized by genomic instability and a high incidence of cancer, whereas Werner's syndrome causes premature ageing. The BLM protein was recently identified as a component of a large complex that includes tumor suppressor proteins, DNA repair and checkpoint proteins that localize to nuclear foci when cells are treated with HU.57 Indeed, cultured cells from Bloom's syndrome patients have S-phase defects, but it is not clear whether these abnormalities include checkpoint abrogation. In general, there are mammalian homologues of all the budding yeast checkpoint proteins, but their potential roles in S-phase checkpoint control have not been thoroughly investigated.
The Mec1 homologs are ATM and ATR.58,59 ATM, the gene mutated in ataxia telangiectasia patients who have an increased incidence of cancer, is a nuclear protein kinase. ATR (ataxia telangiectasia and rad3 related) is also a protein kinase and is structurally more homologous to Mec1 than is ATM. Both ATM and ATR are clearly involved in DNA damage checkpoint signaling,60,61 but do not seem to be required for preventing the onset of mitosis during S-phase. Whether these proteins have roles in regulating cohesion has not been addressed. In the context of the DNA damage checkpoint, the tumor suppressor proteins p53 and Brca1 (breast cancer gene 1) seem to be targets of ATM/ATR, but again there is little evidence for roles in S-phase.62-68 Brca1 is, however, phosphorylated by ATR in response to HU treatment.69
The mammalian Rad53 homologue, kinase Chk2 (Checkpoint kinase),70 and the mammalian homologue of budding and fission yeast Chk1 (also named Chk1), are required for the ATM-dependent DNA damage checkpoint.71,72 After γ-irradiation, mammalian Chk2 phosphorylation (on Thr-68, within the serine/threonine cluster domain of Chk2) and activation is ATM-dependent.73 Chk2 kinase also becomes phosphorylated and activated upon HU treatment, but in an ATM-independent manner, and not on Thr-68.70,73,74 If the HU-induced phosphorylation is relevant for S-phase checkpoint control, it might be that the mammalian S-phase checkpoint operates by a kinase distinct from ATM.
Chk1 is not needed for S-phase checkpoint control in budding yeast but does seem to be required in some higher eukaryotes. Xenopus Chk1 is activated in post-MBT (mid-blastula transition) embryonic cells treated with HU.75 Similarly, there is good evidence for a role of Drosophila Chk1 (named Grapes) in coordinating embryonic DNA replication with the onset of mitosis.76,77 In Xenopus egg extracts, immunodepletion of Chk1 impairs an ability to delay cell cycle progression in response to replication blocks.78 Immunodepletion of ATR has the same effect since Chk1 activity depends on phosphorylation by ATR when unreplicated DNA is present.79 Human ATR has been implicated in Chk1 phosphorylation in response to HU treatment, but it is not known if cells lacking Chk1 or ATR have defective S-phase checkpoint controls.80 In Xenopus, Chk1 activation also depends on a protein named Claspin which has a close human homolog. Xenopus egg extracts depleted of Claspin are S-phase checkpoint deficient.81
Both Chk1 and Chk2 can phosphorylate Cdc25C on Ser-216 in humans and it is thought that this phosphorylation prevents Cdc25C from activating the mitotic kinase, cyclinB1/Cdc2.70,71 Cdc25C is a protein phosphatase that promotes entry into mitosis by dephosphorylating Cdc2. This phosphorylated residue creates a binding site for a 14-3-3 protein, resulting in Cdc25C inhibition.72 Interestingly, expression of a mutant Cdc25C that cannot be phosphorylated on Ser-216 induces mitosis in the presence of unreplicated DNA, suggesting that this pathways may be important for S-phase checkpoint control.72
Topoisomerase II-Dependent Checkpoint
DNA topoisomerase II (topo II) is required for chromosome condensation and segregation in eukaryotes.3,82-87 Although these are mitotic processes, their successful completion depends partly on topo II activity during DNA replication and in G2 phase (Fig. 4). Chromosome replication creates two identical sister DNA molecules that are knotted together (catenated). Topo II removes the catenations; in higher eukaryotes, the majority must be resolved before entry into mitosis to allow accurate chromosome condensation.3,88 A G2 checkpoint ensures that DNA catenations have been sufficiently resolved before mammalian cells enter mitosis.3

It had been known for some time that topo II inhibitors block or delay mammalian cell cycle progression in G2, but the inhibitors used were also known to cause DNA damage; it was assumed that the G2 arrest was due to activation of the DNA damage checkpoint. That cells also need to monitor topo II activity in G2 was an attractive hypothesis, however, and the characterization of novel topo II inhibitors that do not damage DNA allowed this theory to be tested. A variety of assays that measured DNA damage were used to assess the effects of bisdioxopiperazine topo II inhibitors on mammalian cells and on isolated DNA. Bisdioxopiperazines were found not to induce DNA breaks in vivo or in vitro3 (and Refs. therein) but did block mammalian cells in G2. In these studies, entry into mitosis was assessed based on the onset of chromosome condensation. Yet, topo II is needed for chromosome condensation, albeit a requirement at late steps in the process.3,88 It was therefore necessary to prove that cells were physically capable of initiating chromosome condensation when topo II activity was absent. Such evidence could not be sought by isolating loss-of-function budding or fission yeast mutants, since the topo II-dependent checkpoint seems to be absent in yeast.85,89 Therefore the checkpoint was first described in mammals, by demonstrating checkpoint bypass induced with caffeine or kinase and phosphatase inhibitors.3 Cells treated with ICRF-193, the most potent of the bisdioxopiperazines, could be forced into mitosis with caffeine; the cells began to condense chromosomes without delay. Fully condensed chromosomes were not formed, consistent with the essential role of topo II late in the condensation process. Thus, the topo II-dependent checkpoint prevents the onset of chromosome condensation, a process which the cells can begin, but cannot complete in the absence of topo II activity.
Although the evidence for this checkpoint in mammals is substantial, its absence in yeast has prevented the checkpoint components from being rapidly identified. Indeed, it is not known whether topo II levels are sensed directly, or if physical structures within chromosomes, such as catenations (Fig. 5), are monitored. Some data suggest the latter is more likely. Replicative catenations are introduced between daughter DNA duplexes during S-phase. Disentangling daughter duplexes is of crucial importance since they otherwise could not separate and segregate during mitosis. Most replicative catenations are resolved in G2, when cellular topo II activity increases, but since the decatenation reaction is reversible, topo II activity inevitably promotes some catenation. This generates non-replicative catenations, that can involve distant regions of chromatin, and can join different chromosomes together.90,91 Cells inhibited for topo II activity late in G2 and forced to enter mitosis with caffeine, have striking chromosome aberrations caused by persistent non-replicative catenations that join chromosomes together and create W-figures within individual chromosomes (Fig. 4 and Fig. 5).92 Circumstantial evidence suggests that the removal of non-replicative catenations in G2, the process that promotes chromosome individualization, may be monitored by the topo II-dependent checkpoint.92

Another question is what is the nature of the topo II-dependent checkpoint sensor? It might be that sensors bind to sites of DNA catenation. Though purely speculative, there is a precedent for such a proposal, that a signaling cascade might be activated by protein complexes at sites of DNA crossover. In bacteria, stable maintenance of the natural multicopy plasmid CoIE1 requires a cer sequence element (Fig. 6). cer is necessary for recombination that converts unstable plasmid multimers to monomers.93,94 The expression of Rcd, a transcript encoded from within cer, is specifically expressed in cells containing multimers. Rcd1 enforces a cell cycle checkpoint that inhibits cell division when multimers are present,95 thus allowing time for site-specific recombination to occur. An interesting observation is that the Rcd1 promoter resides within the cer sequence, and that the topology of cer is likely to be altered in multimers that assemble recombination complexes at the cer sites.96 This difference in topology might influence Rcd1 transcription, providing an elegant mechanism to activate the checkpoint in the presence of multimers. The parallel between this phenomenon in bacteria and the topo II-dependent checkpoint signal generated by persistent DNA catenations in mammalian cells is remarkable.

