

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Osteoporosis is "…a disease characterized by low bone mass, microarchitectural deterioration of bone tissue, and a consequent increase in fracture risk"—Copenhagen Consensus Conference (1990).
Osteoporosis is "…a bone density 2.5 standard deviations below the mean for young white adult women at lumbar spine, femoral neck or forearm"—The World Health Organization (1994).
Osteoporosis is "…a systemic skeletal disease characterized by low bone mass and microarchitectural deterioriation of bone tissue with a consequent increase in bone fragility and susceptibility of fractures"—The Hong Kong and Amsterdam Consensus Conferences (1996).
Osteoporosis is"…characterized by a failure to maintain bone architecture sufficiently robust to withstand the loading of everyday life without substantial risk of fracture"— Ehrlich and Lanyon (2002).
Osteoporosis is "…a skeletal disorder characterized by compromised bone strength predisposing to an increased risk of fracture"—National Institutes of Health (cited by Pearson et al., 2002).
Osteoporotic postmenopausal women don't need to fall or hit something to break their fragile bones. Their, hips, ribs, wrists and especially vertebrae are apt to be broken or crushed by the muscle pulls or bumps of ordinary daily activities. In fact, the greatest bone breakers are the person's muscles, because as R.B. Martin et al. (1998) pointed out "....the muscle forces acting on the skeleton are quite large relative to the forces exerted on it by the outside world." Those women who were lucky enough to have put enough into their bones when young to keep them above the "spontaneous" fracture threshold during the postmenopausal years of accelerated bone loss may only become "osteopenic" or pseudo-osteoporotic. Their osteopenic bones are more likely to be broken by falls and other bumps and blows due to poor eyesight and balance (Boxsein, 1999). But these bones are still strong enough to resist being broken by the shrinking muscles of older people during their shrinking activities. However, shrinking muscles are not unmixed blessings for bones, because there has to be enough muscle loading and strain to placate the bones' thrift-minded mechanostatic mechanism, which is programmed to destroy understrained bone rather than waste hard-earned resources to maintain it (Noble and Reeve, 2000; Skerry, 1999).
Osteoporosis in women is the most debilitating of the many consequences of the disappearance of estrogen at menopause. Perhaps surprisingly, the osteoporosis that develops more slowly in men is also at least partly due to an estrogen decline, in this case to the declining amounts of estrogen being made from the gradually dropping testosterone by the bone cells' enzyme called "aromatase" (Bilezekian, 2002; Khosla, 2002; Klein, 1999; Troen, 2003). Of course, the estrogen made by the fat cells' aromatase might also moderate the debilitating effects of the lack of ovarian estrogen on the bones of those postmenopausal women whose adrenal glands still make a significant amount of androgens.
The message is loud and clear—aging populations urgently need something that can stimulate bone growth to significantly improve deteriorated bone microstructure and to restore lost bone mass and strength. PTH, the parathyroid hormone, and some of its fragments, given along with a potent osteoclast inhibitor such as osteoprotegerin (OPG) or, as we are about to see, leptin, are currently the most promising answers to this need. But as we shall see further on, there is some very shaky evidence that the cholesterol-lowering, heart attack-preventing statins might be as good as the PTHs (reviewed by Whitfield, 2001a).
There is another kind of bone fragility or osteoporosis that will not greatly affect humankind in the near future but could prevent a few of us from exploring our neighboring planets. But it would be hugely important in the unlikely event that Homo "sapiens" survives its suicidal inter-tribal wars, Snowball Earths (Walker, 2003), and other climatic and plate tectonic challenges. Then H. "sapiens" would eventually have to flee the incinerating embrace of a bloated, dying Sun. Unfortunately, our load-bearing bones are designed to use gravitation-imposed strain to maintain them and at the same time avoid wasting expensive metabolic resources by hanging onto unused bone. The design of the strain-sensing device is elegantly simple. Cells called osteocytes, equipped with tiny bendable, flow-metering antennae and moored to the walls of the bones of our lower limbs with sensitive, tuggable integrin signal "wires" (i.e., matricrine signaling), need the constant ebb and flow of extracellular fluid caused by the bone-squeezing strains of terrestrial heel strikes to get the oxygen and nutrients cells need to function and to carry away their waste. If this gravitation-driven pumping and extracelluar fluid sloshing should fall below a critical level, the now-starving and -suffocating osteocytes, drowning in their own sewage, self-destruct, and in their death throes send signals that summon osteoclasts to get rid of their unneeded bone. So unless we develop effective chemical gravity, astronaut/cosmonaut explorers/emigrants will be stepping out onto another planet after many months or years in the microgravity of space with fracture-prone bones but without the facilities to fix them.
To try to understand how these new bone-making tools might work in the complex world of bones, we must first learn how bones are made and maintained.
BMUS—The Microcrack Fixers
"…If we increase the resolution of our investigation and focus on scales of microns to millimeters, we see that bone is a highly dynamic tissue, continually adjusting to its physiologic and mechanical environment by changes in its composition and microscopic architecture. An important principle of skeletal physiology is that bones are able to sense the mechanical loads which they bear and modify their structures to suit changes in these loads."—R.B. Martin et al. (1998; p. 31).
Strains, Microcracks, Macrocracks, and Molecular Screams
At first sight, bones are deaf, stupid, rock-like things consisting of a hard shell (cortical bone) enclosing a lattice of struts and plates (cancellous or trabecular bone), the spaces of which are filled with red (hemopoietic or blood-forming) marrow or yellow fatty marrow (Jee, 2001) (fig. 1). They are the body's calcium bank (they hold 99% of the body's calcium), they provide protective armour plate (e.g, the skull), or they are "simple" muscle-driven levers. However, they tell you they are very much alive by energetically protesting when they are broken. But then they only slowly mend themselves.
But wait! Look more closely and you may be surprised to see that, like the more charismatic and mercurial networking neurons in the brain, bone cells also live in networks and can talk to each other, share and store memories of past strains, and modify their responsiveness to strain by engraving their past experiences in their bones' structures (Turner et al., 2002). As I will show below, they are equipped like neurons to secrete, reaccumulate, and respond to neurotransmitters such as CGRP (calcitonin gene-related peptide), GABA (γ-amino butyric acid), glutamate, and serotonin (Mason, 2004; Spencer et al., 2004; A.F. Taylor, 2002). Indeed, it now seems likely that bone cells respond in direct proportion to intermittent strain frequency with the same CaMKII (calmodulin-dependent protein kinase II)-driven, long-term potentiation of mechanotransduction (bone formation) as neurons respond to high-frequency stimulation with long-term potentiation of synaptic transmission (Mason, 2004; Spencer and Genever, 2003; Spencer et al., 2004). In other words, bone cells carve their loading experience in bone through a mechanism just like the one used by neurons in the brain to drive the long-term synaptic changes that underly memory storage (Mason, 2004; Spencer et al., 2004). Bone cells have to be smart enough to know where they are and adjust their responsiveness to strain accordingly. If not, they would label relatively unstrained skull bones or ear bones, such as the stapes, as unused and destroy them while putting too much bone onto ribs and lower leg bones that are continuously subjected to breathing and high-strain, high-frequency heel, hoof, or paw strikes. The sophistication of the strain memory is illustrated by Turner et al. (2002), who found that the size of the osteogenic response of the cells of rat ulna to mechanical loading depended on the straining history of the part of the bone where they live. The cells in the habitually high-strain distal region were less responsive to loading than cells in the proximal region, which is habitually exposed to much less strain. In other words, the bone cells are not as stupid as they seem to be. Incredibly, they actually know where they are!! They know how much strain to expect in order to not overload load-bearing bones or destroy other, seemingly under-loaded but in fact essential, bone. Thus, it seems that bones, exactly like brains, record their experience, i.e., their loading history, in their structure, for example in the density of bone signaling connections and networks.
Bone seemingly masochistically harbors cells whose sole job description is simply—destroy bone! And they are good at their job. Obviously, it must have a good reason for harboring such cellular piranhas. And of course it does! A load-bearing bone is like a busy interstate highway, which cracks with the constant pounding by traffic and will eventually crumble unless the cracks are detected, dug out, and the holes refilled in a timely fashion by road maintenance crews. Bones too have maintenance crews—the bone-remodeling BMUs (Basic Multicellular Units). A BMU crew is activated every 10 seconds in an adult human bone, and at any time about 35,000,000 of them are at work removing cracks and digging out and replacing about 500 mg of calcium from the skeleton each day. However, the remodeling rate varies widely throughout the adult skeleton according to the level of microfracturing (R.B. Martin et al., 1998; S. Mori and Burr, 1993) and whether the bone, such as the endocortical surface of the ilium or femur, is bathed in red hemopoietic (blood cell-making) marrow or fatty yellow marrow (Parfitt, 2002). For example, the remodeling rate in adult human cortical bone can be as low as 2% per year in the distal radius and as high as 50% per year in ilial trabecular bone (Noble and Reeve, 2000). This difference is due to the trabecular bone being weaker (i.e., it has a lower elastic modulus—it takes less stress to deform) than cortical bone (R.B. Martin et al., 1998). Because of this, it microcracks more easily and must be remodeled more often than cortical bone, which results in its mineralization being less mature on the average and therefore less hardened than that of cortical bone (R.B. Martin et al., 1998). But as always with bone, things are not so simple (Parfitt, 2002). In cortical bone, remodeling can start anywhere along the so-called Havsersian (osteonal) tunnels running through the bone carrying the dense bones' plumbing (blood vessels) and electrical wiring (nerves), but trabecular remodeling starts only on the surface; therefore, the likelihood of remodeling a trabecula drops off the farther it is from the surface. However, trabeculae thicker than 200 μm do have a central osteonal canal. This means that the usual claim of a very high trabecular remodeling level (i.e., bone formation rate (BFR)/volume) is an overestimate, because it relies only on the surface remodeling level (surface BFR/V) without taking account of the very much lower intra-trabecular remodeling level (Parfitt, 2002).
Why Remodeling?
It appears that this continuous remodeling is characteristic of the big, densely "Haversian" bones of long-lived humans, horses, and elephants and ornithischian, saurischian, and certain therapsid dinosaurs (Bakker, 1986; Enlow and Brown, 1957, 1958; Rensberger and Watanabe, 2000), but not the tiny non-"Haversian" bones of short-lived mice and rats. The reason for this is simple. Big bones have more flaws than small bones and will therefore have shorter fatigue lives than smaller bones equally loaded/unit volume (R.B. Martin, 2003; D. Taylor et al., 1999). R.B. Martin (2003) has given a dramatic example of this size effect. He has estimated that the volumes of the femurs of a shrew, a human, and an elephant are in the relative proportions of 1:900:30,000. If these bones couldn't remodel, the fatigue life of the elephant's femur would be extremely short compared to the shrew's femur when repeatedly subjected to the same physiological strain. In the same paper, R.B. Martin gave another example of the relation between the probabilities of failure of equally loaded, equidiameter iron wires 1, 2, and 3 units in length. The failure probabilities were .63, .87, and .95, respectively. Therefore, the progressively larger and longer bones of the vertebrates that evolved in the Dinosauria and later in the Mammalia were intrinsically more prone to continuous fatigue damage. On the other hand, despite increasingly heavy musculature, the skeletons of these larger animals were selected to be as gracile as possible to give their owners the maximum mobility for their size and thus the maximum possible survivability (R.B. Martin, 2003). But there was an obvious and dangerous downside to this failure to increase bone size in step with muscle mass—increasing bone microdamage during physiological activity. The solution to this problem was the increasing use of the remodeling mechanism during normal activity. Thus, they had to routinely mobilize BMUs to repair the microcracks continuously appearing at high strain sites in the big bones caused by their owners' heel, hoof, and paw strikes (Burr, 1993; R. B. Martin, 2000a, 2000b, 2003; Tami et al., 2002; Verborgt et al., 2000; Whitfield et al., 2002b). Actually, the continual pulling on the ribs by muscles just during breathing causes them to have more microcracks, and thus the greatest remodeling rate and production of Haversian canals, of all the cortical bone in the body (Frost, 1960)! This of course would explain Enlow and Brown's comment (Enlow and Brown (1958)) that "…if a form does possess Haversian tissue in any part of its skeleton, the rib is, with very few exceptions, Haversian". However, the BMU remodeling mechanism was probably available for exploitation, at least in the small mammalian ancestors, because rat bones do resort to Haversian remodeling when needed. If a rat bone such as the ulna, which has no osteons and normally no microdamage or remodeling, is cyclically super-fatigue-loaded enough to produce linear microcracks and more diffuse patches of damage, typical intracortical remodeling starts in a few days to remove the linear microcracks (Bentolila et al., 1998).
But as with all physiologically good things, we shall soon see that remodeling can be dangerous when its level rises above a certain point, as does in postmenopausal women because, for example, the remodeling-driven loading of cortical bone with new BMU-produced Haversian canals increases bone porosity and hence fragility (R.B. Martin et al., 1998).
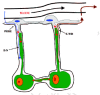
The Road into the Boney Interior, Sloshing Fluids, Flow-metering Cilia, Tugged Integrins, Osteointernet Chattering
The "Haversian" bones are so named because their cortical walls are made up of bundles of individually branching osteons arranged generally along the main loading direction that look like tubular onions, each consisting of layers (or lamellae) of bone wrapped around a central, Haversian canal (named after Clopton Havers (1691). The central canal is a pipeline for nerves and blood vessels from the marrow that convey messages from the hypothalamus, carry food supplies to, and waste from, the network of osteocytes locked in the lamellae (Cowin, 2000; Kerr, 2000; R.B. Martin et al., 1998; S. Takeda et al., 2002) (fig. 2). Blood is pumped into a bone, such as a femur, first through the superior and inferior epiphyseal and metaphyseal arteries and the diaphyseal feeding artery, then through marrow vasculature and only then into the endosteal (i.e., internal) surface and then through the Haversian canals (reviewed by Knothe Tate, 2000). Since the marrow vascular and extravascular compartments equilibrate rapidly, and since the trabeculae and endosteum are very close to the marrow vessels, their osteocytes and lining cells receive any blood-borne agent such as PTH (parathyroid hormone to be discussed at length further on) first, fully and faster than osteocytes in the cortical bone (Knothe-Tate, 2001; Knothe-Tate et al., 2004). A cortical osteocyte in a long bone receives the agent only after the blood vessel carrying it enters the cortex and then branches into the local Haversian canal and then travels through the lacunocanalicular network to the osteocyte. So the agent can be substantially depleted before the cortical osteocytes, especially those farthest from their Haversian capillary, can get their chance to be affected by it. In other words, the trabecular bone is a prefilter or affinity column.
The osteocytes are the bones' professional strain monitors that are programmed to respond when the strain is above or below a particular bone's expected range (Burger, 2001; Klein-Nulent et al., 2003). Osteocytes are the chosen few—the 4% of the osteoblast teams that made the bone but did not have to self destruct after a cortical tunnel or trabecular trench was filled and the big, no longer needed, osteoblast crew had to be laid off. In other words they are retired osteoblasts that were saved from apoptotic self destruction by being locked up in the new bone. But they had to undergo a career change and shrink down from plump osteoblasts into lacuna-locked, spider-shaped chemical and strain sensors as they put away their bone-making tools.
The osteocytes are directly connected to other osteocytes, surface lining cells and less directly to the blood vessels and nerves coursing through the Haversian and Volkmann's canals. They become parts of the bone's 3-D osteointernet (Burger, 2001; R.B. Martin et al., 1998; Moss and Cowin, 1997; L. Wang et al., 2000) (fig. 2). They live in tiny cells called lacunae and extend processes through narrow canals known as canaliculi to plug directly into the other members of the internet with so-called gap junctions, which by directly joining the cells' cytoplasms form the "internal pathway" (fig. 2). (Wonderful methacrylate-divinlybenzene replicas of the awesomely interlacing lacunocanalicular network of a mandibular bone's osteointernet can be seen in Figures 6a,b of Atkinson and Hallsworth (1983)). The canaliculi end in tiny openings or ports in the lining cell-covered osteonal wall, and the processes may set up gap junctions with the lining cells. High molecular weight nutrients from the osteonal blood vessels are pushed through the ports and along the extracellular channel, the "external pathway", in the canaliculi, and wastes are sucked out of them into the osteonal lumen and ultimately the blood by the cycles of compression and expansion of the network during various body movements (Knothe-Tate, 2001,2003; L. Wang et al., 2000) (fig. 2). In trabecular bone, the canalicular ports also open onto lining cell layers, but these lie directly on the vascular elements (sinusoids) of the bone marrow, which are the equivalents of the blood vessels of the cortical bone's osteonal plumbing system except any injected agent, such as a growth factor or hormone, arrives there before the osteonal plumbing (e.g., Knothe-Tate, 2001, 2003; L. Wang et al., 2000).
The osteocytes' processes tend to be directed perpendicularly to the bone surface, i.e., the vascular surface, which means that their prime communication is with lining cells, and moreover some processes can extend beyond the bone surface into the adjacent blood vessels and in the case of adjacent marrow to send information about loading directly to nests of early osteoblast and osteoclast progenitors (Kamioka et al., 2001; Klein-Nulent et al., 2003; Palumbo et al., 1990). In other words, they are part of a giant cellular syncytium gated by large gap junctional pores. However, molecules that are too big to pass through the gap junctions can diffuse along or, in an actively bending bone, be swept along in the sloshing extracellular fluid, via the external pathway at a rate depending on canalicular diameter and pushing and sucking intensities (Knothe-Tate, 2001; Tami et al., 2002; L. Wang et al., 2000). However, the efficiency of the nutrient and factor delivery and waste removal falls off with the distance of the osteocytes from the osteonal or marrow canalicular ports, and these distant cells may therefore be more likely to be more vulnerable to stress and less responsive to any injected or swallowed factor (L. Wang et al., 2000). However, it must be noted that osteocytes do try to compensate for this long distance (or "remoteness") factor by having more processes and canaliculi than those nearest the Haversian canal, much as trees increase their root systems to get enough water and nutrients in dry regions (Knothe-Tate, 2001).
Let's now follow molecules, such as one of the PTHs, that will figure so prominently further on from syringe in skin to the nutrient artery of a femur. Their itinerary has been supplied by Knothe-Tate (2003). The molecules in the marrow blood vessels first see the cells lining the trabeculae, where some will stay and stimulate while others pass through into the trabeculae. The rest are carried by the capillaries into the Haversian channels of the cortical bone. The PTHs are pushed through the vascular walls under a pulsing head of pressure as cyclic bone loading periodically squeezes the bone marrow and its blood vessels. The peptides pass through the layer of bone-lining cells that serve as endosteal and osteonal wall "tiles", regulators of the movements of Ca2+ and K+ into and out of the canalicular fluid, and reversibly retired osteoblasts that can be a rapidly mobilizable "National Guard" to provide osteoblasts for crack-repairing BMUs. The fluid carrying the PTHs then passes through the 500-600-nm canalicular ports. The interlamellar matrix is also a molecular sieve, a slow pathway, through which the 3-10 kDa PTH fragments (but not molecules larger than 40 to 70 kDa) can diffuse. The fluid then flows along the space between the osteocyte surface and the canaliculus wall. The space is not empty—it contains collagens and proteoglycans that impede the PTH progression. However, in the case of the PTHs, they can help their progression along the canaliculus by increasing the porosity of the collagen-proteoglycans by stimulating osteocytes to line the lacuocanalicular walls with hyaluronan, a macroporous (big-pored) molecule (Midura et al., 2003). The osteocye also pushes the fluid along its canaliculi with its pulsing microvilli (Knothe-Tate, 2003). When the fluid and its peptide passengers near the lacuna containing the osteocyte's cell body with its sensory cilium that we will talk about later, they hit a kind of gate or valve formed by the cell processes which favors flow out of, rather than into, the lacuna (Knothe-Tate, 2003).
While the cells and their fluid-filled lacunocanalicular network make up only about 1% of the bone volume, the total surface area of these pipelines is immense! In an adult male skeleton, it is 1200 m2, while the surface areas of the Haversian and Volkmann's canals and trabeculae are only 3.2 and 9m2, respectively (Johnson, 1966; R.B. Martin et al., 1999). Thus, bone is really a stiff, dense, fluid-filled, calciferous sponge. And the rate of flow of fluids and their cargoes into, through, and out of the lacunocanalicular sponge is determined by how often and how tightly the sponge is squeezed by breathing in the case of the ribs and by walking, running, and other activities (Burger et al., 2003). This immense osteocyte internet is the "cell phone" with which they tell each another and lining cells about strains and microcracks and mobilize the appropriate responses.
Unnecessary digging and patching of undamaged bone is prevented by the signals from the cyclic squeezing of the sponges by the owner's breathing and variously moving around. The resulting inhibitory signals in the normal bone sent from the osteocytes and spread though the osteointernet via gap junctions to lining cells on the bone surface and between the lining cells to the adjacent endothelial cells of the osteonal blood vessel or marrow to talk to osteoclast precursors and osteoprogenitors (R.B. Martin, 2000a, 2000b, 2002; Klein-Nulent et al., 2003; Marotti, 1996; Whitfield et al., 2002) (fig. 2).
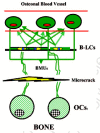
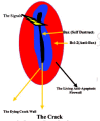
A microcrack or an orthopedic implant screw cuts osteointernet connections and produces a layer of dying cells bordering the crack, which triggers BMU formation by stopping the flow of inhibitory signals from the osteocytes to the lining cells and cuts off the flow of fluid and the oxygen and essential nutrients it contains to surrounding cells (Dodd et al., 1999; Marotti, 1996; Noble, 2000; Noble and Reeve, 2000; Skerry, 1999; Schaffler, 2000; Tami et al., 2002) (fig. 3). The alarm-sounding osteocytes surrounding the microcrack self-destruct by making the deadly, apoptosis-driving Bax protein and things that attract "vulturing" osteoclasts, but the wave of apoptotic damage is stopped from spreading too far beyond the crack by the more distant cells raising an anti-apoptosis "firewall" by making the anti-apoptosis Bcl-2 protein (Verborgt et al., 2002) (fig. 4).
The normal patterns of squeezing and stretching of the bones of an active skeleton, particularly in the hip and distal leg bones by heel striking and in the ribs by breathing, pushes and sucks the fluid in the osteocytes' lacunas and canaliculi back and forth and into and out of the canaliculi's pores in the osteonal walls to produce pulsing shear forces on the cells and their processes (Cowin, 1999; Burr et al., 2002; Ehrlich and Lanyon, 2002; Gooch and Tennant, 1997; Klein-Nulent et al., 2003; Kufahl and Saha, 1990; Knothe-Tate, 2001,2003; Knothe-Tate et al., 2004; L. Wang et al., 1999,2000; You et al., 2001). According to Burger et al. (2003), Knothe-Tate (2003), Knothe-Tate et al. (1998, 2004), Piekarski and Munro (1977), Smit et al. (2002) and L. Wang et al. (2000) strain pulses in long bones, such as tibia and femur from heel strikes during the walking cycle or in the ribs caused by muscle pulling during breathing cycles, cause fluid to flow through the 3-D lacunar-canalicular network to the bone surface and back again—it sloshes back and forth. Static strain is a one-shot suck or push with a change in position without the subsequent pumping action needed to drive osteocyte signaling—the fluid in the lacunas and canaliculi must slosh back and forth within a certain frequency range to keep the osteocytes informed of the level of bone function and signaling appropriately to its fellows in the osteointernet (Burr et al., 2002; Ehrlich and Lanyon, 2002; Gooch and Tennant, 1997). As I said above, cyclic pumping is what keeps food and oxygen coming to the osteocytes and their wastes being carried away. Thus, stopping the pumping strangles the osteocytes. This is why Lanyon and Rubin (1984) found that cyclically loading turkey wing ulna induced new bone formation while stopping the pumping by static loading caused holes to appear in the cortex.
But there is an interesting aspect of strain cycles that affects osteogenic signaling in long bones. Srinivasan and Gross (2000), using the mid-diaphysis of the avian wing ulna, reported that flow rates are maximum during the first load cycle in which fluid is squeezed out of the osteocyte lacunae into the canaliculi. When the load is lifted, the fluid flows back into the lacunae. But if the next loading happens before the lacunae are refilled, there is less fluid to be squeezed into the canaliculi. If this continues, fluid flows and with them osteocyte signaling drop off to produce a steady state equilibrium. This means that if fluid pulsing and signaling are to be maintained at an optimal level, there must be rest periods between the loadings to allow the lacunae to be fully refilled. Srinivasan et al. (2002, 2003) confirmed this by measuring the effects of cyclic loading with and without rest periods on bone formation in avian ulna and the tibias of young, adult, and aged mice. In all cases, inserting rest periods between load cycles enhanced the osteogenicity of low-level loading protocols. For example, 5 consecutive days of 100 low-level loading did not affect the tibial periosteal bone formation rate, but putting 10 seconds of rest between every 10 cycles increased the signaling so that the periosteal bone formation rate increased 8-fold (Sirinvasan et al., 2002).
How do osteocytes sense fluid sloshing? Donahue et al. (2001, 2003) for example have shown that fluid-flow triggers Ca2+ oscillations in rat osteoblastic cells. One thing they, like most other nonproliferating cells, have is a strain-sensing gadget that could produce such oscillations (see www.wadsworth.org/BMS/SCBlinks/Cilia1.html for a long list of cells and publications describing it) which was first noticed in almost every tissue by Zimmermann in 1898 and until now (Whitfield, 2003a) has either not been noticed or has been ignored by the Bone World. This is the nonmotile primary (or solitary) cilium which, unlike a motile cilium with 9 peripheral microtubule doublets, each with attached ciliary dynein motors to move them, and 2 central singlet microtubules, consists of 9 peripheral doublets but no central tubules (Alieva et al., 1999; Alvarez-Buylla et al., 2001; Cameron, 1972; Cohen and Meininger, 1987; Hellio de Graverand et al., 2001; Matthews and Martin, 1971; Pazour and Whitman, 2003; Poole et al., 1985, 1997, 2001; Praetorius and Spring, 2001, 2003a, 2003b; Schwartz et al., 1997; Tonna and Lampen, 1972; Wheatley et al., 1996; Zimmermann, 1898). It is the cell's eyes, nose, and flowmeter. It grows from the mother centriole of the mother-daughter pair of centrioles embedded in the cell's microtubule-organizing centrosome lying beside, and orienting, the nucleus with the Golgi apparatus. Of course the cilium is at its most glorious complexity in the light-sensing retinal rods and cones. Osteoblasts, osteocytes, and their chondrocyte cousins also have them (Doxsey, 2001; Matthews and Martin, 1971; Poole et al., 1997, 2001; Scherft and Daems, 1967; Tonna and Lampen, 1972). The primary cilium is a retractable antenna covered by the cell membrane containing stretch-activatable Ca2+ channels and studded with various receptors, for example those for serotonin (the 5-HT6 receptor) and somatostatin (the SST3 receptor) in neurons (Pazour and Whitman, 2003; Whitfield, 2004). A striking example provided by Dr. Sam Bowser of the antenna-like primary cilium sticking out of a PtK kangaroo rat kidney epithelial cell can be seen in Figure 5. An osteocyte might also use its cilium to assess the levels of key components in its surroundings and measure fluid sloshing caused by heel strikes and tell other cells in the network about them (Whitfield, 2003a).

A model of how osteocytes in a femur, for example, might use their primary cilia to translate fluid sloshing in their lacunae during walking and running into Ca2+ oscillations has been found in MDCK dog kidney cells or mouse embryo kidney cells (Nauli et al., 2003; Praetorius and Spring, 2001, 2003a, 2003b; Whitfield, 2003a). Bending the kidney cell's 8-μm cilium lights up the cell by causing Ca2+ to surge into the cell and then sending a Ca2+-triggered signaling wave traveling though gap junctions into the cell's neighbors (Nauli et al., 2003; Praetorius and Spring, 2001, 2003a, 2003b) (fig. 6). If the cilium is a flowmeter, then the volume and frequency of the signaling would depend on how much and how often the cilium is bent by the extracellular fluid. And it is (Praetorius and Spring, 2001)! Therefore, the cilium would be a reliable flowmeter for measuring the sloshing of extracellular lacunar fluid caused by the squeezing and stretching of bone during a walking or running cycle for example. It is as if the bending of the cilium by sloshing lacunar fluid acts like a signal lamp that sends flashes of light coursing through the osteocyte fiberoptic network to lining cells on the bone surface and beyond.
How could bending a cilium send a signal spreading from an osteocyte in its lacuna in the depths of femoral cortical bone through a lacunocanalicular labyrinth to cells lining the osteonal canal or endosteal surface? The answer may have been found in kidney cells which also monitor fluid flow. A kidney cell's primary cilium has in its membrane a G-protein-coupled mechanosensor, PC-1 (polycystin-1), that is tethered to a somewhat selective Ca2+-permeable cation channel, PC-2 (polycystin-2) to form PC-1—PC-2 complexes. Bending the cilium with a micropipette or by increasing culture fluid perfusion rate, for example from 2 to 8 μl/second, stretches PC-1 which pulls open the attached PC-2 to let Ca2+ flow into the cell (Nauli et al., 2003; Praetorius and Spring, 2001, 2003a, 2003b). The result of this is a surge of Ca2+ into the cilium and stimulation of phospholipase C that releases IP3 from the cilium membrane's phosphoinositides (fig. 6). The IP3 in turn releases Ca2+ from internal stores in MDCK dog kidney cells (Praetorius and Spring, 2001, 2003a, 2003b) (fig. 6), but not in mouse embryo kidney cells (Nauli et al., 2003). However, in both the dog and mouse cells, the result is a cascade of events spreading through the cytoplasm and nucleus and then into the neighboring cells. But, surprising as it may seem, it can't be the little Ca2+ ions that move through the big gap junctions, because they would cause the gap junctions to slam shut. Instead, it is the Ca2+-releasing IP3 in MDCK cells and something else in mouse embryo kidney cells that pass though gap junctions (Nauli et al., 2003; Praetorius and Spring, 2001, 2003) (fig. 6). Thus, bending a primary cilium armed with what appears to be just one PC-1•PC-2 complex triggers a spreading wave of Ca2+- and Ca2+•CaM (calmodulin)-triggered events, such as bursts of adenylyl cyclase activity, NO (nitric oxide) production, nuclear CaMKIV (calmodulin-dependent kinase IV) activity, and gene activations first in the cilium's owner and then in its neighbors (Nauli et al., 2003; Praetorius and Spring, 2001, 2003a, 2003b) (fig. 6). Since osteocytes also have these little antennae, the sloshing canalicular fluid in their lacunae in an active skeleton (Tami et al., 2002) is very likely to keep the antennae bending back and forth and sending streams of Ca2+-triggered signals through the network that would keep the members of the network humming with bone-maintaining or growth-inducing activity (Whitfield, 2003a).
Disrupting the kidney cell's cilium eliminates its ability to sense changes in extracellular fluid flow rate (Praetorius and Spring, 2003a). The horrendous consequence of this "deafening" in both humans and mice—is polycystic kidney disease (PKD) caused by disrupting the kidney tubule cells' gene for PC-1 or PC-2 (Calvert, 2002; Pazour and Witman, 2003; Yoder et al., 2002). If I am right and the osteocyte's primary cilium is used to measure bone strain, and if the osteoblasts (remember they too have them) use their cilia for detecting and initiating the reponse to various things such as growth factors and hormones in their extracellular fluid, PKD should be accompanied by severe skeletal deformation. And this is precisely what happens when the mouse's PC-1-encoding Pkd1 gene is disabled (Boulter et al., 2001; W. Lu et al., 2001)!
Signals from the osteocytes' primary cilia would be accompanied especially at higher strains by the tugging on the signal-generating integrins and associated cytoskeleton that attach the cell and its processes to the osteopontin lining the walls of the lacunae and canaliculi like waves cause a ship to pull on its mooring lines (matricrine signaling). But in this case, the cells' mooring lines are linked to sophisticated signaling instruments (Denhardt et al., 2002; Miranti and Brugge, 2002). This tugging causes the clustering of integrins with signalers and periodic collection of the actin cytoskeleton into thick, oriented stress fibers with a consequent activation of signaling enzymes on actin-linked cell membrane rafts and mechanosensitive Ca2+ channels (Burr et al., 2002; N.X. Chan et al., 2000; Duncan and Misler, 1989). The upshots of this normal pumping and shearing are Ca2+ ions surging through mechanosensitive channels and spiking signalers such as NO from the Ca2+-stimulated NO-synthase, prostacyclin (PGI2) and prostaglandin-E2 (PGE2) produced specifically by the mechosensitive COX-2 (cyclooxygenase-2), and, as we will soon see, glutamate (the well-known excitatory neurotransmitter until recently believed to be used only by central neurons) and serotonin, that passes from osteocyte to osteocyte through gap junctions an/or bounces from receptor to receptor to keep the bone lining cells from unnecessarily ordering vascular endothelial cells to start the repair process by grabbing passing osteoclast precursors (A.D. Bakker et al., 2003; Bliziotes et al., 2001; N.X. Chan et al., 2000; Chenu, 2002; Duncan and Misler, 1989; Edlich et al., 2001; Mason, 2004; Ehrlich and Lanyon, 2002; Kufahl and Saha, 1990; Manolagas, 2000; R.B. Martin, 2000; Noble, 2000; Noble and Reeve, 2000; Noda et al., 1996; Nomura and Takano-Yamamoto, 2000; Parfitt, 1998; Pavalko et al., 1998, 2003; Ryder and Duncan, 2001; Schaffler, 2000; Skerry, 1999; Skerry and Genever, 2001; A.F. Taylor et al., 2002; Westbroek et al., 2001).
The signals from bending cilia, stretched cell membranes, and tugged cadherins and integrins turn on osteogenic genes such as the gene for the strongly osteogenic PTHrP, which will be a very important part of things to come further on in our story (X. Chen, C. Macica et al., 2003). How? The stimulation of the PTHrP gene is mediated by the opening of stretch-activated TREK-family K+ channels (X. Chen, C. Macica et al., 2003). The ciliary bending and membrane deformations also open the cilium's PC-2 channel and another mechanosensitive ion channel in the cell membrane known as ENaC (SA-CAT-stretch-activated cation channel) through which Na+ surges into the cell to depolarize the membrane and thus open L-type VSCCs (voltage-sensitive Ca2+ channels) (Kizer et al., 1997; Pavalko et al., 2003). The result of this ionic bombardment is a cascade of Ca2+-driven and other events that create complexes called mechanosomes that convert the mechanical deformation into biochemical responses and gene expressions and the generation of osteogenic factors such as PTHrP (Pavalko et al., 2003).
The mechanism goes something like this. The nonreceptor tyrosine kinase FAK and cSrc in the tugged integrin-associated focal adhesion plaques are activated and phosphorylate the Crk and p130Cas in Crk•p130Cas complexes in the plaques. The phospho-Tyr-p130Cas attaches to Nmp4/CIZ and kicks Crk out of the complex to form one of the "mechanosomes"— phospho-Tyr-p130Cas•Nmp4/CIZ (Kirsch et al., 2000; Nakamoto et al., 2002; Pavalko et al., 2003; Yamada and Even-Ram, 2002). The activated FAK kinase triggers the Ras → Raf → MEK 1/2 → ERK 1/2 cascade, the last of which phosphorylates and thus enhances the activity of the Cbfa1/Runx-2 transcription factor that drives the expression of key osteoblast genes (Franceschi and Xiao, 2003). Meanwhile, the tugged cadherins in a lining cell's adherens attachment junctions with its neighbors also release β-catenins, which associate with LEF-1 protein to form a β-catenin•LEF1 mechanosome.
The basic "take-home" message from this complicated cluster of cell surface deformation-triggered events is really quite simple to remember. Fluid tugging enhances the activity of the master osteoblast transcription factor Cbfa1/Runx-2 and generates at least two mechanosomes that are fired into the nucleus. There they bind to target sites on the nuclear matrix and then together with Cbfa1/Runx-2 grab, bend and twist the promoters of their bone-making target genes into shapes that enable them to be switched on. One of these mechanosensitive genes codes for PTHrP which, as we shall see further on, is a towering figure in osteoblast differentiation (X. Chen, C. Macica et al., 2003; Daifotis et al., 1992; Pirola et al., 1994; Steers et al., 1998). The steady strain-driven maintenance of a supply of PTHrP by osteocytes could, for example, facilitate crack repair by promoting the flow of osteoprogenitors into the mature BMU osteoblast pool.
To make the strain-response story more complicated, stretching does something rather astonishing—it can activate MLO-Y4 osteocytic cells' nongenomic (membrane-based) estrogen receptors, the signals from which send ERK2 into their nuclei and oppose the initiation of apoptosis by etoposide (Aguirre et al., 2003). In other words, strain can activate estrogen receptors without estrogen (i.e., ligand-naive, receptors)!
In general terms, it seems as if moderate normal body movements keep signals flowing steadily through the extensive osteocyte network (which because of gap junctions is really a giant gated syncytium) from bending cilia. But more strenuous body movements add signals from cadherin and integrin tugging.
As we shall see in the next section, in addition to reshaping gene promoters the intercellular chattering caused by mechanical strain looks a lot like neural network crosstalking. Maybe osteocytes could be called "honorary neurons". An activated neuron sends a wave of Na+ influx-induced membrane depolarization along its axon, which when it hits the axon terminus, opens Ca2+ channels. This triggers the release into the synaptic cleft of packets of a neurotransmitter such as L-glutamate, which activates receptors on a post-synaptic neuron. Amazingly, the same Na+/Ca2+ shifts caused by the punching and pulling of an osteocyte and the tweaking of its cilium by its owner's heel strikes may also cause the cell to release glutamate (Mason, 2004; A.F. Taylor, 2002)!
If the battering be too severe it can actually break the local connections of the osteointernet as well as trigger apoptotic self destruction (Noble, 2000; Noble and Reeve, 2000; Skerry, 1999; Schaffler, 2000). The death of the overstrained osteocytes would lift the restraints on lining cells as effectively as a microcrack (fig. 3). This would enable the lining cells to start making osteoclastogenesis stimulators such as M-CSF (macrophage-colony-simulating factor) and RANKL (for Receptor-Activator of NF-κB Ligand) and cut off a source of the principal physiological osteoclastogenesis suppressor, osteoprotegerin (OPG, formerly called OCIF, osteoclastogenesis inhibitory factor), that the osteocytes and lining cells had been making since its gene was turned on under the influence of the Cbfa1/Runx-2 transcription factor when they became osteoblasts (Atkins et al., 2003; Ducy, 2000; Karsenty, 2000; Komori, 2000; Noble and Reeve, 2000; Thirunavukkarnasu and Halladay, 2000). Thus are recruited the first, or shall we say pathfinder, BMUs to start fixing the damaged bone.
Calling the Diggers
"So how can we explain that osteoclasts ‘know’ their way while they are eating through the bone tissue?"—Burger et al. (2003).
As in a highway repair crew, the first on the job are the diggers—multinuclear osteoclasts armed with a protein shredder (protease) called cathepsin K to chop up the demineralized matrix proteins and heavily loaded with mitochondria to make the large amounts of ATP fuel needed to feed the pump which sprays HCl (hydrochloric acid) to dissolve the bone mineral and the garbage disposal pump which picks up matrix debris at the digging (apical) end of the cell and dumps it out of the other (basal) end of the cell (Blair, 1998; Noble, 2000; Noble and Reeve, 2000; Skerry, 1999; Troen, 2003; Whitfield et al., 1998b). And one of the first responses of osteocytes to the strain around the microcrack is to make osteopontin to help attract the diggers and to glue them to the damaged patch (Nomura and Takano-Yamamoto, 2000). But how the osteocytes summon these diggers to the damaged patch is still a mystery, although we shall see further on that it may be the "smell" of the osteocytes' corpses that does it.
Osteocytes can be induced to self destruct and in the process trigger BMU activation either by very low or by very high strain, but they function best when the strain is within a site-appropriate range of frequencies and strengths produced by normal body movements such as heel or paw strikes during walking or running cycles or the incessant tugging on the ribs by the muscles of breathing (Noble and Reeve, 2000). The osteocytes may be prevented from self-destructing by basal amounts of NO made probably by Ca2+ pulses triggered by the waving of their cilia in the fluid sloshing in their lacunae during normal movements (Klein-Nulent et al., 2003; Whitfield, 2003a) (fig. 6). The pulsing strain pumps blood and extracellular fluid throughout the network of blood vessels and the osteocytes' lacunocanalicular network. This delivers food and oxygen to the osteocytes and flushes out their waste. If the pumping slows and/or weakens because of disuse or the lack of gravity and heel strikes, the starving and oxygen-deprived osteocytes, drowning in their own uncollected wastes respond suicidally by making osteopontin which brings on the osteoclasts to get rid of the apparently redundant bone and, unfortunately, themselves with it (Dodd et al., 1999; Gross et al., 2001). On the other hand, if the matrix strain rises above a certain level, as happens at the tip of a microcrack where it can be 15 times above average (e.g., 30,000 microstrain as opposed to the normal 2,000-3,000 microstrain), osteocytes around the crack self destructively start making the pro-apoptosis Bax protein, probably because of being torn from the walls of their cubicles and the cutting off of the flow of nutrients and oxygen by the severing of their canalicular lifelines (Tami et al., 2002; Verborgt et al., 2000) (fig. 4). However, osteocytes further away from the crack respond to a less severe interruption of nutrient deliveries and prevent the damage from spreading by erecting an anti-apoptosis "firewall" of the anti-apoptosis Bax protein (Verborgt et al., 2000) (fig. 4).
But let's have a look at what happens to gene expressions when osteocytes' oxygen supply is cut off and they rapidly become hypoxic by the failure of the bone fluid pumping action due to the stopping of loading cycles either by disuse or the severing of canaliculi by a microcrack (Dodd et al., 1999; Gross et al., 2001). All of our cells can respond to an oxygen shortage by turning on or turning up two dozen or more so-called hypoxia-inducible genes, the products of which are meant to maintain at least a minimal supply of ATP fuel and other components for at least a short-term survival. When an osteocyte's nutrient-supplying and waste-eliminating pumps are working normally, the components of the hypoxia-inducible transcription factor, HIF-1α and HIF-1β, are constantly being made, but HIF-1α has an oxygen-dependent degradation domain (ODD) that is ubiquitinated and thus dumped into the cell's proteaseome shredder when there is enough oxygen because the oxygen drives the hydroxylation of the domain's key proline residues. The ODD with its hydroxylated prolines sticks into a pocket in the von Hippel-Lindau factor, which causes the von Hippel-Landau factor to ubiquitinate the HIF-1α, which marks the HIF- 1α for delivery to the shredder (L. E. Huang and Bunn, 2003; Latchman, 2004; Marx, 2004; Wenger, 2002). But when the oxygen supply is cut off, HIF-1α's ODD is no longer hydroxylated by the oxygen-driven hydroxylase, there is no hydroxylated proline to be shoved into the von Hippel-Landau factor's pocket, and HIF-1α is thus not ubiquitinated and shredded. HIF-1α then accumulates and flows into the nucleus, where it mates with HIF-1β to form HIF-1α·HIF- 1β transcription factor complexes. One of the incoming HIF-1α's aparagine residues has also not been hydroxylated, and because of this the aparagine can bind to the p300 co-activator on the promoter-switch boxes of two dozen or more hypoxia-inducible genes. The p300 can then interact with components of the basal RNA polymerase II gene-transcriber complex for recruiting the polymerase transcriber to these genes's promoters (Latchman, 2004). Thus the now stabilized HIF-1α·HIF-1β transcription factor activates the genes for glucose transporter-1 (GLUT1) and the components of the glycolytic machinery, such as phosphofructose kinase I, phosphoglycerate kinase, and lactate dehydrogenase A, that will increase the glucose uptake and the anaerobic production of ATP fuel from this additional glucose to try to compensate, albeit less efficiently, for the lack of oxygen-driven mitochondrial fuel production (Ebert, et al., 1995; Gross et al., 2001; L. E. Huang and Bunn, 2003; Schipani et al., 2001). But another gene, the gene for the anti-apoptosis Bcl-2 protein, is turned down or off, and the gene for the pro-apoptosis Nip3 protein is turned up. This means that the struggling osteocyte will be able to hang on only for a while by making more ATP by the glycolytic mechanism, but it is now vulnerable to such things as a reoxygenation, in which the sudden surge of accumulated substrates into the mitochondria and the consequent intermediate "traffic jams" in the chain of oxidative enzymes can cause a lethal spray of ROS (reactive oxygen species) from the mitochondria. Of course, the glycolytic mechanism is less efficient at making ATP fuel than the oxygen-driven mitochondrial ATP-making machinery, so the osteocytes need more glucose, and the glycolytic end product pyruvate will be reduced (protonated) to lactate instead of being processed into acetylCoA and fed into the mitochondrial machinery. The accumulating lactate increases the cellular acidity (i.e., drops the pH). Since the glucose supply is reduced to a trickle or completely cut off, the osteocyte will soon run out of fuel unless the supply lines are reopened, perhaps by the resumption of loading but not if the fuel lines have been severed. To make matters worse, an apoptosis-triggering leakage of agents such as cytochrome c from the mitochondria can no longer be stopped by the Bcl-2 protein, and the cell finally, with the help of the apoptogenic p53 protein, does the "honorable" thing and kills itself. This is the honorable way to go, because an apoptosing cell does not perforate its membrane and release dangerous enzymes that would kill any viable bystanders in the osteointernet.
The death of the osteocytes around the crack cuts off the signals they were sending to the local lining cells in the osteonal central canals to NOT call up BMU digging crews (R.B. Martin 2000a, 2000b; Marotti, 1996). The local lining cells alerted to the damage tell immature osteoblastic stromal cells to make osteoclast recruiters such as M-CSF (macrophage colony-stimulating factor) and RANKL (receptor activator of NF-κB ligand). The specifically RANKL-competent immature osteoblastic marrow stromal cells (they will lose this ability as they mature) put out on their surfaces RANKL ligands which can bind to osteoclast precursors' RANK receptors, signals from which can stimulate the precursors to differentiate. Another possibility is that inflammatory agents seeping into the blood from the injured bone patch would attract these developing osteoclast precursors to the site, where vascular endothelial cells start grabbing them from the passing blood.
At this point, we must try to see what controls the ability of immature osteoblastic cells in the marrow to make the RANKL (mature osteoblasts can't do this) needed to drive the development of osteoclast precursors (Atkins et al., 2003; Kitazawa et al., 1999; G.P. Thomas et al., 2001). As we shall learn further on, the immature stromal preosteoblast owes its promising "osteoblastness" to expressing the master gene transcription factor Cbfa1/Runx-2 we met above. It targets and stimulates a group of genes with the so called OSE2 (osteoblast-specific 2) AACCACA nucleotide sequences in their promoters (i.e., their "on-off switches") (Otto et al., 2003). Two of these genes code for collagen 1 and RANKL. But while the RANKL gene does have OSE2 in its promoter (Kitazawa et al., 1999) and switching it on requires Cbfa1/Runx-2 expression, it does not in fact depend directly on Cbfa1/Runx-2 being on the promoter (O'Brien et al., 2002). Instead, switching on the RANKL gene requires that the immature osteoblastic stromal cell have integrin receptors to bind to a collagenous matrix, the making of which directly depends on Cbfa1/Runx-2 expression (O'Brien et al., 2002). The matricrine signals from the collagen-bound integrins generate the ERK 1/2 kinase activity which phosphorylates and enhances Cbfa1/Runx-2 activity (Franceschi and Xiao, 2003).
The upshot of all of this is the arrival of BMU diggers at the damage site to dig out the microcrack and prepare the site for the making of a new canal by osteoblasts (Marotti, 1996). As the osteoclasts are tunneling through the wall of a Haversian canal in cortical bone or digging a trench on or in a trabecula (fig. 1), the bone's owner is walking, playing tennis, running, breathing, etc. The tunnel being dug in cortical bone has a cutting front or cone, an intermediate resting or pause zone, and a closing or filling tail, along which is a gradient of strains produced by pulse-loading by walking, running, etc. (Smit et al., 2002). Smit et al. (2002) have analyzed the distribution of strain along the advancing tunnel. It is important to remember for things to come later that at the cutting front, this activity-loading stops the lacunocanalicular extracellular fluid sloshing, which, of course, to the ever-alert osteocytes means "bone-not-in-use" and accordingly tells the diggers to get rid of it while further along the tunnel the strain rises and generates signals which mean "bone-in-heavy-use" and thus calls for osteoblasts to make more bone to reduce strain (Klein-Nulent et al., 2003; Smit et al., 2002). More of this later.
Calling Diggers with the Neuron-Like Signaling Machinery of Osteocytes, Osteoblasts, and Osteoclasts
"It is clear that neurotransmitters have profound effects on bone, influencing the differentiation, proliferation, activity, and apoptosis of osteoblasts and osteoclasts."—Spencer et al. (2004).
You may remember that I pointed out at the beginning of this chapter that bone cells unexpectedly share with neurons such things as memory and the ablility to exchange information with each other through extensive networks. Well, this sharing looks as if it runs much deeper than expected. It seems that the major neurotransmitters, such as glutamate and serotonin, may be major osteoctye→osteoblast "osteotransmitters" as well as nerve→lining cell "neuro-osteotransmitters" in the osteonal canals that help initiate the response to loading and microcracking (Bliziotes et al., 2001; Chenu, 2002; Hinoi et al., 2004; Skerry and Genever, 2001; Serre et al., 1999; Skerry and Taylor, 2001; Spencer et al., 2004; Westbroek et al., 2001). This raises the possibility of there being osteosynapses that are similar to neuronal synapses and T-lymphocye-dendritic (Langerhans) cell synapses (Dustin and Colman, 2002). But so far no such synapses have yet been seen, although osteocyte processes have been found terminating "suggestively" on osteoblasts during bone formation (Menton et al., 1984).
Osteocytes, with their primary cilia flowmeters (incidentally neurons also have primarty cilia (Whitfield, 2004)) waving back and forth in the sloshing extracellular fluid, are not glutamate targets. They don't have glutamate receptors, but they make and release it to signal other cells, and they have GLAST (glutamate aspartate transporter)/EAAT1 (excitatory amino acid transporter 1) transporters to reaccumulate it in response to Ca2+ surging through the ciliary PC-2 channels opened by the stretched PC-1 mechanosensors (Mason, 2004; Nauli et al., 2003; Praetorius and Spring, 2001, 2003; Skerry and Genever, 2001; Skerry and Taylor, 2001; Whitfield, 2003a) (fig. 6). In other words, they can send, but not receive, glutamate signals. But osteoblasts and their precursors can receive glutamate signals from nerve terminals and osteocytes, because osteoblast precursors and primary rat osteoblasts have AMPA(DL-α-amino3-hydroxy-5-methylisoxasole-4-propionic acid)/kainic acid andneuron-like NMDA (N-methyl-D-aspartic acid) "ionotropic" (i.e., ion-passing) receptor/channels, and mature osteoblasts have both of these plus the G-protein-coupled, metabotropic glutamate receptors that are related to the CaRs (Ca2+-sensing receptors) and like the CaRs would likely be activated by the extremely high Ca2+ concentrations in the osteoclast excavation sites (Brown and MacLeod, 2001; Chattopadhyay and Brown, 2003; Gu et al., 2002; Jensen et al., 2002). Not only do osteoblasts have NMDA receptors, but amazingly what subunits these receptors have depends on the location of the osteoblast—calvarial (skull) osteoblasts' NMDA receptors/channels have NR2A, NR2B, NR2D subunits but no NR2C subunits, while femoral osteoblasts' NMDA receptor channels have NR2C subunits (Itzstein et al., 2001). Moreover, as Laketic-Ljubojevic et al. (1999) have reported, glutamate causes Ca2+ transients in osteoblastic cells and these cells also have what were once believed to be brain-specific Na+-dependent inorganic phosphate transporters that are involved in the receptor-triggered release of glutamate from its sequestration in VGLUT storage vesicles just as in neurons (Hinoi et al., 2001, 2004). Mature osteoblasts also have GLAST (EAAT 1) transporters needed to recharge their glutamate stores, to clear their surroundings of released glutamate to avoid prolonged signaling from their glutamate receptors and prevent cell death caused by excessively prolonged glutamate signaling, and to maintain a high glutamate signal-to-noise ratio as exactly the same transporters in glial cells do in neuronal synaptic clefts (Carmignoto, 2000; Chenu, 2002; Fields and Stevens-Graham, 2002; Hinoi et al., 2002, 2004; Mason, 2004; Skerry and Taylor, 2001). Indeed, this is particulary important for bone cells because of the large amount of glutamic acid circulating in the blood which would otherwise saturate the receptors and "jam" signaling (Hinoi et al., 2002, 2003). To recharge glutamate stores, maybe they and osteocytes do the same thing as glial cells in the brain. They might sweep up glutamate released by neighboring osteocytes or nerves with their GLAST transporters, convert it to glutamine, and send it back to the osteocytes or the Haversian nerves' axon terminals to be reconverted to glutamate and refill the secretory vesicles (Chenu, 2002).
These receptors and the synapse-like connections they rather loudly suggest are not mere osteoblast or preosteoblast ornaments. Inhibiting the AMPA/kainate and NMDA receptor signaling reduces the production and differentiation of osteoblasts and causes precursor cells to become adipocytes rather than osteoblasts (Dobson and Skerry, 2000; Skerry and Genever, 2001; Taylor et al., 2000). All of this evokes the image of a network of nerves, precursor cells, osteoblasts, osteocytes, and maybe lining cells talking to each other in fluent glutamate!
O.K., but what has all of this to do with "CALLING THE DIGGERS", the title of this section? While a strain-induced glutamate surge stimulates osteoblast generation, it also stimulates osteoclast generation via the baby osteoclasts' wet nurses—immature bone marrow osteoblastic cells (Atkins et al., 2003; Kitazawa et al., 1999; G.P. Thomas et al., 2001). It stimulates the immature, osteoblastic marrow stromal cells to put RANKL on their surfaces while mature, matrix-making osteoblasts cannot make RANKL apparently because its gene's promoter is locked shut by having its CpG sequences methylated at some point during maturation (Atkins et al., 2003; Corral et al., 1998; Kitazawa et al., 1999; G.P. Thomas et al., 2001). RANKL is the ligand for the osteoclast precursors' RANK (Receptor Activator of N F-κB transactivator) receptors the signals from which stimulate osteoclast differentiation (A. Taylor et al., 2000). And the osteoclasts' glutamate-activated NMDA receptor/channels also stimulate differentiation via the NF-κB transactivator exactly as do the signals from RANKL-activated RANK receptors, (Black et al., 2002, 2003; Chenu, 2002; Espinosa et al., 1999; Hinoi et al., 2004; Itzstein et al., 2000; Laketic-Ljubojevic et al., 1999; Mentaverri et al., 2003; Merle et al., 2003; Patton et al., 1998; Peet et al., 1999; Skerry and Genever, 2001). Osteoclasts seem to need the Ca2+ flowing through these activated NMDA receptor/channels in order to mature and later to assemble the actin ring needed to seal off the excavation site (Itzstein et al., 2000; Mason et al., 1997). And the osteoclasts, short-lived cells that they are, also need the Ca2+ surges to stimulate their nNOS (neural nitric oxide synthase) to make the NO they need to hold off self-destructive apoptosis (Mentaverri et al., 2003). Indeed, blocking the NMDA receptors with MK801 or DEP (±-1-[1,2-dipenylethyl]piperidine) cause the cellular Ca2+ content to drop and the cells to self-destruct (Mentaverri et al., 2003).
The Arrival of the Diggers at the Work Site
Signals such as glutamate, NO, PGE2 (prostaglandin E2), and VEGF (vascular endothelial cell growth factor) from the overstrained, hypoxic, and self-destructing (apoptosing) osteocytes with their normal supply lines cut off in a microcracked patch of cortical bone and transmitted to the cells lining the walls of the nearest Haversian or Volkmann's canal start the remodeling/repair job by causing the VEGF-stimulated endothelial cells of a blood vessel to sprout a loop and by dumping osteoclast—mobilizing cytokines such as M-CSF into the blood to stimulate the osteoclast nurseries in the nearest available hemopoietic red marrow sites. The flow of blood from the arterioles into the sprouting capillaries is increased by the vasodilating NO and the membrane-associated guanylyl cyclase and guanylyl cyclase-dependent protein kinase (PKG) it stimulates (Krainock and Murphy, 2000; Lincoln et al., 1997; Zabel et al., 2002; Zaragoza et al., 2002). The NO is initially made in the strained osteocytes from L-arginine by the nitric oxide synthase stimulated by Ca2+ flowing through the cells' stretch-activated Ca2+ channels and then sustained by the vascular endothelial cells' nitric oxide synthase (eNOS) turned on by the increased shear strain from the increased blood flow in what will be a new Haversian canal and vascular pipelines running through it (reviewed by Krainock and Murphy, 2000; Lincoln et al., 1997; R.B. Martin et al., 1998; Whitfield et al., 1998b). The cells at the leading edge of the vascular loop switch on a set of genes, the products of which make the cells' surfaces selectively sticky—"velcroize" them—to snatch appropriately addressed preosteoclasts from the passing blood (Parfitt, 1998, 2000a, 2000b, 2002; Ruoslathi and Rayotte, 2000; Springer, 1994).
A model for osteoclast grabbing has been proposed by Parfitt (1998, 2000a, 2000b, 2002). According to this model, all appropriately addressed preosteoclasts, be they summoned to cortical bone or to trabecular bone, are plucked from the blood circulating, respectively, through the Haverserian tunnels or the marrow sinusoids. When preosteoclasts flying along in the blood hear the osteocytes' distress signals and feel the altered blood vessel walls near the damaged site they, like neutrophils sailing into an infected tissue battle zone (see Sompayrac (2003) for a wonderful description of this " rolling-sniffing-stop-exit" sequence), start putting selectin-binding (selectin ligand) proteins out on their surfaces. After several passes through the region they ultimately become sufficiently and selectively velcroized by putting enough selectin ligand on their surfaces to be grabbed rather loosely by the selectin on the blood vessels' lining (i.e., endothelial) cells. This slows them down, and they start rolling along the vessel wall "sniffing" for damage signals. One of the more important attractants is the increasingly loud signaling from the cells' CaRs by the rising Ca2+ level as they approach the excavation site. When they find them, they put integrin hooks out on their surfaces to grab hold of ICAMs (intercellular adhesion moleculse) on the endothelial cell surfaces like descending aircrafts' tail hooks grabbing the arresting wires on an aircraft carrier's deck. This stops them, and they are lured from the blood vessel and into the bone by various chemoattractant chemokines released by the stressed osteocytes and lining cells.
The preosteoclasts squeeze ("diapedese") between the vascular loop's cells to leave the blood and fuse with others to form large active multinuclear (5 or more nuclei) osteoclasts, each of which digs for the next 2-3 weeks. These pathfinding osteoclasts start tunneling through the damaged patch at a speed of about 25-40 μm/day. The osteoclasts first dig into the wall of the osteon at a right angle and then turn to tunnel parallel to the osteonal columns. As they tunnel along, they are followed by the vascular loop with new preosteoclasts squeezing out of its tip, along with the nutrients and the lots of oxygen needed by the osteoclasts to do their job. Actually, there are only about 10 osteoclasts in a BMU (R.B. Martin et al., 1998), but each osteoclast is a fusion of many individuals. Indeed, an osteoclast is a very strange and often a very large beast—a sort of cellular collective in which nuclei actually come and go (e.g., for about 11 days in dog osteoclasts), while the collective with its ever-changing nuclei keeps tunneling ahead (R.B. Martin et al., 1998).
But what kind of "pheromone" keeps the osteoclasts tunneling through a microcracked patch like "truffle hound" pigs after mushrooms? What are they digging for? What are the "mushrooms"? Remember that osteocytes are driven to self destruct by apoptosis when a microcrack cuts off their supply lines and they are hit by the huge strain spreading from the tip of the crack (fig. 4). Moreover, remember that according to Klein-Nulent et al. (2003) and Smit et al. (2002), once the osteoclasts have started tunneling, the fluid sloshing of the nutrient-supplying/waste-disposing mechanism stops at the tip of the cutting cone. This means that the osteocytes need oxygen. Thus, even the osteocytes that were not immediately cut off or hyperstrained by the microcrack are also gasping and may start making inducible NOS-2, which in turn makes dangerously large amounts of NO that could induce neighbors to kill themselves. But of all things, the most important for answering this question is that deep down in their tiny souls, osteoclasts are "professional" macrophages with a vulture's taste for apoptotic carrion.
The answer to what drives osteoclast tunneling might go something like this, starting with a dying osteocyte. The killer caspase 3 in an apoptosing osteocyte stimulates Ca2+-independent phospholipase A2, which causes the cell to make lysophosphatidylcholine (LPC) and dump it out into the canalicular channel, which, when it seeps out of the mouth of the channel into the cutting cone, excites the osteoclast's tracking and digging machinery by activating the LPC receptors, GPR4 and G2A, on the osteoclast's surface just as it would on the surface of any macrophgage (Grimsley and Ravichandran, 2003). The dying osteocye also inactivates the "flippase" that has been keeping phosphatidylserine on the inner leaflet of the cell membrane but now activates a nonspecific, bidirectional phospholipid "scramblase" that loads phosphatidylserine onto patches or scaffolds on the membrane's outer leaflet (Grimsley and Ravichandran, 2003). Along with this, annexin I is translocated from the cytosol to the outer surface, where it associates with the phosphatidylserine patches to produce an "eat-me! " sign. This is what the relentlessly digging osteoclast, driven by a rising "scent" of LPC, bumps into at the head of the trail. Then its PSRs (phosphatidylserine receptors) bind to the osteocyte corpse's phosphatidylserine annexin I complexes. The signals from the vulturing osteoclast's PSRs trigger the mechanism with which the osteoclast "eats" the osteocye corpse.
Things are different in trabecular (cancellous bone) and the endocortical surfaces facing the bone marrow (fig. 1). There, the osteoclasts dig trenches instead of tunnels. Something remarkable happens when the lining cells on a trabecula are uncoupled from the underlying osteocytes. A kind of blister—the "bone-remodeling compartment (BRC)—forms on the trabecular surface (Hauge et al., 2001; Parfitt, 2001)! It appears that the aroused lining cells start making collagenase to remove the matrix cover and lift off the surface to form a kind of tent over the future work site. This blister forms a pseudoblood vessel with a wall of osteoprogenitors and lining cells instead of true CD34-bearing endothelial cells (Hauge et al., 2001). The blister vessel plugs into the local marrow blood sinusoids to bring preosteoclasts to the worksite. At first sight, it might be assumed that the trabeculae and endocortical bone would enjoy a direct supply of preosteoclasts from the red marrow nursery. But while this may be true in children, the marrow in most peripheral bones in adults is nonhemopoietic fatty yellow marrow so preosteoclasts must be shipped in, sometimes from distant places such as the residual islands of hemopoietic marrow in the upper femurs and the red marrow in the bones of the central skeleton including the ilium.
The Fillers
"Normally, bone cells respond to mechanical loading by increasing their metabolism, activating genes, producing growth factors, and synthesizing bone matrix"—Ehrlich and Lanyon (2002).
"Osteoblasts don't do anything by themselves; they do their job in vivo as sheets of cells, they're all connected"—A. Caplan (in Davies, 2000, p. 255).
What Summons the Fillers?
As the the vulturing "truffle hound" osteoclasts are tunneling through the cortical bone looking for dying osteocytes and in the process digging out the microcrack and a large patch of bone around it by spraying the protein-shredding cathepsin K and mineral-dissolving HCl onto the bone, they release a pack of factors (e.g., bone morphogenic protein (BMP)-2, fibroblast growth factor (FGF)-2, insulin-like growth factors (IGFs)-I and -II, IGF-binding protein (IGFBP)-5, TGF-βs) that were deposited in the matrix 2 to 5 years earlier by the osteoblasts of an ancient BMU. It is widely assumed in the Bone Community that these liberated factors, particularly the FGF-2, and the IGFs, form an immediately accessible store of osteoblast generators that can be drawn on by a subsequent BMU to locally increase osteoblast generation by stimulating osteoprogenitor proliferation to start new bone growth for repairing a microcrack. The TGF-βs would assist by stimulating osteoblastic stromal cells via Cbfa1/Runx-2 and TGF receptor-activated Smad proteins to reduce osteoclast generation by stimulating OPG expression and reciprocally reducing RANKL expression (Takai et al., 1998; Thirunavukkarnasu et al., 2000, 2001; Troen, 2003). However, they may merely be litter from a past repair job—the remnants of the autocrine/paracrine factors that the past osteoblasts were using to stimulate themselves—that may not be able to withstand the osteoclasts' cathepsin K and acid spray.
But the osteoclasts leave a trail of chemokine droppings that attract osteoblastic cells. This cytokine is Mim-1 (myb-induced myeloid protein 1) (Falany et al., 2001; Ponomareva et al., 2002; Troen, 2003). This peptide lures the preosteoblasts to the excavation site and at the same time stimulates the ability of the Cbfa1/Runx-2 to get at, and stimulate the expression of, key osteoblastic genes such as osteocalcin and then drive the mineralization of the new matrix (Ponomareva et al., 2002). Mim-1 can facilitate Cbfa1/Runx-2's access to the osteoblast-specific genes' promoters ("switch boxes") because it is a chromatin-decondensing, gene-accessing histone acetyltransferase.
Serotonin may also be put into the matrix by osteoblasts and subsequently released by the diggers from a future BMU. Osteoblasts and periosteal fibroblastic osteoblast precursors express serotonin receptors and have the machinery to release and reaccumulate it (Bliziotes et al., 2001; Westbroek et al., 2001). And Chopra and Anastassiades (1992) have reported that one of the matrix components—bone sialoprotein (BSP)—binds serotonin, receptors for which might be on the osteoblast primary cilium as they are in neurons in certain parts of the brain (Brailov et al., 2000; Hamon et al., 1999). Although what serotonin does in bone is unknown, it may be another of the several factors that stimulate the proliferation of serotonin-receptor-bearing osteoprogenitor cells which in turn generate crack-filling osteoblasts (Westbroek et al., 2001).
A lot of Ca2+ is also pumped out of the hole when the osteoclasts dissolve the bone mineral (hydroxyapatite) by directing a stream of H+ into the hole with their massively expressed vacuolar H+-ATPase and Cl- counterions though Cl- channels in the ruffled border (Blair, 1998)—as I said a couple of paragraphs ago, they actually spray the bone mineral with hydrochloric acid (HCl)! They ingeniously get the H+ of the HCl from water by first using their carbonic anhydrase to make H+ (HCO3)- (carbonic acid) by combining CO2 with water, then they use their membrane (HCO3)-/Cl- exchanger to pump out the (HCO3)- through their upper surfaces away from the hole and bring in Cl- in exchange for the (HCO3)- and finally pump the H+ out with the electrogenic ATPase, which to maintain electroneutrality pulls Cl- through its channels into the hole and onto the hydroxy apatite. This plume of Ca2+ is a very important two-way switch—it is an "off" switch for the osteoclasts and an "on" switch for osteoprogenitors coming behind them.
When the Ca2+ concentration in a tunnel or trench gets high enough, it activates Ca2+ sensors on the osteoclast's apical H+- and protease-pumping, finger-like ruffles sticking into the hole (Blair et al., 2002; Kameda et al., 1998; T. Yamaguchi, 2003; Zaidi et al., 1999). The resulting Ca2+ surges and signals cause the osteoclast to pull its acid-dripping fingers out of the hole, pull up the actin sealing ring it had put around the hole, and glide over to another part of the patch and start digging again. But if it cannot find another place to attach itself by its integrins, in time to generate the matricrine signals neeed to hold off apoptosis, it will self-destruct—a drastic response to unemployment and homelessness known as anoikis or homelessness-induced apoptosis (Frisch and Screaton, 2001; Lorget et al., 2000; Sakai et al., 2000; Stupack and Cheresh, 2002).
By contrast, the increasingly loud signaling from the Ca2+-sensing receptor on the oncoming osteoblast precursors, as they feel their way along the surface with their integrins (Brown and MacLeod, 2001; Chattopadhyay et al., 2004; Farzaneh-Far et al., 2000; Hinson et al., 1997; Pi et al., 1997; T. Yamaguchi et al., 1998a, 1998b, 2000, 2001), directs the cells to the high-Ca2+excavation site, stimulates their proliferation by triggering a MAP kinase/JNK cascade, and induces them to express BMP-2 and -4, which, in turn, stimulates the expression of Cbfa1/Runx-2, the master osteoblast-specifying transcription factor that starts the post-proliferative stages of the bone-making process (fig. 7; Brown and MacLeod, 2001; Canalis et al., 2003; Chattopadhyay et al., 2004; Nakade et al., 2001; T. Yamaguchi, 2003; T. Yamaguchi et al., 1998a, 1998b, 2000, 2001). And the Cbfa1/Runx-2 in turn feeds back to further stimulate BMPs-2 and-4 because their promoters have Cbfa1/Runx-2 sites in their promoters (Canalis et al., 2003) (fig. 7).
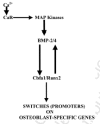
As the osteoclast-attracting osteocyte killing stops and the load-driven sloshing resumes, the Ca2+ sensor-driven stimulation of ERK (extracellular signal regulated kinases) 1/2 in the cells stimulates the expression of eNOS and the production of NO (J. Rubin et al., 2003), the gaseous signaler which diffuses out of the cells to prevent local osteocytes from killing themselves and becoming osteoclast-attracting corpses. This NO also stimulates the enzyme, guanylyl cyclase, in nearby vascular cells by attaching to the enzyme's heme group (Lincoln et al., 1997). The cyclic GMP produced by the cyclase relaxes arterioles, the overall effect of which is to increase the delivery of blood-borne nutrients through the advancing blood capillaries to the filler crews (Lincoln et al., 1997). The NO-stimulated cyclic GMP in the osteoblastic cells also stimulates the cells' PKG (protein kinase G) which in turn may, like the Ca2+ sensor signals, also activate the ERKs 1/2, which in turn activate the cFos/Jun (AP-1) transcription factor complex and the various matrix-related genes it switches on (Pfeilschrifter et al., 2001; Zaragoza et al., 2002). And this in turn also shuts down the expression of RANKL, which turns off osteoclast generation (Rubin et al., 2003).
Where Do the Fillers Come From and Where Do They Go?
This is all very interesting, but where do the osteoblasts come from to make a new Haversian canal surrounded by concentric layers of bone? They come from a group of constantly proliferating, spindle-shaped mesenchymal cells that follow the osteocyte cadaver-hunting osteoclasts along the walls of the cutting cone, driven by the various mitogenic and chemotactic factors, mixed with the large amounts of Ca2+ and phosphate released from the bone by the osteoclasts to make a tasty osteogenic "soup" (Bianco et al., 1993; Doherty and Canfield, 1999; R.B. Martin et al., 1998; Parfitt, 2000a, 2000b, 2001; Schaffler, 2000). The band of proliferating mesenchymal cells advancing along the tunnel wall throw off preosteoblasts that dismantle their mitogenic machinery and become osteoblasts which start depositing layers or lamellae of bone around the tunnel walls. Unlike their progenitors, the new osteoblasts stay in place to lay down concentric 15-μm-thick strips of bone around the tunnel wall, while the proliferating progenitors move on behind the osteoclasts, throwing off new bands of osteoblasts as they go. The result of this is the layering of lamellae around the new blood vessels and nerves.
The question remains as to whether one generation or band of osteoblasts fills the tunnel (except of course the vascular "right-of way"), taking rest breaks between each pair of lamellae or whether several generations of osteoblasts are needed do the job, which would mean that osteoblasts are still being generated by progenitors on the new surfaces. Probably several osteoblast generations are needed for the refill job (Martin et al., 1998). As the refilling progresses, the rate of formation drops for at least two reasons. First, as the lamellan build-up approaches the central blood vessel, the shrinking space somehow interferes with osteoblast differentiation, and second, the progressive drop in stresses with the increasing lamellar build-up results in a fading of the strain signals needed for osteoblast differentiation that were at their peak in the microcrack and at the head of the cutting cone (R.B. Martin et al., 1998).
At a repair site on the trabecular or endosteal surface, the osteoblasts come from the preosteoblasts from the adjacent marrow and the lining cells that make up the canopy of the repair "blister" mentioned above (i.e., the BRC canopy) (Hauge et al., 2001).
If any of the BMP-2 liberated by the osteoclasts from the matrix has escaped being shredded by the osteoclasts' proteases, it would kick osteoprogenitors along the osteoblast differentiation pathway (Chen et al., 1998). They would then be stimulated to start working by the signals from other receptors which include the CaRs in the caveolar pouches in their cell membranes (Brown and MacLeod, 2001; Kifor et al., 1998) and the type 1 PTH/PTHrP (PTHR1) receptors (Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002; McCauley et al., 1996), the expression of which shoots up about 10-fold during the transition from mature osteoprogenitor to preosteoblast.
Another factor that drives the osteoprogenitor proliferation and maturation comes from the cells of the advancing blood vessels. That factor is endothelin-1 which binds to, and activates, the osteoprogenitors' endothelin-A receptors (Mohammad et al., 2003; von Schroeder et al., 2003).
Signaling by glutamate from the dying or hypoxic osteocytes or from invading or adjacent nerves is also needed. However, the osteoblasts also release glutamate using exactly the same vesicular release machinery as neurons (Bhangu et al., 2001). As we learned above, the osteoblasts are also equipped with neuron-like NMDA-type glutamate receptor/channels (Gu et al., 2002; Hinoi et al., 2003; Skerry and Taylor, 2001; Spencer et al., 2004), AMPA/kainate ionotropic receptor channels as well as the G-protein-coupled metabotropic mGluR1b receptors with their 7 transmembrane α-helices, which belong to the same GPCR (G-protein-coupled receptor) family as the Ca2+ receptors (Gu and Publicover, 2000; Hinoi et al., 2001; Skerry and Taylor, 2001; Spencer et al., 2004). Moreover, they have Na+-dependent GLAST (EAA1) transporters to suck the glutamate back into the cell, just as they do in neurons to keep the external glutamate low, which keeps the signaling noise down and prevents dangerously prolonged receptor activation by released glutamate (Chenu, 2002; Mason et al., 1997). The activated mGluRs, like activated Ca2+ sensors and the related PTHR1 receptors, trigger a burst of PLC (phospholipase C) activity, which triggers a prompt release of Ca2+from internal stores followed by the flow of Ca2+ through opened membrane channels and a surge of PKCs (protein kinase Cs) activity (Gu and Publicover, 2000).
The importance of activated NMDA receptor/channels at a critical point in the osteoblast maturation program for driving post-proliferative osteoblast differentiation by stimulating the expression and possibly the transport of the master osteoblast differentiation driver Cbfa1/Runx-2 to its target genes has been demonstrated by Hinoi et al. (2003) using freshly isolated rat calvarial osteoblastic cells. Thus, NMDA receptor/channel inhibitors interfere with the binding of the master osteoblast differentiation driver Cbfa1/Runx-2 to its DNA targets and thus the expression of key osteoblast genes such as alkaline phosphatase and osteocalcin and ultimately the arrival of the cell at the stage where it accumulates Ca2+ for matrix mineralization (Hinoi et al., 2003). The NMDA receptor/channels on the calvarial osteoprogenitors in Hinoi et al.'s experiments (A.F. Taylor, 2002) must have been continuously stimulated by the 500-μM glutamate in the α-MEM culture medium, with no relief being possible from the GLAST transporters, as would be the case in a bone. (This incidentally suggests the extremely important possibility that glutamate has been a covert signaler for osteoblasts in standard culture media.) In a bone, the osteoblastic cells' NMDA receptor/channels would only be stimulated by glutamate boluses from glutamatergic nerves responding to strain or to glutamate released by damaged and dying osteocytes in microcracks.
Despite this, the importance of glutamate as an osteogenic osteotransmitter is still uncertain, because although Dobson and Skerry (2000) and Taylor et al. (2000) have found that inhibiting the glutamate receptors reduce bone formation in vivo and in vitro, Gray et al. (2001) have reported that high doses of NMDA receptor/channel inhibitors did not affect bone formation by cultured rat osteoblasts and bone formation in GLAST transporter knock-out mice was normal. But Skerry et al. (2001) have listed the serious shortcomings of Gray et al.'s GLAST knockout mice which I will not repeat here. Osteoblasts also have AMPA/kainate receptors, which also activate glutamate receptor/channels (Skerry and Genever, 2001), and thus might take over from blocked NMDA receptor/channels, and we do not know whether GLAST activity and glutamate reuptake are as critical for bone making as they are for neurotransmission. One of the most serious errors in Gray et al.'s experiments is their use of competitive NMDA receptor/channel antagonists. These antagonists simply could not compete with the high levels of glutamate in a culture medium that are needed to support the growth of osteoblastic cells (A.F. Taylor, 2002).
The upshot of all of this is that osteoblast differentiation and osteocyte functions, such as sensing and memorizing strain pulse frequency, involves glutamate, and to do their job these cells use exactly the same signaling machinery and transmitters as central neurons (Skerry and Taylor, 2001; Spencer et al., 2004; Turner et al., 2002). In other words, it looks as if we have been looking at "osteoneurons"—what a fantastic change in our view of these cells and the smart bones they make!!
These unexpected neural similarities are not restricted to glutamate receptors and transporters! Active osteoblasts also make BDNF (brain-derived neurotrophic factor) and the TrkB receptors needed to bind it (Yamashiro et al., 2001). Of course, you might say that this BDNF is simply used to stimulate the innervation of new bone. However, the osteoblasts' TrkB receptors mean that they too use their BDNF to stimulate themselves and their neighbors for some aspect of bone making.
Before the osteoblasts arrive on the scene to start filling in the trench or tunnel, the lining cells must sweep up the litter left by the untidy osteoclasts on the resorption cavity floor (Everts et al., 2002). There are collagen "bristles" sticking out of the cavity floor. The lining cells then move onto the surface to give it a good shave and make a smooth surface upon which they slap a layer of osteopontin to glue the new collagen that will be made by the incoming osteoblasts (Everts et al., 2002; McKee and Nanci, 1996). As we shall see below, the residual phosphate from the dissolved bone mineral in the resorption cavity could be the stimulator of this first burst of osteopontin expression (Beck, 2003; Beck and Knecht, 2003; Beck et al., 2000, 2003). These lining cell janitors are in fact reversibly retired osteoblasts who when stimulated by agents such as PTHs can revert to full osteoblasthood to give a first wave of osteoblasts to "kindle" bone building.
As mentioned above, the strained osteocytes in the microcracked site make and release NO gas, but it cannot save those Bax-makers closest to "ground zero" (fig. 4). But it increases arteriolar dilation and the movement of osteoclast precursors into the cortical tunnels and trabecular and endocortical trabecular blisters; NO also stimulates the migration of osteoblasts into the site (Afzal et al., 2000).
While there were as few as 10 osteoclasts doing the digging, there may be hundreds of osteoblasts working in a tunnel or trench (R.B. Martin et al., 1998). Despite their numbers, the osteoblasts take about 4-8 times longer to fill the cortical tunnels and trabecular "blister"-covered trenches than the osteoclasts took to dig them. Therefore, at any moment in the microfracture sites, a mature bone has holes that are being dug or have been recently dug by the microcrack-repairing BMUs—these holes together make up the remodeling space. As the osteoblasts are slowly filling these holes with new factor-loaded matrix (‘osteoid’), some of them will be trapped in it like insects in amber, as it is gradually mineralized with apatite-like Ca-phosphate. When the large amount of Ca2+ in the excavation site has been used by the osteoblasts to mineralize the new matrix and the filling has been finished at the endocortical and trabecular worksites, the blister canopy collapses, and the old lining cells, together with new ones from a lucky few osteoblast survivors, connect to the entrapped osteocytes and cover the repaired patch (Hauge et al., 2001). Walled up in their matrix-lined cells, they reduce their PTHR1 receptor density (Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002) and become osteocytes that connect to the osteointernet and start sending messages from their bending cilia and tugged and twisted integrins to the overlying lining cells and from them to adjacent marrow stromal cells and Haversian blood vessels to again summon BMUs to repair damage when needed or mobilize Ca2+ if needed to restore the circulating level (Marotti, 1996; Martin, 2000a, 2000b, 2002; Hauge et al., 2001; Skerry, 1999; Whitfield et al., 1998b; Yellowley et al., 2000).
When the new patch is finally in place 3-9 months later and the osteocytes start sending the signals needed to prevent lining cells from unnecessarily summoning osteoclast precursors from the blood vessels running through the cortical Haversian canals or from forming a BMU-activating blister on endosteal or trabecular surfaces, the members of the last osteoblast crew are now out of work—they have become redundant! Those that have failed to find a free space on which to settle as lining cells or osteocytes to keep their surface-sensing integrins emitting their survival-promoting signals trigger apoptosis and self-destruct like the unemployed osteoclasts before them (Frisch and Screaton, 2001; Stupack and Cheresh, 2002; Whitfield et al., 1998b, 2000a). However, the doomed, self-destructing osteoblasts make one final contribution to bone formation, specifically mineralization, by dumping alkaline phosphatase into the new matrix in vesicles released from their blebbing surfaces (Farley and Stilt-Coffing, 2001). Some of this alkaline phosphatase will get into the blood to tell the outside World of bone being made and osteoblasts dying.
Most of the self-destructing (apoptosing) cells in bone are located in microcrack sites being repaired by BMUs. The large amount of inorganic phosphate (Pi), released along with Ca2+ from the apatite crystals dissolved by the osteoclasts (Gupta et al., 1996), may increase the chance of osteoblasts triggering apoptosis, unless they are attached and protected by survival factors such as Bcl-2 (Allen et al., 2002; Meleti et al., 2000). The reason for this is likely to be the Na+ gradient-powered pumping of Pi into the osteoblasts by their Pit 1 and 2 type III Na+/Pi transporters, particularly the Pit 1 transporter that they selectively upregulate during maturation for matrix mineralization (Adams et al., 2001; Meleti et al., 2000; Nielsen et al., 2001; Takeda et al., 1999). When the phosphate reaches a critical level, it would be carried into the mitochondria by the Pi-/OH- exchanger and trigger the assembly of the huge mitochondrial permeability transition pore and through this the leakage of cytochrome c, AIF (apoptosis-inducing factor), procaspase-9, and Diablo into the cytoplasm from the intermembrane space. The appropriately named and deadly Diablo neutralizes a group of caspase inhibitors, and cytochrome c forms a complex with APAF-1, which causes caspase-9 to auto-activate and trigger a lethal cascade of so-called "executioner" caspase protein shredders (Crompton, 2000; Finkel, 2001). (This ability of Pi to greatly promote apoptogenesis was first noticed by me nearly 40 years ago, when I was searching for a way to enhance the radiation-induced apoptosis of rat thymic lymphocytes and human peripheral blood lymphocytes for an ultra-sensitive radiation bio-dosimeter (e.g., Whitfield et al., 1967). Meleti et al. (2000) have shown that inhibiting the transporter with low concentrations of phosphonoformic acid (PFA) prevents Pi from killing primary human osteoblasts, and Mansfield et al. (2001) have shown that PFA also prevents Pi from triggering apoptosis in chondrocytes. It follows from this that inhibiting the Na+/Pi transporter might prolong the lifetime of osteoblasts by shielding them from the high Pi in their workplace. However, as we shall see later on, this could be dangerously counterproductive, because the transporter is also a key player in osteoid mineralization.
The apoptogenic action of Pi in cultured human osteoblasts from bone fragments and murine MC3T3-E1 preosteoblasts is greatly enhanced by a small, itself harmless, increase (0.1-1.0 mM) in the Ca2+concentration in the culture medium (Adams et al., 2001). Indeed, it has been shown that both proliferation and the phosphate transporter in osteoblastic cells are stimulated by Ca2+ (Schmid et al., 1998). Therefore, it is likely that the high Ca2+ concentrations in the excavation sites would stimulate the proliferation of osteoprogenitors as well as promote Pi-induced apoptogenesis of both them and mature post-proliferative osteoblasts, unless the cells are protected by some anti-apoptosis protein such as autocrine and paracrine IGF-I, the expression of which is stimulated by the signals from the osteoblasts' PTHR1 receptors activated by the PTHrP that is normally expressed alongside the receptors (fig. 8 Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002) or by an injected PTH (Allen et al., 2002). It seems likely that the burst of phospholipase C activity caused by activated CaRs would produce a surge of IP3 from membrane phospholipid breakdown and a highly localized release of Ca2+ (Ca2+ "hotspots") from IP3 receptor-bearing endoplasmic reticulum Ca2+ stores lying within or beside clusters of mitochondria (Hajnóczky et al., 2000, 2003a, 2003b; Mannella, 2000; Pozzan et al., 2002; Rodland, 2003). This released Ca2+ would enhance Pi action by jumping on the Ca2+ uniporters of the mitochondria clustered around the mouths of the IP3—opened channels in the endoplasmic reticulum and riding on the uniporters into the mitochondria to help Pi stimulate the formation of permeability transition pores through which cytochrome c leaks into the cytoplasm to trigger the lethal apoptogenic caspase cascade, unless the cell is sufficiently protected by a protein such as Bcl-2 (Hajnóczky et al., 2000, 2003).
Finally, osteoblasts are recruited in load-bearing mature human bones only for remodeling BMUs to repair microcracks. But the osteoblast recruitment and osteoclast activity are not coupled in growing bone. Clusters of osteoblasts operate independently from osteoclasts in the growing, so-called modeling, bones of rat pups and human children (Frost, 1997; R.B. Martin et al., 1998; Selye, 1932). And as we shall see below, PTHs can stimulate a massive layering of osteoblasts on trabeculae and bone formation without a prior activation of osteoclasts. Indeed, growing mutant mice, which cannot generate functional osteoclasts, become osteopetrotic due to unopposed bone building by osteoblasts; and mutant mice that cannot generate functional osteoblasts become the opposite, osteoporotic, due to the unopposed osteoclast activity (Karsenty, 1999; Whitfield et al., 1998b).
Dangerous Osteoblastic Transvestites
At this point, we should take a quick look at osteoblasts which don't live in bone, but instead appear in and ossify stressed and injured arteries and aortic valves and thus cause heart attacks and death of atherosclerotics and persons with end-stage kidney disease as well as cause the vascular impairment and consequent amputation of diabetics' limbs (reviewed by T.M. Doherty et al., 2004; Whitfield, 2005). They come from a subpopulation of redifferentiation-competent vascular smooth muscle cells known as "calcifying vascular smooth muscle cells" or CVCs (T.M. Doherty et al., 2004; Whitfield, 2005). They lay down sheets of bone and deposit bone nodules in the walls of affected vessels—they literally make stiff armored blood vessels (T.M. Doherty et al., 2004; Whitfield, 2005).
Factors from LDL particle-stimulated plaque/atheroma endothelial cells and macrophage invaders diapedesing through the excited endothelium into the vascular intima, high concentrations of circulating inorganic phosphate, and the intranuclear action of the cells' hyperexpressed PTHrP stimulate the proliferation and migration of vascular smooth muscle cells from the vascular media into the intima and the redifferentiation of the CVCs among the migrating cells into functioning osteoblasts (T.M. Doherty et al., 2004; Jono et al., 2000; Steitz et al., 2001; Whitfield, 2005). Thus, the CVCs lose their specific smooth muscle lineage markers and α-actin and then go about making bone just like normal osteoblasts (T.M. Doherty et al., 2004; Stein et al., 1996). They express the master osteoblast transcription factor Cbfa1/Runx-2, make type I collagen, alkaline phosphatase, and then ostocalcin and osteopontin when mineralizing the matured matrix (Jono et al., 2000; Steitz et al., 2001). A very strong support for what follows further on in our discussion of PTH's cyclic AMP (cyclic adenosine-3',5'-monophosphate)-triggered bone-building action is the ability of a short exposure to cyclic AMP to cause CVCs to stop proliferating and assume a classical cuboidal osteoblast shape and functions (Tintutt et al., 1998).
The Decline and Fall of Bone Strength and BMU Efficiency But a Rise in Remodeling Rate with Age
With advancing age and decades of microcrack remodeling repair, the density of osteons and their Haversian canals in the compact bone rises with the replacement of primary bone with secondary bone—the bone becomes increasingly "worm-eaten". For example, the osteon density in a human femur rises from about 3/mm2 at 5 years of age to about 25/mm2 at 70 years of age (Kerley, 1965). Since osteons are effectively hollow (i.e., like worm holes), the unavoidable price of microcrack repair is increasing cortical porosity and microarchitectural weakening of the bone (R.B. Martin et al., 1998). And of course with this weakening comes a higher frequency of microdamage and remodeling at prime loading sites—a vicious cycle has begun operating (fig. 9)!
