

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Research Council (US) Committee on Revealing Chemistry through Advanced Chemical Imaging. Visualizing Chemistry: The Progress and Promise of Advanced Chemical Imaging. Washington (DC): National Academies Press (US); 2006.

Visualizing Chemistry: The Progress and Promise of Advanced Chemical Imaging.
Show detailsThe case studies in Chapter 2 underscore the power of chemical imaging to provide insights into a wide variety of problems in the chemical sciences. In this chapter,1 the current capabilities of chemical imaging are examined in detail, as are areas in which basic improvements in imaging capabilities are needed. However, the chapter is not intended to be an exhaustive review of all chemical imaging techniques. It is assumed that the reader has a basic knowledge of the imaging techniques described. The objective of this chapter is to provide an overview of the state of the art in chemical imaging and to identify those areas that would most likely provide breakthroughs.
The imaging techniques described are divided into three main categories. In addition, a section on image processing and computation—which has bearing on virtually all chemical imaging techniques—is also included:
- Optical imaging (Raman, infrared [IR], and fluorescence) and magnetic resonance
- Electron microscopy, X-rays, ions, neutrons
- Proximal probe (force microscopy, near field, field enhancement)
- Processing analysis and computation
OPTICAL IMAGING AND MAGNETIC RESONANCE
Imaging techniques that utilize low-energy resonant phenomena (electronic, vibrational, or nuclear) to probe the structure and dynamics of molecules, molecular complexes, or higher-order chemical systems differ from approaches using higher-energy radiation (X-rays, electrons, etc.) in that they are largely nondestructive and can be performed under in vivo or in situ conditions, even with soft matter. However, these techniques lack the inherent spatial resolution of the higher-energy approaches.
Although similar in these respects, magnetic resonance and optical spectroscopy (electronic and vibrational spectroscopy) have different strengths and weaknesses. Magnetic resonance is the lowest-energy method and as such uses the longest-wavelength radiation. Exquisite detail in molecular structure can be defined due to the fact that atomic interactions can be measured. However, this detail about the atomic interactions is accompanied by a low inherent sensitivity, thus requiring extensive averaging over many molecules and limiting the inherent temporal and spatial resolution. In contrast, optical spectroscopy utilizes radiation at an energy level high enough to allow individual photons to be measured relatively easily with modern equipment at a detection sensitivity almost matched by the mammalian eye. As a result, imaging data are acquired at the sensitivity of individual molecules. The inherent temporal and spatial resolution is also increased proportionately, but the resonance itself is broad because environmental influences are not averaged out within the inherent time scale of interaction between the molecules and this frequency of radiation. As a result, the structural information content of optical spectra is considerably lower than that of magnetic resonance, particularly in the electronic region of the spectrum.
The long-term technical challenge is to extract the maximum possible information from each type of resonance, ultimately providing a detailed structural picture of the chemistry at the molecular level with the spatial resolution of individual molecules and a temporal resolution on the time scale of chemical bonding.
Nuclear Magnetic Resonance
Over the past 50 years, nuclear magnetic resonance (NMR) has grown into an essential tool for chemists in determining structures of newly synthesized compounds, for scientists interested in the structure of solids, and for biochemists in determining structure-function relationships in biomolecules. NMR also forms the basis for magnetic resonance imaging (MRI). The incredible breadth of NMR and its impact on chemical, biological, and medical sciences have created a vibrant and innovative community of scientists working to increase the scope and usefulness of NMR. Many books are dedicated to subsets of the techniques involved in NMR and MRI: thus, the goal here is to give a small taste of the types of information available and to point out areas in which progress would impact a large subset of NMR and MRI experiments. In addition, there is an equally rich field, which is not discussed explicitly, that applies electron spin resonance to many of the same problems to which NMR and MRI are applied.
Recent advances have pushed the limits of molecular structure determination, including applications of NMR to larger and larger molecules and new ways to enhance the detection limits of NMR. MRI has also undergone a major transition from a tool that provides primarily anatomical information to one that can measure a number of aspects of tissue function. Indeed, active areas of the human brain can now be mapped at unprecedented resolution using functional MRI. However, there is much room for improvement, and there are a number of fruitful areas for development. Higher-magnitude magnetic fields, more sensitive detection strategies, and an ever-growing list of MRI contrast agents will continue to expand the usefulness of NMR and MRI, rendering them essential in chemical imaging. This section provides a general outline of the present state of the art of NMR and MRI, describes some exciting new developments in the area, and finally points out some opportunities for future work that can impact NMR and MRI broadly.
Present State of the Art
Nuclear Magnetic Resonance Spectroscopy: Molecular Structure and Dynamics. NMR is the only tool that provides detailed three-dimensional information at angstrom (Å) resolution of molecules both in solution and in noncrystalline solids. NMR is thus important in imaging molecules not only for the organic chemist but also for materials scientists and biochemists. Its exquisite sensitivity to molecular structure is due to the ability to monitor interactions between atoms that report on structure and dynamics. Chemical shift and J-coupling information obtained from NMR is the result of specific chemical bonds and bond angles. Through-space interactions, such as dipole-dipole interactions, are sensitive to short range (1-5 Å) nonbonded information. Thus, rather than using diffraction of radiation as in X-ray crystallography, NMR builds up structures from a large number of specific interatomic distances and bond angles. Over the past 30 years, the development of complex multidimensional NMR experiments on molecules isotopically labeled with 15N, 13C, and 2H has made routine the probing of detailed structures of molecules in solution up to a molecular weight of approximately 40,000. Similar developments in solid-state NMR now allow a number of structural constraints to be obtained for much larger molecules. The awarding of the Nobel Prize in chemistry in 1991 to Richard Ernst for his work in developing fundamental strategies in NMR and in 2002 to Kurt Wuthrich for his work in using NMR to solve protein structures testifies to the impact of NMR.2
In addition to structural information, dynamic information can also be obtained through NMR. Time scales of both fast (picoseconds) and slow (seconds and longer) processes can be followed. Slow processes such as chemical reactivity are probed by following a change in an NMR property such as chemical shift or transfer of magnetization from one spectral site to another. Detailed kinetic information can be extracted in well-established experiments. Faster processes influence the NMR spin relaxation properties, such as T1 or T2, with kinetic information linked to the specific structure being examined. Model-independent ways of analyzing relaxation data have enabled very efficient procedures for determining which parts of a molecule are more dynamic and over what time scales the fluctuations occur. Thus, NMR is unmatched in the detailed structural and dynamic information it offers.
The main limitation of NMR continues to be its relatively low sensitivity, requiring homogeneous (or heterogeneous mixtures with only a few components) samples of relatively high concentrations (e.g., a milliliter of 10 mM concentration) to be studied. Separation techniques such as high-performance liquid chromatography (HPLC) can be performed prior to NMR to help study complex mixtures, but the ability to obtain detailed structural information about complex mixtures that vary at high spatial resolution requires large gains in sensitivity. Three major directions are being pursued to increase sensitivity. First, higher-magnitude magnetic fields increase anywhere from linearly to quadratically in sensitivity with an increase in field strength, depending on the sample. Magnets with fields up to about 20 Tesla operating at 900 MHz frequencies are becoming available at a few dedicated research sites. A second pursuit has been the improvement of detectors for NMR. One such strategy that has become widely available over the past five years is cooling of the NMR detectors to reduce noise, which has increased sensitivity by a factor of 2 to 4. Work is progressing to miniaturize NMR detectors and use detector arrays to increase sensitivity and throughput. Furthermore, work is aimed at using innovative approaches to detect magnetic resonance signals, such as magnetic force microscopy,3 which borrows concepts from near-field imaging, and other classes of detectors continue to be developed, such as superconducting quantum interference devices (SQUID) for NMR.4
A third approach to increase sensitivity is to increase the signal available from a molecule using hyperpolarization techniques. Indeed, hyperpolarization techniques are leading to large increases in sensitivity from 100- to 100,000-fold. Techniques to transfer polarization were pioneered by physicists such as Albert Overhauser, who was awarded the National Medal of Science in 1994 for his work predicting that electron spin polarization could be coupled to nuclear spin polarization, and Alfred Kastler, who was awarded the Noble Prize in physics in 1966 for his work demonstrating that optical pumping could lead to hyperpolarization. These techniques are now beginning to find widespread application. When samples are placed in the magnets typically used for NMR, at least a million spins are required to generate enough of a population difference between ground and excited states to give a signal. In practice, many more molecules are needed for a sufficient signal to be generated for detection. There is a class of techniques that rely on transferring polarization from molecules that have greater population differences to molecules that one would like to detect with NMR and in this way generate a larger population difference with much fewer spins. There are numerous ways to transfer polarization and increase signal. Three specific techniques that have found growing use are transfer of polarization from unpaired electrons in stable free radicals to nuclear spins,5 laser-induced polarization of noble gases such as xenon and helium,6 and chemical formation of molecules from parahydrogen that can be produced in a polarized state.7 These hyperpolarization strategies are being used to increase sensitivity for application to a wide range of problems in physics, chemistry, biochemistry, and medical imaging.
In addition to increasing the sensitivity of NMR, much work is being done to improve the specificity and accuracy of information available from NMR. Perhaps this is most evident in work on biological macromolecules, which is an active area of development for NMR. An exciting recent example shows that partial orientation of molecules in solution greatly increases the strength of dipole-dipole interactions that are important for obtaining distance information. The strategy of partial alignment has led to structural information about molecules (such as proteins) at very high resolution and with very high accuracy.8 There are also a variety of new NMR techniques to measure dynamics of complex molecules in solutions. In general, these techniques rely on measuring NMR relaxation times and interpreting them in the context of a model of the motion. Recent work measuring the relaxation time of deuterium has enabled the measurement of side chain motion of proteins in solution, with molecular weights up to about 100,000 daltons.9 Indeed, a variety of sophisticated NMR pulse sequences enable motion to be analyzed on the picosecond through millisecond time scale. Development of these pulse sequences continues to be an active area of research. Finally, much of the information about structure and dynamics obtained in the solution state by NMR can also be obtained using solid-state NMR for molecules of much higher molecular weight. Detailed structural and dynamic information can be obtained even if the material being studied defies crystallization.10 The exciting area of solid-state NMR is rapidly developing for determining structures of novel materials important for nanotechnology as well as for proteins that do not readily crystallize.
Magnetic Resonance Imaging: Noninvasive Measurement of Anatomy, Function, and Biochemistry. In 1974, Paul Lauterbur introduced a gradient field strategy to obtain images based on NMR. Today, MRI is being employed in more than 10 million scans per year in the United States and is thus having a great impact on the diagnosis and treatment of a wide variety of diseases. Its importance was recognized when the 2003 Nobel Prize in medicine was awarded to Drs. Lauterbur and Mansfield.11 The basis for MRI is the change in chemical shift that an atom undergoes in an applied magnetic field. With proper calibration of the magnetic field gradient, a change in chemical shift can be related to a specific location—a process known as frequency encoding of spatial information. In addition, controlling the applied magnetic field gradients in combination with specific radio-frequency pulses to excite specific regions enables signals to come from these specific regions—a process known as slice selection. Finally, the time evolution of the NMR signal during a series of radio-frequency excitation pulses can be modulated by the chemical shift of the nucleus being detected. Because the chemical shift can be altered by applied magnetic field gradients during these evolution times, spatial information can be obtained—a process known as phase encoding. There is a wide variety of techniques that use innovative combinations of frequency encoding, slice selection, and phase encoding to generate images.
Any nucleus that can be detected by NMR can be imaged with MRI. The most widely used atom is the hydrogen in water because the high concentration of water enables high-resolution images and a large amount of information can be obtained about the environment of water from changes in its NMR relaxation times, T1 and T2. However, much work has been done detecting other nuclei such as 23Na, 31P, and compounds labeled with 13C, to name a few. In most cases, MRI is performed on the hydrogen atoms in water and detects the single NMR peak from water. However, strategies referred to as spectroscopic imaging or chemical shift imaging enable a series of images to be obtained that represent every resonance in an NMR spectrum. In this way, images of complex metabolite distributions have been obtained and applied to get a metabolic fingerprint of normal and diseased tissue.
Interaction of the hydrogen on a water molecule (or any other NMR active nucleus) with an applied magnetic field gradient enables MRI to create images at much higher resolution than the wavelength of the applied radiation, leading to images with resolutions in the range of 0.2-3 mm in humans and as low as 0.05 mm in animals. With small samples at high magnetic fields, resolution as low as a few microns has been achieved. This is a key factor in the ability of MRI to obtain high resolution of tissues nondestructively using long-wavelength, and thus low-energy, nonionizing radiation. The second reason behind the usefulness of MRI is the remarkable degree of specificity and sensitivity to disease. Water reports on changes in its environment, and the relaxation times of water are sensitive to specific tissues, enabling unparalleled anatomical information to be obtained from soft tissues in the body. In addition, spectroscopic imaging gives information about a large range of metabolites that can be affected early in disease processes. The largest application of MRI has been to biomedical problems, but there is a growing list of problems from characterization of solids to understanding fluid flow in complex media that have been addressed with MRI. Indeed, funding to translate developments of MRI in the biomedical arena to other areas central to chemical imaging would have a major impact.
The past decade has seen a rapid growth in the use of MRI to obtain anatomical information and functional information about tissues. Strategies have been developed that enable MRI to generate images of flowing water, enabling angiography to be performed on the circulatory system. MRI can also be used to measure bulk flow of water, allowing regional blood flow to be measured from a number of tissues. NMR has been used for decades to measure the magnitude and direction of molecular diffusion in solution, and it is possible to extend these techniques to MRI. Techniques for measuring regional blood flow and diffusion are having a major impact on assessing ischemic disease such as heart attacks and stroke. Indeed, at an early stage, diffusion and perfusion MRI can be used to decide therapeutic strategies for stroke victims. In addition, MRI can be sensitized to blood oxygenation levels to assess the degree of metabolic activity in a region of a tissue. When a region of the brain becomes active, the increases in blood flow and metabolism lead to changes in blood oxygenation that can be detected by MRI. This oxygenation-dependent, functional MRI contrast has revolutionized cognitive psychology and is leading to a detailed understanding of the regions of the brain that are responsible for complex cognitive functions.12 Finally, NMR spectroscopy can be combined with MRI to generate detailed spectroscopic images of a range of metabolites. The entire range of functional MRI tools is poised to have a major impact on the diagnosis and management of disease.13
The Cutting Edge and Future Directions in NMR and MRI
Higher Magnetic Fields. The sensitivity of magnetic resonance increases with higher magnetic fields. Indeed, in the range where detector noise dominates, sensitivity increases as approximately the square of the increase in field. In practice, this is hard to realize, particularly because many samples of interest contribute noise, leading to an increase in sensitivity that is linearly proportional to magnetic field strength. Nonetheless, much interest has been focused on producing higher magnetic fields for NMR. Most of this work occurs in industry where fields as high as 20 Tesla (T) can be produced for routine analytical chemistry and biochemistry. In MRI, magnets up to 9.4 T that are large enough for humans are becoming available. These high fields should increase the resolution of MRI of hydrogen as well as be a great boost to MRI of nuclei less sensitive than hydrogen, such as 23Na, 31P, and 13C. The cutting edge for development of high-field magnets is at the National Magnet Laboratory at the University of Florida, where magnets as high as 40 T are available for use.14 In France, a new project is proceeding to increase the strength of magnetic fields available for MRI on humans to 12 T.15 Transforming these exciting projects into commercially viable products would have widespread impact and enable the development of new technologies that allow even higher magnetic fields to be created. This major challenge is in need of creative thinking to move forward without the very great expenditures that these projects currently require. For example, with present magnet technology, significant space is required to house a high-strength magnet. Work to decrease the siting requirement of high-field magnets, for example by employing innovative designs for superconducting wire that can carry higher current densities, could decrease the size of magnets, enabling very high field NMR and MRI to transition from dedicated laboratories to widespread use.
There is some work indicating that NMR can become a more portable modality. For example, in the oil industry the NMR system is attached directly to the exploration drill to mine for petroleum sources. A generalization of this portability of NMR could lead to applications in a range of environmental studies as well as in medical contexts, where a handheld MRI device might be available to clinicians working far from a hospital's radiology department. Recently, the use of a SQUID detector has been demonstrated to lead to excellent NMR spectra at very low magnetic fields, pointing to the possibility of making NMR more portable.16 Thus, there is much room for innovative work, both to enable higher magnetic fields and to make NMR more portable with lower magnetic fields.
Development of New MRI Detectors. Another important strategy for increasing sensitivity in NMR and MRI is the development of new detectors. For NMR, an increase in sensitivity from two- to fourfold has occurred by decreasing the temperature of the detector. These advances, using either high-temperature superconducting materials or traditional materials, are now being implemented widely. There have been similar sensitivity gains in MRI due to the widespread availability of high magnetic fields (3-9 T) for human use and the development of parallel detector arrays. Five years ago, for example, an effective scan of a human head was achieved with an MRI detector containing only one element. Today detectors with 8 to 32 elements are becoming common,17 with preliminary data obtained from arrays with up to 90 elements. These arrays increase sensitivity from two- to fivefold and also enable MRI to be performed at much faster speeds.18 When these arrays are dense enough for the coil noise to dominate over the sample noise, cooling arrays should increase the sensitivity of MRI further. The challenge is to insulate the detectors so that very cold temperatures can be achieved while keeping the detectors close to the body so that sensitivity gains can be realized. With the rapid increase in detector density, it is critical to develop strategies that enable miniaturization of the electronics necessary to perform MRI. A concerted effort to miniaturize NMR components not only will enable engineering of dense detector arrays, but also should increase the portability of NMR in general.
There is much to gain by focusing research efforts to increase sensitivity in NMR and MRI. At present, MRI on humans is performed at resolutions of about a millimeter, with recent results pushing these limits to about 300 microns. A factor of 100X gain in sensitivity would place MRI on the brink of detecting single cells in any organ within the human body. This would also enable chemical imaging for a larger variety of problems where the unmatched chemical sensitivity of NMR can be combined with the spatial resolution afforded by MRI. Research on other detector strategies besides those commonly used should be encouraged, for example developing SQUID detectors for NMR or other innovative approaches to detecting signals. Indeed, it is only the lack of sensitivity that at present limits widespread application of MRI as a chemical imaging tool to the full range of problems discussed throughout this report.
Increasing the NMR Signal with Hyperpolarization. A very promising avenue for increasing sensitivity in NMR and MRI is to increase the signal from the molecules being detected. The low radio-frequency energy used for NMR means that specific nuclei in molecules are as likely to be in the excited state as in the ground state. Signal detection is proportional to the population difference between the two states. Typically, it takes a million molecules to generate a larger ground state than excited state population. There are a number of ways to alter this population difference and polarize the sample to obtain more signals. As discussed previously, increasing the magnetic field for NMR and MRI is one way to achieve incremental gains. Another alternative is to decrease the temperature, which is useful only if the sample is amenable to lower temperatures. A final and very dramatic way is to couple the nuclear spins being detected by NMR to other spins with a higher polarization. As mentioned earlier, transferring polarization from electrons, optically pumping to achieve increased nuclear polarization of noble gases, and using parahydrogen have all been successful in increasing the signal by as much as 100- to 100,000-fold. For example, so-called dynamic nuclear polarization experiments coupling a stable free radical to NMR-detectable nuclei have demonstrated great gains in sensitivity for solid-state NMR, enabling experiments that would ordinarily last days to be performed in minutes.19 Furthermore, clever strategies allow the solid to be thawed to a liquid and prepared in a manner such that it can be injected, which enables hyperpolarization to be used in vivo for MRI. Hyperpolarized MRI of 13C-labeled compounds has been shown to increase sensitivity more than 100,000-fold; this offers exciting possibilities to trace specific metabolic pathways to identify diseases such as cancer.20 One major drawback is that these techniques cannot be applied generally to all molecules. Optical pumping of the noble gases xenon and helium can also lead to very large gains in sensitivity. Recent work has demonstrated the potential for producing biosensors from optical-pumped xenon to enable detection to about 200 nM.21 Hyperpolarized noble gases are also finding increasing use for MRI of the air spaces in lungs.22
A major shortcoming of these hyperpolarization studies is that they are applicable to only a few molecules. Generation of new materials optimized for hyperpolarization is very important to enable a large range of molecules to be hyperpolarized. Another major limitation is that the hyperpolarized signal lasts for a time defined by the nuclear spin lattice relaxation time. In the molecules being developed this means that the increased signal lasts for about a minute. Innovative approaches to making the best use of the polarization while it lasts and procedures for replenishing the signal are critical to a broader range of application. Ideally, a new generation of physicists, chemists, and biochemists would be trained to conduct this truly interdisciplinary work.
Detection of Single Spins with Scanning Force Magnetic Resonance. Within the last year the detection of a single electron spin was accomplished with a scanning magnetic resonance experiment using cantilevers similar to those used for scanning force microscopy.23 This was the culmination of many years of progress to detect increasingly fewer electron or nuclear spins using the magnetic resonance phenomenon. The experiment relied on measuring the force generated when the electron spin orientation was flipped by application of the appropriate radio frequency in a magnetic field. Because the electron spin is 1,000 times stronger than a nuclear spin, this result opens the possibility of detection of single nuclei and thus single-molecule detection by magnetic resonance. As a result, one can envision the use of a small cantilever to scan a molecule or molecular assembly to determine its detailed chemical composition and three-dimensional structure. Such an advance will take years of development to realize and requires advances similar to those needed in other scanning near-field imaging techniques, including (1) the development of more sensitive cantilever strategies to measure increasingly smaller forces and (2) a deeper theoretical understanding of single-molecule behavior with respect to magnetic resonance.
Quantitative Understanding of Chemical Shifts. A great triumph for NMR has been the ability to obtain detailed three-dimensional information from molecules with weights up to about 40,000 grams per mole with accuracy to a few angstroms. It is well known that NMR chemical shifts are sensitive to very small bond length and bond angle changes and can thus probe chemical potentials at very short distances. This is due to the exquisite sensitivity of nuclear spins to their electronic environment. One of the great challenges of modern chemistry is to develop quantum mechanical calculations that can predict chemical interactions and chemical reactions of large molecules. A great hurdle to this work is developing analytical tools that can measure potential changes over short distances. Analysis of the chemical shift of nuclei is one of the few techniques that can probe these potentials over short distances. Thus, a critical frontier in work in NMR is to develop computational approaches that enable prediction of chemical shifts in large molecules. Indeed, if this work is successful it will be possible to determine molecular structures of very complex molecules in a time-efficient manner to an unprecedented level of resolution.
Novel Contrast Agents for MRI. Contrast agents have played an important role in the development of MRI. For example, simple gadolinium chelates are critical for the usefulness of MRI in detecting brain tumors, performing angiography, and measuring regional blood flow and metabolism. With the rapid developments in molecular genetics identifying a large number of potential indicators of disease and therapeutic targets, there is increasing interest in developing MRI contrast agents that are specific for particular cells, molecules, or biochemical processes. This emerging area of molecular imaging depends on the marriage of (1) chemical synthesis of new labels to add specificity to the agent and (2) MRI acquisition and processing to optimize strategies to detect these new agents. Recent work has demonstrated that MRI can be used to specifically target cell surface molecules, image gene expression, detect enzymatic reactions, and follow the migration of cells in intact organs.24 These developments are a long way from routine clinical use, and the realization of this potential will take the concerted efforts of a multidisciplinary team of chemists, molecular biologists, radiologists, and MRI physicists. Particularly lacking are chemists with a commitment to work in this highly multidisciplinary area. Furthermore, the general strategies being offered are applicable to a broad range of problems outside the field of medicine, such as detection of sparse molecules of environmental interest or characterization of complex materials. Funding to translate developments in the biomedical area to broader use in chemical imaging would have a great impact.
Conclusions
NMR and MRI represent mature technologies that have widespread impact on the materials, chemical, biochemical, and medical fields. Recent results in determining the structures of key biological macromolecules and the transformation of the cognitive sciences due to functional MRI exemplify this tremendous influence. Despite these achievements, there is much progress yet to be made. Research aimed at improving magnet technology to achieve higher field strengths in smaller footprints will advance the sensitivity and applicability of NMR. Developments to miniaturize NMR electronics will greatly aid the rapid progress in parallel detection for MRI and increase the portability of NMR. Investment in the exciting area of hyperpolarization has an excellent chance to greatly increase the sensitivity and applicability of NMR and MRI. Investments in the theoretical aspects of NMR, especially those that enable the prediction of structural information from chemical shifts and the optimization of approaches to increase sensitivity using hyperpolarization, will pay large dividends. Finally, funding toward development of new materials can impact NMR on many levels. New superconducting materials can impact magnet and detector design, and new approaches to generating sensitive cantilevers will usher in the era of single-molecule detection by magnetic resonance. A new generation of chemists can impact NMR and MRI research by focusing on the development of new molecules amenable to hyperpolarization strategies as well as new contrast agents to contribute to the rapidly growing field of molecular imaging. Funding mechanisms that can lead to faster translation of developments made in the biomedical area to other areas of chemical imaging should be pursued. It is clear that in the coming years, NMR and MRI will continue to expand rapidly and continue to be key tools for chemical imaging.
Vibrational Imaging
A vibrational spectrum provides something like a structural “fingerprint” of matter because it is characteristic of chemical bonds in a specific molecule. Therefore, imaging based on vibrational spectroscopic signatures, such as Raman scattering and IR absorption, provides a great deal of molecular structural information about the target under study.
Raman Scattering and Infrared Absorption Imaging
In particular, because of their high structural selectivity, Raman and IR imaging techniques also have the capability to monitor chemical structural changes that occur in chemical and physical processes. Both IR and Raman imaging techniques benefit from recent developments of array detectors, which allow the rapid collection of both spectral and positional data.
Infrared absorption spectroscopy is a straightforward technique for vibrational imaging. Infrared Fourier transform (FT) microscopy with scanning options allows “chemical mapping” with lateral resolution on the order of tens of microns. The integration of IR absorption spectroscopy into near-field scanning optical microscopy is a promising approach to in situ, nondestructive, high-spatial-resolution imaging, with applications in the chemical characterization of materials and nanotechnology that improve the spatial resolution of IR spectroscopy to 300- 500 nm attainable in the near field.25 Due to the Raman effect, inelastically scattered light is shifted in wavelength relative to the excitation frequency by the characteristic molecular vibrational frequency of the probed material. Therefore, Raman scattering can be applied noninvasively under ambient conditions in almost every environment, including those in which water is present. Today, laser photons over a wide range of frequencies from the near-ultraviolet to the near-infrared region are used in Raman scattering studies, allowing selection of optimum excitation conditions for each sample. By choosing wavelengths that excite appropriate electronic transitions, resonance Raman imaging of selected components of a sample or parts of a molecule can be performed.
The range of excitation wavelengths has been extended to the near-infrared (NIR) region, in which background fluorescence is reduced and photoinduced degradation from the sample is diminished. Moreover, high-intensity diode lasers are easily available, making this region attractive for compact, low-cost Raman instrumentation. Furthermore, the development of low-noise, high-quantum-efficiency multichannel detectors (charge-coupled device, CCD), combined with high-throughput spectrographs and used in combination with holographic laser rejection filters, has led to high-sensitivity Raman spectrometers.
The main advantage of Raman spectroscopy is its capability to provide rich information about the molecular identities of the sample. Sophisticated data analysis techniques based on multivariate analysis have made it possible to exploit the full information content of Raman spectra and draw conclusions about the chemical composition of very complex systems such as biological materials.26 The downside of vibrational imaging techniques comes from relatively small IR absorption cross sections and also from the extremely small cross section for Raman scattering (typically 10−30-10−25 cm2 per molecule), with the larger values occurring only under favorable resonance Raman conditions. The small cross sections result in very weak imaging signals. For comparison, effective fluorescence cross sections can reach about 10−16 cm2 per molecule for high-quantum-yield fluorophores. On the other hand, particularly under ambient conditions, the amount of molecular structural information that can be obtained from fluorescence imaging is limited.
In terms of the high content of chemical structural information at desired spatial and temporal resolutions, Raman spectroscopy would be a very useful technique for chemical imaging. A disadvantage, however, in many applications of Raman imaging results from relatively poor signal-to-noise ratios due to the extremely small cross section of the Raman process, 12 to 14 orders of magnitude lower than fluorescence cross sections. New methodologies such as surface-enhanced Raman scattering and nonlinear Raman spectroscopy can be used to overcome this shortcoming.
Surface-Enhanced Raman Scattering. In the 1970s, a discovery that showed unexpectedly high Raman signals from pyridine on a rough silver electrode attracted considerable attention.27 Within a few years, strongly enhanced Raman signals were verified for many different molecules, which had been attached to various “rough” metal surfaces; the effect was called “surface-enhanced Raman scattering” (SERS). The discovery of SERS showed promise to overcome the traditionally low sensitivity of Raman spectroscopy.
It soon turned out that enhanced Raman scattering signals are associated mainly with nanoscale roughness structures on the silver electrode, and similar and even stronger enhancement factors were observed both for small silver and gold colloidal particles in solution and for evaporated island films of silver and gold. Enhancement of Raman signals occurs due to resonances between the optical fields and the collective motion of the conduction electrons (surface plasmons) in metallic nanostructures. This resonance effect leads to strongly enhanced and spatially confined local optical fields in the close vicinity of metallic nanostructures where spectroscopy takes place, resulting in strongly enhanced Raman spectra. Enhancement of excitation and scattered field results in an increase in Raman scattering signal intensity equal to the fourth power of the field enhancement. In addition to this “electromagnetic” field enhancement effect, electronic interactions between the Raman molecule and the metal (e.g., charge transfer) can result in an increase of the Raman cross section itself; this is known as “chemical or electronic enhancement.”28 Strong enhancement factors that should be associated with a “chemical effect” have been observed recently for small metal clusters.29 Although the Raman shifts, relative peak intensities, and line widths with SERS may differ slightly from those in normal Raman spectra due to a combination of the molecular interaction with the metal, high local confinement of the effect, and large field gradients, a SERS spectrum still provides a clear “fingerprint” of a molecule. Moreover, SERS is an analytical technique that can give information on surface and interface processes, such as charge-transfer processes at the nanoscale.30
The task of imaging single molecules while simultaneously identifying their chemical structures and monitoring structural changes poses a challenge that is of both basic scientific and practical interest in many fields. At present, SERS is the only way to detect a single molecule and simultaneously identify its chemical structure.
Extremely high SERS enhancement factors can bring effective Raman cross sections to the level of fluorescence cross sections and allow Raman spectroscopy of single molecules. Single molecule Raman spectra can be measured with nonresonant NIR excitation,31 as well as with resonant excitation exploiting molecular resonance Raman enhancement in addition to SERS.32 SERS provides a method to detect and identify a single molecule without requiring any label because it is based on the intrinsic surface-enhanced Raman scattering of the molecule. Moreover, it provides structural chemical information and thus the capability to image chemical and physical processes at the single-molecule level without ensemble-averaging effects.
Nonlinear Coherent Raman Spectroscopy. In addition to imaging based on single-photon excited or linear Raman scattering, vibrational images can also be generated using nonlinear coherent Raman spectroscopies. The most prominent nonlinear Raman process for imaging is coherent anti-Stokes Raman scattering (CARS), where molecular vibrations are probed by two incident laser beams, a pump beam at a frequency of ω1 and a Stokes beam at a lower frequency of ω2. When the difference between these two frequencies, ω1 − ω2, matches the frequency of a particular molecular vibration, a strong CARS signal is generated at a new frequency, ω3 = 2 ω1 − ω2, higher than both ω1 and ω2. This CARS signal depends quadratically on the pump beam intensity and linearly on the Stokes beam intensity and therefore requires picosecond or femtosecond laser pulse trains with high peak powers but only moderate average power for excitation (~0.1 mW).33 A CARS spectrum of a sample, often similar to the spontaneous Raman spectrum, can be obtained by tuning the frequency of the Stokes beam and using a broadband Stokes beam.
Like spontaneous Raman microscopy, CARS microscopy does not rely on natural or artificial fluorescent labels, thereby avoiding issues of toxicity and artifacts associated with staining and photobleaching of fluorophores. Instead, it depends on a chemical contrast intrinsic to the samples. However, CARS microscopy offers two distinct advantages over conventional Raman microscopy:
- It is orders of magnitude more sensitive than spontaneous Raman microscopy due to the excitation of coherent molecular vibration in the sample. Therefore, CARS microscopy permits fast vibrational imaging at moderate to average excitation powers (i.e., up to ~10 mW average power) tolerable by most biological samples. It was found that the peak powers of picosecond laser pulses used for CARS microscopy create minimal nonlinear (multiphoton) damage. Overall, the radiation damage is significantly less for CARS than for spontaneous Raman, especially when one is interested in following a dynamic process with short data collection time.
- It has three-dimensional sectioning capability because the nonlinear CARS signal is generated only at the laser focus where laser intensities are highest. Three-dimensional images can be reconstructed by raster scanning the sample layer by layer. This is particularly useful for imaging thick tissues or cell structures.
More Recent Promising Developments in Vibrational Imaging
CARS Microscopy. Although the first CARS microscope was reported in 1982,34 it was not until the development of a new detection scheme in 1998 that high-quality, three-dimensional images of biological samples became possible.35 Since then there has been a continuous evolution of the detection schemes and laser sources for CARS microscopy, and the sensitivity has been significantly improved.36 Many applications in biology and medicine are emerging. For examples, CARS microscopy is used to monitor lipid metabolism in living cells in real time (Figure 3.1) and to image live skin tissue at the video rate.37