The target of the topo II-dependent checkpoint is presumed to be mitotic cyclin/Cdk activity: recent work has shown that a topo II-dependent checkpoint exists in plant cells that can be overridden by overexpressing a mitotic cyclin (J.F.G.A., unpublished data). How the mitotic kinase is regulated in response to topo II-dependent checkpoint activation is not known, although some data give clues as to the upstream components of the pathway. The topo II inhibitor genistein arrests mammalian cells in G2 by activating Chk2 kinase, which in turn leads to the inhibition of Cdc25C-dependent Tyr-15 dephosphorylation of Cdk1.97 Activation of Chk2 occurs very efficiently at genistein doses that inhibit topo II but cause minimal DNA damage compared to other topo II inhibitors such as etoposide. In the case of genistein, ATR might be the kinase upstream of Chk2, since caffeine overrides the G2 arrest whereas Wartmannin does not. In contrast to ATM, which is inhibited by caffeine and Wartmannin, ATR is only efficiently inhibited by caffeine.97 The topo II-dependent checkpoint and DNA damage checkpoint might be regulated primarily by ATR and ATM, respectively. Although these checkpoints are distinct, the possibility remains that they are closely linked pathways. One way to address this issue will be to test whether other components of the DNA damage checkpoint, such as Chk1, p53 and 14-3-3s, are activated in the context of ICRF-193-induced G2 arrest.
Checkpoint Control in Prophase
A recent study identified a novel mammalian checkpoint protein Chfr (Checkpoint with FHA and Ring finger) apparently acting to slow chromosomal condensation in prophase and prometaphase when microtubule polymerization is perturbed.98 In a cohort of 8 human tumor cell lines, three were identified that failed to express Chfr at the transcriptional level, although this was not due to loss of both gene copies. Furthermore, a mutation was identified in a fourth cell line leading to loss of Chfr function. In tumor cell lines lacking Chfr function, mitotic chromosome condensation occurred at the same rate in the presence or absence of nocodazole (or taxol). In cells with functional Chfr, or cells lacking Chfr but transiently transfected with a functional copy, chromosome condensation occurred at a reduced rate in nocodazole-treated cells, relative to cell cycle progression from G2 to metaphase based on the accumulation of cyclin/Cdk activity and prophase separation of centrosomes. Examination of nuclear morphology and DNA content following 48 hours of microtubule depolymerizing treatment demonstrated that Chfr-defective cells undergo aberrant mitosis, implicating this checkpoint in chromosome instability. While there is no definitive yeast homolog for Chfr, two S. cerevisiae open reading frames and one S. pombe gene, defective in mitotic arrest (Dma1),99 appear closely related. Clearly, study of a larger cohort of tumor cell lines and further mechanistic studies need to be performed to fully assess the import of this checkpoint in tumorigenesis. These may also substantiate the authors claims that Chfr is inactivated more frequently than ‘all known spindle checkpoint proteins combined’.98
Spindle Assembly Checkpoint
The spindle assembly checkpoint is an example of how a combination of yeast genetics and cell biology in higher eukaryotes has rapidly expanded our knowledge of a biological system.26,100-104 Eukaryotic cells arrest in metaphase when microtubule polymerization is disturbed. Higher eukaryotic cells, with normal mitotic spindles, also arrest if chromosomes fail to become bioriented on the spindle and have not congressed to the metaphase plate.105 Other defects such as spindle pole body (SPB), kinetochore and centromere abnormalities also activate the checkpoint,100 and it is known that incorrect spindle orientation (relative to the cell axis) can delay the onset of anaphase.106 Therefore, the spindle assembly checkpoint monitors the process of bipolar attachment of all the chromosomes to the mitotic spindle and ensures that the spindle is correctly positioned.
Chromosomes become bioriented by amphitelic attachment of their kinetochores to spindle microtubules.100,107 The process of chromosome capture by the spindle occurs more or less randomly;107 within the same species and cell type, it is accomplished quickly in some cells, but takes much longer in others.105 Therefore, in animals and in yeast, the checkpoint is needed in every cell cycle.108-110 However, biorientation of the chromosomes on the mitotic spindle forms a stable structure,107,111 thus the correct alignment of chromosomes, creating the metaphase plate, is favored. Once the last chromosome becomes bioriented, the spindle assembly checkpoint signal diminishes and anaphase is initiated in a highly regimented manner.105
At least one facet of the checkpoint signal emanates from kinetochores that have not attached to the spindle.112 In higher eukaryotes, a phospho-epitope (recognized by the 3F3/2 antibody) is present on unattached kinetochores (Fig. 7).107,113 Attachment of microtubules to a kinetochore induces dephosphorylation of the 3F3/2 phospho-epitopes at that kinetochore. As chromosomes attach to the spindle, the 3F3/2 epitopes are dephosphorylated, and the checkpoint becomes inactive . The molecular basis of this phosphorylation is not understood, but mechanistically, it is thought that checkpoint sensors, that are tension-sensitive complexes residing within the kinetochores, control the kinetochore phosphorylation status.105,107,112,114 Elegant studies have shown that tension exerted on kinetochores, applied by manipulating chromosomes with a micro-needle, induces loss of the kinetochore 3F3/2 epitopes.115 Therefore, it appears as though a lack of tension generates the checkpoint signal.
The identity of the kinase which creates the 3F3/2 epitope is not known, but recent work indicates that it is an integral component of kinetochores. Cells lysed in detergent do not contain kinetochores that are reactive against the a-3F3/2 antibody, but the a-3F3/2 reactivity can be reinstated by the addition of ATP.116,117 The activity of the kinase must be tightly associated with kinetochores. Furthermore, the substrate and kinase are likely to be associated. In theory, this “in vitro” system could be used as a biochemical assay to identify the kinase.
Genetic studies have revealed components of the yeast spindle assembly checkpoint (Fig. 8). Several groups of checkpoint proteins were identified in genetic screens designed to find mutants sensitive to microtubule antagonists. These are Mad1, Mad2, Mad3 (Mitotic Arrest Defective),109 and Bub1, Bub2, Bub3 (Budding Uninhibited by Benzimidazole).118 In addition, Mps1 is required.119 Many of these proteins have homologs in higher eukaryotes (see Table 1). One of these proteins, Mad2, was shown to bind selectively to phosphorylated kinetochores in vertebrate cells.117 Conversely, Mad2 binding was inhibited by kinetochore-microtubule attachment.120 Therefore, phosphorylated components of attachment-sensitive or tension-sensitive complexes might be recognized by Mad2. The current hypothesis is that Mad2 binding to 3F3/2-positive epitopes leads to the formation of an active checkpoint complex. In this model, kinetochores are catalytic sites for formation of the checkpoint signaling element, namely the activated Mad2 complex.
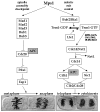
Table 1
Mitotic checkpoint components.