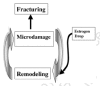
Contributing to this weakening with advancing age is the fact that BMUs are not as good at patching as interstate highway repair crews. (But to be fair to the BMUs, the highway repair crews have a great advantage over the bone repair crews—when there has been too much patching, they can tear up the whole road and replace it, which cannot be done with an old patched-up skeleton.) First, the availability of osteoblasts for BMUs drops with advancing age as the number of osteoprogenitor cells (and possibly stem cells?) in the bone marrow declines (Nishida et al., 1999). Osteoblasts working on the walls of the osteon tunnels in endocortical bone and the trabecular trenches in the cancellous (trabecular) compartment of bone do not completely refill the osteoclasts' excavations, but periosteal osteoblasts tend to overfill the holes (Eriksen, 1994; Eriksen et al., 1994; Frost, 1997). Therefore, with advancing age the cortical shells become thinner as the endocortical and trabecular parts of the bones waste away (R.B. Martin et al., 1998). Fortunately the periosteal overfilling increases the diameters of load-bearing bones (such as the femur), which somewhat compensates for the overall thinning and loss by resisting an increase in the bone's vulnerability to bending and breaking (Einhorn, 1996). But the cortex of the femoral neck becomes thinner without increasing in diameter because there are no periosteal BMUs (Einhorn, 1996). This combination of a thinning cortical shell with perforated plates and severed struts in the cancellous (trabecular) compartment without an increase in the neck's diameter makes the aging hip especially vulnerable to bending and breaking by the huge loads, as much as 5 times the total body weight, that are put on it during a normal walking cycle (Einhorn, 1996; R.B. Martin et al., 1998).
The amount of bone removed by the osteoclasts of remodeling BMUs depends on the number of preosteoclasts that can be grabbed from the passing blood as well as the lifespans of the osteoclasts into which they fuse (Manolagas, 2000; Parfitt, 2000a). When the signaling from a repaired patch stops, i.e., when the "smell" of dying osteocytes fades, osteoclast recruitment also stops and so should the digging. However, depending on the number of osteoclasts in the excavation, less focused digging may go on for some time after the signaling has stopped (Parfitt, 1998, 2000a, 2000b). Indeed once a BMU starts tunneling, it tends to keep going for a while (R.B. Martin et al., 1998). Therefore, any antiresorptive or antiremodeling agent that lowers the number of osteoclasts by reducing their recruitment or killing them can reduce or even stop resorption and remodeling. But as is true for so many good things, there is a rub—reducing remodeling reduces microcrack repair. This would result in a limited amount of bone growth as the unaffected osteoblasts continue filling the existing holes without having to contend with osteoclasts digging more holes faster than they can be filled. When they have finished, the osteoblast crews disband and, except for those 4% who are lucky enough to become osteocytes or lining cells, obligingly self destruct. But the owner continues to move and crack bone, the accumulated microarchitecural deformation of which has not been reversed, and the continuing microdamage is not as easily repaired (i.e., remodeled).
The amount of new bone put into the osteoclast excavations is a function of the number of mature osteoblasts and how long they can work. Therefore, bone formation beyond the amount removed by osteoclasts can be stimulated by agents that increase the number and/or active lifespan of PTHR1 receptor-expressing mature osteoblasts (Dempster, 1997; Manolagas, 2000) (fig. 8). As we shall see, definitely the PTHs, probably leptin, and possibly lipophilic statins, are just such anabolic bone-builders, some of which, like the PTHs, can stimulate bone growth without compromising crack repair.
Menopause and Bone Loss
Leptin, Fat, Brains, and Bones
So far, it has seemed that estrogen is the primum inter pares of an ever-growing number of agents that control bone growth and strength in both women and, perhaps surprisingly, men (Baylink et al., 1999; Klein, 1999; Stevenson and Lindsay, 1999; Vanderschueren et al., 2000). Now it appears that bones are also the direct and indirect targets of one of the principal operators of the mechanism that was originally discovered managing the white fat energy reserves in mice (Ducy et al., 2000a,b; Fleet, 2000; Himms-Hagen, 1999; Karsenty, 2000b; S. Takeda et al., 2003) (fig. 10).
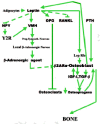
What Is Leptin?
"The leptin endocrine system is like a dynamic puzzle. As more additional pieces of the puzzle are found, more perplexities arise and more extra pieces are needed."—Jean Himms-Hagen (1999).
At first, it was white fat cells in mice and then a lot of other cells, including brain cells and osteoblasts, were found to make a helical cytokine hormone belonging to the IL (interleukin)-6 family of cytokines, the 167-amino acid, 16.7-kDa leptin (from the Greek word leptos meaning thin) (Ahima and Flier, 2000; Bradley et al., 2001; Friedman, 2000; Grasso et al., 1999; Gordeladze et al., 2001, 2002a, 2002b; Himms-Hagen, 1999; Wiesner et al., 1999). The production of leptin by fat cells is a function of their fat load. This means that the amount of circulating leptin tells the neurons in the hypothalamic arcuate nucleus of the size of the fat stores—it's a kind of fuel gauge needle. From the mouse model, leptin got the reputation of being a satiety signaler—a "Fat-O-Stat"—that inhibits the secretion of the orexigenic (eating-stimulator) neuropeptide Y (NPY) by arcuate neurons (Ahima and Flier, 2000; J. Friedman, 2000; Himms-Hagen, 1999; Pedrazzini et al., 2003). If the fat load is at the body's optimal set point, the circulating leptin holds NPY secretion from the arcuate nucleus at the appropriate level. However, if the fat load and with it leptin production drops below a critical limit, NPY secretion, and eating, rise to refill the fat fuel tanks and raise the circulating leptin concentration, which puts the brakes on eating.
But there is a problem with this popular mouse story. In her hard-hitting review, Himms-Hagen (1999) has presented a convincing case for leptin not being being a significant satiety hormone in humans. As she has pointed out, the very name leptin, which was of course coined for the mouse model, is wrong for humans—it is not a "Fat-O-Stat" in humans. Leptin's main role in humans is to serve as an energy monitor, the loss of which, when food becomes scarce enough to critically deplete the leptin-generating fat stores, sets off alarm bells that unleash a set of responses that try to maintain energy reserves without reducing activity (e.g., looking for food) as happens in rodents, but as in rodents to shut down ovarian cycling to avoid a pregnancy that would quickly consume any remaining energy reserves.
But leptin doesn't just monitor energy store and couple the level of this store to ovarian cycling. It also stimulates sympathetic activity that may be responsible for the deadly hypertension of obesity, and injecting it into the brain can activate sympathetic nerves in various regions of the body, including the rat hindlimb (Rahmouni and Haynes, 2002). This driving of sympathetic nerve activity is separable from the arcuate-based metabolic effects, but of course sympathetic activity drives the burning of fat to extract the energy needed to try to replenish the fat stores by hunting for food (Rahmouni and Haynes, 2002). And most important for our story—it can also affect bone growth by modulating sympathetic nerve activity.
What Does Leptin Do to Bone?
It isn't the arcuate nucleus through which leptin operates on bone from the brain. Karsenty's group have recently found the boney consequences of leptin stimulating the neurons of the ventral medial hypothalamic nuclei (VMHN), which projects to the sympathetic nervous system (S. Takeda et al., 2003). The hypothalamus is really the head nucleus of the autonomic nervous system, and leptin keeps the β-adrenergic nervous system functioning by stimulating VMHN neurons, which project to the preganglionic sympathetic neurons of the lateral cell columns of the spinal cord. And directly tweaking the VMHN neurons by squirting leptin into the cerebral ventricles of a mouse affects the beast's bones. But I am getting ahead of myself.
Of course bones are affected by leptin's stimulation of GnRH (gonadotropin-releasing hormone) secretion and the estrogen-dependence of the Lep gene (originally called the Ob gene for the obese mouse in which it was discovered) gene expression (Ahima and Flier, 2000; Brann et al., 1999; Chu et al., 1999; J. Friedman, 2000; Himms-Hagen, 1999). Therefore, when either the Lep (Ob) gene or the LepRb receptor (Sweeny, 2002) is disabled, the animal will have reduced sympathetic activity and thus be hypotensive (Mark et al., 1999). And it will also make too much cortisol and too little estrogen and therefore should be osteopenic. This should be very bad for bones!
But instead of being severely osteopenic as expected, Ob(Lep)-/- obese mice that cannot make leptin and Db-/- mice with disabled LepR receptors have a high vertebral bone mass (HBM) (Ducy et al., 2000a, 2000b; Karsenty, 2000b; S. Takeda et al., 2003). Since the lack of estrogen increases osteoclast production, they do have more osteoclasts than normal, lean animals but only a normal number of hyperactive osteoblasts that override the diggers by making twice as much bone matrix as the osteoblasts in lean mice (Ducy et al., 2000a, b; Karsenty, 2000b).
This supernormal bone production, despite the soaring osteoclast population, is due to a hypothalamic response to the lack of leptin production throughout the body in Ob(Lep)-/- obese mice or the incompetence of leptin receptors in Db-/- obese mice, and it can be stopped in Ob(Lep)-/- mice (which have functional leptin receptors) by injecting leptin into the animals' cerebral ventricles (Ducy et al., 2000a, 2000b; Karsenty, 2000b). Moreover, bone loss can be caused by intracerebroventricular leptin injection into normally boned, lean mice—leptin seems to cause the release from the brain of an osteoblast suppressor. Perhaps the osteoblast hyperactivity is due to the dramatic surge of NPY gene expression in the leptin-lacking Ob(Lep)-/- mouse's hypothalamus (Wilding et al., 1993). But no—cerebrally injected NPY does the same thing as leptin—it causes bone loss (Ducy et al., 2000; Karsenty, 2000)!
Thus, it seems that in mice, a set-point level of leptin might stimulate the neurons in some hypothalamic nucleus to produce a hypothalamic osteoblast inhibitory factor, a HOBIF, that somehow holds osteoblast activity at some optimal level (Ducy et al., 2000a,b; Karsenty, 2000b; Whitfield, 2002b; Whitfield et al., 2002) (fig. 10). It appeared at one point that the nNOS (neural nitric oxide synthase) might be part of this brain-based leptin signaling mechanism that dampens osteoblast activity, because knocking out the nNOS gene causes large increases in bone mass (van't Hof et al., 2002b). But this is not the HOBIF. At any rate, without signals from leptin-activated receptors on hypothalamic neurons, there is no osteoblast restraining HOBIF production, and bone growth climbs along with the food consumption and body weight (fig. 10).
But is all of this just much ado about nothing?! The bone growth in a fat mouse could simply be driven by the escalating loading by the massive increase in body weight and the consequent operation of the body's "mechanostat" to make more bone to reduce the strain as we have learned in the last chapters. But the bone mass starts rising before the weight rises. Furthermore, it also rises in A-ZIP/F-1 mice that do not have the leptin-making, white-fat adipocytes, without which the animals cannot gain weight (Ducy et al., 2000a; Karsenty, 2000b). But readers should beware of this model—all of the other sources of leptin must still be functioning in the A-ZIP/F-1 mice, and as we shall see further on, this bone growth may not be due to a lack of leptin and HOBIF.
The Karsenty group (S. Takeda et al., 2002,2003) now knows what HOBIF is. First, it is not a circulating agent, because when the circulations of two high-bone-mass Db-/- mice are connected (parabiotic pairing) and one receives intracerebral ventricle leptin, only the injected mouse loses bone—the factor does not spread via the circulation (S. Takeda et al., 2002,2003). Second, selectively destroying a normal mouse's ventromedial hypothalamic nuclei (VMH) with gold-thioglucose, but not destroying the arcuate nuclei with monosodium glutamate, drives the bone mass up to the high level in Db-/- mice and prevents the bone mass from dropping after intracerebroventricular leptin injection (S. Takeda et al., 2002,2003). Third, knocking out dopamine β-hydroxylase, and with it the ability of β-adrenergic nerves in the bones to make epinephrine and norepinephrine, does not increase body weight, but it does increase bone mass and blunts the bone-reducing response to intracerebroventricular leptin (S. Takeda et al., 2002,2003). Therefore, HOBIF is a noncirculating, i.e., within-bone, product of dopamine β-hydroxylase in the bones' nerves.
In mice, and probably humans, leptin operates on bone independently from the appetite and body weight controlling hypothalamic neurons of the arcuate nuclei (which, as noted above, can be destroyed without affecting bone mass) by stimulating the sympathetic nervous system via the LepRb receptor-bearing cells of the ventromedial hypothalamic nucleus (Funahashi et al., 2003; S. Takeda et al., 2002,2003), which increase catecholamine secretion by peripheral nerves (S. Takeda et al., 2002,2003). It appears that it is the VMHN-dependent, nonmyelinated sympathetic nerves in the bone that terminate near the trabeculae and follow the osteoblasts filling osteonal tunnels and locally bombard these osteoblasts with β-adrenergic agonists. And osteoblasts are certainly well-armed with β1- and/or β2-adrenoreceptors (Kellenberger et al., 1998; Majeska et al., 1992; Moore et al., 1993; Spencer et al., 2004; Takeuchi et al., 2000; Togari et al., 1997). For example, in my experience the β-adrenergic agonist isoproterenol very strongly stimulates adenylyl cyclase activity in ROS 17/2 rat osteoblasts. Activation of β-adrenoreceptors increases the expression of the pro-osteoclastogenic RANKL in MC3T3-E1 preosteoblasts and mouse bone marrow cells, and chemical sympathectomy inhibits preosteoclast differentiation and impairs bone resorption in adult rats (Cherruau et al., 1999; Spencer et al., 2004; Takeuchi et al., 2000). Also, we know that β-adrenergic stimulators, such as clenbuterol and salbutamol, decrease bone mass in leptin-deficient mice and female Wistar rats by reducing bone cell proliferation; that a β-blocker such as propranolol increases bone mass in intact and ovariectomized wild-type mice; and that taking β-adrenergic blockers reduces bone fracturing in women (Bonnet et al., 2003; Pasco et al., 2004; S. Takeda et al., 2002,2003).
To summarize—injecting leptin into the brain of a mouse stimulates the sympathetic nervous sytem and the adrenergic nerves in the bones via the lepRb-receptor-bearing cells of the ventromedial hypothalamic nuclei, and this raises the level of β-adrenergic transmitters in the bone that slow the proliferation of osteoprogenitor cells (certainly not the terminally post-proliferative osteoblasts as claimed by S. Takeda et al. (2002)) (fig. 10). But there is something inconsistent with their earlier paper. According to Ducy et al. (2000a), the high bone mass in the Ob(Lep)-/- is not due to there being more osteoblasts making bone as claimed by S. Takeda et al. but to the same number of osteoblasts making more bone.
On the basis of leptin's indirect, hypothalamically mediated, osteoblast-suppressing action, Karsenty (2000b) has called it the osteoblast-suppressing equivalent of the osteoclast-suppressing OPG. But it isn't (Gordeladze et al., 2001; Reseland and Gordeladze, 2002; Gordeladze et al.,2002a, 2002b; Whitfield, 2001, 2002)!! While the primary adult mouse osteoblasts in the experiments of Ducy et al. (2000a) apparently did not express the fully signaling Lep receptor—LepRb (Funahashi et al., 2003; Sweeny, 2002; Zabeau et al., 2003)—and thus could not respond directly to leptin, others have found that mouse bone cells can directly and positively respond to the cytokine both in culture and, most importantly, in the mouse. MC3T3-E1 mouse preosteoblasts and MCC-5 mouse chondrocytes both have LepRb receptors, make leptin, and dump it out into the culture medium (Kume et al., 2002). Liu et al. (1997) have reported that noncerebrally injected leptin stimulates endocortical bone formation in obese mice. Steppan et al. (1998, 2000) have also reported the results of experiments showing that intraperitoneally injected, instead of intracerebrally injected, leptin directly and potently stimulates bone growth in Ob(Lep)-/- mice. And could a direct action of the leptin be responsible for the high bone mass in A-ZIP/F-1 mice, which do not have white fat cells, but must have all of the other leptin makers?
As could have been expected from these other reports of leptin's bone-stimulating action in mice, it is now becoming clear that the Karsenty story is misleading because of an inadequate examination of their Ob(Lep)-/- mice and a failure to pay attention to the earlier reports of Steppan et al. (1998, 2000). But Hamrick et al. (2004) have now properly looked at Ob(Lep)-/- mice. It turns out that Steppan et al. (1998, 2000) were right for the appendicular skeleton! Leptin lack does indeed shorten femurs and reduce BMD, bone mineral content, cortical thickness, trabecular thickness, and trabecular volume in the femur. The expression of TGF-β1, an osteoprogenitor stimulator and strong anti-adipogenesis agent, is also reduced, which is consequently associated with a massive increase in femoral adipocytes and a drop in the osteocyte population density. However, the Karsenty group focused on an increased lumbar vertebral bone mass in their Ob(Lep)-/- mice. And according to Hamrick et al. (2004), they were also right! Hamrick et al. found increased bone mineral content, BMD, and trabecular bone volume in the lumbar vertebrae of their Ob(Lep)-/- mice. The Ob(Lep)-/- lumber vertebrae were also longer and broader than in normal lean mice. This suggested to Hamrick et al. that the chondrocytes of the vertebral growth plates respond differently from the femoral growth plate—maybe they are more sensitive to, or more controlled by, leptin/VMNH-dependent β-adrenergic activity. The loss of long bone (appendicular skeleton) mass is likely due in part to severely reduced loading because of a significant drop in muscle (e.g., quadriceps) mass as well as the basic inactivity of such obese beasts. As discussed in detail in Chapter 1, this would stress osteocytes and ultimately trigger their self-destruction and osteoclast scavenging of the cadavers by restricting the transcanalicular provision of nutrients and clearance of waste. And of course, another contributor would be the lack of leptin's direct anabolic action discovered in mice by Liu et al. (1997) and Steppan et al. (1998, 2000). Therefore, all that remains of the Karsenty mouse story is the ability of an intracerebroventricular shot of leptin to stimulate the VMHN-neurons and through them the preganglionic sympathetic neurons of the lateral cell columns of the spinal cord and the bones to which they project.
Cornish et al. (2001) have reported that leptin directly stimulates fetal rat osteoblasts as strongly as IGF-I and increases bone strength in mature male mice by 20% or more. And Picheret et al. (2003) have shown that obese female and male Zuker fa/fa rats with their disabled LepR receptors that cannot respond to the large amounts of leptin pouring out of their fat cells have significantly lower femoral bone density and osteoblast activity (as indicated by a significantly lower blood osteocalcin concentration) than lean control rats. This is supported by Tamasi et al. (2003) who have reported that in these rats tibial trabecular bone volume, trabecular number, and trabecular thickness are also lower than in normal Zuker rats, although the formation rates in the normal and Zuker fa/fa rats were not different. Moreover, Maor et al. (2002) have reported that leptin stimulates endochondral bone formation in cultured murine mandibular condyle and humeral growth plate by causing the cells to express and secrete IGF-I which, as we shall see further on, is also stimulated by PTHs.
Leptin is also one of the drivers of endochondral bone formation in the mouse. And Dumond et al.(2003) have found that leptin stimulates rat chondrocyes to grow and make cartilage by making IGF-I and TGF-β1. Large amounts of the cytokine are made specifically by hypertrophic chondrocytes (see Chapter 5v to find out what these cells do) near invading capillaries and osteoblasts in the primary spongiosa extending from the growth plate of long bones (Kerr, 1999). It seems that the chondrocytes use leptin to get the blood vessel cells moving and proliferating and making matrix metalloproteinases-2 and —9 which they use as drills for tunneling through the collagenous matrix to deliver blood-borne osteoprogenitor cells and other essentials to the construction sites (Kume et al., 2002).
Freshly isolated human osteoblasts (e.g., from trabecular bone in the head of the femur or the iliac crest), normal rat osteoblasts, and ROS 17/2.8 rat osteoblastic osteosarcoma (bone cancer) cells express the all-important big, signaling LepRb receptors (Bassilana et al., 2000; Enjuanes et al., 2002; Evans et al., 2001; Gordeladze et al., 2001; Y.-J. Lee et al., 2002; Reseland and Gordeladze, 2002a, 2002b; Reseland, et al., 2002; Steppan et al., 1999, 2000). Signals from these receptors obviously must stimulate bone growth because Thomas et al. (2001) have reported that intraperitoneally infusing human leptin prevents disuse ("pseudo-microgravity")-induced bone loss in tail-suspended rats. And Burguerera et al. (2001) and Gordeladze et al. (2002a, 2002b) have shown that leptin promotes rat bone growth by suppressing the osteoclast-promoting RANKL expression and stimulating osteoclast-suppressing OPG expression in osteoblastic stromal cells (fig. 10). Iwaniec et al. (1998) have reported that low concentrations (1-100 ng/ml of medium) of leptin increase bone nodule formation in human bone cultures. Leptin also makes human marrow stromal cells differentiate into osteoblasts instead of adipocytes (Thomas et al., 1999), and it stimulates the proliferation of cultured (i.e., probably incompletely matured simply because normal mature osteoblasts cannot proliferate) human osteoblastic cells and causes human marrow stromal osteoprogenitor cells to express alkaline phosphatase, collagen I, osteocalcin and to mineralize matrix (Gordeladze et al., 2002a, 2002b; Evans et al., 2001; Thomas et al., 1999). Leptin also enhances the expression of the genes for the major osteoblast differentiation driver, the master Cbfa1/Runx-2 gene transactivator, as well as alkaline phosphatase and osteocalcin, and it protects the human osteoblasts from self destructing by suppressing the expression of the proapoptotic Bax protein and stimulating the expression of anti-apoptotic Bcl-2 protein (Gordeladze et al., 2002a, 2002b). Leptin also facilitates the transition of cultured osteoblasts into preosteocytes (Gordeladze et al., 2002). Leptin's ability to facilitate the osteoblast→osteocyte transition and stimulate matrix mineralization by human osteoprogenitors fits in nicely with the fact that that the LepRb receptor-bearing human osteoblasts start making and secreting autocrine (self-stimulating)/paracrine (neighbor-stimulatimg) leptin when they are either in the mineralization or early osteocyte stage (Reseland et al., 2001) (fig. 10).
Most important for our story is the very exciting possibility that signals from type 1 PTH/PTHrP receptors (the so-called PTHR1s we will soon be getting to know rather intimately) stimulate the expression and secretion of leptin which, from the accumulating evidence, could at least partly mediate PTH's stimulation of bone growth (fig. 10). This has been suggested by Torday et al.'s (2002) report that PTHrP stimulates the leptin gene, leptin production, and leptin secretion in lung lipofibroblasts and by Gordeladze et al.'s (2002a) report that the effects of leptin on human osteoblasts mimic those of hPTH-(1-84).
The amazing story emerging from all of this goes something like this. Leptin is expressed first in mesenchymal stem cells to steer them onto the osteoblastic pathway, but this stops in proliferating osteoprogenitors. Then it starts again, after proliferation stops, and PTHrP and PTHR1 receptors appear and osteoblasts have matured, in order to help drive matrix deposition and mineralization. The leptin from the osteoblasts stimulates more osteoprogenitors to advance along the walls of the osteoclast tunnels and trenches and stimulates the activity and increase the longevity of their osteoblast friends and neighbors.
All of this means that human osteoblasts, like the cells of blood vessels, muscles, and many other organs throughout the body, are controlled, in the particular case of the osteoblasts negatively controlled, by the body-wide leptin/hypothalamic VMH-promoted β-adrenergic nerve activity. But they are also directly and positively controlled by leptin itself from the white fat adipocytes, local yellow marrow adipocytes, and most importantly from the late-stage osteoblasts themselves (Laharrague et al., 1998; Reseland et al., 2001, 2002; S. Takeda et al., 2003; Whitfield, 2002b) (fig. 10). Therefore, the question arises whether there is an adrenergic inhibition of bone formation or a strong direct stimulation by leptin of osteoblast activity in people with common (as opposed to the much rarer genetic (e.g., Ob(Lep)-/-) obesity who make a lot of the peptide that can't slam the brakes on eating but can still stimulate VMH-driven β-adrenergic activity and cause obesity-dependent hypertension (Rahmouni and Haynes, 2002). The reader should stay tuned in order not to miss the exciting things coming in the Leptin World, especially if an osteogenic leptin analog can be made that cannot pass the blood-brain barrier and trigger the negative VMH-driven β-adrenergic action.
But how does all of this interesting stuff fit into a postmenopausal story? The postmenopausal estrogen drop might turn down both of the leptin mechanisms (Brann et al., 1999; Chu et al., 1999), which would cause a rise in weight and help promote the osteoclast population explosion caused by the lack of estrogen. Thus, for example, in the rat model ovariectomy silences Ob(Lep) gene expression, and leptin consequently vanishes from their circulation (Chu et al., 1999). And the animals get hungry and heavier, while their trabecular bone is being chewed up by osteoclasts and their marrow is getting fattier (Whitfield et al., 1995). The leptin drop and the resulting surge of NPY expression in the hypothalamus cause the hunger and weight gain. At first sight, this seems to conflict with the amount of circulating leptin expression being positively correlated to the body fat load—leptin should rise. But Ob(Lep) gene expression is estrogen-dependent, and giving estrogen to the OVXed rats prevents the leptin drop, overeating, and weight gain (Chu et al., 1999). Therefore, without enough estrogen to maintain the Ob(Lep) gene expression, leptin virtually vanishes from the circulation by 7 weeks after OVX, but the increasing accumulation of white fat in its swelling adipocytes eventually overrides the estrogen lack, leptin production rebounds, and its level soars above the sham OVX level between 9 and 13 weeks after the operation (Chu et al., 1999). Therefore, the bones of an overeating OVXed rat do not at first have to face the debilitating double whammy of leptin-induced, VMH-driven β-adrenergic-suppressed osteoblast activity as well as an osteoclast feeding frenzy, but the direct stimulation of osteoblasts by the eventual leptin surge should slow or stop the bone loss. Alternatively, any positive action of the leptin surge might be counterbalanced by an adrenergic surge. It follows from this that it would be important to find out whether a leptin loss might also affect bone loss in postmenopausal women and whether leptin injection would stimulate bone growth in these women as it does in rats. Indeeed, Ke et al. (2001, 2002) have anticipated leptin's admission into the exclusive Bone Growers Club by patenting it and its analogs for treating osteoporosis and all other conditions causing bone loss and for accelerating fracture healing.
But the hypothalamis has a lot of jobs to do, and there seems to be another player in the brain-and-bones saga—NPY's Y2 receptor. Why do we think so? Well, if the Y2 receptor gene is selectively knocked out in mouse hypothalamic neurons, osteoblasts make more matrix just as they do in Ob(Lep)-/- mice (Baldock et al., 2002; Herzog, 2002)—signals from the Y2 receptors in hypothalamic neurons somehow lead downstream to the reduction of osteoblast activity. And of course, this why Ducy et al. (2000a) found that directly injecting NPY into the cerebral ventricles caused bones to lose mass in their Ob(Lep)-/- mice, just as did leptin injection. Thus, when the leptin level is "normal", NPY secretion and Y2 receptor signaling are minimal or suppressed. But any possible upsurge of osteoblast activity caused by shutting down NPY-Y2 receptor activity is prevented by leptin-driven VMH-mediated β-adrenergic secretion. However, the direct osteogenic action of leptin would still be working in the bones.
But things are complicated by the fact that NPY also directly targets osteoblasts. Indeed, osteoblasts have receptors for the NPY released into the bone by the same sympathetic nerves that are driven by leptin via the VMHN neurons (Bjurholm, 1991; Ekblad et al., 1984; Togari et al., 1997). But we don't yet know how NPY receptors signals directly affect osteoblast activity. All we know at the moment is that NPY inhibits the abilities of noradrenaline and PTH to stimulate adenylyl cyclase in osteoblastic cells (Bjurholm, 1991). However, it is clear that there is a complex control system operating at several levels running from the brain and spinal cord to the bones and osteoblast surfaces to control bone formation.
But before moving on to other things, it must be clearly understood that as matters stand in late 2004, leptin is a direct bone anabolic agent which may indirectly, but only under severely nonphysiological conditions, limit bone growth by increasing local bone β-adrenergic activity via the VMHN neurons. Thus, it would seem to be unreasonable to try to treat osteoporotic patients without cardiovascular disorders with potent β-blockers as suggested by S. Takeda et al. (2003). But Pasco et al. (2004) have presented data indicating that women with cardiovascular disease who have been taking such drugs do have higher bone mineral densities and fewer fractures.
How Does Estrogen Loss Cause an Osteoclast Population Explosion?
The ability of osteocytes to know when the strain exceeds a site-appropriate level and to react to this by signaling through the osteointernet the need to make the various factors such as PGE2, PGI2, IGFs, and TGF-βs as well as our new friend leptin to mobilze osteoblasts to make more bone to bring the strain to a subthreshold level depends on estrogen (Ehrlich and Lanyon, 2002). But as the estrogen level starts falling, estrogen receptor-α (ERα) signals start fading, as must leptin on the approach to menopause, the osteocytes become less responsive to strain, i.e., they become less able to feel the sloshing of the canalicular fluid. This results in fewer calls for resources to maintian bone and increasing uncompensated strain from muscle pulling and limb impacts with a preferential loss of minimally strained bone. But along with the osteocytes becoming "strain-deaf", the estrogen drop also causes an upsurge of osteoclast generation.
However, the vigilant reader will challenge this blanket assertion that the signals from estrogen receptors fade as the estrogen level drops because strain by itself can activate estrogen receptors without estrogen (Aguirre et al., 2003). Thus, I must now qualify this by saying simply that estrogen seems to be replacable in some cells such as osteocytes by strain.
Osteoclast progenitors respond to estrogen through their conventional "genomic" estrogen receptors, which work on the chromosomes in the nucleus to activate target genes with so-called EREs (estrogen response elements) in their "signal boxes" as well as through what appear to be "nongenomic (genotropic)" receptors that are translated into protein from the same primary transcript as the genomic receptors and fire signals into the cell from little caves (caveolae) or pouches in the plasma membrane (Chambliss et al., 2000; Coleman and Smith, 2001; Falkenstein et al., 2000; Fionelli et al., 1996; Kelly and Levin, 2001; Kim et al., 1999; Krishnan et al., 2001; Levin, 2000; Manolagas et al., 2002; Morley et al., 1992; Razandi et al., 1999, 2002; Segars and Driggers, 2002; Toran-Allerand, 2000; C.S. Watson, 2003).
Signals from these receptors in a young woman might limit the size of her preosteoclast population by a TGF-β-mediated killing of the cells by apoptosis (Manolagas, 2000; Whitfield et al., 1998b; Zecchi-Orlandini et al., 1999) (fig. 11 and fig. 12). Therefore, as the estrogen level falls with approaching menopause, less TGF-β is made, and osteoclast precursors live longer to make bigger osteoclast crews for more extensively and deeper-digging BMUs (fig. 12). It is also likely that the lack of estrogen could cause a drop of caveolin production and loss of osteoblastic cells' caveolae, which would normally hold together and restrain groups of signaling enzymes (Chambliss et al., 2000; Couet et al., 2001; Okamoto et al., 1998; Razandi et al., 1999, 2002; Razani and Lisanti, 2001; Razani et al., 1999, 2001; Solomon et al., 2000). This would contribute to the surge of osteoclastogenic signaling. The number of osteoclast progenitors in the microenvironment created by marrow stromal cells also rises because of a surge into the stromal microenvironment of factors such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α from the IL-1β-stimulated marrow monocytes and perhaps most importantly from TNF-α from an increasing population of T-lymphocytes (Cenci et al., 2000; Hofbauer et al., 2000; Jilka, 1998; Katagiri and Takahashi, 2002; Kobayashi et al., 2000; Martin et al., 1998; Pilbeam et al., 1997; Roggia et al., 2001) (fig. 11 and fig. 12). Indeed in mice, TNF-α from a growing, OVX-induced population of marrow T-lymphocytes is required for the estrogen drop to cause bone loss (Roggia et al., 2001). This TNF-α stimulates marrow stromal cells to make M-CSF that puts comitted macrophage-family members on the road to osteoclasts (Hofbauer et al., 2000; Roggia et al., 2001). In the estrogen-rich youthful koric (from the Greek for "youth") bones, the responsiveness of the osteoclast progenitors to the TNF-α-inducing IL-1β is blunted by the estrogen-promoted, functionless type 2 IL-1 decoy receptors on their surfaces that compete with the functional receptors for IL-1β (Hofbauer et al., 2000; Manolagas, 2000; Pilbeam et al., 1997; Suda et al., 1999; Sunyer et al., 1999) (fig. 11 and fig. 12). But as estrogen declines, so do the decoy receptors, and IL-1β is no longer diverted from driving the development of the osteoclast progenitors, which it can do by stimulating the expression of the transcription factor NF-κB (Xing et al., 2003). The estrogen-deprived osteoblastic stromal cells also start making the potent osteoclast differentiation stimulators, IL-6 and IL-11 that have been suppressed by estrogen (Jilka, 1998; Manolagas, 2000; Whitfield et al., 1998b) (fig. 11).
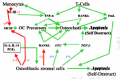

Figure 12
Summary of some of the many consequences of an estrogen crash on bone.
These several interleukins, prostaglandins, and TNF-α focus on immature osteoblastic marrow stromal cells to persuade them to make more RANKL and put it out on their surfaces (Atkins et al., 2003; Aubin and Bonnelye, 2000; Blair et al., 2002; Greenfield et al., 1999; Hofbauer et al., 2000; Karsenty, 1999; Suda et al., 1994) (fig. 11 and fig. 12). However, the TNF-α also tends to increase osteoblast apoptosis and at the same can itself stimulate the differentiation of osteoclast precursors by activating TNFR1 and TNFR2 receptors and the receptors'-associated TRAF(TNF-receptor-associated factor)2 factors, which switch on osteoclast-differentiating genes via the NF-κB transcription factor (Hill et al., 1997; Katagiri and Takahashi, 2002). In fact, TNF-α is actually a better stimulator of osteoclast activity than differentiation from precursors (Fuller et al., 2002). Moreover, TNF-α can very strongly synergize with RANKL, so that even a slight increase in TNF-α that is too small to increase osteoclast generation in the presence of normal levels of the differentiation-driving RANKL would greatly increase osteoclast activity without an increase in osteoclast numbers (Fuller et al., 2002).
The RANKL surfacing on immature osteoblastic marrow stromal cells binds to and activates the differentiation-stimulating RANK receptors on preosteoclasts when they dock on the stromal cells (Atkins et al., 2003; Aubin and Bonnelye, 2000; Hofbauer et al., 2000; Suda et al., 1999). (However, cell-cell contact is not absolutely necessary because free RANKL is also secreted by osteoblastic cells and other cells, such as activated T-lymphocytes, which stimulate the generation of the osteoclasts that, for example, dig holes in rheumatoid arthritic joints (Karsenty, 1999; Kong et al., 1999) (fig. 11). The RANKL-activated RANK receptors on osteoclast precurors and beyond drive maturation, bone-digging activity and survival via receptor-associated TRAF proteins that activate transcription factors such as AP-1, NFAT, and NF-κB that switch on the genes for osteoclast differentiation (Katagiri and Takahashi, 2002; Z.H. Lee and H.-H. Kim, 2003). The TRAFs-mediated RANK signals also stir up a swarm of other kinases that turn on the digging machinery and hold-off the self-destruct apoptogenic mechanism (Z.H. Lee and H.-H. Kim, 2003).
The ability of osteoblastic precursor cells to make RANKL falls as the rankl gene's promoter-switch box is locked shut by methylation of its CpG domains while the anti-osteoclast opg gene stays active as the progeny differentiate into mature bone-making osteoblasts—a wise stratagem to protect the new bone they are making (Atkins et al., 2003; Gori et al., 2000; Kitazawa et al., 1999; G.P. Thomas et al., 2001). To understand the control of osteoclast differentiation, we must know that the expressions of the genes for RANKL and OPG in the individual cultured cells are logically linked by a reciprocal control mechanism, which will be discussed in Chapter 5. Suffice it to know here that when one gene's expression is turned up, the other's expression is turned down (e.g., Ma et al., 2001; Yamagishi et al., 2001).
Estrogen controls the OPG/RANKL expression ratio, which determines the level of osteoclast generation. Thus, a young woman's estrogen stimulates the production of TGF-βs which, beside directly stimulating osteoclast apoptosis, increases the osteoblastic stromal cells' OPG/RANKL expression ratio which probably reflects the relative fractions of RANKL-expressing immature mature cells and OPG-expressing mature cells in the marrow stroma and restrains osteoclast generation (Atkins et al., 2003; Aubin and Bonnelye, 2000; Hofbauer et al., 1999, 2000; Murakami et al., 1998; Takai et al., 1998) (fig. 11). And estrogen also hits osteoclast production directly by preventing the RANKL-triggered RANK signals in osteoclast progenitors in the marrow from driving differentiation by blocking the AP-1 (c-Fos•c-Jun) transcription factor complex's activity by lowering the expression and activity of the c-Jun component (Shevde et al., 2000). OPG is another decoy receptor, like the nonsignaling type 2 IL-1 receptor that is designed to restrain osteoclast production. OPG is actually a floating piece of the RANK receptor that is made by mature osteoblasts and binds to and thus covers up RANKL molecules sticking out of the surfaces of immature osteoblastic stromal cells, which prevents RANKL from binding to the differentiation-driving RANK receptors on osteoclast precursors (Aubin and Bonnelye, 2000; Hofbauer et al., 1999, 2000; Makhluf et al., 2000; Moonga et al., 1998; Murakami et al., 1998; Oyajobi et al., 2001; Takai et al., 1998).
The estrogen decline doesn't only increase the numbers of longer-lived, bone-excavating osteoclasts. The increased number of osteoclasts associated with immature stromal osteoblastic cells and cytokine-stimulated T-lymphocytes probably produce soluble and/or membrane-bound FasL (Fas ligand or binder), which cause osteoblastic cells with Fas receptors on their surfaces to self destruct apoptotically (Kawakami et al., 1997, 1998) (fig. 11).
The TGF-β stores in the bone matrix are also depleted (reviewed by Jilka, 1998; Whitfield et al., 1998b). Therefore, there is even less of it that can evade the proteases and reduce osteoclast generation and kill mature osteoclasts when they dig it out of the matrix (Jilka, 1998). And there is also less of it to attract immature osteoblasts into the excavation sites, although TGF-β receptor signals may be less important for this than the migration-stimulating signals from the osteoblastic cells' Ca2+-sensing receptors stimulated by the very large amount of Ca2+ in the excavation sites (Brown and MacLeod, 2001; T. Yamaguchi et al., 1998).
Since the endothelial cells of the capillary loops following behind the trench-digging and tunneling osteoclasts express estrogen receptors and secrete osteoprogenitor-stimulating factors such as endothelin-1 (Brandi et al., 1993; Mohammad et al., 2003; Soerensen and Eriksen, 1998; von Schroeder et al., 2003), a lack of estrogen receptor signaling could impair the ability of the capillary's endothelial cells to attract and stimulate the development of accompanying osteoprogenitors which would reduce the size of the BMU osteoblast crews (fig. 12).
Since estrogen also lengthens the working lives of osteoblasts and the microcrack-detecting osteocytes by suppressing their apoptotic self destruct mechanism, the estrogen loss unleashes this mechanism and reduces the number of osteocytes (Manolagas, 2000; Manolagas et al., 1999; Tomkinson et al., 1998). This loss of osteocytes would reduce the inhibitory signaling through the osteocyte-lining cell network, which would be equivalent to severing the signaling network by microcracks (R.B. Martin, 2000, 2002). This pseudo-microcrack signaling would increase BMU activation and therefore bone turnover and the remodeling space—functionally it would be equivalent to a widespread microcracking, like the shattering of an automobile windshield.
Perhaps counterintuitively, such BMU activation should, and in my experience does, increase the number of osteoblasts. However, the hole-filling efforts of these osteoblasts can't keep up with the hole-digging zeal of the large osteoclast crews!
Under these low-estrogen conditions, osteoblast working life and activities, such as laying down bone matrix and secreting various factors, may also be reduced, and the BMU replacement deficit thus increased, by the loss of signals from the nongenomic estrogen receptors such as those found on the surfaces of cultured female rat osteoblasts (Falkenstein et al., 2000; Lieberherr et al., 1993; Manolagas, 1999). These surface receptors appear either to be the products of the gene for the nucleus-seeking, gene-activating ERα genomic receptor that have been modified to stay at the cell surface and fire Ca2+ and cyclic AMP signals into the cell when activated by 17β-estradiol (Chambliss et al., 2000; Falkenstein et al., 2000; Fiorelli et al., 1998; Kelly and Levin, 2001; Lieberherr et al., 1993; Razandi et al., 1999; C.S. Watson, 2003; Whitfield et al., 1998b). Or they might be the entirely unrelated ER-X receptors (Toran-Allerand, 2000) or the "γ-adrenergic" receptors that are activated by dopamine, epinephrine, and norepinephrine as well as 17β-estradiol and various xenoestrogens (Benten et al., 2001; Nadal et al., 1998, 2000).
The loss of Ca2+ influx-driving signals from these nongenomic estrogen receptors on the surfaces of intestinal epithelial cells may further reduce intestinal Ca2+ uptake and increase the bone loss due to the vitamin D deficiency that commonly develops in older persons (Chen and Kalu, 1998; Doolan et al., 2000; Picotto et al., 1999) (fig. 12). Adding to this vitamin deficiency may be a drop in the estrogen-dependent expression of the 1α,25-dihydroxyvitamin D3 receptor that drives Ca2+ uptake and transcellular transport to the blood by colon epithelial cells (Schwartz et al., 2000).
In summary, all of this boils down to the menopausal estrogen decline producing more BMUs with bigger osteoclast and smaller osteoblast crews that cannot fill the larger number of deeper holes dug by the swollen osteoclast crews. The results are weakening bones and increasing remodeling, for example because of the escalating number of osteonal tunnels in remodeled cortical bone, and a dramatic upsurge of microcracking and remodeling (Bouxsein, 1999). But the BMU crews answering the calls are getting ever shorter of effective osteoblastic cell power. Thus, the vicious microdamage-driven remodeling cycle of aging is accelerated (fig. 9). The cause of osteoporosis is basically escalating remodeling by osteoclast-overmanned, osteoblast-undermanned BMUs, just as road repair crews with over-manned digging teams and under-manned filling teams would produce an increasingly fragile, crack-prone road.
How to Stop (or at Least Slow) Menopausal Bone Loss
To properly stop menopausal bone weakening and fracturing, it is necessary to break the vicious remodeling↔microdamage cycle (fig. 9). Ideally, to do this we should restore the optimal composition of BMUs by reducing the osteoclast and increasing the osteoblast crews. This would reduce the number of osteonal pores produced during the repair of microdamaged cortical bone and lower the amount of cutting and perforating of trabecular struts and plates by osteoclasts. It would also prolong the mineralization and hence strengthen new bone.
Obviously, bone loss can be stopped by giving the post-menopausal woman estrogens or one of the partially estrogen-mimicking SERMs (selective estrogen receptors modulators) such as raloxifene (Eli Lilly's Evista™) (Roe et al., 2000). But this does not cause a sustained increase in bone formation—it works by reducing remodeling. Giving one of these, raloxifene, to OVXed rats does not affect the expression of bone formation-related genes such as alkaline phosphatase, biglycan, collagen I, IGF- II, collagen-binding protein, and osteocalcin in their femurs, but it actually reduces the expression of decorin and IGF-I (Onyia et al., 2002). On the other hand, it reduces osteoclast generation by reducing the expression of CSF-1 (colony-stimulating factor-1).
Bone loss can also be reduced or even stopped with one of the many bisphosphonates such as alendronate (Merck & Co.'s very popular Fosamax™) (Fleisch, 1997, 1998), or either oral or nasally sprayed calcitonin peptide that shuts down osteoclast activity by activating receptors on the cells of the osteoclast lineage (Roodman, 1996; Suda et al., 1999).
The bisphosphonates are Ca-apatite-loving, bone-binding analogs of pyrophosphate (P-O-P) in which the oxygen has been replaced by a carbon atom, the so-called geminal carbon, which makes them nonmetabolizable. They preferentially attach to osteoclast excavation sites and attack the working mature osteoclast crews (Rogers, 2003). Then, upon the arrival of osteoblasts where the bone mineral is exposed, they are buried in the new bone. There they lie in wait, unchanged for months to years, until the poisonous bone patch is once again cracked and marked for removal by osteoclasts. The osteoclasts inadvertently release the deadly bisphosphonates and suicidally gobble them up along with other debris they want to expel from the hole by carrying them from their apical acid-protease-dripping fingers poking into the holes to their upper basal surfaces by a process called "transcytosis". (The terms apical (upper end) and basal (bottom end) may be a bit confusing here because osteoclasts stick their apical fingers into the bone and their basal tails in the "air".) Because the bisphosphonates lock onto the exposed mineral surfaces of osteoclast-excavating microcrack repair sites, and since trabecular bone reputedly has the highest turnover rate (though see Parfitt (2002)), the bisphosphonates are best able to prevent resorption and remodeling and increase the strength of trabeculae-rich bones such as vertebrae and femoral trochanter, which have the largest numbers of attractive remodeling excavation sites at any given time (Rodan and Reszka, 2002).
The bisphosphonates can persuade osteoclasts to self destruct by apoptosis in one of two ways (Amin et al., 1992; Benford et al., 1999, 2001; Reszka et al., 1999; Rodan and Reszka, 2002; Rogers, 2003; Rogers et al., 2000; van Beek et al., 1999) (fig. 13). The nitrogen-containing bisphosphonates (N-BPs) such as alendronate (Fosamax™), ibandronate, incandronate, risedronate, and zolendronate disable and kill osteoclasts as well as lower cholesterol, just like the cholesterol-lowering statins you are going to meet in Chapter 8 (Rogers, 2003). Both the N-BPs directly, and statins indirectly, inhibit FPP (farnesyl pyrophosphate) synthase (Coxon and Rogers, 2003; Dunford et al., 2002; Luckman et al., 1998; Rodan and Reszka, 2002; Rogers, 2003; van Beek et al., 1999) (fig. 13). This prevents the isoprenylation by farnesyl-PP or geranylgeranyl-PP transferase that small GTPases—Cdc 42, Rac, the Rabs, Rases, and Rhos—need to anchor themselves to cell membrane scaffolds and rafts and cytoskeleton-shaping devices. Thus, for example, on its way to the cell membrane Ras is first isoprenylated by having farnesyl-PP diphosphate tacked onto its C-terminal cysteine residue by farnesyl-PP transferase in the cytosol, then it is delivered to the endoplasmic reticulum and from there to the Golgi membrane system, where it is also palmitoylated and either plugged into a new scaffold and sent to the cell membrane or separately plugged into the cell membrane (Bertiaume, 2002). If farnesylation fails, the Cdc 42, Rabs, Racs, Rases, and Rhos will not be plugged into signaling scaffolds (Coxon and Rogers, 2003). This will disrupt and disable signaling rafts, scaffolds, and their attached actin structures, effects that will reverberate throughout the cell to prevent the formation and operation of the osteoclasts' sealing rings and the proton pumps in the ruffled membrane that sprays the mineral-dissolving HCl and matrix-shredding proteases onto the bone. All of this metabolic mayhem can be prevented simply by feeding the cells geranylgeraniol, an analog of geranylgeranylpyrophosphate that can pass through the cell membrane (Coxon and Rogers, 2003; Benford et al., 1999, 2001; Fisher et al., 1999; Reszka et al., 1999).

Figure 13
The ways statins and N (nitrogen)-bisphosphonates (N-BPs) disable and kill osteoclasts. See fig. 14 and the text for further details of how N-BPs and other bisphosphonates kill osteoclasts.
Let's summarize what we have learned so far. A mature osteoclast, the specific target of an N-BP, does its job with its cytoskeletal machinery. It lays down an actin ring—a bit like a micro-octopus's suction cup. Inside the ring it sets up its ruffled border loaded with molecular hoses for spraying the HCl and matrix-chewing enzymes for digging the hole. These cytoskeletal creations are driven and shaped by the various isoprenylated small GTP-ases. When a N-BP sits down on a bone patch and is swallowed (by fluid-phase endocytosis) by the osteoclast, the whole cytoskeletal machinery grinds to a stop as the cell runs out of its supply of isoprenylated GTPases. So the first effect of a N-BP is to seek out and disable mature osteoclasts wherever they are digging. But something even more sinister follows the shutdown.
One of the effects of the N-BPs towers above the others—the stimulation of the caspase-3 protease that will kill the osteoclast unless blocked in time by an inhibitor such as zVAD-fmk or SB-281277 (Benton et al., 1999, 2001; Reszka et al., 1999). Caspase 3 slices the 50-kDa Mst 1(Krs2) kinase into a regulatory C-terminal fragment and an active 34-kDa kinase fragment, which in turn activates more caspases (Reszka et al., 1999) (fig. 14).
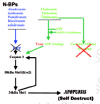
Figure 14
The mechanisms by which N-bisphosphonates drive osteoclasts to self destruction by causing them to switch on their apoptosis mechanism.
The nonnitrogen-containing bisphosphonates (clodronate, etidronate, tiludronate) do not disable the osteoclasts by inhibiting FPP, but they combine with ATP to form toxic ATP analogs (e.g., the forgettably named adenosine-5'(β,γ-dichloromethylene)triphosphate) that kill the cell (Benford et al., 2001; Frith et al., 1997, 2001; Reszka et al., 1999; Rogers, 2003) (fig. 14). Tiludronate can also directly disable the diggers by inhibiting their mineral-dissolving, protease-activating, acid-spraying proton pump (David et al., 1996).
But alendronate (Fosamax™) unfortunately does not leave osteoblasts alone, at least in rats. Far from it—the orally delivered bisphosphonate actually turns down the expression of genes for bone formation in the proximal femoral metaphysis of ovariectomized rats (Onyia et al., 2002)! The inhibited genes include the ones for alkaline phosphatase, biglycan, collagen I, decorin, IGFs-I and II, collagen-binding protein, and osteocalcin. This strong suppression of osteoblast genes explains the strong suppression of bone formation activity by alendronate in rats (Onyia et al., 2002). And this happens not only in rats. Black et al. (2003), Finkelstein et al. (2002), and Neer et al. (2002) have found that alendronate also inhibits alkaline phosphatase and prevents hPTH-(1-34)(Forteo™) and hPTH-(1-84) from stimulating it and bone growth in hip and vertebrae in osteoporotic men and postmenopausal women.
But as I said at the start of this section, alendronate does appear to do something more positive to osteoblasts and osteocytes. It can protect osteoblasts and osteocytes from apoptotic self destruction. The cytoplasms of these cells are normally directly interconnected in a functional syncytium by gap junctions which are pipes made of end-to-end joined halves ("half-pipes") or connexons, each of which is a bundle of six, 4-pass (tetraspanning) transmembrane proteins, such as Cx (connexin) 43, through which passes a 1.5-μm channel through which ions (except Ca2+, which causes the channel to slam shut) and a wide variety of small molecules such as cyclic nucleotides and ATP can pass (Goodenough and Paul, 2003). But these cells also have uncoupled, or extra-junctional (EJ), "half- pipe" connexons that independently stick out of the cell and serve as nonspecific, large channels which slam shut when the external Ca2+ concentration rises above a critical level(Goodenough and Paul, 2003). It seems that alendronate operates by binding to and opening these "1/2 pipes", then passing through them into the cell and activating the cSrc protein-tyrosine kinase, which triggers an apoptosis-blocking MAP/ERK kinase cascade that closes the pipes by phosphorylating their Cx43 proteins (Goodenough and Paul, 2003).
Because these antiresorptives (or antiremodelers) suppress osteoclasts and may also reduce osteoblast action, they only cause, at the most, a limited amount of growth in mature human bones as the osteoblasts continue filling in the microcrack repair holes (the remodeling space), which account for 10% or more of the current total bone volume at any given moment (Dempster, 1995, 1997; Fleisch, 1997, 1998). But this is the limit—the gains in bone mineral density (BMD) in lumbar spine caused by optimal doses of alendronate, etidonrate, HRT, raloxifene, and residronate range only from 2.6 to 6.8% and from 1.0 to 4.8% in the femoral neck by 3 years (Pearson and Miller, 2002).
Because they stop osteoclast development and therefore micocrack-triggered remodeling, bisphosphates such as alendronate at least temporarily reduce fracture rates by prolonging the so-called secondary mineralization (i.e., by hardening the remaining bones and increasing the bone mineralization density level and therefore strength), but they do so without affecting trabecular thickness or number, i.e., without increasing the amount of bone tissue (Boivin and Meunier, 2002; Boivin, et al., 2000; T.J. Martin, 2004; Pearson and Miller, 2002). They may also improve hole-filling by making osteoblasts live and work longer (Allen et al., 2002; Manolagas, 2000) by preventing them from self-destructing by apoptosis by sucking up the large amounts of phosphate in the osteoblast excavations with their Pi transporters (Mansfield et al., 2001). Nevertheless, they can't raise bone mass above the normal level in rats or reduce the fracture risk in the mature human skeleton by more than 50% of the baseline risk (Rosen and Bilezekian, 2000; Seeman, 2001). However, a wrong impression of anabolic ability can be obtained by extrapolating the responses to antiresorptives of the continuously growing bones of mice and rats to the nongrowing mature remodeling human bones, where, because of osteoblast generation's tight coupling to osteoclast activity, the reduction of osteoclast generation ultimately reduces osteoblast generation. Thus, rodents with their always growing, low-microcracking bones treated with antiresorptives are like mutant mice, which cannot produce functional osteoclasts and become osteopetrotic as their contiuously and independently generated osteoblasts continue their unopposed bone making (Karsenty, 1999).
There is a potentially very serious problem with bisphosphonate therapy. Microcracking (i.e., microcrack density) increases exponentilally with age in bones such as the femur, probably because declining bone mass increases strain on the remaining bone and thus cracking (R.B. Martin et al., 1998; Schaffler et al., 1995). Therefore, murdering mature osteoclasts and preventing their replacement might be counterproductive because it will impede repair of the rising microcracks, which could with time lead to a resurgence of fracturing, the seriousness of which would be mitigated of course by the weakening muscle-imposed strain on the aging body by its shrinking muscles (Mashiba et al., 2001; R.B. Martin et al., 1998).
The Amazing Bone-Anabolic PTHs
Anabolics—The Answers to the Osteoporotics' Prayers
Clearly, the antiresorptives are far from being the "Holy Grails" of osteoporosis therapy, although they do break the vicious cyle of escalating remodeling and microdamage (fig. 9). Since by the time of their first fracture, the bones of osteoporotic postmenopausal women have undergone considerable microarchitectural deterioration (e.g., cortical osteonal porosity (R.B. Martin et al., 1998)) and have lost about 30% of their bone mass because of escalating microfracturing and resulting remodeling activity, it is essential to find an agent that can increase bone mass a lot more than the 6-10% which is the most that can be got from a 3-year treatment with antiresorptives/antiremodelers. The ideal drug for treating osteoporosis and accelerating fracture healing in both women and men would be a true anabolic that directly stimulates osteoblast production which actually makes strong new bone instead of just permitting the unopposed filling and supermineralization of existing remodeling holes by murdering osteoclasts and lowering the remodeling activity to a "normal" or even subnormal level.
In osteopenic ovariectomized animal models such as rats or monkeys, such a true osteoblast-stimulating anabolic would push the bone mass well above the normal, sham-operated mass without interfering with microcrack repair, which otherwise could lead to an accumulation of microdamage as may happen with long-term treatment with antiresorptives/antiremodelers such as bisphosphonates (Mashiba et al., 2001). It would largely bypass the BMU remodeling/repair cycle by directly and strongly stimulating bone formation. It would directly stimulate a buildup of longer-lived, less apoptosis-prone osteoblasts, which would restore the mechanical strength of bones such as vertebrae and femurs by thickening their remaining trabeculae and thus strengthen their connectivity (Dempster, 1997; Manolagas, 2000) (fig. 15).
Another property expected of an anbolic agent is a much greater stimulation of trabecular and endosteal growth than cortical growth because growth can only be started on bone surfaces, and the total trabecular surface in an adult human male is 9 m2, while the total surface area of the Haversian and Volkmann's canals inside the cortex is only about 3.2 m2 (Johnson, 1966; R.B. Martin et al., 1999). However, as I pointed out in Chapter 2i the greatest total surface area, 1200 m2, is in the 3-D osteocyte lacunar-canalicular network, but it is obviously inaccessible—it is stuffed with cell bodies and processes (Johnson, 1966; R.B. Martin et al., 1999).
As we shall now see, the PTHs—the big native parathyroid hormone and certain of its small fragments and at least one of the fragments' lactamized (cyclic) analogs—are the first and currently the foremost bone anabolics (Whitfield et al., 1998b, 2002a, 2002b, 2002c).
The Ancient Origins of PTH and Its Receptor
The receptor for PTH and the PTH-like N-terminal 13 amino acids of PTHrP (PTH-related protein), which we will soon be meeting, belong to an ancient family that likely appeared hundreds of millions of years ago in the first animals in the Precambrian seas. The descendents of the ancient Mother Receptor are the modern receptors for calcitonin, corticotrophin-releasing hormone, gastric-inhibitory protein, glucagons, glucagons-like peptide-1, growth hormone-releasing hormone, an insect (the tobacco hornworm Manduca sexta) diuretic hormone, insect (Drosophila) calcitonin-like hormone and vasopressin-like hormone, pituitary adenylyl cyclase-activating protein, secretin, vasoactive intestinal peptide (Brody and Cravchik, 2000; Whitfield et al., 1998b; Wong, 2003). This family is an example of Nature using a basic design to do many different jobs. Thus, although these receptors have different amino acid sequences, they have similar higher order structures. Indeed, they are so alike that changing only one amino acid enables the secretin receptor to be activated by PTH and switching the equivalent amino acid in the PTH/PTHrP receptor to the secretin receptor's amino acid enables the receptor to be activated by secretin!
Hints of the antiquity of the PTH family and their receptors are the ability of mammalian PTH to increase the Ca2+ concentration in crustacean hemolymph by mobilizing the ion from the exoskeleton just as they do from our endoskeletons; the presence of a molecule in the neuroendocrine cells of the pond snail that cross-reacts with anti-mammalian PTH antibodies; and the ability of PTH to stimulate Ca2+ influx into snail neurons (Whitfield et al., 1998b). Is it possible that the Family was emerging in trilobites and Anomalocaris? Certainly our Cambrian ancestors, the notochord-bearing Myllokunmingia fengjiaoa and Pikaia (from Chengjian, China (Xian-Guang et al., 2004) and Alberta, Canada (Long, 1995)), respectively, must have been able to swim 500,000 years ago in the warm Ca2+-rich (ca. 10 mM) Panthalassic and Iapetus oceans, because their Precambrian ancestor(s) had found a way to avoid lethal hypercalcemia from Ca2+ flowing in through their gills, guts, and other portals. The solution was proto-calcitonin, which was made and secreted by the first ultimobranchial bodies that later became the thyroid parafollicular cells of the terrestrial animals and the installation of calcitonin—specific offspring of the ancient mother receptor on the surfaces of gill and proto-kidney cells (Whitfield et al., 1998b). A later invention, specifically by ray-fin fishes and our lobe-fin fish ancestors, was stanniocalcin which was designed to prevent Ca2+ from getting through the gills'chloride cells (Whitfield et al., 1998b).
But things changed radically during the Devonian period when the rapidly evolving lobe fins, sporting their new many-toed limbs instead of fins, left the ocean and invaded weedy coastal lagoons and river deltas (Zimmer, 1998). Eventually, they invaded fresh water and crawled out onto dry land. Then the challenge switched from preventing hypercalcemia to preventing hypocalcemia because it is harder to get this essential, yet dangerous, ion if mismanaged, in fresh water and on dry land (Whitfield and Chakravarthry, 2001). They had already prepared themselves for invading dry land and losing the protection of "anti-gravity" underwater buoyancy by arming themselves with dramatically strengthened endoskeletal ribs and vertebrae made of true "mesodermal cellular calcified bone" (Smith and Hall, 1990). These pre-adaptations enabled them to breathe without being crushed by their own weight like modern beached whales as well as to serve as large Ca2+ storehouses (bones have 99% of the body's calcium) from which the ion could be withdrawn when needed. But they also had to redesign their kidneys so that they could dump water without losing Ca. Around this time, calcitonin was fired from its job as the prime controller of Ca2+ levels. Its place was taken by PTH.
The two great engines of evolution have been gene duplication and gene fusion, the first of which gives the opportunity for safely getting novel mutant genes (i.e., new "ideas") while keeping at least one copy of the original gene information and letting the duplicates mutate freely and safely. The second of which produces novel proteins with new functions. Fusions would put a string of once individually controlled genes or gene duplicates under the command of one promoter as in the case of PTHrP. But where did PTH come from? To try to answer this, we must travel back maybe 540 million years to Cambrian Pikaia and Myllokunmingia.
The first PTH was probably what we now know as the N-terminal part of PTH and PTHrP. The gene for this peptide duplicated and perhaps fused with others to make the string of different factors—"Ur" (German for original)-PTHrP—a modern version of which, like the mammalian PTHrP, is produced by brain cells and various other organs, but, unlike the current mammalian PTHrP which doesn't circulate except in cancer patients, is a true hormone that normally circulates in the blood of fish from lampreys and sharks to sea bream (Sparus aurata) and Puffer fish (Fugu rubripes) (Danks et al., 1998; Ingleton, 2002; Ingleton et al., 2002; Trivett et al., 2002). The ancestral hormone was probably designed first to drive the development of many different tissues of the early beasts in the Precambrian seas, just as its descendent does in modern beasts. It was meant to drive Ca2+-dependent epithelial differentiation in various organs, later the formation by neural crest cells of tough mineralized epidermal structures such as the ancient "jawless fishes" boney head shields, dermal denticles (skin teeth), and their then-new mesenchymal cartilagenous skeletons. It also used its N-terminal region in kidneys, gills, brains, and nerves to internal control Ca2+ and phosphate levels in the very high, indeed the dangerously high, Ca2+ (e.g., 10 mM) seawater.
"Ur-PTHrP" might have been a much slimmer molecule than today's mammalian molecule (Ingleton, 2002). Indeed judging from the modern fish PTHrPs, it was rather like the mammalian PTH. It had the same N terminus, nuclear transport, and RNA-binding domains as the "modern" mammalian PTHrP (Ingleton, 2002). But it did not have the mammalian "osteostatin" domain with the 107-111 (TRSAW) sequence. On the other hand, the mammalian molecule does not have the modern fish and frog PTHrPs' functionally mysterious 38-54 domains (Ingleton, 2002). The ancient PTHrP's gene was located beside the thymopoietin gene, and the mammals have kept their PTHrP gene there during the subsequent hundreds of millions of years of evolution. This enduring linkage seems to be due to thymus epitlelial cells and parathyroid gland cells being derived from the same group of pharyngeal endodermal cells in the embryonic third branchial pouch (Gordon et al., 2001; Günther et al., 2000; Su et al., 2001).
Then something very big happened in a certain line of fish—something needed to avoid a debilitating, if not lethal, Ca2+ shortage which would kill anything armed only to avoid hypercalcemia that tried to leave the Ca2+-rich ocean for fresh water or dry land. If this had not happened, we would not be here. There was another duplication of the N-terminal part of the Ur-PTHrP gene, which was to give rise to PTH. Then there appeared another gene, gcm2 (one of the two known homologs of the Drosophila's "glial cells missing", gcm, gene (J. Kim et al., 1998)), the product of which, when the gene was switched on, staked out a part of the thymus gland's primordium for cells that made the new PTH. This gcm2-expressing part of the combined thymus-parathyroid structure was the forerunner of the separate, encapsulated parathyroid gland of terrestrial vertebrates (Gordon et al., 2001; Su et al., 2001). Echoes of the parathyroid glands originating from an ancient thymic primordium are the fact that although knocking out gcm2 in mice eliminates the parathyroid glands, the thymic epithelial cells can take over and make PTH and, of course, the fact that the mammalian PTHrP gene is still located beside the thymopoietin gene (Günther et al., 2000).
With time, the emerging pharyngeal PTH became the circulating Ca2+ and phosphate managing hormone. Big PTHrP then lost the job of being the circulating Ca2+ controller, but it kept its many other jobs of being a multipurpose, widely expressed and acting autocrine, paracrine, "intracrine", and nuclear gene-activaing polyprotein—a noncirculating string of cytokines instead of a single circulating hormone—that mediates various mesenchymally driven epithelial differentiation programs, endochondral bone formation, smooth muscle relaxation, placental Ca2+ transport, and neural Ca2+ channel repression (Chatterjee et al., 2002; Macica and Broadus, 2003; Nissenson, 2000; Re, 2002a, 2002b; Whitfield and Chakravarthy, 2001). In fact, the various add-ons (gene fusions and/or mutant duplicates of the original gene) to the big new multiple cytokine PTHrP molecule, marked out by 8 endoproteolytic consensus sites, could be cut out of the cytokine chain by processing protease scissors and then used separately to activate different receptors and stimulate the functions of the cells expressing them. On the other hand, the smaller, new PTH took over as the circulating hormonal controller of the body's all-important Ca2+ and phosphate levels via the bone reserves, kidney tubules, and gut epithelium. But it could also functionally overlap with big PTHrP's ancient N-terminal domain because it could still target and affect cells with the PTHR1 receptor which had been used by the Ur-PTHrP. Thus, when the first Devonian vertebrates invaded dry land 350,000,000 years ago, they already had parathyroid glands, the cells of which have Ca2+ sensors/receptors housed in the flask-shaped caveolae of their surface membranes with which they monitor the blood Ca2+ concentration (Kifor et al., 1998, 2002, 2003). If the signals from these sensors should start fading, the cells try to restore the volume of sensor signaling by releasing enough PTH to raise the circulating Ca2+ concentration by increasing the outflow of phosphate, stemming the outflow of Ca2+ through the kidney, increasing Ca2+ uptake through the gut, and commanding the members of the osteocyte internet and bone lining cells to pull Ca2+ out of the bone stores if the other two fail to do the job (Kifor et al., 2002, 2003).
PTHs Induced Bone Growth and Fracture Mending in Animals
Seventy-five years ago, the Gospel carved in stone or perhaps the Gospel-carved-in-Bone was that PTH's job in bone was to extract Ca2+ and dump it into the blood when needed. But Bauer et al. (1929) got a big surprise when they treated female rats with the only available PTH (a bovine parathyroid gland extract from Eli Lilly) to assess the role of trabecular bone as a readily resorbable, accessible Ca2+ reserve, but instead they saw—bone growth! But they didn't know what to make of it. There was an excellent reason for their reluctance to believe that PTH could be osteogenic. Even the intermittent injections that we now know strongly stimulate bone growth still stimulate osteoclasts with their acid hoses and cathepsin K shovels, but their destructive digging can be overridden by the bone-building arm, provided the injections are sufficiently far apart. However, if the PTH is injected at close intervals or continuously infused into mice or rats, the response is net bone resorption instead of net growth (Schneider et al., 2003; Tam et al., 1982).
With all due respect to Bauer et al., the bone-growing PTHs story was really kick-started by a Parisian group who boldly went where no one had gone before—they suggested that a young boy's ultimately lethal bone overgrowth (osteopetrosis) was due to the PTH secreted by his parathyroid tumor (Péhu et al., 1931)! Hans Selye was fascinated and fathered the field of PTHs as anabolics when he announced a year later (Selye, 1932) that daily injections of the Eli Lilly extract into rat pups caused a massive pile up of bone-making (osteogenic) osteoblasts on the surfaces of the trabeculae in cancellous bone, just as had happened in the unfortunate French boy. The Selye experiment gave birth to a paradox—PTH was supposed to be a potent bone destroyer, not a potent bone builder. As Riordan (1987) says on page 31 of his book "The Hunting of the Quark"—"A paradox is a sure sign that nature is trying to tell us something completely new and different about herself".
At this point, we should remember from Chapter 2i that when something like a PTH is injected, it first enters the bone via the marrow blood vessels, which means that the cells in the cortical shell will see it only after cells in the trabeculae and endosteum have had the first grab at it. Therefore, a recurring theme will be the PTHs' apparent "trabeculophilia"—its ability to stimulate trabecular bone growth more than cortical bone growth.
An incredible thing about the dramatic accumulation of osteoblasts induced by the parathyroid extract in Selye's rat pups was that it happened directly without being primed by osteoclasts as happens in human BMUs—there was a direct stimulation of osteoblast precursors in the little beasts' rapidly growing bones which we now know would have been loaded with PTH receptor-loaded, bone-building osteoblasts! But who would have believed it? In those days, people had to use a bovine parathyroid gland extract. The extract undoubtedly contained intact bovine PTH-(1-84) and some of its various proteolytic fragments, but it almost certainly contained other things (Bringhurst, 2002). And it took almost 40 years for the bone people to get around to proving that it was indeed the PTHs that did the job (Kalu et al., 1970; Walker, 1971)! Since then, the basic features of PTH's osteogenic actions described by Selye have been confirmed by the results of scores of experiments on cynomolgus monkeys, dogs, ferrets, rabbits, and sheep, but most often on OVXed rats, using a synthetic piece of PTH, h(human)PTH-(1-34), that has been wrongly believed for decades to be fully bioactive (comprehensively reviewed by Dempster et al. (1993) and Whifield et al. (1998a, 2002a, 2002b, 2002c)).
Actually, hPTH-(1-34) is not fully bioactive—the rest of the native hormone is not just functionless filler as has been generally believed! It now appears that following the gene duplication that put PTH and PTHrP on their separate evolutionary pathways, PTH acquired an additional target and functions. In the mid-1980s, T.M. Murray and his group in Toronto, Canada published almost completely ignored (by me included) evidence for there being another receptor for a region of the native hormone's C-tail (McKee et al., 1985; Rao et al., 1985). Then, in 1998, Erdmann et al. (1998) reported that the big native PTH's residues 73-76 form the core of a region that activates a receptor, the signals from which cause Ca2+ to surge into growth plate chondrocytes. Divieti et al. (2001, 2002a, 2002b) have also found more evidence of this receptor, which they have named the CPTHR (C-terminal PTH receptor), that sees C-terminal PTH fragments. The binding affinity is maximum (20-30 nM) for hPTH-(24-84), but drops 25-fold when residues 25-27 are missing, i.e., the affinity for hPTH-(28-84) is 500-700 nM (Bringhurst, 2002). This receptor is apparently maximally expressed by the spidery osteocytes, i.e., the locked-in-bone cells with extended processes that make mRNAs for gap junctions' connexin 43 and the mineralization-modulating osteocalcin but not the osteoblast-specific alkaline phosphatase or Cbfa1/Runx-2 (Bringhurst, 2002; Divieti et al., 2001). And the CPTHR signals promote the formation of gap junctions, which are such an important part of the intercellular connections in the osteocyte-osteointernet signaling syncytium that registers strain and microfracture (Burger, 2001; D'Ippolito et al., 2002). If the big PTH reaches the osteocytes in their lacunocanalicular hideaways, the signals from this receptor can cause these osteocytes to turn on their self-destruct apoptogenic mechanism (Bringhurst, 2002; Divieti et al., 2001). But this CPTHR-driven osteocyte killing is counterbalanced by the reduction of osteoclast generation by the activation of the CPTHRs on osteoclast progenitors (Divieti et al., 2002a, 2002b). However, the differentiating osteoclasts can be protected from lethal CPTHR signaling by having their CPTHR expression suppressed when the differentiating osteoclasts bind to immature osteoblastic stromal cells' PTH-stimulated RANKLs (Divieti et al., 2002b). Thus, the intact PTH-(1-84), unlike hPTH-(1-34), has many targets—preosteoblasts, osteoblasts, osteocytes, and of course, the many other cells in the body that have the N-terminal PTH-binding PTHR1 receptors as well as osteocytes and osteoclast precursors that have CPTHRs.
The intact PTH that has not clamped onto its target cells is promptly broken down into C-terminal but not N-terminal fragments in the liver by Kupffer cells—the half-life of circulating exogenous or endogenous intact hPTH-(1-84) is just 2 minutes (Bringhurst, 2002; Bringhurst et al., 1988; Kronenberg et al., 2001). Indeed, N-terminal fragments seem to be selectively destroyed (Bringhurst, 2002; Bringhurst et al., 1988; Kronenberg et al., 2001). One of the large circulating C-terminal fragments is hPTH-(7-84) (Kronenberg et al., 2001), which can promote osteocyte apoptosis and impair osteoclastogenesis (Bringhurst, 2002; Divieti et al., 2002). However, as we shall see, a bolus of normally rare N-terminal PTH fragments by subcutaneously injecting the hPTH-(1-34) fragment can obviously override any destructive, apoptogenic action of CPTHR signaling from the circulating large C-terminal breakdown products of the big native PTH and strongly stimulate bone formation.
These observations suggest that the selective destruction of N-terminal fragments (e.g., amino acids 1-34) of the native PTH-(1-84) could be meant to prevent circulating PTH from interfering with differentiation mechanisms driven in the cells of various tissues, including bones, by signals from the cells' PTHR1 receptors activated by the hPTH-(1-34)-like N-terminal domain of locally produced and secreted PTHrP.
The native PTH is a string of 84 amino acids (reviewed by Morley et al., 1999 and Whitfield et al., 1998a). Injecting one small dose (e.g., 1-50 nmoles/100 g of body weight) of the native hPTH-(1-84) or certain of its N-terminal fragments ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-28)NH2 (mini-C), hPTH-(1-30)NH2, hPTH-(1-31)NH2 (Ostabolin™), [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™), b(bovine)PTH-(1-34), hPTH-(1-34)NH2, recombinant hPTH-(1-34) (Forteo™), [Leu27]cyclo(Glu22-Lys26)hPTH-(1-34)NH2, recombinant [Asp35]hPTH-(1-35), hPTH-(1-36), or hPTH-(1-38) (Preos™) into OVXed mice and rats or orchiectomized (castrated) male rats each day stops the bone loss, stimulates cortical bone growth, acts synergistically with external loading to increase cortical bone strength, and dramatically raises the bone mineral density (BMD) and mean trabecular thickness in trabeculae-rich bones such as the distal femur, jaw, tibia, and vertebrae or subcutaneous ectopic "ossicles" as much as 2 or more times above the level in normal or still intact sham-OVXed or orchiectomized (i.e., operated on but the ovaries or testicles are not removed) animals (Table 1; Akhter et al., 2001; Andreassen et al., 1999; Arita et al., 2002; Barry et al., 2002; Brommage et al., 1999; Cosman and Lindsay, 1998; Dempster, 1997, 2000; Dempster et al., 1993; Dobnig and Turner, 1995; Gabet et al., 2003; Gasser, 1997; Hagino et al., 2001; Hodsman et al., 1999a, 1999b; Jerome et al., 1994, 1999, 2001; Jilka et al., 1999; Kawane et al., 2002; Kneissel et al., 2001; Kostenuik et al., 1999, 2000, 2001; Lane et al., 1995; Leaffer et al., 1995; Liang et al., 1999; Ma et al., 2000; Mohan et al., 2000; Manolagas, 2000; Morley et al., 1999, 2001a, 2001b; Mosekilde, 1997; Mosekilde et al., 1997; M. Nakajima et al., 2000; Nishida et al., 1994; Opas et al., 2000; Rixon et al., 1994; Samnegard et al., 2001; Schneider et al., 2003; Shirota et al., 2003; Sogaard et al., 1997; Strein, 1994; Sung et al., 2000; Tam et al., 1982; Tormanoff et al., 1997; von Stechow et al., 2003; Walker, 1971; Whitfield et al., 1995, 1996b, 1997a, 1997b, 1997c, 1997d, 1998a, 1998b, 1998c, 1999a, 1999b, 2000a, 2000b, 2000c, 2000d, 2001, 2002a, 2002b, 2002c; Wronski and Li, 1997; Wronski et al., 2001).
Table 1
The anabolic/osteogenic PTHs.
Starting daily subcutaneous injections of 30 or 75 μg of hPTH-(1-34)/kg of body weight into a young female rat when she is only 5-8 weeks old and continuing for the rest of her life will produce big, grossly abnormal bones (Sato et al., 2002). The PTH injections will keep stimulating bone growth throughout the rat's life. The injections will push the growth of bones such as the femur far above normal values—indeed, so far that the marrow spaces will be nearly filled with bone, the difference between cortical and trabecular bone will disappear in a kind of fusion, and the vertebrae and femora will be greatly stiffened. Moreover, although PTH normally stimulates only the growth of lining cell-covered, marrow-bathed endosteal and trabecular bone (Andreassen and Oxlund, 2000), the lifelong massive stimulation will eventually include periosteal apposition. But, as we shall see further on, this relentless, lifelong battering by daily PTH injections into Fisher rats and the excessive amounts of PTH constantly being made in humans with primary hyperparathyroidism can have a deadly consequence—osteostarcomas.
According to the results of experiments on rats, humans, and cultured human and rat bone cells, the PTHs owe their ability to stimulate bone growth by dramatically stimulating the proliferation of marrow osteoprogenitor cells, followed by the layering of osteoblasts on quiescent bone surfaces without prior signaling from osteoclast activity exactly as Selye reported 72 long years ago (Hodsman and Steer, 1993; Kostenuik et al., 1999). Indeed a contribution of osteoprogenitor proliferation to this layering is indicated by PTH-induced stimulation of DNA synthesis in the the tibias of female rats, a response that is nearly doubled by mechanically loading the tibias (Barry et al., 2002). But there is also a direct and reversible stimulation of the ‘reversion’ of retired osteoblasts—the quiescent lining cells covering the osteonal walls, the endosteum and trabeculae—to active osteoblasts, an increase in osteoblast activity, and an increase in the osteoblast lifespan by preventing apoptotic self-destruction (Dobnig and Turner, 1995; Hock, 1999; Manolagas, 2000; Jilka et al., 1999; Leaffer et al., 1995).
The PTHs are most potently anabolic for the bones of the fast-growing fetal and newborn animals (Walker, 1971) simply because the marrow is still red and loaded with stem cells and osteopogenitor cells, and the growing bones are covered with gangs of mature osteoblasts bristling with PTHR1 receptors (Fermore and Skerry, 1995). Because the growing bones are in this super-responsive state, PTHs can so greatly stimulate trabecular bone growth that it fills the marrow spaces and strangles blood formation in these young animals (and, as we shall soon see, in at least one historically-very-significant young French boy) (Walker, 1971).
Of course, this is why Selye (1932) found that the PTH in the Lilly bovine parathyroid extract so greatly stimulated osteoblast accumulation and bone growth in his rat pups without first stimulating osteoclast activity. As fatty yellow marrow replaces red marrow (Bianco and Riminucci, 1998) and bone growth slows with age, so does the responsiveness of bone to PTH in rats (Walker, 1971). In big animals such as humans with microcracking-prone bones at their loading sites (R.B. Martin, 2002), the availability of PTHR1 receptor-bearing preosteoblasts and mature osteoblasts for an immediate or first response to PTH injection in the mature skeleton is a function of the number of BMUs (Manolagas, 2000), which is in turn linked to osteoclast activation.
None of the PTHs can restore the original bone structure. In rats, they cannot generate new trabeculae de novo, increase the trabecular number, or make broken trabeculae rejoin if they have drifted too far apart (Dempster, 2000; Jerome et al., 2001; Lane et al., 1995; Whitfield et al., 1998b). Nor, it seems, can hPTH-(1-34) stop or reverse the drop in the number of central metaphyseal trabeculae in the distal femur when its injections are started 2 weeks after OVX, because it stimulates osteoclast generation as well as osteoblast bone-building (Whitfield et al., 1998b). In other words, while the OVX- and PTH-(1-34)-stimulated osteoclasts are busily destroying central trabeculae in the femur, PTH-stimulated osteoblasts are thickening the more lateral, load-transmitting trabeculae (reviewed by Whitfield et al., 1998b) (fig. 1). The awesome, marrow-obliterating trabecular growth that can be stimulated by a potent osteogen such as hPTH-(1-31)NH2 (Ostabolin™) is shown in fig. 16. The net result of this is an increased trabecular thickness (but not number) and increased total cancellous volume and a disproportionate strengthening of the OVX rat's load-bearing bones (Dempster et al., 1997; Whitfield et al., 1998b, 1998c) (fig. 1). However, [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™) does not allow the same loss of trabeculae as does hPTH-(1-34)(Forteo™) (Whitfield et al., 1998c). This adds to the other observations that intermittent injections of hPTH-(1-31)NH2 either do not cause, or cause far less, resorption than hPTH-(1-34) in the bones of humans and mice (Fraher et al., 1999; Mohan et al., 2000). This has been strikingly confirmed by results of preclinical trials on cynomolgous (Macaca fascicularis) monkey that intermittent injections of even very high doses of [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™) did not cause hypercalcemia and in fact seemed actually to reduce osteoclast activity while at the same time strongly stimulating bone growth (Jolette et al., 2003)! The failure of [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™)-injected, OVXed rats to lose as many trabeculae as the untreated OVXed or hPTH-(1-34) (Forteo™)-treated rats and the apparent ability of the peptide to reduce osteoclast activity in monkeys could be explained by [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™) preferentially stimulating the maximally receptor-loaded mature osteoblasts, which according to Atkins et al. (2003) express OPG-instead of RANKL, as do immature osteoblastic cells.
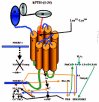
Daily injections of hPTH-(1-34) also stimulate the layering of new bone, primarily on the inner cortical tunnels and trabecular trenches, and thus raise the bone mass and bone strength in ovariectomized nonhuman primates, such as Macaca fascicularis cynomolgous monkeys, above the normal level in sham-operated monkeys (Turner et al., 2001; Sato et al., 2000). But this PTH does something in OVXed monkeys that it can't do in rats—it increases the number of trabeculae (Jerome et al., 2001). But it does not do this by making new trabeculae from scratch. Instead, it does this by increasing the thickness of existing trabeculae, which are then sliced in two by osteoclastic ‘tunneling’ when they reach a certain size (Jerome et al., 1994, 2001). Trabecular tunneling is thus a primate's ingenious way of restoring the original trabecular thickness and number without making new trabeculae from scratch.
Without estrogens, the new bone in a PTH-treated OVXed rat should be, and apparently is in some cases, as much a victim of overdigging BMUs as was the old bone, when the PTH injections are stopped (reviewed by Whitfield et al., 1998b, 2000c, 2000d). But according to the results of most of the relevant experiments on rats, the new "PTH bone", be it endocortical or trabecular, can be effectively protected by an osteoclast suppressor such as a bisphosphonate, calcitonin, or an estrogen (Samnegard et al., 2001; reviewed by Whitfield et al., 1998b, 2000c, 2000d). However, the new bone's need for protection would depend on its rate of turnover (i.e., the size and activity of the osteoclast population). Thus, for example, Mosekilde et al. (1997) found that the new "PTH bone" in the sluggishly turning over bones of aged osteopenic OVXed rats did not need protection by the bisphosphonate, risedronate. And the positive effect of a 12-month treatment with PTH-(1-34) on the cancellous bone of femoral neck, iliac crest, and vertebrae of OVXed cynomolgus monkeys was maintained for 3-6 months after the injections were stopped (Jerome et al., 2001). But new ‘PTH bone’ can face another threat. If it should have grown beyond mechanical needs, it will be under-strained and consequently contain a growing number of dying osteoclasts and then be destroyed by a flock of vulturing osteoclasts (Dempster et al., 1997).
Cotreatment with an antiresorptive to block the resorption component theoretically should enhance the effectiveness of the PTH. But the maximum increase in trabecular and cortical bone mass in rats treated with an optimal dose of hPTH-(1-34) or hPTH-(1-38) alone is the same as in rats cotreated with PTH and a bisphosphonate, calcitonin, or an estrogen (Hodsman et al., 1999a, 1999b). However, cotreatment with the SERM raloxifiene reduces the dose of the PTH fragment needed to get this maximum effect. But Boyce et al. (1996) found that cotreating beagles with hPTH-(1-34) and a bisphosphonate, risedronate, was osteogenically better than treating with hPTH-(1-34) alone. On the other hand, Delmas et al. (1995) found that a bisphosphonate, tiludronate, eliminated the otherwise strong bone-building action of hPTH-(1-34) in old sheep with their slowly remodeling, hence sparsely BMUed, bones. Evidently, the only source of PTH receptor-bearing preosteoblasts and mature osteoblasts in these old beasts were BMUs. So getting rid of BMUs with the bisphosphonate eliminated the waves of osteoclasts needed to kindle an osteogenic response. In other words, there was no prior prompting from osteoclasts, few osteoblasts, hence few PTH targets to start bone growth.
OPG (osteoprotegerin), the physiological inhibitor of osteoclast generation, should potently enhance PTH osteogenicity by selectively stopping the PTH from stimulating osteoclast generation without affecting the stimulation of osteoblast accumulation and activity (Capparelli et al., 2003; Hofbauer et al., 2000; Kostenuik and Shaloub, 2001; Kostenuik et al., 2000a, 2000b). Nor would it have any of the side effects of the other less selective artificial antiresorptives. This expectation has been confirmed by Kostenuik and his colleagues (Kostenuik and Shaloub, 2001; Kostenuik et al., 2001). They OVXed 3-month-old rats and began treating them with PTH and/or OPG 15 months later. OVX significantly reduced the bone mineral density (BMD) in the distal femurs and lumbar vertebrae. Subcutaneously injecting hPTH-(1-34) (0.08 mg/kg) three times a week for 5-1/2 months into the aging rats increased the osteoclast-gnawed surface by 50%, but it also significantly increased the BMD in the distal femurs and lumbar vertebrae. Subcutaneously injecting recombinant OPG (10 mg/kg of body weight) stopped OVX from increasing osteoclast activity. The femoral and vertebral mineral density then rose as the unaffected osteoblasts continued making bone. But the OPG was less effective than the PTH injections in increasing the BMD. Injecting the recombinant OPG along with the PTH prevented the PTH from increasing osteoclast activity without affecting its osteogenic activity and consequently caused a greater increase in BMD than did the injections of either peptide alone. These observations again underscore Selye's original observations that PTH does not need a prior activation of osteoclasts to stimulate bone growth (i.e., it stimulated bone growth despite an essentially total osteoclast shut down) and that OPG strongly enhances a PTH's bone-building action. PTH only needs enough receptor-bearing preosteoblasts and osteoblasts to start the process.
It is important for managing postmenopausal bone loss to know that OPG can prolongedly inhibit bone resorption in postmenopausal women. One subcutaneous injection of an OPG dose, such as 3.0 mg/kg of body weight, reduced the urinary levels of collagen (i.e., bone) breakdown products such as N-terminal telopeptides by as much as 80% by the 4th day after injection, and the levels were still down by as much as 14% as long as 6 weeks after the injection (Bekker et al., 2001). This prolonged suppression of osteoclast activity reduced the blood Ca2+ concentration. This drop turned off the parathyroid glands' Ca2+ sensors, which caused a prolonged elevation of the circulating PTH concentration (from a mean normal 40 pg/ml to about 80 pg/ml at 5 days and still about 55 pg/ml by 30 days) to try to bring the blood Ca2+ concentration back to the normal level. Of course, the hormone couldn't do this by stimulating the osteoclasts to chew up more bone, so it had to rely on reducing phosphate uptake by proximal kidney tubules and increasing the Ca2+ uptake by distal kidney tubules, which resulted in an 80% drop in the urinary Ca2+ concentration by 5 days, a 50% drop by 20 days, and a lingering 10% drop by 30 days.
As expected, the osteogenic PTHs accelerate fracture mending in both normal and osteoporotic bones. Indeed, a fracture enhances the effectiveness of PTH by triggering a shower of BMUs with their PTH receptor-bearing, hence PTH-stimulable, osteoblasts (R.B. Martin et al., 1998). For example, daily subcutaneous injections of 200 μg of hPTH-(1-34)/kg of body weight increased both the ultimate load that could be tolerated before breaking and the callus volume of rat tibial fractures by as much as 75% and 95%, respectively, over the control values by 20 days after fracturing and by 175% and 72%, respectively, over the controls during the next 20 days (Andreassen et al., 1999). A similar acceleration of fracture mending by Ostabolin-C™ ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2) has been reported by Andreassen et al. (2004). Kim and Jahng (1999) have also reported that a daily injection of recombinant hPTH-(1-84) for 30 days significantly accelerated the mending of surgically produced bilateral tibial shaft fractures in OVXed rats. A. Nakajima et al. (2002) have found that daily injections of 10 μg of hPTH-(1-34)/kg of body weight into male Sprague-Dawley rats with unilateral femoral fractures stimulated the probably IGF-I-mediated proliferation of subperiosteal osteoprogenitor cells in the callus, as indicated by the fraction of cells expressing PCNA (proliferating cell nuclear antigen), the DNA polymerase-δ processivity clamp (Whitfield and Chakravarthy, 2001)). By 28 days, the PTH injections had increased BMC (bone mineral content) 61%, BMD 46%, and load to failure 32% more than in control vehicle (the solution in which the PTH was dissolved)-treated rats. By 42 days, these values had risen, respectively, 119%, 74%, and 55% above those in the control untreated rats. Finally, Alkhiary et al. (2003) have reported a similar enhancenment of femoral fracture healing in male Sprague-Dawley rats.
In a different and very elegant way to accelerate fracture mending, Bonadio and colleagues implanted a degradable GAM (gene activated collagen matrix) sponge loaded with a plasmid containing DNA encoding hPTH-(1-34) into surgical breaks in rat femurs as well as beagle femurs and tibias that were so wide that they normally would have healed only very slowly or not at all (Bonadio, 2000; Bonadio et al., 1999; Fang et al., 1996; Goldstein and Bonadio, 1998). But the fibroblasts in the fractures' granulation tissue picked up the plasmid DNA and briefly became mini-bioreactors, making and secreting hPTH-(1-34) that dramatically stimulated fracture healing. Implanting a second GAM with DNA coding for BMP-4 (bone morphogenic protein-4), along with the PTH-GAM, accelerated mending even more.