FIGURE 3.1
Image (background) of lipid domains in a single giant unilamellar vesicle (GUV) visualized with CARS microscopy. (Radius of the vesicle = 14 μm.) The high sensitivity of CARS microscopy allows visualization of the GUV's single lipid bilayer when (more...)
Raman Imaging of Single Molecules. Single-molecule Raman spectroscopy requires extremely high SERS enhancement factors of at least 12-14 orders of magnitude. The origin of such a level of SERS enhancement is still under debate, but it can be understood as a superposition of an extremely strong electromagnetic field enhancement at factors of about 1012 associated with local optical fields and a so-called chemical enhancement effect on the order of ten- to a hundredfold. Due to the mainly electromagnetic origin of the enhancement, it should be possible to achieve a strong SERS effect for each molecule. However, there is still a molecular selectivity of the effect that cannot yet be explained.
A limitation of SERS spectroscopy is that target molecules have to be in the close vicinity of so-called SERS-active substrates such as nanometer-sized silver or gold structures. On the other hand, performing spectroscopy in the local optical fields of the nanostructures provides exciting capabilities for achieving nanoscale resolution in imaging based on Raman contrast. Highly enhanced optical fields are confined at the small probe volume near a metal tip, which is mounted on a cantilever and scanned across a sample surface by atomic force microscopy (AFM). In this tip-enhanced Raman spectroscopy (TERS), the signal enhancement factor is about three orders of magnitude, which is relatively small compared to other SERS experiments. However, the technique allows imaging of single carbon nanotubes with a 25 nm spatial resolution38 and shows the promise of high-sensitivity Raman microscopy beyond the diffraction limit. The development of new SERS-active nanostructures tailored and optimized for high sensitivity and resolution by nanofabrication techniques is a key goal for future developments of SERS-related chemical imaging.
New Labels Based on SERS. Design labels with chemical specificity are crucial to imaging.39 Fluorescence dyes or quantum dots are very common labels, but labels based on SERS signatures for characterizing DNA fragments and proteins have also resulted in high spectral specificity, multiplex capabilities, and photostability.40 Recently suggested SERS labels made from gold nanoparticles and an attached reporter molecule can provide interesting alternatives to fluorescence tags also for imaging. Figure 3.2 demonstrates simultaneous imaging of the indocyanine green (ICG) gold label based on the SERS signal of ICG along with chemical characterization of the environment of the label by surface-enhanced Raman spectra of the cell components in the vicinity of the gold nanoparticles.41 The large effective scattering cross section in SERS allows application of very low laser powers (<4 mW) and very short data acquisition times of 1 second or less per spectrum.