The target of the activated checkpoint complex was revealed in key experiments demonstrating that cell cycle arrest is brought about by inhibition of APC activity, which in turn prevents Pds1 ubiquitination and subsequent degradation. Yeast Mad2 was shown to bind to Cdc20, a component of the APC required for Pds1 degradation,121,122 and this binding can inhibit ubiquitination of APC substrates.121 Overexpression of CDC20, or expression of a cdc20 mutant that cannot bind to Mad2, bypasses the spindle assembly checkpoint arrest.122 In the catalytic kinetochore model, unattached kinetochores might form an active site at which Mad2-Cdc20 complexes are assembled, then released, thereby excluding Cdc20 from APCs. Alternatively, active Mad2 complexes might be released from kinetochores allowing them to inhibit APCCdc20 in other cellular locations. The latter model is supported by measurements of Mad2 localization dynamics in living cells; Mad2 is a transient component of unattached kinetochores, having a t1/2 of roughly 25 seconds.123
But what is the nature and function of the active Mad2 complex? Studies in yeast have shown that spindle defects activate kinase Mps1, resulting in Mad1 hyperphosphorylation (perhaps directly by Mps1).119,124 Overexpression of Mps1 alone can activate the checkpoint, and this arrest is (at least partly) dependent on Mad1, Mad2, Mad3 and Bub1, Bub2, Bub3. Mad1 phosphorylation also requires Bub1, Bub3 and Mad2.124,125 This modified form of Mad1 is required to mediate metaphase arrest. Significantly, Mad1 has been shown to bind to Mad2, and in this complex, Mad1 is a better substrate for Mps1 kinase than is unbound Mad1. At least in Xenopus egg extracts, Mad1 is required for the association of Mad2 to kinetochores that are not attached to the mitotic spindle.126 Together, the yeast genetic data and studies in higher eukaryotes indicate that checkpoint activation relies on the recognition of unattached kinetochores by Mad2, and the formation of an activated Mad1-Mad2 complex in which Mad1 is hyperphosphorylated. But how do the other checkpoint components fit into this scheme?
Somewhat parallel to the case of the Mad1-Mad2 complex, yeast Bub1 and Bub3 are tightly associated.127 This is also the case in mammalian cells, and the Bub1 domain required for Bub3 binding is also needed for localization of Bub1 to kinetochores.128 The implication is that Bub3 drives localization of Bub1 to kinetochores, as is the case for Mad1 and Mad2. A recent study of the budding yeast proteins sheds some light into how these complexes might be related.108 Mad1 was shown to associate with Bub1 and Bub3 in unperturbed cell cycles, and the amount of the complex in cells was increased in response to spindle checkpoint activation. Mad2 and Mps1 are required for the formation of the Bub1-Bub3-Mad1 complex in yeast.108 A Mad1 mutation that abolished Bub1-Bub3-Mad1 complex formation also led to a defective spindle checkpoint.
But how are the Mad1-Mad2 and Bub1-Bub3 complexes related? It may be the case that each complex becomes localized to kinetochores under slightly different conditions, in order to broaden the scope of defects that the checkpoint can detect. However, since deletion mutants of any one of these components results in a fully defective checkpoint, it is hard to argue that the complexes play entirely redundant roles. Instead, the different complexes might well detect different aberrations, but still all be needed for generating the active checkpoint complex that inhibits APCCdc20. Recent studies have allowed a working model to explain such an interconnection between the Mad1-Mad2 and the Bub1-Bub3 complexes, and how APCCdc20 might be inhibited.108 Although Bub1-Bub3-Mad1 complexes exist in yeast, and this complex forms in a manner dependent on Mad2 and Mps1 kinase activity, Mad2 is not found in this complex.108 Additionally, the Bub1-Bub3-Mad1 complex does not seem to be able to bind to Cdc20.108 This might suggest that an exchange mechanism is necessary to generate the active checkpoint complex (Fig. 9). In such a model, Mad2 is displaced from Mad1-Mad2 complexes, induced by Mps1 kinase, simultaneously stimulating the formation of the Bub1-Bub3-Mad1 complex on the one hand and an active Mad2 complex on the other.108,129 Mad1 phosphorylation, dependent on Mps1, Bub1, Bub3 and Mad2, may also be involved in this exchange. The nature of the activated Mad2 complex in not known but may include Mad3.129

In support of this model, animal homologues of Mad1, Mad2, Mad3, Bub1 and Bub3 are found at the kinetochores of prophase and prometaphase (not yet bioriented) chromosomes. Following congression to the metaphase plate, these proteins seem to dissociate.130 These localization studies suggest that formation of an active checkpoint complex within kinetochores is likely to be a conserved mechanism that activates the checkpoint pathway. But how is the checkpoint signal mobilized? How does a single, unattached kinetochore generate a signal that inhibits anaphase spindle elongation and prevents loss of sister chromatid cohesion of all the other chromosomes? One study has revealed important information that should help to resolve this question. Rieder et al. examined the timing of anaphase onset in cells that contain two functional and independent spindles.131 Such polykaryons are generated by cell fusion. When two cells at different stages of the cell cycle are fused, cell cycle progression of their nuclei soon becomes synchronized, allowing measurements of anaphase timing in independent spindles that share a common cytoplasm. This analysis revealed that the inhibitor emanating from a single unattached kinetochore is not freely diffusible, but rather is likely to be associated with the spindle itself. Therefore activated complexes might track from kinetochores along spindles.
Checkpoint Control Of Mitotic Exit
Many of the components of the mitotic exit machinery have been identified by the cell cycle phenotype of budding yeast mutants which arrest as a large dumbbells with elongated spindles (see Table 1 and 2). This phenotype is consistent with arrest at the anaphase/telophase transition. Mutants are unable to pass this arrest, and the proteins are therefore essential for exit from mitosis. These genes appear to define a GTP-dependent kinase signaling cascade, ultimately releasing a phosphatase that induces spindle disassembly, cytokinesis and mitotic exit. Control of mitotic exit therefore resides in the inhibition of this essential pathway.
Table 2
Function/localization of mitotic checkpoint components.
Exit from mitosis absolutely requires inhibition of B-type cyclin/Cdk activity. Under normal circumstances this is mediated by both inhibition of Cdk activity and by degradation of mitotic cyclins. Study of the S. cerevisiae spindle checkpoint has revealed that the ‘spindle assembly’ checkpoint is branched, inhibiting both the transition from metaphase to anaphase and mitotic exit (Fig. 8). The different functions of the two branches begs the question as to whether it is erudite to continue calling both branches by the term “spindle assembly checkpoint”. Others have begun to call the two branches the “spindle assembly” and “spindle position” checkpoints.132 For the purposes of this review we continue to use the term spindle assembly checkpoint for the inhibition of the metaphase-anaphase transition and the generic term “mitotic exit control” for the branch that regulates the activity of the B-type cyclin/Cdk activity. The mammalian machinery for mitotic exit control is currently being elucidated while substantial in-roads into understanding the mechanism has been achieved in S. cerevisiae. Here we describe current knowledge in the S. cerevisiae checkpoint control of mitotic exit (Fig. 10). Comparison to mammalian homologues and discussion of their possible clinical importance in tumorigenesis is left for the next section.

Evidence that Bub2 operates in a separate checkpoint pathway to the Bub1, 3, Mad1-3 pathway (hereafter collectively referred to as the Mad2 pathway) came from studies of double mutants treated with antitubulin drugs.133-138 Double mutant combinations that included bub2D failed to arrest in nocodazole whilst double mutant combinations that did not include Bub2 retained a mitotic delay. However, in mad2D cells treated with nocodazole the metaphase-anaphase transition occurs with kinetics comparable to those of untreated cells while bub2D cells delay the metaphase-anaphase transition. In addition, delay of the cell cycle in ctf13 mutants (limited for a key kinetochore component) requires Bub1 and 3 and Mad 1, 2 and 3 but is independent of Bub2.139 In cdc20 mutants, the mitotic arrest caused by maintained Pds1 levels is dependent on Bub2 but independent of the Mad2 pathway genes.140 Inhibition of Dbf2 in late mitotic arrest requires Bub2 but not the Mad2 pathway proteins.141 Together, these studies define distinct pathways. The Mad2 pathway ultimately targets Pds1, preventing spindle elongation and loss of sister chromatid cohesion at the metaphase to anaphase transition. The Bub2 pathway inhibits mitotic cyclin/Cdk activity and thus prevents spindle disassembly and exit from mitosis. Since the Bub2 pathway also monitors spindle integrity and is triggered by microtubule depolymerizing agents, there is a common upstream element, kinase Mps1. However, in contrast to the inhibition of anaphase onset via APCCdc20 regulation mediated by the Mad2 pathway, the Bub2 pathways appears to primarily act by inhibition of mitotic cyclin degradation and maintenance of mitotic cyclin dependent kinase activity. This is achieved by suppression of APCCdh1 and Sic1 activity which promote Clb1/Clb2 degradation and inhibit mitotic cyclin-dependent kinase activity, respectively.