PTHs can also dramatically accelerate bone formation in and around an implant—an ability of immense orthopedic importance. Indeed, screwing an implant into a bone, like a fracture, should make more PTHR1 receptor-bearing PTH targets and therefore increase the primary response to a PTH in humans by driving squadrons of osteoblast-spawning BMUs out in all directions from the screwholes (R.B. Martin et al., 1998). Skripitz et al. (2000a, 2000b) showed the implant-fixing power by inserting perforated hollow titanium chambers into the cortices of the proximal tibias of male Sprague-Dawley rats and followed the penetration of endosteal cells into the chambers and the growth of bone in the chambers. In untreated rats by 6 weeks, the bone in the chamber had been hollowed out by osteoclasts to form a marrow space with few trabeculae, but in rats which had been injected daily and subcutaneously with hPTH-(1-34), the chamber was filled with thick trabecular struts and plates. Then Skriptz and Aspenberg (2001a, 2001b) showed that subcutaneously injecting hPTH-(1-34) only on Mondays, Wednesdays, and Fridays stimulated the fixation of stainless steel screw implants to rat tibias. In just 2 weeks, the PTH injections so strongly stimulated the growth of dense fibrous bone-anchoring tissue around the screws that it took twice as much force to pull them out of the tibias than to pull the implants out of the control rats' tibias. Shirota et al. (2003) found that hPTH-(1-34) reversed the thinning of bone around titanium screw implants on ovariectomized rats. All of these may herald a new era for people needing new joints because a long lifetime of an implant needs, at the outset, a lot of new bone to build up around and cling to the implant surface without spaces that could fill up with wear debris (Davies, 2000,). This could herald a new era for people needing new joints. More recently, M. Allen et al. (2004) have reported that [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™) is significantly better than hPTH-(1-34) (Forteo™) at stimulating the growth of anchoring bone around a methacrylate pin in rat tibias.
So far, we have been considering the effects of injected PTHs. What would happen if we blinded the parathyroid cells to circulating Ca2+ by disabling their CaRs? The cells would interpret the CaR silence as being due to a large drop in the blood Ca2+ concentration that urgently warrants PTH release. NPS 2143 is a CaR silencer (a "calcilytic"), which when given orally in a dose of 100 μmol/kg causes the PTH concentration in the blood of OVXed Sprague-Dawley rats to shoot up from 25 pg/ml to about 110 pg/ml in 30 minutes and then stay there for at least 4 hours (Gowan et al., 2000; Nemeth and Fox, 2003). This prolonged, indeed continuous, release of endogenous PTH dramatically increases both bone turnover and bone formation in the proximal tibia, but without a net bone loss or gain or without significantly affecting the OVX-induced trabecular crash. However, bone mineral density as well as trabecular thickness and area, but of course not trabecular number, can be increased by giving 17β-estradiol along with the calcilytic to suppress osteoclasts and resorption. But the NPS 2143/estrogen combination is still nowhere nearly as osteogenic as the subcutaneous injections of 10 nmoles/kg of hPTH-(1-34)NH2 or [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™) into OVXed rats (Whitfield et al., 1998b). However, maybe the osteogenicity of the calcilytics can be improved by shortening their circulating half-life and therefore the duration of the PTH surge, which would shift the balance from resorption to bone-building. Remember the Principal Golden Rule for PTHs—they are most effective when given in well-separated brief boluses.
While it is clear that exogenous PTHs have strongly stimulated bone growth in every one of many animal models, the question remains as to whether normal circulating PTH affects bone growth and maintenance. According to Chow et al. (1998), the answer is a resounding—Yes! First, they found that removing the thyroid-parathyroid complexes from young female Wistar rats dramatically reduced bone growth in the caudal vertebrae. And they also showed that circulating PTH is required for the mechanical responsiveness. Mechanical stimulation of the caudal vertebrae in normal rats induced osteogenesis. But the vertebrae in the thyro-parathyroidectomized rats could not respond to the mechanical stimulation.
PTHs Induced Bone Growth in Humans
It was Bauer et al.'s 1929 passing mention of the Lilly bovine parathyroid extract's unexpected stimulation of bone formation in rats, but much more the case of a 8-year-old osteopetrotic boy who was killed by the aplastic anemia caused by wildly growing bone clogging his marrow spaces and stifling his blood cell production (Péhu et al., 1931), that prompted Selye's dramatic demonstration of the awesome osteoblast-generating, osteogeneic action of the Eli Lilly bovine parathyroid extract in rat pups that was to be repeated hundreds of times in the next 72 years (Selye, 1932). The thing that riveted Selye's attention to the French paper was the authors' iconoclastic suggestion that the cause of the unfortunate boy's lethal bone growth was the abnormal amount of PTH pouring out of the boy's parathyroid adenoma—a PTH-producing tumor. But as Enlow and Brown warned 46 years ago: "A great many experimental studies, conclusions, and generalizations concerning bone tissue are based on observations of laboratory animals which do not possess typical human-like bone tissue" (Enlow and Brown, 1958). However, with the advantage of 2005 hindsight, PTH's osteogenicity was already staring the Bone Community in its collective face—postmenopausal women with mild (and mild is the key word) primary hyperparathyroidism do not lose trabecular bone mass. Indeed, these women have more trabecular bone packets with greater wall width, higher bone apposition rates, and active formation periods than their healthy postmenopausal peers (Dempster et al., 1999).
The main reasons for the long delay in extending the animal results to humans were the faith in the then current paradigm and the lack of a reasonably cheap, pure PTH. Forty-four years later, the reasonably cheap synthetic peptide, hPTH-(1-34), which would some day be named Forteo™ and be the first anabolic to reach the clinic, appeared on the market and became available for finding out whether PTH is as osteogenic in humans as it is in animals (Andreatta et al., 1973; Potts et al., 1971; Tregear et al., 1974) (Table 2).
Table 2
Clinical studies and trials with PTH.
To start the story of PTHs and humans, let's look at the rise and fall of the hPTH-(1-34) fragment in the circulation after it is injected subcutaneously. For example, Lindsay et al. (1993) have reported that injecting 25 μg of this peptide once a day into women with osteoporosis (20 μg is the recommened daily dose today for treating osteoporosis) caused a peak of circulating fragment at 30 minutes later at a level which averaged 10-times the basal level followed by an exponential drop in the level with a mean half-time of 75 minutes. Of course, this brief surge of the fragment caused the level of circulating endogenous, native hPTH-(1-84) to drop immediately by 35% as the parathyroid gland cells responded to the sudden loading of the blood with normally very rare N-terminal fragments. All of the many studies, starting in 1976, using this fragment have shown that such a daily bolus of a PTH can potently stimulate bone formation in humans as it does in dogs, mice, monkeys, and rats.
But let's go back and start at the beginning. In 1976, Reeve et al. (1976a, 1976b) reported that single daily injections of 100 μg/kg of hPTH-(1-34) stimulated iliac trabecular bone growth, but they did not affect femoral BMD. In a subsequent uncontrolled, multicenter study, they injected 16 osteoporotic women and 5 osteoporotic men once each day with 50 to 100 μg/kg of hPTH-(1-34) for 6 months to 2 years (Reeve et al., 1980). The treatment produced a significant amount of new, normally mineralized iliac lamellar trabecular bone.
Since 1980, the results of twenty or so studies have been published (reviewed by Cosman and Lindsay, 1999; Dempster et al., 1993; Hodsman et al., 1997; Meunier, 1999; Netelenbos, 1998; Whitfield et al., 1998b, 2000b, 2000d and summarized in Table 2), but I will mention only the more recent examples. By far the most often used measure of a PTH's osteogenic effectiveness in these human studies has been the increase it causes in the overall BMD as determined by dual-energy X-radiation absorptiometry (DEXA) or more recently quantitative computed tomography (QCT) with which the cortical and cancellous (trabecular) compartments can be separately measured (Genant et al., 1982). Remember in reading what follows that a low BMD means a higher fracture risk (Faulkner, 2000).
However, before continuing, I must also note Heaney's warning that BMD (areal density) is the "misbegotten surrogate of bone mass" (Heaney, 2003). Actually as discussed above, the level of remodeling activity is a better indicator of bone fragility than bone mass or BMD.
In Lindsay et al.'s 3-year study, single daily subcutaneous injections of 25 μg of hPTH-(1-34) increased the vertebral BMD by 12.8% in 27 postmenopausal osteoporotic women who also received estrogen, which by itself did not affect the BMD in the 25 women of the control group (Lindsay et al., 1997). As expected, the vertebral BMD rise was accompanied by a drop in vertebral fracturing, which was indicated by the PTH-treated women not losing as much radiographically measured vertebral height as the untreated women. Just as in the animal models, the increases were less in bones with smaller first-to-get-PTH cancellous (trabecular) compartments, such as hip (4.4%) and forearm (1.0%). The whole body BMD increased by 8%.
In a follow-up to this study, Cosman et al. (2001a) reported that the BMD did not change during the 3-year treatment and for 1 year afterwards in the control group receiving HRT (hormone (estrogen) replacement therapy). In the PTH-HRT group, the biochemical markers of both bone formation (bone-specific alkaline phosphatase) and resorption (urinary crosslinked collagen N-telopeptides)—peaked at 6 months and returned to baseline levels by 30 months. During this period, the high cancellous spinal bone mass increased 13.4%, the lower cancellous hip bone mass increased 4.4%, and total body bone mass increased 3.7%. The bone masses did not change significantly during the first year after stopping the PTH injections but continuing HRT. The PTH-HRT treatment dramatically and significantly reduced the number of "incident" (in other words "spontaneous" or muscle-pull or bending-crush) vertebral fractures during the follow-up year. The frequency of incident fractures in the PTH-HRT group was reduced to as little as 0% or 25% of the frequency in the HRT-only group, depending on the choice of height reduction (their measure of vertebral crushing) ‘cut-off point’.
A larger increase in vertebral BMD was seen by Hesch et al. (1989) in a smaller group of 13 osteoporotic women. After 14 months of single daily subcutaneous injections of 54 μg (about 700-750 Units of activity) of hPTH-(1-38), the mean vertebral BMD had increased by 20%. This large response might have been due to the post-PTH prevention of a resumption of resorption by the nasally sprayed anti-osteoclast calcitonin that was also given to the women.
In a randomized, controlled 2-year trial with 30 osteoporotic women receiving 6 treatment cycles, each one of which consisted of 1 month (28 days) of single daily subcutaneous injections of 800 U (or about 60 μg)/day of hPTH-(1-34) followed by a 3-month rest period (to restore full responsiveness to the PTH for the next round of injections), there was a 10.1% increase in the BMD of lumbar vertebrae, a nonsignificant 2.4% increase in the femoral neck BMD, and a 80% drop in the vertebral fracture incidence (Hodsman et al., 1997). However, in this study treating the osteoporotic women with the antiresorptive calcitonin during the rest periods did not enhance PTH's osteogenicity.
Rittmaster et al. (2000) have reported the results of a study in which 75 postmenopausal women were treated for 1 year with daily injections of 50, 70, or 100 μg of recombinant hPTH-(1-84) and then with a daily dose of 10 mg of alendronate for the next year. This treatment stopped cortical bone loss and increased the spinal bone mass by 14% as compared to 6.9 to 9.2% increase in patients without subsequent alendronate treatment.
In a much larger study, Fujita et al. (1999) treated 220 osteoporotic patients with weekly subcutaneous doses of 50 U (3.7 μg; 0.8 nmole), 100 U (7.4 μg; 1.6 nmole), or 200 U (14.8 μg; 3.2 nmole) of hPTH-(1-34) for 48 weeks. The highest dose caused a significant 8.1% increase in the lumbar vertebral BMD, without affecting cortical thickness or metacarpal BMD. The increase in vertebral BMD was accompanied by a 30-40% reduction in "backache".
In another study (Roe et al., 1999), osteoporotic women injected themselves with a placebo or 400 U (about 30 μg) of hPTH-(1-34) subcutaneously once each day for 2 years and also took an oral estrogen (0.625 mg Premarin/day) with or without methoxyprogesterone. They all received 800 U of vitamin D and 1500 mg of calcium/day. By the end of 2 years, the overall (cortical plus trabecular) lumbar vertebral BMD in the PTH/estrogen-treated group was a dramatic 28.3% higher than in the placebo/estrogen-treated control group. The femoral neck BMD in the PTH/estrogen-treated group was 10.8% higher than the femoral neck BMD in the placebo/estrogen control group. The density of the trabecular (cancellous) compartments of the L1 and L2 vertebrae in the PTH/estrogen-treated women (as measured selectively by QCT) had increased even more dramatically—it had increased by 74% during the 2 years while the mean density in the vertebrae of the placebo/estrogen-treated controls had dropped 2.1%.
At the end of 1998, Lindsay et al. (1998) summarized the results of a one-year, Phase II, multicenter (18 centers in Canada and the USA), placebo-controlled, double-blind study of the effects of recombinant rhPTH-(1-84) (Preos™; NPS Pharmaceuticals Inc.'s ALX-1-11 from the now-defunct Allelix Biopharmaceuticals) on BMD and serum indicators of bone resorption and formation in 217 postmenopausal osteoporotic women between 50 and 75 (average, 64.5) years of age. The results of the prior Phase I study had indicated that one subcutaneous injection of 0.02 to 5.0 μg of ALX-1-11/kg into healthy postmenopausal women did not produce a frank hypercalcemia, even at the highest dose (though you will learn further on that this is not the case), which indicated that the hormone was safe and well-tolerated in this dose range (Schweitert et al., 1997). The failure to cause hypercalcemia could be due to the killing of osteoclasts by the stimulation of their CPTHR receptors for a C-terminal domain of the large native PTH (Divieti et al., 2002). The Phase II patients were given single daily injections of 50, 75, or 100 μg of hormone (≈ 0.8 to 1.6 μg/kg). The largest dose increased the vertebral BMD by 6.9%, while the densities of the vertebrae in the placebo-treated women did not change. However, the BMD dropped in the arms and legs by 0.3% (75 μg) and 0.9% (100 μg), respectively. The serum levels of both formation indicators (bone-specific alkaline phosphatase, osteocalcin) and resorption indicators (deoxypyridinolines, N-terminal cross-linked collagen peptides) increased 100 to 200%. There were no serious side effects, nor did the patients make antibodies to the recombinant human hormone. The authors believed that the drops in arm and leg BMDs were only transient, and that PTH appeared to be a promising novel treatment for osteoporosis.
Transient though they might have been, the drops in the BMDs of the arms and legs of Lindsay et al.'s patients are reminiscent of the results of an earlier study by Neer et al. (1991) that nearly kicked the PTHs off the list of credible therapeutics for osteoporosis. As expected, giving hPTH-(1-34) and calcitriol (1α,25(OH)2vitamin D3) to 15 osteoporotic women for 1 to 2 years increased the lumbar vertebral BMD by a whopping 32%! But at the same time there was a net 4% drop in the radial BMD, which was what set off the alarm bells. The great fear was that although PTH can reduce the unpleasant vertebral crushing, it might increase the far more crippling hip fracturing. To explain this unsettling result, they suggested that PTH somehow steals cortical bone to make trabecular bone. Fortunately for the PTHs, this ‘Cortical Steal’ hypothesis as it was called has mercifully died, although there is a reason why there is often a transient drop in cortical BMD. So the common, though not universal (e.g., Lindsay et al.'s (1998) study) experience has been virtually the same as the experience with nonhuman bones—PTHs strongly stimulate trabecular bone growth but either do not affect or less strongly stimulate cortical bone growth. To understand this, remember that the trabecular cells get the first grab at an injected PTH coming into the bone in the nutrient blood vessels.
Neer et al. (2001) and Zanchetta et al. (2003) have reported the effects on bone geometry and fracturing of daily subcutaneous injections of a placebo, or 20 or 40 μg of Eli Lilly's recominant hPTH-(1-34) (Forteo™) into 1637 post-menopausal women who had already suffered vertebral fractures. These numbers were from this venerable, 33-year-old peptide's phase III clinical trial. The relative risk of vertebral fracturing was reduced to 0.35 (95% CI, 0.22-0.55) in women receiving daily injections of 20 μg of the peptide and to 0.31 (95% CI, 0.19-0.50) in women receiving daily injections of 40 μg of the peptide. The relative risk of nonvertebral fracturing was 0.47 in women receiving 20 μg of the PTH (95% CI, 0.25-0.88) and 0.46 in women receiving 40 μg (95% CI, 0.25-0.86). Both doses of the PTH increased the BMD of hip and spine by 2 to 4%, but again there was the usual drop in the BMD of the radial shaft (i.e., cortical bone) during the first year. However, according to Zanchetta et al. (2003), using pQCT instead of DEXA, hPTH-(1-34) actually significantly increased the BMD and area of the distal radius. Thus, hPTH-(1-34), Lilly's Forteo™, improves bone geometry as indicated by increased axial and polar cross-sectional moments of inertia, decreases the risk of wrist, vertebral and nonvertebral fracturing, increases femoral and vertebral BMDs, and is well tolerated, although there was some (29%) hypercalcemia with 40 μg, which, because it causes such unpleasant things as muscle weakness, fatigue, nausea, vomiting, and kidney stones, unfortunately limits the dose that can be used. Indeed, according to Lane et al. (2003), a daily injection of 40 μg of hPTH-(1-34) caused the circulating level of the osteoclastogenic soluble RANKL to peak broadly around 250% of the baseline value between the 3rd and 9th month and the osteoclastogenic IL-6 to peak at about 150% of the baseline value at the 3rd month. Obviously, the new potently anabolic PTHs such as oral Ostabolin™ or Ostabolin-C™ (Jolette et al., 2003) that apparently do not cause hypercalcemia, even at very high doses, will have a considerable clinical advantage over Forteo™ when they complete their clinical trials.
The answer to the crucial question of what the PTH treatment does to cortical bone seems to have been provided by Cann et al. (1999). They used 3D-QCT to separately track the hPTH-(1-34) (Forteo™)-induced changes in the density and mass of the cortical and trabecular (cancellous) parts of the proximal femurs of the osteoporotic women in Roe et al.'s (1999) study. By the end of the second year, the PTH injections had increased the spinal BMD by a very substantial 74% and the BMD of the trabecular compartments of the proximal femur from 10.5% in the trochanter to 12.4% in the whole hip. On the other hand, the Density Devil reappeared here again—the cortical BMD dropped by less than 3.5% during the first year. But, as in Lindsay et al.'s (1998) study, the drop then stopped. So were Neer et al. (1991) right? Yes, but PTH treatment is certainly not hazardous for femoral necks. Despite the small drop in cortical bone density, the use of 3D-QCT enabled Cann et al. to see that the cortical bone mass in the proximal femurs had actually risen 10.9% in the trochanter and by an awesome 20.7% in the femoral neck by the end of the second year. These large increases in cortical mass took place on the endosteum (just as Andreassen and Oxlund (2000) found in the femurs of hPTH-(1-34)-treated OVXed rats) because the endosteum, unlike the periosteum, is both bathed in marrow and covered with PTH receptor-bearing lining cells, which like the equally marrow-bathed trabecular lining cells, can throw PTH-induced growth factors directly at osteoblastic progenitors in the adjacent marrow stroma (Kostenuik et al., 1999; Pun et al., 2001). Thus, as expected in a mature skeleton, PTH rapidly stimulated osteoclast generation by RANKL-producing immature PTHR1 (the common PTH receptor)-poor osteoblastic stromal cells, which would reduce the cortical density by increasing the porosity (i.e., the number of osteoclast-dug tunnels). But this was far more than balanced by a delayed, large production of endocortical bone by increased numbers of osteoblasts resulting from the PTH's three osteoblast-generating actions that will be unveiled in the next chapter.
The same changes have been seen by Burr et al. (2001) in our OVXed relatives, cynomolgus monkeys (Macaca fascicularis), that received one daily subcutaneous injection of 1 or 5 μg of hPTH-(1-34)/kg for 12 or 18 months. The injections increased the cortical porosity, especially in the inner third of the mid-diaphysis (the middle part (see Netter, 1997) for beautiful drawings) of the left humerus, where it was increased 5 to 16 times above the porosity in the sham-operated monkeys. (Ostabolin-C™ causes far less porosity in cynomolgous monkeys (Paul Morley, personal communication).) Surely this must have reduced the bone's mechanical strength. But it did not! The strength actually increased, because although the osteoclasts were digging more holes in the endocortical zone, the PTH-stimulated osteoblasts were busily increasing the mineralizing surface 11-fold more than in the controls and making bone 4 times faster than their peers in control monkeys.
Sato et al. (2004) have also seen the same thing in 9-year-old, OVXed cynolmolgous monkeys that had been subcutaneously injected daily for 18 months with 1 or 5 mg of hPTH-(1-34) (Forteo™). The PTH increased osteon activation, osteonal bone formation, and cortical width and area, but it also increased bone turnover and cortical porosity. However, again the increased bone growth more then compensated for the increased porosity and the bone strength (as indicated by the ultimate load to failure) ranged above the weakened OXV value and the normal sham value.
A similar conclusion was reached by Hirano et al. (2000) from a study of the effects of an osteogenic dose of hPTH-(1-34) on cortical bone porosity and bending rigidity of rabbit tibias. They found that here too the PTH greatly increased the porosity, mostly on the endocortical surface, but this was more than offset by the formation of new endocortical bone as well as periosteal bone, which increased the tibial bending rigidity. The same coincidence of bone loss and large bone building has been seen by Whitfield et al. (1998b) in OVXed rats where hPTH-(1-34) injections did not stop the estrogen-deprived, stimulated osteoclasts from destroying the femoral central metaphyseal trabeculae but they increased thickness of the surviving, laterally sited trabeculae 1.65 times.
At this point, the reader might ask whether preventing the PTH-induced surge of osteoclasts boring holes in the cortex would increase the potency of the anabolic response to a PTH. And the answer is almost certainly yes. "Yes" if one uses the potent, long-acting, and safe anti-osteoclastogenic osteoprotegerin (Kostenuik et al., 2001; Smith et al., 2003) instead of the bisphosphonate alendronate (Fosamax™), which does kill osteoclasts but unfortunately as we shall see below also interferes with the expression of osteoblast-specific genes (Onyia et al., 2002).
Accumulating mature osteoblasts should release an indicator of their presence, such as osteocalcin into the blood. Indeed, this is what happens in PTH-treated osteoporotic humans. The hPTH-(1-34) injections in the study of Lindsay et al. (1997) caused the serum osteocalcin content to rise for the first 6 months, after which it slowly subsided. Cosman et al. (1998, 2000) found that the PTH injections in these experiments caused the serum osteocalcin and pro-collagen I-C-terminal propeptide (released during collagen fiber processing and assembly) to rise during the first 4 to 6 weeks, which indicated a rapid build-up of osteoblasts. There should also have been evidence of PTH-stimulated osteoclasts. But the osteoclast response, as indicated by the release of cross-linked N-telopeptides from osteoclast-cleaved collagen, was not detectable until 6 months and then dropped to baseline values by 24 months.
Reeve et al. (2001) have reported the effects of treating severely osteoporotic women with hPTH-(1-34) or hPTH-(1-38). They found that the PTHs improved the "body calcium balance", "impressively" increased the spinal BMD, and stimulated smaller increases in proximal femur and radius. They concluded "hPTH or comparable PTH receptor activators remain the most promising anabolic treatment for osteoporosis currently under clinical evaluation …".
In mid-2003, Cosman et al. reported the preliminary results of a study on osteoporotic patients that could revolutionize the PTH treatment protocol. Normally, an osteoporotic patient self-injects 20 μg of hPTH-(1-34) (Forteo™) once daily for no more than 2 years. However, Cosman et al. (2003) have tested in osteoporotic women the cyclic protocol first tried by Hodsman et al. (1997). The first reason for cyclic treatment is based on the fact that daily injection of hPTH(1-34) (Forteo™) initially stimulates bone formation more than resorption, but resorption eventually catches up by 6 months. (This might be due to target cells such as the anti-osteoclast, OPG-producing, bone-building osteoblasts and potential osteoblast lining cells having enough receptors to make them more immediately responsive than the pro-osteoclast, RANKL-producing progenitor cells (Atkins et al., 2003)). So if there is a sufficiently long rest period between injections, ostoclast production and resorption would not have a chance to catch up. The second reason is that if cyclic treatment works at least as well as continuing daily injections, it could be only half as expensive/year for the patient. The preliminasry results, like those of Hodsman et al. (1997), are encouraging. When hPTH-(1-34) (Forteo™) was "on" for 3 months, the serum markers of bone formation rose, dropped when it was "off " for the next 3 months, but then rose robustly when turned on again during the next 3 months. By the end of 9 months (one cycle), there was the same approximately 4% increase in the patients' lumbar vertebral BMD that had been stimulated by either the costly, continuing-daily treatment or the cheaper, cyclic treatment.
At the end of 2003, Hodsman et al. reported the effects of 12 months of treatment with NPS Therapeutics's recombinant full-length hPTH-(1-84) (Preos™). Postmenopausal women self-injected 50, 75, or 100 μg of the peptide every morning for 12 months and swallowed one or two CaCO3 (500 mg elemental calcium) tablets and 400 IU of vitamin D along with it. The most effective dose was 100 μg, which by 3 months had increased the lumbar (i.e., L1-L4) spine BMD by a significant 2% and then 7.8% by 12 months. The latter number was actually underestimated because of a 2% areal increase by 12 months. There were also transient dose-dependent incidences of hypocalcemia, which the authors did not consider particularly worrying. At the 25th meeting of the American Society for Bone and Mineral Research in September 2003, Paul Miller (from the University of Colorado in Denver) also summarized (without providing a record of his talk) some of the results coming out of this full-length PTH's Phase III trial. The results he cited were got from 50 post-menopausal women who injected themselves subcutaneously every day with 100 μg of the PTH, which in terms of molecular weight was approximately equivalent to the 20 μg of hPTH-(1-34) (Forteo™) that osteoporotic women are now allowed to self-inject each day. And the anabolic effectiveness of this full-length PTH is about the same as that of hPTH-(1-34) (Forteo™). But with all due respect to Hodsman et al. (2003), there is a very good reason not to use this PTH—as many as 22% of the women suffered from one or more hypercalcemic experiences. So why would a physician prescribe the intact molecule when the patient would get exactly the same anabolic punch/nmole with hPTH-(1-34) (Forteo™) that is already on the pharmacy shelves or with the even smaller, less hypercalcemogenic, osteogenic PTHs such as Ostabolin-C™ that are in the pipeline (Table 1)?
Glucocorticoid therapy, like aging, wandering around in space, or lying immobilized in bed, can also cause osteoporosis. It does this by inhibiting osteoblastogenesis (apparently by suppressing Cbfa1/Runx-2 and Cbfa1/Runx-2-stimulated TGF-β type I receptor expression (Chang et al., 1998; Ducy, 2000)), promoting osteoblast and osteocyte apoptosis, and inhibiting intestinal Ca2+ uptake (Lane et al., 2000; Weinstein et al., 1998). Injecting a PTH can overcome the glucorticoid action and stimulate bone growth. This is shown in a recent experiment involving 51 women (mean age of 63 years) receiving glucocorticoids and estrogens (Lane et al., 2000). Twenty-eight injected themselves with 40 μg (400 U) of hPTH-(1-34) (Forteo™) once each day for 12 months and continued taking estrogen, while 23 just continued taking estrogen. Here again as expected, PTH preferentially stimulated growth of cancellous (trabeculae-rich) bone. By 12 months, the PTH injections had increased the trabecular mass (as measured by QCT) mainly in cancellous lumbar vertebrae by about 35%, which continued rising to 45% during the next 12 months as the PTH-enhanced remodeling space (remember that PTH also increases osteoclast generation and thus excavations) continued to be filled up after the injections were stopped. By 24 months, the overall BMD (as measured by dual energy X-radiation absorptiometry instead of QCT), which is only a function of the extent of mineralization and the amount of remodeling space in the increasingly massive vertebral bone, had increased by only 12.6%. The hip mass rose by 4.7% over the 24-month period. However, the treatment did not affect the cortical bone mass in the forearm.
Rehman et al. (2003) have also found that treating women with glucocorticoid-induced osteoporosis for 1 year with hPTH-(1-34) (Forteo™) plus hormone-replacement therapy (HRT) increased the cross-sectional areas of their L1 and L2 vertebrae by 4.8% over the areas in patients given HRT only. hPTH-(1-34) (Forteo™) also increased the compressive strength of the vertebrae by 200% over the baseline levels, while HRT alone had no effect.
PTH can also treat idiopathic (a fancy word for "cause is unknown") osteoporosis in middle-aged men (Kurland et al., 2000). Unlike postmenopausal osteoporosis in women, which is associated with high bone turnover due to osteoclastic overactivity, male idiopathic osteoporosis is associated with low bone turnover and thus is not as amenable to the drugs designed to suppress osteoporotic postmenopausal women's demonic bone diggers. Kurland et al. (2000) reported the results of an experiment on 23 men (50 ± 1.9 years of age) with idiopathic osteoporosis. They gave 10 of the men a daily subcutaneous injection of 400 IU (about 30 μg of hPTH-(1-34)(Forteo™) for 18 months. While the lumbar spine bone mass did not change in the 13 placebo-treated control patients, the PTH increased it increased by a significant 13.5%.
A very important important question at this point is—how does the human skeletal response to Lilly's hPTH-(1-34) (Forteo™) compare with the response to the Merck-Frosst's bisphosphonate alendronate (Fosamax™)? Body et al. (2002) treated 73 postmenopausal osteoporotic women with 10 mg of oral alendronate plus subcutaneously self-injected placebo (instead of PTH) and oral calcium (1000 mg) and vitamin D (400-1200 IU) each day for 14 months. Another 73 postmenopausal osteoporotic women injected themselves subcutaneously once daily with 40 μg hPTH-(1-34) (Forteo™), took an oral placebo (instead of alendronate), and the same calcium and vitamin D supplements for the same period of time. The greater effectiveness of PTH appeared as early as 1 month, and by 14 months alendronate had increased the BMD of the lumbar spine by only 5.6%, while hPTH-(1-34) had increased it by 12.2% (p < 0.001). The PTH also increased the femoral neck BMD by about 6%, while alendronate increased it by only about 2%. And by 14 weeks, PTH had increased total hip BMD by about 4.5%, while alendronate had increased it by about 2.5%. And the nonvertebral (ankle, foot, radius, ribs, toe) fracture incidence was 4.1% in the PTH-treated group compared with 13% in the alendronate-treated group. Interestingly, the PTH promptly started to increase bone alkaline phosphatase, an indicator of bone growth, by about 50 to 100%, while alendronate equally promptly caused the alkaline phosphatase level to drop by 50%. This would be expected because of alendronate's ability to inhibit the genes for bone formation such as alkaline phosphatase, collagen I, and IGFs-I and II in the proximal femoral metaphyses of OVXed rats (Onyia et al., 2002). Of course, the Density-Loss Devil appeared again—there was the usual small drop (about 4%) in the distal radial BMD in the PTH-treated group, which was expected from other studies in humans, monkeys, and rabbits, and was probably transient and not reflective of the actual increase in bone strength (Burr et al., 2001; Cann et al., 1999; Hirano et al., 2000; Neer et al., 1991, 2001). Clearly, hPTH-(1-34) (Forteo™) is much better than alendronate at increasing BMD at various skeletal sites and decreasing nonvertebral fractures in humans (D.M. Black et al., 2003; Finkelstein et al., 2003).
Alendronate cotreatment actually blunts the anabolic actions on hips and vertebrae of daily injections of 40 μg of hPTH-(1-34) (Forteo™) into osteoporotic men (Finkelstein et al., 2003) and 100 μg of hPTH-(1-84) into osteoporotic women (D.M. Black et al., 2003). The reason for this has been found in rat femurs by Onyia et al. (2002)—alendronate prevents hPTH-(1-34) (Forteo™) from stimulating key bone-growth-driving genes.
Finally, an equally important question is—Does prior exposure to bisphosphonates or SERMs compromise the anabolic action of a subsequent treatment with hPTH-(1-34) (Forteo™)? This is very important because most if not all osteoporotic patients coming to the clinician's office for PTH treatment will have been receiving one of these antiresorptives. Ma et al. (2003) have reported that prior treatment of OVXed Sprague-Dawley rats for 10 months with alendronate (Fosamax™), 17α-ethinyl estradiol, or raloxifene before switching over to hPTH-(1-34) (Forteo™) did not affect the responsiveness of the rats' skeletons to the PTH. But this result would be expected because rats retain a substantial pool of PTH-responsive osteoblastic cells for their continuously growing bones. However, this might not be the case with old humans in whom an antiresorptive-induced lack of BMUs and microcrack repair would blunt the response to a PTH, as happened in Delmas et al.'s (1995) antiresorptive-treated old sheep. And this appears to be the case for the potent alendronate (Fosamax™) in humans.
Ettinger et al. (2004) measured the effects of prior treatment with alendronate (Fosamax™) or raloxifene on the anabolic action of daily injections of hPTH-(1-34) (Forteo™) in postmenopausal, 60-87-year-old women for 18 months after stopping the bisphosphonate treatment. Briefly, raloxifene pretreatment did not affect the ability of the PTH peptide to increase the BMD in hip and spine, but the BMDs in the alendronate-pretreated women increased later and peaked at lower levels than in the raloxifene-pretreated women. The authors suggested that the much longer-lasting suppression of remodeling by alendronate pretreatment than by raloxifene pretreatment would more severly reduce the pool of PTHR1-bearing target osteoblasts for stimulation by the PTH injections in the alendronate-pretreated women as probably happened in Delmas et al.'s antiresorptive-treated old sheep.
How Might PTHs Stimulate Bone Growth?
"…I shall need a herd of elephants, I thought, and a wilderness of spiders, desperately referring to the animals that are reputed longest lived and most multitudinously eyed, to cope with all of this. I should need claws of steel and beak of brass to penetrate the husk. How shall I ever find the grain of truth embedded in this mass of paper?"—Virginia Woolf (1975).
"The whole system is controlled by a vastly complex "signaling" system. Membrane-bound receptors themselves do not float freely in the membrane, but are tied to scaffolding proteins. This locates them precisely with respect to the other proteins ….in the cascades with which they interact…"—John Smythies (2002; p. 2-3) speaking of neuronal synaptic signaling but applicable to bone.
The Starter Gun: The PTHR1 Receptor and the Signals It Fires
"…the receptor protein sticks its molecular equivalent of head and arms out to catch passing…molecules and keeps its foot on the molecular gears inside the cell."—L.H. Caporale (2002; p. 139).
"…it is getting more and more complex and we are frequently overloaded with information. In addition, we have moved from absolute fidelity (one ligand, one receptor, one G protein, one effector, etc.) to almost complete promiscuity (multiple ligands, receptors, G proteins, effectors, and all interacting among them)."—J. Vazquez-Prado et al. (2003; p. 556).
To tackle the formidable job of understanding how the PTHs stimulate bone growth in humans, rodents, and other animals, we must know where and how things start. What signals do they send into their target cells via the PTHR1 receptors to trigger osteogenesis? However, before going on I must warn the reader that most people call the receptor PTH1R. Since there is no type 1 PTH (i.e., PTH1) but there is a type 1 PTH receptor, I use what surely must be the more accurate name for the receptor—PTHR1 (i.e., PTH's receptor No.1).
But aside from the semantics, how do I know that the PTHR1 receptor is the molecular gun which the PTHs use to fire their osteogenic bullets into the bone cells? To show this, Calvi et al. (2001) and Schipani et al. (2002) coupled a mutant human PTHR1 gene (HKrk-H223R) encoding a permanently switched-on receptor to the mouse a(I) collagen gene promoter in order to restrict the gene's expression to the osteoblasts in a "transgenic" mouse, otherwise there would have been widespread chaos in the many other PTHR1-expressing tissues. The receptor encoded by this gene was "frozen" in an active configuration—it didn't need a PTH to activate it. The switched-on PTHR1, like normal PTH-activated PTHR1s, increased endosteal bone formation, caused an accumulation of inter-trabecular preosteoblastic marrow stromal cells and a build-up of hyperactive trabecular osteoblasts in both proximal tibia and cranial bones, and dramatically increased trabecular bone volume. As must happen with PTH infusion or, as in this case, nonstop PTHR1 signaling, there was also a large increase in cortical and trabecular osteoclasts, a lack of periosteal growth, increased cortical porosity, and a resulting drop in cortical bone mass. However, there was no bone loss, and the trabecular volume was even higher in doubly-mutant mice making collagenase-3-resistant a(I) collagen along with the switched-on PTHR1 (Schipani et al., 2002). In these resistant beasts, the diggers couldn't reduce the bone build up by chopping up the collagen matrix.
The PTHR1 receptor (fig. 16 and fig. 17) is one of the 1200 to 1300 members of the G-protein-coupled receptor (GPCR) superfamily (Karnik et al., 2003; Schoneberg et al., 2002; Wong, 2003). According to the latest classification scheme (Karnik et al., 2003), it belongs to the S Family of GPCRs, which has the familial 7-transmembrane α-helices (i.e., like barrel staves inserted through the membrane) and, like all S-family GPCRs, it has a middle-size extracellular "nose" or N-terminus (i.e., about 160 amino acids as compared to less than 100 for R-family receptors and as many as 360 for a G-family receptor such as the Ca2+ receptor (Brown and MacLeod, 2001; Bockaert et al., 2002; Hoare and Usdin, 2001; Hoare et al., 2001; Schoneberg et al., 2002; Whitfield et al., 1998a). PTH-(1-34) dispenses most of its binding energy when it attaches the 15-31 region of its C-terminal tail first to the receptor's N-terminus sticking out of the cell from the first barrel stave and then sticking its N-terminal "nose" onto the Leu232-Lys240 region of the receptor's second barrel stave (TM2) (Chorev, 2002; Gensure et al., 2002; Hoare and Usdin, 2001; Hoare et al., 2001) (fig. 17). The key players in this interaction are Arg233 and Lys240, and replacing one or both of them with alanine prevents the interaction.
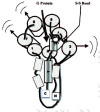
According to Karnik et al. (2003), when a PTH settles down on the receptor's N terminus, it nudges the extracellular loop 2 connecting staves 4 and 5 (fig. 16). This nudge stretches the S-S bond linking staves 3 and 4, which causes the staves to open the way for a GDP•Gs three-component complex, for example, to slip into the bottom of the barrel (fig. 17). According to Wong (2003), the shifting staves cause the three intracellular loops and C-terminal tail to make a pocket that grabs an appropriate G-protein dangling nearby from the membrane (fig. 17). The G-protein is probably hanging beside and somehow linked with the receptor in the inactive GDP•Gα-β? form tethered by the βγ complex to the cell membrane (Petsko and Ringe, 2004). When the nesting PTH jiggles the receptor staves, the GDP•Gα-βγ complex is pulled into the "pocket" of the clustering intracellular loops, where its GDP is pulled off and replaced by a GTP to form active GTP•Gα, which is separated from the βγ pair which may go off on their own to stimulate different signalers. The active GTP•Gα is then changed back to inactive GDP•G, either slowly by its own GTPase activity or much faster by an associated GAP (GTPase activating protein) protein and reassociate with βγ. Of course, the final signal mix is determined by whether the receptor tail is attached to a NHERF and what other signalers are also on the NHERF (fig. 16)
The binding of hPTH-(1-34)'s N-terminal nose specifically generates active GTP•Gsα subunits from the inactive GDP•Gsα-βγ protein complexes. GTP•Gsα then stimulates adenylyl cyclase. Adenylyl cyclase makes cyclic AMP from ATP. At the same time, the binding of the PTH's tail to the receptor's N-terminus generates active GTP•Gq/11α subunit from the GDP• Gq/11complex that stimulates phospholipase-Cβ1 (PLC-β1). PLC-β1 in turn chops a minor, but an incredibly potent, event-driving, membrane phospholipid with the daunting name, phosphatidylinositol (4,5)bisphosphate (or PIP2 for short), into diacylglycerols and inositol (1,4,5)trisphosphate (IP3) (Hoare and Usdin, 2001; Hoare et al., 2001; Morley et al., 1999; reviewed by Bilezekian et al., 2001; Whitfield et al., 1998a) (fig. 16).
The PTHR1 signaling must not be allowed to continue beyond a certain time if it is to be optimally effective. As we shall see further on, one way of doing this is to simply drag the PTHR1•PTH complex into the cell on a β-arrestin platform, pull the PTH off and destroy it, and put the cleaned up receptor back to work on the surface. However, the job of being the cell's general GPCR receptor receptor off-switch has been given to RGS-2, a small member of the B/R4 clan of the 26-member family of RGS (regulators of G-protein signaling) proteins (Hollinger and Hepler, 2002; Petsko and Ringe, 2004). PTHs rapidly stimulate RGS-2's gene via adenylyl cyclase/cyclic AMP (but we don't know what happens when PLC but not adenylyl cyclase is stimulated), and the resulting RGS-2 protein shuts the signaling off by stimulating the conversion of active GTP•Gαs and GTP•Gαq to inactive GDP•Gαs and GDP•Gaq as well as by directly inhibitng AC (Hollinger and Hepler, 2002; Miles et al., 2000; Thirunavukkarnasu et al., 2002; Tsingotjidou et al., 2002). However, RGS-2, like RGS-4, another small member of the B/R4 clan, might have a more positive role, such as triggering Ca2+ oscillations (Hollinger and Hepler, 2002). But it appears that RGS-2 might either require help from β-arrestin to shut the receptor signaling off or it is not really needed for this, because knocking out the β-arrestin-2 gene in mouse osteoblasts prolongs PTH-triggered cyclic AMP signaling, which increases cortical (but not trabecular) bone growth (Pierroz et al., 2003).
So far, the effort to find out how a PTH such as hPTH-(1-34) causes the PTHR1 barrel loops and staves to tumble into an active G-protein-grabbing state has focused only on the stimulation of AC as the activation indicator. Peptides such as hPTH-(3-34), hPTH-(7-34), hPTH-(13-34), hPTH-(28-32), hPTH-(28-42), or hPTHrP-(5-36), which have been N-terminally truncated (i.e., have had their normal N-terminal noses cut off ) are very wrongly labeled antagonists only because they cannot activate AC. But they can activate PTHR1 receptors and stimulate PLC-β1 and PKCs (osteoblasts variously express PKCs-α -β, -δ, -ε, -η, -θ, -ζ, -ι/λ!) as strongly as hPTH-(1-34) in some cells, such as freshly isolated human foreskin fibroblasts, OK opossum kidney cells, ROS 17/2 cells, UMR-106 cells, and S49 murine T-lymphoma cells (Azarani et al., 1995a, 1995b; Chakravarthy et al., 1990; Jouishomme et al., 1992; Whitfield et al., 1998b, 2001). Because of this persistent mistake, there has been a large gap in our understanding of PTHR1 signaling. But this gap is now being narrowed by the discovery in the last couple of years of PTH's "magic tail" to use Bockaert et al.'s (2003) happy term.
The PKCs activation story has now become even more complicated. It appears that hPTH-(1-34) has two separable ways to stimulate PKCs (Yang et al., 2004). When wt9 murine osteoblasts with 800,000 to 1,000,000 PTHR1 receptors/cell are exposed to hPTH-(1-34), they activated AC and phosphorylated and activated their PKC-d. [G1, R19]hPTH-(1-28), which lacks both the PLC-activating and the 29-34 PLC-independent PKC-activating regions (Takasu et al., 1999; Whitfield, Isaacs et al., 2001), did not persuade the wt9 cells to activate PLC or phoshorylate PKC-δ (Yang et al., 2004). On the other hand, the AC-activating [G1, R19]hPTH-(1-34), which still lacks the PLC-activation region, caused the cells to activate PKC-d because it has the 29-34 PLC-independent PKC-activating region that was discovered in my laboratory by Jouishomme et al. (1992)).
It seems that the N-terminally truncated PTHs can't stimulate adenylyl cyclase because they can't get down on the receptor TM2's (barrel stave 2) Arg233 and Lys240 to bump the extracellular loop 2 and start the adenylyl cyclase-activating stave tilting (Gensure et al., 2002) (fig. 17). Nevertheless, they can still bind to TM2's Leu232-Lys240 region with the barrel stave's critical interacting amino acid being Phe238 (Gensure et al., 2002). This interaction, along with the binding of their tails to the receptor's N-terminus, must still cause the receptor's tail to wag and in suitably equipped cells in order to activate PLC-β1 as shown in fig. 16. This PLC-activating tail wag, unappreciated though it has been among PTHR1 buffs, is very important because PTHR1, like the β2-AR (β2-adrenergic receptor), has an atypical PDZ-binding domain (the PDZ ligand)—a Glu-Thr-Val-Met (ETVM) sequence—in its C-tail (fig. 16). The PTHR1 C-tail attaches its PDZ ligand to the PDZ-II domain in the C-tail of NHERF-2 (although the β2-AR binds to the PDZ-I domain), the Na+-H+ exchanger's inhibitory regulatory factor-2 (Bockaert et al., 2002, 2003; Harris and Lim, 2001; Mahon and Segre, 2002; Mahon et al., 2002; Voltz et al., 2001).
Because of its PDZ patch, PTHR1's tail, like a dog's tail, is a sophisticated signaling device. The signaling by the PTHR1 receptor, like several other GPCR receptors, such as the β2-AR, is in some cells mediated by a large particle called a "transducosome" consisting of a core scaffold and several attached and interacting signal components (Kreienkamp, 2002). These transducosomes can be awfully complex—the 5-HT2c receptor's tail, for example, can associate with as many as 15 different proteins (Becamel et al., 2002)! And PTHR1 plugs into a transducosome with its tail. One of the cores of PTHR1's transducosomes is NHERF-2 , a kind of scaffold or circuit board attached to the cell membrane, loaded with a set of interconnected signaling enzymes, and is tethered to underlying cytoskeletal actin cables via an "ERM" (ezrin, radixin, moesin) bridge (Harris and Lim, 2001; Kreienkamp, 2002; Mahon and Segre, 2002; Ponting et al., 1997; Voltz et al., 2001). Thus, if the cell has NHERF-2s , each of its PTHR1 receptors can be part of a signal-transducing supercluster on the NHERF-2 circuit board perhaps along with another GPCR receptor as well as several membrane-associated G-proteins and other components such as PLC-β1.
The NHERF-2 scaffold in cells such as PS120 fibroblasts grabs PLC-β1 by the tail with its PDZ-I domain and puts the phospholipase within easy reach of the active PTHR1 tail, and subunits from the associated membrane-anchored Gi/o or Gq/11 heterotrimers (Mahon et al., 2002; Suh et al., 2001). It appears that when the target cell has NHERF-2 (and it is very important to know that not all cells do!) and PTH-activated PTHR1 attaches to the scaffold with its cytoplasmic C-tail's PDZ-II-binding patch, AC stimulation is suppressed, and PLC-β1 stimulation is greatly amplified. Why?
The reactions triggered by the activated PTHR1 are taking place in a dense cluster of enzymes, G proteins, and receptors on the underside of the cell membrane. In PS120 cells, the mechanism seems to be due to the NHERF-2-connected PTHR1 receptor generating active GTP•Gi/oa subunits that find, bind, and inhibit adenylyl cyclase and the NHERF-2 complex abetting this cyclase suppression by hindering the coupling of the AC—activating Gs in the pocket formed by the intracellular loops to the adenylyl cyclase. Meanwhile, the βγ subunits released from the Gi/o or Gq/11 complexes stimulate the PLC-β1 sticking to the NHERF-2 circuit board's PDZ-I domain (Mahon et al., 2002).
This means that cells with lots of NHERF-2s will respond to a PTH very differently from those with no NHERF-2. Thus, ROS 17/2.8 rat osteosarcoma cells without NHERF-2 respond to PTH-(1-34) with as much as a 30-fold burst of adenylyl cyclase activity but no PLC-β1 activity, but if NHERF-2 be put into these cells, the PTH can stimulate PLC and PKC (Mahon et al., 2002). By contrast hPTH-(1-34) can stimulate PLC activity 3-4 fold and adenylyl cyclase by only 1.2-fold or less in ECV304 cells, which have lots of NHERF-2 (and NHERF-1) (Mahon et al., 2002). And then there are the lymphocytes with lots of NHERF-2s that respond to hPTH-(1-34) with a burst of PLC-β1 and membrane-associated PKC activity but with no increase in AC activity (Mahon et al., 2002; Whitfield et al., 1998b, 1999a). Of course, this could explain why PTH cannot directly stimulate adenylyl cyclase in lymphocytes (Whitfield et al., 1971, 1999a).
This could also explain why N-terminally truncated peptides such as hPTH-(3-34), hPTH-(13-34), hPTH-(28-34) can significantly stimulate PKC in OK opossum kidney cells and ROS 17/2 rat osteosarcoma cells (the parents of the NHERF-2-less ROS 17/2.8 cells), but not in HKRK-B7 cells which have only a low level of NHERF-2 (Azarani et al., 1995a, 1995b; LiChong et al., 1998; Whitfield et al., 2001; M.J. Mahon, personal communication to F.R. Bringhurst).
The take-home message from all of this is that the PTHR1 is basically an adenylyl cyclase activator, a cyclic AMP pulse generator that must be plugged into a NHERF-2 circuit board if it is to activate PLC-β1 and its downstream effectors such as PKCs. Put in another way, PTHR1s by themselves are adenylyl cyclase stimulators, but they are PLC/PKC stimulators only when plugged into a NHERF-2 cluster. Moreover, there is also a revolutionary postscript to this take-home message—N-terminally truncated PTHs can still stimulate PLC/PKC, though not adenylyl cyclase, if the PTHR1 is connected to a NHERF-2 transducosome, but they will not signal at all in cells without a NHERF transducosome.
Another example of the daunting complexity of PTH signaling imposed on it by the transducosome on the NHERF-2 scaffold has been reported by Mahon and Segre (2002). As long as PTHR1 is attached to NHERF-2 in PS120 fibroblasts, hPTH-(1-34) can also stimulate extracellular-regulated kinases (ERKs; Pearson et al., 2001)-1 and -2 within 5 minutes (fig. 16). Since this stimulation was unaffected by inhibiting PKAs (the protein kinases activated by cyclic AMP) and PKCs, the authors concluded that the ERK pathway must have been activated by a novel mechanism. But it still might be cyclic AMP itself, not cyclic AMP-activated PKA, which directly stimulated ERKs via the cyclic AMP•Epac → GTP•Rap1 → B-Raf protein kinase pathway to be discussed further on. However, since NHERF-2 attachment prevents adenylyl cyclase stimulation, disconnecting PTHR1 from NHERF-2 should have enabled rather than prevented a direct cyclic AMP stimulation of the ERK kinases.
A model for this action might be what happens when β2-AR receptor sticks to NHERF's PDZ-I domain (Ahn et al., 1999). The activated β2-AR receptor in this cluster causes the dimerization (pairing) and transactivation of the EGF receptor. The β2-AR receptor also activates the c-Src tyrosine protein kinase, which causes the β2-AR receptor to cluster with the EGF receptor pair. This huge β2-AR •[EGFR]2 cluster is then put by a β-arrestin phosphoprotein into a tear drop-shaped clathrin-coated pit, the neck of which is grabbed and pinched off by a dynamin "pinchase" and the pinched-off "tear drop" vesicle is delivered into the cytoplasm (Smythies, 2002). But to get to the point–the active c-Src and the still-functioning pair of EGF receptors trigger the ERK1/2 cascade and its many effects (Maudsley et al., 2000; Friedman et al., 2002). So if PTHR1 attached to NHERF-2 can do the same things, this would be how hPTH-(1-34) could stimulate ER1/2 phosphorylation/activation.
The attachment of the PTHR1 to the NHERF-2 scaffold in some cells means that there might be yet another signal—an internal pH spike—when PTHR1 is activated as happens when the β2-AR is activated. NHERF-2 inhibits the Na+/H+ exchanger when the exchanger is sitting on NHERF's PDZ1 domain. But the activated β2-AR knocks the exchanger off NHERF's PDZ-I (Barber and Ganz, 1992; Barber et al., 1992; Bockaert et al., 2002). The displaced and rejuvenated exchanger then causes a pH spike in the cell by pumping protons (H+s) out of the cell. So PTH might also stimulate the Na+/H+ exchanger, because when the activated PTHR1 receptor's tail attaches to PDZ-II , though this is not the PDZ-I to which β2-AR attaches, it might also displace Na+/H+ exchanger from its inhibitiory attachment to NHERF (Voltz et al., 2001). Indeed, PTH fragments do trigger a PKA-mediated extracellular acidification response (ECAR; a squirt of protons out of the cell) in neonatal mouse calvaria and ROS 17/2 rat cells, but an apparently PKC-mediated ECAR in human SaOS-2 cells (Belinsky and Tashjian, 2000; Belinsky et al., 1999). But we don't know whether these different cells have different amounts of NHERFs. However, this is probably a "false trail" because proton-squirting by human SaOS-2 osteosarcoma cells cannot be the work of the Na+/H+ exchanger because it is not affected by the exchanger's specific inhibitors (Barrett et al., 1997).
Also among the several PTHR1 signal transducers is phospholipase D (PLD), which chops the major membrane phospholipid—phosphatidylcholine (PtdCh)—into choline and phosphatidic acid (PA), which is converted by phosphatidic acid phosphohydrolase into PKCs-stimulating diacylglycerol (Friedman et al., 1999; Radeff et al., 2002; Singh et al., 1999) (fig. 16). But the phosphatidic acid could also be converted to autocrine (self-stimulating)/paracrine (neighbor-stimulating) LPA (lysophosphatidic acid) by PLA2 (phospholipase A2). LPA is the ligand (binder/activator) for G-protein-coupled LPA receptors (LPARs), which can generate various signalers such as the mitogen-activated protein kinases ERK1/2, p38 MAPK, and MSK 1 that phosphorylate the CREB transcription factor like the cyclic AMP-activated PKA and generate small GTPases such as GTP•Rho A (C.-W. Lee et al., 2003; Xie et al., 2002) (fig. 16). This GTP•RhoA in turn can feed back to activate the PLD-stimulating, RhoPK with a consequent escalation of PKCs (e.g., PKCα)-activity by diacylglycerols (Radeff et al., 2002; Xie et al., 2002). We will meet the "Rho Gang" lurking in caveolar scaffolds in Chapter 8ii when discussing how the erratic statins might stimulate bone formation. This propensity for scaffolding means that RhoPK may be another member of the NHERF-2 signaling pack.
As if this is not enough, there is another NHERF involved in PTH actions! At the 24th meeting of the American Society for Bone and Mineral Research in San Antonio in September 2002, J. M. Mahon announced (without any written record of his talk) that the C-tails of PTHR1 receptors in kidney proximal convoluted tubule cells, specifically OK (opossum kidney) cells, attach to NHERF-1 instead of NHERF-2 (fig. 16). In these cells, PTHR1 signals prevent phosphate reuptake by causing the type II Na/Pi-cotransporters to be dragged from caveolae ("little caves") in the apical surface into the depths of the cell and destroyed (Pfister et al., 1997; Taketani et al., 2003). Disrupting the gene for NHERF-1 prevents the type II cotransporter from being plugged into the apical cell membranes of kidney proximal convoluted tubule cells (Shenolikar et al., 2002). And Mahon reported that phosphate uptake by mutant OK cells lacking NHERF-1 was unaffected by PTH, but giving the cells back their NHERF-1 restored the ability of PTH to inhibit phosphate uptake. So we are faced with yet another PTHR1-coupled scaffold with more signaling complexes, though maybe not in bone cells (fig. 16).
However, NHERF-1 can also couple PTHR1 activation to stimulation of phospholipase C as does NHERF-2 (Mahon and Segre, 2003). This has been shown using cells of the OKH substrain of opossum kidney cells. Exposing these cells to hPTH-(1-34) stimulates their adenylyl cyclase, but there is no effect on phosphopliase-C activity, no inhibition of phosphate uptake by the Na-phosphate cotransporter, and no change in the intracellular Ca2+ concentration. But putting NHERF-1 into these cells results in the localization of PTHR1 to the NHERF-1 and enables the PTH to stimulate the release of βγ subunit complexes from Go/i, which then activate phospholipase-Cβ, which in turn opens a type of Ca2+ channel exactly as would happen with the NHERF-2•PTHR1 complex in fibroblasts and osteoblasts (fig. 16).
But there could be even more! Maybe, just maybe, PTHR1 receptor homodimerizes (twins with another PTHR1) or far more important heterodimerizes (couples with different kinds of receptors). It has long been an article of faith that GPCR receptors like PTHR1 don't do either of these. They were believed to be solitary signalers that would never pair with each other or with any others, as do receptors such as the EGF receptor and its several kin. But there is now a growing number of examples of GPCR homodimers and heterodimers that profoundly affect signaling. For example, there is the adenosine A1 receptor—dopamine 1 receptor heterodimer, the dopamine D2R receptor—somatostatin 5 receptor heterodimer, and the opioid ? receptor —opioid δ receptor heterodimer (Filizola et al., 2002; Jordan et al., 2000, 2001; Marshall, 2001; S.P. Lee et al., 2000; Rocheville et al., 2000a, 2000b; Vazquez-Prado et al., 2003). And of course we must not forget the ability of β2-adrenergic GPCR receptors to activate the entirely different EGF tyrosine kinase receptors by physically joining them in a heteromultimeric transducosome that produces active EGF dimers (Maudsley et al., 2000)! If PTHR1 should have the same unsuspected gregarious tendencies as other GPCRs, its pairings with other family members, along with clusters of various signalers on NHERFs, could produce dazzling displays of multi-track signaling fireworks, depending on the target cell's make up that could be ignited by PTH and, here is the important thing—the ligand of the other member of the couple.
In summary, what PTHR1, like other GPCR receptors, surrounded by a crowd of hangers-on and their widely ramifying actions, is telling us through the fog of ignorance is actually very clear—there is no such thing as standard PTH receptor signals! The kinds of membrane scaffolds and the signal transmitters and trannsducers on them determine what signals a PTHR1 (or, for that matter, any other receptor) gives its cell. But what determines the kind of G-protein the activated receptor couples with? There are several ways that a GPCR receptor like PTHR1 might couple to more than one kind of G-protein and activate different signal mechanisms. There is the sequential model in which the activated receptor initiates a signal cascade that in the process switches back and modifies the receptor to activate a different G-protein and start a second cascade (Schoneberg et al., 2002). As an example of this, phosphorylation of a β-adrenergic receptor by PKA can shift its coupling from Gs to Gi (Daaka et al., 1997; Zamah et al., 2002). Likewise, the activated prostacyclin receptor couples first to Gs, which through its Gsa subunit stimulates adenylyl cyclase, cyclic AMP production, and the cyclic AMP activates PKA, which then turns around and phosphorylates the receptor on its Ser357, which enables the receptor to couple to Gi and Gq and trigger other signal cascades (Lawler et al., 2001). Then there is the parallel model in which the receptor is in equilibrium with two possible activation states (Schoneberg et al., 2002). Thus, in one state PTHR1 without access to a NHERF-2 "chip" would couple to and activate Gs and the Gs would activate adenylyl cyclase, while activated PTHR1 attached to a NHERF-2 "chip" would activate GTP•Gi/o or GTP•Gq complexes, which would prevent Gs from activating adenylyl cyclase and at the same time stimulate PLC-β1 on the NHERF's PDZ-I domain with the βγ subunits libertated from the GTP•Gi/o or GTP•Gq/11 complexes. Perhaps PTHR1 behaves like the β2-adrenergic receptor, which when activated unfurls a motif on its C-tail with which it binds to the appropriate PDZ domain on NHERF-2's tail and thus displaces and releases the Na+/H+ exchanger from its inhibitory linkage to NHERF (Schoneberg et al., 2002; Voltz et al., 2001). The relative responses of adenylyl cyclase and PLC-β1 to a PTH would depend on how many receptors the cell has and how many of these can attach to NHERF-2.
In other words, the adenylyl cyclase/PLC-β1/??? contents of the primary signal from PTHR1s in a cell and whether bone growth will be started by this signal will depend on the cell's PTHR1:NHERF-2 ratio.
Now we should hurry to catch the bone-forming signal train before all of this signal complexity becomes mental mush. But I can't resist sadistically adding one last bit to PTH receptor complexity, which I hope will not break the proverbial camel's (reader's) back. In Section v, I will briefly outline the PTHs' surprising (though actually not when you look more closely) ability to eliminate psoriatic skin lesions that are caused by the dysregulated proliferation and differentiation of epidermal keratinocytes. Keratinocytes, like osteoblasts, can be induced by either hPTH-(1-34) or hPTH-(1-84) to increase their membrane-associated PKC activity and generate a Ca2+ transient, but unlike osteoblasts, they refuse to activate adenylyl cyclase (Orloff et al., 1995; Whitfield et al., 1992, 1996a). The reason for this difference could be that that they do not have PTHR1 receptors, as indicated by their failure to make the gene's 2.3-kb transcript (Hanafin et al., 1995; Orloff et al., 1995; Sharpe et al., 1998). The receptor they make might be smaller than PTHR1—its gene's mRNA transcript seems to be only 1 kb instead of PTHR1's 2.3-kb transcript (Orloff et al., 1995). The keratinocyte receptor, which could be called PTHRK, is partially homologous to PTHR1's 155-281 region, which includes the immediately juxtamembrane part of the N-terminal extracellular region, the first 2 transmembrane α-helices, and the first extracellular loop linking transmembrane α-helices 2 and 3 (fig. 16 and fig. 17) (Orloff et al., 1995). Thus, there may be another—pthrk—gene (or perhaps an alternative transcript of the PTHR1 gene) that progeny of basal keratinocyte stem cells turn on instead of the PTHR1 gene when they become rapidly proliferating, transit-amplifying cells (Errazahi et al., 1998; Whitfield, 2004; Whifield and Chakravarthy, 2001). Or the cells might alternatively translate PTHR1's gene.
But wait! As usual there is a problem. According to Errazahi et al. (2003), keratinocytes from newborn rats seem to make functional PTHR1s. However, there is something peculiar about them because they do not respond to bPTH-(1-34) as do other rat cells such as ROS 17/2 cells. Unfortunately, these authors also don't say whether PTHrP also stimulated adenylyl cyclase in their keratinocytes. Thus, rat keratinocytes may have a modified PTHR1 which just cannot activate adenylyl cyclase or perhaps is prevented from doing so by associating with NHERF-2 (if keratinocytes have NHERF-2). So stay tuned to the so far slowly unfolding saga of keratinocytes and their adenylyl cyclase-shunning PTH/PTHrP receptor.
Of course, we must not ignore other members of the PTH receptor clan—yes, indeed, there are others! There is PTH2, which, unlike PTHRK, is a potent adenylyl cyclase stimulator designed to be activated by the PTH-related, 39-residue tuberoinfundibular peptide, TIP39, and is expressed most abundantly in the nervous system and to a lesser extent in lung, the pancreatic islets' somatostatin-producing D-cells and placenta (Usdin et al., 2002). However, the human PTHR2, but not the rat PTHR2, can also be strongly activated by PTH but not by PTHrP, while TIP39 binds to, but doesn't activate, PTHR1 (Usdin et al., 2002). Then there is the zebrafish's zPTHR3, which shares 67% of its amino acid sequence with hPTHR1 but has a 50-fold greater affinity for hPTHrP than hPTH (Rubin and Jüppner, 1999). Like zPTHR1, zPTHR3 can activate adenylyl cyclase, but unlike zPTHR1, it cannot stimulate PLC (i.e., it cannot stimulate IP3 accumulation) (Rubin and Jüppner, 1999). Then there is a mysterious receptor in the brain (specifically the supraoptic nucleus) that is activated by PTHrP-(1-34) but not by hPTH-(1-34) or hPTHrP-(7-34) (Yamamoto et al., 1998). The signals from this receptor stimulate the expression and secretion of arginine vasopressin by the supraoptic nucleus cells (Yamamoto et al., 1998).
Now enough is enough! Let's see what all of this receptor lore means to bone making.
What Signals Switch on the Bone-Making Machine?
If the adenylyl cyclase response is not muffled by a high NHERF-2:PTHR1 ratio, the burst of cyclic AMP synthesis directly activates certain GEFs (guanosine nucleotide exchange factors) to be discussed further on and whatever cyclic AMP-dependent protein kinase isoforms (PKAs) are around as well as the B-Raf/ERK protein kinase system. The diacylglycerols from PLC-β1-induced PIP2 and PLD-induced PtdCh breakdown activate the available and responsive protein kinase-C isoforms (PKCs), and the IP3 from PIP2 stimulates the release of Ca2+ from internal endoplasmic reticulum stores (fig. 16). These PTH-induced signalers stir up a storm of events. For example, waiting at the the mouths of some of the endoplasmic reticulum's IP3-activated Ca2+ channels are mitochondria, the Ca2+ uniporters or transporters of which carry the Ca2+ released from these channels into the mitochondria to fill the cell's ATP fuel tanks by stimulating key components of the ATP-making machinery such as pyruvate dehydrogenase, NAD+-isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase (Ashby and Tepikin, 2001; Hajnóczky et al., 2000, 2003; Pozzan et al., 2002). Perhaps the most important of these PTH-triggered events is the expression and secretion of FGF-2 and IGF-I, which will also send Ca2+, IP3, PKCs, and PLD signals from their receptors attached to their specifc scaffolded superclusters in the membranes of both the PTHR1-bearing producer cells and in the "juvenile" osteoprogenitor cells which are not yet mature enough to have started expressing PTHR1 receptors (fig. 8). At this point, I must point out that cells with the appropriate receptors can stimulate themselves (cellular masturbation) with growth factors they produce—these are called "autocrine" factors. They can also stimulate appropriately receptored neighboring cells with factors they produce—when they do this, they are called "paracrine" factors. So the FGF-2 and IGF-I made by PTH-stimulated osteoblasts are both autocrine and paracrine factors in bone.
If it is cyclic AMP, bursts of cyclic AMP-stimulated PKA activity and the direct targets of cyclic AMP-activated GEFs (guanine nucleotide exchange factors) such as Epacs 1 and 2 (de Rooij et al., 2000) that trigger bone growth, and if a PTH fragment could be made that needs only to activate adenylyl cyclase to stimulate bone growth, it could have potentially fewer side effects resulting from PKCs' activity in the many different cells expressing PTHR1 receptors. Such a fragment could be made if the multi-signaling hPTH-(1-34) could be cut into smaller, still partly functional, pieces each of which stimulates either one or the other signal mechanism. And this we seemed to have been done in the early '90s when PTHR1 signaling seemed to be so direct and simple without the complications of NHERFs and receptor heterodimers.
The potently osteogenic (in OVXed rats and cynomolgous monkeys) Ostabolin™ (hPTH-(1-31)NH2) and Ostabolin C™ ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2) are as effective adenylyl cyclase stimulators as hPTH-(1-34)NH2 , but unlike hPTH-(1-34)NH2, they do not stimulate PKC activity in cultured nontransformed murine MC3T3-E1 calvarial (skull bones) preosteoblasts, rat ROS 17/2, osteoblast-like osteosarcoma cells, UMR 106-01 rat osteoblast-like osteosarcoma cells, as well as primary rat osteoblasts (Barbier et al., 1997; Jouishomme et al., 1992, 1994; Ryder and Duncan, 2001; Singh et al., 1999; Swarthout et al., 2000; Whitfield et al., 1996b, 1999a). Cultured human fetal osteoblasts (hFOB cells) appear to respond to hPTH-(1-31)NH2 in the same way. In these human osteoblasts, hPTH-(1-31)NH2 does not stimulate the PKC-dependent expression of TGF-β1 (and presumably not PKC activity) in these human fetal osteoblasts, although it stimulates the cyclic AMP-dependent expression of TGF-β2 as strongly as hPTH-(1-34) (Wu and Kumar, 2000).
As might be expected from what the PTHR1 receptor has been trying to tell us, whether such C-terminally truncated fragments can also stimulate PKC activity depends on which cells' membranes the PTHR1 receptors find themselves. For example, hPTH-(1-31)NH2 can increase membrane-associated PKC activity in pig kidney cells engineered to put a huge number (i.e., about 950,000) of human PTHR1 receptors on their surfaces and in normal human newborn foreskin fibroblasts, which have only a very few (in fact not detectable by competitive binding assay) human PTHR1 receptors (Whitfield et al., 2001). The failure of hPTH-(1-31)NH2 (Ostabolin™) and [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™) to increase membrane-associated PKC activity in ROS 17/2 osteoblast-like cells and fetal human osteoblasts means that while PTHR1 receptors on cultured osteoblasts can't activate PLC or PLD, they can activate one or both of these enzymes in densely human-receptored pig kidney cells and very sparsely receptored cultured human foreskin fibroblasts that are differently equipped with the necessary messengers, kinase isoforms, and NHERF scaffolds.
At this point, the reader may be muttering enough's enough with this horrendous signaling complexity. But there's even more! Even the once sort-of-simple and all-important adenylyl cyclase-cyclic AMP mechanism has become formidably complicated (Chin et al., 2002). There is a very large number of possible cell type-specific combinations of different types of PKA R (regulatory) and C (catalytic) subunits in or on the cell membrane or in or around the nucleus. There are cyclic AMP-binding G-protein-activating guanine nucleotide exchange-stimulating factors (GEFs) factors. There are ion channels and transcription factors, such as the very important NF-κB that directly bind and respond to cyclic AMP independently from PKA activity. And R subunits can function independently from the catalytic subunits. For example, the cyclic AMP•RIIβ complex can bind to the CREs (cyclic AMP-responsive elements) in the promoters (the transcription switch boxes) of some genes. Therefore, the cell's response to the cyclic AMP pulses triggered by activated PTHR1 receptors depends on the whether the cells have NHERF scaffolds, what kinds of cyclic AMP-binding targets are available, and what PKA R and catalytic subunit isoforms the cell has and where they are in the cell.
GPCRs such as PTHR1 do not directly activate receptor tyrosine kinase receptors (RTKs). But surprisingly they can indirectly transactivate RTK receptors such as the EGF receptor (Cole, 1999; Daub et al., 1997; Iwamoto and Mekada, 2000; Prenzel et al., 1999)! At first, it seemed that in these cases the EGF receptor was not activated by its EGF ligand, but it now appears that the activated GPCR stimulates a transmembrane metalloproteinase that cuts soluble sHB-EGF (a heparin-binding protein with EGF-like motifs) from membrane-anchored pro-HB-EGF (precursor of heparin-binding EGF) (Iwamoto and Mekada, 2000; Prenzel et al., 1999). The liberated sHB-EGF then activates EGF receptors on its own cell or adjacent cells. When the PTH/PTHR1-"transactivated" EGF receptors phosphorylate themselves, they become sticky flypaper-like or Velcro-like scaffolds for collecting and clustering the components of the Ras/Raf-1-mechanism that triggers the multipurpose MAP kinase cascade (Cole, 1999; Daub et al., 1997; Pearson et al., 2001). However, we don't know what the Ras/Raf/MAP kinase cascades triggered by PTHR1-transactivated RTKs, or whatever other receptor with which PTHR1 might have paired with, might contribute to osteogenesis. Nevertheless, the important message is that the signals triggered by a PTH could come from both PTHR1 receptors and any transactivatable RTKs the cell might be expressing.
PTHR1 signaling is far more complicated, busy, and far-reaching than in the simple schemes of the early 1990s. After having sent its first signals, the PTH•PTHR1 complex is rapidly silenced and pulled into the cell (endocytosed) with a half-time of 3-5 minutes, but then PTHR1 slowly makes its way back to the surface, ready to work again, with a half-time of about 2-4 hours (Chauvin et al., 2001) (fig. 18). But it appears that if the endocytosis of the PTH•PTHR1 complex is prevented in human embryonic HEK 293 kidney cells by having them express β-arrestin-2's C-terminus along with the normal β-arrestin, the receptor is still switched off when the PTH•PTHR1 complex binds to normal β-arrestin, but the lack of endocytosis does not stop the receptor from being put back to work (Bisello et al., 2002). However, if cells such as osteoblasts have their arrestin gene knocked out, PTH(PTHR1) signaling is sustained (Pierroz et al., 2003). But under normal conditions the PTH•PTHR1 complex's endocytosis starts when the receptor's C-tail binds to the β-arrestin-2 scaffolding protein (Chauvin et al., 2001; Pouysségur, 2000) (fig. 18). The PTH•PTHR1•β-arrestin-2 complex is wrapped in a clathrin-coated pit, and c-Src is brought into the complex to phosphorylate and switch on the tiny dynamin motors that pull the packaged receptor complex down into the cytoplasm (Ahn et al., 1999; Chauvin et al., 2001; Pouysségur, 2000). By analogy with the type IA angiotensin II receptor, while RGS-2 and the endocytosis of the PTHR1 receptor stop adenylyl cyclase and PLC-β1 signaling, it might at the same time trigger a second tier of signaling because the β-arrestin-2 scaffold has an RRSLHL (Arg-Arg-Ser-Leu-His-Leu) motif which brings Raf-1, ASK-1, MAP Kinase Kinase-4 and a JNK (c-Jun kinase) together with the PTH•PTHR1 complex (Keenan and Baldassare, 2001; Luttrell et al., 2001; Miller et al., 2001; Pouysségur, 2000). When β-arrestin-2 attaches to the PTHR1 of the PTH•PTHR1 complex, ASK1 could activate MAP Kinase Kinase-4, which in turn activates JNK, which is released from the scaffold, surges into the nucleus, and phosphorylates and activates other gene transactivators such as c-Jun, which forms part of the c-Fos•c-Jun AP-1 gene transcription complex (Keenan and Baldassare, 2001) (fig. 18). However, the c-Src, which turns on the endocytosis motor, may also activate the MAPK mechanism (Mahon and Segre, 2002).
The already challenging tangle of PTHR1 signals (fig. 19) is increased still further by PTHR1's nuclear localization signal! This means that either newly made PTHR1 might be transported into the nucleus without reaching the cell membrane or after being released from the β-arrestin-2 complex, the endocytosed receptor might go into the nucleus along with JNK to stimulate some as yet unknown nuclear functions (fig. 18). The surprising possibility of a receptor directly stimulating genes after being transported from the cell surface into the nucleus after binding and activation by its ligand has recently been demonstrated with EGF and the ErbB-1 and ErbB-4 receptors (Lin et al., 2001; Ni et al., 2001). The EGF receptor, with a strong gene transactivation domain in its C-tail, gets into the nuclei of a variety of cells where it can bind to AT-rich sequences, such as the consensus site in the cyclin D1 gene's promoter-switch box (Lin et al., 2001). When the ErbB-4 receptor on a mammary carcinoma cell surface binds its ligand, heregulin, and starts signaling, its cytoplasmic tail is snipped off by ?-secretase, and it travels to the nucleus where it activates a gene(s), the product(s) of which inhibits the growth and stimulates the differentiation of the cell (Ni et al., 2001). However, at second glance this should really not be too surprising because estrogen receptors and Notch receptors can be activated on the cell surface and then be carried wholly or partly into the nucleus to stimulate suitably marked genes.
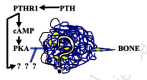
In the late 1980s and early 1990s, we set out to find whether the osteogenic signal from PTHR1 receptors required the stimulation of adenylyl cyclase, membrane-associated PKCs, or both to start making bone. So we needed fragments that stimulate membrane-associated PKC but not adenylyl cyclase. Unlike the C-terminally cropped fragments, no N-terminally truncated PTH fragment can stimulate AC. However, as we learned above, the ability of a N-terminally truncated PTH to stimulate PKC activity also depends on the kind of cell expressing the PTHR1 receptors. For example, b(bovine)PTH-(3-34)NH2, hPTH-(3-34)NH2, hPTH-(13-34)OH, hPTH-(28-48)OH, hPTH-(8-84)OH, and even the tiny hPTH-(28-34)OH, hPTH-(29-32)OH can increase membrane-asociated PKC activity in cultured ROS17/2 osteoblastic cells, UMR-106 rat osteoblastic cells, human fetal osteoblasts, and human dermal fibroblasts, and hPTH-(8-84) can strongly stimulate membrane-associated PKC activity in spleen cells in rats, but they are at best only very weak stimulators of membrane-associated PKC activity in the pig cells with nearly a million human PTHR1 receptors (Fujimori et al., 1992; Jouishomme et al., 1992, 1994; LiChong et al., 1998; Neugebauer et al., 1995; Swarthout et al., 2001; Whitfield et al., 2001; Wu and Kumar, 2000). This of course could be due to the artificially engineered pig cells having only very little NHERF-2, which is required for PTHR1 to activate PLC and thus stimulate PKC activity (M. Mahon, personal communication to F.R. Bringhurst).
hPTH-(1-31)NH2 (Ostabolin™), its lactam derivative, [Leu27]cyclo[Glu22-Lys26]hPTH-(1-31)NH2 (Ostabolin-C™ ), as well as [Leu27]cyclo[Glu22-Lys26]hPTH-(1-28)NH2, which stimulate AC but not membrane-associated PKC activity in cultured rat and human fetal osteoblasts, strongly stimulate bone growth in OVXed rats (Rixon et al., 1994; Whitfield et al., 1996b, 1997b, 1997c, 1997d, 1998b, 1999c, 2000a, 2000b, 2000c). On the other hand, the N-terminally disabled 1-desamino-hPTH-(1-34) and the N-terminally truncated hPTH-(28-48) and hPTH-(8-84) that strongly stimulate membrane-associated PKC activity, but not adenylyl cyclase, in cultured osteoblasts and in rats do not stimulate bone growth in the OVXed rats (Rixon et al., 1994; Strein, 1994). Therefore, as things stand at the moment, activating adenylyl cyclase and the cascades it triggers seems to be all a PTH needs to do to start, but maybe not subsequently drive, bone formation (at least in rats). This is supported by the ability of selective cyclic AMP-elevating, cyclic nucleotide phosphodiesterase inhibitors (Pentoxyfylline and Rolipram) to stimulate bone growth in normal male mice (Kinoshita et al., 2000). But (that caveating word again!) Locklin et al. (2003) have found that hPTH-(3-34), which cannot stimulate adenylyl cyclase or bone formation, nevertheless can stimulate the expression of that master conductor of osteoblastic genesæ bfa1/Runx-2æ in cultured C57BL/6 mouse marrow cells as effectively as the cyclase-stimulating, osteogenic hPTH-(1-34) and hPTH-(1-31).
However, AC stimulation might not be enough to start osteogenesis in ovariectomized rats and mice. MPTH-(1-21) (i.e., [Ala,3,12Nle,8Gln,10 Har,11Trp,14Arg,19Tyr21]rPTH-(1-21)) can bind to PTHR1 and strongly stimulate adenylyl cyclase, but it cannot stimulate bone formation in OVXed mice and rats and was unexpectedly much less potent in affecting alkaline phosphatase activity in human HOB-03-C5 and Saos bone cells (Murrills et al., 2002). Thus, for some reason the cyclic AMP signals from PTHR1 activated by this short, AC-stimulating PTH can't start osteogenesis. But wait! Did the fragment reach the bones in an active state? Rixon et al. (1994) had to answer the same question when they found that hPTH-(3-34) and hPTH-(13-34) did not stimulate bone formation in OVXed rats. Was this because they couldn't stimulate bone formation or they couldn't reach the bone in an active state? In this case, both turned out to be the case. The inability of Rixon et al.'s PTHs to reach their bone targets in the rat was shown by their abilities to strongly stimulate membrane-associated PKC activity, as did hPTH-(1-34) when added to spleen lymphocytes suspensions just a few minutes after they were taken from the rat, but they did not increase membrane-associated PKC activity in spleen lymphocytes still in the rat as did hPTH-(1-34). So one could ask Murrills and colleagues whether their little peptide actually reached bone cells?
The failure of small (less than 34 amino acids) N-terminally truncated PTH fragments to stimulate PKC activity in spleen cells in the living, breathing rat while strongly stimulating PKC activity in freshly isolated spleen cells suggests the existence of something that sees and trashes such small fragments but ignores a larger N-terminally truncated fragment such as rhPTH-(8-84) or a 34-amino acid fragment such as 1-desamino-hPTH-(1-34), with its quasi-normal N-terminus. This nemesis of small "nose-less" fragments might be the 230-kDa protein in rat and human serum that Kukreja et al. (1994) found to inactivate bPTH-(7-34)NH2 and hPTHrP-(7-34)NH2, but not hPTHrP-(1-34). As we learned above, the unselected rhPTH-(8-84) would be able to stimulate osteocytes having both the CPTHR (Bringhurst, 2002; Divieti et al., 2001, 2002) and in my experience (Rixon et al., 1994) the PLC-β1/PKC arm of the PTHR1 response. However, one must be careful of this because Bringhurst (2002) has said that hPTH-(7-84) does not bind to PTHR1 receptors. If this were true, it would mean that spleen lymphocytes responded by either the CPTHR or another receptor, as suggested by Whitfield et al. (1999a). Remember (if by now you need reminding) that nothing is simple with PTHs and their receptors.
There are reasons to believe that hPTH-(1-31)NH2 (Ostabolin™) may also only stimulate adenylyl cyclase in adult human osteoblasts as it seems to do in cultured fetal human osteoblasts (Wu and Kumar, 2000). If hPTH-(1-31)NH2 were a dual-signaler like hPTH-(1-34) in adult human bones, it should affect adult human bone metabolism like hPTH-(1-34). But it doesn't! While the two fragments equally stimulate adenylyl cyclase in human volunteers, hPTH-(1-31)NH2 is a much weaker stimulator of bone resorption than hPTH-(1-34) as indicated by a failure to cause hypercalcemia and increase urinary collagen breakdown products (Fraher et al., 1999). hPTH-(1-31)NH2 is also as strong a stimulator of adenylyl cyclase and bone growth as hPTH-(1-34) in OVXed mice, but it is only half as strong a stimulator of osteoclast activity as hPTH-(1-34) (Mohan et al., 2000). There seems to be some support for this in the observations of Whitfield et al. (1998b) (fig. 1). They found that daily injections of a low dose (0.6 nmole/100 g of body weight) of hPTH-(1-34) into 3-month-old rats, starting 2 weeks after OVX, did not reduce the loss of femoral trabeculae (56% fewer boney hits/mm of metaphyseal scan in the treated OVXed rats compared to 52% fewer in the untreated OVXed rats), particularly the central metaphyseal trabeculae, although they raised the mean thickness of the surviving lateral trabeculae by about 1.7 times above the level in the sham-operated control rats. On the other hand, while injections of the same dose of Ostabolin C™ ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2) raised the trabecular thickness 1.5 times above the sham-operated value they also significantly reduced the OVX-induced loss of central trabeculae from 56% to 29% (fig. 1). And Jolette et al. (2003) have found that Ostabolin-C™ can stimulate bone growth while reducing osteoclast activity in cynomologous monkeys. However, when the trabecular number in Whitfield et al.'s rats was allowed to drop for 9 weeks before starting the 6 weeks of daily injections, neither fragment could appreciably stop any further drop, although both still strongly increased the mean thickness of the remaining lateral trabeculae to 1.6-1.7 times the mean thickness of the trabeculae in the femurs of the sham-operated control animals. The responses to a higher dose of Ostabolin C™ or hPTH-(1-34) (2 nmoles/100 g of body weight) were identical.
As we shall see further on, this difference between the effects of hPTH-(1-34) and the two C-terminally truncated fragments in humans and rodents just might be due to lower doses of hPTH-(1-31)NH2 (Ostabolin™) and Ostabolin-C™ not having hPTH-(1-34)'s ability to stimulate osteoblast progenitors to express the osteoclast differentiation stimulator RANKL and reduce the expression of the osteoclast-inhibiting OPG (Hofbauer et al., 2000; Suda et al., 1999). Indeed maybe Ostabolin™ and Ostabolin C™ increase the OPG/RANKL expression ratio to a level that actually inhibits OVX-induced osteoclast activation, as suggested by the findings of Jolette et al. (2003) with monkeys.
Before going any further, I must repeat that we do not have the full measure of the signals that the PTHs send into their target bone-making cells in the various provinces of the Real Bone World. For example, what NHERFs do the cells in the various stages of osteoblastic development in a PTH-treated bone have, and what are plugged into their NHERFs that can be tweaked by activated PTHR1s? In other words what do the PTHR1-bearing cells have that can be targeted by the primary, osteogenesis-kick-starting cyclic AMP signal?
PTHs Speak to Bones with Forked Tongues—One Prong Says Make It!
Generalities
Obviously, we must find out how the adenylyl cyclase-stimulating (i.e., the N-terminally intact) PTHs and the signals from their PTHR1 receptor stimulate bone growth in order to make even better anabolic drugs for the future. But this is not easy. The first problem is trying to identify the key players in the horde of genes and other things that can respond directly to PTHR1's promiscuous or "shotgun" signaling. Indeed, the PTHR1 signals with their many target genes, transcription factors, enzymes, enzyme regulators, cytoskeletal components and motors set off a barrage of bone-relevant as well as bone-irrelevant reactions. Within this tangle of events are the few that actually lead to bone formation. The challenge is to find the bone-building needles in this tangled haystack of responses (fig. 19).
As a tiny tantalizing taste of things to come, Qiu et al. (2003), starting by characterizing the promoters of 8 "training" genes known to be regulated by PTHs, have identified 31 transcription factors and 80 genes that could be affected directly by PTHs. And Onyia et al. (2001a) have found that 158 genes associated with bone growth are stimulated in rat metaphyseal bone by PTH. Added to these are those in the differently equipped and scaffolded members of the osteoblastic lineage in different parts of the bones that belong to the vast signaling "syncytium" stretching from the cilium-waving strain-sensing osteocytes locked in their lacunae, to their dendrites snaking through canaliculi, to the retired osteoblasts lining the bone surfaces, and from there to the osteoblastic and osteoclastic progenitors and precursors in the bone marrow (Marotti, 1996). We see osteoblastic cells signaling and, amazingly, remembering strain frequencies using the same machinery as neurons in the central nervous system (Skerry and Taylor, 2001; Spencer et al., 2004; Turner et al., 2002). But we are being swamped by an avalanche of reports of relevant, but undoubtedly many more irrelevant, gene transactivators, hundreds of gene expressions, dozens of enzymes and mechanisms in osteoblastic cells not working in their native bones but that have been ripped from their native signaling "syncytium" and planted on plastic or in cells of established or semi-established lines in which cell cycle controls, PTHrP and PTHR1 expressions, and the maturation program have been more or less disconnected. Indeed, such dysregulated cells are like crazy computers simultaneously running many normally mutually exclusive programs. Therefore, what follows can only be the vague outline of the mechanism(s) by which the PTHs cause bones to grow as seen from inside the swirling stream of events.
When an adenylyl cyclase-stimulating PTH is injected subcutaneously, or, as may soon be happening, swallowed in a pill (fig. 20), it first meets and is grabbed by trabecular bone cells in the bone marrow and only later and probably more or less depleted does it reach cells in the Haversian canals and the lacunocanalicular network (see Chapter 2i; Knothe-Tate, 2001). The target cells in the various regions will have their PTHR1s in different superclusters, and some cells will have unconventional PTH/PTHrP receptors. But how many target cells with PTHR1 receptors are available to kindle an osteogenic response? This depends on the age of the rat or human. In a growing rat or human, there are packs of osteoblasts and osteoclasts working independently to build and shape the growing bones. But when growth stops in humans (and other big-boned animals), there are few or no independently working osteoblasts. The only osteoblasts will be in BMUs. So there won't be many PTHR1-bearing first responders in older humans. But if, for example, a long bone is broken and an implant is screwed into it, BMUs will be activated and fan out from the screwhole in all directions along the diaphysis on their several-millimeter journeys and affect the whole length of the bone for months afterwards (R.B. Martin et al., 1998). In other words, these BMUs will generate PTH targets—PTHR1-bearing osteoblasts—and thus enhance the responsivenss to PTH.
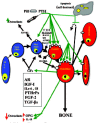
The potently osteogenic hPTH-(1-34) (Forteo™), but possibly not so much other equally potent bone-building PTHs such as hPTH-(1-31)NH2 (Ostabolin™) and [Leu27]cyclo[Glu22-Lys26]hPTH-(1-31)NH2 (Ostabolin-C™), increases osteoclast generation like an estrogen crash and hence the number of holes being dug in the bone at any given moment (i.e., the remodeling space). It stimulates (probably via cyclic AMP) the proliferation of the pluripotent, spleen colony-forming, CFU-S, stem cells and other members of the rat hematopoietic cell populations, which need the microenvironment provided by docking on immature osteoblastic stromal cells (Byron, 1977; Gallien-Lartigue and Carrez, 1974; Perris et al., 1971). And Calvi et al. (2003) have much more recently confirmed that either hPTH-(1-34) or a permanently switched-on mutant PTHR1 receptor, which does not need PTH to activate, it, can enlarge a mouse's hemopoietic stem pool. It seems that PTHR1 receptor signals cause the osteoblastic bone-lining cells to make large amounts of surface Jagged 1, which in turn binds to Notch receptors on adjacent hemopoietic stem cells attached by N-cadherin to the osteoblastic lining cells (Calvi et al., 2004; Ohishi et al., 2003; Schweisguth, 2004; Zhu and Emerson, 2004). The resulting Notch signals in the stem cells increase the pool of hemopoietic stem cells. When cells from the enlarged stem cell pool are mobilized and leave their osteoblastic niche, they can generate increased numbers of "rookie"osteoclast recruits, which carry on the stem cell tradition of making PTHR1 mRNA and receptors, but hold these in their cytoplasm and around their nuclei and maybe even in their nuclei without putting them out onto their surfaces as they mature (Faucheux et al., 2002; Kanatami et al., 1998; Langub et al., 2001; Watson et al., 2002). The PTH-induced burst of cyclic AMP formation also stimulates osteoclast formation by causing some immature marrow osteoblastic stromal cells with PTHR1 receptors to express and deploy on their surfaces the osteoclastogenesis-stimulating RANKL (Atkins et al., 2003; Fu et al., 2001; Galvin et al., 2001; Kanazawa et al., 2000; K. Lee et al., 1994; Stilgren et al., 2001). The PTH also induces cultured osteoblasts and possibly some immature osteoblastic stromal cells to make autocrine (autostimulating)/paracrine (neighbor-stimulating) IL-6 which causes the immature osteoblastic stromal cells to make RANKL (Greenfield et al., 1999; Tamura et al., 1993) (fig. 20).