FIGURE 3.2
A hybrid SERS label made from the Food and Drug Administration-approved dye indocyanine green (ICG) on gold nanoparticles and the application of this label inside living cells. (a) Examples of SERS spectra measured from single living cells incubated with (more...)
Infrared Fourier Transform Microscopy. Infrared Fourier transform (FTIR) microscopy with scanning options allows “chemical mapping” with lateral resolutions of 20 to 60 microns when classical globar light sources are used for broadband illumination. Major advances in recent years in imaging detector technology and step-scan methods have continued to increase the number of applications of IR imaging in materials and biological research.42 Synchrotron light is a nearly perfect light source for IR spectroscopy because it combines very high brightness and a broad energy range.43 This results in a considerable improvement in lateral resolutions for synchrotron light, where aperture settings smaller than the wavelength of light can be used and diffraction controls the lateral resolution. For typical IR absorption lines, this means nearly one order of magnitude improvement in resolution compared to a classical light source. This allows the examination of very small dimension structures such as misfolded proteins at very high resolution. For example, the prion protein (PrP) aggregates in scrapie consist of β-sheet structure and are similar to Alzheimer's neuritic plaques; thus, they should be detectable by IR microscopy. However, compared to Alzheimer's disease, PrP aggregates are very small and/or microdisperse in most prion strains. High resolution of these aggregates can be achieved using synchrotron light to monitor this misfolded protein.44
Terahertz Imaging. Recent developments of new light sources, particularly free-electron lasers, have led to a rapidly growing interest in using the terahertz range (3-300 cm−1) for imaging.45 There is considerable evidence that this energy range gives important information on modes related to hydrogen bonds and other weak interactions and can be used for imaging and discrimination among different materials. Moreover, resonances in the terahertz range detected in large biomolecules such as proteins and DNA polymers can provide unique information about the structure of these molecules complementary to that provided by vibrations in the IR frequency ranges and X-ray crystallography. Terahertz transmission spectroscopy of proteins has demonstrated the sensitivity of the technique for monitoring folding-unfolding processes, particularly in a realistic aqueous environment.46
At present, free-electron lasers are excellent light sources for basic studies on imaging in the terahertz range. The further development of this technique as a powerful tool for imaging will also depend on the development of convenient new terahertz light sources.
Future Methodological Developments in Vibrational Imaging
Advances in vibrational imaging techniques are possible in a number of areas. The following is a brief list of the most promising areas of research:
- sensitive vibrational imaging at the single-molecule level, taking advantage of enhanced and confined local optical fields of tailored nanostructures, especially in combination with scanning probe techniques;
- exploration of nonlinear Raman scattering techniques, such as CARS or sum frequency microscopy, in order to enhance vibrational sensitivity in reduced probe volume;
- use of new light sources for IR and Raman imaging that provide higher brightness and tunability over wide wavelength ranges;
- extension of IR imaging to the terahertz range for probing complex macromolecule dynamics and structures and utilizing specific low-frequency modes for high-contrast imaging.
Fluorescence Imaging
Since it began in the seventeenth century, optical microscopy has evolved in capability as a ubiquitous tool of chemistry, biology, materials science, and engineering. From the early days, wide-field microscopy, in which a magnified image of the plane of focus is viewed with visible light through an eyepiece and recorded on film, has been an essential research tool. In particular, the early growth of cell biology, microbiology, and their associated biological chemistry depended on optical microscopy. The inherent physical limits of resolution have frequently been limiting factors, leading to continuing developments to surpass resolution limits in the focal plane and avoid the out-of-focus background. The contrast in early imaging depended on a variation of refractive index, anisotropic polarizability, light absorption, or scattering. Innumerable advances in capability have followed and are continuing to present major opportunities for advances in chemical imaging that can meet the challenges to solve ever more difficult problems.
Fluorescence Techniques
Fluorescence Microscopy. Unlike NMR spectroscopy and vibrational spectroscopy, electronic spectroscopy involves interactions with electromagnetic waves in the near-infrared, visible, and ultraviolet (UV) spectral regions. While electronic spectroscopy is less enlightening about structural information than NMR and vibrational spectroscopy, the shorter wavelengths involved allow higher spatial resolution for imaging, and its stronger signal yields superb sensitivity. Fluorescence detection, with its background-free measurement, is especially sensitive and makes single fluorescent molecules detectable.
Fluorescence has provided the power and diversity for chemical markers, now frequently designed to bind to particular targets or to be genetically expressed in biological systems. The recent discoveries of green fluorescent proteins (GFPs) and red fluorescent proteins (RFPs) have enabled genetic labeling of particular targets in living systems. This is performed by incorporation of a fluorescent protein gene—from a selection now including many available colors47 and with chemical properties such as pH48 or calcium sensitivity49—for chemical imaging of physiological functions in vivo.
The continuing development of available labels remains one of the most promising avenues for advances in chemical imaging in both biological and soft materials applications. Figure 3.3 illustrates the GFP-RFP varieties available today. However, they are not optimal because they require slow oxidative activation within the cells in which they are expressed and are therefore not sufficient indicators for many dynamic measurements of gene expression. Current research aims to escape this kind of problem by the development of unnatural amino acids that can be expressed as intracellular markers. This difficult area requires sustained and concerted support.
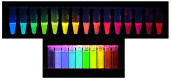
FIGURE 3.3
Top: Various colors of fluorescent proteins now available. These proteins can be expressed in almost any cell and have provided in vivo spectroscopic markers for following the production, degradation, localization, and movement of many different proteins. (more...)
Detecting and tracking individual macromolecules and their nanoscopic tracking in living cell membranes and tissues provide a powerful approach to understanding the biochemical dynamics of life processes. This approach requires bright fluorescent markers and accurate optical imaging to provide nanoscopic spatial resolution over many orders of time ranging from microseconds to several minutes. However, detection is limited by the number of fluorescence photons that can be captured from each marker before it photobleaches. About 103 detected photons are needed at each point in time for a precise location of several nanometers of sparse, and therefore resolvable, markers in an optimized microscope. The central position of a marker is obtained by computation of the centroid of the diffraction-limited microscopic image, which is about 450 nm in diameter for a point source.50 The uncertainty of the position measurement improves roughly as the reciprocal square root of the number of photons detected. Thus, analysis of molecular patterns, mobility, and interactions—important in biological and materials science research—depends on the development of brighter, more durable, chemically specific markers of nanoscopic size in order to label target molecules and follow the time course of their trajectories with nanoscopic precision.
Semiconducting nanocrystals, usually CdSe-ZnS crystals a few nanometers in diameter, called “quantum dots,” can be useful for in vivo imaging of biochemical dynamics but still suffer from three limitations.51 The most severe problem is that quantum dots blink at probability distributions that lead to loss of continuity in keeping track of individual molecules, limiting the ability to measure the dynamics and mechanisms of biophysical chemistry in vivo and ex vivo. Some significant fraction of fabricated quantum dots appear to be totally dark, reducing average quantum yield.52 Another issue arises from protecting quantum dots from the aqueous biological environment. Various chemical coatings of these hydrophobic particles have been tried, and some thin coatings are temporarily effective. However, the only reliable results to date depend on multiple amphiphilic coatings that increase hydrodynamic diameters to >30 nm, which is too large for many biochemical applications.
To avoid the blinking problem with nonblinking markers and the potential toxicity of semiconducting quantum dots, it is possible to aggregate about 20 to 30 organic dye molecules in protective environments by innovative chemistry. The first success was achieved by labeling low-density lipoprotein (LDL) particles with about 30 carbocyanine dye molecules. These particles can be brightly labeled and photobleach slowly, but they are not durable. A more robust alternative is the sequestration of 20 or so organic dye molecules into a silica shell about 30 nm in diameter using established emulsion techniques. Properly bound to the silica, fluorophores such as rhodamine are protected from photobleaching and interactive quenching, providing a marker about 20 times as bright as present quantum dots but unfortunately also still too large for many applications.53 However, these two examples demonstrate the potential of utilizing innovative chemistry in the development of more effective chemical imaging tools.
Fluorescence Correlation Spectroscopy and Fluorescence Burst Analysis. Several nanoscopic chemical imaging approaches work very well for measurements of chemical kinetics, interactions, and mobility in solution. Fluorescence correlation spectroscopy (FCS) measures the temporal fluctuations of fluorescent markers as molecules diffuse or flow in solution through a femtoliter focal volume.54 Their average diffusive dwell times reveal their diffusion coefficients, and additional faster fluctuations can reveal chemical reactions and their kinetics if the reaction provides fluorescence modulation. Cross-correlation of the fluorescence of two distinguishable fluorophore types can very effectively reveal chemical binding kinetics and equilibria at nanomolar concentrations.
These methods work best at nanomolar chemical concentrations so that the focal volume contains typically 1 to 100 molecules on average. Because the method is so sensitive, it is susceptible to perturbation by background fluorescence and instrumentation fluctuations. These problems have become quite tractable during the last decade, such that FCS now supports more than 100 publications per year. A current challenging application is analysis of protein folding kinetics, protein structure fluctuations, and ultrafast chemical kinetics by new methods yet to be published.
Fluorescence burst analysis, a variation of FCS procedures that has an optimum configuration for simple presentation, uses a uniform nanoscopic flow channel with an optically perfect ceiling, uniform cross section, and periodic electrodes that can now be constructed by careful electron lithography techniques.55 By application of controlled electric fields, uniform plug flow of solution through the channel is achieved by controlled electrophoresis. This avoids the parabolic flow velocity profile of pressure-driven flow. The channel cross section is uniformly illuminated by sufficiently large laser beam flows to provide identical illumination pathways for molecules flowing anywhere in the cross section. Thus, all molecules of a given brightness, such as a particular length of DNA labeled with an intercalating dye, yield the same fluorescence burst size, thereby providing a burst size parameter that characterizes the DNA double helix length to approximately ±5 percent over at least three orders of magnitude. Clearly, future applications of this “flow imaging” burst analysis scheme offer potential for development of analytical techniques in medicine such as the elusive counting of the concentration of beta-amyloid clusters and their sizes in cerebral-spinal fluid.
Single-Molecule Fluorescence Spectroscopy and Imaging
Current Technology. In the past decade, rapid developments have made it possible to detect, identify, track, and manipulate single molecules on surfaces, in solutions, and even inside living cells. The ability of single-molecule experiments to avoid ensemble averaging and to capture transient intermediates make them particularly powerful in elucidating mechanisms of molecular machines in biological systems: how they work in real time, how they work individually, how they work together, and how they work inside live cells. New knowledge from single-molecule experiments continues to generate novel insights in a variety of scientific fields.
Single-molecule fluorescence detection in an ambient environment is achieved in part through reduction of the probe volume in order to suppress the background signal.56 This is accomplished by a confocal or total internal reflection microscope and as well as by the high sensitivity of the detectors. Aside from the fluorescence intensity, optical properties such as polarization, fluorescence lifetime, and excitation and emission spectra have been used as contrast mechanisms for acquiring images to follow the temporal behavior of certain molecules. In particular, fluorescence resonance energy transfer has been widely used as a dynamic variable, dubbed a “molecular ruler,” to measure intermolecular distances between two fluorophores. These advances allow one literally to record movies of molecular motions and biochemical reactions.
One of the exciting areas of research with fluorescence microscopy is the study of dynamic behaviors of individual enzyme molecules. Conventional measurements of chemical kinetics rely on determining concentration changes following a perturbation (such as a temperature jump or rapid mixing of reactants). On a single-molecule basis, a chemical reaction, if it occurs, takes place on the subpicosecond time scale. However, the “waiting time” prior to such an action during which the molecule acquires energy to reach the transition state via thermal activation is usually long and stochastic. Stochastic events of chemical changes can be monitored and the histogram of waiting times can be measured for a single enzyme molecule undergoing repetitive reactions. The advantages of single-molecule studies of biochemical reactions include the following: (1) to measure the distributions and fluctuation of enzymatic activities; (2) to unravel reaction mechanisms; and (3) to observe in real time the transient intermediates that are otherwise difficult to capture in conventional experiments due to their low steady-state concentrations.
For example, enzymatic turnover of flavin enzyme molecules was monitored in real time by viewing fluorescence from an active site of the enzyme.57 Cholesterol oxidase, a 53-kilodalton flavoprotein, catalyzes the oxidation of cholesterol by oxygen with the enzymatic cycle shown in Figure 3.4. The active site of the enzyme, flavin adenine dinucleotide (FAD), is naturally fluorescent in its oxidized form but not in its reduced form. Confined in agarose gel containing 99 percent water, the enzyme molecules are immobilized at the laser focus. On the other hand, the small substrate molecules are essentially free to diffuse within the gel. The single FAD emission exhibits on-off behavior, with each on-off cycle corresponding to an enzymatic turnover. This experiment demonstrated that an enzyme molecule is a dynamic entity with a fluctuating catalytic rate constant, a phenomenon that was hidden in ensemble studies.