The mitotic exit branch of the checkpoint is essential if microtubule polymerization is perturbed during anaphase, i.e. after APCCdc20-dependent degradation of Pds1. Normal progression of the cell cycle ensures that Cdc20 remains active and bound to the APC until after the onset of anaphase, when Cdh1 replaces Cdc20 as the APC specificity factor targeting B-type cyclins for degradation. However, deletion of CDH1 is not sufficient to prevent mitotic exit since inactivation of mitotic cyclin-dependent kinase activity by Sic1 is sufficient to allow exit from mitosis. The redundancy of APCCdh1 and Sic1 activity ensures that cells may exit mitosis in the absence of checkpoint stimulation. In the presence of checkpoint stimulation, the activity of both Sic1 and APCCdh1 is inhibited by nucleolar sequestration of the phosphatase Cdc14. Indeed, it appears that release of Cdc14 from the nucleolus is a key event in mitotic exit. The mechanism of Cdc14-mediated exit from mitosis appears to be three-fold. First, by dephosphorylating Cdh1 the APC targets the B-type cyclins for degradation. Ordinarily, phosphorylation of Cdh1 by Cdc28 is inhibitory and thus is self protecting,142 but when Cdh1 is dephosphorylated by Cdc14, this self-protection is removed. Second, by dephosphorylating Swi5, activating the transcription of Sic1, and third by directly dephosphorylating Sic1 itself. Thus Cdc14 both inhibits the activity of cyclin/Cdk activity and induces the destruction of the cyclin components.143
How does the “mitotic exit” checkpoint inhibit the release of Cdc14 from the nucleolus? Throughout most of the cell cycle Cdc14 is held inactive within the nucleolus in complex with Net1/Cfi1 (Fig. 11)144,145 termed the RENT complex (regulator of nucleolar silencing and telophase). This inactive localization appears to be dependent upon Tem1,145 a GTP binding protein localized to the daughter-bound (SPB). Cdc14 may also play a structural role in the nucleolus.146 A recent paper proposes a mechanism for monitoring the completion of anaphase and presents a compelling model.147 When SPB-associated Tem1-GDP locates to the bud at the end of anaphase, it interacts with a cortical protein Lte1, which is a GDP/GTP exchange factor (GEF). Tem1-GDP is activated by conversion to Tem1-GTP triggering the release of Cdc14 via a kinase cascade termed the mitotic exit network (MEN see below). Even when localized to the bud cortex, Tem1 activation could be inhibited by GAP (GTPase activating protein) activity of Bub2, preventing exit from mitosis. There are several compelling reasons for supposing this to be the checkpoint mechanism. Bub2 has considerable sequence homology to cdc16 in S. pombe, which is known to form a two-component GAP with byr4. Together they activate the GTPase encoded by spg1, the S. cerevisiae homolog of Tem1. Furthermore, deletion of the cerevisiae homolog of byr4 (Bfa1) causes a phenotype similar to bub2D, as does overexpression of Tem1. Finally, localization of Bub2 to the SPB and preferentially to that destined for the daughter cell,138,148,149 provides strong circumstantial evidence in support of this model.

The mechanism by which activation of Tem1 leads to the release of Cdc14 from the nucleolus via the MEN involves a number of key components including Cdc15, Cdc5, Dbf2, Dbf20 and Mob1.150 Most appear essential for mitotic exit since single mutants are lethal, while in the case of Dbf2 and Dbf20, it is only the double mutant that is synthetically lethal.151 Localization and phosphorylation of these proteins appear to be important factors during the cell cycle and probably contribute to their function (Table 2). In particular, many of the components localize to the nucleolus or are asymmetrically distributed between the SPBs, being preferentially bound to that destined for the daughter cell. Cdc15 localizes to the SPB during mitosis and relocates to the bud neck after telophase.152,153 Furthermore, Cdc15 phosphorylation increases gradually during the cell cycle until it is rapidly dephosphorylated in late mitosis.152,154 Like Cdc15, Dbf2 localizes to the SPB and moves to the bud neck in telophase.155 While asymmetric localization to the daughter-bound SPB is true for some components, others such as Mob1 (or at least the S. pombe homolog), localize symmetrically to both spindle pole bodies. Localization to the SPB and the relocation of these components to the bud neck in telophase mimics the localization in S. pombe in which their homologs principally act by regulating cytokinesis rather than mitotic exit per se.
While mechanistic details of the MEN/RENT complex are being reported, much work has still to be completed before a clear picture can be drawn. Some details of physical interactions and co-localization can at least allow some description of the cascade (Fig. 10). The rapid dephosphorylation of Cdc15 in late mitosis appears to be mediated by Cdc14.153 However, there also appears to be a role for Cdc15 as an activator of Cdc14, and it is thus both an activator and substrate.154 Also there appears to be a role for Pds1 as an inhibitor of B-type cyclin degradation independent of its role as a securin156 thus forming potential “crosstalk” between the Mad2 and Bub2 checkpoint branches. What is less clear is the role of Cdc5 which physically interacts with Dbf4 (part of the DNA replication machinery) but as yet has no clearly defined role in the mitotic exit pathway. As Cdc5 is a target of DNA damage checkpoint control, this component offers an attractive link between the damage checkpoint and mitotic exit control. Direct interactions have been established for a number of MEN components while the exact nature of many interactions remain unclear. For example, physical interaction between Mps1 and Mob1 has been demonstrated. Furthermore, interactions between Mob1 and Dbf2 and Dbf20 have been demonstrated. However, there has been no direct link established between these components and the remaining MEN components to date. Similarly, the role of Cdc5 in the MEN has yet to be elucidated. Nonetheless, we present an attempt to order the events of mitotic exit regulation based upon physical interactions and localization in Fig. 10. In this scheme we present Cdc15 “upstream” of Cdc14 as the former localizes to the SPB coincident with other upstream elements.
Evidence suggests that Bub2, and its associated partner Bfa1, participate in an essential checkpoint that is also activated by DNA damage.157,158 Thus the maintenance of B-type cyclin/Cdk activity by the Bub2 pathway may represent a universal mechanism that can respond to stress at any stage of G2 and M-phase. Other genetic interactions of Bub2 include synthetic lethality of a bub2D arc35-1 mutant159 and that Bub2 is essential for arrest in tub4-1 cells.160 Thus, the Bub2 pathway seems to respond to defects in spindle orientation, spindle localization, spindle damage and DNA damage.
Oncological Implications of Mitotic Checkpoint Homologs
The existence of numerous mitotic exit and spindle assembly checkpoint protein homologs in S. pombe and higher eukaryotes suggests that similar mechanisms regulate mitotic exit in all eukaryotes despite the fact that the asymmetric cytokinesis in S. cerevisiae appears to have fundamentally different spatial and temporal strategies. As aberrant mitosis frequently results in asymmetric distribution of the genetic material and aneuploid daughter cells, dysfunctional regulation of these checkpoints has become an attractive hypothetical mechanism for chromosomal instability in mammalian tumorigenesis. Indeed, some established tumor cell lines and tumors appear to have dysfunctional checkpoint controls23,161,162 and some of the checkpoint proteins appear to be targets of oncogenic viral proteins.163 However, screening of aneuploid colorectal tumor panels for such mutations revealed only mutations in the human hBUB1/hBUBR1 genes.164 A similar study of 31 aneuploid lung, and head and neck, tumors showed no such mutations.165 One hBUB1 somatic mutation that led to an amino acid substitution was found among 30 human primary lung cancer tumors.166 hBUB1 and hBUBR1 mutants have also been found in adult T-cell leukemias/lymphomas167 and some colorectal tumor cell lines.161 Perhaps significantly, one study has implicated Brca2, which is responsible for a fraction of the inherited susceptibilities to breast cancer, in the spindle assembly checkpoint.168 Brca2 was found to interact with hBubR1 and was phosphorylated by hBubR1 in vitro, though no direct role in the checkpoint was demonstrated. Although inactivation of Bub1 appears to confer chromosomal instability,161 more studies are required to determine whether mutations in BUB1 and other mitotic checkpoint proteins represent significant causative events or whether other checkpoints may account for aneuploid tumorigenesis.