The Target Cells
The basic requirement for a PTH to stimulate bone growth is that there must be enough PTHR1-bearing cells with which to start building up a crew of active PTHR1 receptor-bearing, osteoblasts. The response to a PTH will initially depend on the size of this receptored team. As stated above, in the bones of human children or rat pups, the mature osteoblast teams will be large and located in osteoclast-free, bone-modeling clusters, but in an adult human, it should be much smaller because most of the cells will belong to microcrack-filling BMUs. But there are also PTH-responsive, BMU-independent retired osteoblasts in mature bones which have fewer but still effective PTHR1 receptors (fig. 20). These are the lining cells, covering the walls of the osteons' central canals as well as the endosteal and trabecular surfaces. The PTH will increase the number of the mature bone-making osteoblasts by increasing their generation, their lifespan, or persuading the retired lining cells to come out of retirement (fig. 20). A mature osteoblast is at the end of a long line of cells stretching back to an ALP (alkaline phosphatase)-expressing "grandmother" cell in the bone marrow stroma or the periosteum and ultimately a mesenchymally-derived "great-great-great-grandmother" stem cell, which had unlimited self-renewing potential and the options to found lines of adipocytes, chondrocytes, fibroblasts, or myoblasts (Aubin, 2000, 2001; Aubin and Triffitt, 2002). As is the case for all stem cells (Whitfield and Chakravarthy, 2001), these mesenchymally-derived stem cells normally proliferate only when needed to maintain a pool of extensively proliferating osteoprogenitors (i.e, transit amplifying cells) for growing bone in children and and removing muscle-inflicted microcracks in adults (fig. 20).
But let's look more closely at the PTH targets in the cortical bone of a mature skeleton. As we learned in the first chapter, a pack of continuously proliferating spindle-shaped immature osteoprogenitor cells (i.e., juvenile osteoblasts) capable of expressing RANKL chases the BMU osteoclasts as the osteoclasts are pushing the PTHR1-bearing lining cells aside and tunnelling through the exposed bone, and getting rid of, a patch of microcracked bone. The osteoprogenitor cells put a layer of mature osteoprogenitors around the tunnel wall. These mature osteoprogenitors switch off and dismantle their cell-cycle machinery and become PTHR1-bearing preosteoblasts that mature into "smart" receptor-bearing osteoblasts with primary cilia to measure, and respond to fluctuations in, the components and movements of the extracellular fluid (Tonna and Lampen, 1972) and start laying down the first of 7 or 8 pairs of lamellae around the tunnel wall to fill it in but they save space for the blood vessel's channel which makes a new osteonal central canal. These nearly mature and mature osteoblasts plus the surrounding lining cells are the targets of injected PTH or pill-delivered PTH diffusing from the blood vessel in the center of the tunnel (fig. 20). Presumably, the several growth factors from the PTH-stimulated osteoblasts will "contaminate or dope" the new bone, diffuse down through the stack of cells and stimulate the proliferation of the receptorless outer immature osteoprogenitors, and get into the blood to attract more progenitors to the construction site. PTH-induced factors diffusing out of the stimulated lining cells into the more extensive bone marrow and marrow sinusoids on the endosteal and trabecular surfaces should have greater overall impact because the products of the PTH-stimulated, PTHR1-expressing osteoblastic cells and bone lining cells on endosteal and trabecular surfaces will immediately reach and stimulate immature osteoprogenitor cells in the adjacent marrow which have not yet started deploying PTHR1 receptors. The fact that endosteal surfaces but not periosteal surfaces have lining cells with PTHR1 receptors (though fewer than when they were maximally receptored osteoblasts) explains why PTH stimulates endosteal but not periosteal bone growth as does growth hormone (Andreassen and Oxlund, 2000).
Autocrine PTHrP and Emerging PTHRs1—Proliferation Suppressors and Drivers of Normal Osteoblast Differentiation
And now let's look at the drivers of osteoblast differentiation. A menagerie of factors including hedgehogs (Indian and/or Sonic), Bapx 1, Msx-2 and signals from FGFR1 receptors promote a BMP-2 → BMP1B•II heterodimeric(2-component)receptor → phospho-Smad-1/5 second messenger-mediated surge of the type 2 Cbfa1/Runx-2 expression to close off the adipocyte option and start a clone of extensively proliferating "transit amplifying cells" with limited self-renewing abilitiy—the immature osteoprogenitors (Aubin, 2000, 2001; Aubin and Triffitt, 2002; Banerjee et al., 2001; Bidwell et al., 2001; Chen et al., 1998; Chung et al., 2001; Ducy, 2000; Ju et al., 2000; Karsenty, 2000; Lian and Stein, 2003; Spinella-Jaegle et al., 2001) (fig. 8 and fig. 20). It is essential to know that for the BMPR-triggered surge of phosphoSmads to switch target genes on or off, they must interact with Cbfa1/Runx-2 on the genes' promoters (Canalis et al., 2003). A premature stimulation of the normally cycle-stopping mature osteoblast genes by the type 2 Cbfa1/Runx-2 is prevented and the progenitors kept proliferating by the "homeobox" protein, Msx2, which is expressed during the skeletal growth phase and binds to and shuts down type 2 Cbfa1/Runx-2 (Dodig et al., 1999; Lian et al., 1998; Ryoo et al., 1997; Shirakabe et al., 2001). Indeed, MC3T3 preosteoblasts throw their Cbfa1/Runx-2 into the proteasome garburettor barrel before they start replicating DNA and forced hyperexpression of the Cbfa1/Runx-2 protein reduces proliferation (Galindo et al., 2003). The level of Msx-2 might be determined by the expression of antisense Msx1-AS which downregulates the constitutively expressed Msx1 and maybe also Msx2 (Blin-Wakkach et al., 2001). Overexpression of Msx-2 would keep the osteoprogenitors proliferating to maintain an optimally sized precursor pool and prevent type 2 Cbfa1/Runx-2 from driving their maturation into post-proliferative preosteobolasts and osteoblasts whereas disabling the gene would reduce osteoprogenitor proliferation and cause premature maturation (Dodig et al., 1999). But at a critical point the signals from collagen-stimulated integrins and other factors cause the appearance of another "homeobox protein", Dlx5, which downregulates Msx1 and pulls Msx2 off type 2 Cbfa1/Runx-2 which can then stimulate several key genes with the type 2 Cbfa1/Runx-2-binding, osteoblast-specifc element 2 (OSE2; the AACCACA DNA nucleotide sequence) in their promoter-signal boxes that include the osteoblastic-specific zinc finger protein transcription factor Osterix without which there can be no osteoblast maturation or bone formation (Blin-Wakkach et al., 2001; Ducy and Karsenty, 1995; Katagiri and Takahashi, 2002; Marijanovic et al., 2002; Nakashima et al., 2002; Zhang et al., 2002a). But Dlx5 also inhibits the expression of the osteocalcin gene by binding to a single "homeobox"-binding site in the gene's promoter-switch box (Ryoo et al., 1997). Why this seeming anti-differentiation action? As in all differentiation programs, timing of appearance is all-important. So this is one of two strategies (we will meet the other one soon) to prevent type 2 Cbfa1/Runx-2 from prematurely stimulating the appearance of osteocalcin, which is expressed later on in the ostoblast maturation process to switch off and prevent excessive mineralzation (Ducy et al., 1996).
At this point, we must not forget the surprising likelihood of signals from the osteoblastic cells' neuron-like NMDA glutamate receptors being involved in driving the pivotal expression of Cbfa1/Runx-2 (Hinoi et al., 2003). This possibly hugely important role of glutamate receptors has been hidden by the large amounts of glutamate that saturate the glutamate receptors of osteoblastic cells in the standard cell culture media (Hinoi et al., 2003, 2004). If we can somehow peel away the other functions of this multifunctional amino acid, we can expect to see another part of the machinery that starts and drives osteoblast maturation.
One of the type 2 Cbfa1/Runx-2's major targets is the PTHR1 receptor gene, specifically its P3 promoter (it has three such switch boxes) to which the protein binds as effectively as it does to the osteocalcin promoter-switch box (Karpieren et al., 2000). The now mature osteoprogenitors start expressing PTHrP and putting PTHR1 receptors out on their surfaces (Aubin, 2000, 2001; Aubin and Triffitt, 2002) (fig. 8 and fig. 20). The appearance of PTHrP and PTHR1 receptors coincides with the secretion of autocrine/paracrine PTHrP, the PTH-(1-34)-like N-terminus ("nose") of which both activates and stimulates the further expression of PTHR1 receptors. The signals from the PTHR1 receptors coincide with the shut down of the proliferative machinery, and start the procession of mature osteoblast-specific gene expressions (Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002 (fig. 8 and fig. 20).
To become a mature osteoblast, a progenitor may use its PTHrP and PTHR1s to make the promoter-switch boxes of key osteoblast-specific genes accessible to transcription factors by bending the DNA to bring originally separated regions together to make functional switch boxes (Alvarez et al., 1998). This is done with "architectural" transcription factors, such as NmP4, which we met in Chapter 2i. They can bind to AT-rich regions of the minor groove of DNA and twist and/or bend the DNA to make functional gene promoters (switch boxes), for example by binding to an AT-rich region (between nucleotides 1971-1574 and -1555) which is upstream from the coding region of the collagen-I gene (Alvarez et al., 1998; Feister et al., 2000). Indeed PTH, and therefore autocrine PTHrP, causes NmP4 to get into the nucleus where it bends and twists the collagen-I gene's promoter-switch box into a functional switch for the transcribing machinery (Alvarez et al., 1998).
Since signals from activated PTHR1 receptors cause substantial cytoskeletal shifts as well as potentiate cells' responses to strain, they may use the same c-Src-triggered, Cas-Crk-mediated, nucleus-seeking mechanosomes that we talked about in Chapter 2i that are assembled at the surface of strained osteocytes and then go to the nucleus to stimulate osteoblast-specific genes (Carvalho et al., 1994; Eagan et al., 1991; Goligorsky et al., 1986; Krempien et al., 1978; Lomri and Marie, 1990; Pavalko et al., 2003). In other words, PTHR1 signaling driven by autocrine PTHrP in preosteoblasts mimics strain deformation by sending architectural transcription factors such as NmP4 into the nucleus to bend and twist the promoters of osteoblast-specific genes into functional configurations that can receive and be activated by classical transcription factors.
A remarkable example of the ability of the PTH-triggered "shotgun" signals (fig. 19) to switch on some of the same genes they trigger in osteoblasts even in completely inappropriate cells has been reported by Hefferan et al. (2002). They found that blood leukocytes from patients given 400 IU (activity units) of hPTH-(1-34)/day were expressing typical osteoblast genes such as those for alkaline phosphatase, BMPs-2,-4,-6, collagen I, and IGF-I. This is a striking example of the leukocyte genome being turned on by PTH exactly like the peptide turns on the osteoblast's genome, but, of course, without being able to establish a functioning, osteoblast proteome in such an alien cell body. It also confirms my Group's now ancient, very pioneering, but equally very premature demonstrations of the ability of PTH to control and stimulate hemopoiesis and thymic lymphocyte generation (e.g., Perris et al., 1967, 1971; Whitfield et al., 1973, 1998b).
Zuscik et al. (2002) have shown that the expression of PTHrP's gene is negatively affected by the nuclear Ca2+ content. So the expression of the gene starting in preosteoblasts requires that the nuclear Ca2+ level be low enough. Unfortunately, we don't know how the nuclear Ca2+ level changes during the the progression from immature osteoprogenitor to osteoblast. But maybe when something such as APRO (TIS21), whom I will introduce further on, finally stops a mature osteoprogenitor proliferating, the Ca2+ transients which drive transit through the key stages of the cell cycle would stop, the nuclear Ca2+ level would drop, and the expression of the PTHrP gene would be unleashed. But it also seems that the secretion of the emerging whole PTHrP molecule and its cleaved components would be driven by the very high Ca2+ concentration (e.g., 40 mM!) that hits the preosteoblasts and osteoblasts when they start working in the osteoclast tunnels and trenches. This Ca2+ would activate the osteoblastic cells' Ca2+-sensing receptors, which have their C-tails tied along with MAPK to filamin-A in signaling clusters in caveolar "message centers" (Awata et al., 2001; Hjälm et al., 2001). The signals from these clusters stimulate the PKCs/MEK1,2/p38MAP kinases-mediated synthesis of PTHrP (MacLeod et al., 2003; Sanders et al., 2001; Tfelt-Hansen et al., 2002). Besides driving osteoblast differentiation, signals from PTHR1 receptors activated by the secreted PTHrP would protect the cells from being killed by the large amount of phosphate that accompanies the Ca2+ (Adams et al., 2001; Allen et al., 2002; Meleti et al., 2000).
In Chapter 2i, we learned that strain turns on the PTHrP gene, possibly by opening TREK stretch-sensitive K+ channels in osteoblastic cells (X. Chen et al., 2003). Therefore, the osteogenic PTHrP (Whitfield et al., 1997d) is very likely a principal mediator of loading-induced bone growth and maintenance. In other words, straining osteoblastic cells causes them to make and secrete PTHrP, much like pulling a skunk's tail triggers a spray of pungent perfume.
Indeed, the autocrine stimulation of preosteoblasts by their own PTHR1 signaling and secreted PTHrP is certainly a critical factor driving a progenitor along the road to functional osteoblasthood. Knocking out the PTHrP gene in mice impairs osteoblast development and bone formation (Miao et al., 2002). Schiller et al. (1999) have given an example of the coincidence of the emergence of PTHR1 receptors with proliferative shutdown, which was followed about 5 hours later by the initiation of bone nodule formation in growing cultures of MC3T3-E1 murine calvarial (skull bone) preosteoblasts. The cyclic AMP signals from the PTHR1 receptors activated by the emerging autocrine/paracrine PTHrP shut down the cell cycle machinery and further increase the expression of PTHR1 (Lu et al., 2002). They also push Cbfa1/Runx-2 into the cell nucleus (Fujita et al., 1999, 2001a) and there into the arms of pRb, in this case a cotransactivator. After being pushed into the nucleus by the PTHR1 signals, Cbfa1/Runx 2 doesn't just wander aimlessly around looking for pRb and client genes. It has a nuclear matrix-targeting sequence (NMTS) in its C-tail (amino acids 397-434) that cause it to seek out specific nuclear matrix sites, where it can form transcription complexes with suitably addressed osteoblast genes (Zaidi et al., 2001).The Cbfa1/Runx-2•pRb complex then promotes the effectiveness of the antiproliferative, cycle-blocking p21CIP and p27KIP 1 proteins and stimulates the parade of gene expressions which ends with a mature fully mineralized matrix, loaded with various growth factors from the earlier stages (Lian et al., 1998; Stein et al., 1996; Thomas et al., 2001). The absence of the functional proliferation-restraining hypophosphorylated pRb that would otherwise work with Cbfa1/Runx-2 cotransactivator to open the mature post-proliferative osteoblast gene file explains the 500-times higher incidence of osteosarcoma associated with the disabled pRb in retinoblastoma patients (Gurney et al., 1995; Thomas et al., 2001).
This pivotal PTH/PTHrP- and Cbfa1/Runx-2-driven step in the progression of an osteoprogenitor cell such as MC3T3-E1 toward bone-making osteoblast includes a brief expression of the APRO1 (Antiproliferation1 or TIS21(mouse)/BTG2 (human) or PC3(rat)) gene (Matsuda et al., 2001; Raouf and Seth, 2002; Tirone, 2001) (fig. 8,fig. 20, and fig. 21), the product of which blocks the G1 build-up to chromosome replication (Matsuda et al., 2001; Guardavaccaro et al., 2000; Tirone, 2001). The highly labile APRO1 (TIS21) protein, together with the Cbfa1/Runx-2•pRb complex's enhancement of the effectiveness of the p21CIP and p27Kip cell cycle blockers, prevents the transcription of the cyclin D1 gene and also directly interacts with, and inhibits, the CDK4•cyclin D1 protein kinase (Guardavaccaro et al., 2000; Matsuda et al., 2001; Tirone, 2001). Thus, there is no CDK4•cyclin D1 to start the G1 build up to chromosome replication by hyperphosphorylating and breaking the pRb lock on the cell cycle machinery. Unphosphorylated, pRB locks the cycle machinery by shutting down the activator of cell cycle genes—the E2F protein—and joins with Cbfa1/Runx-2 to drive post-proliferative osteoblast gene expressions (Harbour and Dean, 2000; Whitfield and Chakravarthy, 2001). If there is no APRO1, pRb can be hyperphosphorylated by CDK4•cyclin D1, which makes it let go of E2F, which then collaborates with CDK4•cyclin D1's successor, the CDK2•cyclin E protein kinase, to turn on the genes for key chromosome-replicating proteins (Harbour and Dean, 2000; Matsuda et al., 2001; Tirone, 2001; Whitfield and Chakravarthy, 2001). However, APRO1 is doubly effective—it can also reduce the expression of cyclin E and consequently the CDK2•cyclin E protein kinase (Tirone, 2001).
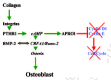
This cycle-stopping expression of APRO1 (TIS21) is by no means unique to preosteoblasts. Still in the context of bone, it is also involved in osteoclast differentiation, where it is induced by RANKL-induced RANK receptor signaling to stop osteoclast precursors proliferating as a prelude to terminal maturation (S.W. Lee et al., 2002). But it also seems to be part of a widespread mechanism for shifting a proliferating precursor cell's progeny into a post-proliferative stage of development. Thus, for example, a mouse or rat neuroectodermal cell in the ventricular zone of the neural tube that has reached the point of generating a post-proliferative neuron switches on its gene for the APRO1 (TIS21) protein (Iacopetti et al., 1994, 1999; Tirone, 2001). One of its daughters will shut her (TIS21) gene off, but will still have some of mother's APRO1 (TIS21) protein and keep it during migration and the initial stage of neuronal differentiation. The appearance of APRO1 (TIS21), the resulting lack of CDK4•cyclin D1 kinase and hence the failure of cycle-suppressing pRb's hyperphosphorylation and inactivation, marks a neuron's birthday which over the subsequent decades the cell would celebrate as its "APRO1 (TIS21) day" (Iacopetti et al., 1994; Tirone, 2001). Knocking out the cycle-locking pRB gene prevents neuronal differentiation and keeps the permanently, cycle-unlocked neuroblasts proliferating, which ultimately results in a massive apoptosis of accumulating neuroblasts (Guardavaccaro et al., 2000; Lee et al., 1994). APRO1(TIS21) also associates with mCaf1, a component of the cell's mRNA deadenylase (Rouault et al., 1998; Tucker et al., 2001). This could mean that when the brief reign of APRO1(TIS21) stops the cell cycling, it ensures that the cycle block will continue by driving the deadenylation and breakdown of the cycle-related mRNAs, which clears the way for reloading the cell with mRNA messages from differentiation-related genes. By the time the APRO1 (TIS21) protein has gone, the cell, be it a neuron or osteoblast, is committed to becoming an irreversibly differentiated neuron or osteoblast.
One might ask whether preosteoblasts use the recently discovered gene-censoring siRNA (small interfering RNA) mechanism to sustain the shut down of the cycle engine. Such siRNAs could selectively cause the cleavage, or at least block the translation, of the mRNA transcripts of key cell cycle genes (Lau and Bartel, 2003).
APRO1(TIS21) is only one of a widely used set of genes the products of which, the maturation or "M" team, turn off the cell proliferation-drivers and irreversibly lock MC3T3-E1 cells into terminal maturation (fig. 20). The quiescence-induction-associated Q (quiescin) 6 sulfhydryl oxidase protein also peaks and drops along with APRO1 (TIS21) (Raouf and Seth, 2002). Indeed, Q6 expression is suppressed in tumor cells (Coppock et al., 1993, 2000). Another member of the M team is HAX-1, which is part of the EDC (epithelial differentiation gene) complex (e.g., involucrin, loricrin, S100s, trichohyalin) on human chromosome 1q21-22 (Marenholz et al., 2001; Raouf and Seth, 2002; Whitfield and Chakravarthy, 2001) (fig. 20). The Ca2+-triggered expression of this EDC gene complex is also at the heart of the terminal, apopotosis-like differentiation (i.e., diffpoptosis) of skin keratinocytes into keratin-loaded cadavers (Whitfield and Chakrvarthy, 2001).
In summary, the initiation of normal osteoblastic maturation might go as follows. The signals from collagen-stimulated integrins trigger the expression of PTHR1 receptor and PTHrP to stimulate the emerging PTHR1 receptors to give cyclic AMP surges for triggering the brief appearance of the very labile APRO1 (Fletcher et al., 1991; Tirone, 2001), which inhibits the expression of both cyclins D1 and E and the activity of any residual CDK4/6•cyclin D1 and CDK2•cyclin E kinases (fig. 21). And they drive Cbfa1/Runx-2 into the nucleus (fig. 21). The Ca2+ surge also caused by PTHR1 signaling may also stimulate the expression of HPX-1, which is another part of the differentiation machinery. The lack of CDK4/6•cyclin D1 prevents the phosphorylation of pRB, which, because of this won't let go of the replication genes' transactivator, E2F, but it can grab Cbfa1/Runx-2 by its C-tail to form a complex that promotes the expression of Cbfa1/Runx-2's target osteoblast-specific genes. Meanwhile, the briefly appearing APRO1 also de-adenylates and thus eliminates the mRNAs for the various parts of the cell cycle machinery.
The take-home message from all of this is—when APRO1 vanishes, the cell's key replication-driver genes are silent with their promoter-switch boxes buried in chromatin condensed by having CpG dinucleotide islands methylated and/or their histones deacetylated, but the cell emerges with a reconfigured nucleus with bone-making genes, such as osteocalcin, with their promoter-switch boxes now accessible for switching by having their chromatin decondensed by demethylation of CpG islands and their histones acetylated (Vilagra et al., 2002).
But (that ominous word again!) Fujita et al. (2002) have claimed that PTHR1 signaling does stimulate osteoblastic cell proliferation because they found that hPTH-(1-34) strongly stimulated cyclic AMP accumulation but only slightly stimulated (about 50%) proliferation of ATDC5, MC3T3-E1, and B-Raf-transformed MG63 bone cells. The proliferogenic mechanism appeared to have to be triggered by the cyclic AMP binding directly to and activating Epac, a cyclic AMP-GEF (Guanosine nucleotide Exchange Factor) (de Rooij et al., 2000; Stork and Schmitt, 2002; Zwartkruis and Bos, 1999). Cyclic AMP•Epac stimulates the exchange of GTP for GDP on GDP•Rap1, a Ras-like GTPase (Zwartkruis and Bos, 1999). GTP•Rap1 stimulates B-Raf protein kinase that sets off the mitogenic MEK1/2•ERK1/2 cascade (Fujita et al., 2002; Pearson et al., 2001; Stork and Schmitt, 2002; Zwartkruis and Bos, 1999). And Tawfeek (2002) has since shown that a PTH such as [Gly1,Arg 19]hPTH-(1-28) which only stimulates adenylyl cyclase (Yang et al., 2004) does indeed stimulate the MEK•ERK cascade, which we would expect from a generation of cyclic AMP•Epac rather than a burst of PKA activity, which would have instead inhibited the MEK•ERK cascade. On the other hand, Mahon and Segre (2002) have found that the PTHR1 receptor's C-tail connection to NHERF-2 is needed for hPTH-(1-34) to stimulate the MEK mechanism in PS120 fibroblasts by a pathway not involving cyclic AMP (i.e., it is not stimulated by the AC-stimulator forskolin) or PKC. Significant though this variously triggered proliferogenic MEK-ERK cascade seems to be for PTH's actions in vitro, it is probably irrelevant to how PTH, PTHrP, and PTHR1 signalling actually stimulate normal osteoblast accumulation in a bone. Neoplastic or semi-neoplastic cells of various established bone cell lines, such as those used by Fujita et al. (2002), have their normally reciprocally controlled, mutually exclusive proliferogenic and maturation mechanisms only more or less disconnected. It is far more likely that the complex interactions of the cyclic AMP from activated PTHR1 receptors collaborate with APRO1 to permanently shut down the cell cycle machinery in normal bone cells.
PTHR1 Signals Turn on the Genes for Making Osteoblasts
Back in Section iiib, we learned that signals from PTHR1 can, like strain, load the nucleus with architectural transcription factors such as Nmp4 that bind target sites on chromosomes to produce genes with functionally reconfigured promoter-switch boxes accessible to being switched on by appropriate classical transcription factors. The accessibility of genes to these transcription factors driven into the nucleus by short bursts of PTHR1 signaling is also likely to need the brief (6-24 h) bursts of the enzyme UBP41 that could snip the ubiquitin off the nucleosomes' histone H2B, which has until now enhanced gene-silencing chromatin coiling by promoting the methylation of certain lysines equivalent to the lysine 79 in the "non-tail" core of the yeast nucleosomes' H3 histones (Briggs et al., 2002; Latchman, 2004; Miles et al., 2002; Osley, 2004; Sun and Allis, 2002). (There is also evidence for deubiquitination of lysine 4 in the H3 tail being involved in turning off certain genes (Osley, 2004)). As a result, the chromatin-uncoiling by the PTHR1-stimuated UBP41 exposes to transcription factors the promoters of the genes for factors such as FGF-2, FGF receptors and IGF-I, and further stimulate the expression of the whole PTHrP chain and one or more of its chopped-out cytokine components (Amizuka et al., 1996; Goltzman and White, 2000; Kartsogiannis et al., 1997; Walsh et al., 1997; Zhang et al., 1995) (fig. 20), and TGF-βs (TGF-β1 and β2 if the PTH is hPTH-(1-34) that stimulates adenylyl cyclase and membrane-associated PKC activity or only TGF-β2 if the PTH is hPTH-(1-31)NH2 that seems only to stimulate adenylyl cyclase in human osteoblasts (Wu and Kumar, 2000)). I call these PTHR1-induced factors "missionaries" because they carry the PTH/PTHrP message, to multiply and get ready for bone making, to progenitors that are too young to have PTHR1 receptors. Some of these factors will be locked up in the new matrix and will not be released until the osteoclasts of a future microcrack-repairing BMU dig them out while digging a new tunnel or trench.
The production of mRNA for PTHrP-(1-139), -(1-141), and -(1-173) by alternate splicing (i.e., cutting and pasting) of the PTHrP gene transcripts in primary human osteoblasts increases a dramatic 38-fold by 45 to 90 minutes after hPTH-(1-34) is added to the culture medium (Walsh et al., 1997). While we know almost nothing about the contributions of PTHrP and its autocrine/paracrine components to mature osteoblast activity and the osteogenic response, there are reasons to believe that they are important. PTHrP's PTH-like 1-34 N-terminal region would bind to and activate the PTHR1 receptors, and through them adenylyl cyclase, as efficiently as a PTH (see Goltzman and White (2000), Lam et al. (1999), and Whitfield et al. (1998a) for reviews) (fig. 18). Indeed, because circulating PTH fragments with N-termini are normally rare, autocrine PTHrP must be the osteoblasts' main AC activator. But unlike PTH, the whole PTHrP molecule or a PTHrP fragment with the 85-107 NTS (nuclear-targeting sequence (Steggerda and Paschal, 2002)) that is either made in the cell or rides into the cell from the surface on its PTHR1 receptor can function as an ‘intrakine’ or "intracrine" factor that using its NLS and NES (nuclear export sequence) signal sequences is shuttled back and forth between the cytoplasm and nucleus (Aarts et al., 1999, 2001; Goltzman and White, 2000; Jans et al., 2003; Lam et al., 1999, 2000, 2002; Re, 2002a, 2002b; Watson et al., 2000).
But let's take a closer look at this ancient string of cytokines. When its mRNA is translated into a protein with a so-called secretory signal sequence, it is directed to the Golgi apparatus, where it is put into the secretory machinery. But the mRNA can also be translated by the ribosomes from an alternative translation-start site into a protein without a secretory signal sequence (Jans et al., 2003). This protein is not fed into the secretory machinery. Instead, it binds by its NLS sequence to the nuclear cargo carrier importin-β1 to form a complex that links to a microtubular trackway, where it is pulled by a dynein motor to the mouth of a pore in the nuclear envelope and handed over to the pore channel transport machinery. The complex is then ratcheted along the pore channel by series of dockings to so-called nucleoporins ("nups") lining the pore channel and then dumped into the nucleus, where it is separted from the importin carrier when the importin binds avidly to Ran•GTP and homes especially onto the nucleolus, where it can directly affect nuclear functions such as the ribosome-making machinery (Fried and Kutay, 2003; Jans et al., 2003; Lam et al., 2002).
It seems likely that this nuclear intrakine/intracrine stimulation would happen only in the mature, postmitotic preosteoblast or osteoblast because the cyclin-dependent protein kinases such as CDK4•cyclin D1 and CDK2•cyclin E that drive the cell cycle would disable the NTS by phosphorylating Thr85 (Goltzman and White, 2000; Lam et al., 1999, 2000). Once in the nucleus and nucleolus, the peptide targets ribosome production and does its ancient job of coordinating the protein-synthesizing machinery with the signals from its activated surface receptors (Amling et al., 1997; Lam et al., 2000; Re, 2002a, 2002b). But it also seems likely that the nuclear PTHrP stimulates certain genes, among them the genes for the anti-apoptosis Bcl-2 and Bcl-XL proteins (Tovar-Sepulveda et al., 2002), that would prolong the cell's matrix-making activity by preventing it from killing itself by apoptosis (Antonsson and Martinou, 2000; Fadeel et al., 1999) (fig. 20). But in murine MC3T3-E1 osteoblasts, the PTHR1 receptor itself is also loaded into the nucleus during the late G1 phase of the cell cycle (when the cyclin-dependent protein kinases are active and would stop NTS-bearing PTHrP from entering the nucleus) and then stays there throughout the rest of the cycle (Watson et al., 2000) (fig. 18). Therefore, the PTHR1 receptor either alone or with a N-terminal fragment lacking the Thr85-containing region seems to be able to get into the preosteoblast's nucleus to carry out some job related to DNA replication, and in this it resembles the transcriptional activity of the EGF receptor (Lin et al., 2001; Ni et al., 2001).
PTHrP is not a hormone (i.e., it does not circulate throughout noncancer-bearing mammalian bodies as it does in fish, for example) or a single cytokine. It is really a string of different cytokines with 8 endoproteolytic consensus sites marking their borders where prohormone thiol protease and prohormone convertases can cut them out to operate as individual cytokines, each with its own receptor and specific mission (Deftos et al., 2001; Hook et al., 2001). One of these cytokines is the phospholipase-C-activating PTHrP-(67-86), the role of which in bone is unknown (Orloff et al., 1995). But another of these is hPTHrP-(107-139) ("osteostatin"), with its highly conserved 107-111 (i.e., Thr(T)107-Arg(R)108-Ser(S)109-Ala(A)110-Trp(W)111) region, that by itself can stimulate the proliferation of cultured fetal rat osteoblasts (Cornish et al., 1999) and possibly bone formation in 7- to 8-month-old OVXed rats (Roufflet et al., 1994). Moreover, concentrations of PTHrP-(107-139) and PTHrP-(107-111) as low as 10-15 and 10-13 M inhibit osteoclast generation and activity in vivo and in vitro (hence the name "osteostatin") (Fenton et al., 1991; Cornish et al., 1997; Zheng et al., 1994). And PTHrP-(107-139) is angiogenic—it could collaborate with the angiogenic action of leptin pouring out of hypertrophic chondrocytes (Kume et al., 2002) by stimulating human osteoblasts to make the angiogenic VEGFs, which would provide the blood vessels needed to feed the new bone being made in a trabecular/endocortical blister or cortical tunnel (Esbrit et al., 2000). And it might be the PTHrP-induced modulator of vascular invasion into cartilage that does not work through the PTHR1 receptor (Lanske et al., 1999). These fragments operate by stimulating PKCs on membrane scaffolds, not by activating PTHR1 but by activating an apparently very high-affinity "TRSAW" (Thr(T)107-Arg(R)108-Ser(S)109-ALA(A)110-Trp(W)111) receptor (Cuthbertson et al., 1999; Moonga and Dempster, 1995; Whitfield et al., 1996a, 1998a).
But there is something else lurking in the PTHrP string of factors which when it is cut out can drive bone formation. PTHrP's 8-11 sequence (LHDK) is virtually identical to endothelin-1's residues 6-9 (LMDK), which means that PTHrP-(8-11) can stimulate the endothelin A receptor (Mohammad et al., 2003). And endothelin-1 concentrations as low as 10-10 do indeed stimulate both proliferation and differentiation of the endothelin-A receptor-expressing osteoblastic cells in rat calvarial cell cultures (von Schroeder et al., 2003). Moreover, stimulating the endothelin A receptor on mouse calvarial cells with either endothelin-1 or PTHrP-(8-11) strongly stimulates bone formation (Mohammad et al., 2003). Since endothelin-1 is made by vascular endothelial cells, this means, for example, that the blood vessels trailing behind the osteoclasts tunneling through bone can use it to stimulate osteoblasts to mature and start plastering new bone on the tunnel wall.
You may remember that stromal cells are put on the osteoblast pathway when sonic hedgehog (Shh) stimulates them via FGF-4 to express BMP (bone morphogenic protein)-2 that stimulates them to express type 2 Cbfa1/Runx-2 (Ducy, 2000; Karsenty, 2000a; Katagiri and Takahashi, 2002; Komori, 2000) (fig. 22). As we shall see below, Cbfa1/Runx-2 is also involved in the driving of chondrocyte maturation and run-down of the prehypertrophic and hypertrophic chondrocytes cells to mineralization and coordinating this with the induction of osteoblasts in perichondrium/periosteum to make bone on calcified cartilage scaffolds (de Crombrugge et al., 2001). Cbfa1/Runx-2 interacts with the R-Smads specifically from activated BMPR-1B receptors to switch on the Osterix gene, which then steers the cell irrevocably onto the osteoblast pathway (Katagiri and Takahashi, 2002; Nakashima et al., 2002; Yagi et al., 2003). Another hedgehog—Indian hedgehog (Ihh)—produced by prehypertrophic and hypertrophic chondrocytes together with BMP-2, BMP-6, and Cbfa1/Runx-2 stimulates differentiation of a distinct population of perichondrial cells into osteoblasts in the collar of the developing bone (Chung et al., 2001; Karsenty, 2001). The osteoblast-determining potency of the Cbfa1/Runx-2 gene product is illustrated by the fact that forcing nonosteoblastic, primary fibroblasts to make it causes them to switch on their osteoblastic genes (Ducy, 2000; Lian and Stein, 2003). And just overexpressing the Cbfa1/Runx-2 gene in an adenovirus construct is enough to stimulate bone formation in a neonatal rat metatarsal organ culture (Krishnan et al., 2003).
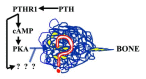
As expected from this, one of the osteogenically determining things an injected adenylyl cyclase-stimulating osteogenic PTH does is push progenitor cells along the osteogenic path by stimulating them to express more PTHR1 receptors (Lu et al., 2001; von Stechow et al., 2003), and via bursts of PTHR1-driven adenylyl cyclase activity turn up the volume of Cbfa1/Runx-2 gene expression and the loading of the cells' nuclei with the Cbfa1/Runx-2 transactivator (Fujita et al., 2001a, 2001b; Krishnan et al., 2003; Moore et al., 2000; Selvamurugan et al., 2000) (fig. 22 and fig. 23). Cbfa1/Runx-2 has a NMTS (nuclear matrix-targeting sequence) domain in its C-tail (Zaidi et al., 2001) and a DNA-binding ‘Runt’ domain (Ducy, 2000; Ito, 1999). With its tail stuck to the matrix, Cbfa1/Runx-2 grabs the so-called OCE2 upstream sites of still silent osteoblast-specific genes such as the genes for ALP (alkaline phosphatase), BSP (bone sialoprotein), osteocalcin, osteopontin, OPG, proα1(I) and proa2(I) procollagen, PTHR1, and type I TGF-β receptor (Franceschi, 1999; Karperien et al., 2000; Kern et al., 2001; Komori, 2000) (fig. 22). Thus, Cbfa1/Runx-2 is one of the DNA-benders and twisters that pins these key genes to sites on the nuclear matrix containing the RNA-polymerase II-gene-transcribing machine (Lian and Stein, 2003; Turner, 2001; Vilagra et al., 2002). These now primed genes with their configured promoters exposed by being pinned down by Cbfa1/Runx-2 stay quiet until pRb and other classical stage-specific transcription factors appear to form transcription-activating multicomponent enhanceosomes that can switch on the transcription printing press (Turner, 2001). And the expression of another gene, the gene encoding the zinc-finger-containing transactivator OSX (osterix), which, as you will remember, is a "must" for the subsequent maturation of the cell into an osteoblast (Katagiri and Takahashi, 2002; Nakashima et al., 2002; Yagi et al., 2003; Zhang et al., 2002a) (fig. 22).
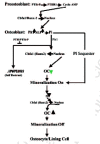
The "mature" or late osteoprogenitors emerging from the inter-trabecular pool of osteoprogenitors and just starting their spurt of PTHR1 receptor expression (one of the global markers of osteoblasts in all parts of the skeleton) and PTHR1-activating PTHrP (fig. 8) can go either directly or from the red marrow's sinusoidal vasculature into trenches dug into trabecular surfaces and the nearby cortical endosteum (Bianco and Riminucci, 1998). Presumably much less bone will be made at fatty marrow sites, as happens with FGF-2 treatment (Pun et al., 2001). However, they can reach cortical bone only from the marrow blood vessels after the cells in the trabeculae have had the first grab at them, which is one of the reasons why the adenylyl cyclase-stimulating PTHs are best at stimulating the growth of trabeculae-rich bones such as the vertebrae. Another reason for the PTHs' "trabeculophilia" besides the peptide first reaching the trabeculae after its injection or ingestion is that, except for their PTHR1 receptors, trabecular osteoblasts differ from cortical osteoblasts in their repertoire of expressed genes and their transcripts (i.e., their "transcriptomes") (Aubin, 2000, 2001; Aubin and Triffitt, 2002; Candeliere et al., 2001).
When the osteoprogenitor cells have arrived at the swept-up tunnel wall or trench and laid enough collagen on it, the signals from autocrine/paracrine PTHrP-activated PTHR1 receptors, the expressions of which have been stimulated by collagen-bound/activated integrins, have led the cells' "M teams" to shut down the cycle-driving machinery and enabled Cbfa1/Runx-2 to up-regulate the differentiation-related genes (fig. 22). But although they may look alike and make bone, the resulting post-mitotic mature osteoblasts do not have the same gene expression profile. The cells' gene expression profiles are specifically tailored to meet the different needs for working in different regions of the skeleton and even in different parts of a particular bone. Thus, for example, the relative expressions of bone sialoprotein, osteopontin, osteocalcin, and PTHrP at the mRNA and protein levels depend on which part of a developing bone (e.g., endocranial or ectocranial surface of mouse calvaria) the cell is located as well as who its immediate neighbors are and consequently the microenvironment in which it finds itself. In another example, osteoblasts in rat calvaria express glutamate NMDA receptor/channels with subunits NR2A, NR2B and NR2D, but not NR2C, while femoral osteoblasts express NR2C (Itzstein et al., 2001). But all osteoblasts, no matter where they are or who their neighbors are, express the genes for the PTHR1 receptor, type I collagen, and alkaline phosphatase (Candeliere et al., 2001). The products of these three genes are absolutely needed—they are the sine qua nons for becoming a mature, post-proliferative, matrix-making, and mineralizing osteoblast.
The PTHR1-expressing preosteoblasts and mature osteoblasts in modeling clusters making new bone and filling excavated microcracks in mature skeletons are the first or prime targets of injected PTH (K. Lee et al., 1994) (fig. 8). But it must be remembered that the signals from the PTHR1 receptors cannot stimulate the proliferation of maturing preosteoblasts and mature osteoblasts that have been proliferatively disabled by their PTH/PTHrP-induced M-teams' APRO1(TIS21)s. By the time they have got rid of their cell cycle machinery and become working osteoblasts, they have greatly (10-fold) increased the expression of PTHR1 receptors, which probably enables the autocrine/paracrine N-terminal PTHrPs that they have made and secreted to give themselves and their neighbors the cyclic AMP signals needed to drive their further maturation and bone-building (Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002).
Each daily PTH-triggered cyclic AMP transient releases active PKA catalytic subunits from inactive R•C holoenzymes (i.e., regulatory subunit–catalytic subunit) that surge from the cytoplasm into the target cell's nucleus through the nuclear pore complexes and turn on a set of early response genes with a CRE (cyclic AMP-responsive element) in their promoters and CREB•CBP/p300 complexes (cyclic AMP responsive element-binding protein•CREB-binding protein complexes) on the CREs (Ptashne and Gann, 2002; Quinn, 2002). However, after the nuclear catalytic subunit surge peaks around 15-30 minutes in cells such as MC3T3-E1 mouse preosteoblasts and ROS 17/2.8 osteoblastic cells, the catalytic subunits are no longer personae gratae. They are grabbed by the protein kinase A inhibitor PKIγ, which, like the various PKI isoforms, has a NES (nuclear export signal) domain, and are dumped out of the nucleus (X. Chen et al., 2002, 2003; Wen et al., 1995a, 1995b; Wiley et al., 1999). This, of course, stops the immediate-early gene expressions that the catalytic subunits were stimulating (X. Chen et al., 2003). The evicted catalytic subunits reassemble inactive R•C holoenzymes in the cytoplasm. Cyclic AMP transients may also feed back to stop the PTHR1 receptor attached to β-arrestin-2 from activating adenylyl cyclase by rapidly and briefly stimulating the expression of the gene for RGS-2, a GTPase activator, which converts receptor-generated adenylyl cyclase-activating GTP•Gsα to inactive GDP•Gsα (Pierroz et al., 2003; Thirunavukkarnasu et al., 2002).
One of the earliest of the many hundreds of genes directly and indirectly booted by the cyclic AMP signals from PTHR1s are genes of the Fos family (c-fos, fosB, fra1, fra2) the products of which can associate with the products of members of the Jun family (c-jun, junB, junD) of genes to form AP-1 (activator protein-1) transcription factor complexes that target an array of genes (Demiralp et al., 2001; K. Lee et al., 1994; McCauley et al., 1997, 2001; Onyia et al., 2001a; Tyson et al., 1999). The turning on of at least one of these genes, c-fos, is essential for PTHs to stimulate bone growth (at least in mice). Thus, knocking out the c-fos gene in mice also knocks out the ability of hPTH-(1-34) to stimulate femoral bone growth (Demiralp et al., 2001).
The switching on of the c-fos gene has been used to mark the sequence of responses of the various bone cells in 4-week-old rats to subcutaneously injecte full-length hPTH-(1-84) (K. Lee et al., 1994). The c-fos gene expression in trabecular osteoblasts, and the few spindle-shaped stromal cells that made PTHR1 receptors, peaked between 15 and 30 minutes and stopped by 60 minutes. Most stromal cells did not make PTHR1 receptors, nor could they, and the osteoclasts express their c-fos genes until 60 minutes by which time the osteoblasts and the few PTHR1-expressing stromal cells had stopped. This delayed c-fos switch-on in the receptorless stromal cells is best explained by missionaries sent to the PTHR1-lacking cells by PTHR1-loaded osteoblasts. Only after the PTHR1-receptorless stromal cells had stopped expressing c-fos at 2 hours were the osteoclasts expressing the gene. The very late burst of c-fos expression in osteoclasts is likely due to RANKL flowing from the immature osteoblstic stromal cells which, unlike the densely PTHR1-receptored mature osteoblasts with their shut-down rankl genes, are the only ones that can make this osteoclastogenic factor (Atkins et al., 2003; Corral et al., 1998).
These brief PTHR1-induced pulses of cyclic AMP and both cytoplasmic and nuclear PKA catalytic subunits and the c-Fos•Jun transcription factor complexes they generate only start the bone-making machine. Daily PTH-induced cyclic AMP spikes can't directly make immature marrow osteoprogenitors or preosteoblasts and mature osteoblasts proliferate (Kostenuik et al., 1999; K. Lee et al., 1994; Y. Wang et al. 2003; reviewed by Whitfield et al., 1998b). Indeed, a striking example of this has been given by Y. Wang et al. (2003), who has reported that PTH stimulated the progression of preosteoblasts to bone nodule-making mature osteoblasts in primary calvarial osteoblast cultures without stimulating proliferation. However, PTH can stimulate the PTHR1-expressing osteoblasts that have been proliferatively switched off by the "M-team" (fig. 20) to make factors that indirectly stimulate the proliferation of the immature osteoprogenitors, which do not yet have PTHR1 receptors.
Speading the PTH Message—The Missionary Factors That Suppress Cell Suicide and Stimulate Growth to Swell the Osteoblast Workforce
It is the products of the cascades of events triggered by the primary drivers such as amphiregulin (AR), FGF-2, IGF-I growth factors, and TGF-βs that grab the wheel and steer the machine in the receptor owners as well as start the generation of osteoblasts by stimulating the receptorless osteoprogenitors (Nishida et al., 1994; Whitfield et al., 1998b) (fig. 20).
But the kick-starting PKA pulses also lengthen the working lifespans of mature osteoblasts by preventing them from committing scheduled apoptotic suicide (fig. 20). This increases the amount of bone they can make. Thus, as Gubrij et al. (2003) have shown, when osteoprogenitors can be made to constitutively overexpress an antiapoptotic protein such as Bcl-2 more of them survive to make more PTH-responsive, super-productive osteoblasts which make thicker trabeculae. The PTH/PTHR1-triggered PKA pulses' anti-apoptotic action results in part from phosphorylation and inactivation of the cells' apoptosis-initiating BAD protein, which makes BAD release the anti-apoptosis Bcl-XL from its repressive grip and the phosphorylated BAD is trapped in the cytosol bound to 14-3-3 protein and thus unable to cause cytochrome c to leak from the mitochondria and trigger apoptosis-executing caspase proteases such as caspase-3 (J.M. Adams, 2003; Bellido et al., 2001; Chao and Korsmeyer, 1998; Stanislaus et al., 2000; Yin et al., 2003). Also they can be protected from suicidal thoughts and responses, maybe by the stimulation of sphingosine kinase that produces the anti-apoptosis sphingosine phosphate, but most definitely by the stimulation of the expression of the very important IGF-I, a stimulator of the expression of the apoptosis-preventing Bcl-2 and Bcl-XL family proteins (Calvi et al., 2001; Hill et al., 1997; Johanson and Rosen, 1997; Locklin et al., 2003; Machwate et al., 1998; Pfeilschrifter et al. 1995; Pugazhenthi et al., 1999; Tovar-Sepulveda et al., 2002; Tumber et al. 2000; Virdee et al., 2000; Watson et al., 1995, 1999) (fig. 24).
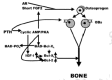
The Bcl-2 gene is also turned on directly by the PTH-triggered cyclic AMP pulses. And its Bcl-2 protein product seems to be the sine qua non for PTH's anti-apopotsis action. Thus, preventing the translation of Bcl-2 gene transcripts with Bcl-2-specific, transcript-censoring siRNA (see the popular review of siRNAs' actions by Lau and Bartel, 2003) eliminates PTH's ability to prevent dexamethasone, etoposide, or the deprivation of substrate adhesion from triggering self destruction by apoptosis in OB-6 osteoblastic cells (Ali et al., 2003). Incidentally, this anti-apoptotic action of PTH plus its ability to stimulate the proliferation of hemopoietic CFU-S stem cells, which are normally clustered close to the PTHR1-bearing endocortical lining cells and responsive to factors such as Jagged1 produced by PTH-stimulated osteoblastic cells (Calvi et al., 2004; Cui et al., 1996; Gallien-Lartigue and Carrez, 1974; Lord, 1990; Perris et al., 1971), are probably why a subcutaneous injection of 50 to 200 USP units of Eli Lilly parathyroid extract (the same extract used by Selye in the 1930s) given 5 minutes to 1 hour after whole-body irradiation with 8.0 Gys of 2000 kvp x-rays increased the 30-day survival of 1296, 300-g male hooded rats from 33% to 73% (p<<<0.001) (Rixon and Whitfield, 1961).
How does PTH stimulate the Bcl-2 gene? The gene has one cyclic AMP-targeted CREB and several Cbfa1/Runx-2 sites in its promoter, and because of these, this anti-self-destruct gene is stimulated directly by the cyclic AMP boluses from PTHR1 (Plotkin et al., 2002). Thus, a cyclic AMP spike would phosphorylate and activate CREB and drive Cbfa1/Runx-2 into the nucleus and onto the Bcl-2 gene's promoter along, with the activated phospho-CREB (Fujita et al., 2001a). The PKA catalytic subunits that also surge into the nucleus would phosphorylate, and activate and thus increase the affinity of Cbfa1/Runx-2 for its target sites on the Bcl-2 promoter (Franceschi and Xiao, 2003; Selvamurugan et al., 2000) (fig. 24 and fig. 25). The PTH-induced, apoptosis-supressing phosphorylations of BAD by PKA and the stimulation of cyclic AMP/CREB/Cbfa1/Runx-2-dependent Bcl-2 expression are transient, i.e., self-limiting (Bellido et al., 2001, 2002). One reason for this transience is the silencing and expulsion of CREB-activating PKA catalytic subunits from the nucleus by PKI, which we met three paragraphs ago. Another reason is that the PTHR1 signaling also causes Cbfa1/Runx-2 to be dumped into the proteasome shredder (Bellido et al., 2002). Thus, only well-separated short bursts of PTHR1 can maintain the anti-apoptotic action. It follows from this that turning off the shredder with anti-protease, such as lactacystin, or overexpressing and swamping the system with Cbfa1/Runx-2 should, and in fact does, prolong PTH's anti-apoptotic action (Bellido et al., 2002).
By now the reader may be getting used to de-generalizing generalizations. In the case of PTH's anti-apoptosis action, two exceptions have been reported. The first exception has been found in murine C3H10T1/2 and MC3T3-E1 cells. As expected, hPTH-(1-34) triggered cyclic AMP-protected preconfluent cells from dexamethasone-induced apoptosis, but, and this is a very big but, the cyclic AMP surge triggered apoptosis in postconfluent C3H10T1/2 and MC3T3-E1 cells (H.-L. Chen et al., 2002). The second exception was found in the distal femurs of intact young Fisher 344 and Sprague-Dawley rats. Single daily injections of hPTH-(1-34) (Forteo™) into male rats that stimulate bone growth also stimulate a wave of osteoblast apoptosis in the proliferating zone just below the growth plate and osteocyte apoptosis in the osteocyte death zone that starts promptly during the first day, peaks around days 3-7, and then gradually drops off by 28 days (Stanislaus et al., 2000). There was no detectable increase in Bcl-2 expression. However, there does seem to be an anti-apoptosis effect in other regions of the femur, as indicated by a large drop in the activities of the apoptosis-driving caspases 2, 3, and 7 in the whole metaphysis. In fact, these drops are so large that they can obscure any more limited regional increase in the caspase activities. In other words, PTH stimulates apoptosis in certain zones of the rat femur, but it inhibits it elsewhere in the bone.
The IGF-I secreted by the PTH-stimulated, longer-lived, working osteoblasts also increases the osteoblast population by serving as a missionary that delivers PTH's call to proliferate to nearby proliferatively turned-on juvenile osteoprogenitors on the walls of new osteonal tunnels under construction or to osteoprogenitors in the bone marrow or trabeculat trenches, none of which have PTHR1 receptors or are not ready yet to make their own IGF-I (Calvi et al., 2001; Kostenuik et al., 1999). The importance of IGF-I for PTH's anabolic action is dramatically indicated by the fact that "knocking out" the IGF-I gene also knocks out the ability of hPTH-(1-34) to stimulate femoral bone growth in mice as well as the fact that PTH doesn't stimulate the proliferation of osteoblasts isolated from these IGF-I-less mice, unless the cells are given exogenous IGF-I (Miyakoshi et al., 2001).
But neither PTH, nor the IGF-I it induces, can stimulate osteoprogenitor proliferation or the growth of unloaded bones, such as hindlimb tibias, (Kostenuik et al., 1999), because the lack of loading disables the responsiveness of the osteoprogenitor cells to IGF-I (Bikle et al., 1994; Kostenuik et al., 1999)! In fact, if this missionary IGF-I is to carry the PTH message to proliferate and migrate to osteoprogenitor cells through the cells' IGF-1 receptors, the cells' aVβ3 integrin signaling tethers must be being tugged if the IGF-I receptors are to be activatable (Sakata et al., 2004). It seems that the activated integrins drive the collection of separately signaling-incompetent IGF-I receptors into signaling-competent clusters at the cell surface (Miyamoto et al., 1996; Sakata et al., 2004; Zheng and Clemmons, 1998). In other words—no loading, no integrin signaling, and so no signaling-competent IGF-1 receptors! As if this is not enough, stopping the waving of the cells' solitary cilial flowmeters and silencing the squeals from tugged integrins reduce the accessibility and responsiveness of osteoblast-specific genes to Cbfa-1/Runx2 and hinder classical transcription factors from stimulating MAP kinases and pumping architectural transcription factors like NMP4 into the nucleus to reconfigure and prime these key genes for responsiveness (Franceschi and Xiao, 2003; Latchman, 2004; Parvalko et al., 2003; Whitfield, 2003a). This reduced the ability of IGF-I to stimulate the Ras-ERK1/2-Akt mechanism and with it the expression of the genes for alkaline phosphatase, a-I collagen and osteocalcin.
This need for loading and the gene accessibility and responsiveness it creates is also seen on the cellular level. In MC3T3-E1 mouse calvarial preosteoblasts, simulated low or microgravity reduces the ability of hPTH-(1-34) to stimulate the expression of the c-Fos and c-Jun transcription factors (Ontiveros et al., 2002). Unloading also stresses and thus increases the incidence of apopototic suicide in the osteoprogenitor cells from unloaded bone (Sakata et al., 2002). But osteoprogenitors are not the only cells affected. As we learned in Chapter 2, the stress of canalicular fluid stasis drives osteocytes to commit apoptotic suicide and release lysophosphatidylcholine, which attracts carrion-hungry osteoclasts to their corpses. This means that there is an overwhelming surge of osteoclastic vultures that eliminate PTH/IGF-I targets and override any possible PTH action.
However, once again we must heed the terrible "BUT" word. Sakai et al. (1999) have found that unloaded tibias in the paralyzed, neurectomized hindlimbs of ddY male mice can still respond to hPTH-(1-34) as effectively as the tibias in the sham-neurectomized contralateral limbs!! Here, of course we are dealing with a particular strain of male mice, instead of Kostenuik et al.'s female rats.
As expected, A. Nakajima et al. (2002) have found that the cells in the PTH-stimulated calluses of rat femoral fractures express IGF-I, with the peak expression occurring between 4 and 7 days. But they have concluded that IGF-I was not responsible for the stimulation of osteoprogenitor proliferation because the surge of DNA-replicating cells expressing PCNA, the DNA polymerase-δ's processive replication factor (Whitfield and Chakravarthy, 2001), did not coincide with the surge of IGF-I expression. Unfortunately, they did not look at the expression of FGF-2, which, as we are about to see, could well have been the PTH-induced missionary mediator of osteoprogenitor proliferation.
IGF-I may also mediate the seemingly strong stimulation of DNA replication in the trabecular cells and chondrocytes of the rapidly developing bones of neonatal (2-day-old) mice injected with hPTH-(28-48) (Rihani-Bisharat et al., 1998). This PTH fragment binds to PTHR1, but as stated in sections i and ii of this chapter, it does not stimulate adenylyl cyclase or bone growth in OVXed rats, but it does stimulate PLC and PKCs (Jouishomme et al., 1992; Strein, 1994). And it is these signals that trigger IGF-I expression in the receptor-bearing, still proliferatively competent, late progenitors and mature, nonproliferating preosteoblasts and osteoblasts.
But cyclic AMP-stimulated IGF-I might not be as important for stimulating bone growth in humans as it seems to be in rodents (Johanson and Rosen, 1997). Human osteoblasts make much more IGF-II than IGF-I (Mohan and Baylink, 1999). Moreover, PTHs do not stimulate human osteoblasts to make IGF-II. Indeed, cyclic AMP actually seems to inhibit IGF-II expression. But the important thing is that the PTHs and cyclic AMP do trigger some production of IGF-I in these human cells (Mohan and Baylink, 1999). It would be interesting to know whether the cells have IGF receptors on their primary cilia to detect IGFs in their surroundings.
IGF-I collaborates with the TGF-βs that are also stimulated by the PTHs (Wu and Kumar, 2000) to strongly stimulate the procollagen α1[I] gene (Vermes et al. 2001). Therefore, these autocrine (self-stimulating)/paracrine (neighbor-stimulating) factors together vigorously drive osteoblasts' matrix production.
The cyclic AMP surges also cause osteoblasts to make two very important things besides IGF-I. One of these is the long (i.e., extended), 24-kDa, NLS (nuclear localization sequence)-bearing, isoform of FGF-2 (also known as bFGF (Okada-Ban et al., 2000; Ornitz and Itoh, 2001)) (Hurley et al., 1999; Liang et al., 1999; Montero et al., 2000; Power et al., 2002; Zhang et al., 2002b) (fig. 26). But this long FGF-2 is no traveling missionary. It is an intracrine factor that stays in the cell, where it operates mainly in the nucleus of rapidly proliferating cells, such as immature osteoprogenitor cells (Olsnes et al., 2003; Re, 2002a, 2002b) (fig. 26).
How does the long FGF-2 get its NLS? The FGF-2 mRNA transcript has three CUG start codons upstream from the "canonical" or conventional AUG start codon (Vagner et al., 1995). Usually, a ribosome binds to the transcript's 5' cap and "sniffs" along the transcript until it finds an AUG start codon and then settles down to translate the message. But upstream from the first start codon is a so-called IRES (internal ribosome entry sequence) with which the transcript plugs directly into the ribosome, which forthwith starts translating the message into the long FGF-2 without looking further on for a AUG start codon. The important point here is that between the transcript's three start sites and the downstream conventional AUG, there is a NLS before the secretion signal sequence (Jans et al., 2003; Stachowski et al., 2003). So the importins-α and -β override the secretion signal sequence, grab the long, FGF-2 by its NLS, and drag it into the nucleus. There it directly or indirectly stimulates the expression of a set of genes which includes the genes for Cbfa1/Runx-2, collagen-I, IGF-I, osteocalcin, and PTHR1, as well as the ribosome genes and its own fgf-2 gene, products of which can also inhibit apoptosis (Bouche et al., 1987; Debiais et al., 2002; Olsnes et al., 2003; Re, 2002a, 2002b) (fig. 26).
The other FGF-2 is the short (18-kDA) FGF-2, which stays mainly in the producer cell's cytoplasm and, like PTHrP, is exported as an intracrine/autocrine/paracrine factor that can activate both its own cell's and neighboring cells' receptors (Olsnes et al., 2003). The short FGF-2 is translated from the transcript's conventional downstream AUG start codon and so does not have the upstream NLS and therefore cannot be kidnapped by the importins and dragged into the nucleus. But it can still get into the nucleus because it is small enough to simply diffuse through the nuclear pores (Stachowski et al., 2003). This is the missionary FGF-2, which when secreted can activate FGF receptors on its producer cell as well as on immature (i.e., PTHR1-lacking) osteoblastic progenitors, or it can ride into the cell and into the nucleus on its receptor (fig. 26). The key point here is that the signals from the activated FGF protein kinase receptors drive the differentiation of osteoprogenitors, not necessarily by increasing the expression and production of Cbfa1/Runx-2, but by stimulating the phosphorylation and therefore the activity of existing Cbfa1/Runx-2 by stimulating ERK 1/2 protein kinases (Franceschi and Xiao, 2003).
The likelihood of FGF-2 being one of the prime mediators, if not the prime mediator, of PTH's bone-forming action is indicated by hPTH-(1-34)'s failure to stimulate bone formation in mice which have had their fgf-2 gene knocked out (Hurley et al., 2002). FGF-2 also stimulates cell migration, which means that it can lure osteoprogenitors to clusters of active FGF-2-producing osteoblasts in osteonal tunnels for example. It should be also be noted that while PTH works largely in OVXed rats by increasing trabecular thickness, injecting FGF-2 by itelf into such animals can also increase trabecular number and connectivity, probably by affecting a broader group of osteoblastic cells (Lane et al., 2003).
Therefore, PTH-induced surges of FGF-2 and IGF-I drive osteoblast accumulation and bone formation in part by stimulating the proliferation of juvenile osteoprogenitors and their ultimate differentiation into PTHR1-receptor-bearing osteoblasts with extended working lives and all thoughts of committing apoptotic suicide suppressed (fig. 20). As would be expected if FGF-2 mediates PTH action, pretreating OVXed rats for 2 weeks with daily intravenous boluses (250 μg/kg) of FGF-2 before 8-weeks of daily subcutaneous injections (80 μg/kg ) of hPTH-(1-34) and (of course a further round of FGF-2 boluses, this time from the PTH-treated cells) increases trabecular thickness and connectivity more than PTH injections alone (Iwaniec et al., 2002). This is probably due to the FGF-2 pretreatment increasing the pool of PTH-responsive osteoblastic cells.
PTHR1-induced bursts of PKA activity in UMR-106-01 rat osteosarcoma cells also rapidly (e.g., within 1 hour) and massively increase (e.g., by 12 times) the expression of the gene encoding amphiregulin (AR), a member of the EGF family, without affecting the other family members (EGF itself, betacellulin, epiregulin, HB-EGF, TGF-α) of the EGF and ErbB2 receptor-activating family (Qin et al., 2003) (fig. 21). Clearly such a potent proliferogen could collaborate with the other missionaries, such as short FGF-2, to stimulate osteoprogenitor proliferation (fig. 25).
Obviously, bone formation would also be enhanced by blocking osteoclast generation. As we have seen, mature osteoblasts have their RANKL genes shut off and instead make the anti-osteoclastogenic osteoprotegerin. But these amazing cells, when stimulated by PTH, give osteoclastogenesis a double whammy by also making the anti-osteoclast IL-18 , the gene for which is stimulated by cyclic AMP/protein kinase A signals from the activated PTHR1 receptors (Raggatt et al., 2003).
But RANKL has unexpectedly, indeed astonishingly, joined the ever-growing mob of factors (fig. 19) clamoring for precedence in the PTHs' osteogenetic action. It has been suggested that RANKL, or more precisely, oligomerized (aggregated, clustered) RANKL mediates PTH's cyclic AMP-driven anabolic action on primary mouse osteoblastic cells!! Why? Well, a cluster (oligomer) of RANKLs fused to the enzyme GST (glutathione-S-transferase) as a platform stimulates bone formation when injected into mice (Faccio et al., 2003). This cluster of RANKLs bound to RANK receptors stays on the surface of murine calvarial osteoblasts much longer than single molecules of RANKL bound to RANK receptors (Faccio et al., 2003)!!! The oligomerized RANKL stays there long enough to drive RANK receptor signaling that is sufficiently prolonged to stimulate collagen production via the ERK1/2 mechanism, which according to Franceschi and Xiao (2003) would among other things, phosphorylate and activate Cbfa1/Runx2. The mediation of PTH's anabolic action in mice by oligomeric RANKL•RANK complexes is also strongly indicated by the ability of osteoprotegerin to prevent PTH action (Faccio et al., 2003). But the question is what cells are stimulated by a PTH to make RANKL oligomers? Since mature osteoblasts' RANKL genes are locked shut by having CpG islands in their promoter-switch boxes methylated, the RANKL-making targets of PTH would have to be immature osteoblastic cells. However, both mouse osteoblasts and human osteoblasts (Reinholz et al., 2002) appear to have the RANK receptors needed to respond to RANKL, wherever it comes from.
There may be another unexpected source of missionary growth factors with which PTH boluses could drive osteoblast accumulation—developing osteoclasts! Kubota et al. (2002) have found that RANKL-stimulated developing osteoclasts to make PDGF (platelet-derived growth factor)-BB, which prevents MC3T3-E1 semi-transformed preosteoblasts from maturing by keeping them in the cycling mode. Therefore, the PDGF-BB from osteoclasts that have been induced to develop by RANKL from PTH-stimulated immature osteoblastic stromal cells could stimulate the proliferation of nearby osteoprogenitors. But this interaction would not last very long because the stimulated osteoprogenitors would increasingly make and release OPG and turn off their RANKL genes and RANKL production, which would combine to reduce osteoclast development and with it PDGF-BB production (Atkins et al., 2003; Kubota et al., 2002).
As if all of this is not enough, the reader may remember that the signals from PTH- or PTHrP-activated PTHR1 receptors probably stimulate the expression of leptin, which we met in Chapter 3i (Torday et al., 2002) (fig. 10). This autocrine and paracrine cytokine does a lot of things to promote bone formation (fig. 10). It opposes osteoclast generation by stimulating osteoblasts to make the anti-osteoclastogenic OPG and reduce the pro-osteoclastogenic RANKL production by immature osteoblastic cells, and it dissuades osteoprogenitors from choosing the adipocyte option, stimulate osteoblast maturation, stimulate the proliferation and migration of vascular endothelial cells to the bone construction site, and finally promote matrix mineralization.
The striking PTH-induced layering of rat bones with mature osteoblasts may not be due only to the stimulation of preosteoblast proliferation and maturation into apoptosis-resistant osteoblasts. In rats, it seems to be at least partly, if not wholly, due to the reversible, reversion of the thin, flat, post-mitotic, osteogenically inactive bone-lining cells to plump, bone-making, but of course still proliferatively disabled, osteoblasts (Dobnig and Turner, 1995; Leaffer et al., 1995). In other words, PTH summons lining cells from retirement to retool themselves for reentering the bone-making workforce. This transformation is likely due to a direct stimulation by PTH because, although the lining cells reduce PTHR1 receptor expression and other traces of their past osteoblast lives, they still express some receptors (at least in humans) (Langub et al., 2001). Presumably, the reverting cells would raise their PTHR1 receptor display to the level in mature osteoblasts and once again start making missionary growth factors such as FGF-2, IGF-I, and TGF-β(s) and releasing them into the adjacent bone marrow to expand the pool of osteoprogenitor cells. When the PTH injections stop, the ex-lining cells relax, stop making bone, and return to being flat lining cells doing their retirement job in the osteocyte-lining cell signaling network (Dobnig and Turner, 1995; Leaffer et al., 1995). The resulting drop in IGF-I production and secretion by the reverting lining cells and aging osteoblasts would restore their vulnerability to apoptosis.
But while the flat, resting lining cells are not "tooled up" to make the huge amounts of bone matrix they made when they were osteoblasts, they can still make some. They sweep up the collagenous litter left by the BMU osteoclasts on the floor of the excavated microcrack repair site (i.e., the blister) (Everts et al., 2002). While doing this, they must be exposed to a lot of inorganic phosphate as well as Ca2+. When enough of this phosphate is brought into the cells by their transporters, it directly activates the phosphate-responsive promoter-switch box of the osteopontin gene (Beck, 2003; Beck and Knecht, 2003; Beck et al., 2000, 2003), the product of which the cells use to mix up a layer of cement to glue the large amount of matrix made by the incoming mature osteoblasts to the old bone (Denhardt and Noda, 1998; Denhardt et al., 2001; McKee and Nanci, 1996). The janitorial lining cells also deposit a thin priming layer of collagen on top of the osteopontin cement to prepare for the arrival of the mature osteoblasts.
Matrix Mineralization
Needless to say, inorganic phosphate and the mature osteoblast's Na+ gradient-driven, type III Pit1 phosphate transporter that ships it into the cell are key players in bone formation (Caverzasio and Bonjour, 1996; Nielsen et al., 2001; Selz et al., 1989; Takeda et al., 1999). Moreover, we are now learning that inorganic phosphate is a major signaler, an activator of certain gene promoters (Beck, 2003; Beck and Knecht, 2003; Beck et al., 2003). The PTHrP-triggered signals from the emerging PTHR1 receptors on the young matrix-making cells drive Cbfa1/Runx-2 into the nucleus to stimulate the ALP (alkaline phosphatase) gene, the product of which is plugged into the cell membrane with its catalytic subunit (i.e., its "business end") sticking out into the extracellular space (Beck and Knecht, 2003; Beck, 2003; Beck et al., 1998, 2000, 2003). But the cell won't immediately start mineralizing matrix because the Cbfa1/Runx-2 pushed into the nucleus by PTHR1 signaling (Fujita et al., 2001a, 2001b) stimulates the expression of the mineralization-blocking osteocalcin (Ducy et al., 1996) (fig. 23). The ALP then snips phosphate out of external substrates such as the β-glycerophosphate that is added to culture media for bone nodule formation and in the Real World out of matrix components. The rising phosphate switches on the gene for the Pit 1 transporter (Beck et al., 2003) to carry phosphate into the cell. The incoming phosphate stimulates the expression of a group of genes whose products drive the transition from matrix-making to mineralization (Beck and Knecht, 2003; Beck et al., 2003). First, the incoming phosphate downregulates the expression of the major matrix components, collagen-1α1 and collagen-1a2, because the emphasis must now shift to mineralization—enough collagen has been made (Beck and Knecht, 2003; Beck et al., 2003). The incoming phosphate also stimulates the gene for Nrf2, the transcription factor that homes on the antioxidant response element (ARE) and the battery of genes that have it in their promoter-switch boxes (Nguyen et al., 2003). Presumably the Nrf2/ARE-driven genes and their products will help the cell survive the formidable challenges of a lot of phosphate and a blebbing membrane (Beck et al., 2003). Of course, the phosphate also stimulates the expression of calcyclin and annexin V which, as we shall see in the next paragraph, are needed for the mineral formation (Beck et al., 2003). And the phosphate also stimulates the gene for cyclin D1, which has some role in differentiating osteoblasts besides its more familiar role of triggering the build-up to DNA replication in proliferatively competent cells (which the mature osteoblast most certainly is not). However, when the Pit-1-pumped phosphate reaches a critical level it stimulates the nuclear export machinery, which empties the nucleus of its Cbfa1/Runx-2 (Fujita et al., 2001a, 2001b) (fig. 23). This dumping of Runx2/Cbfa1 out of the nucleus shuts down osteocalcin expression (Fujita et al., 2001b), which lifts the block to mineralization (Ducy et al., 1996; Fujita et al., 2001a) (fig. 23). (Of course, the block will have to be reimposed later on to avoid runaway mineralization.) Again one must remember that timing is everything.
To mineralize the freshly made matrix, the osteoblast loads Pit transporters, alkaline phosphatase, and annexin V Ca2+ channels into prevesicle patches of their cell membrane that will be budded off to make vesicles that are detached calcium-apatite-making engines. To start out, the large amount of CaR-activating Ca2+ released by the osteoclasts is sucked up, and the osteoblasts' CaRs are silenced when the cells combine the Ca2+ with phosphate to mineralize the osteoid matrix they have just finished making. The Cbfa1/Runx-2-stimulated alkaline phosphatase and Pit1 transporter (the gene for which has been stimulated by incoming phosphate) are now plugged into the prevesicle patches (Beck, 2003). But it seems that the transporter's operation must be protected by the cell's PHEX endopeptidase, which would destroy any transport-inhibiting FGF-23 that the cell might be making or be exposed to (Bowe et al., 2001; Shih et al., 2002). The new osteoid is mineralized when alkaline phosphatase-generated, Pit1 transporter-delivered phosphate and Ca2+ flowing through the channels formed in the vesicle walls by the AnxV, the gene for which has also been stimulated by incoming phosphate, combines to form needles of impure hydroxyapatite (Ca10[PO4]6[OH]2) in the matrix-bound vesicles (Bandorowicz-Pikula et al., 2001; Beck, 2003; Caverzasio and Bonjour, 1996; Hoshi and Ozawa, 2000; Selz et al., 1989). These vesicles armed with alkaline phosphatase, Pit1 transpoprters, and annexin V channels in their walls are now budded from the surfaces of the osteoblasts, and when they have loaded themselves to bursting with calcium apatite-like needles, they spill the needles out onto the osteoid (Caverzasio and Bonjour, 1996; Nielsen et al., 2001). Since PTH-induced cyclic AMP transients stimulate type III transporters, well-separated boluses of AC-stimulating PTHs promote mineralization by selectively increasing the Vmax (the transporter's top carrying speed) of the transporter, which persists even after the vesicles bud from the osteoblast's surface (Caverzasio and Bonjour, 1996).
At this point, we might ask what happens to the PTHR1 receptors when the mineralizing osteoblast's surface is budding off vesicles. Are they budded off along with the vescles? Certainly it appears that the post-mineralization osteocytes have downregulated their PTHR1 receptor expression.
If an osteoblast is lucky enough to survive surging Ca2+ and phosphate and a roiling membrane to become an osteocyte, the incoming phosphate will have triggered a second burst of osteopontin gene expression and osteopontin synthesis to glue the cell and its processes to the walls of its cubicle and its dendritic processes to the walls of their canaliculi (Beck and Knecht, 2003; Beck et al., 1998, 2000, 2003; Denhardt and Noda, 1998; Denhardt et al., 2001; McKee and Nanci, 1996). The emerging osteopontin also prevents the incoming phosphate from inducing apoptosis (Koyama et al., 2003).
The incoming phosphate stimulates osteopontin expression by somehow causing the biphasic activation (by phosphorylation) specifically of ERK 1/2 (Beck and Knecht, 2003). In other words, there is an early surge of phosphorylated ERK 1/2 between 15 and 45 minutes after adding 10 mM phosphate to the MC3T3-E1 cells' culture medium and a second surge beginning at 8 hours and building up at least until 32 hours. Other members of the MAP kinase family such as p38 are not involved, but a separate stimulation of PKCs activity also seems to be needed.
PTHs and Mechanosensitive Ca2+ Channels
Again we turn to to the seemingly intimate relation between PTH and strain responsiveness which surfaced in Section iiib. PTH might also sensitize the strain-responsive osteocytes (which still express PTHR1 receptors, though at much lower levels than when they were osteoblasts (Aubin, 2000; Langub et al., 2001) to strain (Duncan et al., 1992). Thus, adenylyl cyclase-stimulating PTHs phosphorylate the mechanosensitive Ca2+ channels (MSCCs) of MC3T3-E1 mouse preosteoblasts and UMR 106.01 rat osteoblasts by cyclic AMP-stimulated PKA kinase (Duncan and Misler, 1989; Duncan et al., 1992; Ryder and Duncan, 2001). This causes the MSCCs to open wider (increase their "conductance") and stay open longer when they are pulled and stretched by shear stress. However, at high levels of shearing, both the adenylyl cyclase and PLC pathways may be required to increase the MSCCs' responsiveness (Ryder and Duncan, 2001). A need for PTH to maintain MSCC responsiveness to strain could be why thyro-parathyroidectomy has been reported to eliminate the responsiveness of rat bones to mechanical stress (Chow et al., 1998). This shearing also stimulates the cells to pump out ATP and UTP, which by stimulating their P2 nucleotide receptors, sensitize the cells to signals from PTHR1 receptors (Bowler et al., 2001). Since these nucleotides are very short-lived, they accumulate at high levels only around the producing cells—they are autocrine (self-stimulating) but not paracrine (neighbor-stimulating) (Bowler et al., 2001). Thus, they selectively focus and magnify PTH's action on the strained cells and their MSCCs. A practical use of this sensitization of MSCCs by PTH would be to use the peptide to increase bone formation upon return to macro-gravity or remobilization after prolonged inactivity (Chow et al., 1998; Ma et al., 1999).
A Very Quick Overview
Now let's take a short time out and briefly summarize how PTH stimulates bone growth in an adult bone such as a femur or tibia (fig. 20). At the time of the first PTH injection, there are osteoblasts in microcrack-repairing BMUs, busily laying layers of bone around the walls of recently dug tunnels in cortical bone and filling trabecular trenches. But then there is the large population of lining cells—retired osteoblasts—still with some PTHR1 receptors, covering the walls of the cortical canals, the endosteum, and trabeculae. The PTH diffusing from the blood vessels in the canals and the marrow sinusoids will hit all of these cells and cause them to come out of retirement and retool themselves with more PTHR1 receptors and the various other devices for making bone. And they will be kept from slipping back into retirement by periodic kicks from the daily PTH boluses. The reverted lining cells on the endosteal and trabecular surfaces are strategically placed for stimulating various progenitors—they are bathed in bone marrow, which means that the growth factors that the daily PTH boluses cause them to make and secrete can directly reach and stimulate the proliferation of immature osteoprogenitors. Of course, the PTH's antiapoptotic action could delay the inevitable mass (95%) self-destruction of the BMU osteoblast crews, but this would not apply to the stimulated osteoblast/lining cells, which would just stay where they are on the bone surface and slip back into the role of being lining cells connected to osteocytes and monitoring the continuous chattering of osteocytes for signs of fracture (fig. 2 and fig. 3).
PTHs Speak to Bones with Forked Tongues—The Other Prong Says Remove It!
While the osteogenic action of single daily injections of an adenylyl cyclase-stimulating PTH predominates over their osteoclast-stimulating, bone-demolishing action, the balance shifts to net resorption if the PTH is continuously infused for longer than 1 hour or injected too frequently (Dobnig and Turner, 1997). But it appears that although intermittent injections of hPTH-(1-34) have been found to at least transiently increase cortical porosity in humans, monkeys, and rabbits, it has been found in mice and rats that anabolic intermittent injections of medium and high doses of the peptide actually decrease osteoclast surface in OVX rats (Lane et al., 1996) and suppress the final steps of osteoclast differentiaition in male ddY mice (Sakai et al., 1999).
Why and How Does Prolonged PTH Infusion Destroy Bone?
First, Watson et al. (1999) found part of the answer in rats. As expected, intermittent injections of the full-size hPTH-(1-84) caused a massive accumulation of IGF-I-making osteoblasts (Watson et al., 1995, 1999). Continuously infusing the PTH still caused osteoblasts to accumulate, but now things were different. The cells made less autocrine/paracrine IGF-I and start making IGF-binding proteins, IGFBP-3, IGFBP-4, and IGFBP-5 (fig. 27). The IGF-I-trapping IGFBPs, particularly IGFBP-4 (Mohan et al., 1999), would have prevented the smaller amounts of IGF-I from stimulating bone formation. Then, Locklin et al. (2003) reported that intermittent exposure of cultured primary mouse marrow cells to hPTH-(1-34) caused an IGF-I-dependent stimulation of osteoblast differentiation markers while continuous exposure could not stimulate these markers and along with this reduced IGF-I mRNA production. Added to the IGF-I blockade is the switching on of the accumulating osteoblasts' matrix-destroying collagenase-3 by overproduction of FGF-2 (Hurley et al., 1995, 1999) (fig. 27). PTH itself would also have stimulated collagenase-3 expression by stimulating Cbfa1/Runx-2 expression (Locklin et al., 2003; Selvamurugan et al., 2000), but as noted in the next paragraph, this might not happen with excessive exposure to PTH. This combination of IGF-I blockage and a surging, matrix-destroying collagenase should be enough to tip the balance over to bone demolition.

Figure 27
The various things that happen to stimulate osteoclast activity and bone resorption rather than bone growth when a PTH is continuously infused instead of being injected once a day or swallowed once a day in a pill.
Intermittent exposure of mouse marrow to adenylyl cyclase-stimulating PTHs stimulates Cbfa1/Runx-2 that drives osteoblast differentiation and bone building (Locklin et al., 2003). Single daily boluses of adenylyl cyclase-stimulating PTHs actually trigger the loading of nuclei with Cbfa1/Runx-2 and the expression of about 158 genes in rat femoral metaphyseal bone cells (Fujita et al., 2001a, 2001b; Moore et al., 2000; Onyia et al., 2001). Among these genes is the one encoding a still functionally mysterious, 145-amino acid cytoplasmic protein PAIGB—in the tibial cells of Sprague-Dawley rats (Robinson et al., 2003).
These daily boluses also promptly trigger only brief (6-24 h) bursts of ubiquitin-specific protease UBP41, expression of which could save key gene transactivators by removing any attached ubiquitin chains that would mark them for the proteasome shredder (Chung and Baek, 1999; Huang et al., 1995; Miles et al., 2002). However, as I said in section iiid, it seems likely that these brief bursts of UBP41 activity could promote the reconfiguration of chromatin that makes genes more accessible to appropriate transcription factors (Latchman, 2004). Ubiquitination of histone H2B facilitates the methylation on lysine residues 4 and 79 of histone H3, which promotes a more tightly packed chromatin structure and gene silencing (Latchman, 2004). Therefore, removing these ubiquitins with PTH-triggered, cyclic AMP-driven bursts of UBP41 could result in the demethylation of H3 histones and the unlocking of dozens of genes, such as the osteogenically key genes that are responsive to the Cbfa1/Runx2 that is, at the same time, being loaded into the nucleus by the PTH boluses.
But continuously infusing these PTHs into rats selectively switches on a different and very much larger, set of 759 genes in the metaphyseal cells, which, for example, no longer includes the gene for the PAIGB protein, but instead includes genes for bone matrix-shredding proteases (Onyia et al., 2001a, 2001b; Robinson et al., 2003). Continuous PTH exposure also locks on UBP41 expression (Miles et al., 2002; Murray et al., 1998), which, by increasing the free ubiquitin pool along with a cyclic AMP-induced stimulation of the proteasomal endopeptidases, would shift the cell's proteome (its protein complement) from one for bone making to one for bone destruction. This prolonged activity of UBP41 and increased proteasomal enzymes drives a PKA-stimulated ubiquitination and destruction of Cbfa1/Runx-2 with a consequent shutdown of Cbfa1/Runx-2-dependent osteoblast-specific genes such as the genes for Bcl-2 and OPG (Bellido et al., 2002; Tintut et al., 1999) (fig. 25). The upshot of these changing gene expressions during continuous PTH infusion are drops in the expression of the antiosteoclastogenesis OPG and osteoblasts' bone-making genes, such as those for BSP, collagen I, and osteocalcin, as well as a reciprocal increase in RANKL and the products of other genes that drive osteoclast differentiation and activation and bone resorption (Ma et al., 2001).
How could continuous PTH infusion turn on or off so many more genes than single daily boluses of the peptide? Evidently, continuous infusion activates additional transcription factors. According to Chauvin et al. (2001), spacing the boluses 24 hours apart would enable an osteoblast's PTHR1 receptors to fully recycle once and enable the cell to present a fully receptored surface to the next PTH bolus. Each bolus would cause a brief burst of cyclic AMP/PKA signaling from the full complement of PTHR1s on the various target cells. But a continuously infused PTH might cause a continuous, β-arrestin-2-mediated endocytic cycling of PTHR1s with reduced steady-state level of receptors and very low-level adenylyl cyclase/cyclic AMP signaling (Locklin et al., 2003) but with a steady, β-arrestin-2-mediated pumping of MKK4-activated JNK kinase (and maybe PTHR1 receptor itself ) into the nucleus (fig. 18). Such a sustained injection of a transcription factor(s) activator such as JNK into the nucleus could massively stimulate the expression of a much larger set of genes and their resorption-driving products.
The most important arbiter of whether a PTH builds or destroys bone is the RANKL/OPG expression ratio it establishes. Halladay et al. (2002) have used UMR106 rat osteoblast-like cells transfected with an opg gene promoter construct pOPG5.9βgal (-5917 to +19) to see the effects of short and long exposures to hPTH-(1-38) and a PTH of the Ostabolin™ Family, hPTH-(1-31) (Ostabolin™), on the activity of the opg gene's promoter as indicated by the extent of stimulation of a β-galactosidase "reporter gene" attached to the promoter construct. Exposing the UMR106 cells to the PTHs or to cyclic AMP-elevators such as the potent adenylyl cyclase-stimulating forskolin or to a cyclic AMP turnover inhibitor, the cyclic nucleotide phosphodiesterase inhibiting, IBMX, or to cyclic AMP itself for 8 hours nearly doubled the opg promoter-switch box activity, with the hPTH-(1-31) being slightly more effective than hPTH-(1-38). However, exposure to either PTH or one of the cyclic AMP elevators for 24 or 48 hours turned the promoter off. (Unfortunately, they did not look at the effects of the peptides at shorter intervals, such as the physiologically relevant 1 or 2 hours.) On the other hand, a PKC stimulator such as TPA (12-O-tetradecanoyl phorbol-13 acetate) (at 10-8 or 10-7 M) reduced the opg gene promoter activity in cells exposed to the drug for 8 hours but strongly stimulated the opg promoter in cells exposed for 24 or 48 hours. Thus, the stimulatory effect of a short exposure to an adenylyl cyclase-/PKC-stimulating PTH (remember that this of course depends on the cell's NHERF-2 content) on opg gene activity would be reduced by PKC stimulation, while the inhibitiory effect of a prolonged exposure would be blunted by PKC stimulation. This intervention of PKC into the process could at least partly explain why intermittent injections of members of the Ostabolin™ family (hPTH-(1-31NH2 (Ostabolin™) and [Leu27]Glu22-Lys26hPTH-(1-31)NH2)hPTH-(1-31) (Ostabolin-C™)), which tend not to stimulate PKC, are poor stimulators of osteoclast generation and do not cause hypercalcemia in various animal models and humans, even at very high doses (Fraher et al., 1999; Jolette et al., 2003; Mohan et al., 2000; Whitfield et al., 1998c).
Added to direct opg gene suppression by prolonged cyclic AMP pulsing is the ability of a prolonged PTH exposure to prevent osteoblastic cells from making OPG by stimulating the proteasome that destroys Cbfa1/Runx-2 which normally keeps the opg gene working by binding to the opg promoter's 12 so-called OSE2 sites (Bellido et al., 2002; Fu et al., 2001; Galvin et al., 2001; Hofbauer et al., 1999, 2000; Kanazawa et al., 2000; Miles et al., 2002; Murray et al., 1998; Onyia et al., 2000; Stilgren et al., 2001; Thirunavukkarnasu et al., 2000; Tintut et al., 1999).
But what about the other player, the pro-osteoclast RANKL? Within an hour of a single injection of the adenylyl cyclase/PKC-stimulating hPTH-(1-38) into rats, RANKL mRNA production surges and OPG mRNA reciprocally crashes in the distal femoral metaphysis and diaphysis, but OPG expression soon rebounds as its gene's promoter-switch box is turned on and rankl gene expression is reciprocally turned off by 3 hours, which means that the RANKL/OPG ratio must be low by this time (Ma et al., 2001; Onyia et al., 2000). Such intermittent OPG peaks could be why daily injections of hPTH-(1-34) (Forteo™) or [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™) decrease osteoclast activity or kill osteoclasts in OVX rats and cynomolgus monkeys (Jolette et al., 2003; Lane et al., 1996).
According to the observations of Locklin et al. (2003) on mouse marrow cells, if the PTH treatment is continuous, rankl gene expression should rise while the opg gene's promoter and thus its expression should be inhibited in the rat. Indeed, as expected, continuous infusion of hPTH-(1-38) into rats causes a sustained rise in rankl expression, a sustained drop in OPG expression, and increases in serum Ca2+ concentration and osteoclast generation (Ma et al., 2001). Locklin et al. (2003) have also found that although a continuous exposure to hPTH-(1-31) (Ostabolin™) is a 2 to 3 times more effective stimulator of rankl gene expression than continuous exposure to hPTH-(1-34) in their mouse marrow cultures, the two peptides only equally increased osteoclast generation. This indicates a large difference between the actions of factors controlling message processing. And it could mean that hPTH-(1-31) signaling lacks something needed to get the additional number of rankl gene transcripts translated into active RANKL. But as we will see further on, we are being confused by data from "pure" and multicomponent cultures and the whole animal.