FIGURE 3.4
(A) Fluorescence image of single cholesterol oxidase (COx) molecules immobilized in a thin film of agarose gel of 99 percent buffer solution. (B) Enzymatic cycle of COx that catalyzes the oxidation of cholesterol by molecular oxygen. The enzyme's naturally (more...)
The conformational dynamics of enzymes is intimately related to enzymatic activity and can now be probed at the single-molecule level. Fluorescence resonant energy transfer (FRET) is used widely in biochemical and biophysical studies of conformational motions.58 The efficiency of FRET between a donor and an acceptor pair is E = 1/(1 + (R/R0)6), where R is the distance between the pair and R0 is the Forster radius, which is dependent on the spectral overlap between the donor emission and acceptor absorption spectra and the relative orientations of the donor and acceptor dipoles. FRET between a single donor and acceptor pair within a single biomolecule has been used to probe conformational dynamics.59 For example, a small RNA enzyme called the hairpin ribozyme has been studied.60 The ribozyme's two domains were labeled with a FRET pair, and the FRET time traces showed striking heterogeneity in docking and undocking kinetics, suggesting the presence of a large number of stable conformational states under functional conditions.
For experiments with fluorescent substrates, substrate concentrations must be kept low to avoid a strong fluorescent background. At millimolar substrate concentrations where many enzymatic reactions occur, conventional FCS would not work at the usual femtoliter focal volumes. To escape this limitation, it has been possible to provide attoliter focal volumes in electron lithographically formed zero-mode waveguides. For example, these structures have allowed the tracking of the formation of the complementary DNA sequence for a template sequence by high-processivity function of a single DNA polymerase.61 Essentially, this geometry provides the opportunity for virtual single-molecule enzyme kinetics at appropriate fluorescent substrate concentrations wherever a non-interfering signal can be created.
Parallel with and complementary to single-molecule studies by optical means has been tremendous work on mechanical manipulation of single molecules accomplished through the use of either optical tweezers62 or magnetic tweezers.63 These techniques offer the possibility of actively controlling the behavior, or even chemical reactions, of single molecules and have yielded much new knowledge about the mechanisms of enzymatic machineries such as molecular motors64 and nucleic acid enzymes.65
Cutting-edge Technology. Integrating chemical and biological labels with advanced microscopes and detectors is the focus of many research activities. Other contrast mechanisms of single-molecule imaging, in addition to fluorescence, are also being pursued.
The spatial resolution of fluorescence microscopy has been limited to about half the wavelength of light due to the diffraction limit associated with the wave nature of light. However, if one has a single isolated molecule with bright fluorescence, the accuracy of determining the center position of its diffraction-limited image can be as high as 1 nm. In this way, nanometer movements of a molecular motor can be followed in real time.66
At high concentration, when molecules are no longer isolated in space, a conventional optical microscope is unable to resolve them within the diffraction limit. Efforts have been made to circumvent the diffraction limit by engineering the point spread function using nonlinear optical techniques. Spatial resolution of 20 nm in a cell has been demonstrated without using a proximal probe.67
Future Technology. Recent innovations of single-molecule fluorescence imaging and dynamical studies have lead to unprecedented sensitivity, molecular specificity, time resolving power, and spatial resolution. In the future, the challenges and opportunities in optical imaging will lie with biology. Single-molecule sensitivity for three-dimensional imaging in a living cell with specific and noninvasive labeling of a macromolecule of interest, along with millisecond time resolution and nanometer spatial resolution, will provide answers to many biological questions. The integration of these elements will come with time, and a motion picture of a living cell should prove possible with continuing developments.
Laser Scanning Microscopies
Confocal Microscopy
The availability of laser scanning confocal fluorescence microscopy,68 first commercially offered in the 1980s, enabled a major advance in chemical microscopy imaging; convenient summaries are available in the Handbook of Biological Confocal Microscopy.69 Confocal fluorescence microscopy works by focusing continuous-wave laser illumination to a diffraction-limited spot in a focal plane within the specimen and collecting the excited fluorescence through a confocal optical aperture that excludes most of the out-of-focus fluorescence background. Images are formed by scanning the laser beam in a video raster and recording the photomultiplier-detected fluorescence in a computer array. Lateral resolutions are comparable with wide-field microscopy, and effective axial resolution is enhanced by exclusion of out-of-focus background. The technique allows imaging to depths of about 50 microns in soft biological tissue but is limited by the background-scattered fluorescence able to pass through the confocal aperture. This technology is used widely today in cell biology and soft-matter materials using fluorescent markers for chemical imaging.
Multiphoton Microscopy
Laser scanning fluorescence microscopy entered a new generation in 1990 when the nonlinear optical physics of two-photon molecular excitation (first analyzed by Maria Goeppert-Meyer in 1931 but not demonstrated until sufficiently bright lasers were created in the 1960s) was finally formulated for useful multiphoton laser scanning fluorescence microscopy.70 Multiphoton excitation of fluorescence provides several critical advantages over wide-field and confocal microscopy. Because multiphoton excitation of a molecule requires that it “simultaneously” absorb two or more excitation photons, fluorescence excitation is typically limited to the focal volume where concentration of the laser power provides sufficient photon flux density. Since the two-photon excitation rate depends on the square of the illumination intensity, the out-of-focus background excitation falls as the reciprocal fourth power of distance above and below the focal volume, thus generating negligible out-of-focus fluorescence along the out-of-focus beam path. Photodamage is also negligible since the long laser wavelengths needed for multiphoton excitation (nearly invisible infrared photons) are not significantly absorbed by tissue. This nonlinear microscopy can productively image fluorescence signals to depths in living tissue of approximately 500 microns (about the thickness of human skin).
Multiphoton microscopy (MPM) utilization has grown rapidly and continuously since then, with more than 200 refereed publications per year citing the use of MPM or two-photon microscopy. Commercial sources for MPM instruments did not become available until several years later, but adequate titanium sapphire (Ti:sapphire) 100-femtosecond lasers were (and are) available, albeit at exorbitant costs. Many MPM instruments were and still are assembled by the scientists using them, a point that may become relevant in future specialized chemical imaging opportunities. Convenient laboratory instruments for MPM imaging are now available from Zeiss Microscopy.
The earliest, fastest-growing, and possibly most productive area of application of MPM is in the imaging of neuronal functions in ex vivo functional brain slices and protracted imaging of function in intact brains of living animals over extended times as the neural circuits develop.71 The most popular chemical applications of MPM have been based on fluorescent molecular indicators of calcium ion activity, a ubiquitous intracellular signal, and of membrane potential. Recently, GFP gene labels of specific receptors and ion channels and fluorescent labels of protein active sites involved in the molecular mechanisms of biological functions have provided additional powerful research tools.72 The development of three-photon infrared excitation of the intrinsic UV excitable indoleamines, serotonin and melatonin allows research to be conducted on secretory kinetics and mechanisms for neuromodulator release in cell cultures and in living tissue.73 This capability has yet to be fully realized for in vivo or ex vivo studies of the secretion of these important neuromodulator molecules in brain.
Deeper Multiphoton Fluorescence Imaging in Living Tissue Through GRIN Lenses. It is possible to translate the focal volume of MPM imaging by up to 0.5 cm distances using gradient refractive index (GRIN) lenses. These lenses consist of small-diameter rods of exotic, rare-earth-containing glasses of graded concentrations that provide a radial gradient of refractive index, thereby acting as a lens with flat ends. Multiphoton images at depths up to about 0.5 cm in the brains of living mice have been obtained with access to the intact mouse hippocampus and negligible tissue damage en route.74 The longer GRIN rods for focusing transfer are a few millimeters in diameter and are capped by a short, higher-numerical-aperture objective lens. It is possible to miniaturize these devices by using extensions of current techniques and delivering the femtosecond laser pulses with suitable fiber optics using vibrational scanning.75 This technique appears to have great promise for deeper in vivo chemical imaging.
Intrinsic Biological Fluorescence and Potential Applications in Medicine. The most recent and potentially most important advances in MPM are based on chemical imaging of the intrinsic fluorescence of crucial molecular species. One, in particular, images the long-known fluorescence of nicotinamide adenine dinucleotide (NADH) to measure the metabolic pattern in brain, recognizing oxidative exhaustion of neurons caused by their electrical signaling activity and the slower contribution to restoration of their metabolism by astrocytic glycolysis.76 Other possible chemical signals that supposedly couple astrocytes with neurons in brain function were entertainingly but significantly summarized in Scientific American in April 2004.77 An interesting challenge of metabolic imaging is the chemistry of NADH-NAD(P)H and their binding to protein cofactors in the mitochondria and cytoplasm, which modulates the fluorescence quantum yield and confounds the accuracy of quantitative measurements of metabolic state.78 In addition, this topic will be a chemical imaging challenge that must be solved in coming years, since MPM imaging of brain metabolism in living animals, including neurodegenerative disease models, now appears to be approaching feasibility.
Second Harmonic Generation (SHG) Imaging
Another nonlinear optical technique, known as SHG, can be imaged with bright-pulsed laser illumination of optically noncentrosymmetric materials with nonlinear (intensity-dependent) dielectric properties. Certain amphiphilic or hydrophobic electropolarizable dye molecules that are lipid soluble can be aligned in parallel by the electric fields commonly present across cell membranes. These fields routinely reach up to 250,000 volts per centimeter, a signal of great significance for controlling cellular behavior in neurons. The demonstrated effectiveness of SHG fast imaging of neuronal signals motivates efforts to develop further improvements of the noncentrosymmetric electric field-sensitive indicator molecules. There have been sustained international efforts to develop membrane potential sensitive fluorescent molecules, but SHG electric field indicators are a relatively fertile photochemical challenge.
SHG has been found to provide a selective marker for imaging neuronal axons through SHG generation by the parallel-oriented bundles of their microtubules, which provide the selective tracks for movement of cargo in vesicles to and from synapses by the molecular motors dynein and kinesin.79 The parallel polarization of microtubules in axons and their random orientation in neuronal dendrites had previously been detectable only by tedious multistage electron microscopy. This new capability may be useful in diagnosing the effects of aggregation of the microtubule-associated tau protein (imageable by its MPM intrinsic fluorescence) that induces neurofilamentary tangles in Alzheimer's disease.
SHG imaging of collagen structures has been very effectively achieved and is now rather well understood.80 Combining SHG and MPM fluorescence appears feasible for creation of a simple optical label of collagen chemical assembly structure type and anomalies in orthopedic surgery, discussed further below.
Other Multiphoton Coherent Optical Microscopy
In a manner similar to SHG imaging, third harmonic generation (THG) imaging has been demonstrated.81 CARS microscopy (discussed earlier) is another form of MPM, providing chemical information via vibrational spectroscopy. As in two-photon fluorescence microscopy, SHG, THG, and CARS techniques have small probe volumes and offer three-dimensional resolution. However, unlike two-photon fluorescence, SHG, THG, and CARS signals (like those of NMR) are coherent. Therefore, the contrast interpretations in SHG, THG, and CARS microscopy are more complicated than two-photon fluorescence microscopy. In recent years, these techniques have been studied experimentally and theoretically in great detail.82 The spectral specificity and high sensitivity of CARS microscopy are particularly attractive for chemical imaging of living cells.
Ultrafast Spectroscopy and Imaging
The advent in recent years of pulsed light sources that generate femtosecond pulsed lasers that are easily operated, are computer controlled, and can be integrated into imaging systems has provided new capabilities in imaging spectroscopy in two respects. First, and thus far most important, the high peak power that such pulses provide has made it possible to perform nonlinear imaging spectroscopy routinely. The most popular version of this, and the only commercially available approach, is multiphoton fluorescence microscopy. Other nonlinear imaging techniques such as sum-difference spectroscopy of surface molecules have been developed as well but are not yet commonplace. Another current use of ultrafast pulses in imaging is the measurement of excited state dynamics. In particular, fluorescent lifetime imaging has become popular in the biological community because the fluorescent lifetime is sensitive to the environment but does not depend on the concentration of the label. Finally, ultrafast pulses are being used for tomography of deep tissue, primarily in the realm of analyzing light transmitted through a scattering sample.
Current Capabilities
Fluorescence Lifetime Imaging (FLIM). Commercial instruments are now available for FLIM of surfaces or three-dimensional objects in conjunction with either multiphoton or confocal microscopy. At each point in the image, an excited state dynamics trace is measured, typically on the few hundred picosecond to nanosecond time scale. This type of imaging is particularly useful in conjunction with FRET systems. When the donor and acceptor become close to one another, the fluorescence lifetime of the donor decreases as the energy transfer rate becomes comparable to, or faster than, the inherent donor excited state lifetime (Figure 3.5).83 Other labels have lifetimes that are particularly sensitive to specific environments. For example, DNA intercalating dyes such as thiazole orange have very short lifetimes in solution, but much longer and somewhat variable lifetimes when intercalated into DNA. This allows one to determine not only whether the signal comes from DNA but also the state of the DNA (e.g., base content and structure).84 Finally, almost all organic dyes have inherent environmentally sensitive fluorescent lifetimes and can be used to report on conformational changes in proteins or other molecules in an imaged fashion.85 FLIM provides another dimension to distinguish between the signal from the label under study and background fluorescence. In the case of biological imaging, autofluorescence is usually relatively short-lived compared to most of the organic dyes employed. In addition, FLIM provides increased resolution between labels that have similar spectra but different lifetimes, allowing the ability to separate many fluorophores from one another in a sample simultaneously using a two-dimensional (time versus spectrum) approach.

FIGURE 3.5
Example of the use of FLIM to investigate molecular interactions in cells. Two-photon FLIM was performed on GFP-tagged protein kinase C (GFP-PKC) coexpressed with DsRed-tagged caveolin (DsRed-cav) in Chinese hamster ovary (CHO) cells. Coexpression of (more...)
Ultrafast Tomography. Another developing imaging technique is tomography using ultrafast near-infrared pulsed lasers.86 This can be done in several ways. One basic approach involves detecting transmission of ultrafast pulses through tissue. Here, one is looking specifically at light that travels directly through the tissue and therefore is not delayed in its arrival at the detector. This is performed by gated detection methods; in this way, it is possible to generate a two-dimensional projection image that includes information about absorption and scattering density in the tissue. Ultrafast pulses can also be used specifically to detect light scattered from a particular depth in the tissue by timing the round-trip.
Enhanced penetration capabilities of wide-field microscopy87 have been obtained by elegant application of interferometry in a technique called optical coherence tomography88 that images optical backscatter contrast in tissue at millimeter depths. This powerful technique is already being applied in medicine, particularly to image the retina within the eye, an especially favorable application for deep imaging since the optical path has little scattering. Interferometry, beginning with early designs of Zernike and Nomarski, has been extended to picometer displacement measurements of the transduction mechanisms of audition and their biochemical modulation.89 Image resolution enhancement by about twofold of thin biological preparations has been obtained by computational convolutions of stacks of images.90
Cutting-edge Technology
Ultrafast spectroscopic approaches are increasingly being applied to imaging in terms of both nonlinear imaging and dynamics. This is driven partially by the improvement in and ease of use of ultrafast laser systems and detectors. It is not possible to review all of the methods currently under development, but several examples are given below.
Spectral Lifetime Imaging. Because of improvements in detector sensitivity, multidimensionality, and speed, a number of different ultrafast methods have been converted into imaging methods. A particularly interesting example is the use of two-dimensional streak cameras in fluorescence imaging. The streak camera allows one to record real-time signals with subpicosecond resolution in one dimension while simultaneously resolving another dimension such as wavelength. This provides an opportunity to record the fluorescence decay time and the spectrum with high resolution at each point in a three-dimensional image using confocal microscopic techniques. This approach, coupled with hyperspectral analysis in both dimensions, has the potential for extraordinary resolution of different fluorescent targets in a complex sample mixture.91 The addition of a spectral dimension to a typical fluorescence microscope can provide an increase in sensitivity, throughput, and data accuracy.
Reverse Imaging. In imaging, one normally thinks of obtaining information about a system that is spatially and/or temporally resolved. However, similar approaches can also be used to project patterned information into a chemical system. This, for lack of a better phrase, is referred to as “reverse imaging” in this document. The idea is to use the same kinds of methods and machines that we use to record images to also control chemistry or biochemistry in a patterned fashion.
Current Applications
Chemistry. Photolithography has been refined for several decades by the electronics industry. Three-dimensional sculpting of photopolymerizable materials using nonlinear excitation has allowed the production of objects with resolutions below 100 nm. Applications to patterning of chemical systems can be seen in the photoprocessing of DNA chips (see Figure 3.6, for example) by companies such as Affymetrix (Santa Clara, CA). Light is used to build large arrays of DNA oligonucleotides layer by layer in the horizontal plane. Similar approaches are now being used to pattern chemistry using electrochemical methods on electrode arrays (Combimatrix, Seattle WA). However, this technology has been limited largely to polymerization of homogeneous photopolymers and production of DNA. The potential for developing chemical systems that can be modulated in time and space by imaged radiation is huge.
FIGURE 3.6
Example of a DNA array and fluorescent detection. Synthesis of chips such as these is valuable in determining changes in gene expression under various physiological conditions with high throughput. SOURCE: Photograph courtesy of Peter R. Hoyt, Oklahoma (more...)
Biology. In biology, studies have been conducted using imaged killing of cells either for directed evolution or for tissue engineering. Also, laser-based photoablation of cells has been used in fate-mapping studies of development. Subcellular surgeries have been performed in which specific features of a cell have been ablated. However, these are crude approaches since the method used in these procedures is typically photodestruction rather than specific manipulation.
An important technique in current use is image-active photochemistry. This is employed in the use of chemical caged derivatives of neurotransmitters and biological activators such as adenosine 5′-triphosphate (ATP), which can be used as powerful optical tools for photoactivated research pharmacology. Effective chemical cages for one-photon photoactivation are numerous and widely used. The continuing advance in development of these photochemical tools indicates the potential for further chemical advances. Unfortunately, two-photon excitation of these same caged reagents has not worked as well as the best reagents for one-photon excitation, thus excluding the pharmacology envisioned in vivo.92 These obstacles could be overcome by utilizing new cage designs or three-photon excitation of the UV-activated cages to avoid both parity selection rules and pulse pairs for multiple-stage cage release kinetics. This is a challenge for future fast chemical imaging activated approaches.
Laser tweezers can also be used to manipulate specific parts of cells in ways much more subtle than photodestruction. It has also become possible to design genes in which expression is controlled by light. This opens the door for patterned gene expression at cellular resolution in both two and three dimensions. In the chemical world, the future holds many opportunities for conducting two- and three-dimensional solid-phase synthetic chemistry using light. A fairly large arsenal of photoprotective groups now exists, allowing a wide variety of chemical couplings to be performed. This may revolutionize the search for new drugs, sensors, or materials because very large libraries of molecules can be synthesized rapidly. The advantage over standard combinatorial chemistry is that these are not random combinations of pieces but specific molecular structures, allowing one to combine rational design driven by computation with very high throughput screening and selection during molecular evolution.
Prospectus for Future Optical Microscopy
Which aspects of chemical imaging by optical microscopy will be most important in coming years? What needs to be done to take advantage of its powers? In vivo and ex vivo chemical imaging in the brain and nervous system will continue to be important. Current applications are being extended to chemical diagnostics of biochemical signaling between dendritic spines and presynaptic patches on axons that defines the development of neural circuits during growth and learning of the brain.93 The principal challenge of neuroscience research in the coming decades will be discovery of the chemical signals governing the development of nervous system organization and the mapping of its consequent geometry and function.
Intrinsic fluorescence of tissue components is imaged efficiently by MPM in living animals and tissues, with results that demonstrate substantial capability for instant recognition of cancer in several organs.94 Direct comparisons between the incipient tumor MPM images in organs at risk with the corresponding pathologists' images of absorption-stained fixed tissues show that MPM is quite functional for disease diagnosis and study.
Medical applications for use in surgery and diagnosis are already being developed in various collaborations at several institutions and will certainly expand worldwide. Two basic challenges must be met: (1) the need for increasing knowledge of biological photochemistry and (2) the development and engineering of suitable optical physics for devices compatible with the restrictions, demands, and safety of medical and surgical procedures.
The crucial biological photochemistry issue is to understand in greater detail all of the origins of intrinsic tissue fluorescence and its changes with disease. It is clear that NADH (an indicator of metabolic state) and the many flavin compounds that vary greatly in tissue types will be among the most conspicuously imaged indicators of disease. Other fluorescence contributors may include carotinoids and oxidized indoleamines. Imaging and macroscopic mapping of different types of collagens using second harmonic generation also appears promising as a medical tool, especially for orthopedic diagnostics and surgery.
General medical applicability of MPM calls for more compact, rugged, and user-friendly instrumentation. Ideally, designs could be built onto the observation optics currently used in surgery, particularly the endoscopic optics of laparoscopic surgery. The possibility of achieving this adaptability is advanced by (1) the discovery of rugged fiber optics capable of single-mode propagation by the high-power femtosecond laser pulses needed for MPM,95 and (2) the development of vibrating optical-fiber scanners for illumination scanning (Optiscan, Inc., Australia). Endoscopic MPM microscopy, which effectively provides in situ chemical and structural imaging, appears capable of instantly providing image information equivalent to conventional fixed-tissue pathology imaging.
Fundamental advances in research on complex chemical process are being enabled by new nanoscopic imaging developments that enable measurements of (1) chemical kinetics of protein folding and misfolding (e.g., amyloid aggregate formation in neurodegenerative diseases); (2) fluctuations in single-molecule enzyme kinetics to understand multistep enzymatic reactions; and (3) ultrafast chemical kinetics. These processes and their possibilities for application represent opportunities to be exploited via new developments in nanoscopic and nonlinear optical imaging discovery.
ELECTRON, X-RAY, ION, AND NEUTRON SPECTROSCOPY
This section covers techniques that probe samples with wavelengths much smaller than that of visible light and that provide high-resolution chemical and structural information below surfaces of materials. For example, X-rays are able to penetrate materials so deeply it is possible to determine the identity and local configuration of all the atoms present in a sample. Further attributes and limitations of these techniques are provided below.
Electron Microscopy
Since its invention nearly 100 years ago, electron microscopy (EM) has developed into a remarkably versatile imaging tool. Nearly all major R&D organizations (universities, national laboratories, industrial labs) have EM facilities that can accommodate well-established imaging techniques. Despite its apparent maturity, new techniques continue to be developed that not only push the resolution limits of EM but also expand the range of specimens and environments in which it can be used.
The usefulness of electrons for imaging comes from the fact that an electron wavelength is about 1000 times smaller than that of visible light, providing a much higher-resolution probe. In addition, higher-energy electrons (on the order of hundreds of kiloelectronvolts, or keV) can penetrate materials, thus providing access to imaging below their surfaces. Similarly the use of light, energy-resolved electron detection is able to provide chemical information in parallel to structural imaging. However, significant limitations to its use do exist, such as the need for a vacuum to produce and transmit electrons and electron beam damage to samples. Nevertheless, ingenious development of a variety of EM techniques has had tremendous impact in fields ranging from condensed matter physics to structural biology.
One of the most important forms that EM technology takes is the transmission electron microscope (TEM). The TEM operates much like a slide projector in the sense that electrons of sufficiently high energy (usually in the range of a few hundred kiloelectronvolts) are passed through a thin sample (usually less than a micrometer thick) to a detector where a variety of imaging schemes can be implemented. A number of applications of TEM are described below.
Cryo-Electron Microscopy for Biological Structure Determinations
The use of electron microscopy to image biological systems is a fast-growing area. One advance is the use of rapid freezing to transfer hydrated specimens into an electron microscope, a process known as cryo-electron microscopy (cryo-EM). After such a transfer, new techniques of imaging may be employed to display three-dimensional structures over a length scale ranging from single biomolecules to synapses and neurons.96 Three-dimensional structure determinations based on cryo-EM have become a standard tool of structural biology in recent years.97 As in crystallography,98 the technique of freezing samples in vitreous ice for EM analysis99 has made it possible to obtain two-dimensional projected images with minimal distortion or artifacts. If there is a plentiful supply of nearly identical frozen particles in random orientations, these projections can be combined to form three-dimensional images. The resolution of these images has been improving rapidly due to improvements in reconstruction techniques.100 In particular, efforts have centered on (1) the accurate determination of the contrast function that corrects the two-dimensional images for the experimental out-of-focus distance; (2) the accurate determination of the relative orientation of the projected images; and (3) the use of a far greater number of particles. As a result, it is now routine to obtain cryo-EM image reconstructions to have an estimated resolution of 10 Å (sometimes down to 7 Å) with expectations of reconstructions as low as 4 Å.101
Recent advances in instrumentation, data collection, and data analysis have resulted in cryo-EM maps of the ribosome with significantly higher resolution, as shown in Figure 3.7. For the first time, molecular signatures can be recognized: these include RNA helices with distinctly visible major and minor grooves, and protein domain structure with clearly defined shapes. The high degree of definition of these features has made it possible to fit known atomic structures with an accuracy of 3 Å.