In addition to the MAD2 and BUB2 pathway components, a number of other S. cerevisiae genes appear to have mammalian homologs which have been implicated as either proto-oncogenes or as tumor suppressor genes (see Table 1). Notably S. cerevisiae Pds1 may have two mammalian homologs, at least one of which is associated with pituitary tumors.169-172 Most of the human genes have been localized at least to the chromosome level, and recent publication of the human genome will therefore facilitate further study. Furthermore, many mouse homologs have been identified, and murine models of tumorigenesis may further elucidate the contribution of these genes to tumorigenesis. At the present time, only two knockout mouse models have been reported, MAD2173 and BUB3,174 both of which are early embryonic lethals. In the BUB3 mouse, from day 3.5 onwards, embryonic cells display mitotic aberrations such as micronuclei, anaphase chromosome laggards and bridging. In the presence of microtubule antagonists, the cells fail to arrest in metaphase.
References
- 1.
- Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225:159–162. [PubMed: 5409962]
- 2.
- Downes CS, Musk SR, Watson JV. Caffeine overcomes a restriction point associated with DNA replication, but does not accelerate mitosis. J Cell Biol. 1990;110:1855–9. [PMC free article: PMC2116112] [PubMed: 2161852]
- 3.
- Downes CS, Clarke DJ, Mullinger AM. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature. 1994;372:467–70. [PubMed: 7984241]
- 4.
- Ghosh S, Schroeter D, Paweletz N. Okadaic acid overrides the S-phase check point and accelerates progression of G2-phase to induce premature mitosis in HeLa cells. Exp Cell Res. 1996;227:165–9. [PubMed: 8806464]
- 5.
- Musk SR, Downes CS, Johnson RT. Caffeine induces uncoordinated expression of cell cycle functions after ultraviolet irradiation.Accelerated cycle transit, sister chromatid exchanges and premature chromosome condensation in a transformed Indian muntjac cell line. J Cell Sci. 1988:591–9. [PubMed: 3253296]
- 6.
- Schlegel R, Pardee AB. Caffeine-induced uncoupling of mitosis from the completion of DNA replication in mammalian cells. Science. 1986;232:1264–1266. [PubMed: 2422760]
- 7.
- Steinmann KE, Belinsky GS, Lee D. Chemically induced premature mitosis: differential response in rodent and human cells and the relationship to cyclin B synthesis and p34cdc2/cyclin B complex formation. Proc Natl Acad Sci U S A. 1991;88:6843–7. [PMC free article: PMC52185] [PubMed: 1830667]
- 8.
- Tolmach LJ, Jones RW, Busse PM. The action of caffeine on X-irradiated HeLa cells. I. Delayed inhibition of DNA synthesis. Radiat Res. 1977;71:653–65. [PubMed: 897091]
- 9.
- Tolmach LJ, Busse PM. The action of caffeine on X-irradiated HeLa cells. IV. Progression delays and enhanced cell killing at high caffeine concentrations. Radiat Res. 1980;82:374–92. [PubMed: 7375638]
- 10.
- Moser BA, Brondello JM, Baber FB. Mechanism of caffeine-induced checkpoint override in fission yeast. Mol Cell Biol. 2000;20:4288–94. [PMC free article: PMC85796] [PubMed: 10825192]
- 11.
- Blasina A, Price BD, Turenne GA. Caffeine inhibits the checkpoint kinase ATM. Curr Biol. 1999;9:1135–8. [PubMed: 10531013]
- 12.
- Sarkaria JN, Busby EC, Tibbetts RS. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–82. [PubMed: 10485486]
- 13.
- Zhou BB, Chaturvedi P, Spring K. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 275:10342–8. [PubMed: 10744722]
- 14.
- Yamamoto A, Guacci V, Koshland D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J Cell Biol. 1996;133:99–110. [PMC free article: PMC2120772] [PubMed: 8601617]
- 15.
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J Cell Biol. 1996;133:85–97. [PMC free article: PMC2120769] [PubMed: 8601616]
- 16.
- Cohen-Fix O, Peters JM, Kirschner MW. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–93. [PubMed: 8985178]
- 17.
- Cohen-Fix O, Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci U S A. 1997;94:14361–6. [PMC free article: PMC24978] [PubMed: 9405617]
- 18.
- Ciosk R, Zachariae W, Michaelis C. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067–76. [PubMed: 9635435]
- 19.
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. [PubMed: 9335333]
- 20.
- Uhlmann F, Nasmyth K. Cohesion between sister chromatids must be established during DNA replication. Curr Biol. 1998;8:1095–101. [PubMed: 9778527]
- 21.
- Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400:37–42. [PubMed: 10403247]
- 22.
- Jensen S, Segal M, Clarke DJ. A novel role of the budding yeast separin Esp1 in anaphase spindle elongation: evidence that proper spindle association of Esp1 is regulated by Pds1. J Cell Biol. 2001;152:27–40. [PMC free article: PMC2193664] [PubMed: 11149918]
- 23.
- Zou H, McGarry TJ, Bernal T. Identification of a Vertebrate Sister-Chromatid Separation Inhibitor Involved in Transformation and Tumorigenesis. Science. 1999;285:418–422. [PubMed: 10411507]
- 24.
- Allen JB, Zhou Z, Siede W. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994;8:2401–15. [PubMed: 7958905]
- 25.
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–65. [PubMed: 7926756]
- 26.
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–72. [PubMed: 8939848]
- 27.
- Weinert T. DNA damage checkpoints update: getting molecular. Curr Opin Genet Dev. 1998;8:185–93. [PubMed: 9610409]
- 28.
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. [PubMed: 7813016]
- 29.
- Sugimoto K, Shimomura T, Hashimoto K. Rfc5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast. Proc Natl Acad Sci U S A. 1996;93:7048–52. [PMC free article: PMC38933] [PubMed: 8692942]
- 30.
- Araki H, Leem SH, Phongdara A. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci U S A. 1995;92:11791–5. [PMC free article: PMC40488] [PubMed: 8524850]
- 31.
- Wang H, Elledge SJ. DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1999;96:3824–9. [PMC free article: PMC22379] [PubMed: 10097122]
- 32.
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article: PMC316339] [PubMed: 10640278]
- 33.
- Masumoto H, Sugino A, Araki H. Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously replicating sequence region of budding yeast. Mol Cell Biol. 2000;20:2809–17. [PMC free article: PMC85497] [PubMed: 10733584]
- 34.
- Lee SK, Johnson RE, Yu SL. Requirement of yeast SGS1 and SRS2 genes for replication and transcription. Science. 1999;286:2339–42. [PubMed: 10600744]
- 35.
- Michael WM, Ott R, Fanning E. Activation of the DNA Replication Checkpoint Through RNA Synthesis by Primase. Science. 2000;289:2133–2137. [PubMed: 11000117]
- 36.
- Tercero JA, Labib K, Diffley JF. DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. Embo J. 2000;19:2082–93. [PMC free article: PMC305696] [PubMed: 10790374]
- 37.
- Sanchez Y, Desany BA, Jones WJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–60. [PubMed: 8553072]
- 38.
- Frei C, Gasser SM. RecQ-like helicases: the DNA replication checkpoint connection. J Cell Sci. 2000:2641–6. [PubMed: 10893179]
- 39.