At this point, the reader might ask why the expressions of RANKL and OPG are reciprocal in the same cell? The answer might lie in the phosphorylation of the CREB protein's Ser133 by PKA activated by the cyclic AMP signals coming from PTH-activated PTHR1 (Fu et al., 2002). The PKA-phosphorylated/activated CREB plus a CBP/p300 (Ptashne and Gann, 2002; Quinn, 2002) on the rankl gene promoter's CRE turns on the rankl gene (fig. 28). At the same time, CREB•CBP/p300 on the c-fos gene's promoter stimulates this gene's expression (Tyson et al., 1999). The c-Fos product turns off the opg gene via the AP-1 complexes (Latchman, 2004) it forms with c-Jun on the opg gene's promoter (Fu et al., 2002) (fig.28). (Interestingly, the hPTH-(1-34)-induced increase in AP-1 activity and suppression of opg gene expression in osteoblasts could be enhanced by signals from serotonin receptors activated by serotonin coming from strain-stimulated osteocytes (Bliziotes et al., 2001). This mechanism explains the prompt stimulation of rankl and suppression of opg by the brief bolus of hPTH-(1-38) in the rats of Ma et al. (2001). But why did opg expression surge upwards and rankl crash during the next hour or so? Obviously the inhibitory AP-1 complexes on the opg promoter must rapidly disappear leaving a still activated opg promoter. According to this sort of scenario, a continuing supply of AP1 and suppression of opg, activation of rankl and the resulting increased osteoclast generation would require a sustained barrage of PTH boluses. But there is a problem here. I suspect that this reciprocity mechanism at the single-cell level may actually be irrelevant in the whole bone or primary mixed bone cell cultures, because immature osteoblastic cells express their rankl gene, but then turn the gene off and switch the opg gene on when they become functioning osteoblasts (Atkins et al., 2003; Corral et al., 1998).
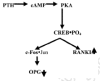
But in mice the responses of OPG and RANKL expressions to PTH injections appear to depend on whether the animal is OVXed or not (Lu et al., 2001). They have found that OVX alone caused the level of RANK expression in the tibias of C57BL/36 mice to double by 4 weeks and then drop back to only one-third of the starting level during the next 7 weeks and the OPG expression dropped to only 20% of the starting level by 11 weeks. Injecting hPTH-(1-34) into intact mice once daily for 5 days a week caused RANKL expression to increase 4 times and PTHR1 expression to triple. But, OVX inverted the responses of RANKL and OPG to PTH. The same PTH injections into OVXed mice reduced RANKL mRNA production to 37% of the starting level and caused a 2- to 3-fold increase in the level of OPG mRNA and PTHR1 expression, probably because the PTH-stimulated the estrogen-lacking RANKL-competent immature cells to mature into RANKL-incompetent, OPG-making osteoblasts without being replaced by new RANKL-making immature cells. The estrogen lack could indeed have caused a shortage of immature osteoblastic stromal cells by diverting the flow of bi-potential precursor cells from the osteoblastic to adipocyte pathway (Okazaki et al., 2002). This suggests that PTHs might be less able to stimulate bone growth in ovary-intact than in OVXed rats. And this is indeed the case. The abilities of hPTH-(1-34) and the two members of the ostabolin family of PTHs to stimulate femoral trabecular growth in ovary-intact rats are only about half of their abilites to stimulate trabecular growth in OVXed rats (unpublished observations in my laboratory). It is obviously not a little important to know whether this inversion can be repeated and whether it happens in postmenopausal women.
Yet another factor will come into play when the PTH boluses are too closely spaced. Adenylyl cyclase-stimulating PTHs increase the top speed (Vmax) of the osteoblasts' Pit1 phosphate transporter (Caverzasio and Bonjour, 1996; Nielsen et al., 2001; Selz et al., 1989). This transporter normally provides the inflow of phosphate needed to drive the expression of osteopontin and the accumulation of mineral in the matrix vesicles budded off the osteoblasts (Beck et al., 2000; Caverzasio and Bonjour, 1996; McKee and Nanci, 1996; Selz et al., 1989). Well-spaced cyclic AMP/PKA pulses would promote osteoid mineralization by causing phosphate to surge into the matrix vesicles. However, continuous infusion or more closely spaced boluses of a PTH might boost the flow of phosphate into the cell to an apoptosis-triggering level (Allen et al., 2002; Mansfield et al., 2001; Meleti et al., 2000).
The ability of PTHs to stimulate resorption has bedeviled the clinical application of these peptides since the 1930s. So the dream of all "PTH buffs" has been to make a PTH that could stimulate bone growth, but not bone resorption. Higher doses of such a PTH could be safely used to maximally accelerate fracture healing and get a bigger anabolic bang for the osteoporotics' buck. It appears that two such PTHs have at last been made. These PTHs belong to the Ostabolin™ family—hPTH-(1-31)NH2 (Ostabolin™) and [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™ i.e., cyclized Ostabolin™). The potently anabolic Ostabolin™ has been shown to be both orally administrable and a very poor stimulator of resorption in humans and mice (Fraher et al., 1999; Mehta et al., 2000; Mohan et al., 2000). And Jolette et al. (2003) have shown that the potent stimulation of bone formation in cynomologous monkeys, even by daily injections of such a large dose of Ostabolin-C™ as 80 μg/kg of body weight for 7 days, decreased the indices of bone resorption!
Moreover, Paul Morley (Zelos Terapeutics) has informed me that Ostabolin-C™ does not cause the burst of cortical porosity in cynomologus monkeys that is caused by hPTH-(1-34) (Forteo™) (Burr et al., 2001). Intermittent injections of Ostabolin-C™ may simply be better at suppressing osteoclast differentiation than Forteo™ (Lane et al., 1996; Sakai et al., 1999). Indeed during the first 8 weeks after OVX, the metaphyseal trabecular number in distal femurs of Sprague-Dawley rats fell 52% from the normal (sham-operated) 4.6 ± 0.1/mm to 2.2 ± 0.1(p<0.01). When hPTH-(1-34) (Forteo™) was injected once daily into the OVXed rats starting 2 weeks after the operation and ending 6 weeks later, the mean number of metaphyseal trabeculae had dropped from the normal (sham-operated) value of 4.6 ± 0.1 to 2.0 ± 0.1/mm (p<0.01), which was not significantly different from the 2.2 ± 0.1 in the untreated OVXed rats. However, when Ostabolin-C™ was injected instead of hPTH-(1-34) (Forteo™) the trabecular number dropped by only 28% to 3.3 ± 0.1/mm, which was 1.7-fold higher than the 2.0 ± 0.1/mm (p<0.01)in the hPTH-(1-34) (Forteo™)-treated rats. If the PTH injections were delayed for 9 weeks after OVX when the maximum osteoclast activity had passed, there was no difference between the trabecular numbers in the animals treated daily with either peptide between 9 and 15 weeks. In both kinds of experiment, the two peptides equally and greatly increased the mean trabecular thickness to a value about 1.7 times higher than in the femurs of the sham-operated animal. Thus, it seems from these numbers that Ostabolin-C™ and hPTH-(1-34) (Forteo™) are equally osteogenic, but Ostabolin-C™ is the better osteoclast suppressor or killer, at least in rats and monkeys.
Why would these Ostabolin™-family peptides be such poor stimulators of bone resorption? On the basis of the available information from cultured cells, it simply makes no sense! To try to answer this question, we must look more closely at what we have learned in the past few paragraphs. RANKL expression is unquestionably stimulated by cyclic AMP/PKA. And all of the osteogenic peptides, including the Ostabolin™ family peptides, activate PTHR1, which stimulates adenylyl cyclase which stimulates RANKL. So why would Ostabolin™ family members be poorer osteoclast stimulators than hPTH-(1-34) (Forteo™)? Why would the potent adenylyl cyclase-stimulator and potently anabolic [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™) actually reduce osteoclast activity in OVXed rats as wells as monkeys?
The reason may stem from two things. First, the density of PTHR1 receptors on an osteoblastic cell depends on its "maturity" (Aubin, 1998, 2000, 2001; Aubin and Triffitt, 2002) (fig. 20). Second, immature progenitors and mature osteoblasts differently express RANKL and OPG (Atkins et al., 2003). RANKL is made by immature marrow osteoblastic cells with relatively few emerging PTHR1 receptors, while the mature osteoblasts with maximum PTHR1 receptor densities do not make RANKL but can make OPG (fig. 8 and fig. 20). As I said back in Chapter 2ii, this maturity-dependent expression of RANKL and OPG is a smart move on Nature's part because the two populations live in different places with different neighbors and jobs to do in the crowded spaces of an osteonal tunnel or a trabecular trench. This means that a PTH will stimulate RANKL expression in immature osteoblasts and OPG rather than RANKL in mature osteoblasts, and the relative levels of expression will depend on the population's maturity/receptor density profile. Thus, PTH-stimulated immature, RANKL-competent osteoblastic cells following osteoclasts in a new osteonal tunnel or in a trabecular trench would help the osteoclasts, but PTH-stimulated, mature, OPG-expressing, working osteoblasts with their RANKL genes shut down (Kitazawa et al., 1999) would stop osteoclasts with OPG to protect the bone they are making.
Now we come to the point. The Ostabolin™ family peptides have lower affinities for PTHR1 than does hPTH-(1-34) (Forteo™), with Ostabolin™ having the lowest value (Whitfield et al., 2000c). This means that during their brief appearance in a single injected bolus, the Ostabolin™ family peptides would be less able than hPTH-(1-34) (Forteo™) to grab onto enough of the fewer PTHR1s to stimulate the immature RANKL makers. But they would have enough time and not be significantly less effective than hPTH-(1-34) (Forteo™) in grabbing enough PTHR1s to stimulate the OPG-making preosteoblasts and mature osteoblasts with many more PTHR1 receptors on their surfaces. However, the advantage should vanish when the peptides are given much more time to grab PTHR1s during continuous infusion. This should be the case for Ostabolin-C™, which has a higher affinity for PTHR1 than Ostabolin™ but still a lower affinity for PTHR1 than hPTH-(1-34)NH2 (Whitfield et al., 2000c). Indeed, that is exactly what happens. When Ostabolin™ is continuously infused (at a dose of 4 nmoles/kg per day) into a Sprague-Dawley rat, the blood Ca2+ concentration rises at about the same rate as in rats receiving hPTH-(1-34)NH2 but then stops rising during the next 5 days while the blood Ca2+ concentration continues rising in the animals receiving hPTH-(1-34)NH2. However, in striking contrast to its total inability to raise the blood Ca2+ level when injected intermittently in monkeys (Jolette et al., 2003), continuous Ostabolin-C™ infusion causes the blood Ca2+ concentration to rise as steadily and at least as high as, or even higher than, in the rats receiving hPTH-(1-34)NH2.
A differential response of RANKL-expressing immature and OPG-expressing mature osteoblastic cells could also account for the strange observation of Fujita et al. (1999) that a once-a week injection of 60 μg of hPTH-(1-34) (Forteo)™ significantly increased spinal BMD but at the same time significantly reduced the level of collagen breakdown bits in osteoporotic patients. In this case, a weekly brief bolus of PTH would target most strongly the mature, receptor-rich, OPG-making osteosteoblasts, but it would be too ephemermal to significantly stimulate the immature RANKL-makers.
How an Odd Couple—PTHrP and an Indian Hedgehog—Build Bones
An odd couple, PTHrP and a hedgehog, actually an Indian hedgehog, controls the growth of a long bone such as the femur. Indian hedgehog is one of a family of several proteins that are called hedgehogs, because disabling the single hedgehog gene in the fruit fly Drosophila causes the fly's larvae to have a pattern of cuticular denticles that make it look like a prickly hedgehog (Bier, 2000).
The femur begins as a bone-shaped template, a condensation of mesenchymal cells expressing the master cartilage-inducing transcription factor known as Sox 9 (de Crombrugge et al., 2001). Sox 9 probably stimulates the expression of the genes for the cell surface components needed for chondrocytic condensation (de Crombrugge et al., 2001). Then Sox 9 causes the cells to express L-Sox 5 and Sox 6 that are "architectural" transcription proteins that reconfigure nuclear chromatin structures to enable Sox 9 and other classical transcription factors to turn on chondrocye-specific genes (de Crombrugge et al., 2001). The condensed mesenchymal cells differentiate into chondrocytes that lay out a cartilage model of the future bone and perichondrial cells that surround and enclose the emerging model. The cells at the center of the model stop proliferating, stop expressing Sox 9, and start maturing into terminal hypertrophic (swollen) cells, which calcify the matrix to enable it to be resorbed by osteoclasts and replaced by bone. While this is going on in the center, the model's perichondrium is invaded by capillaries and changes from a perichondrium into a periosteum. In other words, the perichondrial layer spawns osteoblasts that deposit bone on the inner surface of the periosteum to make a hollow cylinder—the bone collar. Meanwhile, the hypertrophic cells' matrix calcification reduces their already low oxygen and nutrient supplies, which causes them to trigger the suicidal apoptotic mechanism. The calcified matrix is removed by "chondroclasts", leaving only trabeculae-like calcified cores or spicules in a primary marrow space. Vascular and perivascular cells from the periosteum then invade this marrow space. The blood vessels produce the marrow sinusoids, and the perivascular invaders generate the bone marrow stromal cells, osteoprogenitors, and the first osteoblasts, which layer bone onto the calcified cartilage cores to make the first trabeculae. The intramembranous (i.e., directly from mesenchymal cells rather than from mesenchymal cells via cartilage) ossification by mesenchymal cells to make the bone collar and the endochondral (i.e., indirect via cartilage) bone formation in the center of the cartilage model together make up the so-called primary ossification center (Kerr, 1999).
As the bone lengthens, the cartilage layers attached to the inner surfaces of the bony epiphyses at both ends of the growing bone are reduced to thin growth plates (also called physes). Now the bone has proximal (nearer the body) and distal (farther from the body) growth plates, which contain chondrocyte stem cells and are the engines that drive the lengthening of the bone. The round, moderately sized chondrocytes at the top of a growth plate nearest the epiphysis, the reserve or resting zone, are the stem or chondroprogenitor cells (R.B. Martin et al., 1998). They are fed with nutrients diffusing down through the cartilage matrix from the blood vessels in the overlying epiphyseal bone. Thus, the core of the growth plate and the columns of cells extending down from it are hypoxic, and to do their jobs they have to turn to HIF-1α and HIF-1β (which we met in Chapter 1) and the large number of oxygen-regulated genes they control to stimulate the genes for the the glucose transporter 1 and the enzymes of the anerobic glycolysis machinery to replace the mitochondrial oxygen-driven ATP production (Schipani et al., 2001; Wenger, 2002). They must stop trashing HIF-1α. When the oxygen level in the core drops below a critical level, the cells' oxygen-driven protein hydroxylases stop working, which means that prolines in their HIF-1αs' oxygen-dependent destruction domains are no longer hydroxylated, and the peptides are no longer polyubiquitinated and dumped into the cells' protein-shredding proteasome carburetor barrels (Marz, 2004; Schipani et al., 2001; Wenger, 2002). The accumulating HIF-1α moves into the nucleus together with HIF-1β and stimulates the transcription of the genes for the components of the glycolytic machinery.
When a stem cell gets the signal to start its proliferative engine and divide, one of its two daughter cells stays home to keep up the self-renewing stem cell tradition, while the other daughter sheds it and leaves home to join the parallel columns or stacks of flat or disk-like "transit amplifying cells", cells in the proliferative zone. Eventually the great granddaughters of the original stem cell daughter reach the end of the proliferative column and plunge into the deadly ionic turbulence of the hypertophic zone (R.B. Martin et al., 1998), where they switch off and dismantle their proliferative engines and start their apoptosis-terminated hypertrophic maturation program in which they increase their volume (i.e., hypertrophy) 5-to 10-fold while increasing their expression of alkaline phosphatase, annexin V, BMP-6, PTHR1 receptors, type X collagen and, at this point, the very important Indian hedgehog (e.g., Zuscik et al., 2002). The purpose of all of this activity is to lay down cartilagenous matrix primed for calcification, which is necessary for osteoclasts to attach to the surface, remove it, and thus enable its ultimate relacement by bone.
Finally, the cells start depositing Ca2+-hydroxyapatite crystals on the matrix between the cell columns. The calcification of the cartilage drives the hypertrophic cells to apoptotic self destruction by impeding the diffusion of food supplies and disposal of wastes (there is no lacunocanalicular network in cartilage to efficiently distribute nutrients and dispose of waste via the marrow vasculature as in cortical bone). The dying (apoptosing) cells release proteases, which shred the cartilaginous matrix in the distal segments of the cell columns but leave the calcified matrix between the columns intact. The resulting proximal calcified "stalactites" and distal calcified "stalagmites" are targeted by osteoclasts and osteoblasts and become the foundations of the primary proximal and distal metaphyseal trabeculae. The invasion of these osteoclasts and osteoblasts is enabled by the proliferation of blood vessels driven by leptin-induced proteases and the HIF-1α-induced expression of the chondrocytes' vascular endothelial growth factor (VEGF) genes (Wenger, 2002).
But what do PTHrP and its pet Hedgehog do in this process? The Sox 9-driven cells of the embryonic bone's initial cartilage model make Indian hedgehog (Ihh), but Ihh production is eventually confined to the prehypertrophic stack cells (fig. 29). Ihh makes its way from the producer cells (Bitgood and McMahon, 1995; de Crombrugge et al., 2001; Minina et al., 2001; Karaplis, 2001; T. Kobayashi et al., 2002; Vortkamp et al., 1996, 1998) to the resting peri-articular (around the joint) cells, which have receptors called "Patched or Ptch for short" which grab and suppress, other receptors called "Smoothened" or just "Smo" to their friends (summarized in Whitfield and Chakravarthy, 2001). When Ihh binds to the peri-articular cells' Ptch receptors, they let go of Smo, which then emits signals that send the transcription factor Gli into the nucleus to stimulate the expression of TGF-β2, which in turn stimulates the expression of the PTHrP gene (Alvarez et al., 2002; Karaplis, 2001; Stark, 2002; Whitfield and Chakravarthy, 2001) (fig. 29). This Ihh → TGF-β → PTHrP sequence also stimulates the peri-articular cells to start proliferating by expressing cyclin D1, the first of the parade of stage-specific, cell cycle-driving, cell cycle-engine kinases (Beir et al., 2001; Whitfield and Chakravarthy, 2001), and join the parallel columns of dividing chondrocytes (T. Kobayashi et al., 2002). And Ihh stimulates the appearance of BMP-2, which in turn stimulates the expression of Cbfa1/Runx-2 in mesenchymally derived perichondrial cells, which commits the cells to the osteoblast lineage and formation of the periosteum (Canalis et al., 2003; de Crombrugge et al., 2001) (fig. 7).
But remember what Zuscik et al. (2002) said about Ca2+ and PTHRP gene expression. The intracellular Ca2+ concentration must be low enough to enable PTHrP expresson. This Ca2+ sensitivity resides in the -1498 to the -863 region of the PTHrP gene's promoter (Zuscik et al., 2002). This part of the PTHrP gene's promoter has consensus sites for eleven transactivators (Zuscik et al., 2002). One of these is YY1, a possible silencer of the Ca2+-driven binding of which to the PTHrP gene promoter interferes with a positive factor (Roy et al., 1996; Zuscik et al., 2002). Alternatively, one of these eleven may be a transcription promoter, the binding of which to its target site on the PTHrP gene promoter is reduced by nuclear Ca2+ just as nuclear Ca2+ reduces the binding of the transcription enhancer, CBF/NF-Y, to its target site on the grp78/BiP gene (Roy et al., 1996). In either case, the Ca2+ concentration in the peri-articular region must be kept low enough, or the cells' Ca2+-sequestering pumps must work hard enough, to keep the nuclear Ca2+ level low enough to allow PTHrP to be made in response to Ihh stimulation. The PTHrP from the peri-articular cells then percolates along the stacks of proliferating cells, which deploy increasing numbers of PTHR1 receptors on their surfaces as they get near the deadly, seething, hypertrophic zone (Amizuka et al., 1994; Karaplis, 2001; Lee et al., 1995; Weisser et al., 2002).
The cyclic AMP signals from the growing numbers of PTHR1 receptors and probably PTHrP's intracrine nuclear action keep the cells from switching off their proliferative machinery and tearing open the "HT (hypertrophic)" gene envelope, which contains the genes for alkaline phosphatase, annexin V, BMP-6, collagen X, PTHR1, and of course, Ihh, and eventually killing themselves (e.g., Zerega et al., 1999) (fig. 29). The PTHR1 signals do this via PKA, which by phosphorylating two sites on the Sox 9 protein increases Sox 9's DNA-binding ability and its ability to prevent the transition from proliferation to hypertrophy (de Crombrugge et al., 2001) (fig. 29). This prevention is ultimately due to the suppression of the expression and production of p57Kip2, an inhibitor of the cyclin-dependent protein kinases that drive the build-up to DNA replication (MacLean et al., 2004; Whitfield and Chakravarthy, 2001). As we have learned above, PTHR1-induced bursts of cyclic AMP-dependent PKA activity should prevent the proliferating chondrocytes from triggering apoptosis by phosphorylating and inactivating their pro-apoptotic BAD and Bax proteins and stimulating them to make the anti-apoptosis Bcl-2 protein (Amling et al., 1997; Bellido et al., 2001, 2002). Indeed, PTHrP does stimulate Bcl-2 expression in growth plate chondrocytes, and knocking out the Bcl-2 gene causes premature maturation and apoptosis of the chondrocytes and shortened long bones in mice (Amling et al., 1997). The PTHrP might also hold off maturation by intracranially using its NTS to get into the nucleus and localize to the fibrillar component of the nucleolus, where it can inhibit ribosome production (Aarts et al., 2001).
Leptin also has an important role in cartilage function. As we learned in Chapter 3i, large amounts of leptin are made specifically by hypertrophic chondrocytes near invading capillaries and osteoblasts in the primary spongiosa extending from the growth plate in the ever-growing mouse long bones (Kerr, 1999). It appears that the chondrocytes use their leptin to get vascular endothelial cells moving and proliferating and make proteases to tunnel through the matrix to enable the delivery of osteoprogenitor cells to the new bone sites (Kume et al., 2002). A much more recent report by Kishida et al. (2003) also indicates that leptin is one of the controllers of chondroctye maturation and apoptosis in the mouse bones. But they have found that it is the prehypertrophic chondrocytes in mice that make leptin, and the swollen hypertrophic chondrocytes have the Lep Rb receptors to respond to it. The importance of the chrondrocyte leptin is indicated by the growth plates in leptinless Ob/Ob mice, being nearly twice as fragile as the growth plates in normal mice (Kishida et al., 2003). When they turned to ATDC5 chondrocytes that express LepRb receptors, they found that a normal serum concentration of leptin (i.e., 1-10 ng/ml of medium) acted like PTHrP to stop matrix calcification and delay or prevent apoptosis (fig. 29). This leptin-inhibitory action might be partly mediated by a gradual increase in PTHrP expression, because the effect was partly eliminated by neutralizing anti-PTHrP antibody. In humans, there is marked expression of leptin in osteoarthritic cartilage and osteophytic bone outgrowths, but many fewer chrondoytes made leptin in normal cartilage (Dumond et al., 2003). Dumond et al.(2003) also reported that leptin stimulates the anabolic actions of rat chondrocytes and induced genes for, and the syntheses of, IGF-I and TGF-β1. Thus, it appears that in mice, prehypertophic chondrocytes make leptin, which causes hypertrophic cells to make PTHrP, which restrains prehypertrophic cells from opening their HT envelopes and calcifying their cartilage and killing themselves. However, perhaps confusingly, this restraining of the prehypertrophic-hypertrophic transition by leptin delays PTHrP expression (Kishida et al., 2003). In humans, hyperexpression of leptin is responsible for osteophyte growth and thus plays a key role in osteoarthritis. Obviously, the story of leptin as a controller of chondrocyes is only in the unsettled first of many chapters.
The younger proliferating cells push the older chondrocytes into an increasing external Ca2+ concentration and a strongly increasing, maturation-driving inorganic phosphate concentration on the approaches to the hypertrophic calcification zone (Wuthier, 1993).
The Ca2+ turns on the cell's Ca2+ receptors (the CaRs; W. Chang et al., 2002, 2003; Chattopadhyay and Brown, 2003; Yamaguchi, 2003), the signals from which raise the [Ca2+i]enough to "activate" the Ca2+-sensitive silencer seeking the -1498 to the -863 region of the PTHrP gene's promoter. Once the signals from the Ca2+ receptors become loud enough, the silencer shuts off PTHrP expression, and signaling from the high density of PTHR1 receptors on the mature hypertrophic cells' surfaces fades away (W. Chang et al., 2002; Iannotti et al., 1994; Weisser et al., 2002; Wu et al., 1995; Zuscik et al., 2002). Turning off PTHR1 signaling switches off the proliferation engine and Sox 9 expression, which lifts the restraints on "HT" genes, which can be stimulated by, among other things, Ihh-stimulated Cbfa1/Runx-2 (de Crombrugge et al., 2001) (fig. 29). And perhaps most importantly, the silencing of the PTHR1 receptors by the PTHrP crash removes the anti-apoptosis shield. In other words, when the external Ca2+ concentration reaches a critical level, the progression of the cells to the hypertrophic stage and their doom is triggered.
As we learned above when the inorganic phosphate reaches a critical level in cells such as osteoblasts, it turns on the apoptotic mechanism (Meleti et al., 2000), unless stopped by PTH (Allen et al., 2002). The shutdown of PTHrP expression and PTHR1 signaling would thus enable the inorganic phosphate flowing into the hypertrophying cells through the type III Na-phosphate cotransporters to do its ultimately deadly job in collaboration with Ca2+ (Magne et al., 2003). The phosphate stimulates key genes such as the type II collagen gene and together with Ca2+ drops the anti-apoptosis Bcl-2 level and increases the pro-apoptosis Bax level (Coe et al., 1992; Magne et al., 2003).
The now gasping, hypoxic, hungry HT cells in the increasingly impermeable calcifying matrix, no longer protected by PTHR1 signals and with no longer adequate HIF-1α-driven glycolytic machinery and consequently dwindling glycogen reserves, rapidly emptying ATP fuel tanks, and spluttering ion pumps, are now plunging toward their self-inflicted apoptotic doom in the hypoxia of the lower hypertrophic zone as their internal phosphate level rises menacingly, and they fill their endoplasmic reticular storage vesicles with in-rushing Ca2+ and, in a last futile attempt to avoid a Ca2+ catastrophe, dump it into their leaking mitochondria (Brighton et al., 1976, 1978; Iannotti et al., 1994; Magne et al., 2003; Mansfield et al., 1999; Rajpurohit et al., 1999; L.N.Y Wu et al., 1995). As the hypertrophs start apoptosing, their membranes sprout blebs with annexin V plug-ins. These blebs are released as 100-nm vesicles, loaded with alkaline phosphatase, Ca2+-binding proteins, collagens II and X and proteases, with their membranes studded with channels formed by annexin V through which Ca2+ flows and type III Na-phosphate cotransporters through which phosphate flows to form hydroxyapatite crystals on the inner surface of the vesicle membranes. When the vesicles are loaded, their membranes are ruptured by the proteases and the hydoxyapatite needles are dumped into the extracellular fluid, where they trigger a self-sustaining mineral nucleation on the matrix using the ambient normal levels of Ca2+ and phosphate (H.C. Anderson, 1995; Bandorowicz-Pikula et al., 2001; Boskey, 1992; Hoshi and Ozawa, 2000; Kirsch et al., 1997, 2000; Plate et al., 1996). The cartilage calcifiers also now express collagen I, which bridges the transition from a mineralized type II/IX/XI collagen cartilage matrix to a type I collagen-rich bone matrix made by the progeny of the Ihh-stimulated perichondrial/periosteal cells (Kirsch et al., 1997).
As these hypoxic chondrocytes have been pushed downward, something else may happen that contributes to their injury and death. Their HIF-1αs have been busily stimulating the production of VEGF, which has been stimulating the extension of blood vessels into the bone to deliver osteoclast and osteoblast progenitors to, respectively, clear away the cartilage and replace it with bone. With these vessels comes oxygen, which instead of being a welcome relief for the hypoxic chondrocytes, delivers the coup de grace because by causing a wave of acetyl CoA from the accumulated lactate to surge into the reawakened mitochondria. This causes the mitochondria to spray deadly ROS (reactive oxygen species) into the cytoplasm, because the cytochrome oxidases are overwhelmed and cannot put all of the flood of electrons safely into water.
Interestingly, elasmobranchs (sharks and rays) appear to use part of the same PTHrP-dependent mechanism to make their cartilagenous skeleton (Trivett et al., 2002). But the final stages are missing. There is no cartilage calcification, destruction, and replacement by bone. Trivett et al. (2002) have suggested that ancestral elasmobranchs actually had the full endochondral bone-making process but lost it along the way. Indeed, there is still some calcification of these beasts' outer notochordal sheath.
Why don't cortical osteocytes strangle themselves like chondrocytes when buried in calcified matrix? The answer is really quite simple—that elegant network of canals, the lacunocanalicular osteointernet. The buried osteocytes are continuously supplied with food and oxygen from the blood flowing through the vessels of the cortical osteonal canals and their waste is carried out and dumped into the blood through their lacunocanalicular network that is connected to blood vessels and nerves, while chondrocytes are solitary, anti-social individuals who pay the price of their splendid isolation by having limited access to marrow blood vessels, which forces them to depend entirely on diffusion through the cartilage matrix to get the most of their supplies (Archer and Francis-West, 2003). While diffusion from the bone vascular "railheads" at the growth plate is at first quite adequate in the proliferative zone, because the matrix is largely water and the cells can operate under the hypoxia prevailing in the core regions conditions by upregulating their HIF-1α (R.B. Martin et al., 1998; Schipani et al., 2001), it becomes lethally inadequate when the matrix is mineralized in the hyertrophic zone.
Disabling the genes for Ihh, PTHrP, or PTHR1 releases the brakes on the proliferative "off-switch", and the cells prematurely rip open the "HT" envelope, commit apoptosis, and the bone under construction is abnormally short, and its owner is a dwarf (Amizuka et al., 1994; Karaplis, 2001; Karaplis et al., 1994; Lanske et al., 1996; St-Jacques et al., 1999). However, if the Ihh gene is still working, accelerating maturation, and therefore the production of Ihh-making prehypertrophic cells by disabling the PTHrP or PTHR1 genes, increases Ihh production by the prehypertrophic cells. The increasing Ihh tries to make up for the chondrocyte shortage caused by the premature maturation by increasing the flow of peri-articular cells into the dividing columns (T. Kobayashi et al., 2002). On the other hand, uncontrolled, persistent PTHR1 signaling delays the proliferative switch-off and maturation which would tend to reduce the appearance of Ihh-making prehypertrophic cells and thus slow up the flow of cells into the dividing columns (Schipani et al., 1995, 1997; Weir et al., 1996). Excessive Ihh production also inhibits the initiation of maturation by swiftly turning up the peri-articular cells' PTHrP production, which in turn would tend to feed back and reduce the appearance of Ihh-making prehypertrophic cells (T. Kobayashi et al., 2002; Minina et al., 2001; Vortkamp et al., 1996). Finally, Ihh-stimulated chondrocyte proliferation together with persistently firing, maturation-blocking PTHR1 receptors could be a deadly combination—it could cause tumors. And indeed it does! Hopyan et al. (2002) have found an abnormally signaling PTHR1 receptor in the cells of benign cartilage tumors from a human suffering from endochondromatosis (i.e., Ollier and Maffucci diseases).
Before leaving the cartilage and its chondrocytes, one might ask whether the chondrocytes in a permanent cartilage tissue such as articular (joint) cartilage, as opposed to the ephemeral cartilage in a growing femur, are equipped like osteocytes to measure and appropriately respond to strain. Articular chondrocytes, unlike osteocytes, do not form intercommunicating networks with direct access to blood vessels. They rely on diffusion of nutrients through the matrix. Therefore, they must function with very low oxygen levels. This means that they must use anerobic glycolysis instead of aerobic respiration to make their ATP fuel and thus do not have the large mitochondrial complements of osteoblasts and osteoclasts (Archer and Francis-West, 2003). Nevertheless they obviously do their job. To do this, they express the stress-reducing, function-maintaining, hypoxia-inducible factor compnents (HIF-1α and HIF-1β) and use the HIF-1α•HIF-1β pair and the AP-1 transcription factor complex to drive various gene expressions such as the genes for TGF-βs and for the components of the glycolytic mechanism (Archer and Francis-West, 2003; Bilton and Booker, 2003; L. E. Huang and Bunn, 2003; Lando et al., 2003). Moreover each articular chondrocyte has a single cilium which is pulled back and forth by attached collagen fibers and very likely sends Ca2+ signals to tell the cell to respond appropriately, when the cartilage is bent and stretched by joint bending and twisting (Archer and Francis-West, 2003; Gillespie, 1999; C. Jensen et al., 2004; Meier-Vismara et al., 1979; Poole et al., 1997, 2001; Whitfield, 2003a; Wilsman, 1978; Wilsman and Fletcher, 1978). But there is a huge difference between an articular chondrocyte and an osteocyte. The signal from a collagen-fiber-pulled cilium of an anti-social chondrocyte would not spread to its neighbors as would a signal triggered by the bending cilium flowmeter of an extensively networking osteocyte (Whitfield, 2003a).
Hopefully, the reader would like to take a quick glance at another of the many examples of PTHrP controlling tissue development—in this case, the skin epidermis. (We must not forget that some of our ancient predecessors had boney armorplates in their skin, although these were the products of neural crest epithelial cells (Smith and Hall, 1990).) Here, too, we see the same pattern as in growing bone—stem cells giving rise to rapidly proliferating transit amplifying progeny which stop proliferating, mature, and die in a curious hybrid process we have called diffpoptosis (differentiation and apoptosis) (Whitfield and Chakravarthy, 2001). The cycling transit amplifying basal keratinocytes start making and deploying PTH/PTHrP receptors that respond differently from the PTHR1 receptors of chondrocyte and osteoblast—normal keratinocytes may (Errazahi et al., 2003) or may not (Orloff et al., 1995; Whitfield and Chakravarthy, 2001) express their gene for the conventional PTHR1. At any rate, the signals from this receptor, whatever it may be, do not include adenylyl cyclase activation, although the cells can respond with a large burst of adenylyl cyclase activity to the β-adrenergic receptor-activating isoproterenol or to PTH-(1-34) when they have been artificially engineered to express PTHR1 (Orloff et al., 1995; Whitfield et al., 1992). However, the keratinocytes do not make the PTHrP to stimulate these unconventional PTH receptors until they lift off the basal lamina; otherwise, the PTHrP would interfere with their proliferation. Indeed, Menon et al. (1991) have shown that the proliferating basal cells are the only keratinocytes with substantial amounts of internal Ca2+, which according to the findings of Zuscik et al. (2002) should prevent PTHrP expression. These cells would be generating nuclear Ca2+ transients to start replicating their chromosomes and then to enter and complete mitosis (Whitfield and Chakravarthy, 2001). Then when the cells lift off the basal lamina, they stop proliferating, nuclear Ca2+ drops, and PTHrP is expressed (Juhlin et al., 1992; Menton et al., 1991; Whitfield et al., 2001). The amount of PTHrP coming down from the suprabasal cells tells the basal cells how many suprabasal cells are there and accordingly controls the rate of proliferation—a lot of cells in the stack means a lot of PTHrP coming down and a reduced production of basal cells, while a depleted cell stack means less PTHrP coming down and increased basal cell proliferation (Whitfield, 2004). However, the PTHrP expression in the lifted cells does not last. The external Ca2+ concentration triples or more when the cells rise through the granular layer and into the lower corneum and, as happens at the threshold of the hypertophic cartilage zone, this is enough to shut off PTHrP expression and trigger diffpoptosis, which converts granular keratinocytes into cornified corpses (Whitfield, 2004; Whitfield and Chakravarthy, 2001).
Before going any further, we must quickly look at what causes some basal stem cell daughters to lose their "stemcellness" and start differentiating. When these daughters reach a certain point on the border of the stem cell niche, they stop expressing Jagged or Delta molecules but express a receptor for these molecules known as Notch. This means that the Jagged or Delta on their stem cell sisters will bind to Notch, the signals from which causes the borderland daughters to become transit amplifying cells each of which produces a limited number of terminally differentiating offspring (Whitfield, 2004).
If the PTHrP from the suprabasal cells should also reach the dermal fibroblasts, it would activate their normal adenylyl cyclase-coupled PTHR1 receptors. This could cause the fibroblasts to make and maybe release, among other things, Jagged-1as happens in bone marrow (Calvi et al., 2004). This Jagged-1 (the skin's most expressed Notch ligand) would stimulate the Notch receptors on the overlying keratinocytes and drive their differentiation (Lowell et al., 2000; Nickoloff et al., 2002; Nicolas et al., 2003; Okuyama et al., 2004; Rangarajan et al., 2001; Thélu et al., 2002). This could be another way the amount of suprabasal PTHrP could control keratinocyte production. It would also mean that applying hPTH-(1-34) to the skin could reduce keratinocyte proliferation and drive differentiation (Whitfield, 2004).
It follows that a failure of keratinocytes to express PTHrP would cause a massively dysregulated cell production like that in psoriasis (Juhlin et al., 1992). The beefy red psoriatic patch is produced by the hyperproliferation of keratinocytes, which causes a pile up of cornified squames with an impaired ability to slough off (desquamate). It appears that the reason for the psoriatic hyperproliferation is underexpression of the Jagged-Notch-Fringe signaling system, which is upregulated in the basal and differentiating layers of the normal epidermis and maintains the normal relation between proliferation and differentiation (Thélu et al., 2002). The hyperproliferation of the basal keratinocytes and the suppression of the Delta-Notch control mechanism is somehow triggered locally by bacterially or otherwise activated Langerhans dendritic cells and driven for example by IL-8 from neutrophils streaming out of local blood vessels (Nickoloff et al., 2000). The keratinocytes in turn secrete autocrine (autostimulating)/paracrine (neighbor-stimulating) NGF that prevents them from self destructing while stimulating the T cells, which have been directed to the battle zone by the activated Langerhans cells coming to the lymph nodes from the skin, and recruiting other factor-spewing inflammatory cells (Lebwohl, 2004; Nickoloff et al., 2000). And the keratinocytes also emit NO that causes the local dermal blood vessels to dilate, which gives the patch its swollen redness and enhances the movement of activated Langerhans cells to the nodes, T-cells, and neutrophils to and from the battle zone (Nickoloff et al., 2000). This torrent of cytokines and other factors results in the basal layer having a proliferation-favoring, lower than normal, level of extracellular Ca2+ and the suprabasal cells having an abnormally high extracellular Ca2+ level, which could be why suprabasal psoriatic keratinocytes do not make PTHrP, the proliferative terminator (Holick et al., 1994; Menon and Elias, 1991; Zuscik et al., 2002).
Is this lack of PTHrP the reason for the excessive proliferation? Indeed, it seems to be! Topically applying an adenylyl cyclase-stimulating PTH such as hPTH-(1-34) or one of the newer smaller Ostabolin™ Family PTHs, but not hPTH-(7-34) which cannot stimulate adenylyl cyclase, stops the wild cell production and eliminates psoriatic plaques (Holick et al., 2003). How? Keratinocytes don't have a PTH/PTHrP receptor that activates adenylyl cyclase! The answer may lie with the dermal fibroblasts which do have the adenylyl cyclase-coupled PTHR1 and the ability of the closely related PTHR1-bearing, bone marrow stromal cells to make large amounts of Jagged-1 when treated with hPTH-(1-34) (Calvi et al., 2004; Whitfield, 2004). Such jagged-1 could restore order to the disturbed keratinocytes by reviving their Notch signaling-driven differentiaion mechanism (Lowell et al., 2000; Nickoloff et al., 2002; Nicolas et al., 2003; Okuyama et al., 2004; Rangarajan et al., 2001; Thélu et al., 2002; Whitfield, 2004).
PTHs and Rheumatoid Arthritis
In rheumatoid arthritis, a systemic autoimmune disease, immune-response cells such as macrophages, polymorphonuclear leukocytes, and T-lymphocytes invade the joints and start pumping out cytokines that produce an inflamed synovium, the membrane of the sac that encloses the fluid that lubricates the joints. In mice and rats, this cytokine storm can be started by injecting type II collagen intradermally, along with Freund's adjuvant, to induce an autoimmune reaction to the animal's own cartilage (Mori et al., 2002). Cytokines such as IL-1 and interferon-γ from this autoimmune response cause the formation of clusters of tall endothelial cells in the postcapillary venules of the synovial membrane that serve as the ports of entry of lymphocytes that form perivascular lymphoid aggregates or cuffs around the blood vessels like those of tonsil-like lymphoid tissues (Iguchi and Ziff, 1986; Ziff, 1993). These lymphocytes plus macrophages and plasma cells swarming through the layer of flat endothelial cells of the post-capillary venules release a swarm of cytokines which includes the mighty TNF-α, that pushes the synovial fibroblasts to proliferate and make things such as PTHrP and RANKL, which stimulate osteoclast generation and with it bone destruction (Funk, 2001; Funk et al., 2002; Goldring, 2002; Goldring and Gravallese, 2000, 2002; Gravallese and Goldring, 2000; Iguchi and Ziff, 1986; Mori et al., 2002). The surface of the underlying bone is primed for an osteoclast onslaught by proteases from both the invading leukocytes and the proliferating synovial fibroblasts. It becomes the site for the generation of osteoclasts from invading precursors that is driven initially by RANKL from the invading T-lymphocytes but later mostly from the cytokine-crazed, proliferating synovial fibroblasts (Goldring, 2002; Goldring and Gravallese, 2000, 2002; Gravallese and Goldring, 2000; Mori et al., 2002).
The synovium now relentlessly thickens into a predatory, giant ameba-like, bone-eating blanket or pannus (Latin for cover), a tumor-like blanket of inflammatory factor-secreting granulation tissue made up of polymorphonuclear leucocytes and lymphocyte invaders and the proliferating fibroblasts which vigorously make PTHrP that, like TNF-α, stimulates RANKL expression by the underlying osteoblastic cells (Funk et al., 2002; Goldring, 2002; Goldring and Gravallese, 2000, 2002; Gravallese and Goldring, 2000; Mori et al., 2002).
It must clearly be understood that inflammation and its associated cytokines do not by themselves cause the osteoclast attack and bone loss—something special is needed. This special something is the RANKL from T-cells and synovial fibroblasts as well as bone marrow stromal cells and osteoblasts (Bolon et al., 2002a, 2002b). Therefore, mice without functional RANKL genes can still develop severely inflammed joints in an autoimmune response response, but without RANKL no osteoclasts arrive on the scene to dig holes in the bones (Goldring, 2002; Goldring and Gravallese, 2000). However, the IL-1 cytokine from the immunocytes does stimulate osteoclast generation by its sharing with RANKL the ability to stimulate NF-κB in osteoclast precursors (Xing et al., 2003).
As the pannus grows, it, like a tumor, must make angiogenic factors such as VEGF (vascular endothelial growth factor) to make new blood vessels to get its oxygen, nutrients and along with these more blood-borne cytokine-producing lymphocytes, macrophages, neutrophils, and osteoclast precursors to help drive it forward (Funk et al., 2002). The endothelial cells of the pannus's blood vessels make PTHrP, but, unlike bone vascular endothelial cells, they don't have the PTHR1 receptors to respond to it, although they can ship some of it into their own nuclei instead of outside the cell to stimulate genes the products of which can prevent them from committing apoptotic suicide (Funk et al., 2002; Lam et al., 2002). The PTHrP from the endothelial cells hits the underlying vascular smooth mucle cells, which do have PTHR1 receptors (Funk et al., 2002). The cyclic AMP signals from the PTHR1 receptors cause the smooth muscle cells to relax and the blood vessels to dilate and thus carry more blood into the advancing pannus.
The steady pounding from the rising tide of RANKL-stimulating PTHrP from the pannus cells crawling over the joint causes the excessive osteoclast generation and digging to spread from the superficial subchondral bone to endosteal bone, which can eventually spread systemically to osteoporosis (Fu et al., 2002; Funk, 2001; Funk et al., 2002).
Obviously, the bone erosion by osteoclasts in a rheumatoid arthritic joint can be stopped, for example, with the very long-acting OPG, the RANKL decoy ligand, that prevents mature osteoclasts from setting up their sealing ring and resorbing bone by directly binding to a 140-kDa protein on their surfaces and takes RANKL out of action, which prevents osteoclast generation by depriving osteoclast precursors of signals from their RANK receptors (Bolon et al., 2002a, 2002b; Goldring, 2002; Goldring and Gravallese, 2002; Hakeda et al., 1998). Alternatively, osteoclasts can be disabled and killed by one of the bisphosphonate gang or calcitonin. It seems likely that a short treatment with single daily shots of one of the osteogenic PTHs, particularly hPTH-(1-31)NH2 (Ostabolin™) or [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin-C™), that are very poor stimulators of osteoclasts (Fraher et al., 1999; Jolette et al., 2003; Mohan et al., 2000; Whitfield, 2003b) should be able to do something that OPG or the antiresorptives cannot do—regrow the bone in the osteoclast-gnawed joints and as well as restore bone lost throughout the skeleton because of the spread of osteoclastogenic factors from the damaged joints, the lack of activity due to severe joint pain, and the use of osteoporosis-inducing glucocorticoids (Lane et al., 2000; Whitfield, 2003b). Indeed, the very promising first indications of this have been provided by Fukata et al.'s (2004) report that intermittent hPTH-(1-34) injections (3 times per week for 4 or 6 weeks) can prevent or stop the drop in cortical and trabecular BMD, increase trabecular thickness, and increase the trabecular mineral apposition rate, i.e., increase trabecular bone formation, in the tibias of female Sprague-Dawley rats with collagen-induced arthritis. But the PTH treatment did not affect the arthritis itself.
PTHs and Cancer—A Concern with All Growth Stimulators
Anything that directly or indirectly stimulates proliferation may also drive tumor formation. The level of the risk depends on the intrinsic genomic instability of the target cell population, the mutagenicity of the agent, and the number of cycles it forces its target cells to pass through (i.e, the duration of exposure), as well as the levels of other mutagens in its surroundings (Gibbs, 2003; Greaves, 2000). If there should be a carcinogenesis-initiating mutation in one of the osteoprogenitors stimulated to proliferate by the missionary short FGF-2 or IGF-I from the PTH-stimulated post-proliferative osteoblasts, there would be no danger if the mutant osteoprogenitor or its progeny could still eventually mature into irreversibly post-mitotic preosteoblasts and beyond after only a few additional cycles. But danger arises if the PTH-driven barrage of factors persists and mutant cells appear with disabled cycle suppressor genes, or permanently activated genes for cell cycle drivers, which start the cells independently making their own autocrine growth factors, any one of which locks them into the cycling mode. These mutations force the cells to keep proliferating and accumulating mutations that produce tumor-forming mutant progeny that do not commit apopototic suicide, probably because of the disabling or loss of p53, the mutation-detecting policeman that would normally condemn such mutants to apoptotic self-destruction (Whitfield and Chakravarthy, 2002) (fig. 30). Then in one of these rogue clones, a cell may undergo an additional, deadly mutation, which enables it to found a clone of widely metastasizing osteosarcoma cells, the ultimate release from the shackles of controlled multicellularity (fig. 30).
This appears to be what happens in a rat subjected to a life-long treament with a large dose of rhPTH-(1-34) (Forteo™). At the July 27, 2001 meeting of the FDA Endocrinologic and Metabolic Drugs Advisory Committee, it was revealed that a lifelong treatment with Lilly's rhPTH-(1-34) (Forteo™) is a potent osteosarcoma inducer in both female and male Fisher rats (Center for Drug Evaluation and Research, 2001; Vahle et al., 2002). For example, subcutaneously injecting 75 μg of Forteo™/kg of body weight each day into 60 rats starting when they were 6 to 8 weeks of age and continuing for 2 years increased the incidence of osteosarcoma from the 0.2 to 0.4% in the untreated control animals to 40% in the treated females and 60% in the treated males. At 13 months, tumors began appearing at several sites throughout the skeleton and then spread to various soft tissues in about one-third of the animals.
At first glance, this seems to mean that PTH treatment is downright dangerous. But it really is not! This kind of demonstration is ludicrously unrealistic. The daily dose of the fragment given to the rats (75 μg/0.4-0.6 kg of body weight) was about 400 times the 20 μg/40-60 kg of body weight recommended for treating osteoporosis (Center for Drug Evaluation and Research, 2001; Neer et al., 2001; Vahle et al., 2002). Although the extensively proliferating rat and human osteoprogenitors obey the same rules, they are probably more genomically unstable in an inbred rat than in a "wild-type" human, as indicated by the very high basal osteosarcoma incidence of 2000-4000 x 10-6 in the Fisher rats compared to the 1000-fold lower (3-5 x 10-6) incidence in humans (Gurney et al., 1995). Moreover, the short-lived, small-boned rats keep growing throughout their lives (though at a decelerating rate and with a decreasing osteogenic responsiveness to PTH (Walker, 1971)) and therefore maintain a substantial pool of immature osteoprogenitors for continuous bone making. But because the long-lived, large-boned humans close their bones' growth plates and stop growing, the size of their osteoprogenitor pools are limited to providing osteoblasts for the microcrack-repairing BMUs. Indeed, the osteosarcoma incidence in humans peaks in growing children around 13-15 years of age and then virtually vanishes after growth stops (Gurney et al., 1995), while a rat, especially a male rat, stays equivalent to a human child throughout its life. Neither the humans nor 80 cynomolgus monkeys treated in the trials with 8 times the human dose of Forteo™ for 18 months developed osteosarcoma (Center for Drug Evaluation and Research, 2001). Of course, if osteosarcomas are as rare in monkeys as in humans, the Lilly Forteo™ team would have needed a huge number of monkeys to detect an increased osteosarcoma incidence.
Of course, the fallacy of extrapolating the effects of a lifelong treatment with PTH in rats to humans is that a large dose of the peptide would never be given to a human for 60-90 years—only 20 μg will be given daily for no longer than 2 years! And in a talk (unfortunately unaccompanied by a hard copy record) given at the 24th meeting of the American Society for Bone and Mineral Research in San Antonio Texas, J. Vahle (Eli Lilly and Company) reported that when the rats were given 30 μg of hPTH-(1-34) for 2 years, 27.5% of them developed osteosarcomas, but when they were given the same dose for only 6 months between the ages of 2 and 8 months or between 6 and 12 months, the osteosarcoma incidence was only 3%, which was virtually the same as in untreated rats. In other words, a treatment with hPTH-(1-34) that was short enough not to trigger osteosarcoma was far more than enough to strongly stimulate bone growth. But we did not know whether a clinically realistic PTH treatment can promote or enhance the effectiveness of real carcinogens.
We were briefly reassured by the fact that none of 12,000 chronically hyperparathyroid Swedes has developed osteosarcoma (Center for Drug Evaluation and Research, 2001). But they didn't look hard enough—as of the first half of 2003, there were reports of four women with parathyroid adenomas and high blood concentrations of PTH, two of whom are Swedes! Wiig et al. (1971) reported two cases of hyperparathyroid Swedish women, a 46-year-old with tibial dedifferentiated chondrosarcoma and a 34-year-old with a high-grade mandibular osteosarcoma. Smith et al. (1997) described the case of a 49-year-old woman with a surgically removed parathyroid adenoma and multiple malignant bone tumors. The latest is a recent report by Betancourt et al. (2003) of a 69 year-old black woman who had a parathyroid adenoma that had raised the circulating PTH concentration above 1000 pg/ml (a normal concentration would have been 10-65 pg/ml), and along with this, she had developed a high-grade fibroblastic osteosarcoma. These osteosarcomatous women are very significant because they were all much older than than the usual osteosarcoma patient, who is 15 or less years old.
But there could be another danger—colon cancer! The proliferatively active colon cells, like osteoblasts, also have PTHR1 receptors (Gentili et al., 2001, 2002; Li et al., 1995). Signals from these receptors in rat duodenal cells rapidly (within just 5 minutes) cause a c-Src-kinase-mediated phosphorylation and activation of phosphatidylinositol-3 kinase, which in turn triggers a mitogenic ERK1•ERK2 cascade (see Pearson et al., 2001 for a review of these proliferation-stimulating enzymes) in rat intestinal cells (Gentili et al., 2001, 2002) just as the signals from PTHR1 receptors attached to NHERF-2 scaffolds do in PS120 fibroblasts (Mahon and Segre, 2002). Therefore, boluses of PTH could stimulate colon cell proliferation and at least promote colon carcinogenesis. Indeed, there are scattered reports of primary hyperparathyroidism with its 2-10-fold higher level of circulating PTH-(1-84) being associated with increased colon cancer incidence in humans (Artru et al., 2001; Conteas et al., 1988; Farr, 1976; Feig and Gottesman, 1987; Kawamura et al., 1999; Strodel et al., 1988).
It seems that colon cancer may not be a problem in rats. The lifetime treatment of Lilly's Fisher 344 rats with huge doses of hPTH-(1-34) (Forteo™) seems to have only induced osteosarcomas which could spread to soft tissue (Vahle et al., 2002). But we still had to find out whether a realistic PTH treatment, i.e., one that was just long enough to stimulate bone growth but no longer could either by itself trigger colon carcinogenesis or enhance the triggering of carcinogenesis by a colon carcinogen. In collaboration with Dr R.P. Bird of the University of Manitoba, we (Whitfield et al., 2003) have tested the ability of a strongly osteogenic 6-week treatment (i.e., once-daily injections of 20 nmoles/kg of body weight) of Sprague-Dawley rats with two PTH fragments, hPTH-(1-34) and [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 (Ostabolin C™), starting 9 weeks after OVX, to initiate colon carcinogenesis or increase the initiatory activity of two pretreatment injections (15 mg/kg of body weight) of a potent colon carcinogen—AOM (azoxymethane). The initiation of colon carcinogenesis by AOM is indicated by the appearance within just 6 weeks of as many as 263-273 of strikingly visible and very easily counted ACFs (aberrant crypt foci) per colon, a few of which if we had waited long enough would have sprouted tumors (Bird and Good, 2000; Whitfield et al., 2003). (In other words, one does not have to wait very long to find out whether an agent can trigger colon carcinogenesis, but of course it would be necessary to wait for 2 years to know how many of the aberrant crypt foci could generate tumors (Bird and Good, 2000)). While the daily PTH injections stimulated femoral bone formation, they did not cause the appearance of ACFs or affect the induction of ACFs by AOM or accelerate the progression of primary single ACFs to multiple ACFs (Whitfield et al., 2003). Nor did AOM affect the PTHs' abilities to stimulate bone formation (Whitfield et al., 2003).
While a PTH treatment that is no longer than needed to strongly stimulate bone formation does not by itself initiate colon carcinogenesis or enhance or accelerate the initiation of colon carcinogenesis by a potent carcinogen in rats, we can't lower our guard yet. There is a remote danger that a human (or primate)-type colon may respond differently to PTHs, as indicated by the seemingly increased incidence of colon cancer due to the continuous PTH stimulation in hyperparathyroid humans. Clearly, we must find out whether PTHs stimulate ACFs and ultimately tumor formation, at least in the colons of nonhuman primates.
The Very Bright Clinical Prospects of the PTHs
"Market was the key word here, for at big pharma, marketers, not scientists, decided which drugs the companies should pursue."….M. Shaynerson and M.J. Plotkin (2002).
The osteoporosis market is a rapidly growing "marketer's" dream. And several years ago there was no known bone-growing drug—only the ever-worrisome (for cancerophobics) estrogens and the other antiresorptives. So the marketeers at Eli Lilly decided that the venerable hPTH-(1-34) would be the ideal drug with which to break into this pharmaceutical gold mine. All they had to do was take it off the shelf, brush off the dust, spider webs, and dead flies, and infect Escherichia coli with a suitable expression vector into which the gene had been stitched to make it, and then sell tons of it to the growing hordes of fracturing "greys" to toughen up their fragile bones and strengthen anchorage of the artificial hips and knees they will also need. Thus was born Lilly's recombinant hPTH-(1-34), which was officially approved for clinical use under the trade name Forteo™ by the US FDA in November 2002.
It must clearly be understood that any PTH, such as the expensive hPTH-(1-34) (Forteo™), will never be used as a preventative like the much cheaper antiresorptives. This, of course, does not please the marketers who love "cash-cow" drugs like the diabetics' insulin and osteoclast-killing antiresorptives such as Fosamax™ that must be given continuously and thus produce a steady cash flow. Indeed, even space travelers need not use an expensive PTH to maintain their bones during flight because Fosamax™ would be a cheaper bone-maintaining agent. A PTH will just be used for no more than a couple years to restore severly deteriorated microarchitecture and lost bone strength and then turn the job over to an antiresorptive to protect the new "PTH bone" from still surging osteoclasts in postmenopausal women. It is also likely that the PTHs will be used to accelerate fracture mending not just in osteoporotics, but in people of all ages, especially children with their osteoblast-loaded, hence maximally PTH-responsive, bones (Koch, 2001; Walker, 1971), and they should be used to strengthen the depleted bones of astronauts returning from long space missions during which they had not been protected from bone loss by taking an antiresorptive such as Fosamax™. But the PTH's may be less effective or ineffective during long space missions because of a progressive loss of the ability of the osteoprogenitor cells, genomically unprimed by strain, in unloaded bone to respond to PTH-induced IGF-I (Bikle et al., 1994; Kostenuik et al., 1999)! The PTH's should also be able to strengthen bone eroded by arthritis (Fukata et al., 2004). And last, but by no means least for aging populations, the PTHs will soon be accelerating, greatly strengthening, and prolonging the anchorage of various orthopedic and dental implants (M. Allen et al., 2003; Kawane et al., 2002; Shirota et al., 2003; Skripitz and Aspenberg, 2001a, 2001b; Skripitz et al., 2000a, 2000b).
Beside Eli Lilly's "Forteo™", the first generation of PTHs include the recombinant hPTH-(1-84) (Preos™; NPS Pharmaceuticals Inc.'s ALX 1-11), which, like Forteo™, was also just taken off the shelf several years ago and put into bacteria by the now-defunct Allelix Corporation (Toronto, Canada), has reached the phase III trial level, but in my experience is no better at building bone than the smaller peptides and is virtually certain not to be modifiable for oral delivery as is one of the Ostabolins™ (Mehta et al., 2001; Whitfield et al., 1997c). These will soon be followed into the clinic by the new Ostabolin™ Family of designer PTH peptides such as Zelos Therapeutic's potently anabolic Ostabolin-C™ ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2), high doses of which, as I said in Section iv, actually decrease bone resorption while strongly stimulating bone growth in cynomologous monkeys (Jolette et al., 2003). In this case, there would not be the worrisome possibility of the PTH causing a stressful hypercalcemia in some patients and the huge advantage of being able to safely use higher doses.
Ostabolin™ (hPTH-(1-31)NH2) was made at the National Research Council of Canada during a project to locate the different signaling domains of hPTH-(1-34) using ROS 17/2 rat osteoblastic cells for signal testing (Jouishomme et al., 1992; Whitfield et al., 1996). The fragment, unlike hPTH-(1-34) (Forteo™), had a C-terminal NH2 group instead of an OH group, which gave it the ability to associate with PTHR1 and still stimulate adenylyl cyclase but not PKCs in ROS 17/2 cells as strongly as hPTH-(1-34), to resist serum carboxypeptidases, and to strongly stimulate bone growth in OVXed rats (Whitfield et al., 1996). Ostabolin™ was then made more effective by improving its receptor-binding ability in two ways (Barbier et al., 1997; Whitfield et al., 1998c). First, the peptide's major, but unstable, receptor-binding, α-helix between Ser17 and Gln29 was stabilized by tying Glu22 to Lys26 (a so-called lactam linkage), and second, by increasing the hydrophobicity of the hydrophobic face of the amphiphilic α-helix by replacing the natural polar Lys at position 27 with nonpolar Leu to produce ([Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2) or Ostabolin-C™ (i.e., cyclized Ostabolin) (Barbier et al., 1997; Whitfield et al., 1998a, 1998b, 1998c). The ability of such changes to increase the binding to PTHR1 receptors and adenylyl cyclase stimulation has been vividly demonstrated when hPTH-(1-28) with its very low receptor-binding and adenylyl cyclase-stimulating activities was changed into [Leu27]cyclo(Glu22-Lys26)hPTH-(1-28)NH2 ("mini-C"), and its receptor-binding and adenylyl cyclase-stimulating activities dramatically rose to those of hPTH-(1-34) (Whitfield et al., 2000c).
Then there will come newer oral, nasal, or topical designer analogs or mimetics. Those who are trying to develop orally deliverable insulin and other peptide hormones face the very difficult problem of making a peptide that maintains a functional level of the hormone in the blood. BUT the goal for oral PTH delivery is the exactly the opposite—an orally deliverable PTH that peaks only briefly in the circulation to transiently flood the circulation with normally rare N-terminal fragments and mimic the transient PTH boluses from daily subcutaneous injections. A recent report by Mehta et al. (2001) suggests that a "PTH pill" is possible. They have found that the osteogenic, second-generation hPTH-(1-31)NH2 (Ostabolin™ (Whitfield et al., 1996; Mehta et al., 2002)) incorporated into an orally administered solid capsule formulation is much more more able to get into the blood of dogs than Lilly's hPTH-(1-34) (Forteo™) in the same type of capsule. And what is more, it peaked only briefly in the blood between 120 and 150 minutes after giving the capsule!! In one example, the peak blood concentration of hPTH-(1-31)NH2 was an impressive 2155 ± 456 (SEM) pg/ml compared to a much more modest 359 ± 152 (SEM) pg/ml obtained with an equimolar amount of hPTH-(1-34). (This striking difference is probably due to hPTH-(1-31)NH2 ‘s terminal NH2 group, making it more resistant to gut proteases than hPTH-(1-34)). And both we and Glaxo-SmithKline have confirmed that this recombinant preparation is at least as strongly osteogenic in the OVXed rat as hPTH-(1-34)NH2, and Glaxo-SmithKline have announced in October 2004 that the orally administered peptide can get into the circulation of humans in an intact and active form. The desired brevity of the PTH peak was achieved by constructing the capsule so that it protected the enclosed PTH from the low gastric pH but released it upon contact with the high pH of the duodenum in a "puff " of acid, which along with the terminal NH2 group, would hold off the high pH (i.e., alkaline)-requiring proteases long enough for some of the little PTH peptide to escape from the dangerous intestinal lumen either through (the transcellular route) or between (the paracellular route) the gut-lining cells.
As I have said, the most exciting thing about the new peptides, Ostabolin™ and Ostabolin-C™ , is the inability or considerably reduced ability of daily boluses of even very large doses of them to stimulate osteoclasts. Thus, the label on the bottles will include the statement "does not cause the major side effect of hypercalcemia".
Other agents, such as BMPs, FGF-2, and IGF-I, are being considered as anabolic therpeutics. But as we have learned, the PTHs actually operate through the same Cbfa1/Runx-2 protein as BMP-2 acts and stimulate osteoblastic cells to make FGF-2 and IGF-I to mediate their osteogenic action and stimulate osteoprogenitors that are too immature to make PTHR1 receptors. In other words, the patient gets a triple whammy for the price of one in each PTH injection.
A possible drawback of the PTHs is that a lot of cells stretching from brain to bone to bowels express PTHR1 receptors at critical stages of differentiation, mainly for responding to locally produced PTHrP. However, no widespread effects have yet been seen. Obviously, the best strategy in the long run would be to make a functional PTH analog, perhaps by conjugating it with a bisphosphonate that localizes to bone. Such an analog would then have minmal side effects.
It is also possible that a "calcilytic" with a short cirulating half-life will be invented that can trigger short, osteogenic spurts of endogenous PTH secretion by briefly binding to and blinding the CaRs on the patient's parathyroid cells to circulating Ca2+ and thus fooling the cells into believing that there is a dangerously low level of circulating Ca2+ (Gowan et al., 2000; Nemeth and Fox, 2003). But osteoclasts are normally turned off by Ca2+ stimulating their CaR, so a calcilytic would jam the "off switch". However, if OPG, an estrogen, or a SERM are used to prevent the calcilytic from stimulating osteoclasts, the calcilytic-induced pulses of endogenous PTH might be strongly osteogenic. However, why would we need such a drug when blocking CaRs might be too risky because a lot of other cells use these sensors to control major body functions ranging from brain to gut and kidneys (Brown and MacLeod, 2001; Chattopadhyay and Brown, 2003)? The need for such a drug will vanish upon the arrival of the oral, non-hypercalcemia-causing PTHs such as Ostabolin™.
All of this means that the time has come when female and male osteoporotics or returning space travelers will be given (or, like diabetics, will give themselves) a daily shot of a PTH or swallow a daily Ostabolin™ or Ostabolin-C™ pill to make new bone. Returning space travelers may also take a daily PTH shot unless, of course, they were protected from bone loss by taking an antiresorptive drug such as alendronate (Fosamax™) during their flight. After the PTH treatment, he or she will take a weekly dose of Fosamax™ or maybe a subcutaneous shot of the very long-acting OPG (Bolon et al., 2002a, 2002b; Capparelli et al., 2003; Morony et al., 1999; Bekker et al., 2001) for 1 or 2 years to extend the mineralization and thus the optimal hardening of the new bone. Perhaps some day, the person may be able to take leptin (perhaps which has been modified to not cross the blood-brain barrier), along with the PTH to collaborate with bone building and harden the new bone by promoting mineralization and restraining osteoclasts (Whitfield, 2002b).
OGP—The Osteogenic Growth Peptide
Scooping out the marrow or driving a nail into the marrow cavity of a bone such as the tibia causes the marrow cavity to fill with a blood clot and releases a shower of signalers such as β2-microglobulin, IGF-I, PDGF, TGF-βs, and VEGF from platelets, shocked bone-lining cells, and injured vascular cells (Bab and Einhorn, 1993). They trigger and then drive a burst of blood vessel regeneration and bone formation (without an intermediate cartilaginous stage) that spreads from the cortical periphery to the center of the cavity and eventually fills the cavity with trabecular (cancellous) bone. At the peak of this osteogenic phase, the marrow cavity is filled with busy osteoblasts and both mineralized and unmineralized matrix. It is these that summon the osteoclasts and then the hempoietic cells. The osteoclasts chew up the trabecular bone plug, which is replaced by normal marrow (Bab and Einhorn, 1993). In other words, the primary bone plug and its builders establish the microenvironment for the new bone marrow with its stromal cells and their osteogenic progeny and hemopoietic stem cells and their osteoclastic and various other progeny.
Our hero, OGP, appears when the marrow is dug out or "nailed" and marrow regeneration starts. It comes from the clot and regenerating marrow to locally stimulate osteoprogenitors and bone growth. But it also escapes into the general circulation and stimulates bone formation in such distant places as the other tibia or the lower jaw. It is actually OGP-(10-14) (reviewed most recently by Bab and Chorev, 2002).
OGP starts out as a 19-amino acid piece of histone H4—actually H4's 85-103 C-terminal tail. Histone H4 genes (there are 4 of them) are turned on by the repair signals because repair requires cell proliferation, which in turn requires that all of the H1, H2A, H2B, H3, and H4 histone genes be switched on when the cells start replicating their chromosomes in preparation for later division. An ingenious, cell cycle-coupled, repair-amplifying by-product of the switch-on of the histone H4 genes is the translation of some of the genes' mRNA messages starting, not at the normal first site to make full-length H4 for new chromosomal nucleosomes, but at an alternative site—AUG85—to produce the preOPG-(85-103) fragment, which is then trimmed down to OGP-(90-103). The OGP-(90-103) is then dumped out of the cell along with OGP-(90-103)BP (binding protein) in an OGP•OGPBP complex. Outside the cell, OGP separates from the complex and is activated by being further trimmed down to OGP-(10-14). OGP-(10-14) can then bind to G-protein-coupled (specifically Gi) receptors on the producer cell as well as its neighbors (Bab and Chorev, 2002). Other OGPs make their way into the blood, where they combine with α2-macroglobulin to form a mobile OGP reservoir and travel to distant bones.
OGP stimulates trabecular bone formation in OVXed mice by stimulating the generation and activity of osteoblasts. But it seems to be not nearly as potent as the PTHs. In the presumably best published example provided by Chen et al. (2000), it did not raise the trabecular volume in OVXed mice above the sham value in intact mice as do the PTHs in OVXed rats. It can mildly stimulate the proliferation of osteoblastic precursor cells and fibroblasts, and it can stimulate the proliferation of hemopoietic stem cells (Bab and Chorev, 2002). And it can enhance fracture healing in rabbits under certain unstable conditions (immobilized with a plastic plate), but curiously not under "stable" conditions (a dynamic compression plate), but it does not increase callus size as do rhPTH –(1-34) (Forteo™) and [Leu27]cyclo[Glu22-Lys26]hPTH-(1-31)NH2 (Ostabolin-C™) (Andreassen et al., 1999, 2004; Sun and Ashhurst, 1998). However, OGP can significantly increase callus formation and the bridging of mid-femoral fractures with new bone in rats (Gabet et al., 2004).
The Statins
While the PTHs are the by far leading anabolic agents for treating osteoporosis and mending fractures, another family of drugs has been trying to challenge them but with very mixed results (Mundy, 2000; Whitfield, 2001, 2002a). These now-old drugs were discovered by Endo and colleagues during a search for the weapons of mass destruction that microorganisms use to defend themselves by blocking key sterol syntheses in their enemies (Endo, 1992). They are produced by molds such as Penicillium citrinum (Endo 1972, et al., 1977; Kuroda et al., 1977; Veillard and Mach, 2002). This ability to inhibit sterol production turned out to be useful for reducing blood cholesterol and preventing heart attacks and stroke (Veillard and Mach, 2002).
There are at least eight statins and undoubtedly more on the big pharmas' busy drawing boards in their test tubes. Four (lovastatin, mevaststin, pravastatin, simavstatin) are natural or fermentation-derived, while the other four (atorvastatin, cerivastatin, fluvastatin, rosurvastatin) are synthetic (Veillard and Mach, 2002). The various statins differ in their lipophilicity, which determines their ability to diffuse into the cell through its predominantly lipid plasma membrane. Thus, pravastatin has the lowest lipophilicity of the eight and thus cannot passively get through cell membranes as easily as, for example, cerivastsin or simvaststin.
Although the PTHs are very close to the ideal anabolic drugs, they must still be injected subcutaneously (though not for long), and they affect the many cells that share PTHR1 receptors with bone cells. But the injection drawback will soon disappear with advent of the Ostabolin™ family PTHs such as rhPTH-(1-31)NH2 (Mehta et al., 2001, 2002). The ideal drug would be a small molecule that stimulates bone growth as strongly as a PTH by selectively stimulating bone cells when given orally, or even better, topically.
Such a drug should stimulate osteoblast progenitors to make BMP-2. At this point, I must remind the reader of BMPs' main job—"A fundamental function of the BMPs is to induce the differentiation of mesenchymal cells toward cells of the osteoblastic lineage to promote osteoblastic maturation and function" (Canalis et al., 2003). The BMP in turn, would set up an escalating positive feedback cycle by stimulating the cells to express the BMP-2/4-stimulating Cbfa1/Runx-2 and with this commit their great-granddaughters to becoming mature osteoblasts (Canalis et al., 2003; Ji et al., 2000; Yamaguchi et al., 2000) (fig. 7).
Therefore, Mundy and his colleagues tested more than 30,000 small ‘natural’ compounds for the ability to stimulate BMP-2 expression (Garrett et al., 2001b; Mundy, 2000; Mundy et al., 1999). They used 2T3-BMP-2-LUC mouse osteoblasts that had been immortalized by carrying the SV-40 tumor virus's large T-antigen attached to a BMP-2 gene promoter and a firefly luciferase gene, which was also coupled to a BMP-2 gene promoter (Ghosh-Choudhury et al., 1996; Mundy et al., 1999). Their effort was rewarded when one of the compounds stimulated the luciferse gene's BMP-2 promoter at 0.1-5 μM (Garrett et al., 2001c; Mundy et al., 1999). This compound was lovastatin, a member of a family of very widely used, serum cholesterol-lowering drugs. Later they found that other "lipophilic" statins—atorvastatin, mevastatin, simvastatin—were also effective. Currently, the lipophilic synthetic cerivastatin is the most potent, but the old hydrophilic cardiovascular "work horse", pravastatin, does not stimulate the BMP-2 promoter (Garrett et al., 2001c).
Sugiyama et al. (2000), using human osteosarcoma cells with a BMP-2 or BMP-4 promoter-driven luciferase reporter gene, reported that simvastatin stimulated the BMP-2 promoter but not the BMP-4 promoter. Significantly, the statin did not affect BMP-2 expression in Chinese hamster ovary cells. Evidently osteoblasts, but not Chinese hamster ovary cells, had something statins needed to selectively stimulate a BMP-2 promoter. More recently, Maeda et al. (2001) have reported that simvastatin, at concentrations between 0.01 and 0.1 μM, stimulated MC3T3-E1 mouse preosteoblasts to express BMP-2 and to activate alkaline phosphatase and to mineralize the extracellular matrix they make.
This discovery was, and still is, very exciting because the statins were found at the end of the 1970s and have since then become very familiar drugs that have been given to many thousands of men and women throughout the world to reduce cholesterol levels and the risk of heart attacks and other cardiovascular problems (Herrington and Klein, 2001; Moghadasian and Frohlich, 2001; Whitfield, 2001, 2002a). After being converted in the body from lactone pro-drugs to the β-hydroxy acid drugs, they stop cholestrol synthesis by inhibiting the synthesis of its mevalonic acid precursor from HMG-CoA by HMG-CoA reductase (Fisher et al., 1999; Garrett et al., 2001b; Whitfield, 2001, 2002a) (fig. 15).
Even more exciting (if it can be repeated) is the reduced incidence of Alzheimer's dementia (odds ratio of 0.6 to 0.73) in people treated with the statins, but not other lipid-lowering drugs (i.e., acipimox, benzafibrate, ciprofibrate, clofibrate, fenofibrate, gemfibrozil, colestipol, cholestyramine, niacin/nicotinic acid), probably by reducing the cleavage of the amyloid precursor protein into plaque-forming β42 fragments (Fassbender et al., 2001; Jick et al., 2000; Panegyres, 2001; Wolozin et al., 2000).
Animal Studies
Mundy and his colleagues (Garrett et al., 2001c) first confirmed that the statins, atorvastatin, cerivastatin, fluvaststain, mevaststatin, and simvastatin, but not pravastatin, stimulated bone cells to make BMP-2. Then they showed that 0.25 and 0.5 μM lovastatin and simvaststain stimulated bone formation in mouse calvarial organ cultures by 2.8- and 4.3-fold, respectively, and that the bone-building action could be inhibited by the BMP-2 blocker noggin, which binds to BMP proteins and prevents them from binding to their receptors (reviewed by Garrett et al., 2001c; Mundy et al., 1999). Amazingly, only one 24-hour exposure to cerivastatin was enough to kick-start the osteogenic machine into working for 2 weeks! They also gave simvastatin to ovary-intact and OVXed rats by oral gavage for 35 days. Giving the animals 5 or 10 mg of simvastatin/kg/day nearly doubled trabecular volume and increased the bone formation rate by about 50% (Garrett et al., 2001c; Mundy et al., 1999). Oxlund and Andreassen (2002) have since reported that giving OVXed Wistar rats simvastatin by gastric tube twice each day for 3 weeks significantly increased the cortical mineral apposition rate and the bending strength of the tibial diaphysis. And Skoglund et al. (2003) have reported that simvastatin, which as we shall see below does not stimulate the growth of femoral cortical and trabecular bone in OVXed Swiss-Webster mice (von Stechow et al., 2003), significantly increases callus size and the force required to refracture femurs of mature male BALB/c mice.
Oral delivery of statins is not the best way to stimulate bone growth in mice and rats because the drugs can be converted into inactive metabolites by the cytochrome P450 system during their first passage through the liver (Gutierrez et al., 2000; Shepherd, 2000). In fact, the pharmaceutical industry has designed these drugs to selectively target the liver—the main site of cholesterol synthesis (Garrett et al., 2001c). The ideal would be to initially avoid the liver as much as possible by delivering them through the skin istead of the gut. And this can be done. Gutierrez et al. (2000) have found that lovastatin was, on a per-mg basis, a 50 times better bone maker when applied to rat skin than when given orally. And Whang et al. (2000) reported that lovastatin's potency as a mouse calvarial (skull bones) osteogen was increased as much as 80 times when delivered continuously from a poly[lactide-coglycolide] scaffold implanted into the skin over the mouse skulls.
But, and this is a very big, if not lethal, "but", some groups can't get statins to stimulate bone growth in mice and rats or murine calvarial explants as does the potently anabolic hPTH-(1-34) (Forteo™) or prevent OVX-induced bone loss in mice and rats (Crawford et al., 2001; Gasser, 2001; Sato et al., 2001; Staal et al., 2003; Yao et al., 2001)! For example, von Stechow et al. (2003) found that the mean trabecular bone volume in OVXed mice after 8 weeks of daily injections of 80 μg of hPTH-(1-34)/kg was 4 times higher than in vehicle-injected control mice, but simvastatin did not increase the serum osteocalcin level, did not affect trabecular volume or stimulate bone growth. However, Staal et al. (2003) have shown that cerivastatin does inhibit osteoclastic bone resorption in vitro in proportion to its ability to inhibit HMG-CoA reductase but not in the rat, although it does hit the rat's bone cells. The lack of an effect of statins such as lovastatin and simvastatin in the rat could be due to the ability of the tissues to mount a compensatory upregulation of HMG-CoA reductase that overrides the statin action (Staal et al., 2003). But, as we shall see further on, it could also be due to the statins inhibiting osteoblast proliferation (Reinholz et al., 2002). And Banu and Kalu (2002) have reported that cerivastatin, unlike hPTH-(1-34), does not prevent age-dependent bone loss in Sprague-Dawley rats.
Osteogenic Mechanism
Statins' primary action, from which all others seem to flow, is inhibition of HMG-CoA reductase (i.e., hydroxymethyl glutaryl-coenzyme A) (Fisher et al., 1999; reviewed by Garrett et al., 2001c; Veillard and Mach, 2002; and Whitfield, 2001, 2002a) (fig. 31). Besides preventing cholesterol, FPP (farnesyl pyrophosphate) and GPP (geranygeranyl pyrophosphate) syntheses, the inhibition of HMG-CoA reductase by a statin such as lovastatin will inhibit osteoclast generation and trigger osteoclasts' apoptotic self-destruction exactly like the FPP synthase-inhibiting N-BPs (N-containing bisphosphonates) such as alendronate (Fosamax™) (Benford et al., 1999, 2001; Fisher et al., 1999; Rezka et al., 1999; van Beek et al., 1999) (fig. 13 and fig. 14). However, in some cells, the statins may do something else, because they also selectively stimulate the expression of BMP-2, which in turn mediates the osteogenic response, probably by stimulating the expression of Cbfa1/Runx-2 and the shower of events it enables (fig. 7). This would mean that sometimes in rodents the statins are ‘double-barreled’ bone anabolics that murder osteoclasts and stimulate osteoblasts. To get the same effect with a PTH, it would have to be given along with leptin, OPG, or an antiresorptive such as raloxifene to prevent the PTH from stimulating osteoclasts.
At first sight, it would seem that if, or when, they happen, the stimulation of BMP-2 and osteogenesis should be independent from HMG-CoA-reductase inhibition, simply because the N-BP alendronate that inhibits FPP synthase doesn't stimulate osteoblast-specific genes or bone formation in rats (Onyia et al., 2002). But it isn't—Garrett et al. (2001b) have reported that mevalonate or geranylgeranyl pyrophoshate prevents lovastatin from stimulating BMP-2 expression and bone formation in rat bone cultures.
An exciting abstract by Garrett et al. (2000) suggested that the osteoblast-stimulating mechanism had been found. But this has turned out to be a false alarm. All that came from it was the discovery of different, but clinically useless, kinds of osteoblast stimulator!
The clue to the supposed new stimulators was the resemblance of the statin lactone ring to that of lactacystin, an inhibitor (from Streptomyces) of the cell's protein shredder/garburettor—the 26S proteasome (i.e., multicatalytic endopeptidase complex). They then found that lactacystin and other unrelated shredder inhibitors (the peptide aldehydes PS-1 and MG132; the α-β-epoxyketone epoxomicin) which (according to them) are unlikely to be HMG-CoA inhibitors, were bone anabolics. They all stimulated BMP-2 expression, the growth of cultured neonatal mouse bone and trabecular bone in rats and mice. Moreover, preosteoblasts and osteoblasts could also be made to express alkaline phosphatase and osteocalcin and make bone by disabling the ubiquitin-proteasome system (UPS), either by having the cells over-express a dominant negative mutant of the E3 ubiquitin ligase gene or by inhibiting the E3 ligase with N-Leu-Ala. Significantly, the chymotrypsin of the osteoblasts' shredder was more responsive to the inhibitors than other cells' chymotrypsins. This disabling of osteoblast UPS would be expected to stop the cytoplasmic turnover of the β-catenin transcription enhancer, which would then surge into the nucleus and stimulate genes for BMP-2, Cbfa1/Runx-2, cyclin D1, Myc, and osteopontin (Hershko et al., 2000; Zhao et al., 2002). And Chen et al. (2001) have shown that disabling the osteoblast UPS with epoxomicin or proteoasome inhibitor-1 also stops the Slimb ubiquitin ligase from inducing the cleavage of active big Gli3 into inactive little (the N-terminal short form) Gli3 which enables big Gli3 to accumulate and get into the nucleus to stimulate the bmp-2 gene and the production of autocrine/paracrine BMP-2. Operating through Smad second messengers, BMP-2, like the osteogenic PTHs operating through cyclic AMP (Fujita et al., 2000; Moore et al., 2000; Selvamurugan et al., 2000), would stimulate the cells to express Cbfa1/Runx-2 and drive the maturation of the cells into fully fledged bone-making osteoblasts (fig. 15 and fig. 16). As expected, inhibiting BMP-2 gene expression with Noggin prevented both the UPS inhibitors and statins from stimulating bone formation (Garrett et al., 2001a).
This was all very elegant. But Garrett and his group had not addressed THE KEY POINT—do statins in fact inhibit the shredder and cause a Gli3 build up? If they do stimulate bone formation by inhibiting the UPS, why are their anabolic actions, like the antiresorptive actions of N-bisphosphonates such as alendronate, prevented by mevalonate or geranylgeranyl pyrophosphate (Garrett et al., 2001a)? It would be expected that alendronate, for example, would be a potent stimulator of BMP-2 expression and bone growth, which it is not (Onyia et al., 2002). In fact, alendronate actually inhibits the expression of the genes for several bone formation markers in the proximal femoral metaphyses of OVXed rats (Onyia et al., 2002)! And now at least one lipophilic statin—lovastatin—has been shown to actually stimulate the shredder in MC3T3-E1 mouse calvarial preosteoblasts (Murray and Murray, 2001)! Alas, as so often happens, a beautiful idea has been murdered by an ugly fact. Obviously, although we know that UPS shredder inhibitors can stimulate osteoblasts and maybe bone formation, we must look elsewhere to find out how statins' stimulate bone growth.
Since statins stimulate eNOS, the constitutive Ca2+•calmodulin-activatable endothelial nitric oxide synthase, by extending the lifespan of its mRNA's by blocking the Rho GTPase's geranylgeranylation-dependent, membrane-associated, eNOS-suppressing activity (e.g., Feron et al., 2001; Laufs and Liao, 1998); since knocking out the eNOS gene interferes with osteoblast differentiation and migration to microdamage sites and reduces bone formation in mice and the osteogenic response of calvarial osteoblasts from these eNOS-/- mice (Afzal et al., 2000; Aguirre et al., 2001; Armour et al., 2001; Garrett et al., 2001b; van't Hof and Ralston, 2001; van't Hof et al., 2002); since inhibiting eNOS activity prevents statins from stimulating BMP-2 expression and bone formation in murine calvarial cultures (Garrett et al., 2001b), another place to look for the statin targets are the eNOS-sequestering caveolae ("little caves"), which are small (60-100 mm in diameter) clathrin-free pouches in the membranes of most cells including human and murine osteoblastic cells (Lofthouse et al., 2001; Mineo and Anderson, 2001; Razani et al., 2002; Solomon et al., 2000) (fig. 31).
Caveolar pouches are rich in sphingomyelin and glycosphinglolipids, but most important at this point is that their formation and function require cholesterol (Anderson and Jacobson, 2002; Mineo and Anderson, 2001; Razani et al., 2002). The key protein component of a caveola is one of 3 caveolins, particularly caveolin-1. Cholesterol has an extremely high affinity for caveolin-1, and palmitoylated molecules of caveolin-1 bind to cholestrol in cholesterol-rich lipid rafts floating in the plasma membrane. The cholesterol-attached caveolin-1 molecules then bind to each other with their oligermerization domains and in the process bend the raft down into a pouch. Caveolin-1 is one of a family of proteins known as MORFs (modifiers of raft function) that recruit and organize signal circuit boards or rafts into specifically functioning clusters. A large number of receptors, membrane proteins, nonreceptor tyrosine kinases, other enzymes, GTP-ases, signal protein adapters, nuclear receptors (androgen and estrogen receptors), structural proteins, and others (see Razani et al., 2002 for a recent list) are variously contained and arranged on the bottoms of these little pouches. eNOS is one of these and, like the others, is bound by its caveolin-binding motif consisting of Phe-Ser-Ala-Ala-Pro-Phe-Ser-Gly-Tryp amino acids to the CSDs (caveolin scaffolding domains) of caveolin-1 oligomers hanging down from the bottoms of the caveolae (Chambliss et al., 2000; Chen et al., 2001; Couet et al., 2001; Okamoto et al., 1998; Razandi et al., 1999, 2002; Razani et al., 1999, 2001, 2002; Solomon et al., 2000).
It is a general rule that binding to the CSDs silences various signaling proteins (Anderson, 1998; Couet et al., 2001; Feron et al., 2001; Fielding and Fielding, 2001; Razani et al., 2002; Xu et al., 2001). Thus, when palmitoylated eNOS is bound to the lipid raft domain, it is optimally active, but it shuts down when its CBD (caveolin-binding domain) in or near its catalytic region binds to caveolin-1's CSD. The caveolin-1•eNOS linkage must be broken if the eNOS is to function. The linkage can be broken and a burst of eNOS activity triggered by Ca2+•calmodulin generated by various signaling mechanisms (Razani et al., 2002). Prenylated (i.e.,geranylgeranylated), caveola-bound Rho can also inhibit eNOS (van't Hof et al., 2002a). Since caveolin-1 expression is controlled by the free cholesterol level and the cholestreol level must be at a certain threshold level in order to form caveolae (Fielding and Fielding, 2001; Razani et al., 2002), and since eNOS is bound and repressed by prenylated Rho in the caveolae (van't Hof et al., 2002a), reducing cholesterol production with a statin should, and indeed does, reduce caveolar formation as well as deprenylate and inactivate Rho and thus liberate eNOS which might still be clinging to the raft. This sidelining of Rho and reduction of caveolin-1 should raise the basal and Ca2+-stimulable eNOS activity and NO production that stimulates BMP-2 expression and bone growth (Feron et al., 2001) (fig. 31).
While the lipophilic statins and bisphosphates such as alendronate share the same ultimate osteoclast-killing action—a failure of FPP synthase to hold the deadly apoptosis-triggering caspase-3 in check (Chapter 3iii; fig. 13 and fig. 14)—only the statins can stimulate eNOS and bone growth. And according to van't Hof et al. (2002a), the anabolic action of mevastatin on mouse calvaria was reduced in eNOS-knock-out mice, while the bisphosphonate-like antiresorptive action was unaffected by knocking the eNOS gene out. So while the anti-osteoclast action of statins is mediated by the same lack of FPP synthase that is caused by alendronate and other N-bisphosphonates (fig. 13 and fig. 14), the statins' anabolic action, at least in mice, may be mediated by a stimulation of eNOS activity (fig. 31).
A model for how statins could stimulate eNOS has been found in vascular endothelial cells. In these cells, a statin such as atorvastatin starts by reducing cholesterol and with it caveolin-1. Released from the caveolin's inhibitory grip, eNOS binds to hsp (heat-shock protein) 90 (Brouet et al., 2001a, 2001b). The statin also stimulates PI-3 kinase (Dimmeler et al., 2001; Urbich et al., 2002) and induces the tyrosine phosphorylation of the hsp90 chaperone (Brouet et al., 2000). hsp90 then binds eNOS. PI-3 phosphorylates and thus activates Akt/PKB. The phospho-hsp90•eNOS complex then recruits the activated phospho-Akt/PKB by binding hsp90's 327-340 region to the phospho-Akt/PKB kinase's 229-309 region to form a phospho-hsp90•phospho-Akt/PKB•eNOS complex in which the phospho-Akt/PKB activates eNOS by phosphorylating eNOS's Ser 1177 (Brouet et al., 2001a, 2001b; S. Sato et al., 2000).
But there is a problem with eNOS and its NO product being responsible for statin-induced BMP-2 expression and osteogenesis (when it happens!). While inhibiting eNOs in mouse calvarial cells with L-NAME prevents the osteogenic response to statins, the same does not appear to apply to human cells (fig. 31). Ohnaka et al (2001) have reported that inhibiting Rho-associated protein kinase probably by preventing Rho-GTPase's isoprenylation and tethering to the caveolar scaffold mediated pitavastatin's stimulation of BMP-2 and the turning on of the osteocalcin gene in both normal human osteoblasts from surgically discarded bone chips and MG-63 human osteosarcoma cells (fig. 31). As would be expected if these statin-stimulated gene expressions were due to a failure of Rho-GTPase to be prenylated, plugged into the membrane and from there drive Rho-kinase, they were prevented from stimulating the osteoblasts by giving them geranylgeranyl pyrophosphate. But here's the whammy, the statin's actions were unaffected by inhibiting eNOS with L-NAME or, for that matter, by inhibitng various protein kinases such as PKCs, protein tyrosine kinases, and MAP kinases. Mediation by Rho-kinase was supported by its specific inhibitor, hydroxyfasudil (Nakamura et al., 2001; Shimokawa et al., 1999), also increasing BMP-2 and osteocalcin gene expressions (fig. 31).