FIGURE 3.7
A cryo-EM map of the Escherichia coli ribosome (complexed with fMet-tRNAf Met and mRNA); where fMet = formylmethionine obtained from 73,000 particles at a resolution of 11.5 Å. (a-d) Four views of the map, with the ribosome 30S subunit painted (more...)
Scanning Transmission Electron Microscopy
Conventional imaging using TEM has provided a tremendous capability to image atomic arrangements over a large range of length scales. To investigate the chemical nature of materials, however, elemental resolution is required. Developments in the formation and positioning of atomic-sized electron beams have enabled the development of scanning transmission electron microscopy (STEM) in which the highly focused probe beam is rastered across the sample with atomic level control. This has enabled the development of Z-contrast imaging in which the intensity of the formed image is directly related to the atomic number Z. As a result, elementally sensitive imaging can be performed with atomic-level resolution. Pennycook and colleagues demonstrated 0.6 Å resolution using Z-contrast STEM in imaging columns of silicon atoms (Figure 3.8).102

FIGURE 3.8
(A) Annular dark-field images of a silicon crystal using Z-contrast STEM. The image has been low-pass filtered to reduce the noise, and the small effects of image drift during the scan have been unwarped. This direct, subangstrom-resolution image shows (more...)
Further chemical information can be extracted by the use of an energy analyzer in the STEM. Electron energy loss spectroscopy (EELS) can be performed in conjunction with STEM to provide chemical bonding information. This has been extremely useful for investigating the chemical nature of defect structures such as grain boundaries in ceramics. Klie and Browning have examined the atomic structure, composition, and bonding of grain boundaries in strontium titanate (SrTiO3) and found segregation of oxygen vacancies to the grain boundary that is increased at elevated temperatures and is independent of the cation arrangement.103 These measurements provide direct support for recent experimental and theoretical predictions that nonstoichiometry, and in particular oxygen vacancies, can be responsible for widely observed grain boundary properties.104
In Situ Imaging: Solid-Liquid Interfaces and Epitaxial Growth
The chemistry of solid-liquid interfaces lies at the heart of numerous fields of chemistry, chemical engineering, and biochemistry. The ability to image at this interface has proven a tremendous technical challenge. Despite the apparent incompatibility of electrons with liquids, new advances in TEM have been developed that permit direct imaging of a reacting solid-liquid interface, namely, electrochemical deposition. The chemistry of electrodeposition is vital to a variety of technologies ranging from coatings to microelectronics, and high-resolution imaging of this process would greatly improve our understanding of this chemistry. An important technical challenge has been the development of a working electrochemical cell within the vacuum environment of a TEM that would be thin enough for electron transmission. Ross and colleagues have made progress in this area by manufacturing a micron-scale electrochemical cell that allows them to image the electrodeposition of copper in situ at video rates using TEM.105 The formation, growth, and dissolution of individual, nanometer-scaled clusters were imaged quantitatively, allowing an analysis of the nucleation and growth process for copper plating.
While high-energy, penetrating TEM has proven useful for the in situ imaging of solid-liquid interfaces, lower-energy implementations of electron microscopy are ideal for imaging processes such as thin-film epitaxy. Low-energy electron microscopy (LEEM) is a relatively new technique that takes advantage of the surface sensitivity of electrons in the 0 to 100 eV energy range. Electrons in this range penetrate only a few atomic layers into a specimen before being reflected. The principle of LEEM is directly analogous to that of traditional light microscopy in the sense that low-energy electrons are beamed at the specimen and reflected back, and by using a series of lenses, a real space image is formed. A key aspect of this technique is that video-rate images can be obtained of processes that can occur over a wide range of parameters. Sample temperatures can be varied from that of liquid nitrogen to 1700 K. Exposure to reactive gases and deposition sources can be performed simultaneously with the imaging. Finally, LEEM can be coupled with a photon source (e.g., a synchrotron) for the generation of photoemitted electron microscopy (PEEM), allowing for chemically resolved imaging. An example of the in situ, time-resolved capabilities of LEEM is the investigation of the alloy formation between tin and copper. Schmid and colleagues imaged the incorporation of tin into a copper surface, tracking the nanoscale motion of tin clusters throughout the process. 106
Future Opportunities. Major effort must be brought to bear to improve electron optics, detectors, stage design, and computing power. The Office of Science in the Department of Energy is sponsoring an initiative to develop these advances and create the next generation of electron microscopes that will feature substantial advances in spatial, temporal, and spectral resolution concomitantly with higher brightness and sensitivity, providing unprecedented opportunities for atomic-level characterization of materials. The core of this effort will focus on overcoming the limitations currently imposed by aberrations in the electron lenses in microscopes. New technology will be developed for aberration correction that will lead to greatly improved electron beam characteristics. The benefit to the scientific community will be not only higher spatial resolution but also the ability to greatly expand the in situ environments for electron microscopy that are vital for imaging chemistry. Aberration correction will enable higher energy resolution for spectral and chemical sensitivity; faster analytical mapping; and extension of EM techniques to chemical systems in which, previously, enough signal could not be obtained before radiation damage made the measurement irrelevant. Existing or anticipated instrumental improvements associated with aberration-compensated optics will:
- enhance cryo-tomographic studies of proteins;
- provide chemical analysis based on bonding (through valence and core-level EELS) and elemental analysis (through EELS and energy dispersive X-ray (EDX)) at very high spatial resolution (in favorable cases, atom-by-atom);
- further facilitate application of EM techniques to fully hydrated systems (e.g. environmental-EM and “wet-SEM”), thereby overcoming a major limitation of traditional, vacuum based EM.
With these new capabilities, direct imaging of chemical processes and reactions that to date have only been hypothesized will be possible. Recently, A. H. Zewail and coworkers107 reported preliminary results on the development of 4D ultrafast electron microscopy (UEM). Providing the spatial resolution of TEM, UEM provides the ability to nondestructively image complex structures utilizing femtosecond pulses. Using UEM, it was possible to obtain images of single crystals of gold, amorphous carbon, and polycrystalline aluminum, and cells of rat intestines.
X-ray Spectroscopy and Imaging
X-rays are short-wavelength, high-energy photons. As a result, they can penetrate materials much more deeply than either visible light or electrons, producing chemical images that cannot be obtained by any other means. Chemical imaging using X-rays plays a significant role in science, health, technology development, and national security. Using X-rays, it is possible to chemically image objects as large as a shipping container or significantly smaller than the nucleus of a single cell. When X-rays interact with an atom or molecule, a variety of signals can result, depending on the type of atom and its chemical environment. Taking advantage of this phenomenon and allowing analysis of samples with wide variations in size and character have required the development of a number of different X-ray imaging techniques. These techniques can be organized into three broad categories: spectroscopy, direct imaging, and scattering. Synchrotrons produce X-rays that span a broad spectrum. For clarity, these three imaging categories have been subdivided according to the X-ray wavelength used in the experiment. Relatively long-wavelength X-rays (around 1 to 15 nm) are referred to as “soft X-rays.” X-rays with wavelengths shorter than 1 nm are classified as “hard X-rays,” based on the fact they are much higher in energy than soft X-rays.
Spectroscopy
By examining the energies at which X-rays are absorbed or emitted, it is possible to determine the identity and local configuration of all the atoms present in a sample. These techniques are analogous to identifying the elements involved in a combustion reaction based on the color of the flame. A single X-ray spectrum can unambiguously identify the composition and chemistry of all elements contained in a sample. The spectra also contain finer details, which can reveal chemical and magnetic information specific to the sample being studied. It is the combination of unambiguous determination of the chemistry of all elements present in the sample with subtle details of sample-specific chemical bonding that gives X-ray methods much of their utility. The spectral properties of sample constituents also form the physical basis for the contrast mechanism in direct image formation. Combining this X-ray spectral fingerprint with direct X-ray imaging is termed spectromicroscopy. This measures the local atomic-scale details of the structural and electronic environment of a chosen atomic species. Spectromicroscopy allows the fine-grained mapping of this local structure for all types of atoms throughout the entire sample volume. This provides a type of complete picture that is vital to all high-technology research and development.
Soft X-ray Spectroscopy. The unifying feature of soft X-ray spectroscopy is that some property of a sample is measured as a function of photon energy, measured either at a fixed energy or over a range of energies. Since it is possible to measure a wide range of sample-specific phenomena, many types of soft X-ray spectroscopies have been developed, including soft X-ray absorption spectroscopy (XAS), near-edge X-ray absorption fine structure (NEXAFS) spectroscopy, soft X-ray emission spectroscopy (SXES), resonant inelastic X-ray scattering (RIXS), X-ray magnetic circular dichroism (XMCD), X-ray photoemission spectroscopy (XPS), and Auger spectroscopy.
At the most basic level, the absorption, transmission, or reflectivity of a sample is measured as a function of incident photon energy. XAS and NEXAFS are methods in which the photon energy is scanned during the measurements. NEXAFS contrast soft X-ray spectromicroscopy is much better than elemental or density-based chemical imaging—it easily rivals vibrational spectroscopies with regard to functional group sensitivity. At the most sophisticated level, a “double” spectroscopy measurement can be performed. In the case of photon-in,electron-out, one measures the energy spectrum of photoemitted electrons (XPS) as a function of X-ray excitation energy. In the case of photon-in, photon-out, one measures the spectrum of fluorescent or inelastically scattered photons (SXES, RIXS) and does this for the range of energies of the incident photon. Often, the photon energy in XPS and SXES is chosen specifically to enhance certain absorption cross sections, but it remains fixed during the measurement. Another dimension to the technique is polarization. Certain chiral and magnetic systems respond differently to elliptical polarization produced by special synchrotron beamline insertion devices.
Soft X-ray spectroscopies employ the excitation of electrons in relatively shallow core levels (100-2000 eV) to probe the electronic structure of various kinds of matter. These techniques have been applied to imaging a number of different chemical systems, including strongly correlated materials, magnetic materials, environmental science systems, wet samples at ambient pressure, and catalysis. This type of spectroscopy relies heavily on elemental specificity. Atoms or ions of each element have their own unique set of core-level transitions that occur at characteristic energies. Through the chemical shift in both the core-level energies and the orbital energies of excited electrons, the method is sensitive to the chemical environment, such as the functional group, oxidation state, local bonding geometry, and so forth. In this sense, soft X-ray spectroscopy becomes atom specific, not just element specific. The photon energy tunability of synchrotron radiation is essential for such experiments, due to the extremely small cross sections of many atoms. Therefore, photon-in, photon-out techniques (SXES and RIXS) are viable only at bright synchrotron sources.
Hard X-ray Spectroscopy. Hard X-ray spectroscopy has been applied in a wide variety of scientific disciplines (physics, chemistry, life sciences, and geology) to investigate and image geometric and electronic structures. The method is element, oxidation, state, and symmetry specific. The technique has been particularly useful in the characterization of new materials. It has also been used in the elucidation of chemical speciation in dilute samples of environmental concern.
In the simplest experimental setup, the sample is mounted between two detectors, one that measures the incident radiation and another that measures the transmitted radiation. The ratio of incident and transmitted signals is monitored as the photon energy is swept through element-specific core-level values, or photon absorption edges. There are two main variants of the technique depending on the range of the photon energy sweep.
- EXAFS. A wide sweep of the photon energy above a core-level edge displays small oscillations in the absorption from which it is possible to deduce nearest-neighbor distances and nearest-neighbor numbers. The photoelectron wave released in the absorption process bounces back to the atom of origin, not unlike the “ping” from submarine sonar. In this way, EXAFS probes the local structural and electronic environment
- NEXAFS. A narrow sweep just above the core-level edge displays characteristic peaks in the spectrum that can serve as a “fingerprint” of the chemical bonding around the atom of origin.
The tunability of synchrotron radiation is essential for the sweeps across the core-level edges. The intensity of synchrotron radiation is essential for the detection of dilute species.
Direct Imaging
Direct imaging using X-rays is achieved by one of two basic experimental methods: full-field imaging or scanning. In full-field imaging, the entire sample is imaged in a single “shot.” This is analogous to taking a photograph, where images are recorded on a highly pixilated X-ray-sensitive camera. With scanning imaging, a very small spot of focused X-rays is moved, or “rastered,” across the sample. Images are recorded by a single-pixel detector one point at a time. These “single-point images” are then stitched together to form the full image of the sample. In either method, there has to be a physical means by which contrast is generated. This can be achieved by taking advantage of differences in absorption, refraction, composition, or the spectral properties of the chemical microenvironments being imaged. Either imaging method can, in principle, be combined with the absorption spectroscopies discussed in this section.
Soft X-ray Imaging. The nature of their interaction with matter makes soft X-rays ideal for imaging the interior structure of inorganic nanoscopic systems and biological cells. Consequently, soft X-ray microscopy has been most widely applied to chemical imaging in the fields of cell biology, environmental science, soft matter and polymers, and nanomagnetism.
Synchrotron-generated X-rays cover the entire spectral range, which allows the collection of imaging data at specific X-ray wavelengths. By a judicious choice of wavelength, it is possible to generate contrast mechanisms that differentiate between various chemical and biochemical environments. For example, in biological imaging, data are typically collected in the “water window” (300-500 eV). In this energy range, atoms such as carbon and nitrogen absorb strongly whereas water molecules are mostly transparent. This difference in absorption produces images that show striking contrast among biological structures, cellular solutions, and electron-dense markers. Similarly, in the case of nanomagnetism studies, contrast mechanisms can be generated by collecting data using magnetically sensitive, circularly polarized X-rays at the absorption wavelengths of iron, cobalt, and nickel (600-900 eV).
In recent years, a wide range of soft X-ray microscopies has been developed. These include photoelectron emission microscopy, scanning transmission X-ray microscopy (STXM), and X-ray tomography microscopy (XTM). With PEEM, a small region of the sample is illuminated and the emitted photoelectrons are passed through an EM column to produce an image. With STXM the photon energy is chosen to correspond exactly to excitations at a particular X-ray absorption resonance, leading to images with unique chemical sensitivity. Different carbon functional groups can also be detected and imaged. Vertical and horizontal polarization can be used to further enhance the image. X-ray tomography is performed by taking multiple full-field absorption images of the specimen while tilting it through 180 degrees. The three-dimensional image is then reconstructed using computer algorithms to yield quantitative information about the specimen (Figure 3.9). The resolution of STXM and TXM is on the order of 15-50 nm; that of PEEM is approximately 50 nm. Diffraction imaging microscopy (DIM) and holography are lensless imaging techniques. The future application of these techniques requires the development of ultrabright, ultracoherent sources such as the linear coherent light source (LCLS).
FIGURE 3.9
X-ray tomography produces images of the internal structure of cells that cannot be obtained by any other means. These images are of yeast cells, volume rendered and color coded according to the absorption of X-rays by various molecular environments in (more...)
Hard X-ray Imaging. Similar to the way medical radiographs can reveal a broken bone, chemical imaging with hard X-rays is used to examine internal or hidden components in thick, dense samples. This technique has been applied in virtually every field from life sciences to engineering to archaeology. A few representative uses are:
- two-dimensional mapping of magnetic domains;
- three-dimensional mapping of composite materials;
- determining the properties of individual grains in a polycrystalline material;
- mapping the distribution of elements in cells;
- identifying strains in near-perfect crystals;
- time-resolved imaging of sprays;
- investigations of human and animal physiology.
Since they have highest photon energy, hard X-rays also have the greatest depth penetration. This makes their use essential when imaging the chemistry of buried interfaces, probing the internal microstructure of objects such as bones, or imaging large biological specimens.
X-ray Scattering
Soft X-Ray Scattering. Soft X-ray scattering is a photon-in, photon-out technique. The sample is illuminated with monochromatic soft X-rays, and the scattered photons are detected over a small angular range, in either the elastic or the inelastic mode. In the former, the speckle diffraction pattern is measured, and for the latter, scattered photons are passed through a spectrometer and analyzed. The energy spectrum is essentially a replica of the occupied density of states. Additional information is obtained in the resonant condition when the incident photon is near a core-level absorption edge. Soft X-ray scattering techniques employ the excitation of electrons in relatively shallow core levels (100-2000 eV) to probe the electronic structure and other properties of various kinds of matter. The types of chemistries imaged by soft X-ray spectroscopic techniques include strongly correlated materials, magnetic materials, environmental science, wet samples at ambient pressure, and catalysis.
Every element has its own set of core levels with characteristic energies, a phenomenon that confers on these techniques a high degree of elemental specificity. Consequently, taking advantage of this can be done only by using synchrotron radiation. In addition, since many of the elements being imaged have very low absorption cross sections, many of these scattering techniques can be performed only when using the brilliant light produced at a modern, third-generation synchrotron.
Hard X-ray Scattering. When X-rays interact with matter, they are scattered. This scattering can occur in two modes, elastic or inelastic. In elastic scattering, the energy (wavelength) of the detected X-ray is the same as that of the incident X-ray. With inelastic scattering, however, some energy is lost to other processes, so the energy of the detected X-ray is lower than that of the incident X-ray. This difference in energy is due to vibrational, electronic, or magnetic excitation. The detection system in such cases measures the energy loss.
A large number of variants of hard X-ray scattering have been developed. These include small-angle X-ray scattering (SAXS), wide-angle X-ray scattering (WAXS), grazing incidence small-angle X-ray scattering (GISAXS), X-ray Raman scattering, Compton scattering, inelastic X-ray scattering (IXS), resonant inelastic X-ray scattering, nuclear resonant scattering (NRS), and X-ray photon correlation spectroscopy (XPCS).
Hard X-ray scattering techniques have been applied to the chemical imaging of an enormous range of systems, primarily systems that are not perfectly ordered or static. Problems addressed include:
- short-range order in amorphous materials;
- liquid-vapor, liquid-liquid, and molecular film interfaces;
- colloids, solution-phase proteins, polymers;
- collective dynamics in soft materials;
- phonons and elementary excitations in solids.
Hard X-rays have wavelengths comparable to the interatomic distances. When a crystalline sample is illuminated with X-rays, the X-rays are scattered (diffracted) in very specific directions and with various specific strengths, or intensities. Detectors are used to measure this diffraction pattern, which is then processed to compute the arrangement of atoms within the crystal. There are two principal diffraction modes. In Bragg diffraction, the incident X-rays are monochromatic (single wavelength) and the sample is an oriented single crystal. In Laue diffraction, the incident X-ray beam is white (the entire spectrum of X-ray wavelengths) and the sample can be in the form of a powder or a random ensemble of microcrystals.
Much of the current knowledge regarding the atomic structure of materials has been derived from hard X-ray diffraction. Research problems that this technique can address are
- structural studies of crystalline materials;
- drug design by the pharmaceutical industry;
- biomineralization;
- new microporous materials including natrolites, phosphates, and titanates;
- novel complex oxides: structure-property relationships, phase transitions;
- residual stress determination in situ.
Many materials are impossible to investigate with typical research laboratory X-ray diffraction equipment, especially if the crystals are small and therefore produce diffraction intensities that are too weak to measure. Synchrotron-generated hard X-rays provide significant advantages over such laboratory sources and make these experiments possible. In this regard, one of the major areas of chemical imaging is the field of macromolecular crystallography. In particular, the technique has proven to be an exceptionally powerful tool for imaging the three-dimensional chemical environment in biological molecules and complexes. The technique has also proven amenable to applications such as drug design and ligand-binding studies, where large numbers of distinct chemical interactions must be imaged rapidly.
Macromolecular Crystallography
A major use of macromolecular crystallography is imaging potential new therapeutics in situ at the active site of an enzyme or other biomolecule. This information provides visual indications of how the potential therapeutic can be made more effective and helps cut dramatically the cost of development and the time required to produce a candidate suitable for clinical trials. In the design of a new drug, it is typical to perform a large number of cycles of imaging the drug molecule in the active site and then, based on this information, to synthesize an enhanced variant of the drug. Despite great successes, a major drawback to macromolecular crystallography is that the sample must be crystallized prior to analysis. This can often be difficult, or even impossible, since the crystallization of biomolecules remains an empirical trial-and-error process.
The structural information obtained from a crystallographic analysis has many other uses:
- elucidation of enzyme mechanisms;
- understanding structure-function relationships;
- identifying molecular recognition surfaces and topologies;
- structural genomics and proteomics;
- identification of novel “fold” motifs.
All of these implementations have enormous potential for improving health and contributing to the economy of the nation.
Summary
A plethora of X-ray chemical imaging techniques now exists. These can accommodate almost every conceivable sample type, in terms of both physical size and composition, and give unique insights into the deep internal molecular and atomic structure of most materials. On first inspection, these techniques appear complex and esoteric. The advanced capabilities of the current generation of synchrotron light sources, however, make these techniques much more accessible. Indeed, in many cases these experiments are now relatively simple and produce easily interpretable results. For example, macromolecular crystallography is now highly automated, in terms of both sample handling and postexperiment computation. At a synchrotron facility, it is possible to collect and determine the structure of a macromolecule in less than 30 minutes. This model for high-throughput imaging is being implemented on other types of experiments. The latest X-ray microscopes, especially those aimed at generating tomographic reconstructions, have been designed to function with similar speed and ease of use. Continued development of new synchrotron X-ray sources (particularly those providing soft X-rays) together with the development of plasma-based X-ray sources for use in individual research group laboratories are integral component of chemical imaging. In both soft X-ray spectromicroscopy and lensless imaging, the third-generation light sources in the United States are clearly the world leaders. Further investment could have major differential impact. The key to these areas (as in so much of imaging) is brightness. Investments in third- and fourth-generation light sources, as well as lab-based X-ray lasers and laser-excited plasmas, are likely to accelerate a chemical imaging revolution in this area. X-ray free-electron lasers have the potential to revolutionize the way we study matter at the atomic and molecular levels, allowing atomic resolution snapshots on the ultrafast time scale associated with the intrinsic motions of atoms in matter. A significant part of this task will be the development of new, enhanced X-ray optical systems such as zone plates to permit imaging at higher resolution, and the development of detectors capable of functioning on the femtosecond time scale. Recent results show that lensless imaging is capable of visualizing simple materials in isolation at approximately 10 nm resolution. This technique is applicable to specific sample geometries, (i.e., compact support).
Molecular Imaging by Mass Spectrometry
The two implementations of mass spectroscopy discussed in this section are secondary ion mass spectrometry (SIMS) and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. These techniques produce images by ionizing from a clearly identified point on a flat sample and by moving the point of ionization over the sample surface. SIMS provides information on the spatial distribution of elements and low-molecular-weight compounds as well as molecular structures of these compounds. MALDI yields spatial information about higher-molecular-weight compounds such as peptides and proteins, including their distributions in tissues at very low levels, as well as information about their molecular structures. Application of these methods to analytical problems requires appropriate instrumentation, sample preparation methodology, and data presentation usually in a three-coordinate plot where x and y are physical dimensions of the sample and z is signal amplitude. The unique analytical capabilities of mass spectrometry for mapping material and biological samples are described below.
SIMS Imaging
SIMS was the first mass spectrometry technique used to generate two-dimensional ion density maps or images from a variety of solid materials and thin sections of biological tissues.108 SIMS involves the bombardment of a sample with a pulsed beam of ions (typically Cs+) having energies in the low kiloelectronvolt range (10 to 15 keV). The impact of these energetic ions on the surface of the sample initiates desorption o rionization processes. The bombarding beam can be tightly focused on the surface of the sample in an area smaller than 1 μm2. From a raster of the surface of the sample, ion density maps or images at specific mass-to-charge (m/z) values are generated. A wide range of various, primary ion beams are available such as Cs+, Ga+, Ar+, Xe+, and In+. Cluster ion beams are also being actively developed including Aunn+, O2+, SF5+, Cn+, and C60+. Cluster ion beams tend to be more efficient for desorption and ionization of higher-molecular-weight organic compounds. The primary ion energy is transferred to target atoms via atomic collisions, and a so-called collision cascade is generated. Part of the energy is transported back to the surface, allowing surface atoms and molecules to overcome the surface binding energy. The interaction of the collision cascade with surface molecules is soft enough to allow even large and nonvolatile molecules with masses up to 10,000 atomic mass units (amu) to escape with little or no fragmentation. Most of the emitted particles are neutral, but a small proportion of these are also positively or negatively charged. Subsequent mass analysis of the emitted ions provides detailed information on the elemental and molecular composition of the surface.
SIMS is a very surface-sensitive technique because the emitted particles originate from the uppermost one or two monolayers. The dimensions of the collision cascade are rather small and the particles are emitted within an area of a few nanometers' diameter. Hence, SIMS can be used for microanalysis with very high lateral resolution (50 nm to 1 μm), provided such finely focused primary ion beams can be formed. Furthermore, SIMS is destructive in nature because particles are removed from the surface. This can be used to erode the solid in a controlled manner to obtain information on the in-depth distribution of elements.109 This dynamic SIMS mode is widely applied to analyze thin films, layer structures, and dopant profiles. To receive chemical information on the original undamaged surface, the primary ion dose density must be kept low enough (<1013 cm−2) to prevent a surface area from being hit more than once. This so-called static SIMS mode is used widely for the characterization of molecular surfaces (see Figure 3.10).