- Sun Z, Hsiao J, Fay DS. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science. 1998;281:272–4. [PubMed: 9657725]
- 40.
- Paciotti V, Clerici M, Lucchini G. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 2000;14:2046–59. [PMC free article: PMC316858] [PubMed: 10950868]
- 41.
- Rouse J, Jackson SP. LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. Embo J. 2000;19:5801–5812. [PMC free article: PMC305794] [PubMed: 11060031]
- 42.
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. [PubMed: 9741624]
- 43.
- Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–8. [PubMed: 9783589]
- 44.
- Longhese MP, Paciotti V, Neecke H. Checkpoint proteins influence telomeric silencing and length maintenance in budding yeast. Genetics. 2000;155:1577–91. [PMC free article: PMC1461196] [PubMed: 10924458]
- 45.
- Craven RJ, Petes TD. Involvement of the checkpoint protein Mec1p in silencing of gene expression at telomeres in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:2378–84. [PMC free article: PMC85413] [PubMed: 10713162]
- 46.
- Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–20. [PubMed: 10367890]
- 47.
- Clarke DJ, Segal M, Mondesert G. The Pds1 anaphase inhibitor and Mec1 kinase define distinct checkpoints coupling S phase with mitosis in budding yeast. Curr Biol. 1999;9:365–8. [PubMed: 10209118]
- 48.
- Blat Y, Kleckner N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell. 1999;98:249–59. [PubMed: 10428036]
- 49.
- Clarke DJ, Mondesert G, Segal M. Dosage suppressors of pds1 implicate UBA domains in checkpoint control Mol Cell Biol 2001. In Press. [PMC free article: PMC86795] [PubMed: 11238935]
- 50.
- Ortolan TG, Tongaonkar P, Lambertson D. The DNA repair protein Rad23 is a negative regulator of multi-ubiquitin chain assembly. Nat Cell Biol. 2000;2:601–608. [PubMed: 10980700]
- 51.
- Bartz SR, Rogel ME, Emerman M. Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J Virol. 1996;70:2324–31. [PMC free article: PMC190074] [PubMed: 8642659]
- 52.
- Withers WE, Jowett JB, Stewart SA. Human immunodeficiency virus type 1 Vpr interacts with HHR23A, a cellular protein implicated in nucleotide excision DNA repair. J Virol. 1997;71:9732–42. [PMC free article: PMC230283] [PubMed: 9371639]
- 53.
- Gragerov A, Kino T, Ilyina GG. HHR23A, the human homologue of the yeast repair protein RAD23, interacts specifically with Vpr protein and prevents cell cycle arrest but not the transcriptional effects of Vpr. Virology. 1998;245:323–30. [PubMed: 9636371]
- 54.
- Watt PM, Louis EJ, Borts RH. Sgs1: a eukaryotic homolog of E.coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–60. [PubMed: 7736577]
- 55.
- Yu CE, Oshima J, Fu YH. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–62. [PubMed: 8602509]
- 56.
- Ellis NA, Groden J, Ye TZ. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. [PubMed: 7585968]
- 57.
- Wang Y, Cortez D, Yazdi P. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–39. [PMC free article: PMC316544] [PubMed: 10783165]
- 58.
- Savitsky K, Bar SA, Gilad S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. [PubMed: 7792600]
- 59.
- Bentley NJ, Holtzman DA, Flaggs G. The Schizosaccharomyces pombe rad3 checkpoint gene. Embo J. 1996;15:6641–51. [PMC free article: PMC452488] [PubMed: 8978690]
- 60.
- Kastan MB, Zhan Q, el Deiry WS. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. [PubMed: 1423616]
- 61.
- Wright JA, Keegan KS, Herendeen DR. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci U S A. 1998;95:7445–50. [PMC free article: PMC22645] [PubMed: 9636169]
- 62.
- Tibbetts RS, Brumbaugh KM, Williams JM. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7. [PMC free article: PMC316393] [PubMed: 9925639]
- 63.
- Banin S, Moyal L, Shieh S. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. [PubMed: 9733514]
- 64.
- Canman CE, Lim DS, Cimprich KA. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. [PubMed: 9733515]
- 65.
- Cortez D, Wang Y, Qin J. Requirement of ATM-Dependent Phosphorylation of Brca1 in the DNA Damage Response to Double-Strand Breaks. Science. 1999;286:1162–1166. [PubMed: 10550055]
- 66.
- Bunz F, Dutriaux A, Lengauer C. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. [PubMed: 9822382]
- 67.
- Chan TA, Hermeking H, Lengauer C. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–20. [PubMed: 10524633]
- 68.
- Hermeking H, Lengauer C, Polyak K. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. [PubMed: 9659898]
- 69.
- Tibbetts RS, Cortez D, Brumbaugh KM. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. [PMC free article: PMC317107] [PubMed: 11114888]
- 70.
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7. [PubMed: 9836640]
- 71.
- Sanchez Y, Wong C, Thoma RS. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. [PubMed: 9278511]
- 72.
- Peng CY, Graves PR, Thoma RS. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–5. [PubMed: 9278512]
- 73.
- Matsuoka S, Rotman G, Ogawa A. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–10394. [PMC free article: PMC27034] [PubMed: 10973490]
- 74.
- Chaturvedi P, Eng WK, Zhu Y. Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene. 1999;18:4047–54. [PubMed: 10435585]
- 75.
- Kappas NC, Savage P, Chen KC. Dissection of the XChk1 Signaling Pathway in Xenopus laevis Embryos. Mol Biol Cell. 2000;11:3101–3108. [PMC free article: PMC14978] [PubMed: 10982403]
- 76.
- Sibon OC, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997;388:93–7. [PubMed: 9214509]
- 77.
- Yu KR, Saint RB, Sullivan W. The Grapes checkpoint coordinates nuclear envelope breakdown and chromosome condensation. Nat Cell Biol. 2000;2:609–615. [PubMed: 10980701]
- 78.
- Kumagai A, Guo Z, Emami KH. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J Cell Biol. 1998;142:1559–69. [PMC free article: PMC2141764] [PubMed: 9744884]
- 79.
- Guo Z, Kumagai A, Wang SX. Requirement for atr in phosphorylation of chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in xenopus egg extracts. Genes Dev. 2000;14:2745–56. [PMC free article: PMC317027] [PubMed: 11069891]
- 80.
- Liu Q, Guntuku S, Cui XS. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article: PMC316686] [PubMed: 10859164]
- 81.
- Kumagai A, Dunphy WG. Claspin, a Novel Protein Required for the Activation of Chk1 during a DNA Replication Checkpoint Response in Xenopus Egg Extracts. Mol Cell. 2000;6:839–849. [PubMed: 11090622]
- 82.
- Adachi Y, Luke M, Laemmli UK. Chromosome assembly in vitro: topoisomerase II is required for condensation. Cell. 1991;64:137–48. [PubMed: 1846085]
- 83.
- Hirano T, Mitchison TJ. Cell cycle control of higher-order chromatin assembly around naked DNA in vitro. J Cell Biol. 1991;115:1479–89. [PMC free article: PMC2289196] [PubMed: 1661728]
- 84.
- Hirano T, Mitchison TJ. Topoisomerase II does not play a scaffolding role in the organization of mitotic chromosomes assembled in Xenopus egg extracts. J Cell Biol. 1993;120:601–12. [PMC free article: PMC2119547] [PubMed: 8381118]
- 85.
- Uemura T, Ohkura H, Adachi Y. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell. 1987;50:917–25. [PubMed: 3040264]
- 86.
- Wood ER, Earnshaw WC. Mitotic chromatin condensation in vitro using somatic cell extracts and nuclei with variable levels of endogenous topoisomerase II. J Cell Biol. 1990:2839–50. [PMC free article: PMC2116389] [PubMed: 2176652]
- 87.