The likelihood of such a Rho-kinase-mediated restraint of bone-specific gene expression such as BMP-2 and Cbfa1/Runx2 has very recently been extended to the cells in fetal mouse calvarial cultures by Harmey et al. (2004). They have shown that stimulating Rho-kinase with Pasteurella multocida toxin blocks the progression of the calvarial cells into bone nodule formers, while Rho-kinase inhibitors stimulated BMP-2 and Cbfa1/Runx2 expression and accelerated the differentiation of the calvarial cells into bone nodule-forming osteoblasts.
The identity of the Rho-kinase is unknown. But it seems that geranylgeranylated Rho-protein kinase plugged into a lipid-raft/scaffold and activated by geranylgeranylated GTP•Rho phosphorylates and inhibits a protein that otherwise would stimulate BMP-2 expression and osteoblast maturation. A model for this mechanism has been found in the rabbit basilar artery (Nakamura et al., 2001). Rho-kinase inhibits MLC (myosin light chain) phosphatase and thus promotes myosin light chain phosphorylation and the artery's contraction. But inhibiting the Rho-kinase with hydroxyfasudil releases MLC phosphatase which dephosphorylates myosin light chain and causes the artery to relax and dilate.
Recently another factor has been introduced into the tortured statin story by Reinholz et al. (2002). They reported that, as expected, lipophilic statins (mevastatin and simvastatin) stimulated BMP-2 expression in human fetal osteoblasts. In addition, the statins stimulated the cells to express RANKL and OPG with the induction of RANKL being greater than OPG so that there was a drop in the OPG/RANKL ratio. But despite the BMP-2 stimulation, there were drops in cell proliferation and matrix mineralization! Since anti-RANKL antibody partially prevented the reduction of hFOB cell proliferation but not the reduction of mineralization, it appears that statins reduce hFOB cell proliferation via BMP-2-stimulated RANKL expression and matrix mineralization by a RANKL-independent mechanism. In other words these results, if confirmed, mean that RANKL can substantially affect osteoblasts as well as kill osteoclasts. This suggests that osteoblasts have RANK receptors! And this has been shown by Faccio et al. (2003) to be true for murine primary calvarial osteoblastic cells. This BMP-2-RANKL interaction may be why statin anabolic action is so frustratingly uncertain.
Finally, we must not leave this section without mentioning that statins undoubtedly directly inhibit osteoclast generation as well as disable and murder mature osteoclasts just like N-bisphosphonates (fig. 13 and fig. 14). Also there is something else! You may remember that osteoclast generation is stimulated by signals from RANK receptors on osteoclast progenitors and that these signals activate the NF-κB (the "NK" in RANK) transcription factor. Statins have an anti-inflammatory action by preventing NF-κB activation in immune cells (Veillard and Mach, 2002). They may do this by inhibiting the prenylation (i.e., geranlygeranylation) of Rho-kinase, which prevents the kinase from being plugged into an appropriate membrane scaffold and triggering a kinase cascade that ultimately phosphorylates and switches off a nuclear receptor•PPARα (peroxisome proliferator-activated receptor-α). If not phosphorylated, PPARα combines with the nuclear retinoic acid X receptor (RXR), and the resulting complex activates genes with promoter-switch boxes having peroxisome proliferator response elements (Schoonjans et al., 1996; Veillard and Mach, 2002). But unphosphorylated PPARα also binds to NF-κB, which prevents it from binding to and activating the promoters of its many target genes (Delerive et al., 1999; Veillard and Mach, 2002)! Therefore, without active, membrane-inserted, isoprenylated Rho kinase, the unphosphorylated PPARα in statin-treated osteoclast progenitors can prevent RANK-activated NF-κB from driving their differentiation into mature osteoclasts.
Human Studies
If statins stimulate bone formation in rodents, there should be some indication of this in humans in the hundreds of thousands of people treated with these drugs. But, as we shall see, the anabolic effects of statins in postmenopausal women are just as uncertain as in rodents.
Statins Reduce Fracturing
Bauer et al.(1999) reported the results of an analysis of the data from a study of osteoporotic fractures (8412 women 65 years and older) and a fracture intervention trial (6459 women aged 55 to 80 years). Most of the 599 statin users among the 14871 women in the two studies had been given lovastatin. The relative risk of hip fracture was only 0.30 (95% confidence interval (CI), 0.08-1.18), and the overall relative risk of nonspine fractures was 0.83 (95% CI, 0.61-1.15).
Chan et al.(2000) reported that according to the records from six U.S. HMOs (health maintenance organizations), the relative risk of fracturing for patients who had had more than 13 dispensings of statins was 0.48 (95% CI, 0.27-0.83). Meier et al. (2000) found the same from 300 practices in the UK-based General Practice Research Database. Their base population was 91,611 patients who were at least 50 years old. Of these, 28,340 were taking lipid-lowering drugs. The adjusted odds ratio for fracturing in statin users relative to controls was 0.55 (95% CI, 0.44-0.69). Other lipid-lowering drugs did not affect the odds ratio. L. Wang et al. (2000) found similarly lowered fractured risks in the records of 6,110 New Jersey residents aged 65 years or more. Statin users had a relative hip fracture risk of 0.54 (95% CI, 0.36-0.82). Chung et al. (2000) examined the records of 69 type 2 diabetics, 33 of whom had not been given statins and 36 of whom had been given lovastatin, pravastatin (which would not have affected bone cells), and simvastatin. The spinal BMD rose 2.9% in the statin-treated group during 14 months. During the same time, the BMDs in the different regions of the femur and in the total hip increased by a significant 0.88-2.32% in the statin-treated men but by only 1.8% in the statin-treated women. Finally, M.H. Chan et al. (2001) have reported that giving simvastatin (20 mg/day for 4 weeks) to 17 hypercholesterolemic, nonosteoporotic patients increased one indicator of bone formation, the serum osteocalcin concentration. But it did not increase another formation indicator, the bone-specific alkaline phosphatase activity.
Pasco et al. (2002) have reported the results of the small-scale "Geelong" (Geelong, southeastern Australia) osteoporosis study. In this study, there were 16 statin users in the fracture group and 53 users in the nonfracture group with the fracture odds for the statin users (adjusted for BMD) at the femoral neck, spine, and whole body being, respectively, 0.45(95% CI, 0.25-0.80), 0.42 (95% CI, 0.24-0.75), 0.43 (95% CI, 0.24-0.78) of the values in the nonusers. Adjusting for age, weight, medications, and "life-style" factors did not "substantially" change these facture odds. Surprisingly, in view of the statin users' large, 60% reduction in fracture risk, there were at most only marginal (in fact, nonsignificant) increases in BMD in the femoral neck, spine, and whole body.
Montagnani et al. (2003) have treated 30 postmenopausal, hypercholesterolemic women (of average age, 61.2 ± 4.9 years) with simvastatin for one year. They found small increases in the femoral and lumbar BMDs by the end of the treatment, which was significantly higher than in the postmenopausal control women with normal blood cholesterol levels.
It seems from these reports that the statins are indeed safe, effective bone anabolics or pseudo-anabolics that can reduce the risk of hip and spine fractures by as much as 71%, depending on the extent of use. However, there are other reports, which just as convincingly indicate that lipophilic statins do not stimulate bone growth or affect the fracture risk.
Statins Don't Reduce Fracturing
A problem with retrospectively assessed effects of giving statins on fracturing is that one statin, the popular hydrophilic pravastatin, does not affect cultured bone cells or stimulate bone growth in rodents (Garrett et al., 2001). This explains the findings of Reid et al. (2001) on the effects of long-term pravastatin treatment on fracturing in 9,014 patients with ischemic heart disease. There were 107 pravastatin-treated (40 mg/day) patients admitted to hospital for fractures compared with 101 in the placebo-treated group (fracture risk, 1.05 (95% CI 0.80-1.16)). When fractures not requiring hospital admission were also included, the fracture risk was 0.94 (95% CI, 0.77-1.16).
LaCroix et al. (2003) searched the huge database in the Women's Health Initiative Observational Study (WHI-OS) and found no reducton of fracturing in statin (atorvastatin, lovastatin, pravastatin, fluvastatin)-treated women. In a study funded by Proctor and Gamble, van Staa et al. (2000) also found that cholesterol-lowering doses of atorvastatin, pravastatin, and simvastatin did not significantly lower the fracture risk. This was a much more extensive analysis of the UK General Practice Research database than that of Meier et al., which indicated that statins nearly halved the fracture risk. van Staa et al.'s conclusion that were was no reduction was based on a population of 216,062 fracture patients from 686 general practices, while Meier et al.'s conclusion was based on 91,611 fracture patients from "only" 300 practices. Sirola et al. (2001) and Solomon et al. (2001) could also find no evidence in the clinical databases that statins significantly lower fracture risk. Finally, Cauley et al. (2000) used the WHI-OS database to find out whether statins could enhance the BMDs of post-menopausal women. They found that although statin treatment did not affect fracture risk, treatment with a high-potency (based on cholesterol-lowering ability) lipophilic statin, such as atorvastatin or simvastatin for more than 3 years, only modestly protected hip and lumber vertebral BMD. Edwards et al. (2000) also found that treatment of 41 women with various statins (27 received the high potency simvastatin or atorvastatin) did not reduce fracturing, although there were 8.5 to 12% increases in the BMDs of hip and spine.
Cosman et al. (2001), like M. H. Chan et al. (2001), have carried out a small, short-term experiment on 14 postmenopausal women (mean age of 58 years) designed to find out what a 12-week treatment with 0.4 mg of cerivastatin/day might do to bone instead of serum cholesterol levels. Bone formation markers (type I procollagen propeptide and osteocalcin) did not change, but the resorption markers (urinary N- and C-terminal telopeptides) did drop slightly (<20%) within 6 weeks in the cerivastatin-treated group. They concluded that this statin did not detectably stimulate bone formation, but it might have had a modest bisphosphonate-like antiresorptive action. Moreover, the very small increase Montagnani et al. (2003) found in BMD and bone alkaline phosphatase activity in 30 hypercholesterolemic postmenopausal women treated for 1 year with simvastatin could have been due to as much to osteoclast inhibition as to direct stimulation of bone growth.
Rejnmark et al. (2004) have reported the results of an experiment to determine whether simvaststin can stimulate bone formation. Eighty-two otherwise healthy, osteopenic or osteoporotic postmenopausal women were randomized for daily treatment for 1 year (52 weeks) with 40 mg of simvastatin/day or placebo. None of the women were hyperlipidemic, but simvastatin reduced the serum cholesterol by an average 27%, but by 26 weeks after the end of the treatment, the cholesterol level had returned to the baseline value. This obviously effective dose of the statin did not affect indicators of bone formation (bone-specific alkaline phosphatase or osteocalcin) or indicators of bone resorption. Nor did it significantly affect the BMD of lumbar spine or hip. There was, however, a small increase in the forearm BMD in the treated group. The authors conclude that "our results do not support a general beneficial effect of simvastatin on bone".
However, statins are rather effective anti-osteoclast agents because they, like N-bisphosphonates such as alendronate (Fosamax™), can prevent the synthesis of the so-called isoprenoids farnesyl diphosphate and geranygeranyl diphosphate. Indeed, statins are better than N-BPs at inhibiting bone resorption by rabbit osteoclasts and by osteoclasts in mouse calvarial cultures (Rogers, 2003). These lipids are tacked by appropriate transferases onto the C-terminal cysteine residue of each member of the small GTPases of the Ras, Rho, and Rab families (Coxon and Rogers, 2003). This enables the "isoprenylated" GTPases to drive various osteoclast functions such as podosome formation, formation of the sealing ring, the transport to the apical ruffled border of key components, the fusion of the acid pumps to the ruffled border membrane, and the prevention of apoptosis (Coxon and Rogers, 2003). Obviously, the prevention of the GTPases' isoprenylation when a statin inhibits an osteoclasts's HMG-CoA reductase will throw a very large "monkey wrench" into the digger's machinery with disastrous consequences, including death for the digger but happily respite for the bone.
Statins' Clinical Prospects
As we learned above, some groups but not others have found statins to be potent bone anabolics for rats and mice, and their anabolic potency for human bones is also uncertain at best. There could be two reasons for this uncertainty. First, the statins are antiresorptives like the N-BP alendronate and owe their osteogenic effectiveness in young rodents to their potent osteoclast-disabling and killing abilities, which would leave the still-growing bones to the osteoblasts as happens in RANKL-knock-out mice. The spotty success in showing responses in rats could be due to compensatory surges of HMG-CoA reductase overriding the statin action, but humans do not have such a compensatory response (Staal et al., 2003). In mature humans with their tighly coupled osteoclast-osteoblast crack-repairing remodeling team, killing osteoclasts could counterproductively reduce BMU mobilization and thus impair microfracture repair. Alternatively, data such as those in the large WHI-OS and the UK's General Practice Research Database might be partly misleading because they come from patients treated for various lengths of time with different doses and with statins such as pravastatin that were meant to reduce cholesterol rather than fracturing.
However, there may be another explanation. As we learned above, the anabolic as opposed to antiresorptive actions of statins may be mediated through a burst of eNOS activity. If this be true, the anabolic effectiveness of statins would be affected by the lack of estrogen in OVXed rodents and postmenopausal women, which would drastically drop the eNOS level and increase the eNOS-inhibiting caveolin level (Farhat et al., 1996; Pavo et al., 2000; Stefano and Peter, 2001; Razandi et al., 2002; Tan et al., 1999; Whitfield et al., 2002; Zhu and Smart, 2003). In other words, in an estrogen-depleted OVXed rat or postmenopausal woman, there may not be enough eNOS for an optimally anabolic, fracture-lowering response to statins, but they could still kill osteoclasts and produce a N-BP-like antiresorptive response. But we can't ignore Ohnaki et al.'s (2001) finding that inhibiting eNOS did not affect pitavastatin-induced stimulation of BMP-2 and osteocalcin expression in human osteoblasts.
Clearly, there must be prospective placebo-controlled trials designed specifically to assess the osteogenicity rather than the lipid-lowering abilites of various doses of statins with reduced hepatoselectivity in postmenopausal patients. Moreover, the estrogen and eNOS statuses of the patients should be assessed along with BMD and fracturing. If topical delivery of a potent bone-anabolic statin turns out to be as feasible in humans as it has been in some rats, we will have a double-barreled osteoclast-killing/osteoblast-stimulating drug, like leptin (Whitfield, 2002b), but with the considerable bonus of protecting the patient from a wide variety of other things ranging from Alzheimer's dementia to cardiovascular diseases. However, if it should turn out that the statins require help from estrogens to be reliably anabolic for postmenopausal women, they would be considerably less attractive than the potently anabolic PTHs, which do not need help from estrogen, or anything else for that matter, to stimulate bone growth.
Finally, all of this can become irrelevant if the anti-proliferative action of the BMP-2-induced RANKL in mevastatin- or simvastatin-stimulated human fetal osteoblastic cells (remember mature osteoblasts can't proliferate) (Reinholz et al., 2002) can be confirmed, because this is most likely reason why the statins' anabolic actions are so inconsistent.
Surface Signaling Steroids: Real Anabolics or Pseudo-Anabolics?
Vitamin Ds
In 1998, Sicinski et al. reported the synthesis of new 1α,25-(OH)2-19-nor-vitamin D3 analogs such as 2MD (2-methylene-19-nor-(20S)-1α,25(OH)2D3), which are super-potent derivatives of the natural 1α,25-(OH)2-vitamin D3. Four years later, Shevde et al. (2002) reported that 2MD is a far more potent stimulator of osteoblast activities than 1α,25-(OH)2-vitamin D3. Indeed, Shevde et al. (2002) have written "…there is little evidence that vitamin D plays a direct role in new bone formation, apart from its action to maintain calcium and phosphorus levels in the blood ". But this osteoblast stimulation is compromised by 2MD being a 100-fold better stimulator of the RANKL gene (which has a vitamin D-responsive site in its promoter (Kitazawa et al., 2003)) and down-regulator of osteoprotegrin expression by presumably immature osteoblastic cells than the natural hormone. The result of this is the formation of more, bigger, and better bone-digging osteoclasts, which make 2MD a 30-fold more effective mobilizer of bone Ca2+.
As might be expected from this, orally giving 18.7 pmoles of 2MD/kg of body weight to old OVXed retired breeder rats for 7 days a week for 23 weeks increased total bone mineral density to a mean value 7% higher than in sham-operated rats and 9% higher than in untreated OVXed rats (Shevde et al., 2002). The steroid actually increased femoral trabecular bone volume. And, most important, it didn't do this by decreasing bone resorption, but by actually increasing the rate of bone formation which 1α,25-(OH)2-vitamin D3 can't do (Shevde et al., 2002). However, Shevde et al. did not say, by how much, 2MD increased the formation rate, which would be a surprising omission if the stimulation had been significantly large.
How might 2MD stimulate osteoblast activity? 1α,25-(OH)2-vitamin D3 activates two differently sited receptors (Boland et al., 2002; Farach-Carson, 2001; Farach-Carson and Xu, 2002; Nemere and Farach-Carson, 1998). One of these is the slowly acting, classical nVDR which operates in the nucleus and stimulates various genes and requires 1α,25-(OH)2-vitamin D3's 1α OH group to be activated while the other, somehow related receptor—the almost instantly responding, fast-acting mVDR—is plugged into the osteoblast's cell membrane and does not need the 1α,25-(OH)2-vitamin D3's 1a OH group to be activated. The rapidly triggered signals from the mVDR are similar, if not identical, to those from activated PTHR1—they include G-protein-coupled activations of c-Src protein-tyrosine kinase, adenylyl cyclase and phospholipases A, C, and D, which open Ca2+ channels and stimulate the release of Ca2+ from internal stores and activate PKCs, and set off MAP kinase cascades that stimulate various genes (Boland et al., 2002; Nemere and Farach-Carson, 1998). The fact that 2MD does not have a greater affinity for the nVDR (Shevde et al., 2002) suggests that the analog might have a greater affinitiy for the mVDR than 1α,25-(OH)2-vitamin D3. This would mean that 2MD is actually a PTH mimic! This also suggests that other analogs, such as 25(OH)-16ene-23yne-D3 (Farach-Carson, 2001), which cannot activate nVDR because it lacks 1α (OH), should also be PTH-like bone anabolics.
2MD certainly looks like a good candidate for admission to the exclusive Bone Anabolics Club. It has the great marketing advantage of being an oral drug. But it is also unfortunately a super osteoclast generator in rats, which raises the worrisome specter of hypercalcemia in osteoporotic humans, which has prevented worldwide approval of other vitamin Ds for treating osteoporosis. Indeed, as I said above. the osteoclast-stimulating RANKL gene in osteoblasts has a vitamin D-responsive site in its promoter (Kitazawa et al., 2003). It is important now to find out whether 2MD is a direct stimulator of osteoblast generation and activity like PTH, or whether the anabolic response is just a reaction to strong osteoclast activity. Can 2MD stimulate bone formation when given along with osteoprotegerin to prevent osteoclastogenesis?
Then there is ED-71 (1α,25-(OH)2-2β-(3-hydroxypropoxy)vitamin D3) (Kubodera et al., 2003). This is an excellent example of a pseudo-anabolic agent. In OVXed Wistar-Imamichi rats, ED-71 lowered bone resorption without affecting ongoing bone formation and thus stopped and reversed the OVX-induced BMD drop (Kubodera et al., 2003). This differential effect resulted in a large increase (double the value in sham-operated rats) in the concentration of osteocalcin from the unaffected osteoblasts in the blood of OVXed rats given 0.2 μg/kg of body weight twice a week for 3 months. However, the authors of this report did not tell us what ED-71 did to bone in sham-operated rats.
ED-71 also reduces osteoclast activity and increases the BMD of the L2-L4 vertebrae of osteoporotic men and women without causing hypercalcemia (Kubodera et al., 2003).
Unisex Steroids
It is possible to make politically correct unisex steroid analogs that can activate cell surface receptors in caveolar scaffolds but have no genomic (or genotropic) actions (Kousteni et al., 2001, 2002a, 2002b). One of these gender-nonspecific ligands is estren—4-estren-3α,17β-diol—which does not stimulate the transcription of genes with promoters having EREs (estrogen-response elements) (Kousteni et al., 2002a, 2002b). Remarkably, it can target the ligand-binding domains of androgen as well as estrogen receptors.
Starting continuous estren infusion (by implanting it in slow-release pellets) immediately after OVX into female Webster mice or after orchidectomy into male Webster mice significantly reduces or prevents any post-operative bone loss and prevents frequency of apopotic osteoblasts from rising above the sham level (Kousteni et al., 2002a; Manolagas et al., 1999). As expected from its lack of genotropic activity, the analog cannot affect uterine or seminal vesicle weight or stimulate the proliferation MCF-7 mammary cells (Kousteni et al., 2002a; Manolagas et al., 1999). However, and this is a very important "however", the continuous pounding with estren reduces, but by no means stops, as can estradiol, the loss of femoral trabeculae or raise the bone mass above the sham value as do the PTHs (Kousteni et al., 2002a; Manolagas et al., 1999). Furthermore, the analog even cannot hold the vertebral bone formation rate at the sham-operated value. In the experiment of Kousteni et al. (2002a), the bone formation rate/bone area (%/day) in the estren-treated OVXed mice was only 0.158 compared to 0.457 in the sham-OVXed mice. Nor does estren increase the number of osteoblasts per bone perimeter above the sham value. By contrast, an optimal injection of any of the anabolic PTHs starting immediately after OVX or several weeks after OVX can raise the trabecular thickness far above the sham level in rats (Whitfield et al., 1998c). Indeed, it would be interesting to find out what this unisex steroid analog does to bone loss in OVXed rats as well as mice and directly compare its actions to those of the PTHs.
A potent anabolic agent like a PTH stimulates bone growth in three ways—cause the reversion of lining cells to active osteoblasts, the secretion by PTHR1-expressing osteoblastic cells of missionary factors such as FGF-2, IGF-I, and TGF-β1 that stimulate the proliferation of osteoprogenitor cells, and prevent osteoblast and osteocyte apoptosis. It appears that estren signaling at best prevents osteoblast apoptosis, which is obviously not enough to cause supra-sham bone growth as do the PTHs. However, it is no more effective than dihydrotestosterone or estradiol in preventing osteoblast apopotosis. It seems that the anti-apoptotic action of estrogens on osteoblasts is due to the activation of the membrane-associated ERα, ERβ, or androgen receptors' ligand-binding domain, which triggers a Src/Shc/ERK1/2 signal cascade (Kousteni et al., 2002a, 2002b). Indeed, an analog such as 1,2,5-tris(4-hydroxylphenyl)-4-propylpyrazole that only has a genotropic action does not reduce osteoblastic apoptosis or increase osteoclast apoptosis as do the native sex steroids (Kousteni et al., 2002a).
All that can be said at the time of writing about this journalist-hyped unisex steroid estren is that it is only a weak pseudo-anabolic antiresorptive like the native sex steroids. But it could be a safe osteopenia/osteoporosis preventative or maintainer of new PTH-bone because it has none of the potential breast, seminal vesicle, or uterine cancer-promoting activities of the native steroids (Kousteni et al., 2002a). However, it has clearly shown how little an increase in bone growth can be produced by just blocking apoptosis.
Strontium, Calcium's Big Brother
Strontium (Sr) has been very recently hyped as the latest thing in the treatment of osteoporosis, as indicated by the title of a paper by Reginster et al. (2003)—"Strontium Ranelate: A New Paradigm for the Treatment of Osteoporosis". When two atoms of strontium combine with ranelic acid, they form strontium ranelate, Sr-ranelate, that effectively stimulates the proliferation of fibroblasts and pre-osteoblasts from fetal Sprague-Dawley rat calvaria and less effectively the proliferation of mature osteoblasts from these tissues. (But of course, this should have been expected, because as we have learned mature osteoblasts can't proliferate.) Neither Ca-ranelate nor ranelic acid alone can mimic the effects of Sr-ranelate on these cultured rat cells (Reginster et al., 2003). Sr-ranelate, also inhibits osteoclast activity on culture models without killing the cells or their ability to attach to bone (Reginster et al., 2003). Again, neither Ca-ranelate nor ranelic acid alone mimicked Sr-ranelate. Thus, the stimulation of osteoblast precursor proliferation and the inhibition of osteoclast activity are specific for Sr-ranelate.
It might be expected that Sr ranelate could act via the CaR (the Ca-sensing receptor) (Brown, 2003). And it does activate the CaR (Brown, 2003; Reginster et al., 2003). As we learned in Chapter 2, activating the CaRs on immature osteoblastic cells stimulates their proliferation and their migration to osteoclast excavation sites and at the same time inhibits osteoclast activity (Brown, 2003). But this would seem not to be the case, because Ca-ranelate does not mimic the stimulatory actions of Sr-ranelate on the rat calvarial cells. But wait! There is evidence from experiments on Chinese hamster ovary cells AtT20 cells that the responsiveness of the CaR to Sr-ranelate is enhanced by a high Ca2+ concentration such as 2 mM (Reginster et al., 2003). Therefore, since Sr-ranelate must operate in osteoclast excavation sites with extremely high Ca2+ concentrations (see Chapter 2), the Sr-ranelate action might operate at least in part through the CaRs on osteoblastic cells and osteoclasts. However, Pi and Quarles (2004) have produced evidence for bone cells having a novel Ca2+-sensing receptor that responds to strontium. Maybe this receptor belongs to the CaR-related mGluR (metabotropic glutamate receptor) family (Chattopadhyay and Brown, 2003; Hinoi et al., 2004; A.F. Taylor, 2002).
Of course, these culture models suggested that Sr-ranelate might be a powerful, "double-barreled" osteoblast stimulating/osteoclast-suppressing drug for stimulating bone growth in rats and humans. Indeed, at a dose of 900 mg/kg/day, Sr-ranelate increased the ultimate strength of mishaft femure and lumbar vertebrae of normal rats (Marie et al., 2001). At a dose range of 77-308 mg/kg/day, Sr-ranelate was able to substantially reduce the post ovariectomy loss of trabecular bone from an untreated 45% to 25% (Marie et al., 2001). However, unlike the powerfully anabolic PTHs, which can raise the post-ovariectomy trabecular bone volume to twice the value in the sham-operated control rats, Sr-ranelate couldn't even get the trabecular volume back to the sham value. The Sr-ranelate also reduces the trabecular bone loss in rat long bones caused by immobilization (Marie et al., 2001).
Of course, the big question is—what does Sr-ranelate do to osteoporotic women? After all, young rats, unlike old postemenopausal women, have growing bones, and the inhibition of osteoclasts alone can release the osteoclastic brakes on their bone growth. The answer has recently been published in the New England Journal of Medicine by Meunier et al. (2004). This paper reports the results of Sr-ranelate's Phase II trial. As expected, the results are less dramatic than in rodents. There were 723 placebo-treated and 719 Sr-ranelate-treated postmenopausal women of an average age of 69 years with a relative compliance with therapy of 85 and 83%, respectively. Two g of Sr-ranelate, dissolved in normal drinking water, were swallowed each day for 3 years. The Sr salt reduced the new vertebral fracture rate from a placebo rate of 32.8 to 20.9%. The data indicated that 9 patients would have to be treated in order to prevent 1 patient from having a vertebral fracture. The 41% reduction of the risk of new vertebral fractures by 3 years of treatment with Sr-ranelate, useful though it is, can't match the much higher 65% reduction by 21 months of treatment with hPTH-(1-34) (Forteo™) (Meunier et al. (2004); Neer et al., 2001).
In conclusion, Sr-ranelate can modestly increase bone formation in humans, probably as in rats by stimulating the proliferation of osteoprogenitor cells and somehow incapacitating osteoclasts without killing them. Swallowing Sr-ranelate may well be the the best thing to give astronauts to prevent microgravity-induced bone loss as they set out on long space voyages with initially strong bones. Sr-ranelate might also be a simple oral drug to swallow while self-injecting (or maybe also swallowing) PTHs to enhance the latter's formidable osteogenicity by stopping the peptides from stimulating osteoclasts, which is something osteoclast-killing alendronate (Fosamax™) can't do because it inhibits osteoblast activity (Onyia et al., 2002).
Afterword
We can now see lights, some dazzling but others much less so, at the Ends of the Osteoporotics', Arthritics', and Space Travelers' Tunnels.
Our aging and now space-faring world needs anabolic drugs that can directly stimulate bone growth instead of merely stopping bone loss, useful though this may be. They need such drugs to hasten fracture healing, treat osteoporosis, and strengthen the anchorage to bone and thus extend the working lives of artificial hips and knees. As we have seen, there are now some promisingly potent anabolic drugs. One of the PTHs, the venerable hPTH-(1-34) (Forteo™), was approved by the US FDA in October 2001, and others such as Ostabolin-C™ are starting their trials. They are safe peptides that use two other candidate anabolic agents, FGF-2 and IGF-I, to implement their potent osteogenicities. But they suffer from having to be injected subcutaneously into osteoporotics for a couple of years to grow new bone before the injections can be stopped and the patient be switched over to an oral antiresorptive to keep the new bone. However, the experience from the several trials indicates that self-injection by osteoporotics is not a significant deterrent, and there is one oral Ostabolin™ Family PTH already on the horizon. Close behind the PTHs may be the noninjectable, topically applicable, but erratic, statins with some bone anabolic and antiresorptive promise from experiments on mice and rats and with known beneficial cardiovascular and neurological side effects in humans. Then there is the "sleeper"—leptin—which to everyone's surprise may turn out to be a "star" member of the Bone Builders' Club.
Farewell dear reader, but do stay tuned so as not to miss the exciting things that will be happening at the cutting edge of research and development of bone-growing drugs. As you have seen, this is really a work in progress loaded with rewards for researchers, big pharmas, and most important for aging, fracture-prone, fragile skeletons.
References
- 1.
- Aarts MM, Davidson D, Corluka A. et al. Parathyroid hormone-related protein promotes quiescence and survival of serum-deprived chondrocytes by inhibiting rRNA synthesis. J Biol Chem. 2001;276:37934–37943. [PubMed: 11489898]
- 2.
- Aarts MM, Rix A, Guo J. et al. The nucleolar targewting signal (NTS) of parathyroid hormone-related protein mediates endocytosis and nucleolar translocation. J Bone Miner Res. 1999;14:1493–14503. [PubMed: 10469277]
- 3.
- Adams C, Mansfield K, Perlot RL. et al. Matrix regulation of skeletal cell apoptosis: Role of calcium and phosphate atoms. J Biol Chem. 2001;276:20316–20322. [PubMed: 11278303]
- 4.
- Afzal F, O'Shaughnessy M, Nohadani RM. et al. Osteoblast growth and differentiation is retarded in endothelial nitric oxide synthase knockout mice (abstract). J Bone Miner Res. 2000;15:S217.
- 5.
- Adams JM. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. [PubMed: 14561771]
- 6.
- Aguirre J, Buttery L, O'Shaughnessy M. et al. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 2001;158:247–257. [PMC free article: PMC1850250] [PubMed: 11141498]
- 7.
- Aguirre JL, Plotkin LI, Strotman B. et al. The anti-apoptotic effects of mechanical stimulation in osteoblasts/osteocytes are transduced by the estrogen receptor (ER): A novel ligand-independent function of the ER. J Bone Miner Res. 2003;18:S71.
- 8.
- Akhter MP, Kimmel DB, Recker RR. Effect of parathyroid hormone (HPTH[1-84]) treatment on bone mass and strength in ovariectomized rats. J Clin Densitom. 2001;4:13–23. [PubMed: 11309515]
- 9.
- Alieva IB, Gorgidze LA, Komarova YA. et al. Experimental model for studying the primary cilia in tissue culture cells. Membr Cell Biol. 1999;12:895–905. [PubMed: 10512057]
- 10.
- Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. [PubMed: 10845097]
- 11.
- Ali AA, Plotkin LI, Foote IP. et al. Bcl-2 is a pivotal mediator of the anti-apoptotic effect of PTH on osteoblasts: Evidence from RNA-silencing and Bcl-2-deficient mice (abstract). J Bone Miner Res. 2003;18:S73.
- 12.
- Alkhiary YM, Gerstenfeld LC, Cullimane DM. et al. Parathyroid hormone (1-34; teriparitide) enhances experimental fracture healing (abstract). J Bone Miner Res. 2003;18:S24.
- 13.
- Allen CM, Chakravarthy B, Morley P. et al. hPTH-(1-34) and hPTH-(1-31)NH2 prevent phosphate from killing MC3T3-E1 preosteoblasts and HKRKB7 pig kidney cells (abstract). J Bone Miner Res. 2002;17:S390.
- 14.
- Allen MJ, Schoonmaker JE, Mann KA. et al. PTH analogs enhance bone formation at a weight-bearing cement-bone interface. Trans Orthoped Res Soc. 2004
- 15.
- Alvarez J, Sohn P, Zeng X. et al. TGFβ2 mediates the effects of hedgehog on the hypertrophic differentiation amd PTHrP expression. Development. 2002;129:1913–1924. [PubMed: 11934857]
- 16.
- Alvarez M, Thunyakitpisal P, Morrison P. et al. PTH-responsive osteoblast nuclear matrix architectural transcription factor binds to the rat type I collagen promoter. J Cell Biochem. 1998;69:336–352. [PubMed: 9581872]
- 17.
- Alvarez-Buylla A, Garcia-Verdugo J, Tramontin AD. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001;2:287–293. [PubMed: 11283751]
- 18.
- Amalric F, Baldin V, Bosc-Bierne I. et al. Nuclear translocation of basic fibroblastic growth factor. Ann NY Acad Sci. 1991;638:127–138. [PubMed: 1785798]
- 19.
- Amin D, Cornell SA, Gustafson SK. et al. Bisphosphjonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis. J Lipid Res. 1992;33:1657–1663. [PubMed: 1464749]
- 20.
- Amizuka N, Karaplis AC, Henderson JE. et al. Haploinsufficiency of parathyroid hormone related peptide (PTHrP) results in abnormal postnatal development. Dev Biol. 1996;175:166–176. [PubMed: 8608863]
- 21.
- Amizuka N, Warshawsky H, Henderson JE. et al. Parathyroid hormone-related peptide-depleted mice showabnormal epiphyseal cartilage development and altered endochondral one formation. J Cell Biol. 1994;126:1611–1623. [PMC free article: PMC2290963] [PubMed: 8089190]
- 22.
- Amling M, Neff L, Tanaka S. et al. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. [PMC free article: PMC2132464] [PubMed: 9008714]
- 23.
- Anderson RGW. The caveolar membrane system. Annu Rev Biochem. 1998;67:199–225. [PubMed: 9759488]
- 24.
- Anderson RGW, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296:1821–1825. [PubMed: 12052946]
- 25.
- Anderson HC. Molecular biology of matrix vesicles. Clin Orthop. 1995;314:266–280. [PubMed: 7634645]
- 26.
- Andreassen TT, Ejersted C, Oxlund H. Intermittent parathyroid hormone (1-34) treatment increases callus formation and mechanical strength of healing rat fractures. J Bone Miner Res. 1999;14:960–968. [PubMed: 10352105]
- 27.
- Andreassen TT, Oxlund H. The influence of combined parathyroid hormone and growth hormone treatment on cortical bone in aged ovariectomized rats. J Bone Miner Res. 2000;15:2266–2275. [PubMed: 11092409]
- 28.
- Andeassen TT, Willick GE, Morley P. et al. Treatment with parathyroid hormone hPTH(1-34),hPTH(1-31), and moncyclic hPTH-(1-31) enhances fracture strength and callus amount-after withdrawal fracture strength and callus mechanical quality continue to increase. Calcif Tissue Int. 2004;74:351–356. [PubMed: 15255072]
- 29.
- Anreatta RH, Hartmann A, Kamber JA. et al. Synthese der Sequenz 1-34 von menschlichem Parat-hormon. Helv Chim Acta. 1973;56:470–473. [PubMed: 4721748]
- 30.
- Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–57. [PubMed: 10739651]
- 31.
- Archer CW, Francis-West P. The chondrocyte. Int J Biochem Cell Biol. 2003;35:401–404. [PubMed: 12565700]
- 32.
- Arita S, Ikeda S, Ito M. et al. Intermittent hPTH administration increases bone strength in rat lumbar vertebral body after ovariectomey with cortical bone mass increase (abstract). J Bone Miner Res. 2002;17:S274.
- 33.
- Armour KE, Armour KJ, Gallagher ME. et al. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. 2001;142:760–766. [PubMed: 11159848]
- 34.
- Artru P, Tournigand C, Mabro M. et al. Hypoparathyroïdie primitive associée au cancer colique. Gastroenterol Clin Biol. 2001;25:208–209. [PubMed: 11319452]
- 35.
- Ashby MC, Tepikin AV. ER calcium and the functions of intracellular organelles. Cell Devel Biol. 2001;12:11–17. [PubMed: 11162742]
- 36.
- Atkins GT, Kostakis P, Pan B. et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18:1088–1098. [PubMed: 12817763]
- 37.
- Aubin JE. Advances in the osteoblast lineage. Biochem Cell Biol. 1998;76:899–910. [PubMed: 10392704]
- 38.
- Atkinson PJ, Hallsworth AS. The changing structure of aging human mandibular bone. Gerodontology. 1983;2:57–66.
- 39.
- Aubin JE. The role of osteoblastsIn: Henderson JE, Goltzman D, eds.The Osteoporosis PrimerCambridge: Cambridge University Press,200018–35.
- 40.
- Aubin JE. Regulation of osteoblast formation and function. Rev Endocrine Metab Dis. 2001;2:81–94. [PubMed: 11704982]
- 41.
- Aubin JE, Bonnelye E. Osteoprotegerin and its ligand: A new paradigm for regulation of osteoclastogenesis and bone resorption Medscape Women's Health 20005http//wwwmedscapecom . [PubMed: 10792853]
- 42.
- Aubin JE, Triffitt JE. Mesenchymal stem cells and osteoblast differentiation In: Principles of Bone Biology. 2nd edSan Diego: Academic Press,2002159–91.
- 43.
- Awata H, Huang C, Handlogten ME. et al. Interactions of the calcium-sensing receptor and filamin, a potential scaffolding protein. J Biol Chem. 2001;276:34871–34879. [PubMed: 11390379]
- 44.
- Azarani A, Goltzman D, Orlowski J. Parathyroid hormone and parathyroid hormone-related peptide inhibit the apical Na+/H+ exchanger NHE-3 isoform in renal cells (OK) via a dual signaling cascade involving protein kinase A and C. J Biol Chem. 1995a;270:20004–20010. [PubMed: 7650018]
- 45.
- Azarani A, Orlowski J, Golzman D. Parathyroid hormone and parathyroid hormone-related peptide activate the Na+/H+ exchanger NHE-1 isoform in osteoblastic cells (UMR-106) via a cAMP-dependent pathway. J Biol Chem. 1995b;270:23166–23172. [PubMed: 7559463]
- 46.
- Bab I, Chorev M. Osteogenic growth peptide: From concept to drug design. Biopolymers (Peptide Science). 2002;66:33–48. [PubMed: 12228919]
- 47.
- Bab I, Einhorn TA. Regulatory role of osteogenic growth polypeptides in bone formation and hemopoiesis. Crit Rev Eukaryot Gene Express. 1993;3:31–46. [PubMed: 8439709]
- 48.
- Bakker AD, Klein-Nulent J, Burger EH. Mechanotransduction in bone cells proceeds via activation of COX-2, but not COX-1. Biochem Biophys Res Commun. 2003;305:677–683. [PubMed: 12763047]
- 49.
- Bakker RT. The Dinosaur HeresiesNew York: William Morrow and Compnay, Inc., 1986.
- 50.
- Baldock PA, Sainsbury A, Couzeau M. et al. Hypothalamic Y2 receptors regulate bone formation. J Clin Invest. 2002;109:915–921. [PMC free article: PMC150931] [PubMed: 11927618]
- 51.
- Bandorowicz-Pikula J, Buchet R, Pikula S. Annexins as nucleotide-binding proteins: Facts and speculations. BioEssays. 2001;23:170–178. [PubMed: 11169590]
- 52.
- Banerjee C, Javed A, Choi JY. et al. Differential regulation of the two principal Runx2/Cbfa1 n-terminal isoforms in response to protein morphogenic protein-2 during development of the osteoblast phenoptype. Endocrinology. 2001;142:4026–4039. [PubMed: 11517182]
- 53.
- Banu J, Kalu DN. Effects of cerivastatin and parathyroid hormone in male Sprage-Dawley rats (abstract). J Bone Miner Res. 2002;17:S296. [PubMed: 12110431]
- 54.
- Barber DL, Ganz MB. Guanine nucleotides regulate β-adrenergic activation of Na-H exchange independently of receptor coupling to Gs. J Biol Chem. 1992;267:20607–20612. [PubMed: 1328204]
- 55.
- Barber DL, Ganz MB, Bongiorno PB. et al. Mutant constructs of the β-adrenergic receptor that are uncoupled from adenylyl cyclase retain functional activation of Na-H exchange. Mol Pharmacol. 1992;41:1056–1060. [PubMed: 1319545]
- 56.
- Barbier J-R, Neugebauer W, Morley P. et al. Bioactivities and secondary structures of constrained analogues of human parathyroid hormone: Cyclic lactams of the receptor-binding region. J Med Chem. 1997;40:1373–1380. [PubMed: 9135034]
- 57.
- Bauer DC, Mundy GR, Jamal SA. et al. Statin use, bone mass and fracture: An analysis of two prospective studies (abstract). J Bone Miner Res. 1999;14:S179.
- 58.
- Barrett MG, Belinsky GS, Tashjian Jr AH. A new action of parathyroid hormone, receptor-mediated stimulation of extracellular acidification in human osteoblast-like SaOS-2 cells. J Biol Chem. 1997;272:26346–26353. [PubMed: 9334207]
- 59.
- Barry JA, Tanner SJ, Peyton A. et al. Mechanical loading and PTH stimulation of DNA synthesis (abstract). J Bone Miner Res. 2002;17:S329.
- 60.
- Bassilana F, Susa M, Keller HJ. et al. Human mesenchymal cells undergoing osteogenic differentiation express leptin and and functional leptin receptors (abstract). J Bone Miner Res. 2000;15:S378.
- 61.
- Bauer E, Aub JC, Albright F. Studies of calcium and phosphorus metabolism: V. Study of the bone trabeculae as a readily available reserve supply of calcium. J Exp Med. 1929;49:145–162. [PMC free article: PMC2131520] [PubMed: 19869533]
- 62.
- Baylink DJ, Strong DD, Mohan S. The diagnosis and treatment of osteoporosis: Future prospects. Mol Med Today. 1999;5:133–140. [PubMed: 10203737]
- 63.
- Becamel C, Alonso G, Galeotti N. et al. Synaptic multiprotein complexes associated with 5-HT2c receptors: A proteomic approach. EMBO J. 2002;21:2332–2342. [PMC free article: PMC126011] [PubMed: 12006486]
- 64.
- Beck Jr GR. Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem. 2003;90:234–243. [PubMed: 14505340]
- 65.
- Beck Jr GR, Knecht N. Osteopontin regulation by inorganic phosphate is ERK 1 / 2, PKC and proteasome dependent. J Biol Chem. 2003;278:41921–41929. [PubMed: 12920127]
- 66.
- Beck Jr GR, Moran E, Knecht N. Inorganic phosphate regulates multiple genes during osteoblast differentiation, including Nrf2. Exp Cell Res. 2003;288:288–300. [PubMed: 12915120]
- 67.
- Beck Jr GR, Sullivan EC, Moran E. et al. Relationship between alkaline phosphatase levels, osteopontin expression, and mineralization in differentiating MC3T3-E1 osteoblasts. J Cell Biochem. 1998;68:269–280. [PubMed: 9443082]
- 68.
- Beck Jr GR, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci USA. 2000;97:8352–8357. [PMC free article: PMC26951] [PubMed: 10890885]
- 69.
- Beir F, Ali Z, Mok D. et al. TGFβ and PTHrP control chondrocyte proliferation by activating cyclin D1 expresssion. Mol Biol Cell. 2001;12:3852–3863. [PMC free article: PMC60760] [PubMed: 11739785]
- 70.
- Bekker PJ, Holloway D, Nakanishi A. et al. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16:348–360. [PubMed: 11204435]
- 71.
- Belinsky GS, Morley P, Whitfield JF. et al. Ca2+ and extracellular acidification rate responses to parathyroid hormone fragments in rat ROS 17/2 and human SaOS-2 cells. Biochem Biophys Res Commun. 1999;266:448–453. [PubMed: 10600523]
- 72.
- Belinsky GS, Tashjian Jr AH. Direct measurement of hormone-induced acidification in intact bone. J Bone Miner Res. 2000;15:550–556. [PubMed: 10750570]
- 73.
- Bellido T, Plotkin LI, Davis J. et al. Protein kinase A-dependent phosphorylation and inactivation of the pro-apoptotic protein Bad mediates the anti-apoptotic effect of PTH on osteoblastic cells (abstract). J Bone Miner Res. 2001;16:S203.
- 74.
- Bellido T, Plotkin LI, O'Brien A. et al. PTH-mediated control of proteasome-mediated degradation of Runx2/Cbfa1: A pivotal determinant of the longevity of PTH-initiated anti-apoptosis signaling in osteoblastic cells. J Bone Miner Res. 2002;17:S128.
- 75.
- Benford HL, Frith JC, Auriola S. et al. Farnesol and geranylgeraniol prevent activation of caspases by aminobisphosphonates: Biochemical evidence for two distinct pharmacological classes of bisphosphonate drugs. Mol Pharmacol. 2001;56:131–140. [PubMed: 10385693]
- 76.
- Benford HL, McGowan NW, Helfrich MH. et al. Visualization of bisphosphonate-induced caspase-3 activity in apoptotic osteoclasts in vitro. Bone. 2001;28:465–473. [PubMed: 11344045]
- 77.
- Benten WP, Stephan C, Lieberherr M. et al. Estradiol signaling via sequestrable surface receptors. Endocrinology. 2001;142:1669–1677. [PubMed: 11250949]
- 78.
- Bentolila V, Boyce TM, Fyhrie DP. et al. Intracortical remodeling in adult rat long bones after fatigue loading. Bone. 1998;23:275–281. [PubMed: 9737350]
- 79.
- Bertiaume LG. Insider information: How palmitoylation of Ras makes it a signaling double agent Science's STKE 2002. http//wwwstkeorg/cgi/content/sigtrans2002/152/pe41 . [PubMed: 12359913]
- 80.
- Betancourt M, Wirfel KL, Raymond AK. et al. Osteosarcoma of bone in a patient with primary hyperparathyroidism: A case report. J Bone Miner Res. 2003;18:163–166. [PubMed: 12510819]
- 81.
- Bhangu PS, Genever PG, Spencer GJ. et al. Evidence for targeted vesicular exocytosis in osteoblasts. Bone. 2001;29:16–23. [PubMed: 11472886]
- 82.
- Bianco P, Bradbeer JN, Riminucci M. et al. Marrow stromal cells: Identification, morphometry, confocal imaging and changes in disease. Bone. 1993;14:315–320. [PubMed: 8363874]
- 83.
- Bianco P, Riminucci M. The bone marrow stroma in vivo: Ontogeny, structure, cellular composition and changes in diseaseIn: Beresford JN, Owen ME, eds.Marrow Stromal Cell CultureCambridge: Cambridge University Press,199810–25.
- 84.
- Bianco P, Riminucci M, Kuznetsov S. et al. Multipotential cells in the bone marrow stroma: Regulation in the context of organ physiology. Crit Revs Euk Gene Express. 1999;10:159–173. [PubMed: 10445154]
- 85.
- Bidwell JP, Torrungruang K, Alvarez M. et al. Involvement of the nuclear matrix in the control of skeletal genes: The NMP1 (YY1), NMP2 (Cbfa1), and NMP4 (Nmp4/CIZ) transcription factors. Crit Rev Eukaryotic Gene Express. 2001;11:279–297. [PubMed: 12067068]
- 86.
- Bier E. The Coiled Spring Cold Spring HarborNew York: Cold Spring Harbor Laboratory Press,2000 .
- 87.
- Bikle DD, Harris J, Halloran BP. et al. Skeletal unloading indices resistance to insulin-like growth factor. J Bone Miner Res. 1994;9:1789–1796. [PubMed: 7532347]
- 88.
- Bilezikian JP. Sex steroids, mice, and men: When androgens and estrogens get very close to one another. J Bone Miner Res. 2002;17:563–566. [PubMed: 11918214]
- 89.
- The Parathyroid Hormone In: Bilezikian JP, Marcus MA, Levine MA eds. 2nd ed. San Diego, CA: Academic Press,2001 .
- 90.
- Bilton RL, Booker GW. The subtle side to hypoxia-inducible factor (HIFα) regulation. Eur J Biochem. 2003;270:791–798. [PubMed: 12603312]
- 91.
- Bird RP, Good CK. The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol Lett. 2000;112-113:395–402. [PubMed: 10720758]
- 92.
- Bisello A, Sneddon WB, Friedman PA. PTHR1 endocytosis is not necessary for resensitization of camp signaling. J Bone Miner Res. 2002;17:S287.
- 93.
- Bitgood MJ, McMahon AP. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interactions in the mouse embryo. Dev Biol. 1995;172:126–138. [PubMed: 7589793]
- 94.
- Bjurholm A. Neuroendocrine peptides in bone. Int Orthop. 1991;15:325–329. [PubMed: 1725676]
- 95.
- Black DM, Greenspan SL, Ensrud KE. et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–1215. [PubMed: 14500804]
- 96.
- Black KM, Theriault BL, Anderson GI. Do bones have memory? Glutamate receptor expression in bone (abstract). J Bone Miner Res. 2002;17:S330.
- 97.
- Black TM, Theriault BL, Anderson GI. Mechanotransduction via glutamate receptors: Regulation of osteoblast activity in response to mechanical stimulation. J Bone Miner Res. 2003;18:S133.
- 98.
- Blair HC. How the osteoclast degrades bone. BioEssays. 1998;20:837–846. [PubMed: 9819571]
- 99.
- Blair HC, Zaidi M, Schlesinger PH. Mechanisms balancing skeletal matrix synthesis and degradation. Biochem J. 2002;364:329–341. [PMC free article: PMC1222578] [PubMed: 12023876]
- 100.
- Blin-Wakkach C, Lezot F, Ghoul-Mazgar S. et al. Endogenous Msx1 antisense transcript: In vivo and in vitro evidences, structures, and potential involvement in skeleton development in mammals. Proc Natl Acad Sci USA. 2001;98:7336–7341. [PMC free article: PMC34669] [PubMed: 11390985]
- 101.
- Bliziotes MM, Eshleman AJ, Zhang XW. et al. Neurotransmitter action in osteoblasts: Expression of a functional system for serotonin receptor activation and reuptake. Bone. 2001;29:477–486. [PubMed: 11704501]
- 102.
- Bockaert J, Claeysen S, Becamel C. et al. G protein-coupled receptors: Dominant players in cell-cell communication. Int Rev Cytol. 2002;212:63–132. [PubMed: 11804040]
- 103.
- Bockaert J, Marin P, Dumuis A. et al. The ‘magic tail’ of G protein-coupled receptors: An anchorage for functional protein networks. FEBS Lett. 2003;546:65–72. [PubMed: 12829238]
- 104.
- Boivin GY, Chavassieux PM, Santors AC. et al. Alendronate increases bone strength by increasing the mean degree of mineralization of bone tissue in osteoporotic women. Bone. 2000;27:687–694. [PubMed: 11062357]
- 105.
- Boivin G, Meunier PJ. Effects of bisphosphonates on matrix mineralization. J Musculoskel Neuron Interact. 2002;2:538–543. [PubMed: 15758388]
- 106.
- Boland R, De Boland AR. et al. Nongenomic stimulation of tyrosine phosphorylation cascades by 1,25(OH)2D3 by VDR-dependent and -independent mechanisms in muscle cells. Steroids. 2002;67:477–482. [PubMed: 11960624]
- 107.
- Bolon B, Campagnuolo G, Feige U. Duration of bone protection by a single osteoprotegerin injection in rats with adjuvant-induced arthritis. Cell Mol Life Sci. 2002a;59:1569–1576. [PubMed: 12440777]
- 108.
- Bolon B, Shaloub V, Kostenuik PJ. et al. Osteoprotegerin, an endogenous antiosteoclast factor for protecting bone in rheumatoid arthritis. Arthritis Rheum. 2002b;46:3121–3135. [PubMed: 12483715]
- 109.
- Bonadio J. Tissue engineering via local gene delivery: Update and future prospects for enhancing the technology. Adv Drug Del Rev. 2000;44:185–194. [PubMed: 11072114]
- 110.
- Bonadio J, Smiley E, Path P. et al. Localized, direct plasmid gene delivery in vivo: Prolonged therapy results in reproducible tissue regeneration. Nature Medicine. 1999;5:753–759. [PubMed: 10395319]
- 111.
- Bonnet N, Brunet-Imbault B, Parnaud CJ. et al. β2-Adrenergic agonists have negative effects on bone architecture and density in rat. J Bone Miner Res. 2003;18:S42.
- 112.
- Boskey AL. Mineral-matrix interactions in bone and cartilage. Clin Orthop. 1992;281:244–274. [PubMed: 1323440]
- 113.
- Bouche G, Gas N, Prats H. et al. Basic fibroblast growth factor enters the nucleolus and stimulates the transcription of ribosomal genes in ABAE cells undergoing G0-G1 transition. Proc Natl Acad Sci USA. 1987;84:6770–6774. [PMC free article: PMC299166] [PubMed: 3477808]
- 114.
- Boulter C, Mulroy S, Webb S. et al. Comparison of the cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci USA. 2001;98:12174–12179. [PMC free article: PMC59787] [PubMed: 11593033]
- 115.
- Bouxsein ML. Etiology and biomechanics of hip and vertebral fracturesIn: Marcus R, ed.Atlas of Clinical Endocrinology, OsteoporosisPhiladelphia: Current Medicine19993139–148.
- 116.
- Bowler WH, Buckley KA, Gartland A. et al. Extracellular nucleotide signaling: A mechanism for integrating local and systemic responses in the activation of bone remodeling. Bone. 2001;28:507–512. [PubMed: 11344050]
- 117.
- Bowe AE, Finnegan R, Jan de Beur SM. et al. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284:977–981. [PubMed: 11409890]
- 118.
- Boyce RW, Paddock CL, Franks AF. et al. Effects of intermittent hPTH-(1-34) alone and in combination with 1,25(OH)2D3 or risedronate on endosteal bone remodeling in canine cancellous and cortical bone. J Bone Miner Res. 1996;11:600–613. [PubMed: 9157775]
- 119.
- Brailov I, Bancila M, Brisorgeuil MJ. et al. Localization of 5-HT6 receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 2000;872:271–275. [PubMed: 10924708]
- 120.
- Brandi ML, Crescioli C, Tanini A. et al. Bone endothelial cells as estrogen targets. Calcif Tissue Int. 1993;53:312–317. [PubMed: 8287318]
- 121.
- Briggs SD, Xiao T, Sun ZW. et al. Gene silencing: trans-histone regulatory pathway to chromatin. Nature. 2002;418:498. [PubMed: 12152067]
- 122.
- Brighton CT, Hunt RM. Histochemical localization of calcium in growth plate mitochondria and matrix vesicles. Fed Proc. 1976;35:143–147. [PubMed: 1248647]
- 123.
- Brighton CT, Hunt RM. The role of mitochondria in growth plate calcification as demonstrated in a rachitic model. J Bone Joint Surg. 1978;60A:630–639. [PubMed: 681381]
- 124.
- Brann DW, De Sevilla L, Zamorano PL. et al. Regulation of leptin gene expression and secretion by steroid hormones. Steroids. 1999;64:659–663. [PubMed: 10503725]
- 125.
- Bringhurst FR. PTH receptors and apoptosis in osteocytes. J Musculoskel Neuron Interact. 2002;2:245–251. [PubMed: 15758445]
- 126.
- Bringhurst FR, Stern AM, Yotts M. et al. Peripheral metabolism of PTH: Fate of biologically active amino terminus in vivo. Am J Physiol. 1988;255:E886–E893. [PubMed: 3202165]
- 127.
- Brody T, Cravchick A. Drosophila melanogaster G protein-coupled receptors. J Cell Biol. 2000;150:F83–F88. [PMC free article: PMC2180217] [PubMed: 10908591]
- 128.
- Brommage R, Hotchkiss CE, Lees CJ. et al. Daily treatment with human recombinant parathyroid hormone-(1-34), LY33333, for 1-year increases bone mass in ovariectomized monkeys. J Clin Endocrinol Metab. 1999;84:3757–3763. [PubMed: 10523026]
- 129.
- Brouet A, Sonveaux P, Dessy C. et al. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J Biol Chem. 2001a;276:32663–32669. [PubMed: 11425855]
- 130.
- Brouet A, Sonveaux P, Dessy C. et al. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. 2001b;89:866–873. [PubMed: 11701613]
- 131.
- Brown EM. Is the calcium receptor a molecular target for the action of strontium on bone? Osteoporosis Int. 2003;14(Suppl 3):S25–S34. [PubMed: 12730784]
- 132.
- Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Revs. 2001;81:240–297. [PubMed: 11152759]
- 133.
- Burger E. Experiments on cell mechanosensitivity: Bone cells as mechanical engineersIn: Cowin SC, ed.Bone Mechanics Handbook. 2nd edBoca Raton: CRC Press,200128–1-28-16.
- 134.
- Burger EH, Klein-Nulent J, Smit TH. Strain-derived canalicular fluid flow regulates osteoclast activity in a remodelling osteon-a proposal. J Biomechanics. 2003;36:1453–1459. [PubMed: 14499294]
- 135.
- Burguera B, Hofbauer LC, Thomas T. et al. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142:3546–3553. [PubMed: 11459801]
- 136.
- Burr DB, Hirano T, Turner CH. et al. Intermittently administered human parathyroid hormone (1-34) treatment increases intracortical bone turnover and porosity without reducing bone strength in the humerus of ovariectomize cynomolgus monkeys. J Bone Miner Res. 2001;16:157–165. [PubMed: 11149480]
- 137.
- Burr DB, Robling AG, Turner CH. Effects of biomechanical stress on bone in animals. Bone. 2002;30:781–786. [PubMed: 11996920]
- 138.
- Byron JW. Cyclic nucleotides and the cell cycle of the hematopoietic stem cellIn: Abou-Sabé M, ed.Cyclic Nucleotides and the Regulation of Cell GrowthStroudsburg: Dowden, Hutchinson and Ross Inc.,197781–92.
- 139.
- Calvert JP. Cilia in PKD-letting it all hang out. J Am Soc Nephrol. 2002;13:2614–2616. [PubMed: 12239253]
- 140.
- Calvi LM, Adams GB, Olson DP. et al. Expression of a constitutively active PTH/PTHrP receptor in bone marrow stromal cells leads to expansion of hematopoietic stem cells in vivo and in vitro. J Bone Miner Res. 2002;17:S138.
- 141.
- Calvi LM, Adams GB, Weibrecht KW. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2004;425:841–846. [PubMed: 14574413]
- 142.
- Calvi LM, Sims NA, Hunzelman JL. et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–286. [PMC free article: PMC199196] [PubMed: 11160151]
- 143.
- Cameron DA. The ultrastructure of boneIn: Bourne GH, ed.The Biochemistry and Physiology of Bone. 2nd edNew York: Academic Press,1972191–236.
- 144.
- Canalis E, Economides AN, Gazzerro E. Bone morphogenic proteins, their antagonists, and the skeleton. Endocrine Rev. 2003;24:218–235. [PubMed: 12700180]
- 145.
- Candeliere GA, Liu F, Aubin JE. Individual osteoblasts in the developing calvaria express different gene repertoires. Bone. 2001;28:351–361. [PubMed: 11336915]
- 146.
- Cann CE, Roe EB, Sanchez SD. et al. PTH effects in the femur: Envelope-specific responses by 3DQCT in postmenopausal women (abstract). J Bone Miner Res. 1999;14:S137.
- 147.
- Caporale LH. Darwin in the GenomeNew York: McGraw-Hill, 2002 .
- 148.
- Capparelli C, Morony S, Warmington K. et al. Sustained antiresorptive effects after a single treatment with human recombinant osteoprotegerin (OPG): A pharmacodynamic and pharmacokinetic analysis in rats. J Bone Miner Res. 2003;18:852–858. [PubMed: 12733724]
- 149.
- Carmignoto G. Reciprocal communication systems between astrocytes and neurons. Prog Neurobiol. 2000;62:561–581. [PubMed: 10880851]
- 150.
- Carvalho RS, Scott JE, Suga DM. et al. Stimulation of signal transduction pathways in osteoblasts by mechanical strain potentiated by parathyroid hormone. J Bone Miner Res. 1994;9:999–1011. [PubMed: 7942169]
- 151.
- Caverzasio J, Bonjour JR. Characteristics and regulation of Pi transport in osteogenic cells for bone metabolism. Kidney Int. 1996;49:975–980. [PubMed: 8691747]
- 152.
- Cenci S, Weitzmann MN, Roggia CR. et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-α J Clin Invest. 2000;106:1229–1237. [PMC free article: PMC381439] [PubMed: 11086024]
- 153.
- Center for drug evaluation and research endocrinology and metabolic drugs advisory committeemeeting 2001. Available from URLhttp//wwwfdavideocom .
- 154.
- Chakravarthy BR, Durkin JP, Rixon RH. et al. Parathyroid hormone fragment [3-34] stimulates protein kinase C (PKC) activity in rat osteosarcoma and murine T-lymphoma cells. Biochem Biophys Res Commun. 1990;171:1105–1110. [PubMed: 2171507]
- 155.
- Chambliss KL, Yuhanna IS, Mineo C. et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into functional signaling module in caveolae. Circ Res. 2000;87:E44–E52. [PubMed: 11090554]
- 156.
- Chan K, Andrade SE, Boles M. et al. Inhibitors of hydroxymethylglutaryl-coenzyme a reductase and risk of fracture among older women. Lancet. 2000;355:2185–2188. [PubMed: 10881890]
- 157.
- Chan MH, Mak TW, Chiu RW. et al. Simvastatin increases serum osteocalcin concentration in patients treated for hypercholesterolaemia. J Clin Endocrinol Metab. 2001;86:4556–4559. [PubMed: 11549708]
- 158.
- Chan NX, Ryder KD, Pavalko FM. et al. Ca2+ regulates fluid shear-induced cyto-skeletal reorganization and gene expression in osteoblasts. Am J Physiol Cell Physiol. 2000;278:C989–997. [PubMed: 10794673]
- 159.
- Chang DJ, Ji C, Kim KK. et al. Reduction in transforming growth factor beta receptor I expression and transcription factor CBFa1 on bone cells by glucocorticoid. J Biol Chem. 1998;273:4892–4896. [PubMed: 9478931]
- 160.
- Chang W, Rodriguez L, Tu C. et al. High extracellular Ca2+ enhances the differentiation of growth plate chondrocytes. (abstract). J Bone Miner Res. 2003;18:S290.
- 161.
- Chang W, Tu C, Chen TH. et al. Expression and signal transduction of calcium-sensing receptors in cartilage and bone. Endocrinology. 1999;140:5883–5893. [PubMed: 10579354]
- 162.
- Chang W, Tu C, Pratt S. et al. Extracellular Ca2+-sensing receptors modulate matrix production and mineralization in chondrogenic RCJ3.1C5.18 cells. Endocrinology. 2002;143:1467–1474. [PubMed: 11897705]
- 163.
- Chao DT, Korsmeyer SJ. BCL-2 family: Regulators of cell death. Annu Rev Immunol. 1998;16:395–419. [PubMed: 9597135]
- 164.
- Chatterjee O, Nakchbandi IA, Philbrick WM. et al. Endogenous parathyroid hormone-related protein functions as a neuroprotective agent. Brain Res. 2002;930:58–66. [PubMed: 11879796]
- 165.
- Calcium-Sensing ReceptorIn: Chattopadhyay N, Brown EM, eds.Boston: Kluwer Academic Publishers,2003.
- 166.
- Chattopadhyay N, Yano S, Tfelt-Hansen et al. Mitogenic action of calcium-sensing receptor on rat calvarial osteoblasts. Endocrinology. 2004;145:published April 7 online ahead of print. [PubMed: 15084499]
- 167.
- Chauvin S, Vilardaga J, Benecsik M. et al. Parathyroid hormone receptor recycling: Regulation by specific structural features of the receptor (abstract). J Bone Miner Res. 2001;16:S228.
- 168.
- Chen C, Kalu DN. Modulation of intestinal estrogen receptor by ovariectomy, estrogen and growth hormone. J Pharmacol Exp Ther. 1998;286:328–333. [PubMed: 9655876]
- 169.
- Chen D, Garrett IR, Qiao M. et al. Proteasome inhibitors stimulate osteoblast differentiation and bone formation by inhibiting GLI3 degradation and enhancing BMP-2 expression (abstract). Bone. 2001;28:S74.
- 170.
- Chen D, Ji X, Harris MA. et al. Differential roles for bone morphogenic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J Cell Biol. 1998;142:295–305. [PMC free article: PMC2133031] [PubMed: 9660882]
- 171.
- Chen D, Zangel AL, Zhao Q. et al. Ovine caveolin-1: cDNA cloning, E. coli expression, and association with endothelial nitric oxide synthase. Mol Cell Endocrinol. 2001;175:41–56. [PubMed: 11325515]
- 172.
- Chen H-L, Demiralp B, Schneider A. et al. Parathyroid hormone and parathyroid hormone-related protein exert both pro-and anti-apoptotic effects in mesenchymal cells. J Biol Chem. 2002;277:19374–19381. [PubMed: 11897779]
- 173.
- Chen X, Dai J, Orellana SA. et al. Is PKIγ responsible for termination of immediate-early gene expression after induction by PTH? J Bone Miner Res. 2002;17:S250.
- 174.
- Chen X, Dai J, Orellana SA. et al. PKIγ knock down inhibits termination of immediate-early gene expression induced by PTH. J Bone Miner Res. 2003;18:S75.
- 175.
- Chen X, Macica CM, Liang G. et al. Stretch-induced parathyroid hormone -related peptide (PTHrP) gene expression in bone (abstract). J Bone Miner Res. 2003;18:S218.
- 176.
- Chenu C. Glutamatergic innervation in bone. Microscopy Res Tech. 2002;58:70–76. [PubMed: 12203705]
- 177.
- Cherruau M, Morvan FO, Schirar A. et al. Chemical sympathectomy-induced changes in TH-, VP-, and CGRP-immunoreactive fibers in the rat manible periosteum: Influence on bone resorption. J Cell Physiol. 2003;194:341–348. [PubMed: 12548553]
- 178.
- Chin K-V, Yang W-L, Ravatn R. et al. Reinventing the wheel of cyclic AMP. Novel mechanisms of cAMP signaling. Ann NY Acad Sci. 2002;968:49–64. [PubMed: 12119267]
- 179.
- Chorev M. Parathyroid hormone 1 receptor: Insights into structure and function. Receptors and Channels. 2002;8:219–242. [PubMed: 12529939]
- 180.
- Chow JW, Fox S, Jagger CJ. et al. Role of parathyroid hormone in mechanical responsiveness of bone. Am J Physiol. 1998;274:E146–E154. [PubMed: 9458760]
- 181.
- Chu S-C, Chou Y-C, Liu J-Y. et al. Fluctuation of serum leptin level in rats after ovariectomy and the influence of estrogen supplement. Life Sci. 1999;64:2299–2306. [PubMed: 10374920]
- 182.
- Chung CH, Baek SH. Deubiquiting enzymes: Their diversity and emerging roles. Biochem Biophys Res Commun. 1999;266:633–640. [PubMed: 10603300]
- 183.
- Chung U, Schipani E, McMahon AP. et al. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107:295–304. [PMC free article: PMC199199] [PubMed: 11160153]
- 184.
- Chung YS, Lee MD, Lee SK. et al. HMG-CoA reductase inhibitors increase BMD in type 2 diabetes mellitus patients. J Clin Endocrinol Metab. 2000;85:1137–1142. [PubMed: 10720052]
- 185.
- Coe MR, Summers TA, Parsons SJ. et al. Matrix mineralization in hypertrophic chondrocyte cultures. β Glycerophosphate increases type X collagen messenger RNA and the specific activity of pp60-src kinase. Bone Miner. 1992;18:91–106. [PubMed: 1381978]
- 186.
- Cohen J, Stewart I. What does a Martian look like?Hoboken: John Wiley & Sons,2003 .
- 187.
- Cole JA. Parathyroid hormone activates mitogen-activated protein kinase in opossum kidney cells. Endocrinology. 1999;140:5771–5779. [PubMed: 10579343]
- 188.
- Consensus development conference: Prophylaxis and treatment of osteoporosis. Am J Med. 1991;90:107–110. [PubMed: 1986575]
- 189.
- Consensus development conference: Prophylaxis and treatment of osteoporosis. Am J Med. 1993;94:646–650. [PubMed: 8506892]
- 190.
- Conteas CN, Desai TK, Arlow FA. Relationship of hormones and growth factors to colon cancer. Gastroenterol Clin North America. 1988;17:761–772. [PubMed: 3068140]
- 191.
- Cooper D. Regulation and organization of adenylyl cyclases and cAMP. Biochem J. 2003;375:517–529. [PMC free article: PMC1223734] [PubMed: 12940771]
- 192.
- Coppock D, Kopman C, Gudas J. et al. Regulation of the quiescence-induced genes: Quiescin Q6, decorin, and ribosomal protein S29. Biochem Biophys Res Commun. 2000;269:604–610. [PubMed: 10708601]
- 193.
- Coppock D, Kopman C, Scandalis S. et al. Preferential gene expression in quiescent human lung fibroblasts. Cell Growth Diff. 1993;4:483–493. [PubMed: 8396966]
- 194.
- Cornish J, Callon KE, Bava U. et al. The direct actions of leptin on bone cells increase bone strength in vivo: An explanation of low fracture rates in obesity (abstract). Bone. 2001;28:S88.
- 195.
- Cornish J, Callon KE, Lin C. et al. Stimulation of osteoblast proliferation by C-terminal fragments of parathyroid hormone-related protein. J Bone Miner Res. 1999;14:915–922. [PubMed: 10352099]
- 196.
- Cornish J, Callon KE, Nicholson GC. et al. Parathyroid hormone-related protein-(107-139) inhibits bone resorption in vivo. Endocrinology. 1997;138:1299–1304. [PubMed: 9048639]
- 197.
- Corral DA, Amling M, Takeda S. et al. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci USA. 1998;95:13835–13840. [PMC free article: PMC24916] [PubMed: 9811887]
- 198.
- Cosman F, Lindsay R. Is parathyroid hormone a therapeutic option for osteoporosis? A review of the clinical evidence. Calcif Tissue Int. 1998;62:475–480. [PubMed: 9576972]
- 199.
- Cosman F, Lindsay R. Parathyroid hormone as an anabolic treatmentIn: Stevenson JC, Lindsay R, eds.OsteoporosisLondon: Chapman & Hall,1998293–307.
- 200.
- Cosman F, Nieves J, Formica L. et al. Parathyroid hormone in combination with estrogen dramatically reduces vertebral fracture risk (abstract). Osteoporosis Int. 2000;11(Suppl 2):S176.
- 201.
- Cosman F, Nieves JW, Luckey MM. et al. Daily versus cyclic PTH combined with alendronate versus alendronate alone for treatment of osteoporosis (abstract). J Bone Miner Res. 2003;18:S32.
- 202.
- Cosman F, Nieves J, Woelfert L. et al. Parathyroid hormone added to established hormone therapy: Effects on vertebral fracture and maintenance of bone mass after parathyroid hormone withdrawal. J Bone Miner Res. 2001a;16:925–931. [PubMed: 11341338]
- 203.
- Cosman F, Nieves J, Woelfert L. et al. Alendronate does not block the anabolic effect of PTH in postmenopausal osteoporotic women. J Bone Miner Res. 1998;13:1051–1055. [PubMed: 9626638]
- 204.
- Cosman F, Nieves J, Zion M. et al. Effects of short-term cerivastatin on bone turnover (abstract). J Bone Miner Res. 2001b;16:S296.
- 205.
- Couet J, Belanger MM, Roussel E. et al. Cell biology of caveoli and caveolin. Adv Drug Deliv Rev. 2001;49:223–235. [PubMed: 11551396]
- 206.
- Cowin SC. Bone poroelasticity. J Biomech. 1999;32:217–238. [PubMed: 10093022]
- 207.
- Crawford DT, Qi H, Chisey-Frink KL. et al. Statin increases cortical bone in young male rats by single, local administration but fails to restore bone to ovariectomized (OVX) rats by daily systemic administration (abstract). J Bone Miner Res. 2001;16:S295.
- 208.
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 2000;341:233–249. [PMC free article: PMC1220352] [PubMed: 10393078]
- 209.
- Cui Y-F, Lord BI, Woolford LB. et al. The relative spatial distribution of in vitro-CFCs in the bone marrow, responding to specific growth factors. Cell Prolif. 1996;29:243–257. [PubMed: 8782487]
- 210.
- Cuthbertson RM, Kemp BE, Barden JA. Structure of osteostatin PTHrP[Thr 107](107-139). Biochim Biophys Acta. 1999;1432:64–72. [PubMed: 10366729]
- 211.
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. [PubMed: 9363896]
- 212.
- Daifotis AG, Weir EC, Dreyer BE. et al. Stretch-induced parathyroid hormone-related peptide gene expression in the rat uterus. J Biol Chem. 1992;267:23455–23458. [PubMed: 1385422]
- 213.
- Danks JA, Trivett MK, Power DM. et al. Parathyroid hormone-related protein in lower vertebrates. Clin Exp Pharmacol Physiol. 1998;25:750–752. [PubMed: 9750969]
- 214.
- Daub H, Wallasch C, Lankenau A. et al. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. [PMC free article: PMC1170306] [PubMed: 9384582]
- 215.
- David P, Nguyen H, Barbier A. et al. The bisphosphonate tiludronate is a potent inhibitor of the osteoclast vacuolar H+-ATPase. J Bone Miner Res. 1996;11:1498–1507. [PubMed: 8889850]
- 216.
- Davies JE. Bone Engineering. Toronto: em2inc. 2000.
- 217.
- de Crombrugge B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–727. [PubMed: 11698188]
- 218.
- Debiais F, Lasmoles F, Lefevre G. et al. Glycogen synthase kinase-3 (GSK-3) signaling is involved in the antiapoptotic effect of fibroblast growth factor-2 in human calvaria osteoblasts. J Bone Miner Res. 2002;17:S239.
- 219.
- Deftos LJ, Burton D, Hastings RH. et al. Comparative tissue distribution of the processing enzymes "prohormone thiol protease", and prohormone convertases 1 and 2, in human PTHrP-producing cell lines and mammalian neuroendocrine tissues. Endocrine. 2001;15:217–224. [PubMed: 11720250]
- 220.
- Delerive P, De BosscherK, Besnard S. et al. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-\kappa\B and AP-1. J Biol Chem. 1999;274:32048–32054. [PubMed: 10542237]
- 221.
- Delmas PD, Vergnaud P, Arlot ME. et al. The anabolic effect of human PTH (1-34) on bone formation is blunted when bone resorption is inhibited by the bisphosphonate tiludronate-Is activated resorptionn a prerequisite for the in vivo effect of PTH on formation in a remodeling system? Bone. 1995;16:603–610. [PubMed: 7669436]
- 222.
- Dempster DW. Bone remodelingIn: Riggs BL, Melton III LJ, eds.Osteoporosis: Etiology, Diagnosis, and Management. 2nd edPhiladelphia: Lippincott-Raven Publishers,199567–91.
- 223.
- Dempster DW. Exploiting and bypassing the bone remodeling cycle to optimize the treatment of osteoporosis. J Bone Miner Res. 1997;12:1152–1154. [PubMed: 9258744]
- 224.
- Dempster DW. The contribution of trabecular architecture to cancellous bone quality. J Bone Miner Res. 2000;15:20–23. [PubMed: 10646110]
- 225.
- Dempster DW, Cosman F, Parisien M. et al. Anabolic action of parathyroid hormone. Endocrine Revs. 1993;14:690–709. [PubMed: 8119233]
- 226.
- Denhardt DT, Giachelli CM, Rittling SR. Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol. 2001;41:723–749. [PubMed: 11264474]
- 227.
- Denhardt DT, Noda M. Osteopontin expression and function: Role in bone remodeling. J Cell Biochem. 1998;30-31(Suppl):92–102. [PubMed: 29345811]
- 228.
- de RooijJ, Rehmann H, van Triest M. et al. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. [PubMed: 10777494]
- 229.
- Dimmeler S, Aicher A, Vasa M. et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. [PMC free article: PMC209365] [PubMed: 11489932]
- 230.
- D'Ippolito G, Divieti P, Howard GA. et al. Regulation of gap-junctional communication in bone-derived cells by PTH fragments with different C termini. J Bone Miner Res. 2002;17:S392.
- 231.
- Divieti P, Inomata N, Chapin K. et al. Receptors for the carboxyl-terminal region of PTH(1-84) are highly expressed in osteocytic cells. Endocrinology. 2001;142:916–925. [PubMed: 11159865]
- 232.
- Divieti P, John MR, Jüppner H. et al. Human PTH-(7-84) inhibits bone resorption in vitro via actions independent of the type 1 PTH/PTHrP receptor. Endocrinology. 2002a;143:171–176. [PubMed: 11751606]
- 233.
- Divieti P, Lotz O, Geller A. et al. Inhibition of osteoclast formation by human PTH-(7-84) involves direct actions on hemopoietic cells (abstract). J Bone Miner Res. 2002b;17:1178.
- 234.
- Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632–3638. [PubMed: 7628403]
- 235.
- Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1-34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138:4607–4612. [PubMed: 9348185]
- 236.
- Dobson KB, Skerry TM. The NMDA-type glutamate receptor antagonist MK801 regulates the differentiation of rat bone marrow osteoprogenitors and influence adipogenesis (abstract). J Bone Miner Res. 2000;15:S272.
- 237.
- Dodd JS, Raleigh JA, Gross TS. Osteocyte hypoxia: A novel mechanotransduction pathway. Am J Physiol (Cell Physiol 46). 1999;277:C598–C602. [PubMed: 10484347]
- 238.
- Doherty MJ, Canfield AE. Gene expression during vascular pericyte differentiation. Crit Rev Eukaryot Gene Expr. 1999;9:1–17. [PubMed: 10200908]
- 239.
- Doherty TM, Fitzpatrick LA, Inoue D. et al. Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocrine Rev. 2004;25:629–672. [PubMed: 15294885]
- 240.
- Donahue SW, Donahue HJ, Jacobs CR. Osteoblastic cells have refractory periods for fluid-flow-induced intracellular calcium oscillations for short bouts of flow and display multiple low-magnitude oscillations during long-term flow. J Biomech. 2003;36:35–43. [PubMed: 12485636]
- 241.
- Donahue SW, Jacobs CR, Donahue HJ. Flow-induced calcium oscillations in rat osteoblasts are age, loading frequency, and shear stress dependent. Am J Physiol Cell Phsyiol. 2001;281:C1635–1641. [PubMed: 11600427]
- 242.
- Doolan CM, Condliffe SB, Harvey BJ. Rapid nongenomic activation of cytosolic cyclic AMP-dependent protein kinase activity and [Ca2+]I by 17β-oestradiol in female rat distal colon. Br J Pharmacol. 2000;129:1375–1386. [PMC free article: PMC1571973] [PubMed: 10742293]
- 243.
- Ducy P. Cbfa1: A molecular switch in osteoblast biology. Devel Dynamics. 2000;219:461–471. [PubMed: 11084646]
- 244.
- Doxsey S. Reevaluating centrosome function. Nature Rev Mol Cell Biol. 2001;2:688–698. [PubMed: 11533726]
- 245.
- Ducy P, Amling M, Takeda S. et al. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell. 2000;100:197–207. [PubMed: 10660043]
- 246.
- Ducy P, Desbois C, Boyce B. et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. [PubMed: 8684484]
- 247.
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–1869. [PMC free article: PMC230411] [PubMed: 7891679]
- 248.
- Ducy P, Schinke T, Karsenty G. The osteoblast: A sophisticated fibroblast under central surveillance. Science. 2000;289:1501–1504. [PubMed: 10968779]
- 249.
- Dumond H, Presle N, Terlain R. et al. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003;48:3009–3012. [PubMed: 14613274]
- 250.
- Duncan R, Misler S. Voltage-activated and stretch-activated Ba2+-conducting channels in an osteoblast-like cell line (UMR 106). FEBS Lett. 1989;251:17–21. [PubMed: 2473926]
- 251.
- Duncan RL, Hruska KA, Misler S. Parathyroid hormone activation of stretch-activated cation channels in osteosarcoma cells (UMR 106.01). FEBS Lett. 1992;307:219–223. [PubMed: 1379539]
- 252.
- Dunford JE, Ebetino EH, Rogers MJ. The mechanism of inhibition of farnesyl diphosphate synthase by nitrogen-containing bisphosphonates. J Bone Miner Res. 2002;17:S255.
- 253.
- Dustin ML, Colman DR. Neural and immunological synaptic relations. Science. 2002;298:785–789. [PubMed: 12399580]
- 254.
- Ebert BL, Furth JD, Ratcliffer PJ. Hypoxia and mitochondrial inhibitirs regulate expression of glucose transporter-1 via distinct cis-acting sequences. J Biol Chem. 1995;270:29083–29089. [PubMed: 7493931]
- 255.
- Edlich M, Yellowley CE, Jacobs CR. et al. Oscillating fluid flow regulates cytosolic calcium concentration in bovine articular chondrocytes. J Biomechanics. 2001;34:59–65. [PubMed: 11425081]
- 256.
- Edwards CJ, Hart DJ, Spector TD. Oral status and increased bone mineral density in postmenopausal women. Lancet. 2000;355:2218–2219. [PubMed: 10881898]
- 257.
- Einhorn TA. Biomechanics of boneIn: Bilzekian JP, Raisz LG, Rodan GA, eds.Principles of Bone BiologySan Diego: Academic Press, 1996:25-37, and 2nd Ed., Vol. 1,200217–32.
- 258.
- Egan JJ, Gronowicz G, Rodan GA. Parathyroid hormone promotes the disassembly of cytoskeletal actin and myosin in cultured osteoblastic cells: Mediation by cyclic AMP. J Cell Biochem. 1991;45:101–111. [PubMed: 1848561]
- 259.
- Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569–1582. [PubMed: 1464741]
- 260.
- Endo A, Tsujita Y, Kuroda M. et al. Inhibition of cholesterol synthesis in vitro and in vivo by ML-236A and ML-236B. Eur J Biochem. 1997;77:31–36. [PubMed: 908337]
- 261.
- Enjuanes A, Supervia A, Nogues X. et al. Leptin receptor (OB-R) gene expression in human primary osteoblasts: Confirmation. J Bone Miner Res. 2002;17:1135. [PubMed: 12054170]
- 262.
- Enlow DH, Brown SO. A comparative histological study of fossil and recent bone tissues. Part II. Texas J Sci. 1957;9:186–214.
- 263.
- Enlow DH, Brown SO. A comparative histological study of fossil and recent bone tissues. Part III. Texas J Sci. 1958;10:187–230.
- 264.
- Ehrlich PJ, Lanyon LE. Mechanical strain and bone cell function: A review. Osteoporosis Int. 2002;13:688–700. [PubMed: 12195532]
- 265.
- Ekblad E, Edvinsson L, Wohlestedt C. et al. Neuropeptide Y coexists and cooperates with noradrenaline in perivascular nerve fibers. Regul Pept. 1984;8:225–235. [PubMed: 6379758]
- 266.
- Erdmann S, Burkhardt H, von der Mark K. et al. Mapping of a carboxy-terminal active site of parathyroid hormone by calcium-imaging. Cell Calcium. 1998;23:413–421. [PubMed: 9924633]
- 267.
- Eriksen EF, Axelrod DW, Melsen F. Bone Histomorphometry New York: Raven Press, 1994.
- 268.
- Eriksen EF, Langdahl B, Klassen M. The cellular basis of osteoporosis. Spine: State Arts Revs. 1994;8:23–62.
- 269.
- Errazahi A, Bouizar Z, Lieberherr M. et al. Functional type I PTH/PTHrP receptor in freshly isolated newborn rat keratinocytes: Identification by RT-PCR and immunohistochemistry. J Bone Miner Res. 2003;18:737–750. [PubMed: 12674335]
- 270.
- Errazahi A, Bouizar Z, Rizk-Rabin M. RT-PCR identification of PTHrp PTH/PTHrp receptor mRNAs during the steps of differentiation pathway of rat newborn keratinocytes: A putative autocrine role of PTHrp. Bone. 1998;23:S248.
- 271.
- Esbrit P, Alvarez-Arroyo MV, De Miguel F. et al. C-terminal parathyroid hormone-related protein increases vascular endothelial growth factor in himan osteoblastic cells. J Am Soc Nephrol. 2000;11:1085–1092. [PubMed: 10820172]
- 272.
- Espinosa L, Itzstein C, Cheynel H. et al. Active NMDA glutamate receptors are expressed by mammalian osteoclasts. J Physiol. 1999;518:47–53. [PMC free article: PMC2269403] [PubMed: 10373688]
- 273.
- Ettinger B, San Martin J, Crans G. et al. Differential effects of teriparatide on BMD after treatment with raloxifene or alendronate. J Bone Miner Res. 2004;19:745–751. [PubMed: 15068497]
- 274.
- Evans BAJ, Elford C, Gregory JW. Leptin control of bone metabolism (abstract). Bone. 2001;28:S149.
- 275.
- Everts V, Delaisse JM, Korper W. et al. The bone lining cell: Its role in cleaning Howship's lacunae and initiating bone formation. J Bone Miner Res. 2002;17:77–90. [PubMed: 11771672]
- 276.
- Faccio R, Lam J, Aya K. et al. Oligomeric RANKL induces bone formation via the ERK pathway. J Bone Miner Res. 2003;18:S141.
- 277.
- Fadeel B, Zhivotovsky B, Orrenius S. All along the watchtower: On the regulation of apoptosis regulators. FASEB J. 1999;13:1647–1657. [PubMed: 10506569]
- 278.
- Falany ML, Thames AM, McDonald JM. et al. Osteoclasts secrete the cytokine MIM-1. Biochem Biophys Res Commun. 2001;281:180–185. [PubMed: 11178977]
- 279.
- Falkenstein E, Tillmann H-C, Christ M. et al. Multiple actions of steroid hormones-a focus on rapid, nongenomic effects. Pharmacol Revs. 2000;52:513–555. [PubMed: 11121509]
- 280.
- Fang J, Zhu YY, Smiley E. et al. Stimulation of new bone formation by direct transfer of osteogenic plasmid genes. Proc Natl Acad Sci USA. 1996;93:5753–5758. [PMC free article: PMC39133] [PubMed: 8650165]
- 281.
- Farach-Carson MC, Xu Y. Microarray detection of gene expression changes induced by 1,25(OH)2D3 and a Ca2+ influx-activating analog in osteoblastic ROS 17/2.8 cells. Steroids. 2002;67:467–470. [PubMed: 11960622]
- 282.
- Farhat MY, Lavigne MC, Ramwell PW. The vascular protective effects of estrogen. FASEB J. 1996;10:615–624. [PubMed: 8621060]
- 283.
- Farley JR, Stilt-Coffing B. Apoptosis may determine the release of skeletal alkaline phosphahtase activity from human osteoblast-line cells. Calcif Tissue Int. 2001;68:43–52. [PubMed: 12037623]
- 284.
- Farr HW. Hyperparathyroidism and cancer. CA Cancer J Clin. 1976;26:66–74. [PubMed: 816432]
- 285.
- Farzaneh-Far A, Proudfoot D, Weisberg PL. et al. Matrix gla protein is regulated by a mechanism functionally related to the calcium-sensing receptor. Biochem Biophys Res Commun. 2000;277:736–740. [PubMed: 11062022]
- 286.
- Fassbender K, Simons M, Bergmann C. et al. Simvaststin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vivo and in vitro. Proc Natl Acad Sci USA. 2001;98:5856–5861. [PMC free article: PMC33303] [PubMed: 11296263]
- 287.
- Faucheux C, Horton MA, Price JS. Nuclear localization of type I parathyroid hormone/parathyroid hormone-related protein receptors in deer antler osteoclasts: Evidence for parathyroid hormone-related protein and receptor activator of NF-κB-dependent effects on osteoclast formation in regenerating mammalian bone. J Bone Miner Res. 2002;17:455–464. [PubMed: 11874237]
- 288.
- Faulkner K. Bone matters: Are density increases necessary to reduce fracture risk? J Bone Miner Res. 2000;15:183–187. [PubMed: 10703919]
- 289.