FIGURE 3.10
(a) Surface spectroscopy: The aim of a static SIMS investigation is analysis of the original, nonmodified surface composition. Because SIMS in principle is a destructive technique, the contribution of secondary ions to the spectrum originating from already-bombarded (more...)
The following example of imaging with SIMS illustrates the effectiveness of using “high-mass, modest-spatial-resolution” imaging. In the analysis of photochemically produced defects on a chrome surface (Figure 3.11), imaging was performed in the focused-bunched mode of a gallium ion gun. The defects are approximately 3-5 μm in size but are easily resolved spatially with high-mass-resolution detection. The signal-to-noise ratio and acquisition time are much more efficient with this mode than with the burst mode of the gallium gun. The field of view in the images is 50.8 × 50.8 μm.

FIGURE 3.11
SIMS imaging of photochemically produced defects on a chrome surface. (a, b) High-mass-resolution spectra of defects extracted from the raw data stream of images for the C3H4N3O3+ ion (m/z = 130.03). (c-f) Ion images for C3H4N3O3+, Cr+, NH4+, and Si+ (more...)
Recent developments in sample preparation for SIMS technology have incorporated the principle of using a matrix to enhance secondary ion yield.110 In matrix-enhanced (ME) SIMS, the same matrices routinely in MALDI mass spectrometry (MS) are used for imaging compounds from biological tissue sections.111 With ME SIMS, peptide ion signals can be detected and imaged in a molecular weight range up to 2500 daltons (Da) with a lateral resolution better than 3 μm. This molecular weight range is complementary to those achievable with cluster SIMS and MALDI MS.
Imaging with Laser Microprobes and MALDI Mass Spectrometry
Lasers have been used as microprobes for several decades to investigate both organic and inorganic particles.112 More recently, laser desorption coming directly off porous silicon surfaces has been demonstrated for low-molecular-weight organic molecules, such as drugs and pharmaceutical compounds, as well as for small peptides.113 Biological samples have also been investigated. Imaging tissue sections with laser microprobes has been demonstrated in mapping the distribution of drugs in the mouse brain, dyes from strained eye lens tissue sections, and cations in pine tree root sections.
MALDI time-of-flight (TOF) MS was introduced in the late 1980s. This technique employs the co-crystallizing of matrix (low-molecular-weight organic crystalline compound) and analyte on a target plate. Irradiation of these crystals by short (nanosecond time scale) pulses of UV or IR light initiates desorption and ionization, where predominantly singly protonated intact molecular ions ([M + H]+) are produced. Since ionization is a pulsed process, it is easily compatible with a TOF mass analyzer. MALDI MS is an extremely sensitive tool permitting the detection of sample molecules below the femtomole level with mass accuracies better than 10−4 (in a mass range up to about 30 kDa). Over the past decade, many improvements to MALDI MS instrumentation have been made, and this technology is now accepted as one of the major analytical tools to detect, identify, and characterize peptides and proteins as well as many other polymers of biological interest.114
One of the newest developments in applications of MALDI TOF MS is its use in profiling and imaging peptides and proteins directly from surfaces such as thin-layer chromatograms and thin tissue sections in order to obtain specific information on the local molecular composition, relative abundance, and spatial distribution.115 Results from such tissue imaging experiments yield a great wealth of information, allowing investigators to measure and compare many of the major molecular components of the section in order to gain a deeper understanding of the biomolecular processes involved. In tissue profiling experiments, one is interested in a discrete number of spots or areas in terms of comparing protein patterns. To accomplish this, matrix is homogeneously deposited on a tissue section and analyzed using an ionizing laser that is rastered over the surface of the sample according to a predetermined grid pattern of fixed dimension (Figure 3.12). The distance between two adjacent grid coordinates defines the imaging resolution. With MALDI MS, it is possible to obtain images with resolution as low as 25 μm. At each grid coordinate a full mass spectrum is recorded. Peptide and protein ion images are reconstructed by integrating the signal intensities at chosen mass values.

FIGURE 3.12
Principle of MALDI-based imaging mass spectrometry. Frozen sections can be mounted on a metal plate, coated with an UV-absorbing matrix, and placed in the mass spectrometer. A pulsed UV laser desorbs and ionizes analytes from the tissue, and their m/z (more...)
Beyond peptides and proteins, MALDI MS imaging of tissue section for the detection of low-molecular-weight compounds can also be achieved. Of particular interest is the posttreatment location of pharmaceutical compounds in targeted tissues or organs. Further, in parallel to location, the effects of a drug on the local proteome can be observed as a function of dose or time. Variations in the proteome are indicative of drug efficacy.116
Future Developments and Perspectives
Imaging by MS is currently being developed actively from both a SIMS and a MALDI perspective. Instrumentation is being upgraded constantly to perform imaging faster and at higher resolution. In SIMS MS, the development of cluster primary ion beams such as Aun+, Bin+, and C60+ now allows the analysis of a wider range of organic molecules. ME SIMS allows us to expand the molecular weight range investigated and is valuable for the analysis of low-molecular-weight peptides. In parallel, ion optics allowing the better focusing of these beams are being developed. In the case of laser microprobes, the near future developments of fast lasers (with repetition rates in the kilohertz regime), improved electronics, and acquisition systems will allow significantly reduced image acquisition times (from hours to minutes) even at high resolution.
Efforts to obtain smaller laser beam size are progressing to allow imaging at higher resolution. Very accurate sample stage control systems will have to be added to the mass spectrometer ion sources. Routine imaging with laser microprobes at resolutions better than 1 micron is foreseeable within the next five years. For MALDI MS and ME SIMS, improved protocols for homogeneous matrix deposition on biological samples with the formation of micron-size crystals have to be developed. Matrix-free desorption systems allowing the study of peptides and proteins, as well as other classes of biomolecules under vacuum or at atmospheric pressure, are also needed. One such approach is already being pursued that utilizes an electrospray source as a desorption-ionization probe.117 The rapid progression of computer technologies and informatics allows for fast data and image processing. However, with increasing acquisition and imaging resolutions, data size and volume are expected to increase significantly; thus, processing capabilities will have to keep pace with these increases. Furthermore, improved software will be needed to process and correlate images across experiments and time points. Ultimately, informatics tools will have to be developed to visualize molecular images obtained by multiple imaging technologies from the same samples.
Imaging MS is and will become increasingly critical for many aspects of materials science. One example is in the semiconductor industry, where the ability to provide spatial and chemical information on the length scales of current integrated circuit fabrication (50 nm or better) with depth profiling to provide layer-by-layer maps of the fabricated layers is critical for the continued advancement of the computer industry. Maps of any heterogeneous surface are important in other areas of materials science. For example, using various laser desorption techniques, information about the molecules found in specific inclusions in meteorites or defects in reactive surfaces can be obtained.
Molecular imaging of biological samples by MS is also foreseen to play a pivotal role in understanding numerous biological processes in fields ranging from neuroscience to cancer research. The fundamental contributions of the technology in providing molecular-weight-specific images rapidly, at relatively high resolution and sensitivity, will yield important information in the investigation of cellular processes in both health and disease. While the imaging technology can rapidly distinguish protein markers of interest, their identification is still a slow and labor-intensive process. Progress in the fields of protein identification and characterization by MS will allow a more rapid throughput. Imaging MS is of extraordinary benefit as a discovery tool because one does not need to know in advance the specific proteins that may have changed in a comparative study. For example, comparisons of protein profiles and images between tissues allow researchers to highlight protein markers indicative of the health or disease status of an individual.118 Furthermore, the cellular origins and relative concentrations of markers across the section can be assessed, helping us understand the progression of a disease at the molecular level (e.g., cancer). Clinically, imaging mass spectrometry can provide molecular assessment of tumor staging and progression in biopsies, with the potential to identify subpopulations that are not evident based on the cellular phenotype determined histologically.119 Also, assessment of the efficacy of drug treatment through comparative proteomics is feasible. Perhaps MS's greatest value lies in the fact that it significantly augments, but does not replace, existing molecular tools. Together, these tools promise to promote new discoveries in biology and medicine.
PROXIMAL PROBES
Since the advent of the scanning tunneling microscope in the early 1980s,120,121 a wide variety of related microscopies using similar experimental principles and instrumentation have been developed for imaging samples based on their electronic, optical, chemical, mechanical, and magnetic properties. Today, scanning tunneling microscopy (STM)121,122 atomic force microscopy (SFM),122 near-field scanning optical microscopy (NSOM),123 and scanning electrochemical microscopy (SECM)124,125 all find broad applications in high-resolution chemical imaging experiments. Because of the similarities in the instrumentation employed in each form of microscopy, these different techniques are grouped together in this report under the general class of “proximal probe microscopies.”
Imaging Modalities
By definition, proximal probe microscopes employ a small probe that is positioned very close to the sample of interest for the purposes of recording an image of the sample, performing spectroscopics experiments, or manipulating the sample. All such methods were originally developed primarily for the purposes of obtaining the highest possible spatial resolution in imaging experiments. Since then, many other unique advantages of these techniques have been realized. Proximal probe methods derive their unique capabilities specifically from the close proximity of probe and sample.
In STM, image contrast is derived from spatial variations in current flowing between the proximal probe and the sample.126 Tunneling in an STM relies on the spatial overlap of the tip and sample electronic orbitals. Therefore, the tunneling current falls off very rapidly (on atomic length scales) as a function of distance between the tip and a particular sample feature such as an isolated atom. Tunneling current variations and information on surface chemistry are specifically derived from the associated atomic-scale variations in the density of states near the sample surface.
AFM methods derive image contrast from magnetic, electrostatic, dipolar, dispersion, and quantum mechanical (i.e., as in covalent and hydrogen bonding) interactions between the tip and sample.127 Although magnetic, electrostatic, and dipolar interactions between the tip and sample decay over relatively long distance scales (i.e., micrometers), other forces decay over shorter angstrom ranges. Because of the short interaction lengths of the latter forces, they can be used to obtain high-resolution images of a sample surface or to make measurements of surfaces forces on isolated molecules or surface regions. AFM and variants thereof are perhaps the most widely utilized forms of scanning probe microscopy in existence today. As judged by the number of times the first article describing AFM has been cited (ranking fourth out of the top 10 published articles in Physical Review Letters),128 development of AFM represents one of the most important advances in the recent history of science.
NFOM methods (optical proximal probe methods) provide subwavelength-resolved images via the use of spatially confined evanescent fields.129,130 These decay over length scales determined by the size of the proximal probe. High-resolution information is obtained only when subwavelength-sized probes are employed and maintained at subwavelength distances from the sample. In this near-field regime, optical fields from the proximal probe have not yet had a chance to spread through diffraction.
Scanning electrochemical microscopy relies on similar principles to obtain high resolution. In this case, however, image contrast is usually obtained via the recording of faradaic currents associated with probe and/or surface electrochemical reactions.131 The probe must be kept small and positioned in close proximity to the sample surface to limit degradation of image resolution by diffusion of the electrochemically active species involved in generating image contrast.
In all proximal probe imaging experiments, the probe and sample are moved laterally relative to each other, and the desired signal is recorded as a function of position. In all such instruments, the resulting signal is fed into a servo (or feedback) loop for regulation of the probe-sample separation during both imaging and single-point measurements.132
Scanning Tunneling Microscopy and Spectroscopy
Spatial Resolution
Scanning tunneling microscopes routinely yield atomic resolution because of the extremely steep gradient of current as a function of tip-sample separation. Changes in tunneling current are readily measurable for STM probe tip motions of <0.01 Å for the most stable of such microscopes; this resolution is the result of sensitivity to the tip-sample separation. As noted above, the geometric and electronic structures are convoluted, so that rather than images of atoms, electrons distant from the nuclei (on a chemical scale) are measured with exquisite spatial resolution and significant energy resolution as well. Detailed comparison to theory is able to explain this and other features, but a general method of predicting STM images a priori remains to be developed.
STM probes (e.g., from W or Pt-It wire) are fabricated by either mechanical cutting or electrochemical etching. Further treatments are sometimes used to sharpen them, such as annealing under high fields 133,134 in techniques handed down from the original atomic resolution microscopies—field emission microscopy and field ion microscopy.135,136 Another method employed is to lift an atom or molecule onto the probe tip so as to define the tip precisely. One important issue that can prevent clear interpretation of STM images but can also be used to tremendous advantage is the fact that the atom at the very apex of the STM probe (or rather its electronic orbitals) plays a crucial role in governing tunneling and the appearance of the sample image.137 Figure 3.13 shows an example of this phenomenon in which Ni-O rows are imaged alternately with a clean tungsten tip and again after an oxygen atom was adsorbed to the tip apex.