- Rose D, Holm C. Meiosis-specific arrest revealed in DNA topoisomerase II mutants. Mol Cell Biol. 1993;13:3445–55. [PMC free article: PMC359813] [PubMed: 8388537]
- 88.
- GimÄnez-Abiçn JF, Clarke DJ, Mullinger AM. A postprophase topoisomerase II-dependent chromatid core separation step in the formation of metaphase chromosomes. J Cell Biol. 1995;131:7–17. [PMC free article: PMC2120606] [PubMed: 7559788]
- 89.
- Holm C, Goto T, Wang JC. DNA topoisomerase II is required at the time of mitosis in yeast. Cell. 1985;41:553–563. [PubMed: 2985283]
- 90.
- Koshland D, Strunnikov A. Mitotic chromosome condensation. Annu Rev Cell Dev Biol. 1996;12:305–33. [PubMed: 8970729]
- 91.
- Dietzel S, Jauch A, Kienle D. Separate and variably shaped chromosome arm domains are disclosed by chromosome arm painting in human cell nuclei. Chromosome Res. 1998;6:25–33. [PubMed: 9510507]
- 92.
- GimÄnez-Abiçn JF, Clarke DJ, Devlin J. Premitotic chromosome individualization in mammalian cells depends on topoisomerase II activity. Chromosoma. 2000;109:235–44. [PubMed: 10968252]
- 93.
- Summers DK, Sherratt DJ. Multimerization of high copy number plasmids causes instability: CoIE1 encodes a determinant essential for plasmid monomerization and stability. Cell. 1984;36:1097–103. [PubMed: 6323019]
- 94.
- Summers DK, Sherratt DJ. Resolution of ColE1 dimers requires a DNA sequence implicated in the three-dimensional organization of the cer site. Embo J. 1988;7:851–8. [PMC free article: PMC454402] [PubMed: 3294000]
- 95.
- Patient ME, Summers DK. ColE1 multimer formation triggers inhibition of Escherichia coli cell division. Mol Microbiol. 1993;9:1089–95. [PubMed: 7523831]
- 96.
- Sharpe ME, Chatwin HM, Macpherson C. Analysis of the CoIE1 stability determinant Rcd. Microbiology. 1999:2135–44. [PubMed: 10463180]
- 97.
- Darbon JM, Penary M, Escalas N. Distinct Chk2 activation pathways are triggered by genistein and DNA-damaging agents in human melanoma cells. J Biol Chem. 2000;275:15363–9. [PubMed: 10809772]
- 98.
- Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406:430–5. [PubMed: 10935642]
- 99.
- Murone M, Simanis V. The fission yeast dma1 gene is a component of the spindle assembly checkpoint, required to prevent septum formation and premature exit from mitosis if spindle function is compromised. Embo J. 1996;15:6605–16. [PMC free article: PMC452485] [PubMed: 8978687]
- 100.
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–80. [PubMed: 8939672]
- 101.
- Nishimoto T, Uzawa S, Schlegel R. Mitotic checkpoints. Curr Opin Cell Biol. 1992;4:174–9. [PubMed: 1318057]
- 102.
- Straight AF. Cell cycle: checkpoint proteins and kinetochores. Curr Biol. 1997;7:R613–6. [PubMed: 9368739]
- 103.
- Straight AF, Murray AW. The spindle assembly checkpoint in budding yeast. Methods Enzymol. 1997;283:425–40. [PubMed: 9251039]
- 104.
- Waters JC, Salmon E. Pathways of spindle assembly. Curr Opin Cell Biol. 1997;9:37–43. [PubMed: 9013671]
- 105.
- Rieder CL, Schultz A, Cole R. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127:1301–10. [PMC free article: PMC2120267] [PubMed: 7962091]
- 106.
- O'Connell CB, Wang YL. Mammalian spindle orientation and position respond to changes in cell shape in a dynein-dependent fashion. Mol Biol Cell. 2000;11:1765–74. [PMC free article: PMC14882] [PubMed: 10793150]
- 107.
- Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–7. [PubMed: 9005842]
- 108.
- Brady DM, Hardwick KG. Complex formation between Mad1p, Bub1p and Bub3p is crucial for spindle checkpoint function. Curr Biol. 2000;10:675–8. [PubMed: 10837255]
- 109.
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. [PubMed: 1651172]
- 110.
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–35. [PubMed: 9182760]
- 111.
- Hyman AA, Karsenti E. Morphogenetic properties of microtubules and mitotic spindle assembly. Cell. 1996;84:401–10. [PubMed: 8608594]
- 112.
- Rieder CL, Cole RW, Khodjakov A. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–8. [PMC free article: PMC2199954] [PubMed: 7642709]
- 113.
- Gorbsky GJ, Ricketts WA. Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J Cell Biol. 1993;122:1311–21. [PMC free article: PMC2119849] [PubMed: 7690762]
- 114.
- McIntosh JR. Structural and mechanical control of mitotic progression. Cold Spring Harb Symp Quant Biol. 1991;56:613–9. [PubMed: 1819511]
- 115.
- Nicklas RB, Ward SC, Gorbsky GJ. Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J Cell Biol. 1995;130:929–39. [PMC free article: PMC2199958] [PubMed: 7642708]
- 116.
- Campbell MS, Daum JR, Gersch MS. Kinetochore “memory” of spindle checkpoint signaling in lysed mitotic cells. Cell Motil Cytoskeleton. 2000;46:146–56. [PubMed: 10891860]
- 117.
- Waters JC, Chen RH, Murray AW. Mad2 binding by phosphorylated kinetochores links error detection and checkpoint action in mitosis. Curr Biol. 1999;9:649–52. [PubMed: 10375530]
- 118.
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–17. [PubMed: 1651171]
- 119.
- Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–23. [PMC free article: PMC2120695] [PubMed: 8567717]
- 120.
- Waters JC, Chen RH, Murray AW. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol. 1998;141:1181–91. [PMC free article: PMC2137189] [PubMed: 9606210]
- 121.
- Li Y, Gorbea C, Mahaffey D. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci U S A. 1997;94:12431–6. [PMC free article: PMC24983] [PubMed: 9356466]
- 122.
- Hwang LH, Lau LF, Smith DL. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279:1041–4. [PubMed: 9461437]
- 123.
- Howell BJ, Hoffman DB, Fang G. Visualization of Mad2 Dynamics at Kinetochores, along Spindle Fibers, and at Spindle Poles in Living Cells. J Cell Biol. 2000;150:1233–1250. [PMC free article: PMC2150717] [PubMed: 10995431]
- 124.
- Hardwick KG, Weiss E, Luca FC. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–6. [PubMed: 8688079]
- 125.
- Hardwick KG, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–20. [PMC free article: PMC2120625] [PubMed: 7593191]
- 126.
- Chen RH, Shevchenko A, Mann M. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol. 1998;143:283–95. [PMC free article: PMC2132829] [PubMed: 9786942]
- 127.
- Roberts BT, Farr KA, Hoyt MA. The Saccharomyces cerevisiae checkpoint gene BUB1 encodes a novel protein kinase. Mol Cell Biol. 1994;14:8282–91. [PMC free article: PMC359367] [PubMed: 7969164]
- 128.
- Taylor SS, Ha E, McKeon F. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol. 1998;142:1–11. [PMC free article: PMC2133037] [PubMed: 9660858]
- 129.
- Hardwick KG, Johnston RC, Smith DL. MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol. 2000;148:871–82. [PMC free article: PMC2174553] [PubMed: 10704439]
- 130.
- Nasmyth K. Separating sister chromatids. Trends Biochem Sci. 1999;24:98–104. [PubMed: 10203756]
- 131.
- Rieder CL, Khodjakov A, Paliulis LV. Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage. Proc Natl Acad Sci U S A. 1997;94:5107–12. [PMC free article: PMC24639] [PubMed: 9144198]
- 132.
- Hoyt MA. Exit from mitosis: spindle pole power. Cell. 2000;102:267–70. [PubMed: 10975516]
- 133.