- Feig DS, Gottesman IS. Familial hyperparathyroidism in association with colonic carcinoma. Cancer. 1987;60:429–432. [PubMed: 3594382]
- 290.
- Feister HA, Torrungruang K, Thunyakitpisal P. et al. NP/NMP4 transcription factors have distinct osteoblast nuclear matrix subdomains. J Cell Biochem. 2000;79:506–517. [PubMed: 10972987]
- 291.
- Fenton AJ, Kemp BE, Hammonds RG. et al. A potent inhibitor of osteoclastic bone resorption within a highly conserved pentapeptide region of parathyroid hormone-related protein; PTHrP[107-111] Endocrinology. 1991;129:3424–3426. [PubMed: 1954916]
- 292.
- Fermore B, Skerry TM. PTH/PTHrP receptor expression in osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J Bone Miner Res. 1995;10:1935–1943. [PubMed: 8619374]
- 293.
- Feron O, Dessy C, Desager J-P. et al. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. [PubMed: 11136695]
- 294.
- Fielding CJ, Fielding PE. Caveolae and intracellular trafficking of cholestero. Adv Drug Del Rev. 2001;49:251–264. [PubMed: 11551398]
- 295.
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. [PMC free article: PMC1226318] [PubMed: 12386325]
- 296.
- Finkel E. The mitochondrion: Is it central to apoptosis? Science. 2001. pp. 624–626. [PubMed: 11330312]
- 297.
- Fiorelli G, Gori F, Frediani U. et al. Membrane-binding sites and nongenomic effects of estrogen in cultured human preosteoclastic cells. J Steroid Biochem Molec Biol. 1996;59:233–240. [PubMed: 9010339]
- 298.
- Finkelstein JS, Hayes A, Hunzelman JL. et al. Effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med. 2003;349:1216–1226. [PubMed: 14500805]
- 299.
- Fisher JE, Rogers MJ, Halasay JM. et al. Alendronate mechanism of action: Geranylgeraniol, an intermediate in the mevalonate pathway, prevents inhibition of osteoclast formation, bone resorption, and kinase activation in vitro. Proc Natl Acad Sci USA. 1999;96:133–138. [PMC free article: PMC15105] [PubMed: 9874784]
- 300.
- Fiziola M, Weinstein H. Structural models for dimerization of G-protein coupled receptors: The opioid receptor homdimers. Biopolymers. 2002;66:317–325. [PubMed: 12539260]
- 301.
- Fleet JC. Leptin and bone: Does the brain control bone biology? Nutr Rev. 2000;58:209–211. [PubMed: 10941257]
- 302.
- Fleisch H. Bisphosphonates in Bone diseaseLondon: The Parthenon Publishing Group, 1997.
- 303.
- Fleisch H. Bisphosphonates: Mechanisms of action. Endocrine Revs. 1998;19:80–100. [PubMed: 9494781]
- 304.
- Fletcher BS, Lim RW, Varnum BC. et al. Structure and expression of TIS21, a primary response gene induced by growth factors and tumor promoters. J Biol Chem. 1991;266:14511–14518. [PubMed: 1713584]
- 305.
- Fraher LJ, Avram R, Watson PH. et al. A comparison of the biochemical responses to 1-31 hPTH and 1-34 hPTH given to healthy humans by slow infusion. J Clin Endocrinol Metab. 1999;84:2739–2743. [PubMed: 10443671]
- 306.
- Franceschi RT. The developmental control of osteoblast-specific gene expression: Role of specific transcription factors and the extracellular matrix environment. Crit Rev Oral Biol Med. 1999;10:40–57. [PubMed: 10759426]
- 307.
- Franceschi RT, Xiao G. Regulation of the osteoblast-specific transcription factor , Runx2: Responsiveness to multiple signal transduction pathways. J Cell Biochem. 2003;88:446–454. [PubMed: 12532321]
- 308.
- Fried H, Kutay U. Nucleocytoplasmic transport; taking an inventory. Cell Mol Life Sci. 2003;60:1659–1688. [PubMed: 14504656]
- 309.
- Friedman J, Babu B, Clark RB. β2-Adrenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinases 2 in human embryonic kidney 293 cells: Lack of evidence for Gs/Gi switching. Mol Pharmacol. 2002;62:094–1102. [PubMed: 12391272]
- 310.
- Friedman JM. Obesity in the new millennium. Nature. 2000;404:632–634. [PubMed: 10766249]
- 311.
- Friedman PA, Gesek FA, Morley P. et al. Cell-specific signaling and structureactivity relations of parathyroid analogs in mouse kidney cells. Endocrinology. 1999;140:301–309. [PubMed: 9886839]
- 312.
- Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. [PubMed: 11544023]
- 313.
- Frith JC, Monkkonen J, Auriola S. et al. The molecular mechanism of action of the antiresorptive and anti-inflammatory drug clodronate: Evidence for the formation in vivo of a metabolitie that inhibits bone resorption and causes osteoclast and macrophage apoptosis. Arthritis Rheum. 2001;44:2201–2210. [PubMed: 11592386]
- 314.
- Frith JC, Monkkonen J, Blackburn GM. et al. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5"-(β,γ-dichloromethylene) triphosphate by mammalian cells in vitro. J Bone Miner Res. 1997;12:1358–1367. [PubMed: 9286751]
- 315.
- Frost HM. Presence of microscopic cracks in vivo in bone. Henry Ford Hospital Medical Bull. 1960;8:25–35.
- 316.
- Frost HM. OsteoporosesIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,19971–27.
- 317.
- Fu Q, Jilka RL, Manolagas SC. et al. Parathyroid hormone stimulates receptor activator of NF\kappa\B ligand and inhibits osteoprotegerin expression via protein kinase A activation of CREB. J Biol Chem. 2002;277:48868–48875. [PubMed: 12364326]
- 318.
- Fujimori A, Cheng SL, Avioli LV. et al. Structure-function relationship of parathyroid hormone: Activation of phospholipase-C, protein kinase-A and -C in osteosarcoma cells. Endocrinology. 1992;130:29–36. [PubMed: 1727705]
- 319.
- Fujita T, Inoue T, Morii H. et al. Effect of an intermittent weekly dose of human parathyroid hormone (1-34) on osteoporosis: A randomized double-masked prospective study using three dose levels. Osteoporosis Int. 1999;9:296–306. [PubMed: 10550446]
- 320.
- Fujita T, Fukuyama R, Izumo N. et al. Transactivation of of core binding factor α1 as a basic mechanism to trigger parathyroid hormone-induced osteogenesis. Jpn J Pharmacol. 2001a;86:405–416. [PubMed: 11569614]
- 321.
- Fujita T, Izumo N, Fukuyama R. et al. Phosphate provides an extracelluar signal that drives nuclear export of Runx2/Cbfa1 in bone cells. Biochem Biophys Res Commun. 2001b;280:348–352. [PubMed: 11162521]
- 322.
- Fujita T, Meguro T, Fukuyama R. et al. New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells. Checkpoint of modulation by cyclic AMP. J Biol Chem. 2002;277:22191–22200. [PubMed: 11956184]
- 323.
- Fukata S, Hagino H, Okano T. et al. Effects of intermittent administration of human parathyroid hormone on bone mineral density and arthritis in rats with collagen-induced arthritis. Arthritis Rheum. 2004;50:4060–4069. [PubMed: 15593226]
- 324.
- Fukumoto S. Localization and function of calcium-sensing receptors in bone cells. Nippon Rinsho. 1998;56:1419–1424. [PubMed: 9648458]
- 325.
- Fuller K, Murphy C, Kirsten B. et al. TNF-α potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143:1108–1118. [PubMed: 11861538]
- 326.
- Funahashi H, Yada T, Suzuki R. et al. Distribution , function, and properties of leptin receptors in the brain. Internat Rev Cytol. 2003;224:1–27. [PubMed: 12722947]
- 327.
- Funk JL. A role for parathyroid hormone-related protein in the pathogenesis of inflammatory / autoimmune diseases. Internat Immunopharmacol. 2001;1:1101–1121. [PubMed: 11407305]
- 328.
- Funk JL, Wei H, Downey KJ. et al. Expression of PTHrP and its cognate receptor in the rheumatoid synovial microcirculation. Biochem Biophys Res Commun. 2002;297:890–897. [PubMed: 12359237]
- 329.
- Gabet Y, Kohavi D, Moller R. et al. Human parathyroid hormone 1-34 reverses bone loss in orchidectomized adult rats mainly by vastly increasing trabecular thickness. J Bone Miner Res. 2003;18:S386–S387.
- 330.
- Gabet Y, Muller R, Regev E. et al. Osteogenic growth peptide modulates fracture callus structural and mechanical properties. Bone. 2004;35:65–73. [PubMed: 15207742]
- 331.
- Galindo M, Stein JL, Stein G. et al. Bone-specific Runx2 levels are tightly regulated during the cell cycle to support a novel function in control of the switch from proliferation to quiescence in osteoblasts. J Bone Miner Res. 2003;18:S137.
- 332.
- Gallien-Lartigue O, Carrez D. Induction in vitro de la phase S dans les cellules souches multipotentes de la moelle osseuse par l'hormone parathroïdienne. CR Acad Sci Paris [D] 1974;278:1765–1768. [PubMed: 4368158]
- 333.
- Galvin RJS, Fuson TR, Yang X. et al. hPTH (1-38) stimulation of bone resorption in murine calvariae is mediated in part by its ability to decrease OPG (abstract). J Bone Miner Res. 2001;16:S449.
- 334.
- Garrett IR, Chen D, Zhao M. et al. Statins mediate bone formation by enhancing BMP-2 expression (abstract). Bone. 2001a;28:S75.
- 335.
- Garrett IR, Gutierrez G, Chen D. et al. Specific inhibitors of the chymotryptic component of the proteasome are potent bone anabolic agents (abstract). J Bone Miner Res. 2000;15:S197.
- 336.
- Garrett IR, Gutierrez G, Chen D. et al. Statins stimulate bone formation by enhancing eNOS expression (abstract). J Bone Miner Res. 2001b;16:S75.
- 337.
- Garrrett IR, Gutierrez G, Mundy GR. Statins and bone formation. Curr Pharma Design. 2001c;7:715–736. [PubMed: 11405194]
- 338.
- Gasser JA. Quantitative assessment of bone mass and geometry by pQCT in rats in vivo and site specificity of changes at different skeletal sites. J Jpn Soc Bone Morphom. 1997;7:107–114.
- 339.
- Gasser JA. Fluvastatin and cerivastatin are not anabolic for bone after local or systemic administration of nontoxic doses in mice and rats (abstract). J Bone Miner Res. 2001;16:S295.
- 340.
- Genant HK, Cann CE, Etting B. et al. Quantitative computed tomography of vertebral spongiosa: A sensitive method for detecting early bone loss after oophorectomy. Ann Intern Med. 1982;97:699–705. [PubMed: 6291439]
- 341.
- Gensure RC, Shimizu N, Tsang JC. et al. Evidence that Bpa 19-modified parathyroid hormone-related peptide (PTHrP) agonist and antagonist analogs crosslink to distict sites in transmembrane domain 2 of the PTH/PTHrP receptor. J Bone Miner Res. 2002;17:S286.
- 342.
- Gentili C, Morelli S, Boland R. et al. Parathyroid hormone activation of MAP kinase in rat duodenal cells is mediated by 3',5'-cyclic AMP and Ca2+ Biochim Biophys Acta. 2001;1540:201–212. [PubMed: 11583815]
- 343.
- Gentili C, Morelli S, de Boland AR. Involvement of PI3-kinase and its association with c-Src in PTH-stimulate rat enterocytes. J Cell Biochem. 2002;86:773–783. [PubMed: 12210743]
- 344.
- Ghosh-Choudhury N, Windle JJ, Koop BA. et al. Immortalized murine osteoblasts derived from BMP2-T-antigen expressing transgenic mice. Endocrinology. 1996;137:331–339. [PubMed: 8536632]
- 345.
- Gibbs WW. Untangling the roots of cancer. Sci Amer. 2003;289:57–65. [PubMed: 12840947]
- 346.
- Gillespie BT. The influence of growth factors and cytokines on hypertrophy and primary cilia expression of articular chondrocyteshttp://www.realscience.breckschol.org/upper/research/research97-99pub/GF/gfa.html1999 .
- 347.
- Goodenough DA, Paul DL. Beyond the gap: Functions of unpaired connexon channels. Nature Rev Mol Cell Biol. 2003;4:1–10. [PubMed: 12671651]
- 348.
- Gohel A, Gronowicz G. Glucocorticoids induce apoptosis in osteoblasts in mice by the regulation of BCL-2 and other cell cycle factors (abstract). J Bone Miner Res. 1997;12:S284.
- 349.
- Goldring SR. Bone and joint destruction in rheumatoid arthritis: What is really happening? J Rheumatol Suppl. 2002;65:44–48. [PubMed: 12236623]
- 350.
- Goldring SR, Gravallelese EM. Pathogenesis of bone erosions in rheumatoid arthritis. Curr Opin Rheumatol. 2000;12:195–199. [PubMed: 10803748]
- 351.
- Goldring SR, Gravallese EM. Pathogenesis of bone lesions in rheumatoid arthritis. Curr Rheumatol Reports. 2002;4:226–231. [PubMed: 12010607]
- 352.
- Goldstein SA, Bonadio J. Potential role for direct gene transfer in the enhancement of fracture healing. Clin Orthop. 1998;355(Suppl):S154–162. [PubMed: 9917636]
- 353.
- Goligorsky MS, Menton DN, Hruska KA. Parathyroid hormone-induced changes of the brush border topography and cytoskeleton in cultured renal proximal tubular cells. J Membr Biol. 1986;92:151–162. [PubMed: 3761360]
- 354.
- Goltzman D, White JH. Developmental and tissue-specific regulation of parathyroid hormone (PTH)/ PTH-related peptide receptor gene expression. Crit Rev Eukaryot Gene Expr. 2000;10:135–149. [PubMed: 11186329]
- 355.
- Gooch KJ, Tennant CJ. 1997. Mechanical forces: Their effects on cells and tissues. New York: Springer-Verlag.
- 356.
- Gordeladze JO, Drevon CA, Syversen U. et al. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem. 2002a;85:825–836. [PubMed: 11968022]
- 357.
- Gordeladze JO, Reppe S, Gautvik KM. et al. Direct leptin exposure and mechano-stimulation of human osteoblastic cells promote proliferation and differentiation, while inhibiting apoptosis. J Bone Miner Res. 2002b;17:S444.
- 358.
- Gordeladze JO, Reseland JE, Drevon CA. Pharmacological interference with transcriptional control of osteoblasts: A possible role for leptin and fatty acids in maintaining bone strength and body lean mass. Curr Pharma Design. 2001;7:275–290. [PubMed: 11254890]
- 359.
- Gordon J, Bennett AR, Blackburn CC. et al. Gcm2 and Foxn1 mark early parathyroid and thymus-specific domains in the developing third pharyngeal pouch. Mech Devel. 2001;103:141–143. [PubMed: 11335122]
- 360.
- Gori F, Hofbauer LC, Dunstan CR. et al. The expression of osteoprotegerin and RANKL ligand and thed support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated. Endocrinology. 2000;141:4768–4776. [PubMed: 11108292]
- 361.
- Gowen M, Stroup GB, Dodds RA. et al. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest. 2000;105:1595–1604. [PMC free article: PMC300853] [PubMed: 10841518]
- 362.
- Grasso P, Leinung MC, Lee DW. Epitope mapping of secreted mouse leptin utilizing peripherally administered synthetic peptides. Reg Pept. 1999;85:93–100. [PubMed: 10651062]
- 363.
- Gravallese EM, Goldring SR. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum. 2000;43:2143–2151. [PubMed: 11037873]
- 364.
- Gray C, Marie , Arora M. et al. Glutamate does not play a major role in controlling bone growth. J Bone Miner Res. 2001;16:742–749. [PubMed: 11316002]
- 365.
- Greaves M. Cancer: The evolutionary legacyOxford: Oxford University Press,2000 .
- 366.
- Greenfield EM. ODF/OPGL is increased by IL-1 and IL-6 (abstract). J Bone Miner Res. 1999;14:S361.
- 367.
- Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: Eat-me, don't eat me and come-get-me signals. Trends Cell Biol. 2003;13:648–656. [PubMed: 14624843]
- 368.
- Gros TS, Rabaia NA, Moy NY. et al. Osteopontin: A marker of osteocyte stress in response to loss of mechanical loading (abstract). J Bone Miner Res. 2002;17:S191.
- 369.
- Gross TS, Akeno N, Clemens TL. et al. Selected contribution: Osteocytes upregulate HIF-α in response to acute disuse and oxygen deprivation. J Applied Physiol. 2001;90:2514–2519. [PubMed: 11356821]
- 370.
- Gu Y, Publicover SJ. Expression of functional metabotropic glutamate receptors in primary cultured rat osteoblasts. J Biol Chem. 2000;275:34252–34259. [PubMed: 10950953]
- 371.
- Guaradvaccaro D, Corrente G, Covone F. et al. Arrest of G1-S progression is Rb dependent and relies on the inhibition of of cyclin D1 transcription. Mol Cell Biol. 2000;20:1797–1815. [PMC free article: PMC85361] [PubMed: 10669755]
- 372.
- Gubrij I, Ali AA, Chambers TM. et al. Decreased apoptosis and increased bone formation in implants of marrow-derived osteoblast progenitors overexpressing Bcl-2: In vivo evidence for a pivotal role of apoptosis in bone formation (abstract). J Bone Miner Res. 2003;18:S136.
- 373.
- Günther T, Chen Z-F, Kim J. et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. 2000;406:199–203. [PubMed: 10910362]
- 374.
- Gupta A, Miyauchi A, Fujimori A. et al. Phosphate transport in osteoclasts: A functional and immunochemical characterization. Kidney Int. 1996;49:968–974. [PubMed: 8691746]
- 375.
- Gurney JG, Severson RK, Davis S. et al. Incidence of cancer in children in the United States. Sex-, raceand 1-year age-specific rates by histologic type. Cancer. 1995;75:186–2195. [PubMed: 7697611]
- 376.
- Gutierrez G, Garrett JR, Rossini G. et al. Dermal application of lovaststin to rats causes greater increases in bone formation and plasma concentration than when administered by oral gavage (abstract). J Bone Miner Res. 2000;15:S427.
- 377.
- Hagino H, Okano T, Akhter MP. et al. The effect of parathyroid hormone on cortical bone response to in vivo external loading. J Bone Miner Metab. 2001;19:244–250. [PubMed: 11448017]
- 378.
- Hajnóczky G, Csordαs G. et al. Old players in a new role: Mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium. 2003a;32:363–377. [PubMed: 12543096]
- 379.
- Hajnóczky G, Csordas G, Krishnamurthy R. Mitochondrial calcium signaling driven by the IP3 receptor. J Bioenerg Biomembranes. 2000;32:15–25. [PubMed: 11768758]
- 380.
- Hajnóczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003b;304:445–454. [PubMed: 12729578]
- 381.
- Hakeda Y, Kobayashi Y, Yamaguchi K. et al. Osteoclastogenesis inhibitory factor (OCIF) directly inhibits bone-resobing activity of isolated mature osteoclasts. Biochem Biophys Res Commun. 1998;251:796–801. [PubMed: 9790989]
- 382.
- Hall RA, Ostedgaard LS, Premont RT. et al. A C-terminal motif found in the β2-adrenergic receptor, P2Y1 receptor and cyctic fibrosis transmembrane conductance regulator determines binding to the Na+/H+ exchanger regulatory factor family of PDZ proteins. Proc Natl Acad Sci USA. 1998;95:8496–8501. [PMC free article: PMC21104] [PubMed: 9671706]
- 383.
- Halladay DL, Miles RR, Gornik SA. et al. Identification of signal transduction pathways and promoter sequences that mediate parathyroid hormone 1-38 inhibition of osteoprotegerin gene expression. J Cell Biochem. 2002;84:1–11. [PubMed: 11746511]
- 384.
- Hamon M, Doucet E, LeFevre K. et al. Antibodies and antisense oligonucleotides for probing the distribution and putative functions of 5-HT6 receptors. Neuropsychopharmacol. 1999;21:68S–76S. [PubMed: 10432491]
- 385.
- Hamrick MW, Pennington C, Newton D. et al. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004;34:376–383. [PubMed: 15003785]
- 386.
- Hanes DR, Crotti TN, Loric M. et al. Osteoprotegrin and receptor activator of nuclear factor kappB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint. Rheumatology (Oxford). 2001;40:623–630. [PubMed: 11426018]
- 387.
- Hanafin NM, Chen TC, Heinrich G. et al. Cultured human fibroblasts and not cultured human keratinocytes express a PTH/PTHrP receptor mRNA. J Invest Dermatol. 1995;105:133–137. [PubMed: 7615967]
- 388.
- Harbour JW, Dean DC. The Rb/E2F pathway: Expanding roles in emerging paradigms. Genes Devel. 2000;14:2393–2409. [PubMed: 11018009]
- 389.
- Harmey D, Stenbeck G, Nobes CD. et al. Regulation of osteoblast differentiation by Pasteurella multocida toxin (PMT): a role for the Rho GTPase in bone formation. J Bone Miner Res. 2004;19:661–670. [PubMed: 15005854]
- 390.
- Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. [PubMed: 11591811]
- 391.
- Hauge EM, Qvesel D, Eriksen EF. et al. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblast markers. J Bone Miner Res. 2001;16:1575–1582. [PubMed: 11547826]
- 392.
- Havers C. Osteologia nova, or some new observations of the bones, and the parts belonging to them, with the manner of their accretion and nutrition, communicated to the Royal Society in several discoursesLondon: Samuel Smith, 1691 .
- 393.
- Heaney RP. Is the paradigm shifting ? Bone. 2003;33:457–465. [PubMed: 14555248]
- 394.
- Hefferan TE, Rehman O, Lane NE. et al. Monitoring the response to PTH by analysis of peripheral leukocyte gene expression (abstract). J Bone Miner Res. 2002;17:S214.
- 395.
- Hellio de Graverand M, Rattner J, Eggerer J. et al. Primary cilia are a hallmark of rabbit meniscal cells. Proc 47th Annual Meeting of the Orthopaedic Research Society. 2001:43.
- 396.
- Herrington DM, Klein KP. Statins, hormones, and women: Benefits and drawbacks for athersclerosis and osteoporosis. Curr Atherosclerosis Reports. 2001;3:35–42. [PubMed: 11123846]
- 397.
- Hershko A, Ciechanover A, Varshavsky A. The ubiquitin system. Nature Medicine. 2000;6:1073–1081. [PubMed: 11017125]
- 398.
- Herzog H. Hypothalamic Y2 receptors: Central coordinator of energy homeostasis and bone mass regulation. Drug News Perspect. 2002;15:506–510. [PubMed: 12677189]
- 399.
- Hesch RD, Busch U, Prokop M. et al. Increase of vertebral density by combination therapy with pulsatile 1-38 PTH and sequential addition of calcitonin nasal spray in osteoporotic patients. Calcif Tissue Int. 1989;44:176–180. [PubMed: 2493323]
- 400.
- Hesp R, Hulme P, Williams D. et al. The relationship between changes in femoral bone density and calcium balance in patients with involutional osteoporosis treated with human PTH fragment 1-34. Metab Bone Dis Rel Res. 1981;2:331–334.
- 401.
- Hill PA, Tumber A, Meikle MC. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology. 1997;138:3849–3858. [PubMed: 9275074]
- 402.
- Himms-Hagen J. Physiological roles of the leptin endocrine system: Differences between mice and humans. Crit Rev Clin Lab Sci. 1999;36:575–655. [PubMed: 10656540]
- 403.
- Hinoi E, Fujimori S, Nakamura Y. et al. Group III metabotropic glutamate receptors in rat cultured calvarial osteoblasts. Biochem Biophys Res Commun. 2001;281:341–346. [PubMed: 11181052]
- 404.
- Hinoi E, Fujimori S, Takarada T. et al. Facilitation of glutamate release by ionotropic glutamate receptors in osteoblasts. Biochem Biophys Res Commun. 2002;297:452–458. [PubMed: 12270113]
- 405.
- Hinoi E, Fujimori S, Yoneda Y. Modulation of cellular differentiation by N-methyl-D-aspartate receptors in osteoblasts. FASEB J. 2003;17:1532–1534. [PubMed: 12824297]
- 406.
- Hinoi E, Takarada T, Yoneda Y. Glutamate signaling system in bone. J Pharmacol Sci. 2004;94:215–220. [PubMed: 15037805]
- 407.
- Hinson TK, Damodaran TV, Chen J. et al. Identification of putative trans-membrane receptor sequences homologous to the calcium-sensing G-protein-coupled receptor. Genomics. 1997;45:279–289. [PubMed: 9344650]
- 408.
- Hirano T, Burr DB, Cain RL. et al. Changes in geometry and cortical porosity in adult, ovary-intact rabbits after 5 months treatment with LY333334 (hPTH 1-34). Calcif Tissue Int. 2000;66:456–460. [PubMed: 10821883]
- 409.
- Hjälm G, MacLeod J, Kifor O. et al. Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J Biol Chem. 2001;276:34880–34887. [PubMed: 11390380]
- 410.
- Hoare RJ, Gardella TJ, Usdin TB. Evaluating the signal transduction mechanism of the parathyroid hormone 1 receptor: Effect of receptor G-protein interaction on the ligand binding mechanism and receptor conformation. J Biol Chem. 2001;276:7741–7753. [PubMed: 11108715]
- 411.
- Hoare SRJ, Usdin TB. Molecular mechanisms of ligand recognition by parathyroid hormone 1 (PTH1) and PTH2 receptors. Curr Pharma Design. 2001;7:689–713. [PubMed: 11375776]
- 412.
- Hock JM. Stemming bone loss by suppressing apoptosis. J Clin Invest. 1999;104:371–373. [PMC free article: PMC408531] [PubMed: 10449428]
- 413.
- Hodsman AB, Drost D, Fraher LJ. et al. The addition of raloxifene analog (LY117018) allows for reduced PTH(1-34) dosing during reversal of osteopenia in ovariectomized rats. J Bone Miner Res. 1999;14:675–679. [PubMed: 10320515]
- 414.
- Hodsman AB, Fraher LJ, Watson PH. Parathyroid hormone: The clinical experience and prospectsIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,199783–108.
- 415.
- Hodsman AB, Fraher LJ, Watson PH. et al. A randomized controlled trial to compare the efficacy of cyclical parathyroid hormone versus cyclical parathyroid hormone and sequential calcitonin to improve bone mass in post-menopausal women with osteoporsis. J Clin Endocrinol Metab. 1997;82:620–628. [PubMed: 9024265]
- 416.
- Hodsman AB, Watson PH, Drost D. et al. Assessment of maintenance therapy with reduced doses of PTH(1-34) in combination with a raloxifene analogue (LY117018) following anabolic therapy in the ovariectomized rat. Bone. 1999;24:451–455. [PubMed: 10321904]
- 417.
- Hodsman AB, Watson PH, Fraher LJ. et al. Increased bone turnover without change in cortical thickness or porosity after 2 years of cyclical PTH therapy for postmenopausal osteoporosis (abstract). J Bone Miner Res. 2000;15:799. [PubMed: 10913928]
- 418.
- Hodsman AB, Steer BM. Early histomorphometric changes in response to parathyroid hormone therapy in osteoporosis: Evidence for de novo bone formation on quiescent cancellous surfaces. Bone. 1993;14:523–527. [PubMed: 8363903]
- 419.
- Hofbauer LC, Khosla S, Dunstan CR. et al. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999;140:4367–4370. [PubMed: 10465311]
- 420.
- Hofbauer LC, Khoslab S, Dunstan CR. et al. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. [PubMed: 10646108]
- 421.
- Holick MF, Chimeh FN, Ray S. Topical PTH (1-34) is a novel, safe and effective treatment for psoriasis: A randomized self-controlled trial and an open trial. Brit J Dermatol. 2003;149:370–376. [PubMed: 12932245]
- 422.
- Holick MF, Ray S, Chen TC. et al. A parathyroid hormone antagonist stimulates epidermal proliferation and hair growth in mice. Proc Natl Acad Sci USA. 1994;91:8014–8016. [PMC free article: PMC44535] [PubMed: 8058749]
- 423.
- Hollinger S, Hepler JR. Cellular regulation of RGS proteins: Modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. [PubMed: 12223533]
- 424.
- Hook VY, Burton D, Yasothornsrikul S. et al. Proteolysis of PTHrP(1-141) by "prohormone thiol protease" at multibasic residues generates PTHrP-related peptides: Implications for PTHrP peptide production in lung cancer cells. Biochem Biophys Res Commun. 2001;285:32–938. [PubMed: 11467841]
- 425.
- Hopyan S, Gokgoz N, Poon R. et al. A mutant PTH/PTHrP type 1 receptor in endochondromatosis. Nat Genet. 2002;30:306–310. [PubMed: 11850620]
- 426.
- Hoshi K, Ozawa H. Matrix vesicle calcification in bones of adult rats. Calcif Tissue Int. 2000;66:430–434. [PubMed: 10821879]
- 427.
- Huang LE, Bunn HF. Hypoxia-inducible factor and its biomedical relevance. J Biol Chem. 2003;278:9575–19578. [PubMed: 12639949]
- 428.
- Huang Y, Baker RT, Fischer-Vize JA. Control of cell fate by a deubiquitinating enzyme encoded by the fat facets gene. Science. 1995;270:1828–1831. [PubMed: 8525378]
- 429.
- Hurley MM, Marcello K, Abreu C. et al. Transcriptional regulation of the collagenase gene by basic fibroblast growth factor in osteoblastic MC3T3-E1 cells. Biochem Biophys Res Commun. 1995;214:331–339. [PubMed: 7677739]
- 430.
- Hurley MM, Okada Y, Sobue T. et al. The anabolic effect of parathyroid hormone is impaired in bones of Fgf2 null mice (abstract). J Bone Miner Res. 2002;17:S140.
- 431.
- Hurley MM, Tetradis S, Huang Y-F. et al. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res. 1999;14:776–783. [PubMed: 10320526]
- 432.
- Iacopetti P, Barsacchi G, Tirone F. et al. Developmental expression of PC3 gene is correlated with neuronal cell birthday. Mech Devel. 1994;47:127–137. [PubMed: 7811636]
- 433.
- Iacopetti P, Michelini M, Stuckmann I. et al. Expression of the anti proliferative gene TIS21 at the onset of neurogenesis identifies single neuroepithelial cells that switch from proliferative to neuron-generating division. Proc Natl Acad Sci USA. 1999;96:4639–4644. [PMC free article: PMC16385] [PubMed: 10200315]
- 434.
- Iannotti JP, Naidu S, Noguchi Y. et al. Growth plate matrix vesicle biogenesis. Clin Orthop Rel Res. 1994;306:222–229. [PubMed: 8070200]
- 435.
- Iguchi T, Ziff M. Electron microscopic study of rheumatoid synovial vasculature. J Clin Invest. 1986;77:355–361. [PMC free article: PMC423354] [PubMed: 3944263]
- 436.
- Ingleton PM. Parathyroid hormone-related protein in lower vertebrates. Comp Biochem Physiol B Biochem Mol Biol. 2002;132:87–95. [PubMed: 11997212]
- 437.
- Ingleton PM, Bendell LA, Flanagan JA. et al. Calcium-sensing receptors and parathyroid hormone-related protein in the caudal neurosecretory system of the flounder (Platichthys flesus). J Anat. 2002;200:487–497. [PMC free article: PMC1570735] [PubMed: 12090395]
- 438.
- Ito Y. Molecular basis of tissue-specific gene expression mediated by the Runt domain transcription factor PEBP2/CBF. Genes to Cells. 1999;4:685–696. [PubMed: 10620014]
- 439.
- Itzstein C, Cheynel H, Burt-Pichat B. et al. Molecular identification of NMDA glutamate receptors expressed in bone cells. J Cell Biochem. 2001;82:134–144. [PubMed: 11400170]
- 440.
- Itzstein C, Espinosa L, Delmas PD. et al. Specific agonists of NMDA receptors prevent osteoclasts sealing zone formation required for bone resorption. Biochem Biophys Res Commun. 2000;268:201–209. [PubMed: 10652236]
- 441.
- Iwamoto R, Mekada E. Heparin-binding EGF-like growth factor: A juxtacrine growth factor. Cytokine Growth Factor Rev. 2000;11:335–344. [PubMed: 10959080]
- 442.
- Iwaniec UT, Mosekilde L, Mitova-Caneva NG. et al. Sequential treatment with basic fibroblast growth factor and PTH is more efficacious than treatment with PTH alone for increasing vertebral bone mass and strength in osteopenic ovariectomizrd rats. Endocrinology. 2002;143:2515–2526. [PubMed: 12072383]
- 443.
- Iwaniec UT, Shearon CC, Heaney RP. et al. Leptin increases the number of mineralized bone nodules in vitro. J Bone Miner Res. 1998;13:S212.
- 444.
- Jakoby MG, Semenkovich CF. The role of osteoprogenitors in vascular calcification. Curr Opin Nephrol Hypertens. 2000;9:11–15. [PubMed: 10654819]
- 445.
- Jans DA, Hassan G. Nuclear targeting by growth factors, cytokines, and their receptor: A role in signaling? BioEssays. 1998;29:400–411. [PubMed: 9670813]
- 446.
- Jans DA, Thomas RJ, Gillespie MT. Parathyroid hormone-related protein (PTHrP): A nucleoplasmic shuttling protein with distinct paracrine and intracrine roles. Vitamins and Hormones. 2003;66:345–384. [PubMed: 12852260]
- 447.
- Jee W. Integrated bone tissue physiology: Anatomy and physiologyIn: Cowin SC, ed.Bone Mechanics Handbook. 2nd edBoca Raton: CRC Press,20001–1-1-68.
- 448.
- Jensen AA, Greenwood JR, Brouner-Osborne H. The dance of the the clams: Twists and turns in the family C GPCR homodimer. Trends Pharmacol Sci. 2002:491–493. [PubMed: 12413796]
- 449.
- Jensen CG, Poole CA, McGlashen SR. et al. Ultrastructural tomographic and confocal imaging of the chondrocyte primary cilium in situ. Cell Biol Int. 2004;28:101–110. [PubMed: 14984755]
- 450.
- Jerome CP, Burr DB, Van Bibber T. et al. Treatment with human parathyroid hormone (1-34) for 18 months increases cancellous bone volume and improves trabecular architecture in ovariectomized cynomolgus monkeys (Macaca fascicularis). Bone. 2001;28:150–159. [PubMed: 11182372]
- 451.
- Jerome CP, Carlson CS, Register TC. et al. Bone functional changes in intact, ovariectomized, and ovariectomized, hormone-supplemented adult cynomolgus monkeys (Macaca fascicularis) evaluated by serum markers and dynamic histomorphometry. J Bone Miner Res. 1994;9:527–540. [PubMed: 8030441]
- 452.
- Jerome CP, Johnson CS, Vafai HT. et al. Effect of treatment for 6 months with human parathyroid hormone (1-34) peptide in ovariectomized cynomolgus monkeys (Macaca fascicularis). Bone. 1999;25:301–309. [PubMed: 10495134]
- 453.
- Ji X, Chen D, Xu C. et al. Patterns of gene expression associated with BMP-2-induced osteoblasts and adipocyte differentiation of mesenchymal progenitor cell 3T3-F442A. J Bone Miner Res. 2000;15:132–139. [PubMed: 10783846]
- 454.
- Jick H, Zornberg GL, Jick SS. et al. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. [PubMed: 11089820]
- 455.
- Jilka RL. Cytokines, bone remodeling, and estrogen deficiency. Bone. 1998;23:75–78. [PubMed: 9701464]
- 456.
- Jilka RL, Weinstein RS, Bellido T. et al. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–446. [PMC free article: PMC408524] [PubMed: 10449436]
- 457.
- Johansson A, Rosen CJ. The insulin-like growth factors: Potential anabolic agents for the skeletonIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,1997185–205.
- 458.
- Johnson LC. The kinetics of skeletal remodeling. Birth defects Original Article Series. 1966;2:66–142.
- 459.
- Jolette J, Smith SY, Mayer J. et al. Ostabolin-C™: A novel parathyroid hormone analogue uncouples bone turnover in monkeys (abstract). J Bone Miner Res. 2003;18:S386.
- 460.
- Jono S, McKee MD, Murry CE. et al. Phosphate regulation of vascular smooth muscle calcification (abstract). Circ Res. 2000;87:E10–E17. [PubMed: 11009570]
- 461.
- Jordan BA, Cvejic S, Devi LA. Opioids and their complicated receptor complexes. Neuropsychopharmacol. 2000;23:S5–S18. [PubMed: 11008063]
- 462.
- Jordan BA, Trapaidze N, Gomes I. et al. Oligomerization of opioid receptors with β2-adrenergic receptors: A role in trafficking and mitogen-activated protein kinase activation. Proc Natl Acad Sci USA. 2001;98:343–348. [PMC free article: PMC14592] [PubMed: 11134510]
- 463.
- Jouishomme H, Whitfield JF, Chakravarthy B. et al. The protein kinase-C activation domain of the parathyroid hormone. Endocrinology. 1992;130:53–60. [PubMed: 1727720]
- 464.
- Jouishomme H, Whitfield JF, Gagnon L. et al. Further definition of the protein kinase C activation domain of the parathyroid hormone. J Bone Miner Res. 1994;9:943–949. [PubMed: 8079668]
- 465.
- Ju W, Hoffman A, Verschueren K. et al. The bone morphogenic protein 2 signaling mediator Smad 1 participates predominantly in osteogenic and not in chondrogenic differentiation in mesenchymal progenitors C3H10T1/2. J Bone Miner Res. 2000;15:1889–1899. [PubMed: 11028440]
- 466.
- Juhlin L, Hagforsen E, Juhlin C. Parathyroid hormone related protein is localized in the granular layer of normal skin and in the dermal infiltrates of mycosis fungoides but is absent in psoriatic lesions. Acta Derm Venereol. 1992;72:81–83. [PubMed: 1350411]
- 467.
- Juul A. The effects of oestrogens linear bone growth. Hum Reprod Update. 2001;7:303–313. [PubMed: 11392377]
- 468.
- Kalu DN, Doyle FH, Pennock J. et al. Parathyroid hormone and experimental osteoslerosis. Lancet. 1970;1:1363–1366. [PubMed: 4195059]
- 469.
- Kameda T, Mano H, Yameda Y. et al. Calcium-sensing receptors in mature osteoclasts which are bone-resorbing cells. Biochem Biophys Res Commun. 1998;245:419–422. [PubMed: 9571166]
- 470.
- Kamioka H, Honjo T, Takano-Yamamoto T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone. 2001;28:145–149. [PubMed: 11182371]
- 471.
- Kanazaki M, Zhang YQ, Mashima H. et al. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nature Cell Biol. 1:165–170. [PubMed: 10559903]
- 472.
- Kanatami M, Sugimoto T, Takahashi Y. et al. Estrogen via the estrogen receptor blocks cAMP-mediated parathyroid hormone (PTH)-stimulated osteoclast function. J Bone Miner Res. 1998;13:854–862. [PubMed: 9610750]
- 473.
- Kanazawa M, Sugimoto T, Kanatami M. et al. Involvement of osteoprotegerin/osteoclastogenesis inhibitory factor in the stimulation of osteoclast formation by parathyroid hormone in mouse bone cells. Eur J Endocrinol. 2000;142:61–664. [PubMed: 10822231]
- 474.
- Karaplis AC. PTHrP: Novel roles in skeletal biology. Curr Pharma Design. 2001;7:655–670. [PubMed: 11375774]
- 475.
- Karaplis AC, Luz A, Glowacki J. et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289. [PubMed: 8314082]
- 476.
- Karnik SK, Gogonea C, Patil S. et al. Activation of G-protein coupled receptors: A common molecular mechanism. Trends Endocrinol Metab. 2003;14:431–437. [PubMed: 14580763]
- 477.
- Karperien M, Farih-Sips H, Papapoulos SE. et al. Involvement of Cbfa1 in transcritional regulation of the type I PTH/PTHrP-receptor in KS483 osteoblasts (abstract). J Bone Miner Res. 2000;15:S175.
- 478.
- Karsenty G. The genetic transformation of bone biology. Genes Dev. 1999;13:3037–3051. [PubMed: 10601030]
- 479.
- Karsenty G. Role of Cbfa1 in osteoblast differentiation and function. Seminars Cell Dev Biol. 2000a;11:343–346. [PubMed: 11105898]
- 480.
- Karsenty G. The central regulation of bone remodeling. Trends Endocrinol Metab. 2000b;11:437–439. [PubMed: 11091123]
- 481.
- Karsenty G. Chondrogenesis just ain't what it used to be. J Clin Invest. 2001;107:405–407. [PMC free article: PMC199281] [PubMed: 11181639]
- 482.
- Kartsogiannis V, Moseley J, McKelvie B. et al. Temporal expression of PTHrP during endochondral bone formation in mouse and intramembranous bone formation in an in vivo rabbit model. Bone. 1997;21:385–392. [PubMed: 9356731]
- 483.
- Katagiri T, Takahashi N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis. 2002;8:147–159. [PubMed: 12108759]
- 484.
- Kawakami A, Eguchi K, Matsuoka N. et al. Fas and Fas ligand interaction is necessary for human osteoblast apoptosis. J Bone Miner Res. 1997;12:1637–1646. [PubMed: 9333124]
- 485.
- Kawakami A, Nakashima T, Tsuboi M. et al. Insulin-like growth factor I stimulates proliferation and Fas-mediated apoptosis of human osteoblasts. Biochem Biophys Res Commun. 1998;247:46–51. [PubMed: 9636651]
- 486.
- Kawamura YJ, Kazama S, Miyahara T. et al. Sigmoid colon cancer associated with primary hyperparathyroidism: Report of a case. Surg Today. 1999;29:789–790. [PubMed: 10483759]
- 487.
- Kawane T, Takahashi S, Saitoh H. et al. Anabolic effects of recombinant human parathyroid hormone (1-84) on the mandibles of osteopenic ovariectomized rats with maxillary molar extraction. Horm Metab Res. 2002;34:293–302. [PubMed: 12173069]
- 488.
- Ke HZ, Steppan CM, Swick AG. Treatment of skeletal disorders. United States Patent. 2001;6(352):970 B1.
- 489.
- Ke HZ, Steppan CM, Swick AG. Treatment of skeletal disorders United States Patent 2002. Application Publication 2002/0019351 A1 .
- 490.
- Keenan SM, Baldassare JJ. Molecular scaffold protein and cellular responses. Trends Endocrinol Metab. 2001;12:184–186. [PubMed: 11397637]
- 491.
- Kellenberger S, Muller K, Richener H. et al. Formoterol and isoproterenol induce c-fos gene expression in osteoblast-like cells by activating β2-adrenergic receptors. Bone. 1998;22:471–478. [PubMed: 9600780]
- 492.
- Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrino Metab. 2001;12:152–156. [PubMed: 11295570]
- 493.
- Kempf H, Chen RE, Kerley ER. The microscopic determination of age in human bone. Am J Phys Anthropol. 1965;23:149–164. [PubMed: 5826273]
- 494.
- Kern B, Shen J, Starbuck M. et al. Cbfa1 contributes to the osteoblast-specific expression of type I collagen genes. J Biol Chem. 2001;276:7101–7107. [PubMed: 11106645]
- 495.
- Kerr JB. Atlas of Functional HistologyLondon: Mosby International Limited, 1999.
- 496.
- Khosla S. Oestrogen, bones and men: When testosterone just isn't enough. Clin Endocrinol (Oxford). 2002;56:291–293. [PubMed: 11940038]
- 497.
- Kifor O, Diaz R, Butters R. et al. The calcium-sensing receptor is localized in caveolin-rich plasma membrane domains of bovine parathyroid cells. J Biol Chem. 1998;273:21708–21713. [PubMed: 9705306]
- 498.
- Kifor O, Kifor I, Brown EM. Signal transduction in the parathyroid. Curr Opin Nephrol Hypertens. 2002;11:397–402. [PubMed: 12105389]
- 499.
- Kifor O, Kifor I, Moore FD. et al. m-Calpain colocalizes with the calcium-sensing receptor (CaR) in caveolae in parathyroid cells and participates in degradation of the CaR. J Biol Chem. 2003;278:31167–31176. [PubMed: 12783889]
- 500.
- Kim DW, Kempf H, Chen RE. et al. Characterization of Nkx3.2 DNA binding specificity and its requirement for somatic chondrogenesis. J Biol Chem. 2003;278:27532–27539. [PubMed: 12746429]
- 501.
- Kim HW, Jahng JS. Effect of intermittent administration of parathyroid hormone on fracture healing in ovariectomized rats. Iowa Orthop J. 1999;19:71–77. [PMC free article: PMC1888615] [PubMed: 10847519]
- 502.
- Kim HP, Lee JY, Jeong JK. et al. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem Biophys Res Commun. 1999;263:257–262. [PubMed: 10486286]
- 503.
- Kim J, Jones BW, Zock C. et al. Isolation and characterization of mammalian homologs of the Drosophila gene glial cells missing. Proc Natl Acad Sci USA. 1998;95:12364–12369. [PMC free article: PMC22837] [PubMed: 9770492]
- 504.
- Kinoshita T, Kobayashi S, Ebara S. et al. Phosphodiesterase inhibitors, Pentoxyfylline and Rolipram, increase bone mass mainly by promoting bone formation in normal mice. Bone. 2000;27:811–817. [PubMed: 11113392]
- 505.
- Kirsch T, Harrison G, Golub EE. et al. The roles of annexins and types II and X collagens in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem. 2000;275:35577–35583. [PubMed: 10956650]
- 506.
- Kirsch T, Nah HD, Shapiro IM. et al. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol. 1997;137:1149–1160. [PMC free article: PMC2136219] [PubMed: 9166414]
- 507.
- Kishida Y, Myoui A, Tamai N. et al. Effect of leptin on growth plate and chondrocyte differentiation (abstract). J Bone Miner Res. 2003;18:S59.
- 508.
- Kitazawa R, Kitazawa S, Maeda S. et al. Promoter structure of mouse RANKL/TRANCE/OPGL/ODF gene. Biochim Biophys Acta. 1999;1445:134–141. [PubMed: 10209265]
- 509.
- Kitazawa S, Kajimoto K, Kondo T. et al. Vitamin D3 supports osteoclastogenesis via functional vitamin D response element of human RANKL gene promoter. J Cell Biochem. 2003;89:771–777. [PubMed: 12858342]
- 510.
- Kizer N, Guo K-L, Hruska K. Reconstitution of stretch-activated cation channels by expression of the epithelial sodium channel cloned from osteoblasts. Proc Natl Acad Sci USA. 1997;94:1013–1018. [PMC free article: PMC19631] [PubMed: 9023374]
- 511.
- Klein RF. Osteoporosis in menIn: Marcus R, ed.Atlas of Clincal Endocrinology OsteoporosisPhiladelphia: Current Medicine1999385–99.
- 512.
- Klein-Nulent J, Nijweide PJ. Osteocyte and bone structure. Curr Osteoporosis Rep. 2003;1:5–10. [PubMed: 16036059]
- 513.
- Kneissel M, Boyde A, Gasser JA. Bone tissue and its mineralization in aged estrogen-depleted rats after long-term intermittent treatment with parathyroid hormone (PTH) analog SDZ PTS 893 or human PTH-(1-34). Bone. 2001;28:237–250. [PubMed: 11248653]
- 514.
- Knothe-Tate M. Interstitial fluid flowIn: Cowin SC, ed.Bone Mechanics Handbook. 2nd edBoca Raton: CRC Press,200122–1-22-29.
- 515.
- Knothe-Tate ML. Whither flows the fluid in bone? An osteocyte's perspective. J Biomechanics. 2003;36:1409–1424. [PubMed: 14499290]
- 516.
- Knothe-Tate ML, Adamson JR, Tami AE. et al. The osteocyte. Int J Biochem Cell Biol. 2004;36:1–8. [PubMed: 14592527]
- 517.
- Knothe-Tate ML, Knothe U, Niederer P. et al. Experimental elucidation of mechanical load-induced fluid flow and its potential role in bone metabolism and functional adaptation. Am J Med Sci. 1998;316:189–195. [PubMed: 9749561]
- 518.
- Knothe-Tate ML, Steck R, Forwood MR. et al. In vivo demonstration of load-induced fluid flow in the rat tibia and its potential implications for processes associated with functional adaptation. J Exp Biol. 2000;203:2737–2745. [PubMed: 10952874]
- 519.
- Kobayashi K, Takahashi N, Jimi E. et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–286. [PMC free article: PMC2195746] [PubMed: 10637272]
- 520.
- Kobayashi T, Chung U, Schipani E. et al. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986. [PubMed: 12050144]
- 521.
- Koch CA. Rapid increases in bone mineral density in a child with osteoporsis and autoimmune hypoparathyroidism treated with PTH 1-34. Exp Clin Endocrinol Diabetes. 2001;109:350–354. [PubMed: 11571675]
- 522.
- Komori T. A fundamental transcription factor for bone and cartilage. Biochem Biophys Res Commun. 2000;276:813–816. [PubMed: 11027552]
- 523.
- Kong Y-Y, Felge U, Sarosi I. et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. [PubMed: 10580503]
- 524.
- Kostenuik PJ, Capparelli C, Morony S. et al. OPG and PTH-(1-34) have additive effects on bone density and mechanical strength in osteopenic ovariectomized rats. Endocrinology. 2001;142:4295–4304. [PubMed: 11564687]
- 525.
- Kostenuik PJ, Harris J, Halloran BP. et al. Skeletal unloading causes resistance of osteoprogenitor cells to parathyroid hormone and to insulin-like growth factor-I. J Bone Miner Res. 1999;14:21–31. [PubMed: 9893062]
- 526.
- Kostenuik PJ, Shalhoub V. Osteoprotegerin: A physiological and pharmacological inhibitor of bone resorption. Curr Pharma Design. 2001;7:613–635. [PubMed: 11375772]
- 527.
- Kousteni S, Bellido T, Plotkin LI. et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed: 11257226]
- 528.
- Kousteni S, Chen J-R, Bellido T. et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002a;298:843–846. [PubMed: 12399595]
- 529.
- Kousteni S, Han L, Plotkin LJ. et al. Nongenotropic regulation of CREB-, C/EBPβ-, as well as ELK-1 and AP-1-mediated transcription by estrogens: Downstream effects of ERK and JNK kinase modulation required for anti-apoptosis. J Bone Miner Res. 2002b;17:S169.
- 530.
- Koyama Y, Kitahara K, Ritting SR. et al. Osteopontin plays a role in regulating apoptosis and mineralization under the control of inorganic phosphate. J Bone Miner Res. 2003;18:S137.
- 531.
- Krainock R, Murphy S. Nitric oxideIn: Conn PM, Means AR, eds.Principles of Molecular RegulationTotowa, NJ: Humana Press,2000219–227.
- 532.
- Kreienkamp H-J. Organization of G-protein receptor signaling complexes by scaffolding proteins. Curr Opin Pharmacol. 2002;2:581–586. [PubMed: 12324263]
- 533.
- Krempien B, Friedrich E, Ritz E. Effect of PTH on osteocyte ultrastructure. Adv Exp Med Biol. 1978;103:437–450. [PubMed: 717118]
- 534.
- Krishnan V, Heat H, Bryant HU. Mechanism of action of estrogens and selective estrogen receptor modulators. Vitamins and Hormones. 2001;60:123–147. [PubMed: 11037623]
- 535.
- Krishnan V, Moore TL, Ma YL. et al. Parathyroid hormone bone anabolic action requires cbfa1/runx-2-dependent signaling. Mol Endocrinol. 2003;17:423–435. [PubMed: 12554794]
- 536.
- Kronenberg HM, Bringhurst FR, Segre GV. et al. Parathyroid hormone-Biosynthesis and MetabolismIn: Bilezikian JP, ed.The Parathyroids. 2nd edSan Diego: Academic Press,200117–30.
- 537.
- Kubota K, Sakikawa C, Katsumata M. et al. Platelet-derived growth factor BB secreted from osteoclasts acts as an osteoblastogenesis inhibitory factor. J Bone Miner Res. 2002;17:257–265. [PubMed: 11811556]
- 538.
- Kubodera N, Tsuji N, Uchiyama Y. et al. A new active vitamin D analog, ED-71, causes increase in bone mass with preferential effects on bone in osteoporotic patients. J Cell Biochem. 2003;88:286–289. [PubMed: 12520528]
- 539.
- Kufahl RH, Saha S. A theoretical model for stress-generated fluid flow in the canaliculi-lacunae network in bone tissue. J Biomech. 1990;23:171–180. [PubMed: 2312521]
- 540.
- Kukreja SC, D'Anza JJ, Wimbiscus SA. et al. Inactivation by plasma may be responsible for lack of efficacy of parathyroid hormone antagonists in hypercalcemia of malignancy. Endocrinology. 1994;134:2184–2188. [PubMed: 8156920]
- 541.
- Kurland EH, Cosman F, McMahon DJ. et al. Parathyroid hormone as a therapy for idiopathic osteoporosis in men: Effects on bone mineral density and bone markers. J Clin Endocrinol Metab. 2000;85:3069–3076. [PubMed: 10999788]
- 542.
- Kuroda M, Hazama-Shimada Y, Endo A. Inhibition of sterol synthesis by citrinin in a cell-free system from rat liver and yeast. Biochim Biophys Acta. 1977;486:254–259. [PubMed: 13847]
- 543.
- Kume K, Satomura K, Nishisho S. et al. Potential role of leptin in endochondral ossification. J Histochem Cytochem. 2002;50:159–169. [PubMed: 11799135]
- 544.
- LaCroix AZ, Cauley JA, Pettinger M. et al. Statin use, clinical fracture, and bone density in postmenopausal women: Results from Women's Health Initiative Observational Study. Ann Intern Med. 2003;139:97–104. [PubMed: 12859159]
- 545.
- Laharrague P, Larrouy D, Fontanilles AM. et al. High expression of leptin by human bone marrow adipocytes in primary culture. FASEB J. 1998;12:747–752. [PubMed: 9619453]
- 546.
- Laketic-Ljubojevic I, Suva LJ. et al. Functional characterization of the N-methyl-D-aspartic acid-gated channels in bone. Bone. 1999;25:631–637. [PubMed: 10593407]
- 547.
- Lam MH, House CM, Tiganis T. et al. Phosphorylation at the cyclin-dependent kinase site (Thr 85) of parathyroid hormone-related protein negatively regulates its nuclear localization. J Biol Chem. 1999;274:18559–18566. [PubMed: 10373465]
- 548.
- Lam MH, Thomas RJ, Loveland KL. et al. Nuclear transport of parathyroid hormone (PTH)-related protein is dependent on microtubules. Mol Endocrinol. 2002;16:390–401. [PubMed: 11818509]
- 549.
- Lam MHC, Thomas RJ, Martin TJ. et al. Nuclear and nucleolar localization of parathyroid hormone-related protein. Immunol Cell Biol. 2000;78:395–402. [PubMed: 10947864]
- 550.
- Lando D, Gorman JJ, Whitelaw ML. et al. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and aspariginyl hydroxylation. Eur J Biochem. 2003;270:781–790. [PubMed: 12603311]
- 551.
- Lane NE, Kimmel DB, Nilsson MH. et al. Bone-selective analogs of human PTH-(1-34) increase bone formation in an ovariectomized rat model. J Bone Miner Res. 1996;11:614–625. [PubMed: 9157776]
- 552.
- Lane NE, Sanchez S, Modin GW. et al. Bone mass continues to increase at the hip after parathyroid hormone treatment is discontinued in glucocorticoid-induced osteoporosis: Results of a randomized controlled clinical trial. J Bone Miner Res. 2000;15:944–951. [PubMed: 10804025]
- 553.
- Lane NE, Thompson JM, Strewler GJ. et al. Intermittent treatment with parathyroid hormone (hPTH 1-34) increased trabecular bone volume but not connectivity in osteopenic rats. J Bone Miner Res. 1995;10:1470–1477. [PubMed: 8686502]
- 554.
- Lane NE, Yao W, Arnaud CD. Changes in serum RANKL, OPG ans IL-6 during PTH (1-34) administration in patients with glucocorticoid induced osteoporosis. J Bone Miner Res. 2003;18:S95.
- 555.
- Lane NE, Yao W, Balooch M. et al. Both hPTH (1-34) and bFGF increase bone mass in osteopenic animals with different effects on trabecular bone architecture (abstract). J Bone Miner Res. 2003;18:S95. [PubMed: 14672345]
- 556.
- Langub MC, Monier-Faugere M-C, Qi Q. et al. Parathyroid hormone/parathyroid hormone-related peptide type1 receptor in human bone. J Bone Miner Res. 2001;16:448–456. [PubMed: 11277262]
- 557.
- Lanske B, Amling M, Neff L. et al. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J Clin Invest. 1999;104:399–407. [PMC free article: PMC408525] [PubMed: 10449432]
- 558.
- Lanske B, Karaplis AC, Lee K. et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. [PubMed: 8662561]
- 559.
- Lanyon LE, Rubin CT. Static vs dynamic loads as an influence on bone remodelling. J Biomech. 1984;17:897–905. [PubMed: 6520138]
- 560.
- Latchman DS. Eukaryotic Transcription Factors. 4 edSan Diego: Elsevier Academic Press, 2004.
- 561.
- Lau NC, Bartel DP. Censors of the genome. Sci Amer. 2003;289:34–41. [PubMed: 12884536]
- 562.
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. [PubMed: 9727051]
- 563.
- LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by α-secretase and presenilin / γ-secretase and release signaling fragments. J Biol Chem. 2003;278:34127–34137. [PubMed: 12826675]
- 564.
- Lawler OA, Miggin SM, Kinsella BT. Protein kinase A mediated phos-phorylation of serin 357 of the mouse prostacyclin receptor regulates its coupling to Gs-, to Gi- and to Gq-coupled effector signaling. J Biol Chem. 2001;276:33596–33607. [PubMed: 11443126]
- 565.
- Leaffer D, Sweeny M, Kellerman LA. et al. Modulation of osteogenic cell ultrastructure by RS-23581, an analog of human parathyroid hormone (PTH)-related peptide-(1-34), and ovine PTH-(1-34). Endocrinology. 1995;136:3624–3631. [PubMed: 7628402]
- 566.
- Lebwohl M. Psoriasis. The lancet. 2003;361:1197–1205. [PubMed: 12686053]
- 567.
- Lee C-W, Nam J-S, Park Y-K. et al. Lysophosaphatidic acid stimulates CREB through mitogen- and stress-activated protein kinase-1. Biochem Biophys Res Commun. 2003;305:455–461. [PubMed: 12763014]
- 568.
- Lee E, HU N, Yuan S-SF. et al. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8:2008–2021. [PubMed: 7958874]
- 569.
- Lee K, Deeds JD, Chiba S. et al. Parathyroid hormone induces sequential c-fos expression in vivo: In situ localization of its receptor and c-fos messenger ribonucleic acids. Endocrinology. 1994;134:441–450. [PubMed: 8275957]
- 570.
- Lee K, Deeds JD, Segre GV. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acid during fetal development of rats. Endocrinology. 1995;136:453–463. [PubMed: 7835276]
- 571.
- Lee K, Lanske B, Karaplis AC. et al. Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology. 1996;137:5109–5118. [PubMed: 8895385]
- 572.
- Lee SP, Xie Z, Varghese G. et al. Oligomerization of dopamine and serotonin receptors. Neuropsychopharmacol. 2000;23:S32–S40. [PubMed: 11008065]
- 573.
- Lee SW, Kwak HB, Lee HC. et al. The anti-proliferative gene TS21 is involved in osteoclast differentiation. J Biochem Mol Biol. 2002;35:609–614. [PubMed: 12470596]
- 574.
- Lee Y-J, Park J-H, Ju S-K. et al. Leptin receptor isoform expression in rat osteoblasts and their functional analysis. FEBS Lett. 2002;528:43–47. [PubMed: 12297277]
- 575.
- Lee ZH, Kim H-H. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun. 2003;305:211–214. [PubMed: 12745060]
- 576.
- Levin ER. Nuclear receptor versus plasma membrane oestrogen receptor. Novartis Found Symp. 2000;230:41–55. [PubMed: 10965501]
- 577.
- Lian JB, Stein GS. Runx2/Cbfa1: A multifunctional regulator of bone formation. Curr Pharma Design. 2003;9:2677–2685. [PubMed: 14529540]
- 578.
- Lian JB, Stein GS, Stein JL. et al. Transcriptional control of osteoblast differentiation. Biochem Soc Trans. 1998;26:14–21. [PubMed: 10909750]
- 579.
- Liang H, Pun S, Wronski TJ. Bone anabolic effects of basic fibroblast growth factor in ovariectomized rats. Endocrinology. 1999;140:5780–5788. [PubMed: 10579344]
- 580.
- Liang JD, Hock JM, Sandusky GE. et al. Immunohistochemical localization of selected early response genes expressed in trabecular bone of young rats given PTH 1-34. Calcif Tissue Int. 1999;65:369–373. [PubMed: 10541762]
- 581.
- LiChong KP, Li Dy, Orlowski J. et al. Selective domains of PTH and PTHrP activate the protein kinase A and protein kinase C pathways and differentially influence the Na+/K+ exchanger (NH3) and the type II Na/Pi cotransporter in renal OK cells (abstract). Bone. 1998;23:S356.
- 582.
- Lieberherr M, Grosse B, Kachkache M. et al. Cell signaling and estrogens in female rat osteoblasts: A possible involvement of unconventional nonnuclear receptors. J Bone Miner Res. 1993;8:1365–1376. [PubMed: 8266828]
- 583.
- Lin S-Y, Makino K, Xia W. et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. [PubMed: 11533659]
- 584.
- Lincoln J, Hoyle CHV, Burnstock G. Nitric Oxide in Health and DiseaseCambridge: Cambridge University Press,1997.
- 585.
- Lindsay R, Hodsman A, Genant H. et al. A randomized controlled multi-center study of 1-84hPTH for treatment of postmenopausal osteoporosis (abstract). Bone. 1998;23:S175.
- 586.
- Lindsay R, Nieves J, Formica C. et al. Randomised controlled study of effect of parathyroid hormone on vertebral bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;550:555. [PubMed: 9284777]
- 587.
- Lindsay R, Nieves J, Henneman E. et al. Subcutaneous administration of the amino-terminal fragment of human parathyroid hormone (1-34): Kinetics and biochemical response in estrogenized osteoporotic patients. J Clin Endocrinol Metab. 1993;77:1535–1539. [PubMed: 8263137]
- 588.
- Llinαs RR. I of the Vortex: From Neurons to SelfCambridge, MA: The MIT Press,2001 .
- 589.
- Locklin RM, Khosla S, Turner RT. et al. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89:180–190. [PubMed: 12682918]
- 590.
- Lofthouse RA, Davis JR, Frondoza CG. et al. Identification of caveoli in normal human osteoblasts. J Bone Joint Surg Br. 2001;83:124–129. [PubMed: 11245520]
- 591.
- Lomri A, Marie PJ. Changes in cytoskeletal proteins in response to parathyroid hormone and 1,25-dihydroxyvitamin D in human osteoblastic cells. Bone Miner. 1990;10:1–12. [PubMed: 2168775]
- 592.
- Long JA. The Rise of FishesBaltimore: Johns Hopkins University Press,1995.
- 593.
- Lord BI. The architecture of bone marrow cell populations. Int J Cell Cloning. 1990:317–331. [PubMed: 2230283]
- 594.
- Lorget F, Kamel S, Mentaverri R. et al. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem Biophys Res Commun. 2000;268:899–903. [PubMed: 10679302]
- 595.
- Lowell , Le Roux I, Dunne J. et al. Stimulation of human epidermal differentiation by Delta-Notch signalling at the boundaries of stem cell clusters. Curr Biol. 2000;10:491–500. [PubMed: 10801437]
- 596.
- Lu SS, Ducayen-Knowles M, Dempster DW. et al. Effects of parathyroid hormone on gene expression of RANK ligand (RANKL), osteoprotegerin (OPG) and the cognate receptor for PTH in mice. J Bone Miner Res. 2001;16:S426.
- 597.
- Lu W, Shen X, Pavlova X. et al. Comparison of pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Human Mol Genet. 2001;10:2385–2396. [PubMed: 11689485]
- 598.
- Luckman SP, Hughes DE, Coxon FP. et al. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13:581–589. [PubMed: 9556058]
- 599.
- Luttrell LM, Roudabush FL, Choy EW. et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454. [PMC free article: PMC30158] [PubMed: 11226259]
- 600.
- Ma YL, Bryant HU, Zeng Q. et al. New bone formation with teriparatide [human parathyroid hormone-(1-34)] is not retarded by long-term pretreatment with alendronate, estrogen, or raloxifene in ovariectomized rats. Endocrinology. 2003;144:2008–2015. [PubMed: 12697709]
- 601.
- Ma Y, Jee W, Yuan Z. et al. Parathyroid hormone and mechanical usage have a synergistic effect in rat tibial diaphyseal cortical bone. J Bone Miner Res. 1999;14:439–448. [PubMed: 10027909]
- 602.
- Ma YL, Zeng QQ, Cain RL. et al. PTH induces similar anabolic effects in lumbar vertebrae of adult ovary-intact and osteopenic ovariectomized rats (abstract). J Bone Miner Res. 2000;15:813.
- 603.
- Ma YL, Cain RL, Halladay DL. et al. Catabolic effects of continuous human PTH (1-38) in vivo is associated with sustained stimulation of of RANKL and inhibition of osteoprotegerin in gene-associated bone formation. Endocrinology. 2001;142:4047–4054. [PubMed: 11517184]
- 604.
- Machwate M, Rodan SB, Rodan GA. et al. Sphingosine kinase mediates cyclic AMP suppression of apoptosis in rat periosteal cells. Mol Pharmacol. 1998;54:70–77. [PubMed: 9658191]
- 605.
- Macica CM, Broadus AE. PTHrP regulates cerebral blood flow and is neuroprotective. Am J Physiol Regulat Integr Comp Physiol. 2003;284:R1019–R1020. [PubMed: 12626363]
- 606.
- MacLean HE, Guo J, Knight MC. et al. The cyclin-dependent kinase inhibitor p57Kip2 mediated proliferative actions of PTHrP in chondrocytes. J Clin Invest. 2004;113:1334–1343. [PMC free article: PMC398433] [PubMed: 15124025]
- 607.
- MacLeod RJ, Chattopadhyay N, Brown EM. PTHrP stimulated by the calcium-sensing receptor requires MAP kinase activation. Am J Physiol Endocrinol Metab. 2003;284:E435–E442. [PubMed: 12388158]
- 608.
- Maeda T, Matsunuma A, Kawane T. et al. Simvastatin promotes osteoblast differentiation and mineralization in MC3T3 cells. Biochem Biophys Res Commun. 2001;280:874–877. [PubMed: 11162604]
- 609.
- Magne D, Bluteau G, Faucheux C. et al. Phosphate is a specific signal for ATDC5 chondrocyte maturation and apopotosis-associated mineralization: Possible implications of apoptosis in the regulation of endochondral ossification (abstract). J Bone Miner Res. 2003;18:1430–1442. [PMC free article: PMC2071932] [PubMed: 12929932]
- 610.
- Mahon MJ, Donowitz M, Yun CC. et al. Na+/H+ exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signaling. Nature. 2002;417:858–861. [PubMed: 12075354]
- 611.
- Mahon MJ, Segre GV. Parathyroid hormone-mediated activation of MAPK is dependent on NHERF2 in PS120 cells (abstract). J Bone Miner Res. 2002;17:S215.
- 612.
- Mahon MJ, Segre GV. NHERF-1 is required for phospholipase-C-dependent calcium influx induced by parathyroid hormone in opossum kidney cells. J Bone Miner Res. 2003;18:S29.
- 613.
- Majeska RJ, Minkowitz B, Bastian W. et al. Effects of β-adrenergic blockade in an osteoblast-like cell line. J Orthop Res. 1992;10:379–384. [PubMed: 1349041]
- 614.
- Makhluf HA, Mueller SM, Mizuno S. et al. Age-related decline in osteoprotegerin expression expression by human bone marrow cells cultured in three-dimensional collagen sponges. Biochem Biophys Res Commun. 2000;268:669–672. [PubMed: 10679262]
- 615.
- Mannella CA. Our changing views of mitochondria. J Bioener Biomembranes. 2000;32:1–4. [PubMed: 11768754]
- 616.
- Manolagas SC. Advances in the treatment of osteoporosis Medscape Endocrinology Journal 19991http//wwwmedscapecom .
- 617.
- Manolagas SC. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocrine Revs. 2000;21:115–137. [PubMed: 10782361]
- 618.
- Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409. [PubMed: 12017554]
- 619.
- Manolagas SC, Weinstein RS, Bellido T. et al. Activators of nongenomic estrogen-like signaling (ANGELS): A novel class of small molecules with bone anabolic properties (abstract). J Bone Miner Res. 1999;14:S180.
- 620.
- Mansfield K, Teixeira CC, Adams CS. et al. Phoaphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. 2001;28:1–8. [PubMed: 11165936]
- 621.
- Mansfield K, Rajpurohit R, Shapiro IM. Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J Cell Physiol. 1999;179:276–286. [PubMed: 10228946]
- 622.
- Maor GA, Rochwerger M, Segev Y. et al. Leptin acts as a skeletal growth factor on chondrocytes of skeletal growth centers. J Bone Miner Res. 2002;17:1034–1043. [PubMed: 12054158]
- 623.
- Marie PJ, Ammann P, Boivin G. et al. Mechanism of action and the therpeutic potential of strontium in bone. Calcif Tissue Int. 2001;69:121–129. [PubMed: 11683526]
- 624.
- Marenholz I, Zirra M, Fischer DF. et al. Identification of human epidermal differentiation complex (EDC)-encoded genes by subtractive hybridization of entire YACs to a gridded keratinocyte cDNA library. Genome Res. 2001;11:341–355. [PMC free article: PMC311024] [PubMed: 11230159]
- 625.
- Marie PJ, Jones D, Vico L. et al. Osteobiology, strain, and microgravity: Part 1. Studies at the cellular level. Calcif Tissue Int. 2000;67:2–9. [PubMed: 10908405]
- 626.
- Mark AL, Shaffer RA, Correia ML. et al. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–1953. [PubMed: 10703894]
- 627.
- Marotti G. The structure of bone tissues and the cellular control of their deposition. Ital J Anat Embryol. 1996;101:25–79. [PubMed: 9203871]
- 628.
- Marijanovic I, Kronenberg MS, Erceg I. et al. Dlx3,5 and 6 may have redundant effects on bone development. J Bone Miner Res. 2002;17:S443.
- 629.
- Martin RB. Toward a unifying theory of bone remodeling. Bone. 2000a;26:1–6. [PubMed: 10617150]
- 630.
- Martin RB. Does osteocyte formation cause the nonlinear refilling of osteons ? Bone. 2000b;26:71–78. [PubMed: 10617159]
- 631.
- Martin RB. Is all cortical bone remodeling initiated by microdamage? Bone. 2002;30:8–13. [PubMed: 11792558]
- 632.
- Martin RB. Fatigue microdamage as an essential element of bone mechanics and biology. Calcif Tissue Int. 2003;73:101–107. [PubMed: 14565590]
- 633.
- Martin RB, Burr DB, Sharkey NA. Skeletal Tissue MechanicsNew York: Springer Verlag, 1998.
- 634.
- Martin TJ. Does bone resorption inhibition affect the anabolic response to parathyroid hormone? Trends Endocrinol Metab. 2004;15:49–50. [PubMed: 15080150]
- 635.
- Martin TJ, Romas E, Gillespie MT. Interleukins in the control of osteoclast differentiation. Crit Rev Eukaryot Gene Expr. 1998;8:107–123. [PubMed: 9714893]
- 636.
- Marx J. How cells endure low oxygen. Science. 2004;303:1454–1456. [PubMed: 15001751]
- 637.
- Mashiba T, Hirano T, Turner CH. et al. Suppressed bone turnover by bisphosphonates increases microdamage accumulation and reduces some biomechanical properties in dog rib. J Bone Miner Res. 2001;15:613–620. [PubMed: 10780852]
- 638.
- Mason DJ. Glutamate signalling and its potential application to tissue engineering of bone. Eur Cell Mater. 2004;7:12–25. [PubMed: 15073696]
- 639.
- Mason DJ, Suva LJ, Genever PG. et al. Mechanically regulated expression of a neural glutamate transporter in bone: A role for excitatory amino acids as osteotropic agents? Bone. 1997;20:199–205. [PubMed: 9071469]
- 640.
- Matsuda S, Rouault J-P, Magaud J-P. et al. In search of a function for the TIS21/PC3/BTG1/TOB family. Febs Lett. 2001;497:67–72. [PubMed: 11377414]
- 641.
- Matthews JL, Martin JH. Intracellular transport of calcium and its relationship to homeostasis and mineralization. Am J Med. 1971;50:589–597. [PubMed: 4930267]
- 642.
- Maudsley S, Pierce KL, Zamah AM. et al. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–9580. [PubMed: 10734107]
- 643.
- McCauley LK, Koh AJ, Beecher CA. et al. PTH/PTHrP receptor is temporally regulated during osteoblasts differentiation and is associated with collagen synthesis. J Cell Biochem. 1996;61:638–647. [PubMed: 8806088]
- 644.
- McCauley LK, Koh AJ, Beecher CA. et al. Proto-oncogene c-fos is transcriptionally regulated by parathyroid hormone (PTH) and PTH-related protein in a cyclic adenosine monophosphate-dependent manner in osteoblastic cells. Endocrinology. 1997;138:5427–5433. [PubMed: 9389528]
- 645.
- McCauley LK, Koh-Paige H, Chen CC. et al. Parathyroid hormone stimulates fra-2 expression in osteoblastic cells in vitro and in vivo. Endocrinology. 2001;142:1975–1981. [PubMed: 11316763]
- 646.
- McKee MD, Murray TM. Binding of intact parathyroid hormone to chicken renal plasma membranes evidence for a second binding site with carboxyl terminal specificity. Endocrinology. 1985;117:1930–1939. [PubMed: 2995001]
- 647.
- McKee MD, Nanci A. Osteopontin: An interfacial extracellular matrix protein in mineralized tissues. Connective Tissue Res. 1996;35:197–205. [PubMed: 9084658]
- 648.
- Mehta NM, Gilligan JP, Stern B. et al. Biological activity of recombinant PTH analog UGL7841(abstract). J Bone Miner Res. 2002;17:S273.
- 649.
- Mehta N, Stern W, Sturmer A. et al. Oral delivery of PTH analogs by a solid dosage formulation (abstract). J Bone Miner Res. 2001;16:S540.
- 650.
- Meir CR, Schlienger RC, Kraenzlin ME. et al. HMG-CoA reductase inhibitors and the risk of fractures. JAMA. 2000;283:3205–3210. [PubMed: 10866867]
- 651.
- Meier-Vismara E, Walker N, Vogel A. Single cilia in the articular cartilage of the cat. Expl Cell Biol. 1979;47:161–171. [PubMed: 467766]
- 652.
- Meleti Z, Shapiro IM, Adams CS. Inorganic phosphate induces apoptosis of osteoblast-like cells in culture. Bone. 2000;27:359–366. [PubMed: 10962346]
- 653.
- Menon GK, Elias PM. Ultrastructural localization of calcium in psoriatic and normal human epidermis. Arch Dermatol. 1991;127:57–63. [PubMed: 1986708]
- 654.
- Mentaverri R, Kamel S, Wattel A. et al. Regulaion of bone resorption and osteoclast survival by nitric oxide: Possible involvement of NMDA receptor. J Cell Biochem. 2003;88:1145–1156. [PubMed: 12647297]
- 655.
- Menton DN, Simmons DJ, Chang SL. et al. From bone lining cells to osteocyte-an SEM study. Anat Rec. 1984;209:29–39. [PubMed: 6329038]
- 656.
- Merle B, Itzstein C, Delmas PD. et al. NMDA glutamate receptors are expressed by osteoclast precursors and involved in the regulation of osteoclastogenesis. J Cell Biochem. 2003;90:424–436. [PubMed: 14505357]
- 657.
- Meunier PJ. Evidence-based medicine and osteoporosis: A comparsion of fracture risk reduction data from osteoporosis randomized clinical trials. Int J Clin Pract. 1999;53:122–129. [PubMed: 10344048]
- 658.
- Meunier PJ, Roux C, Seeman E. et al. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. New Engl J Med. 2004;350:459–468. [PubMed: 14749454]
- 659.
- Miao D, He B, Tong XK. et al. Conditional knockout of PTHrP in osteoblasts leads to premature osteoporosis (abstract). J Bone Miner Res. 2002;17:S138.
- 660.
- Midura RJ, Su X, Morcuende JA. et al. Parathyroid hormone rapidly stimulates hyaluronan synthesis by periosteal osteoblasts in the tibial diaphysis of the growing rat. J Biol Chem. 2003;278:51462–51468. [PubMed: 14514685]
- 661.
- Miles RR, Sluka JP, Halladay DL. et al. Parathyroid hormone (hPTH-(1-38) stimulates the expression of UBP41, an ubiquitin-specific protease, in bone. J Cell Biochem. 2002;85:229–242. [PubMed: 11948679]
- 662.
- Miles RR, Sluka JP, Santerre RF. et al. Dynamic regulation of RGS-2 in bone: Potential new insights into parathyroid hormone signaling mechanisms. Endocrinology. 2000;141:28–36. [PubMed: 10614620]
- 663.
- Miller WE, McDonald PH, Cai SF. et al. Identification of a motif in the carboxyl terminus of β-arrestin 2 responsible for activation of JNK3. J Biol Chem. 2001;276:27770–27777. [PubMed: 11356842]
- 664.
- Mineo C, Anderson RGW. Potocytosis. Histochem Cell Biol. 2001;116:109–118. [PubMed: 11685539]
- 665.
- Minina E, Wenzel HM, Kreschel C. et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–4534. [PubMed: 11714677]
- 666.
- Miranti CK, Brugge JS. Sensing the environment: A historical perspective on intergrin signal transduction. Nature Cell Biol. 2002;4:83–90. [PubMed: 11944041]
- 667.
- Miyakoshi N, Kasukawa Y, Linkhart TA. et al. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142:49–56. [PubMed: 11564695]
- 668.
- Miyamoto S, Teramoto H, Gutkind JS. et al. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation; roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–1642. [PMC free article: PMC2133938] [PubMed: 8978828]
- 669.
- Moghadasian MH, Frohlich JJ. Statins and bones. Can Med Assoc J. 2001;164:803–805. [PMC free article: PMC80878] [PubMed: 11276549]
- 670.
- Mohammad KS, Guise TA, Chirgwin JM. PTHrP stimulates new bone formation by molecular mimicry of endothelin- (abstract). J Bone Miner Res. 2003;18:S26.
- 671.
- Mohan S, Baylink DJ. IGF system components and their role in bone metabolismIn: Rosenfeld RG, Roberts Jr CT, eds.The IGF System. Molecular Biology, Physiology, and Clincal ApplicationsTotowa, New Jersey: Humana Press,1999457–496.
- 672.
- Mohan S, Kutilek S, Zhang C. et al. Comparison of bone formation responses to parathyroid hormone(1-34), (1-31), and (2-34) in mice. Bone. 2000;27:471–478. [PubMed: 11033441]
- 673.
- Montagnani A, Gonnelli S, Cepollaro C. et al. Effect of simvastatin on bone mineral density and bone turnover in and bone density in hypercholesterolemic postmenopausal women: A 1-year longitudinal study. Bone. 2003;32:427–433. [PubMed: 12689687]
- 674.
- Montero A, Okada Y, Tomita M. et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093. [PMC free article: PMC300831] [PubMed: 10772653]
- 675.
- Moonga BS, Dempster DW. Effects of peptide fragments of protein kinase C on isolated rat osteoclasts. Exp Physiol. 1998;83:717–725. [PubMed: 9782182]
- 676.
- Moonga BS, Sun L, Corisdeo S. et al. Novel mechanistic insights into the regulation of mature osteoclasts by osteoprotegerin (OPG) and osteoprotegerin-ligand (OPGL). Evidence for resorption stimulation through inhibition of extracellular Ca2+ sensing (abstract). J Bone Miner Res. 1998;14:S363.
- 677.
- Moore RE, Smith CK, Bailey CS. et al. Characterization of β-adrenergic receptors on rat and human osteoblast-like cells and demonstration that β-receptor agonists can stimulate bone resorption in organ culture. Bone Miner. 1993;23:301–315. [PubMed: 7908582]
- 678.
- Moore TL, Krishnan VG, Onyia JE. et al. A mechanism for bone anabolic activity of PTH through Cbfa1/Osf-2 (abstract). J Bone Miner Res. 2000;15:S158.
- 679.
- Mori H, Kitazawa R, Mizuki S. et al. RANK ligand, RANK, and OPG expression in type II collagen-induced arthritis mouse. Histochem Cell Biol. 2002;117:283–292. [PubMed: 11914926]
- 680.
- Mori S, Burr DB. Increased intracortical remodeling following fatigue damage. Bone. 1993;14:103–109. [PubMed: 8334026]
- 681.
- Morley P, Whitfield JF, Willick G. Design and application of parathyroid hormone analogues. Curr Med Chem. 1999;11:1095–1106. [PubMed: 10519915]
- 682.
- Morley P, Whitfield JF, Willick GE. et al. The effect of monocyclic and bicyclic analogs of human parathyroid hormone hPTH-(1-31)NH2 on bone formation and mechanical strength in ovariectomized rats. Calcif Tissue Int. 2001;68:95–101. [PubMed: 11310353]
- 683.
- Morley P, Whitfield JF, Vanderhyden BC. et al. A new, nongenomic estrogen action: The rapid release of intracellular calcium. Endocrinology. 1992;131:1305–1312. [PubMed: 1505465]
- 684.
- Morony S, Capparelli C, Lee R. et al. A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resoption induced by IL-1β, TNF-α, PTH, PTHrP, and 1,25(OH)2D3. J Bone Miner Res. 2002;14:1478–1485. [PubMed: 10469275]
- 685.
- Mosekilde L. Osteoporosis: Mechanisms and modelsIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,199731–58.
- 686.
- Mosekilde L, Thomsen JS, McOsker JE. No loss of biomechanical effects after withdrawal of short-term PTH treatment in an aged, osteopenic, ovariectomized rat model. Bone. 1997;20:429–437. [PubMed: 9145240]
- 687.
- Moss ML, Cowin SC. Mechanosensory mechanisms in boneIn: Lanza R, Chick W, eds.Principles of Tissue EngineeringAustin, TX: RG Landes, Compnay,1997645–659.
- 688.
- Mundy G. Pathogenesis of osteoporosis and challenges for drug delivery. Adv Drug Del Res. 2000;42:165–173. [PubMed: 10963834]
- 689.
- Mundy G, Garrett R, Harris S. et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–1949. [PubMed: 10583956]
- 690.
- Murakami T, Yamamoto M, Ono K. et al. Transforming growth factor-β1 increases mRNA levels of osteoclastogenesis inhibitory factor in osteoblastic /stromal cells and inhibits the survival of murine osteoblast-like cells. Biochem Biophys Res Commun. 1998;252:747–752. [PubMed: 9837778]
- 691.
- Murray EJB, Bentley GV, Grisanti MS. et al. The ubiquitin-proteasome system and cellular proliferation and regulation in osteoblastic cells. Exp Cell Res. 1998;242:460–469. [PubMed: 9683533]
- 692.
- Murray EJB, Murray SS. Lovastatin stimulates rather than inhibits proteasome in vitro and in osteoblasts (abstract). J Bone Miner Res. 2001;16:S206.
- 693.
- Murrills RJ, Bhat BM, Bodine PVN. et al. Dissociation of functional response from cyclic AMP stimulation in a substituted and C-terminally truncated PTH peptide. J Bone Miner Res. 2002;17:S284.
- 694.
- Nadal A, Ropero AB, Laribi O. et al. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane recepor unrelated to estrogen receptor α and estrogen receptor β Proc Natl Acad Sci USA. 2000;97:11603–11608. [PMC free article: PMC17247] [PubMed: 11027358]
- 695.
- Nadal A, Rovira JM, Laribi O. et al. Rapid insulinotropic effect of 17β-estradiol via a plasma membrane receptor. FASEB J. 1998;12:1341–1348. [PubMed: 9761777]
- 696.
- Nakade O, Takahashi K, Takuma T. et al. Effect of extracellular calcium on the gene expression of bone morphogenic protein-2 and -4 of normal human bone cells. J Bone Miner Metab. 2001;19:13–19. [PubMed: 11156467]
- 697.
- Nakajima A, Shimoji N, Shiomi K. et al. Mechanisms for the enhancement of fracture healing in rats treated with intermittent low-dose human parathyroid hormone (1-34). J Bone Miner Res. 2002;17:2038–2047. [PubMed: 12412812]
- 698.
- Nakajima M, Ejiri S, Tanaka M. et al. Effects of intermittent administration of human parathyroid hormone (1-34) on mandibular condyle of ovariectomized rats. J Bone Miner Metab. 2000;18:9–17. [PubMed: 10633271]
- 699.
- Nakamura K, Nishimura J, Hirano K. et al. Hydroxyfasudil, an active metabolite of fasudil hydrochloride, relaxes the rabbit basilar artery by disinhibition of myosin light chain phosphatase. J Cereb Blood Flow Metab. 2001;21:876–885. [PubMed: 11435800]
- 700.
- Nakashima K, Zhou X, Kunkel G. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. [PubMed: 11792318]
- 701.
- Nauli SM, Alenghat FJ, Luo Y. et al. Polycystins 1 and 2 mediate mechnosensation in the primary cilium of kidney cells. Nature Genet. 2003;33:129–137. [PubMed: 12514735]
- 702.
- Neer RM, Arnaud CD, Zanchetta JR. et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. New Engl J Med. 2001;344:1434–1441. [PubMed: 11346808]
- 703.
- Neer R, Hayes A, Rao A. et al. Effects of parathyroid hormone, alendronate, or both on bone density in osteoporotic postmenopausal women (abstract). J Bone Miner Res. 2002;17:S135.
- 704.
- Neer R, Slovik D, Daly M. et al. Treatment of postmenopausal osteoporosis with daily parathyroid hormone plus calcitriolIn: Christiansen C, Overgaard K, eds.Osteoporosis. Copenhagen: Osteopress 19911314–1317.
- 705.
- Nemere I, Farach-Carson MC. Membrane receptors for steroid hormones: A case for specific cell surface binding sites for vitamin D metabolites and estrogens. Biochem Biophys Res Commun. 1998;248:443–449. [PubMed: 9703943]
- 706.
- Nemeth EF, Fox J. Compounds acting on the parathyroid calcium receptor as novel therapies for hyperparathyroidism or osteoporosisIn: Chattopadhyay N, Brown EM, eds.Calcium-Sensing ReceptorBoston: Kluwer Academic Publishers,2003173–202.
- 707.
- Netelenbos C. Osteoporosis: Intervention options. Maturitas. 1998;30:235–239. [PubMed: 9881322]
- 708.
- Netter FH. Atlas of Human Anatomy. 2nd ed. East Hanover, NJ: Novartis. 1997.
- 709.
- Neugebauer W, Barbier J-R, Sung WL. et al. Solution structure and adenylyl cyclase stimulating activities of C-terminal truncated human parathyroid hormone analogues. Biochemistry. 1995;34:8835–8842. [PubMed: 7612624]
- 710.
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. [PubMed: 12359864]
- 711.
- Ni C-Y, Murphy MP, Golde TE. et al. γ-Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001;294:2179–2181. [PubMed: 11679632]
- 712.
- Nickoloff BJ, Schroeder JM, von den Driesch P. et al. Is psoriasis a T-cell disease? Exp Dermatol. 2000;9:359–375. [PubMed: 11016857]
- 713.
- Nickoloff BJ, Qin JZ, Chuturvedi V. et al. Jagged-1 mediated activation of notch signaling induces cmplete maturation of human keratinocytes through NF-κB and PPARγ Cell Death Differ. 2002;9:842–855. [PubMed: 12107827]
- 714.
- Nicolas M, Wolfer A, Raj K. et al. Notch 1 functions as a tumor suppressor in mouse skin. Nat Gen. 2003;33:416–421. [PubMed: 12590261]
- 715.
- Nicolella DP, Bonewald LF, Lankford J. Microcrack matrix strain is 15 times greater than average global strains; implications for osteocyte signaling. J Bone Miner Res. 2002;17:S183.
- 716.
- Nielsen LB, Pedersen FS, Pedersen L. Expression of type III sodium-dependent phosphate transporters/retroviral receptors mRNAs during osteoblast differentiation. Bone. 2001;28:160–166. [PubMed: 11182373]
- 717.
- Nishida S, Endo N, Yamagiwa H. et al. Number of osteoprogenitor cells in human bone marrow markedly decreases after skeletal maturation. J Bone Miner Metab. 1999;17:171–177. [PubMed: 10757676]
- 718.