FIGURE 3.13
Ni-O rows on a Ni (1 × 1) surface imaged with a clean tungsten tip (left) and the same region after adsorption of an oxygen atom at the tip apex (right). SOURCE: Ruan, L., F. Besenbacher, I. Stensgaard, and E. Laegsgaard. 1993. Atom resolved discrimination (more...)
Some of the most dramatic STM images have been recorded for the Si(111) 7 × 7 reconstruction, as depicted in Figure 3.14.138 These images have been recorded at several different biases (see “Spectroscopy and Chemical Selectivity,” below) and provide one of the best examples of how STM can be used to better understand the chemistry of such surfaces (specifically, the electrophilicity and nucleophilicity of the individual surface atoms). Clearly depicted in these results are the orbitals associated with surface atoms, rest atoms, and backbonds. Such studies have continued and been greatly extended into the exploration of a variety of chemical reactions that occur on silicon surfaces. These studies are described in detail in a recent review.135

FIGURE 3.14
Using STM to understand the chemistry of surfaces. STM images of the Si(111) 7 × 7 surface reconstruction showing the spatial location of different electronic orbitals associated with different surface and near-surface atoms. The images depict (more...)
Complete monolayers adsorbed on surfaces can also be observed by STM.139 The separation of monolayer components in self-assembled monolayers on gold was unknown and unexpected before multicomponent films were examined using STM.140 Having the ability to resolve components with molecular resolution,107 this field has since advanced rapidly to exploit intermolecular interactions to produce desired patterns.141 Such advances in other important materials could easily be driven by this ability to observe their structures and functions with atomic resolution. Other environments—liquid, gas, elevated and reduced temperature—are all accessible, and important chemistry and materials problems await the requisite tools.
Dynamics and Time-Resolved Imaging
Not long after the invention of scanning probe microscopes, “noise” in images was attributed to the motion of atoms and molecules at rates higher than the rather slow scanning speed. The highest imaging rates are now >100 Hz,142,143 and while some advances are possible here, much information in images is not relevant to dynamics. By focusing on the dynamic property measured, such as the location of an individual adsorbate, larger dynamic ranges can be reached.144,145 Likewise, remaining in one position and using the STM to measure other properties also can overcome the limits imposed by mechanical scanning.
Another approach is to use pulsed or frequency-based measurements.146 Such methods allow time resolution in the nanosecond to femtosecond regime to be achieved. This area has just begun to be explored, and as spectroscopies are further combined with scanning probes, dynamics will be made accessible by “focusing” on the relevant information.
Spectroscopy and Chemical Selectivity
Many interesting materials systems are chemically heterogeneous on a wide range of length scales down to atomic dimensions. The development of chemically selective proximal probe imaging methods has played a central role in uncovering sample heterogeneity and understanding its origins. However, numerous chemically selective, spectroscopic proximal probe methods continue to emerge from a number of labs around the world. Both the evolution of existing methods and the further development of new ones promise significant advances in our ability to obtain chemical information on heterogeneous samples on a variety of relevant length scales.
Scanning tunneling spectroscopy provides detailed information on the local electronic properties of conducting and semiconducting surfaces. The earliest tunneling spectroscopy experiments were performed by varying the bias potential applied between the proximal probe and the sample, while recording the tunneling current.147 Such data can be recorded in a single point modality or by modulating the bias during imaging such that multiple images of the same sample region are acquired “simultaneously” at several different bias potentials. Data derived from these experiments allow the energies of both filled and unfilled electronic surface orbitals to be assessed, providing a chemically relevant view of the local surface electronic structure. It is this electronic structure that governs the chemical reactivity of such surfaces.
Feenstra and colleagues showed such chemical information in overlaying images of filled and empty orbitals on arsenic and gallium atoms of the stoichiometric GaAs(110) surface.148 This has direct chemical implications because the electrophilic and nucleophilic centers of molecules are attracted to the filled and empty states, respectively. STM data have elucidated this effect further to show that (and which) enhancement of orbitals at specific energies is relevant to guide binding, structures, dynamics, and chemistry. Thus, STM can probe the surface the way a mobile molecule does by tuning the bias voltage to energies relevant to these interactions.149
As with UV-visible spectroscopy in the bulk, such techniques do not yield chemical identification, so that combining other local spectroscopies with STM is typically necessary to identify the atoms and molecules present. Specialized approaches have been developed for this, such as STM photoemission spectroscopy (PESTM) and inelastic electron tunneling spectroscopy to yield vibrational and other information. This area is extremely promising for further work in combining any number of spectroscopies with the exquisite spatial resolution of STM.
PESTM experiments are based on the fact that the tunneling process can produce electronically excited surface species that subsequently relax by radiative decay. This process is analogous to common bulk electroluminescence experiments and can be used to distinguish between chemically different surface species and/or species present in different environments. Gimzewski and coworkers and Alvarado and coworkers, both of the IBM Research Division, were the first to demonstrate PESTM experiments.150,151
Since its initial demonstration, PESTM has been employed in a broad range of interesting experiments, often performed as a means to better understand the local optical properties of a particular sample. An interesting recent application of PESTM is in the excitation of surface plasmon modes by inelastic tunneling processes118 in nanostructured gold corrals.152 PESTM imaging of the local plasmon excitation efficiency in such structures could provide a means for mapping the properties of these materials in both the presence and the absence of molecular adsorbates. Such studies could provide a means to better understand variations in enhancement factors in surface-enhanced spectroscopies employing similar substrates.153,154
Vibrational spectroscopy provides unique and detailed information on the chemical composition and structure of a sample. Vibrational spectra of surface-adsorbed species can also be obtained in some STM experiments using inelastic electron tunneling spectroscopy (IETS).155 This form of spectroscopy is analogous to the sandwich tunneling junction measurements that have been made over the last 30 years.156 Important, using IETS, this information can be obtained at the single-molecule level, with atomic-scale lateral resolution. IETS experiments are performed by measuring the tunneling conductance as a function of applied bias. When the energy of tunneling electrons matches the energy of a vibrational mode of surface-adsorbed molecules, the tunneling conductance changes as energy is deposited into the vibrations. IETS data are recorded under vacuum conditions at cryogenic temperatures. Spectra recorded by this method can be used to identify chemical species on surfaces and to follow surface chemical reactions. Figure 3.15 show example IETS spectra obtained for acetylene and deuterated acetylene on a copper surface.153

FIGURE 3.15
Spectroscopic imaging of the inelastic tunneling observed for acetylene and deuterated acetylene. (A) Regular constant-current STM image of C2H2 and C2D2 molecules (left and right). (B)-(D) Spectroscopic images recorded at 358 mV (showing C2H2), 266 mV (more...)
Future Challenges and Emerging Methods
Like all scanning probe methods, STM experiments are limited by the rate at which images can be recorded. Again, limitations in the imaging rate arise from mechanical instrument design issues (i.e., resonances of the microscope itself). Further limitations arise from the small scan range usually employed in STM experiments. With relatively low scan rates, it is frequently difficult to image more than a tiny fraction of the actual surface area of a particular sample. Increased scanning rates might possibly be obtained in the future via the development of new feedback and scanning electronics. Improvements in both the imaging rate and the imaging area can be achieved via the implementation of multiprobe microscope designs157,158 (i.e., with which several images can be recorded simultaneously).
Further advances will come by combination of STM with other forms of spectroscopy and scanning probe microscopy. One such emerging method that allows researchers to “see beneath the surface” of samples is ballistic electron emission microscopy (BEEM).159
Atomic Force Microscopy
High resolution in AFM imaging experiments is somewhat more difficult to achieve than in STM experiments. The ultimate resolution achievable in many AFM experiments is often limited by the participation of relatively long-range interactions in governing the forces between the proximal probe and sample.160,161 Such long-range forces might arise from capillary interactions between the probe tip and the contamination layer on a sample surface imaged in the ambient environment. Relatively long-range tip-sample interactions also arise from Coulomb and dipolar interactions between probe and sample species. The shortest-range interactions result from quantum mechanical forces associated with chemical bonding. To achieve the highest possible resolution, these latter short-range forces must obviously dominate over the long-range “background” forces also present.
Along with the dominance of short-range interactions, high-resolution AFM imaging requires the use of the smallest possible probe tip. Atomic resolution imaging by definition requires the use of a probe tip with a single atom present on its apex. Pyramidal probes for AFM experiments are often obtained via a variety of well-controlled, wet-chemical and reactive ion etching procedures.162 As such, fabrication of conventional AFM probes is extremely reproducible. Silicon nitride-based probes having nanometer-scale end diameters for use in contact and intermittent contact AFM are readily obtained from several commercial sources. Under relatively routine imaging circumstances, such probes yield resolutions in the nanometer range for relatively smooth samples.
For higher-resolution imaging and imaging of “porous” materials incorporating deep pores of narrow diameter, sharpened probes prepared by etching procedures and ion beam milling are also available. Carbon nanotube-based AFM probes have recently been developed for high-resolution imaging applications.163 Such proximal probes have tremendous potential for use in biological imaging studies and for general materials imaging applications requiring very high aspect ratio probes.164 In the latter case, the continued development of advanced, high-aspect-ratio probes promises to allow improved imaging of topographically complex surfaces, such as those of certain catalysts, and of porous biological membranes and other porous thin-film materials (i.e., allowing experiments to “see within the pores”).
As in STM, true atomic resolution in AFM has been achieved on well-ordered, atomically smooth samples imaged under high vacuum.165 True atomic resolution has also been demonstrated under liquids on well-ordered mineral surfaces.166 Obtaining such high-resolution images is somewhat challenging even under high-vacuum conditions. Employment of frequency modulation AFM methods and more rigid cantilevers can improve the signal-to-noise ratio and minimize the influence of strong probe-sample interactions.
High-resolution AFM imaging is also being used to obtain a new understanding of biologically important surfaces.167 Recent work includes the study of membrane protein arrays, such as the Aquaporin-Z protein crystals shown in Figure 3.16.168 From repeated imaging of such proteins, an average topographical structure can be derived that can then be used along with image analysis methods to develop a detailed picture of the conformational structure of the protein. Extensive effort is also now being devoted to understanding the processes by which prion proteins aggregate and the morphological structures formed by these aggregates. Associated amyloid fibers play an important role in neurodegenerative diseases such as Alzheimer's and Creutzfeldt-Jakob disease. AFM imaging promises to have important implications for our understanding of how these diseases arise (i.e., how misfolded protein structures are “inherited” by properly folded proteins and how they might be treated).169

FIGURE 3.16
Example of high-resolution AFM imaging of a biological surface. Contact mode AFM image of Aquaporin-Z membrane protein crystals showing their surface structure with <1 nm resolution. Scale bar, 10 nm. SOURCE: Horber, J.K.H., and M.J. Miles. 2003. (more...)
AFM Dynamics and Time Resolution
Of equal importance to achieving high spatial resolution in many proximal probe imaging experiments is the ability to acquire time-resolved images of dynamics associated with surface and thin-film chemical processes. All proximal probe methods described herein rely on the raster scanning of a single probe tip in relation to the sample of interest. The rate at which images can be recorded is the limiting factor determining the time resolution that can be obtained in dynamics experiments. Sample and/or probe raster scanning rates are limited primarily by two factors: (1) the bandwidth of the feedback circuit used for maintaining probe-sample separation and (2) the resonance frequency of the microscope.
The feedback-bandwidth limit arises primarily from the requirement that the probe follow the sample topography. In cases where the sample topography exceeds a few nanometers, imaging rates are limited to approximately one per minute or less, using conventional feedback systems. In intermittent-contact AFM and shear-force surface topographic measurements, the feedback bandwidth is limited by the response of the proximal probe cantilever (AFM) and tuning fork (shear force) to changes in the probe-sample interactions.170 The response time in each case is often limited by the sharpness of the sensor resonance frequency (the “quality factor” or Q).171 Higher Q values give longer response times. Improved feedback bandwidths can therefore be obtained by reducing the Q using electronic means.172
When the only topography is of atomic dimensions, constant height imaging methods can be employed and the feedback response is of little consequence. Under these circumstances, images can be recorded using line rates of approximately one-tenth the microscope resonance frequency, allowing tens of images to be recorded per second (i.e., at or near video rates).173 However, faster imaging rates have also been demonstrated. In these experiments, the fast-scan motions are actually driven by one of the microscope mechanical resonances, allowing the recording of as many as 100 images per second in situations where it is not necessary that the surface topography be followed.174
The ultimate goal in time-resolved proximal probe methods, however, is not always faster image acquisition. Rather, the most useful methods provide a large dynamic range, allowing processes that occur on time scales ranging from seconds to picoseconds and even femtoseconds to be studied. Such issues are best defined in relation to the exact form of probe microscopy employed, as described below.
Chemical Force Microscopy. AFM experiments can be performed using probes that have been derivatized with specific chemical functional groups. These proximal probes allow for the detection and utilization of specific probe-sample interactions as a means of obtaining chemical contrast in AFM images, and this technique is commonly known as chemical force microscopy.175 Chemical force microscopy represents an extrapolation to very short (i.e., nanometer) length scales of well-known surface forces measurements.176 In these studies, probe-sample interactions are frequently detected either by measuring “pull-off” forces (force required to separate the tip and sample after contact) or by detection of frictional forces between the sample and the contacting probe.177
The potential for detection of chemically specific interactions between an AFM probe and a sample surface was realized using underivatized probes very early in the development of AFM, as exemplified by the detection of what appear to be discrete hydrogen bonding interactions between proximal probe and sample surface silanol species in studies by the Hansma group.178 The capability of chemically specific AFM imaging using derivatized probes was clearly demonstrated in a later study by the Lieber group (see Figure 3.17, for example).179