- Li R. Bifurcation of the mitotic checkpoint pathway in budding yeast. Proc Natl Acad Sci U S A. 1999;96:4989–94. [PMC free article: PMC21804] [PubMed: 10220406]
- 134.
- Gardner RD, Burke DJ. The spindle checkpoint: two transitions, two pathways. Trends Cell Biol. 2000;10:154–8. [PubMed: 10740270]
- 135.
- Burke DJ. Complexity in the spindle checkpoint. Curr Opin Genet Dev. 2000;10:26–31. [PubMed: 10679385]
- 136.
- Taylor SS. Chromosome segregation: dual control ensures fidelity. Curr Biol. 1999;9:R562–4. [PubMed: 10469560]
- 137.
- Alexandru G, Zachariae W, Schleiffer A. Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. Embo J. 1999;18:2707–21. [PMC free article: PMC1171353] [PubMed: 10329618]
- 138.
- Fraschini R, Formenti E, Lucchini G. Budding Yeast Bub2 Is Localized at Spindle Pole Bodies and Activates the Mitotic Checkpoint via a Different Pathway from Mad2. J Cell Biol. 1999;145:979–991. [PMC free article: PMC2133126] [PubMed: 10352016]
- 139.
- Wang Y, Burke DJ. Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:6838–44. [PMC free article: PMC230938] [PubMed: 8524250]
- 140.
- Tavormina PA, Burke DJ. Cell cycle arrest in cdc20 mutants of Saccharomyces cerevisiae is independent of Ndc10p and kinetochore function but requires a subset of spindle checkpoint genes. Genetics. 1998;148:1701–13. [PMC free article: PMC1460108] [PubMed: 9560388]
- 141.
- Fesquet D, Fitzpatrick PJ, Johnson AL. A Bub2p-dependent spindle checkpoint pathway regulates the Dbf2p kinase in budding yeast. Embo J. 1999;18:2424–34. [PMC free article: PMC1171325] [PubMed: 10228157]
- 142.
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9:227–36. [PubMed: 10074450]
- 143.
- Visintin R, Craig K, Hwang ES. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–18. [PubMed: 9885559]
- 144.
- Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398:818–23. [PubMed: 10235265]
- 145.
- Shou W, Seol JH, Shevchenko A. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233–44. [PubMed: 10219244]
- 146.
- de Almeida A, Raccurt I, Peyrol S. The Saccharomyces cerevisiae Cdc14 phosphatase is implicated in the structural organization of the nucleolus. Biol Cell. 1999;91:649–63. [PubMed: 10668096]
- 147.
- Bardin AJ, Visintin R, Amon A. A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell. 2000;102:21–31. [PubMed: 10929710]
- 148.
- Pereira G, Hofken T, Grindlay J. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell. 2000;6:1–10. [PubMed: 10949022]
- 149.
- Daum JR, Gomez ON, Winey M. The spindle checkpoint of saccharomyces cerevisiae responds to separable microtubule-dependent events. Curr Biol. 2000;10:1375–8. [PubMed: 11084338]
- 150.
- Jaspersen SL, Charles JF, Tinker KR. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2803–17. [PMC free article: PMC25555] [PubMed: 9763445]
- 151.
- Toyn JH, Johnston LH. Spo12 is a limiting factor that interacts with the cell cycle protein kinases Dbf2 and Dbf20, which are involved in mitotic chromatid disjunction. Genetics. 1993;135:963–71. [PMC free article: PMC1205757] [PubMed: 8307336]
- 152.
- Cenamor R, Jimenez J, Cid VJ. The budding yeast Cdc15 localizes to the spindle pole body in a cell-cycle-dependent manner. Mol Cell Biol Res Commun. 1999;2:178–84. [PubMed: 10662594]
- 153.
- Xu S, Huang HK, Kaiser P. Phosphorylation and spindle pole body localization of the Cdc15p mitotic regulatory protein kinase in budding yeast. Curr Biol. 2000;10:329–32. [PubMed: 10744974]
- 154.
- Jaspersen SL, Morgan DO. Cdc14 activates cdc15 to promote mitotic exit in budding yeast. Curr Biol. 2000;10:615–8. [PubMed: 10837230]
- 155.
- Frenz LM, Lee SE, Fesquet D. The budding yeast Dbf2 protein kinase localises to the centrosome and moves to the bud neck in late mitosis. J Cell Sci. 2000:3399–3408. [PubMed: 10984431]
- 156.
- Cohen-Fix O, Koshland D. Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit. Genes Dev. 1999;13:1950–9. [PMC free article: PMC316926] [PubMed: 10444593]
- 157.
- Neff MW, Burke DJ. A delay in the Saccharomyces cerevisiae cell cycle that is induced by a dicentric chromosome and dependent upon mitotic checkpoints. Mol Cell Biol. 1992;12:3857–64. [PMC free article: PMC360258] [PubMed: 1324407]
- 158.
- Wang Y, Hu F, Elledge SJ. The Bfa1/Bub2 GAP complex comprises a universal checkpoint required to prevent mitotic exit. Curr Biol. 2000;10:1379–82. [PubMed: 11084339]
- 159.
- Schaerer BC, Riezman H. Saccharomyces cerevisiae Arc35p works through two genetically separable calmodulin functions to regulate the actin and tubulin cytoskeletons. J Cell Sci. 2000:521–32. [PubMed: 10639338]
- 160.
- Spang A, Geissler S, Grein K. gamma-Tubulin-like Tub4p of Saccharomyces cerevisiae is associated with the spindle pole body substructures that organize microtubules and is required for mitotic spindle formation. J Cell Biol. 1996;134:429–41. [PMC free article: PMC2120879] [PubMed: 8707827]
- 161.
- Cahill DP, Lengauer C, Yu J. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. [PubMed: 9521327]
- 162.
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–8. [PubMed: 8824189]
- 163.
- Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. [PubMed: 9546394]
- 164.
- Cahill DP, da CL, Carson WE. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics. 1999;58:181–7. [PubMed: 10366450]
- 165.
- Yamaguchi K, Okami K, Hibi K. Mutation analysis of hBUB1 in aneuploid HNSCC and lung cancer cell lines. Cancer Lett. 1999;139:183–7. [PubMed: 10395177]
- 166.
- Gemma A, Seike M, Seike Y. Somatic mutation of the hBUB1 mitotic checkpoint gene in primary lung cancer. Genes Chromosomes Cancer. 2000;29:213–218. [PubMed: 10992296]
- 167.
- Ohshima K, Haraoka S, Yoshioka S. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma. Cancer Lett. 2000;158:141–150. [PubMed: 10960763]
- 168.
- Futamura M, Arakawa H, Matsuda K. Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1. Cancer Res. 2000;60:1531–5. [PubMed: 10749118]
- 169.
- McCabe CJ, Gittoes NJ. PTTG--a new pituitary tumour transforming gene. J Endocrinol. 1999;162:163–6. [PubMed: 10425453]
- 170.
- Zhang X, Horwitz GA, Heaney AP. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999;84:761–7. [PubMed: 10022450]
- 171.
- Zou H, McGarry TJ, Bernal T. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 1999;285:418–22. [PubMed: 10411507]
- 172.
- Prezant TR, Kadioglu P, Melmed S. An intronless homolog of human proto-oncogene hPTTG is expressed in pituitary tumors: evidence for hPTTG family. J Clin Endocrinol Metab. 1999;84:1149–52. [PubMed: 10084610]
- 173.
- Dobles M, Liberal V, Scott ML. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–45. [PubMed: 10892650]
- 174.
- Kalitsis P, Earle E, Fowler KJ. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 2000;14:2277–2282. [PMC free article: PMC316933] [PubMed: 10995385]
- DNA Damage-Independent Checkpoints from Yeast to Man - Madame Curie Bioscience D...DNA Damage-Independent Checkpoints from Yeast to Man - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...