- Nishida S, Yamaguchi A, Tanizawa T. et al. Increased bone formation by intermittenet parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone. 1994;15:717–723. [PubMed: 7873302]
- 719.
- Nissenson RA. Parathyroid hormone-related protein. Rev Endo Metab Dis. 2000;1:343–352. [PubMed: 11706748]
- 720.
- Noble BS. Osteocyte death: Its biological significance (abstract). J Bone Miner Res. 2000;15:823.
- 721.
- Noble BS, Reeve J. Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol. 2000;159:7–13. [PubMed: 10687847]
- 722.
- Noda M, Takuwa Y, Katoh T. et al. Mechanical force regulation of vascular parathyroid hormone-related peptide expression. Kidney Int. 1996;55(Suppl):S154–S155. [PubMed: 8743541]
- 723.
- Nomura S, Takano-Yamamoto T. Molecular events caused by mechanical stress in bone. Matrix Biol. 2000;19:91–96. [PubMed: 10842092]
- 724.
- O'Brien CA, Kern B, Gubrij I. et al. Cbfa1 does not regulate RANKL gene activity in stromal/osteoblastic cells. Bone. 2002;30:453–462. [PubMed: 11882458]
- 725.
- Ohishi K, Katayama N, Shiku H. et al. Notch signaling in hematopoiesis. Semin Cell Devel Biol. 2003;14:143–150. [PubMed: 12651098]
- 726.
- Ohnaka K, Shimoda S, Nawata H. et al. Pitavastatin enhanced BMP-2 and osteocalcin expression by inhibition of rho-associated kinase in human osteoblasts. Biochem Biophys Res Commun. 2001;287:337–342. [PubMed: 11554731]
- 727.
- Okada-Ban M, Thiery JP, Jouanneau J. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:263–267. [PubMed: 10716624]
- 728.
- Okamoto T, Schlegel A, Scherer PE. et al. Caveolins, a family of scaffolding proteins for organizing "preassembled signaling complexes" at the plasma membrane. J Biol Chem. 1998;273:5419–5422. [PubMed: 9488658]
- 729.
- Okazaki R, Inoue D, Shibata M. et al. Estrogen promotes ealy osteoblast differentiation and inhibits adipocyte differentiation in mouse bone marrow stromal cell lines that express estrogen receptor (ER) α or β Endocrinology. 2002;143:2349–2356. [PubMed: 12021200]
- 730.
- Okuyama R, Nguyen BC, Talora C. et al. High commitment of embryon- ic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory me- anism. Dev Cell. 2004;6:551–562. [PubMed: 15068794]
- 731.
- Olsnes S, Klingenberg O, Wiedlocha A. Transport of exogenous growth factors and cytokines to the cytosol and to the nucleus. Physiol Rev. 2003;83:163–182. [PubMed: 12506129]
- 732.
- Onyia JE, Dow E, Adams C. et al. Gene array analysis of the bone effects of raloxifene and alendronate show that alendronate strongly inhibits the expression of bone formation marker genes (abstract). J Bone Miner Res. 2002;17:S157.
- 733.
- Onyia JE, Gelbert I, Zhang M. et al. Analysis of gene expression by DNA microarray reveals novel clues to the mechanism of the catabolic and anabolic actions of PTH in bone (abstract). J Bone Miner Res. 2001a;16:S227.
- 734.
- Onyia JE, Ma YL, Galbreath E. et al. ADAMTS-1: A cellular disinetgrin and metalloprotease with thrombospondin motifs is essential for normal bone growth and PTH regulated bone metabolism (abstract). J Bone Miner Res. 2001b;16:S158.
- 735.
- Oniya JE, Miles RR, Halladay DL. et al. In vivo demonstration that parathyroid hormone (hPTH 1-38) inhibits the expression of osteoprotegerin (OPG) in bone with the kinetics of an immediate early gene. J Bone Miner Res. 2000;15:863–871. [PubMed: 10804015]
- 736.
- Ontiveros CS, McCauley LK, McCabe LR. Gravitational force alters intracellular signaling and gene transactivation. J Bone Miner Res. 2002;17:S308.
- 737.
- Opas EE, Gentile MA, Rossert JA. et al. Parathyroid hormone and prostaglandin E2 preferentially increase luciferase levels in bone of mice harboring a luciferase transgene transgene controlled by the elements of the pro-α1(I) collagen promoter. Bone. 2000;27:27–32. [PubMed: 10617154]
- 738.
- Orloff JJ, Kats Y, Urena P. et al. Further evidence for a novel receptor for amino-terminal parathyroid hormone-related protein on keratinocytes and squamous carcinoma cell lines. Endocrinology. 1995;136:3016–3023. [PubMed: 7789327]
- 739.
- Ornitz DM, Itoh N. Fibroblast growth factors Genome Biol 20012 http//genomebiologycom/2001/2/3/reviews/39951 . [PMC free article: PMC138918] [PubMed: 11276432]
- 740.
- Orwoll E, Scheele WH, Paul S. et al. The effect of parathyroid hormone (1-34) therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9–17. [PubMed: 12510800]
- 741.
- Osley MA. H2B ubiquitylation: the end is in sight. Biochim Biophys Acta. 2004;1677:74–78. [PubMed: 15020048]
- 742.
- Otto F, Lübbert M, Stock M. Upstream and downstream targets of RUNX proteins. J Cell Biochem. 2003;91:1239–1247. [PubMed: 12682904]
- 743.
- Oyajobi BO, Anderson DM, Traianedes K. et al. Therapeutic efficacy of a soluble receptor activator of nuclear factor \kappa\B-IgG Fc fusion protein in suppressing bone resorption and hypercalcemia in a model of humoral hypercalcemia of malignancy. Cancer Res. 2001;61:2572–2578. [PubMed: 11289133]
- 744.
- Palumbo C, Palazini S, Zaffe D. et al. Osteocyte differentiation in the tibia of newborn rabbit: An ultrastructural study of the formation of cytoplasmic processes. Acta Anat. 1990;137:350–358. [PubMed: 2368590]
- 745.
- Panegyres PK. The functions of the amyloid precursor gene. Revs Neurosci. 2001;12:1–39. [PubMed: 11236063]
- 746.
- Parfitt AM. Osteoclast precursors as leukocytes: Importance of the area code. Bone. 1998;23:491–494. [PubMed: 9855456]
- 747.
- Parfitt AM. BMU origination and progression: Relationship to targeted and nontargeted remodeling (abstract). J Bone Miner Res. 2000a;15:823.
- 748.
- Parfitt AM. The mechanism of coupling: A role for the vasculature. Bone. 2000b;26:319–323. [PubMed: 10787232]
- 749.
- Parfitt AM. The bone remodeling compartment: A circulatory function for bone lining cells. J Bone Miner Res. 2001;16:1583–1585. [PubMed: 11547827]
- 750.
- Parfitt AM. Misconceptions (2). Turnover is always higher in cancellous than cortical bone. Bone. 2002;30:807–809. [PubMed: 12052445]
- 751.
- Pasco JA, Hemnry MJ, Sanders KM. et al. β-Adrenergic blockers reduce the risk of fracture partly by increasing bonemineral density: Geelong osteoporosis study. J Bone Miner Res. 2004;19:19–34. [PubMed: 14753732]
- 752.
- Pasco JA, Kotowicz MA, Henry MJ. et al. Statin use, bone mineral density, and fracture risk. Geelong osteoporosis study. Arch Intern Med. 2002;162:537–540. [PubMed: 11871921]
- 753.
- Patton AJ, Genever PG, Birch MA. et al. Expression of an N-methyl-D-aspartate receptor by human and rat osteoblasts and osteoclasts suggests a novel glutamate signaling pathway in bone. Bone. 1998;22:645–649. [PubMed: 9626403]
- 754.
- Pavalko FM, Chen NX, Turner CH. et al. Fluid shear-induced mechanical signaling in MC3T3-E1 osteoblasts requires cytoskeleton-integrin interactions. Am J Physiol. 1998;275:C1591–1601. [PubMed: 9843721]
- 755.
- Pavalko FM, Norvell SM, Burr DB. et al. A model for mechanotransduction in bone cells: The load-bearing mechanosomes. J Cell Biochem. 2003;88:104–112. [PubMed: 12461779]
- 756.
- Pavo I, Laszlo F, Morschl E. et al. Raloxifene, an estrogen-receptor modulator, prevents decreased constitutive nitric oxide and vasoconstriction in ovariectomized rats. Eur J Pharmacol. 2000;410:101–104. [PubMed: 11134661]
- 757.
- Pazour GJ, Witman GB. The vertebrate primary cilium is a sensory organelle. Curr Opin Cell Biol. 2003;15:1–6. [PubMed: 12517711]
- 758.
- Clinical Trials in OsteoporosisIn: Pearson D, Miller CG, eds. London: Springer-Verlag,2002.
- 759.
- Pearson G, Robinson F, Beers T. et al. Mitogen-activated protein (MAP)kinase pathways: Regulation and physiological functions. Endocrine Rev. 2001;22:153–183. [PubMed: 11294822]
- 760.
- Pedrazzini T, Prolong F, Grouzmann E. Neuropeptide Y: The universal soldier. Cell Mol Life Sci. 2003;60:350–377. [PubMed: 12678499]
- 761.
- Peet NM, Grabowski PS, Laketic-Ljubojevic I. et al. The glutamate receptor antagonist MK801 modulates bone resorption in vitro by a mechanism predominantly involving osteoclast differentiation. FASEB J. 1999;13:2179–2185. [PubMed: 10593865]
- 762.
- Péhu M, Policard A, Dufort A. L'Ostopétrose ou maladie des os marmoréens. La Presse Médicale. 1931;53:999–1003.
- 763.
- Perris AD, MacManus JP, Whitfield JF. et al. Parathyroid glands and mitotic stimulation in rat bone marrow after hemorrhage. Am J Physiol. 1971;220:773–778. [PubMed: 5545689]
- 764.
- Perris AD, Whitfield JF, Rixon RH. Stimulation of mitosis in bone marrow and thymus of normal and irradiated rats by divalent cations and parathyroid extract. Radiation Res. 1967;32:550–563. [PubMed: 6065249]
- 765.
- Petsko GA, Ringe D. Protein Structure and FunctionLondon: New Science Press Ltd., 2004.
- 766.
- Pfeilschrifter J, Eberhardt W, Huwiler A. Nitric oxide and mechanisms of redox signaling: Matrix and matrix-metabolizing enzymes as prime oxide targets. Eur J Pharmacol. 2001;429:279–286. [PubMed: 11698047]
- 767.
- Pfeilschrifter J, Laukhuf F, Moller-Beckmann B. et al. Parathyroid hormone increases the concentration of insulin-like growth factor-1 and transforming growth factor beta 1 in rat bone. J Clin Invest. 1995;96:767–774. [PMC free article: PMC185261] [PubMed: 7635970]
- 768.
- Pfister MF, Lederer E, Forgo J. et al. Parathyroid hormone-dependent degradation of type II Na+/Pi cotransporters. J Biol Chem. 1997;272:20125–20130. [PubMed: 9242686]
- 769.
- Pi M, Garner SC, Flannery P. et al. Sensing of extracellular cations in CasR-deficient osteoblasts. Evidence for a novel cation-sensing mechanism. J Biol Chem. 2000;275:3256–3263. [PubMed: 10652312]
- 770.
- Pi M, Hinson TK, Quarles LD. Failure to detect the extracellular calcium-sensing receptor (CasR) in human osteoblast cell lines. J Bone Miner Res. 1999;14:1310–1319. [PubMed: 10457263]
- 771.
- Pi M, Quarles LD. A novel cation-sensing mechanism in osteoblasts is a molecular target for strontium. J Bone Miner Res. 2004;19:862–869. [PubMed: 15068510]
- 772.
- Picheret C, Horcajada M-N, Mathey J. et al. Isoflavone consumption does not increase the bone mass in osteopenic obese female Zuker rats. Ann Nutr Metab. 2003;47:70–77. [PubMed: 12652058]
- 773.
- Picotto G, Vazquez G, Boland R. 17β-Oestradiol increases intracellular Ca2+concentration in rat enterocytes. Potential role of phospholipase C-dependent storeoperated Ca2+ influx. Biochem J. 1999;339:71–77. [PMC free article: PMC1220129] [PubMed: 10085229]
- 774.
- Piekarski K, Munro M. Transport mechanism operating between blood supply and osteocytes in long bones. Nature. 1977;269:80–82. [PubMed: 895891]
- 775.
- Pierroz DD, Glatt V, Rizzoli R. et al. Sustained cAMP signaling in primary osteoblasts from β-arrestin2 KO mice leads to altered skeletal response to intermittent PTH (abstract). J Bone Miner Res. 2003;18:S175.
- 776.
- Pilbeam C, Rao Y, Alander C. et al. Downregulation of mRNA expression for the ‘decoy’ interleukin-1 receptor 2 by ovariectomy (abstract). J Bone Miner Res. 1997;12:S433.
- 777.
- Pirola CJ, Wang HM, Strgacich MI. et al. Mechanical stimuli induce vascular parathyroid hormone-related protein gene expression in vivo and in vitro. Endocrinology. 1994;134:2230–2236. [PubMed: 8156926]
- 778.
- Plate U, Tkotz T, Wiesmann HP. et al. Early mineraliztion of matrix vesicles in the epiphyseal growth plate. J Microsc. 1996;183(Pt 1):102–107. [PubMed: 8760406]
- 779.
- Plotkin LI, Bellido T, Ali AA. et al. Runx2/Cbfa1 is essential for the anti-aopotosis effect of PTH on osteoblasts (abstract). J Bone Miner Res. 2002;17:S166.
- 780.
- Ponomareva LV, Wang W, Koszewski NJ. et al. Mim-1, an osteoclast secreted chemokine, stimulates differentiation, matrix mineralization and increased Vitamin D receptor binding at the VDRE of osteoblastic precursor cells. J Bone Miner Res. 2002;17:S155.
- 781.
- Ponting CP, Phillips C, Davies KE. et al. PDZ domains: Targeting signalling molecules to sub-membranous sites. BioEssays. 1997;19:469–477. [PubMed: 9204764]
- 782.
- Poole CA, Flint MH, Beaumont BW. Analysis of the morphology and function of primary cilia in connective tissues: A cellular cybernetic probe. Cell Motil. 1985;5:175–193. [PubMed: 4005941]
- 783.
- Poole CA, Jensen CG, Snyder JA. et al. Confocal analysis of primary cilia structure and colocalization with the Golgi apparatus in chondrocytes and aortic smooth muscle cells. Cell Biol Int. 1997;21:483–494. [PubMed: 9451805]
- 784.
- Poole CA, Zhang Z-J, Ross J. The differential distribution of acetylated and detyrosinated alpha-tubulin in the microtubular cytoskeleton and primary cilia of hyaline cartilage chondrocytes. J Anat. 2001;199:393–405. [PMC free article: PMC1468350] [PubMed: 11693300]
- 785.
- Potts Jr JT, Tregear GW, Keutmena HT. et al. Synthesis of a biologically active N-terminal tetratriacontapeptide of parathyroid hormone. Proc Natl Acad Sci USA. 1971;68:63–67. [PMC free article: PMC391103] [PubMed: 4322265]
- 786.
- Pouysségur J. Signal transduction: An arresting start for MAPK. Science. 2000;290:1574–1577. [PubMed: 11185509]
- 787.
- Power RA, Iwaniec UT, Wronski TJ. Changes in gene expression associted with the bone anabolic effects of basic fibroblast growth factor in aged ovariectomized rats. Bone. 2002;31:143–148. [PubMed: 12110427]
- 788.
- Pozzan T, Magalhaes P, Rizzuto R. The comeback of mitochondria to calcium signaling. Cell Calcium. 2002;28:279–283. [PubMed: 11115367]
- 789.
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membrane Biol. 2001;184:71–79. [PubMed: 11687880]
- 790.
- Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membrane Biol. 2003a;191:69–76. [PubMed: 12532278]
- 791.
- Praetorius HA, Spring KR. The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens. 2003b;12:517–520. [PubMed: 12920399]
- 792.
- Prenzel N, Zwick E, Daub H. et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of pro-HB-EGF. Nature. 1999;402:884–888. [PubMed: 10622253]
- 793.
- Ptashne M, Gann A. Genes & Signals Cold Spring HarborNY: Cold Spring Harbor Laboratory Press,2002 .
- 794.
- Pugazhenthi S, Miller E, Sable C. et al. Insulin-like growth factor-I induces bcl-2 promoter through the transcription factor cAMP-response element-binding protein. J Biol Chem. 1999;274:27529–27535. [PubMed: 10488088]
- 795.
- Pun S, Dearden RL, Ratkus AM. et al. Decreased bone anabolic effect of basic fibroblast growth factor at fatty marrow sites in ovariectomized rats. Bone. 2001;28:220–226. [PubMed: 11182382]
- 796.
- Qin L, Raggatt L, Li X. et al. Amphiregulin: A possible mediator of parathyroid hormone's anabolic action in bone (abstract). J Bone Miner Res. 2003;18:S100.
- 797.
- Qiu P, Qin L, Sorrentino RP. et al. Comparative promoter analysis and its application in analysis of PTH-regulated gene expression. J Mol Biol. 2003;326:1327–1336. [PubMed: 12595247]
- 798.
- Quinn PG. Mechanisms of basal and kinase-inducible transcription activator by CREB. Progr Nucleic Acid Res Mol Biol. 2002;72:269–305. [PubMed: 12206454]
- 799.
- Radeff JM, Singh ATK, Dossing DA. et al. Evidence that activation of phospholipase D is an early event in downstream signaling by PTH in UMR-106 osteoblasts. J Bone Miner Res. 2002;17:S342.
- 800.
- Raggatt LJ, Om L, Partrideg NC. Parathyroid hormone stimulation of IL-18 in osteoblastic cells. J Bone Miner Res. 2003;18:S398.
- 801.
- Rahmouni K, Haynes WG. Leptin and the central neural mechanisms of obesity hypertension. Drugs of Today. 2002;38:807–817. [PubMed: 12582470]
- 802.
- Rao LG, Murray TM. Binding of intact parathyroid hormone to rat osteosarcoma cells: Major contribution of binding sites for the carboxyterminal region of the hormone. Endocrinology. 1985;117:1632–1638. [PubMed: 2992916]
- 803.
- Rangarajan A, Talora C, Okuyama R. et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427–3436. [PMC free article: PMC125257] [PubMed: 11432830]
- 804.
- Raouf A, Seth A. Discovery of osteoblast-associated genes using cDNA microarrays. Bone. 2002;30:463–441. [PubMed: 11882459]
- 805.
- Razandi M, Pedram A, Greene GL. et al. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: Studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;13:307–319. [PubMed: 9973260]
- 806.
- Razandi M, Oh P, Pedram A. et al. ERs associate with and regulate the production of calveolin: Implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100–115. [PubMed: 11773442]
- 807.
- Razani B, Rubin B, Lisanti MP. Regulation of cAMP-mediated signal transduction via interaction of caveolins with the catalytic subunit of protein kinase A. J Biol Chem. 1999;274:26353–23360. [PubMed: 10473592]
- 808.
- Razani B, Lisanti MP. Caveolin-deficient mice: Insights into caveolar function and human disease. J Clin Invest. 2001;108:1553–1561. [PMC free article: PMC201001] [PubMed: 11733547]
- 809.
- Razani B, Woodman SE, Lisanti MP. Caveolae: From cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. [PubMed: 12223531]
- 810.
- Razani B, Zhang XL, Bitzer M. et al. Caveolin-1 regulates transforming growth factor (TGF)-β/SMAD signaling through an interaction with the TGF-β type I receptor. J Biol Chem. 2001;276:6727–6738. [PubMed: 11102446]
- 811.
- Re RN. The origins of intracrine hormone action. Am J Med Sci. 2002a;323:43–48. [PubMed: 11814142]
- 812.
- Re RN. Toward a theory of intracrine hormone action. Regul Pept. 2002b;106:1–6. [PubMed: 12047903]
- 813.
- Re RN. The intracrine hypothesis and intracellular peptide hormone action. BioEssays. 2003;25:401–409. [PubMed: 12655647]
- 814.
- Reeve J, Hesp R, Williams D. et al. Anabolic effect of low doses of a fragment of human parathyroid hormone fragment on the skeleton in postmenopausal osteoporosis. Lancet. 1976;I:1035–1036. [PubMed: 57447]
- 815.
- Reeve J, Meunier PJ, Parsons JA. et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: A multicentre trial. Brit Med J. 1980;280:1340–1344. [PMC free article: PMC1601798] [PubMed: 6992932]
- 816.
- Reeve J, Mitchell A, Tellez M. et al. Treatment with parathyroid peptides and estrogen replacement for severe postmenopausal vertebral osteoporosis; prediction of long-term responses in spine and femur. J Bone Miner Metab. 2001;19:102–114. [PubMed: 11281158]
- 817.
- Reeve J, Tregear GW, Parsons JA. Preliminary trial of low doses of human parathyroid hormone fragment 1-34 in treatment of osteoporosis. Clin Endocrinol. 1976;21:469–477. [PubMed: 782668]
- 818.
- Reginster J-Y, Deroisy R, Jupsin I. Strontium ranelate: a new paradigm in the treatment of osteoporosis. Drugs of Today. 2003;39:89–101. [PubMed: 12698204]
- 819.
- Rehman Q, Lang TF, Arnaud CD. et al. Daily treatment with parathyroid hormone is associated with an increase in vertebral cross-sectional area in postmenopausal women with glucocorticoid-induced osteoporosis. Osteoporosis Int. 2003;14:77–81. [PubMed: 12577188]
- 820.
- Reid IR, Hague W, Emberson J. et al. Effect of pravastatin on frequency of fracture in the LIPID study: Secondary analysis of a randomized controlled trial. Long-term intervention with pravastatin in ischaemic disease. Lancet. 2001;357:509–512. [PubMed: 11229669]
- 821.
- Reinholz GG, Reinholz MM, Getz B. et al. Lipophilic statins increase RANKL and OPG mRNA levels in human osteoblast cells (abstract). J Bone Miner Res. 2002;17:S445.
- 822.
- Rejnmark L, Buus NH, Vestergaard P. et al. Effects of simvastatin treatment in postmenopausal osteopenic women: A randomized controlled trial. J Bone Miner Res. 2003;18:S32. [PubMed: 15068496]
- 823.
- Rejnmark L, Buus NH, Vestergaard P. et al. Effects of simvastatin treatment on bone turnover and BMD: a 1-year randomized controlled trial in postmenopausal osteopenic women. J Bone Miner Res. 2003;19:737–744. [PubMed: 15068496]
- 824.
- Rensberger JM, Watanabe M. Fine structure of bone in dinosaurs, birds and mammals. Nature. 2000;406:619–622. [PubMed: 10949300]
- 825.
- Reseland JE, Gordeladze JO. Role of leptin in bone growth: Central player or peripheral supporter? FEBS Lett. 2002;528:40–42. [PubMed: 12297276]
- 826.
- Reseland JE, Gordeladze JO, Drevon CA. Leptin receptor (OB-R) gene expression in human primary osteoblasts: Reaffirmation. J Bone Miner Res. 2002;17:1136. [PubMed: 12054170]
- 827.
- Reseland J, Syversen U, Bakke I. et al. Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J Bone Miner Res. 2001;16:1426–1433. [PubMed: 11499865]
- 828.
- Reszka AA, Halasy-Nagy JM, Masarachia PJ. et al. Bisphosphoinates act directly on the osteoclast to induce caspase cleavage of Mst1 kinase during apoptosis. J Biol Chem. 1999;274:34967–34973. [PubMed: 10574973]
- 829.
- Riggs BL. The mechanism of estrogen regulation of bone resorption. J Clin Invest. 2000;106:1203–1204. [PMC free article: PMC381441] [PubMed: 11086020]
- 830.
- Rihani-Bisharat S, Maor G, Lewinson D. In vivo anabolic effects of parathyroid hormone (PTH) 28-48 and N-terminal fragments of PTH and PTH-related protein on neonatal mouse bones. Endocrinology. 1998;139:974–981. [PubMed: 9492028]
- 831.
- Riordan M. The Hunting of the QuarkNew York: Simon and Schuster, 198731.
- 832.
- Rittmaster RS, Bolognese M, Ettinger MP. et al. Enhancement of bone mass in osteoporotic women with parathyroid hormone followed by alendronate. J Clin Endocrinol Metab. 2000;85:2129–2134. [PubMed: 10852440]
- 833.
- Rixon RH, Whitfield JF. The radioprotective action of parathyroid extract. Int J Radiat Biol. 1961;3:361–367. [PubMed: 13741987]
- 834.
- Rixon RH, Whitfield JF, Gagnon L. et al. Parathyroid hormone fragments may stimulate bone growth in ovariectomized rats by activating adenylyl cyclase. J Bone Miner Res. 1994;9:1179–1189. [PubMed: 7976500]
- 835.
- Robinson JA, Susulic V, Zhao W. et al. PTH anabolic induced gene in bone (PAIGB), a novel gene which may play an important rone in bone formation. J Bone Miner Res. 2003;18:S174.
- 836.
- Rocheville M, Lange DC, Kumar U. et al. Receptors for dopamine and somatostatin: Formation of heterooligomers with enhanced functional activity. Science. 2000a;288:154–157. [PubMed: 10753124]
- 837.
- Rocheville M, Lange DC, Kumar U. et al. Subtypes of the somatostatin receptor assemble as fuctional homo- and heterodimers. J Biol Chem. 2000b;275:7862–7869. [PubMed: 10713101]
- 838.
- Rodan GA, Reszka AA. Bisphosphonate mechanism of action. Curr Mol Med. 2002;2:571–577. [PubMed: 12243249]
- 839.
- Rodland KD. Calcium receptor-mediated signalingIn: Chattopadhyay N, Brown EM, eds.Calcium-Sensing ReceptorBoston: Kluwer Academic Publishers,200353–67.
- 840.
- Roe EB, Chiu KM, Arnaud CD. Selective estrogen receptor modulators and postmenopausal health. Adv Intern Med. 2000;45:259–278. [PubMed: 10635051]
- 841.
- Roodman GD. Advances in bone biology: The osteoclast. Endocrine Revs. 1996;17:308–332. [PubMed: 8854048]
- 842.
- Rogers MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharma Design. 2003;9:2643–2658. [PubMed: 14529538]
- 843.
- Rogers MJ, Gordon S, Benford HL. et al. Cellular and molecular mechanisms of action of bisphosphonates. Cancer. 2000;88:2961–2978. [PubMed: 10898340]
- 844.
- Rose EB, Sanchez SD, Del Puerto GA. et al. Parathyroid hormone 1-34 (hPTH 1-34) and estrogen produce dramatic bone density increases in postmenopausal osteoporosis (abstract). J Bone Miner Res. 1999;14:S137.
- 845.
- Rosen CJ, Bilezikian JP. Anabolic therapy for osteoporosis. J Clin Endocrinol Metab. 2001;86:957–964. [PubMed: 11238469]
- 846.
- Rouault J-P, Prevot D, Berthet C. et al. Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcription regulatory complex. J Biol Chem. 1998;273:22563–22569. [PubMed: 9712883]
- 847.
- Roufflet J, Coxam V, Gaumet N. et al. Preserved bone mass in ovarectomized rats treated with parathyroid-hormone-related peptide (1-34) and (107-111) fragments. Reprod Nutr Dev. 1994;34:473–481. [PubMed: 7802939]
- 848.
- Roy B, Li WW, Lee AS. Calcium-sensitive transcriptional activation of the proximal CCAAT regulatory element of the grp78/BiP promoter by the human nuclear factor CBF/NF-Y. J Biol Chem. 1996;271:28995–29002. [PubMed: 8910550]
- 849.
- Rubin DA, Juuml;ppner H. Zebrafish express the common parathyroid hormone/parathyroid hormone-related peptide receptor (PTH1R) and a novel receptor (PTH3R) that is preferentially activated by mammalian and fugufish parathyroid hormone-related peptide. J Biol Chem. 1999;274:28185–28190. [PubMed: 10497171]
- 850.
- Rubin J, Murphy TC, Zhu L. et al. Mechanical strain differentially regulates eNOS and RANKL expression via ERK 1 / 2 in primary bone stromal cells (abstract). J Bone Miner Res. 2003;18:S10–S11.
- 851.
- Ruoslahti E, Rajotte D. The address system in the vasculature of normal tissues and tumors. Annu Rev Immunol. 2000;18:813–827. [PubMed: 10837076]
- 852.
- Ryder KD, Duncan RL. Parathyroid hormone enhances fluid shear-induced [Ca2+]I signaling in osteoblastic cells through activation of mechanosensitive and voltage-sensitive Ca2+ channels. J Bone Miner Res. 2001;16:240–248. [PubMed: 11204424]
- 853.
- Ryoo HM, Hoffmann HM, Beumer T. et al. Stage-specific expression of Dlx-5 during osteoblasts differentiation: Involvement in regulation of osteocalcin gene expression. Mol Endocrinol. 1997;11:1681–1694. [PubMed: 9328350]
- 854.
- Sakai A, Sakata T, Ikeda S. et al. Intermittent administration of human parathyroid hormone(1-34) prevents immobilization-related bone loss by regulating bone marrow capacity for bone cells in ddY mice. J Bone Miner Res. 1999;14:1691–1699. [PubMed: 10491216]
- 855.
- Sakai H, Kobayashi Y, Sakai E. et al. Cell adhesion is a prerequisite for osteoclast survival. Biochem Biophys Res Commun. 2000;270:550–556. [PubMed: 10753662]
- 856.
- Sakata T, Wang Y, Halloran BP. et al. Skeletal unloading induces resistance to insulin-like growth factor-I (IGF-I) by inhibiting activation of IGF-I signal pathways. J Bone Miner Res. 2004;19:436–446. [PubMed: 15040832]
- 857.
- Samnegard E, Iwaniec UT, Cullen DM. et al. Maintenance of cortical bone in human parathyroid hormone(1-84)-treated ovariectomized rats. Bone. 2001;28:251–260. [PubMed: 11248654]
- 858.
- Sanders JL, Chattopadhyay N, Kifor O. et al. Ca2+-sensing receptor expression and PTHrP secretion in PC-3 human prostate cancer cells. Am J Physiol Endocrinol Metab. 2001;281:E1267–E1274. [PubMed: 11701443]
- 859.
- Sato M, Schmidt A, Cole H. et al. The skeletal efficacy of statins do not compare with low-dose parathyroid hormone (abstract). Bone. 2001;28:S80.
- 860.
- Sato M, Vahle J, Schmidt A. et al. Abnormal bone architecture and biomechanical properties with near-lifetime treatment of rats with PTH. Endocrinology. 2002;143:3230–3242. [PubMed: 12193534]
- 861.
- Sato M, Westmore M, Clendenon J. et al. Three-dimensional modeling of the effects of parathyroid hormone on bone distribution in lumbar vertebrae of ovariectomized cynomolgus macaques. Osteoporos Int. 2001;11:871–880. [PubMed: 11199192]
- 862.
- Sato M, Westmore M, Ma YL. et al. Teriparatide [hPTH(1-34)] strengthens the proximal femur of ovariectomized nonhuman primates despite increasing porosity. J Bone Miner Res. 2004;19:623–629. [PubMed: 15005850]
- 863.
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp 90. Proc Natl Acad Sci USA. 2000;97:10832–10837. [PMC free article: PMC27109] [PubMed: 10995457]
- 864.
- Schaffler MB. The role of osteocytes in targeting microdamage-induced remodeling (abstract). J Bone Miner Res. 2000;15:823.
- 865.
- Schaffler MB, Choi K, Milgrom C. Aging and matrix microdamage accumulation in human compact bone. Bone. 1995;17:521–525. [PubMed: 8835305]
- 866.
- Scherft JP, Daems W. Single cilia in chondrocytes. J Ultrastructural Res. 1967;19:546–555. [PubMed: 6055783]
- 867.
- Schiller PC, D'Ippolito G, Roos BA. et al. Anabolic or catabolic responses of MC3T3-E1 osteoblastic cells to parathyroid hormone depends on time and duration of treatment. J Bone Miner Res. 1999;14:1504–1512. [PubMed: 10469278]
- 868.
- Schipani E, Chiusaroli R, Maier A. et al. In vivo impairment of collagenase activity does not affect bone formation in transgenic mice expressing constitutively active PTH/PTHrP receptors in osteoblasts (abstract). J Bone Miner Res. 2002;17:S308.
- 869.
- Schipani E, Kruse K, Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. [PubMed: 7701349]
- 870.
- Schipani EE, Lanske B, Hunzelman J. et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA. 1997;94:13689–13694. [PMC free article: PMC28367] [PubMed: 9391087]
- 871.
- Schipani E, Ryan HE, Didrickson S. et al. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. [PMC free article: PMC312800] [PubMed: 11691837]
- 872.
- Schneider A, Taboas JM, McCauley LK. et al. Skeletal homeostasis in tissue-engineered bone. J Orthop Res. 2003;21:859–864. [PubMed: 12919874]
- 873.
- Schoneberg T, Schulz A, Gudermann T. The structural basis of G-protein-coupled receptor function and dysfunction in human diseases. Rev Physiol Biochem Pharmacol. 2002;144:145–227. [PubMed: 11987825]
- 874.
- Schoojans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302:93–109. [PubMed: 8695669]
- 875.
- Schwartz B, Smirnoff P, Shany S. et al. Estrogen controls expression and bioresponse of 1,25-dihydroxyvitamin D receptors in the rat colon. Mol Cell Biochem. 2000;203:87–93. [PubMed: 10724336]
- 876.
- Schwartz EA, Leonard ML, Bizios R. et al. Analysis and modeling of the primary cilium bending response to fluid shear. Am J Physiol. 1997;272:F132–F138. [PubMed: 9039059]
- 877.
- Schweisguth F. Regulation of Notch signaling activity. Curr Biol. 2004;14:R129–R138. [PubMed: 14986688]
- 878.
- Schweitert HR, Groen EWJ, Sollie FAE. et al. Single dose subcutaneous administration of recombinant human parathyroid hormone [rhPTH(1-84)] in healthy postmenopausal volunteers. Clin Pharmacol Ther. 1997;61:360–376. [PubMed: 9084461]
- 879.
- Seeman E. How do antiresorptive agents reduce fracture? IBMS BoneKEy. 2001
- 880.
- Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: Membrane-associated signaling complexes. Trends Endocrinol Metab. 2002;13:349–354. [PMC free article: PMC4137481] [PubMed: 12217492]
- 881.
- Selvamurugan N, Pulumati MR, Tyson DR. et al. Parathyroid hormone regulation of the rat collagenase-3 promoter by protein linase A-dependent transactivation of core binding factor α1. J Biol Chem. 2000;275:5037–5042. [PubMed: 10671545]
- 882.
- Selye H. On the stimulation of new bone formation with parathyroid extract and irradiated ergosterol. Endocrinology. 1932;16:547–558.
- 883.
- Selz T, Caverzasio J, Bonjour J-P. Regulation of Na-dependent Pi transport by parathyroid hormone in osteoblast-like cells. Am J Physiol. 1989;256:E93–E100. [PubMed: 2536233]
- 884.
- Serre CM, Farlay D, Delmas PD. et al. Evidence for a dense and intimate innervation of the bone tissue, including glutamate-containing fibers. Bone. 1999;25:623–629. [PubMed: 10593406]
- 885.
- Sharpe GR, Dillon JP, Durham B. et al. Human keratinocytes express transcripts for three isoforms of parathyroid hormone-related protein (PTHrP), but not for the parathyroid hormone/PTHrP receptor: Effects of 1,25(OH)2 vitamin D3. Br J Dermatol. 1998;138:944–951. [PubMed: 9747354]
- 886.
- Shaynerson M, Plotkin MJ. The killers Within-the deadly rise of drug-resistant bacteria. Boston: Little Brown and Company. 2002:94.
- 887.
- Shenolikar S, Voltz JW, Minkoff CM. et al. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci USA. 2002;99:11470–11475. [PMC free article: PMC123280] [PubMed: 12169661]
- 888.
- Shepherd J. Lipid lowering: Statins and the future. Heart. 2000;84:46–47. [PMC free article: PMC1766539] [PubMed: 10956323]
- 889.
- Shevde NK, Bendixen AC, Dienger KM. et al. Estrogens suppress RANK ligand-induced osteoclast differentiation via stromal cell independent mechqanism invoving c-Jun expression. Proc Natl Acad Sci USA. 2000;97:7829–7834. [PMC free article: PMC16630] [PubMed: 10869427]
- 890.
- Shevde NK, Plum LA, Clagett-Dame M. et al. A potent analog of 1α,25-dihydroxyvitamin D3 selectively induces bone formation. Proc Natl Acad Sci USA. 2002;99:13487–13491. [PMC free article: PMC129700] [PubMed: 12374862]
- 891.
- Shih NR, Jo OD, Yanagawa N. Effects of PHEX antisense in human osteoblast cells. J Am Soc Nephrol. 2002;13:394–399. [PubMed: 11805167]
- 892.
- Shimokawa H, Seto M, Katsumata N. et al. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylation in a swine model of coronary artery spasm. Cardiovasc Res. 1999;43:1029–1039. [PubMed: 10615430]
- 893.
- Shirota T, Tashiro M, Ohno K. et al. Effect of intermittent parathyroid hormone (1-34) treatment on the bone response after placement of titanium implants into the tibia of ovariectomized rats. J Oral Maxillofac Surg. 2003;61:471–480. [PubMed: 12684966]
- 894.
- Singh ATK, Bhattacharyya RS, Radeff JM. et al. Regulation of parathyroid hormone-stimulated phospholipase D in UMR-106 cells by calcium, MAP kinase and small G proteins. J Bone Miner Res. 2003;18:1453–1460. [PubMed: 12929934]
- 895.
- Singh AT, Kunnel JG, Strieleman PJ. et al. Parathyroid hormone (PTH)-(1-34), [Nle(8,18),Tyr34]PTH-(3-34) amide, PTH-(1-31)amide, and PTH-related peptide-(1-34) stimulate phosphatidylcholine hydrolysis in UMR-106 osteoblastic cells: Comparison with effects of phorbol 12,13-dibutyrate. Endocrinology. 1999;140:131–137. [PubMed: 9886817]
- 896.
- Sirola J, Honkanen R, Kröger H. et al. Effects of HMG-CoA reductase inhibitors, statins, on bone loss: A prospective population-based cohort study in early postmenopausal women (abstract). Bone. 2001;28:S220. [PubMed: 12111013]
- 897.
- Skerry TM. Identification of novel signaling pathways during functional adaptation of the skeleton to mechanical loading: The role of glutamate as a paracrine signaling agent in the skeleton. J Bone Miner Metab. 1999;17:66–70. [PubMed: 10084405]
- 898.
- Skerry TM, Genever PG. Glutamate signaling in nonneuronal tissues. Trends Pharmacol Sci. 2001;22:174–181. [PubMed: 11282417]
- 899.
- Skerry TM, Genever P, Taylor A. et al. Absence of evidence is not evidence of absence. The shortcomings of the GLAST knockout mouse. J Bone Miner Res. 2001;16:742–749. [PubMed: 11547846]
- 900.
- Skerry TM, Taylor AF. Glutamate signaling in bone. Curr Pharma Des. 2001;7:737–750. [PubMed: 11375777]
- 901.
- Skoglund B, Forslund C, Aspenberg P. Simvastatin improves fracture healing in mice. J Bone Miner Res. 2002;17:2004–2008. [PubMed: 12412808]
- 902.
- Skriptiz R, Andreassen TT, Aspenberg P. Strong effect of PTH(1-34) on regenerating bone. Acta Orthop Scand. 2000a;71:619–624. [PubMed: 11145391]
- 903.
- Skriptiz R, Andreassen TT, Aspenberg P. Parathyroid hormone (1-34) increases the density of rat cancellous bone in a bone chamber. J Bone Joint Surg [Br] 2000b;82B:138–141. [PubMed: 10697330]
- 904.
- Skripitz R, Aspenberg P. Early effect of parathyroid hormone (1-34) on implant fixation. Clin Orthop Rel Res. 2000a;392:427–432. [PubMed: 11716418]
- 905.
- Skripitz R, Aspenberg P. Implant fixation enhanced by intermittent treatment with parathyroid hormone. J Bone Joint Surg [Br] 2000b;83B:437–440. [PubMed: 11341434]
- 906.
- Skripitz R, Aspenberg P. Parathyroid hormone (1-34) increases attachment of PMM cement to bone. J Orthop Sci. 2001;6:540–544. [PubMed: 11793177]
- 907.
- Smit TH, Burger EH, Huyghe JM. A case for strain-induced fluid flow as a regulator of BMU-coupling and osteonal alignment. J Bone Miner Res. 2002;17:2021–2029. [PubMed: 12412810]
- 908.
- Smith BB, Cosenza ME, Mancini A. et al. A toxicity profile of osteoprotegerin in the cynomologous monkey. Int J Toxicol. 2003;22:403–412. [PubMed: 14555415]
- 909.
- Smith J, Huvos AG, Chapman M. et al. Hyperparathyroidism associated with sarcoma of bone. Skeletal Radiol. 1997;26:107–112. [PubMed: 9060102]
- 910.
- Smith MM, Hall BK. Development and evolutionary origin of vertebrate skeletogenic and odontogenic tissues. Biol Revs. 1990;65:277–373. [PubMed: 2205303]
- 911.
- Smythies J. The Dynmaic NeuronCambridge, MA: The MIT Press,2002 .
- 912.
- Soerensen L, Eriksen EF. Endothelial cells mediate osteoblastic responses to sex steroids and selective estrogen receptor modulators (SERMS) via NO independent pathways (abstract). Bone. 1998;23:S369.
- 913.
- Sogaard CH, Mosekilde L, Thomsen JS. et al. A comparison of the effects of two anabolic agents (fluoride and PTH) on ash density and bone strength assessed in an osteopenic rat model. Bone. 1997;20:439–449. [PubMed: 9145241]
- 914.
- Solomon DH, Finkelstein JS, Wang PS. et al. Statin lipid-lowering drugs and bone density (abstract). J Bone Miner Res. 2001;16:S293.
- 915.
- Solomon KR, Danciu TE, Adolphson LD. et al. Caveolin-enriched membrane signaling complexes in human and murine osteoblasts. J Bone Miner Res. 2000;15:2380–2390. [PubMed: 11127203]
- 916.
- Sompayrac L. How the Immune System Works. Oxford: Blackwell Science Ltd. 2003
- 917.
- Spencer GJ, Genever PG. Long-term potentiation in bone-a role for glutamate in strain-induced cellular memory. BMC Cell Biol. 2003;4(1):9. [PMC free article: PMC179892] [PubMed: 12892570]
- 918.
- Spencer GJ, Hitchcock IS, Genever PG. Emerging neuroskeletal signalling pathways: A review. FEBS Lett. 2004;559:6–12. [PubMed: 14960299]
- 919.
- Spinella-Jaegle S, Rawadi G, Kawai S. et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocyte differentiation. J Cell Sci. 2001;114:2085–2094. [PubMed: 11493644]
- 920.
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76:301–314. [PubMed: 7507411]
- 921.
- Srinivasan S, Gross TS. Canalicular fluid flow by bending of a long bone. Medical Engineering & Physics. 2000;22:127–133. [PubMed: 10854966]
- 922.
- Staal A, Frith JC, French MH. et al. The ability of statins to inhibit bone resorption is directly related to their inhibitory effect on HMG-CoA reductase activity. J Bone Miner Res. 2003;18:88–96. [PubMed: 12510809]
- 923.
- Stachowiak MK, Fang X, Myers JM. et al. Integrative nuclear FGFR1 signaling (INFS) as part of a unigversal "feed-forward-and-gate" signaling module that controls cell growth and differentiation. J Cell Biochem. 2003;90:662–691. [PubMed: 14587025]
- 924.
- Stanislaus D, Yang X, Liang D. et al. In vivo regulation of apoptosis in metaphyseal trabecular bone of young rats by synthetic human parathyroid hormone (1-34) fragment. Bone. 2000;27:209–218. [PubMed: 10913913]
- 925.
- Steers WD, Broder SR, Persson K. et al. Mechanical stretch increases secretion of parathyroid hormone-related protein by cultured bladder smooth muscle cells. J Urol. 1998;160:908–912. [PubMed: 9720586]
- 926.
- Stefano GB, Peter D. Cell surface estrogen receptors coupled to eNOS mediate immune and vascular tissue regulation: Therapeutic implications. Med Sci Monit. 2001;7:1066–1074. [PubMed: 11535959]
- 927.
- Steggerda SM, Paschal BM. Regulation of nuclear import and export by the GTPase Ran. Int Rev Cytol. 2002;217:41–91. [PubMed: 12019565]
- 928.
- Stein GS, Lian JB, Stein JL. et al. Transcriptional control of osteoblast growth and differentiation. Physiol Rev. 1996;76:593–629. [PubMed: 8618964]
- 929.
- Steitz SA, Speer MY, Curinga G. et al. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. [PubMed: 11739279]
- 930.
- Steppan CM, Crawford DT, Chidsey-Frink KL. et al. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Peptides. 2000;92:73–78. [PubMed: 11024568]
- 931.
- Steppan CM, Ke HZ, Swick AG. et al. Leptin administration cause an increase in brain size and bone growth in ob/ob mice (abstract). J Obes Rel Metab Disord. 1999;22(Suppl 1):O131.
- 932.
- Stevenson JC, Lindsay R. Osteoporosis. London: Chapman & Hall Medical, Philadelphia Current Medicine. 1999
- 933.
- Stilgren LS, Reppe S, Abrahamsen B. et al. Differential effects of PTH peptides on OPG and RANK-L mRNA expression in human OHS osteosarcoma cells: A possible pathway of osteoblast dependent bone resorption (abstract). J Bone Miner Res. 2001;16:S545.
- 934.
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. [PMC free article: PMC316949] [PubMed: 10465785]
- 935.
- Stork PJS, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. [PubMed: 12074885]
- 936.
- Strein K. Are animal studies with bisphosphonates and PTH fragments predictive for the clinical studies?In: Russell RG, ed.Bone Diseases and OsteoporosisLondon: IBC Technical Services Limited,1994article 12.
- 937.
- Strodel WE, Thompson NW, Eckhauser FE. et al. Malignancy and concomitant primary hyperparathyroidism. J Surg Oncol. 1988;37:10–12. [PubMed: 3336214]
- 938.
- Stupack DG, Cheresh DA. Get a ligand, get a life: Integrins, signaling and cell survival. J Cell Sci. 2002;115:3729–3738. [PubMed: 12235283]
- 939.
- Su D, Ellis S, Napier A. et al. Hoxa3 and Pax1 regulate epithelial cell death and proliferation during thymus and parathyroid organogenesis. Dev Biol. 2001;236:316–329. [PubMed: 11476574]
- 940.
- Suda T, Takahashi N, Udagawa N. et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocrine Revs. 1999;20:345–357. [PubMed: 10368775]
- 941.
- Sugiyama M, Kodama T, Konishi K. et al. Compactin and simvastatin, but not pravastatin, induce bone morphogenic protein-2 in human osteosarcoma cells. Biochem Biophys Res Commun. 2000;271:688–692. [PubMed: 10814523]
- 942.
- Suh PG, Hwang JI, Ryu SH. et al. The roles of PDZ-containing proteins in PLC-β-mediated signaling. Biochem Biophys Res Commun. 2001;288:1–7. [PubMed: 11594744]
- 943.
- Sun YQ, Ashhurst DE. Osteogenic growth pwptide enhances the rate of fracture healing in rabbits. Cell Biol Int. 1998;22:313–319. [PubMed: 10101048]
- 944.
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. [PubMed: 12077605]
- 945.
- Sung WL, Chan BS, Luk CK. et al. High-yield expression of fully bioactive N-terminal parathyroid hormone analogue in Escherichia coli. IUBMB Life. 2000;49:131–135. [PubMed: 10776596]
- 946.
- Sunyer T, Lewis J, Collin-Osdoby P. et al. Estrogen's bone-protective effects may involve differential IL-1 receptor regulation in human osteoclast-like cells. J Clin Invest. 1999;103:1409–1418. [PMC free article: PMC408450] [PubMed: 10330423]
- 947.
- Swarthout JT, Doggett TA, Lemken JL. et al. Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C dependent. J Biol Chem. 2001;276:7586–7592. [PubMed: 11108712]
- 948.
- Sweeny G. Leptin signaling. Cellular Signalling. 2002;14:655–663. [PubMed: 12020765]
- 949.
- Takai H, Kanematsu M, Yano K. et al. Transforming growth factor-β stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem. 1998;273:27091–27096. [PubMed: 9765225]
- 950.
- Takeda E, Taketani Y, Morita K. et al. Sodium-dependent phosphate cotransporters. Int J Biochem Cell Biol. 1999;31:377–381. [PubMed: 10224663]
- 951.
- Takeda S, Elefteriou F, Karsenty G. Common endocrine control of body weight, reproduction , and bone mass. Annu Rev Nutr. 2003;23:403–411. [PubMed: 12730321]
- 952.
- Takeda S, Elefteriou F, Levasseur R. et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317. [PubMed: 12419242]
- 953.
- Taketani Y, Nashik K, Sawada N. et al. Subcellular localization and PTH-dependent transclation of NaPi-IIa in renal proximal tubular cells. (abstract). J Bone Miner Res. 2003;18:S170.
- 954.
- Takeuchi T, Tsuboi T, Arai M. et al. Adrenergic stimulation of osteoclastogenesis mediated by expression of osteoclast differentiation factor in MC3T3-E‘ osteoblast-like cells. Biochem Pharmacol. 2000;61:579–586. [PubMed: 11239501]
- 955.
- Tam CS, Heersche JN, Murray TM. et al. Parathyroid hormone stimulates the bone apposition rate independently of its resorptive action: Differential effects of intermittent and continuous administration. Endocrinology. 1982;110:506–512. [PubMed: 7056211]
- 956.
- Tamasi JA, Arey BJ, Bertolini DR. et al. Characterization of bone structure in leptin receptor-deficient Zuker (fa /fa) rats. J Bone Miner Res. 2003;18:1605–1611. [PubMed: 12968669]
- 957.
- Tami AE, Nasser P, Verborgt MB. et al. The role of interstitial fluid flow in the remodeling response to fatigue loading. J Bone Miner Res. 2002;17:2030–2037. [PubMed: 12412811]
- 958.
- Tamura T, Udgawa N, Takahashi N. et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin-6. Proc Natl Acad Sci USA. 1993;90:11924–11928. [PMC free article: PMC48097] [PubMed: 8265649]
- 959.
- Tan E, Gurjar MV, Sharma RV. et al. Estrogen receptor-α transfer into bovine aortic endothelial cells induces eNOS gene expression and inhibits cell migration. Cardiovasc Res. 1999;43:788–797. [PubMed: 10690351]
- 960.
- Tawfeek HA. PTH stimulation of mitogen-activated protein kinase does not require PTH/PTHrP receptor phosphorylation and internalization or phospholipse C activation (abstract). J Bone Miner Res. 2002;17:S392.
- 961.
- Taylor AF. Functional osteoblastic ionotropic glutamate receptors are a prerequisite for bone formation. J Musculoskel Neuron Interact. 2002;2:415–422. [PubMed: 15758409]
- 962.
- Taylor AF, Brabbs AC, Peet NM. et al. Bone formation /resorption and osteoblast/adipocytes plasticity mediated by AMPA/kainate glutamate receptors in vitro and in vivo (abstract). J Bone Miner Res. 2000;15:S275.
- 963.
- Taylor D, O'Brien F, Prima-Mello A. et al. Compression data on bovine bone confirm that a "stressed volume" principle explains the variability of strength results. J Biomech. 1999;32:1199–1203. [PubMed: 10541070]
- 964.
- Tfelt-Hansen J, MacLeod RJ, Chattopadhyay N. et al. Calcium-sensing receptor stimulates PTHrP secretion by PKC-dependent p38 pathway. J Bone Miner Res. 2002;17:S494.
- 965.
- Thélu J, Rossio P, Favier B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002;2(1):7. [PMC free article: PMC111189] [PubMed: 11978185]
- 966.
- Thirunavukkarnasu K, Halladay DL, Miles RR. et al. The osteoblast-specific transcription factor Cbfa1 contributes to the expession of osteoprotegerin, a potent inhibitor of osteoclast differentiation and function. J Biol Chem. 2000;275:25163–25172. [PubMed: 10833509]
- 967.
- Thirunavukkarnasu K, Halladay DL, Miles RR. et al. Analysis of regulator of G-protein signaling-2 (RGS-2) expression and function in osteoblastic cells. J Cell Biochem. 2002;85:837–850. [PubMed: 11968023]
- 968.
- Thomas D, Cart SA, Piscopo DM. et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8:303–316. [PubMed: 11545733]
- 969.
- Thomas GP, Baker SUK, Eisman JA. et al. Changing RANKL/OPG mRNA expression in differentiating murine osteoclasts. J Endocrinol. 2001;170:451–460. [PubMed: 11479141]
- 970.
- Thomas T, De VittorisR, David VN. et al. Leptin prevents disuse-induced bone loss in tail-suspended female rats (abstract). J Bone Miner Res. 16:S143.
- 971.
- Thomas T, Gori F, Khosla S. et al. Leptin acts on human marrow stromal cells to enhance differentiation of osteoblasts and to inhibit differentiation of adipocytes. Endocrinology. 1999;140:630–1638. [PubMed: 10098497]
- 972.
- Tintutt Y, Parhami F, Bostrom K. et al. cAMP stimulates osteoblast-like differentiation of calcifying vascular cells. J Biol Chem. 1998;273:7547–7553. [PubMed: 9516456]
- 973.
- Tintutt Y, Parhami F, Le V. et al. Inhibition of osteoblast-specific transcription factor Cbfa1 by the cAMP pathway inotseoblastic cells. Ubiquitin/proteasome-dependent regulation. J Biol Chem. 1999;274:28875–28879. [PubMed: 10506130]
- 974.
- Tirone F. The gene PC3TIS21/BTG2, prototype member of the PC3/BTG/TOB family: Regulator in control of cell growth, differentiation, and DNA repair? J Cell Physiol. 2001;187:155–165. [PubMed: 11267995]
- 975.
- Togari A, Arai M, Mizutani S. et al. Expression of mRNAs for neuropeptide receptors and β-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci Lett. 1997;233:125–128. [PubMed: 9350848]
- 976.
- Tomkinson A, Gevers EF, Wit JM. et al. The role of estrogen in the control of osteocyte apoptosis. J Bone Miner Res. 1998;13:12143–12150. [PubMed: 9718192]
- 977.
- Toran-Allerand CD. Novel sites and mechanisms of oestrogen action in the brain. Novartis Found Symp. 2000;230:56–69. [PubMed: 10965502]
- 978.
- Tonna EA, Lampen NM. Electron microscopy of aging skeletal cells. I. Centrioles and solitary cilia. J Gerontol. 1972;27:316–324. [PubMed: 5046597]
- 979.
- Torday JS, Sun H, Wang L. et al. Leptin mediates the parathyroid hormone-related protein paracrine stimulateion of fetal lung maturation. Am J Physiol Lung Cell Mol Pysiol. 2002;282:L405–L410. [PMC free article: PMC2942763] [PubMed: 11839533]
- 980.
- Toromanoff A, Ammann P, Mosekilde L. et al. Parathyroid hormone increases bone formation and improves mineral balance in vitamin D-deficient female rats. Endocrinology. 1997;138:2449–2457. [PubMed: 9165035]
- 981.
- Tovar-Sepulveda VA, Shen X, Falzou M. Intracrine PTHrP protects against serum starvation-induced apoptosis and regulates the cell cycle in MCF-7 breast cancer cells. Endocrinology. 2002;143:596–606. [PubMed: 11796515]
- 982.
- Tregear GW, van Rietschoten J, Greene E. et al. Solid-phase synthesis of the biologically active N-terminal 1-34 peptide of human parathyroid hormone. Hoppe Seylers Z Physiol Chem. 1974;355:415–421. [PubMed: 4474131]
- 983.
- Trivett MK, Walker TI, Macmillan DL. et al. Parathyroid hormone-related protein (PTHrP) production sites in elasmobranchs. J Anat. 2002;201:41–52. [PMC free article: PMC1570896] [PubMed: 12171475]
- 984.
- Troen BR. Molecular mechanisms underlying osteoclast formation amd activation. Exp Gerontol. 2003;38:605–614. [PubMed: 12814795]
- 985.
- Tsingotjidou A, Nervina JM, Pham L. et al. Parathyroid hormone induces RG-2 expression by a cyclic adenosine 3",5"-monophosphate-mediated pathway in primary neonatal murine osteoblasts. Bone. 2002;30:677–684. [PubMed: 11996904]
- 986.
- Tumber A, Meikle MC, Hill PA. Autocrine signals promote osteoblast survival in culture. J Endocrinol. 2000;167:383–390. [PubMed: 11115765]
- 987.
- Tucker M, Valencia-Sanchez MA, Staples RR. et al. The transcription factor associated Ccr4 annd Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001;104:377–386. [PubMed: 11239395]
- 988.
- Turner BM. 2001. Chromatin and Gene Regulation. Oxford: Blackwell Science.
- 989.
- Turner CH, Burr DB, Hock JM. et al. The effects of PTH-(1-34) on bone structure and strength in ovariectomized monkeys. Adv Exp Med Biol. 2001;496:165–179. [PubMed: 11783618]
- 990.
- Turner CH, Robling AG, Duncan RL. et al. Do bone cells behave like a neuronal network? Calcif Tissue Int. 2002;70:435–442. [PubMed: 12149636]
- 991.
- Tyson DR, Swarthout JT, Partridge NC. Increased osteoblastic c-fos expression by parathyroid hormone requires protein kinase A phosphorylation of the cyclic adenosine 3',5'-monophosphate response element-binding protein at serine 133. Endocrinology. 1999;140:1255–1261. [PubMed: 10067851]
- 992.
- Urbich C, Dernbach E, Zeiber AM. et al. Double-edged role of statins in angiogenesis signaling. Circ Res. 2002;90:737–744. [PubMed: 11934843]
- 993.
- Usdin TB, Bonner TI, Hoare SRJ. The parathyroid hormone 2 (PTH2) receptor. Receptors and Channels. 2002;8:211–218. [PubMed: 12529938]
- 994.
- Vagner S, Gensac MC, Maret A. et al. Alternative translation of human fibroblast growth factor 2 mRNA occurs by internal entry of ribosomes. Mol Cell Biol. 1995;15:35–44. [PMC free article: PMC231905] [PubMed: 7799942]
- 995.
- Vahle JL, Sato M, Long CC. et al. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312–321. [PubMed: 12051548]
- 996.
- van Beek E, Pieterman E, Cohen L. et al. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. 1999;264:108–111. [PubMed: 10527849]
- 997.
- Vanderschueren D, Boonen S, Ederveen GH. et al. Skeletal effects of estrogen deficiency as induced by an aromatase inhibitor in an aged rat model. Bone. 2000;27:611–617. [PubMed: 11062346]
- 998.
- van Staa TP, Wegman SLJ, DeVries F. et al. The use of statins and risk of fractures (abstract). J Bone Miner Res. 2000;15:S155.
- 999.
- van't Hof RJ, Ralston SH. Nitric oxide and bone. Immunology. 2001;103:255–261. [PMC free article: PMC1783253] [PubMed: 11454054]
- 1000.
- van't Hof RJ, Armour KJ, Helfrich MH. et al. The anabolic, but not anti-resorptive, effect of mevaststin in vivo is depenedent on endothelial nitric oxide synthase (abstract). J Bone Miner Res. 2002a;17:1191.
- 1001.
- van't Hof RJ, Armour KJ, Penman S. et al. Mice deficient in nNOS have increased bone mass; possible indications for a central mechanism of regulation for bone turnover(abstract). J Bone Miner Res. 2002b;17:S442.
- 1002.
- Veillard NR, Mach E. Statins: The new aspirins? Cell Mol Life Sci. 2002;59:1771–1786. [PubMed: 12530513]
- 1003.
- Vazquez-Prado J, Casas-Gonzalez P, Garcia-Sainz JA. G protein-coupled receptor cross-talk: Pivotal roles of protein phosphorylation and protein-protein interactions. Cell Signalling. 2003;15:549–557. [PubMed: 12681442]
- 1004.
- Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–67. [PubMed: 10646115]
- 1005.
- Verborgt O, Tatton NA, Majeska RJ. et al. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: Complementary roles in bone remodeling regulation? J Bone Miner Res. 2002;17:907–914. [PubMed: 12009022]
- 1006.
- Vermes C, Glant TT, Hallab NJ. et al. The potential role of the osteoblast in the development of periprosthetic osteolysis. J Arthroplasty. 2001;16(Suppl 1):95–100. [PubMed: 11742458]
- 1007.
- Vico L, Hinsenkamp M, Jones D. et al. Osteobiology, strain, and microgravity. Part II: Studies at the tissue level. Calcif Tissue Int. 2001;68:1–10. [PubMed: 12037617]
- 1008.
- Vilagra A, Gutierrez J, Paredes R. et al. Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblaats. J Cell Biochem. 2002;85:112–122. [PubMed: 11891855]
- 1009.
- Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptosis protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. [PubMed: 10996800]
- 1010.
- Voltz JW, Weinman EJ, Shenolikar S. Expanding the role of NHERF, a PDZ-domain containing protein adapter, to growth regulation. Oncogene. 2001;20:6309–6314. [PubMed: 11607833]
- 1011.
- von Schroeder HP, Veillette CJ, Payandeh J. et al. Endothelin-1 promotes osteoprogenitor proliferation and differentiation in fetal rat calvarial cell cultures. Bone. 2003;33:673–684. [PubMed: 14555273]
- 1012.
- von Stechow D, Fish S, Yahalom D. et al. Does simvastatin stimulate bone formation in vivo? BMC Musculoskelet Disord. 2003;4:8. [PMC free article: PMC156891] [PubMed: 12718758]
- 1013.
- Von Stechow D, Libermann T, Hattersley G. et al. Transcriptionally profiling of estrogen- and PTH-regulated gene during bone formation in OVX mice. J Bone Miner Res. 2003;18:S138.
- 1014.
- Vortkamp A, Lee K, Lanske B. et al. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. [PubMed: 8662546]
- 1015.
- Vortkamp A, Pathi S, Peretti GM. et al. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mech Dev. 1998;71:65–76. [PubMed: 9507067]
- 1016.
- Walker D. The induction of osteopetrotic changes in hyphophysectomized thyroparathyroidectomized, and intact rats of various ages. Endocrinology. 1971;89:1389–1406. [PubMed: 5120639]
- 1017.
- Walker G. Snowball Earth. New York: Crown Publishers. 2003
- 1018.
- Walsh CA, Bowler WB, Bilbe G. et al. Effects of PTH on PTHrP gene expression in human osteoblasts: Upregulation with the kinetics of an immediate early gene. Biochem Biophys Res Commun. 1997;239:155–159. [PubMed: 9345287]
- 1019.
- Wang l, Cowin SC, Weinbaum S. et al. Modeling tracer transport in an osteon under cyclic loading. Ann Biomed Eng. 2000;29:810–816. [PubMed: 11144981]
- 1020.
- Wang L, Fritton SP, Cowin SC. et al. Fluid pressure relaxation depends upon osteonal microstructure modeling an oscillatory bending experiment. J Biomech. 1999;32:663–672. [PubMed: 10400353]
- 1021.
- Wang PS, Solomon DH, Mogun H. et al. HMG-CoA reductase inhibitors and the risk of hip fractures in elderly patients. JAMA. 2000;283:3211–3216. [PubMed: 10866868]
- 1022.
- Wang Y, Liu Y, Lee S. et al. PTH promotes osteoblastic differentiation/commitment but not proliferation in GFP-marked primary osteoblast cultures (abstract). J Bone Miner Res. 2003;18:S77.
- 1023.
- >The Identities of Membrane Steroid ReceptorsIn: Watson CS.ed. Boston: Kluwer Academic Publishers,2003.
- 1024.
- Watson PH, Fraher LJ, Hendy GN. et al. Nuclear localization of the type 1 PTH/PTHrP receptor in rat tissues. J Bone Miner Res. 2000;15:1033–1044. [PubMed: 10841172]
- 1025.
- Watson PH, Fraher LJ, Kisiel M. et al. Enhanced osteoblast development after continuous infusion of hPTH-(1-84) in the rat. Bone. 1999;24:89–94. [PubMed: 9951775]
- 1026.
- Watson PH, Fraher LJ, Natale BV. et al. Nuclear localization of the type 1 parathyroid/parathyroid hormone-related peptide receptor in MC3T3-E1 cells: Assocoation with serum-induced cell proliferation. Bone. 2000;26:221–225. [PubMed: 10709993]
- 1027.
- Watson PH, Kisiel M, Patterson EK. et al. Expression of type I PTH/PTHrP receptors in the rat osteoclast and osteoclast-like RAW 264.7 cells. J Bone Miner Res. 2002;17:S287.
- 1028.
- Watson P, Lazowski D, Han V. et al. Parathyroid hormone restores bone mass and enhances osteoblast insulin-like growth factor I gene expression in ovariectomized rats. Bone. 1995;16:375–365. [PubMed: 7786639]
- 1029.
- Weisser J, Riemer S, Schmidt M. et al. Four distinct chondrocyte populations in the fetal bovine growth plate: Highest expressionlevels of PTH/PTHrP receptor, Indian hedgehog, and MMP-13 in hypertrophic chondrocytes and their suppression by PTH (1-34) and PTHrP (1-40). Exp Cell Res. 2002;279:1–13. [PubMed: 12213208]
- 1030.
- Wen W, Meinkoth JL, Tsien R. et al. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995a;82:463–473. [PubMed: 7634336]
- 1031.
- Wen W, Taylor SS, Meinkoth JL. The expression and intracellular distribution of the heat-stable protein kinase inhibitor is cell cycle regulated. J Biol Chem. 1995b;270:2041–2046. [PubMed: 7836431]
- 1032.
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. [PubMed: 12153983]
- 1033.
- Weinstein RS, Jilka RL, Parfitt M. et al. Inhibition of osteobastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoid. J Clin Invest. 1998;102:274–282. [PMC free article: PMC508885] [PubMed: 9664068]
- 1034.
- Weir EC, Philbrick WM, Amling M. et al. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci USA. 1996;93:10240–10245. [PMC free article: PMC38368] [PubMed: 8816783]
- 1035.
- Westbroek I, van der Plas A, de Rooij KE. et al. Expression of serotonin receptors in bone. J Biol Chem. 2001;276:2896–28968. [PubMed: 11387323]
- 1036.
- Whang K, Zhao M, Qiao M. et al. Administration of lovastatin locally in low doses in a novel delivery system induces prolonged bone formation (abstract). J Bone Miner Res. 2000;15:S225.
- 1037.
- Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biol Int. 1996;20:73–81. [PubMed: 8936410]
- 1038.
- Whitfield JF. Statins; new drugs for treating osteoporosis? Exp Opin Invest Drugs. 2001a;10:409–415. [PubMed: 11424897]
- 1039.
- Whitfield JF. Leptin, brains and bones. Exp Opin Invest Drugs. 2001b;10:1617–1622. [PubMed: 11772271]
- 1040.
- Whitfield JF. Statins and the stimulation of bone growth? Do they or don't they? Geriatric Times. 2002a;3:23–28.
- 1041.
- Whitfield JF. Leptin-A new member of the bone builder's club? Medscape Women's Health eJournal 2002b7(4)http//wwwmedscapecom/viewarticle/439243 . [PubMed: 12466737]
- 1042.
- Whitfield JF. The primary cilium-is it an osteocyte's strain-sensing flowmetere? J Cell Biochem. 2003a;89:33–237. [PubMed: 12704786]
- 1043.
- Whitfield JF. How to grow bone to treat osteoporosis and mend fractures Curr Rheumatol Rep 2003b545–56.& Curr Osteoporosis Rep 2003b 132-40 . [PubMed: 12590885]
- 1044.
- Whitfield JF. The neuronal primary cilium-an extrasynaptic signaling device. Cell Signal. 2004;16:763–767. [PubMed: 15115655]
- 1045.
- Whitfield JF. Taming psoriatic keratinocytes-PTHs' uses go up another notch. J Cell Biochem. 2004;93:251–256. [PubMed: 15368353]
- 1046.
- Whitfield JF. Osteogenic PTHs and vascular ossification-is there a danger for osteoporotics? J Cell Biochem 2005. in press . [PubMed: 15786490]
- 1047.
- Whitfield JF, Bird RP, Morley P. et al. The effects of parathyroid hormone (PTH) fragments on bone formation and thei lack of effects on the initiation of colon carcinogenesis in rats as indicated by preneoplastic aberrant crypt formation. Cancer Lett. 2003;200:107–113. [PubMed: 14568163]
- 1048.
- Whitfield JF, Chakravarthy B. Calcium: The Grand-Master Cell SignalerOttawa: NRC Research Press,2001 .
- 1049.
- Whitfield JF, Chakravarthy BR, Durkin JP. et al. Parathyroid hormone stimulates protein kinase C but not adenylate cyclase in mouse epidermal keratinocytes. J Cell Physiol. 1992;150:299–303. [PubMed: 1310323]
- 1050.
- Whitfield JF, Isaacs RJ, Chakravarthy B. et al. Stimulation of protein kinase-C activity in cells expressing human parathyroid hormone (PTH) receptors by C- and N-terminally truncated fragments of PTH 1-34. J Bone Miner Res. 2001;16:441–447. [PubMed: 11277261]
- 1051.
- Whitfield JF, Isaacs RJ, Jouishomme H. et al. C-terminal fragment of parathyroid hormone-related protein, PTHrP-(107-111), stimulates membrane-associated protein kinase C activity and modulates the proliferation of human and murine skin keratinocytes. J Cell Physiol. 1996a;166:1–11. [PubMed: 8557757]
- 1052.
- Whitfield JF, Isaacs RJ, MacLean S. et al. Stimulation of membrane-associated protein kinase-C activity in spleen lymphocytes by hPTH-(1-31)NH2, its lactam derivative, hPTH-(1-31)NH2 hPTH-(1-31)NH2, and hPTH-(1-30)NH2. Cell Signal. 1999a;11:159–164. [PubMed: 10353689]
- 1053.
- Whitfield JF, MacManus JP, Youdale T. et al. The roles of calcium and cyclic AMP in the stimulatory action of parathyroid hormone on thymic lymphocyte proliferation. J Cell Physiol. 1971;78:355–368. [PubMed: 4334368]
- 1054.
- Whitfield JF, Morley P, Fraher L. et al. The stimulation of vertebral and tibial bone growth by the parathyroid hormone fragments, hPTH-(1-31)NH2, [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2,and hPTH0(1-30)NH2. Calcif Tissue Int. 2000a;66:307–312. [PubMed: 10742450]
- 1055.
- Whitfield JF, Morley P, Langille RM. et al. Adenylyl cyclase-activating agents: Parathyroid hormone and prostaglandins EIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,1998a108–149.
- 1056.
- Whitfield JF, Morley P, Ross V. et al. Restoration of severely depleted femoral trabe-cular bone in ovariectomized rats by parathyroid hormone-(1-34). Calcif Tissue Int. 1995;56:227–231. [PubMed: 7750029]
- 1057.
- Whitfield JF, Morley P, Ross V. et al. The hypotensive actions of opsteogenic and nonosteogenic parathyroid hormone fragments. Calcif Tissue Int. 1997a;60:302–308. [PubMed: 9069170]
- 1058.
- Whitfield JF, Morley P, Willick G. et al. Stimulation of the growth of femoral trabecular bone in ovariectomized rats by the novel parathyroid hormone fragment hPTH-(1-31)NH2 (ostabolin). Calcif Tissue Int. 1996;58:81–87. [PubMed: 8998682]
- 1059.
- Whitfield JF, Morley P, Willick GE. "The Parathyroid Hormone: An Unexpected Bone builder for Treating Osteoporosis". Austin: Landes Bioscience Company. 1998b
- 1060.
- Whitfield JF, Morley P, Willick G. et al. Cyclization by a specific lactam Increases the ability of human parathyroid hormone hPTH-(1-31)NH2 to stimulate bone growth in ovariectomized rats. J Bone Miner Res. 1997b;12:1246–1252. [PubMed: 9258755]
- 1061.
- Whitfield JF, Morley P, Willick G. et al. Comparison of the ability of recombinant humanparathyroid hormone, rhPTH-(1-84), and hPTH-(1-31)NH2 to stimulate femoral trabecular bone growth on ovariectomized rats. Calcif Tissue Int. 1997c;60:26–29. [PubMed: 9030476]
- 1062.
- Whitfield JF, Morley P, Willick G. et al. Comparison of the abilities of human parathyroid hormone (1-31)NH2 and human parathyroid hormone-related protein (1-31)NH2 to stimulate femoral trabecular bone growth in ovariectomized rats. Calcif Tissue Int. 1997d;61:322–326. [PubMed: 9312203]
- 1063.
- Whitfield JF, Morley P, Willick G. et al. Comparison of the abilities of human parathyroid hormone (hPTH)-(1-34) and [Leu27]cyclo(Glu22-Lys26)hPTH-(1-31)NH2 to stimulate femoral trabecular bone growth in ovariectomized rats. Calcif Tissue Int. 1998c;63:423–428. [PubMed: 9799828]
- 1064.
- Whitfield JF, Morley P, Willick G. The bone-building actions of the parathyroid hormone: Implications for the treatment of osteoporosis. Drugs Aging. 1999b;15:117–129. [PubMed: 10495071]
- 1065.
- Whitfield JF, Morley P, Willick G. Stimulation of femoral tabecular bone growth in ovariectomized rats by human parathyroid hormone (hPTH)-(1-30)NH2. Calcif Tissue Int. 1999c;65:143–147. [PubMed: 10430648]
- 1066.
- Whifield JF, Morley P, Willick GE. The parathyroid hormones: Bone-forming agents for treatment of osteoporosis Medscape Women's Health 2000b5 http//womenshealthmedscapecom . [PubMed: 11113778]
- 1067.
- Whitfield JF, Morley P, Willick G. et al. Lactam formation increases receptor binding, adenylyl cyclase stimulation and bone growth stimulation by human parathyroid hormone (hPTH)-(1-28)NH2. J Bone Miner Res. 2000c;15:964–970. [PubMed: 10804028]
- 1068.
- Whitfield JF, Morley P, Willick G. The parathyroid hormone, its fragments and analogs?potent bone-builders for treating osteoporosis. Exp Opin Invest Drugs. 2000d;9:1293–1315. [PubMed: 11060744]
- 1069.
- Whitfield JF, Morley P, Willick G. The control of bone growth by parathyroid hormone (PTH), leptin and statins. Crit Rev Eukaryotic Gene Express. 2002a;12:23–51. [PubMed: 12433064]
- 1070.
- Whitfield JF, Morley P, Willick GE. Parathyroid hormone, its fragments and their analogs for the treatment of osteoporosis. Treat Endocrinol. 2002b;1:175–190. [PubMed: 15799210]
- 1071.
- Whitfield JF, Morley P, Willick G. Bone growth stimulators: New tools for treating bone loss and mending fractures. Vitamins and Hormones. 2002c;65:1–80. [PubMed: 12481542]
- 1072.
- Whitfield JF, Rixon RH, MacManus JP. et al. Calcium, cyclic adenosine 3',5'-monophosphate, and the control of cell proliferation. In Vitro. 1973;8:257–278. [PubMed: 4348032]
- 1073.
- Whitfield JF, Youdale T, Perris AD. Early postirradiation changes leading to the loss of nuclear structure in rat thymocytes. Exp Cell Res. 1967;48:461–472. [PubMed: 6082323]
- 1074.
- Wiig JNS, Bakker T. Hyperparathyroidism with multiple malignant tumors of bone with giant cells. Acta Chir Scand. 1971;137:391–393. [PubMed: 5149362]
- 1075.
- Wilding JP, Gilbery SG, Bailey CJ. et al. Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology. 1993;132:1939–1944. [PubMed: 7682936]
- 1076.
- Wiley JC, Wailes LA, Idzerda RL. et al. Role of regulatory subunits and protein kinase inhibitor (PKI) in determining nuclear localization and activity of the catalytic subunit of protein kinase A. J Biol Chem. 1999;274:6381–6387. [PubMed: 10037729]
- 1077.
- Wilsman NJ. Cilia of adult canine articular chondrocytes. J Ultrastruct Res. 1978;64:270–281. [PubMed: 712881]
- 1078.
- Wilsman NJ, Fletcher TF. Cilia of neonatal articular chondrocytes: Incidence and morphoilogy. Anat Rec. 1978;190:871–890. [PubMed: 637325]
- 1079.
- Wolozin B, Kellman W, Ruosseau P. et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. [PubMed: 11030795]
- 1080.
- Wong SK-F. G protein selectivity is regulated by multiple intracellura regions of GPCRs. Neurosignals. 2003;12:1–12. [PubMed: 12624524]
- 1081.
- Woolf V. A Room of One's Own. Harmonsworth: Penguin Books Ltd. 1975:28.
- 1082.
- World healthorganization. Assessment of Fracture Risk and its Application to Screening fpr Postmenopausal Osteoporosis. Report of a WHO study Group. Geneva: World Health Organization. 1994 [PubMed: 7941614]
- 1083.
- Wronski TJ, Li M. PTH: Skeletal effects in the ovariectomized rat model for OsteoporosisIn: Whitfield JF, Morley P, eds.Anabolic Treatments for OsteoporosisBoca Raton: CRC Press,199759–81.
- 1084.
- Wronski TJ, Ratkus AM, Thomsen JS. et al. Sequential treatment with basic fibroblast growth factor and parathyroid hormone restores lost cancellous bone mass and strength in the proximal tibia of aged ovariectomized rats. J Bone Miner Res. 2001;16:1399–1407. [PubMed: 11499862]
- 1085.
- Wu LNY, Ishikawa Y, Sauer GR. et al. Morphological and biochemical characterization of mineralizing primary culturers of avian growth plate chondrocytes: Evidence for cellular processing of Ca2+ and Pi prior to matrix mineralization. J Cell Biochem. 1995;57:218–237. [PubMed: 7759559]
- 1086.
- Wu Y, Kumar R. Parathyroid hormone regulates transforming growth factor β1 and β2, synthesis in osteoblasts via divergent signaling pathways. J Bone Miner Res. 2000;15:879–884. [PubMed: 10804017]
- 1087.
- Xian-Guang H, Aldridge RJ, Bergström J. et al. The Cambrian Fossils of Chengjiang, China. Oxford: Blackwell Science Ltd. 2004
- 1088.
- Xie Y, Gibbs TC, Meier KE. Lysophosphatidic acid as an autocrine and paracrine mediator. Biochim Biophys Acta. 2002;1582:270–281. [PubMed: 12069838]
- 1089.
- Xing L, Carlson L, Story B. et al. Expression of either NF-κB p50 or p52 in osteoclast precursors is required for IL-1-induced bone resorption. J Bone Miner Res. 2003;18:260–269. [PubMed: 12568403]
- 1090.
- Xu HL, Galea E, Santizo RA. et al. The key role of caveolin-1 in estrogen-mediatedregulation of endothelial nitric oxide function in cerebral arterioles in vivo. J Cereb Blood Flow Metab. 2001;21:907–913. [PubMed: 11487725]
- 1091.
- Yagi K, Tsuji K, Nifuji A. et al. Bone morphogenic protein-2 enhances osterix gene expression in chondrocytes. J Cell Biochem. 2003;88:1077–1083. [PubMed: 12647290]
- 1092.
- Yamagishi T, Otsuka E, Hagiwara H. Reciprocal control of expression of mRNAs for osteoclast differentiation factor and OPG in osteogenic stromal cells by genistein: Evidence for the involvement of topoisomase II in osteoclastogenesis. Endocrinology. 2001;142:3632–3637. [PubMed: 11459812]
- 1093.
- Yamaguchi T. Calcium-sensing receptor in boneIn: Chattopadhyay N, Brown EM, eds.Calcium-Sensing ReceptorBoston: Kluwer Academic Publishers,2003103–124.
- 1094.
- Yamaguchi A, Komori T, Suda T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehog and Cbfa 1. Endocrinology. 2000;21:393–411. [PubMed: 10950158]
- 1095.
- Yamaguchi T, Chattopadhyay N, Kifor O. et al. Mouse osteoblastic cell line (MC3T3-E1) expresses extracellular calcium (Ca2+o)-sensing receptor and its agonists stimulate chemotaxis and proliferation of MC3T3-E1 cells. J Bone Miner Res. 1998a;13:1530–1538. [PubMed: 9783541]
- 1096.
- Yamaguchi T, Chattopadhyay N, Kifor O. et al. Activation of p42/44 and p38 mitogen-activated protein kinases by extracellular calcium-sensing receptor agonists induces mitogenic responses in the mouse osteoblastic MC3T3-E1 cell line. Biochem Biophys Res Commun. 2000;279:363–368. [PubMed: 11118293]
- 1097.
- Yamaguchi T, Chattopadhyay N, Kifor O. et al. Expression of extracellular calcium-sensing receptors in human osteoblastic MG-63 cell line. Am J Physiol Cell Physiol. 2001;280:C382–C393. [PubMed: 11208534]
- 1098.
- Yamaguchi T, Kifor O, Chattopadhyay N. et al. Expression of extracellular calcium (Ca2+o)-sensing receptor in the clonal osteoblast-like cellines, UMP-106 and SAOS-2. Biochem Biophys Res Commun. 1998b;243:753–757. [PubMed: 9501005]
- 1099.
- Yamamoto S, Morimoto I, Zeki K. et al. Centrally administered parathyroid hormone (PTH)-related protein (1-34) but not PTH(1-34) stimulates arginine-vasopressin secretion and its messenger ribonucleic acid expression in supraoptic nucleus of the conscious rats. Endocrinology. 1998;139:383–3888. [PubMed: 9421437]
- 1100.
- Yamashiro T, Fukunaga T, Yamashita K. et al. Gene and protein expression of brain-derived neurotrophic factor and TrkB in bone and cartilage. Bone. 2001;28:404–409. [PubMed: 11336921]
- 1101.
- Yang D, Guo J, Bringhurst FR. PTH(1-34) accelerates osteoblast differentiation via a PLC-independent PKC pathway. J Bone Miner Res. 2004;19:S41.
- 1102.
- Yao W, Li CY, Farmer RW. et al. Simvastatin did not prevent bone loss in ovariectomized rat (abstract). J Bone Miner Res. 2001;16:S294.
- 1103.
- Yellowley CE, Zhongyong LI, Zhou Z. et al. Functional gap junctions between osteocyctic and osteoblastic cells. J Bone Miner Res. 2000;15:209–217. [PubMed: 10703922]
- 1104.
- Yin H, Morioka H, Towle CA. et al. Evidence that HAX-1 is an interleukin-1α N-terminal binding protein. Cytokine. 2001;15:122–137. [PubMed: 11554782]
- 1105.
- Yin X-M, Ding W-X, Zhao Y. Bcl-2 family proteins: Master regulators of apoptosisIn: Yin X-M, Dong Z, eds.Essentials of ApoptosisTotowa, NJ: Humana Press,200313–27.
- 1106.
- Yoder BK, Hou X, Guay-Woodford LM. The polycystic disease proteins, polycystin-1, polycystin-2, and cyctin, are colocalized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. [PubMed: 12239239]
- 1107.
- You L, Cowin SC, Schaffler MB. et al. A model for strain amplification in the actin cytoskeleton of osteocytes due to fluid drag on pericellular matrix. J Biomech. 2001;34:1375–1386. [PubMed: 11672712]
- 1108.
- Zabeau L, Lavens D, Peelman F. et al. The ins and outs of leptin receptor activation. FEBS Lett. 2003;546:45–50. [PubMed: 12829235]
- 1109.
- Zabel U, Kleinschnitz C, Oh P. et al. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nature Cell Biol. 2002;4:307–311. [PubMed: 11887187]
- 1110.
- Zaid A, Adebanjo OA, Moonga BS. et al. Emerging insights into the role of calcium ions in osteoclast regulation. J Bone Miner Res. 1999;14:669–674. [PubMed: 10320514]
- 1111.
- Zaidi SK, Javed A, Choi JY. et al. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J Cell Sci. 2001;114:3093–3102. [PubMed: 11590236]
- 1112.
- Zamah AM, Delahunty M, Luttrell LM. et al. Protein kinase A-mediated phosphorylation of the β2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem. 2002;277:31249–31256. [PubMed: 12063255]
- 1113.
- Zanchetta JR, Bogado CE, Ferretti JL. et al. Effects of teriparatide [recombinant human parathyroid hormone (1-34)] on cortical bone in postmenopausal women with osteoporosis. J Bone Miner Res. 2003;18:539–543. [PubMed: 12619939]
- 1114.
- Zao M, Zhang J, Feng JQ. et al. β-Catenin directly activates bone-specific genes in osteoblasts and chondrocytes. J Bone Miner Res. 2002;17:S182.
- 1115.
- Zaragoza Z, Soria E, Lopez E. et al. Activation of the mitogen activated protein extracellular signal-regulated kinase 1 and 2 by the nitric oxide-cGMP-cGMP-dependent protein kinase axis regulates the expression of matrix metalloproteinase 13 in vascular endothelial cells. Mol Pharmacol. 2002;62:927–935. [PubMed: 12237340]
- 1116.
- Zecchi-Orlandini S, Formigli L, Tani A. et al. 17β-estradiol induces apoptosis in the preosteoclastic FLG29.1 cell line. Biochem Biophys Res Commun. 1999;255:680–685. [PubMed: 10049770]
- 1117.
- Zerega B, Ceremelli S, Bianco P. et al. Parathyroid hormone [hPTH-(1-34)] and parathyroid hormone-related protein [hPTHrP-(1-34)] promote reversion of hypertrophic chondrocytes to a prehypertrophic proliferating phenotype and prevent terminal differentiation of osteoblast-like cells. J Bone Miner Res. 1999;14:1281–1289. [PubMed: 10457260]
- 1118.
- Zhang RW, Supowit SC, Xu X. et al. Expression of selected osteogenic markers in the fibroblast-like cells of rat marrow stroma. Calcif Tissue Int. 1995;56:283–291. [PubMed: 7767839]
- 1119.
- Zhang X, Schwarz EM, Young DA. et al. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002a;109:1405–1415. [PMC free article: PMC151001] [PubMed: 12045254]
- 1120.
- Zhang X, Sobue T, Hurley MM. FGF-2 increases colony formation, PTHreceptor, and IGF-1 mRNA in mouse marrow stromal cells. Biochem Biophys Res Commun. 2002b;290:526–531. [PubMed: 11779203]
- 1121.
- Zheng B, Clemmons DR. Blocking ligand occupancy of the αVβ3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998;95:11217–11222. [PMC free article: PMC21622] [PubMed: 9736716]
- 1122.
- Zheng MH, McCaughan HB, Papadimitriou JM. et al. Tartrate resistant acid phosphatase activity in rat cultured osteoclasts is inhibited by a carboxyl terminal peptide (osteostatin) from parathyroid hormone-related protein. J Cell Biochem. 1994;54:145–153. [PubMed: 8175889]
- 1123.
- Zhou Y-X, Xu X, Chen L. et al. A Pro250Arg substitution in mouse Fgfr 1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet. 2000;9:2001–2008. [PubMed: 10942429]
- 1124.
- Zhu J, Emerson SG. A new bone to pick: osteoblasts and the hematopoietic stem-cell niche. BioEssays. 2004;26:595–599. [PubMed: 15170855]
- 1125.
- Ziff M. Role of endothelium in the pathogenesis of rheumatoid synovitis. Int J Tissue React. 1993;15:135–137. [PubMed: 8188449]
- 1126.
- Zimmer C. At the Water's EdgeNew York: The Free Press,1998.
- 1127.
- Zimmermann KW. Beitroge zur Kenntnis einiger Drosen und Epithelien. Arch Mikr Entwicklungsmech. 1898;52:552–706.
- 1128.
- Zuscik MJ, D'Souza MD, Ionescu AM. et al. Growth plate chondrocyte maturation is regulated by basal intracellular calcium. Exp Cell Res. 2002;276:310–319. [PubMed: 12027460]
- 1129.
- Zwartkruis FJT, Bos JF. Ras and Rap 1: Two highly related small GTPases with distinct function. Exps Cell Res. 1999;253:157–165. [PubMed: 10579920]
- Introduction
- BMUS—The Microcrack Fixers
- Strains, Microcracks, Macrocracks, and Molecular Screams
- Calling the Diggers
- The Arrival of the Diggers at the Work Site
- The Fillers
- The Decline and Fall of Bone Strength and BMU Efficiency But a Rise in Remodeling Rate with Age
- Menopause and Bone Loss
- The Amazing Bone-Anabolic PTHs
- How Might PTHs Stimulate Bone Growth?
- The Very Bright Clinical Prospects of the PTHs
- OGP—The Osteogenic Growth Peptide
- The Statins
- Animal Studies
- Osteogenic Mechanism
- Human Studies
- Statins' Clinical Prospects
- Surface Signaling Steroids: Real Anabolics or Pseudo-Anabolics?
- Strontium, Calcium's Big Brother
- Afterword
- References
- What Is Osteoporosis? - Madame Curie Bioscience DatabaseWhat Is Osteoporosis? - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...