FIGURE 3.17
This image of a patterned self-assembly monolayer surrounded by a CH3-terminated region was obtained using alternately a CH3-terminated probe and a COOH-terminated probe in friction force and intermittent contact imaging studies. (A) Optical image showing (more...)
A number of research groups have since demonstrated chemical force imaging of deliberately patterned surfaces, pointing to the broad applicability of this method to a variety of problems. These methods have been applied in force spectroscopy studies of specific single-molecule interactions (i.e., “molecule-pulling” experiments).180 These experiments are described in the next section. The primary challenge in chemical force imaging has been in extending these procedures to imaging of samples for which the surface chemical composition is not known a priori. Although such studies can be performed on monolayer samples supported on solid substrates,181 further difficulties arise when thicker samples (i.e., phase-separated polymer blends)182 are to be investigated. Under these circumstances, factors such as the mechanical stiffness of the sample can lead to variations in tip-sample interaction area, making it difficult to distinguish variations in probe-sample chemical interactions. Nevertheless, researchers have recently made such measurements on a number of different samples, including oxidized polymeric surfaces.183
Force Spectroscopy. The atomic force microscope is also frequently used to make single-point measurements of specific interaction forces between derivatized AFM probes and sample surfaces. Tip-sample interaction forces as small as piconewtons can be measured readily. As with chemical force microscopy imaging experiments, these studies are often performed under liquids, in inert atmospheres, or in a vacuum to eliminate the strong capillary forces that can dominate tip-sample interactions.184 Force spectroscopy (i.e., pull-off or adhesion force measurements) has been used to probe interactions between individual functional groups,185 as in studies of interactions between carboxylic acid-terminated probe and sample surfaces.186 Additional studies of discrete interactions between peptides and proteins have been reported, 187 and studies of discrete base-pairing (hydrogen bonding) interactions between individual nucleotide bases188 and complementary DNA strands189 have also been described.
Present limitations in chemical forces measurements include the need for detailed knowledge of the cantilever spring constant and the tip-sample interaction area in studies where quantitative values of absolute forces are to be measured. One possible solution to the problem of tip-sample contact area has been described by Beebe and coworkers.190 In these methods, the ratio of the variance to the mean of Poisson-distributed adhesion forces yields a measure of the force associated with a single bond.
Electric Force Microscopy. Long-range forces can also be used to generate contrast in force microscopy imaging experiments. A variety of electric force measurements have been reported and variously described as scanning capacitance microscopy, Kelvin probe microscopy, and electric force microscopy.191 Such methods have been employed to study surface charges and potential in diverse systems. Included are studies of phase separation in ionic thin films,192 change trapping in organic semiconductor films,193 and charge density mapping of bilayer membranes.194 Such experiments are often limited in spatial resolution to about 50 nm, due to the long-range nature of electric forces. Implementation of carbon nanotube-based tips in electric force methods have recently been shown to yield enhanced spatial resolution.195 Because of the long-range forces employed, electric force methods are also promising for depth-dependent sample imaging.
Surface Patterning. Patterning of surfaces using AFM and/or STM has been explored extensively in recent years.196 Lithographic scanning probe methods involve bringing the probe near the surface to be patterned, where it can interact with and modify the local structure. The probe is then scanned laterally in a manner that will produce the desired structure. The resolution of the patterns thus produced can approach the molecular scale. Typically, changes in the surface that have been affected involve either the direct placement of molecules,197 probe tip-mediated replacement or desorption of surface-bound molecules,198 or probe tip-catalyzed surface reactions.199 One limitation of many such applications is the serial nature of the fabrication process. As a result, structure fabrication is quite slow, and much effort is now being devoted to the development of parallel processing through the integration of multiple scanning probe tips.200
Dip-pen nanolithography (DPN) is a variety of scanning probe lithography (direct-write) developed by Mirkin and coworkers, where components of interest are transferred from an AFM tip to a substrate.201 DPN has been used to pattern a wide variety of materials on surfaces, including small organic molecules (most commonly n-alkanethiols), DNA, nanoparticles, proteins, viruses, and precursors for inorganic thin films.
Liu and coworkers have used both AFM and STM to desorb molecules selectively within an alkanethiolate self-assembled monolayer (SAM).202 The desorption mechanism differs between the two instruments: the basis of molecule removal with an AFM is detachment from the surface under an increased loading force that is significantly greater than the usual load employed for imaging. The desorption mechanism in STM lithography is electrochemical, since molecules can be desorbed by passing high-energy electrons through the film (i.e., at bias voltages of ~3-4 V).
The process of nanografting has been developed by Liu and coworkers as a method for creating both positive and negative patterns in a SAM.203 Using AFM, molecules from a preexisting SAM matrix are removed by scanning at a force greater than the threshold displacement force. New alkanethiols are then backfilled from the contacting solution and “grafted” into the bare areas. By using longer-chain alkanethiols as the grafting solution, a positive pattern can be made; conversely, shorter-chain alkanethiols produce a negative pattern. In addition, alkanethiols possessing different functional groups (e.g., OH- or COOH-terminated) can be grafted, thereby creating a patterned SAM with varying degrees of reactivity that can be used in further applications.
Near-Field Scanning Optical Microscopy
Near-field optical microscopy involves optical imaging of a sample using a subwavelength-sized light source positioned in close proximity to the sample surface. Because NSOM employs many of the same light sources, optical elements, and detectors commonly used in conventional spectrophotometers, it is particularly simple to combine NSOM methods with those of conventional optical spectroscopy. Hence, direct spectroscopic evidence of the local chemical composition of a sample can be obtained with nanometer-scale spatial resolution using NSOM methods. Important, the data obtained from such experiments are often similar to a first approximation in form and content to those obtained in conventional far-field optical spectroscopic experiments. As a result, data interpretation is greatly facilitated via the use of well-known principles and methods for interpreting optical spectroscopic data. However, as in all proximal probe methods, NSOM images sometimes incorporate contrast arising from interactions between the probe and the sample, producing image features that are not a direct result of sample properties alone. Such probe-sample coupling poses a challenge to the interpretation of NSOM data.
To date, a number of chemically selective near-field imaging methods have been demonstrated. Near-field contrast mechanisms that rely on electronic spectroscopy (UV-visible absorption and fluorescence),204 vibrational spectroscopy (IR absorption and Raman spectroscopies), dielectric spectroscopy (microwave dispersion), and nonlinear spectroscopy (second harmonic generation) have all been demonstrated at length scales well below the diffraction limit of light.
Fluorescence NSOM experiments have been used to observe the spatial localization of fluorescent species in composites,205 to investigate lipid layer structures206 and their evolution,207 to probe biological membranes and membrane proteins,208 to detect isolated chromophores,209 to monitor variations in sample properties brought about by aggregation phenomena,210 and to investigate the photophysical properties of organic semiconductors.211 Fluorescence resonance energy transfer experiments performed using NSOM have provided particularly dramatic resolution of energy transfer processes occurring across interfaces
One of the significant promises of NSOM is the development of chemical imaging based on vibrational spectroscopic data acquired with nanometer-scale spatial resolution. The advantages of apertureless (versus aperture-based212) methods become particularly important in imaging experiments performed in the infrared spectral region,213 where suitable optical fibers are difficult to obtain and implement in NSOM experiments. The apertureless approach used for the image shown in Figure 3.18 is notable because of the extremely high spatial resolution reported (>λ/200) and the absorption contrast between two polymers, imaging with laser lines that coincide with absorption bands of the polymers.

FIGURE 3.18
Images showing representative data from phase-separated domains of poly(styrene) in a poly(methyl methacrylate) matrix. Left: sample topography. Right: apertureless infrared scattering near-field optical images of poly(styrene) domains in a poly(methyl (more...)
Raman spectroscopy can also be implemented in near-field imaging experiments,214 providing an alternative method for acquiring chemically specific vibration information. Because of the extremely weak signals usually obtained in Raman experiments, Raman NSOM imaging has been greatly facilitated by the development of chemically etched probes with higher throughput.215 Probe-enhanced Raman NSOM imaging has also been extremely important to the advancement of this method.216 In probe-enhanced methods, the proximal probe serves to enhance the electromagnetic fields in the near-field regime in a manner similar to that of more conventional surface-enhanced Raman experiments.217
Although NSOM methods presently provide insufficient spatial resolution to directly resolve individual molecules in organized structures, molecular organization can still be probed using polarization-dependent NSOM methods. At present, polarization-dependent NSOM imaging can be performed only by aperture-based methods. Unfortunately, the polarization characteristics of aperture-based NSOM probes often suffer from imperfections in the metallic coating. The metal coatings on NSOM probes can be roughly on a length scale similar to the aperture size, due to grain formation. These grains alter the polarization state of the optical fields from the aperture, making interpretation of polarization-dependent images difficult. Methods for improving aperture uniformity and hence polarization quality involve focusing on ion-beam milling of the proximal end of the probes.218
Probing Organized Molecular Structures
Polarization-dependent and polarization-modulation NSOM imaging methods have been demonstrated for a number of organized molecular systems and for single-molecule detection.219 Images of the fluorescence excitation (dipole) patterns of single molecules provide a dramatic view of the electric field polarization from the end of an aperture-based NSOM probe, as well as valuable information on molecular orientation (Figure 3.19). Similar methods have been used to probe molecular orientation in phospholipids films.220 Polarization-dependent imaging studies of organized materials have also helped advance our understanding of molecular aggregates,221 semiconducting polymers,222 and metallic, dielectric, and inorganic semiconductor structures.223

FIGURE 3.19
(A) Dipole fluorescence excitation patterns recorded for single molecules of an indocarbocyanine dye using aperture-based NSOM methods. (B) Model of the molecular dipole orientations for the image in A. SOURCE: Reprinted with permission from Betzig, E., (more...)
Some of the most useful polarization-dependent NSOM methods, however, involve modulation of the polarization from the probe (see Figure 3.20),224 coupled with synchronous detection of the near-field signals. Such methods allow for multiple imaging modalities so that topography, absorption dichroism, and/or birefringence information can all be readily obtained.

FIGURE 3.20
(a) NSOM topography, (b) polarization modulation amplitude, and (c) polarization modulation phase images of rhodamine 110 microcrystals. The arrows appended to image c depict the transition dipole orientation in each crystal. SOURCE: Reprinted with permission (more...)
NSOM Dynamics Imaging—Millisecond to Femtosecond Resolution
Studies of topographic and morphological dynamics by NSOM methods are usually limited to (sub)video-rate imaging, although much faster imaging has been reported under special circumstances.225 Imaging rate limitations in NSOM not only arise from feedback bandwidth and microscope resonance issues, but also depend on optical signal levels. In transmission imaging experiments where signal levels can be significant, video-rate imaging is possible on flat (<10 nm topography) samples.226 In other situations (i.e., fluorescence and Raman NSOM experiments), several minutes to hours are often required to acquire each image. As noted above, significant improvements in imaging rates can be achieved when high-throughput, chemically etched NSOM probes are employed.
NSOM experiments performed in the UV-visible spectral region have the unique advantage that well-developed forms of time-resolved optical spectroscopy can be implemented directly in studies of dynamics that can be retriggered at each image pixel. The dynamic range of such experiments is virtually unlimited, spanning the entire range from milliseconds227 to femtoseconds.228 However, in cases where fast dynamics are to be studied (i.e., nanoseconds to femtoseconds), sophisticated laser systems and electronics must be coupled with the near-field microscope. Hence, time resolution in the microsecond regime represents the practical limit for microscopes employing continuous-wave (CW) lasers and conventional optical modulators and electronics.
NSOM experiments depicting local dynamics induced by applied electric fields have recently been reported and have led to better understanding of diverse processes such as charge carrier dynamics in organic semiconductor films229 and field-induced molecular reorientation in polymer-encapsulated liquid-crystal droplets.230
NSOM Photolithography
Well-developed far-field photolithographic methods used routinely in the fabrication of structures tens of micrometers in size can also be employed in near-field optical microscopes for the fabrication of structures having dimensions far below the diffraction limit. In these methods, UV or visible light normally employed for photopatterning can be coupled to aperture-based probe fibers231 or scattered from the probe tip in apertureless configurations.232 Field confinement allows subwavelength-sized features to be produced, and fabrication of features as small as 70 nm has been reported using nonlinear excitation in an apertureless microscope.233 Examples of what has been demonstrated to date include lithographic production of text,234 line structures in photosentive polymers,235 and the photochemical oxidation and removal of thiol species on gold surfaces.236 The same limitations on lithographic speed mentioned in the discussion of AFM- and STM-based methods also apply here.
Future Challenges and Emerging Methods
Several of the existing challenges in NSOM imaging are similar to those in other scanning probe methods: namely, imaging of larger sample regions and faster image acquisition. Although parallel probe NSOM imaging is now possible,237 it requires the use of multiple detectors and/or array detectors. Fast video-rate NSOM imaging using etched NSOM probes has also been demonstrated.238
An important emerging technology that promises to revolutionize near-field imaging by enhancing the spatial resolution and optical signals is the further implementation of field enhancement effects at the end of metallic apertureless probes in imaging experiments.239 To date, field enhancement effects have been demonstrated successfully as a means to excite molecular fluorescence in the near field by both linear and nonlinear optical processes, as well as a means to enhance near-field Raman signals.240 The design of specific proximal probe geometries that provide high field enhancements241 for scattering and sample excitation242 is now under way. The development of probes that are easy to fabricate and implement in NSOM experiments will increase the signals that can be obtained and hence improve imaging rates.
NSOM methods can provide extremely valuable information about the functional properties of optical and optoelectronic materials.243 In many such applications, electrical potentials and/or electric fields are applied to samples as a means to induce changes in the local optical properties of the sample. Frequently, an electrified NSOM probe is employed in these studies.244 The resulting changes in the sample are then detected optically in the near field. Both static and dynamic information on the variations in sample properties is thus obtained. In related methods, the probe itself or external electrodes can be used as a means to detect photocurrents245 and/or photovoltages generated in a sample in a fashion similar to more common far-field methods. Near-field studies, however, provide the distinct advantages of higher-resolution images, coupled with invaluable topographic information that is routinely recorded along with the optical data. In the future, the chemical specificity afforded by spectroscopic NSOM methods will add a new dimension to the data, providing a direct link between local chemical composition and/or structure and materials performance.
Scanning Electrochemical Microscopy
Spatial Resolution
In SECM, a metallic probe is typically employed as a redox electrode at which an electrochemical oxidation (or reduction) process occurs. In many instances the probe-sample separation is regulated using the faradaic current as a feedback signal.246 More recently, tuning-fork-detected shear force methods have also been used to maintain probe-sample separation,247 opening the possibility of improved electrochemical resolution and allowing multiple simultaneous contrast mechanisms to be employed.248 In all situations, the spatial resolution obtained in electrochemical images is limited by diffusion of redox active species between the proximal probe and the sample. Common values reported for SECM resolution are on the order of 1 μm, in cases where the probe-sample separation is large. However, resolution approaching 1 nm has been obtained in situations where a small tip is positioned in extremely close proximity to the sample.249
SECM imaging methods have been used to image heterogeneous surface reactivity on a number of different surfaces. Many such systems are of relevance to energy conversion and utilization. For example, SECM has been used to observe variations in the electrochemical activity of metal oxide surfaces.250 More recently, it has been used as a means to characterize the reactivity of bimetallic catalysts for use as oxygen reduction electrodes in fuel cell systems.251 SECM methods have also been used to image the electrochemical reactivity of biological samples, most notably living cells.252
Dynamics and Time-Resolved Imaging
The dynamics of charge-transfer processes are important in a wide range of biological and technological materials. SECM provides a means to study interfacial charge-transfer dynamics, while minimizing some of the difficulties associated with other methods. The rates of electron transfer can be measured by both steady-state253 and time-resolved chronoamperometric254 SECM methods. Mass transport through films,255 porous membranes,256 and biological membranes (even in living cells)257 can also be probed. In studies of diffusion within films, methods analogous to those used in fluorescence recovery after photobleaching (FRAP) are employed to extract the desired diffusion coefficient. An initial electrochemical potential step is employed to generate a reactant (i.e., an oxidant) at the SECM probe tip. The oxidant then locally oxidizes the film being studied. Charge migration through the film leads to the reduction of the oxidized region of the film in time. After a defined period of time, oxidant is again generated at the SECM probe, and the measured redox current provides information on the extent to which the film region has been reduced during the waiting period.
Electrochemistry and Chemical Selectivity
Scanning electrochemical microscopy is to some degree inherently chemically selective, although this selectivity is not always utilized directly in SECM experiments. Chemical selectivity in electrochemical microscopy arises from the dependence of the faradaic current on the oxidation and reduction potentials of the species being detected. Examples of experiments in which the chemical selectivity of SECM is used to advantage include those in which transient species generated at electrode surfaces are detected. The release of certain chemicals (i.e., neurotransmitters) from cells can also be selectively detected and imaged. In one particularly interesting experiment described recently by the Mirkin group, nonmetastatic and metastatic human breast cells were imaged by SECM. Imaging was based on the measurement of redox currents arising from the products of cellular enzymatic redox reactions.258
Surface Patterning
SECM can be used to electrochemically desorb surface-adsorbed species (i.e., such as self-assembled monolayers)259 or to electrochemically deposit metals260 for the purposes of preparing surface structures of controlled geometry. The production of micrometer-sized features is accomplished by setting (or scanning) the potential of the SECM probe (or substrate) at the appropriate reducing or oxidizing potential, while the probe position is scanned relative to the substrate surface. Limitations of this method include the relatively low spatial resolution that can be achieved at present and the relatively slow writing process. The resolution here again is limited by diffusion of the species being deposited or of those used to induced desorption.
Future Challenges and Emerging Methods
As with the other forms of scanning probe microscopy, SECM experiments could also be improved by the development of methods for faster image acquisition,261 allowing better time resolution in imaging experiments. Improvements of the spatial resolution in SECM will be achieved via the continued implementation of new and/or alternative feedback mechanisms. These feedback mechanisms will rely on signals decoupled from the redox activity of the surface as a means to sense tip-sample proximity (i.e., as in the use of shear force feedback).262 They will also allow the probe-sample separation to be maintained at extremely small values, limiting lateral diffusion of redox active species and, hence, improving spatial resolution.263
Recently, integration of SECM with other scanning probe techniques (STM,264 AFM,265 and NSOM266) has proved to be a valuable means for obtaining detailed, complementary chemical information. Such research should continue. Particular benefits include the ability to exploit the high-resolution imaging capabilities of STM and AFM in particular. New SECM probes will also continue to be developed. The most interesting and promising new probes presently being developed may be those based on carbon nanotubes and other nanoprobes.267
New developments in SECM will also include expansion of the methods available to incorporate new types of electrochemical excitation and more sophisticated data analysis. For example, the SECM probe is now being used for simultaneous amperometric and conductometric measurements, providing additional information about sample impedance on micro- and nanometer length scales.268 Voltammetric methods, in which an entire cyclic voltammogram is recorded using the SECM probe tip at each image pixel, are also being developed for use in SECM experiments, improving their chemical selectivity.
Finally, applications of SECM to understanding the chemistry of biological systems are expanding quickly and will continue to grow. Important studies include measurements of membrane transport and characterization of ion channels and studies of charge-transfer reactions involved in photosynthesis. Imaging of single cells to better understand their function at subcellular levels will continue to expand, and the methods employed will continue to improve. One challenge in this area will be the development of SECM methods for making spatially resolved measurements inside living cells. Particularly important applications of these methods would include the development of approaches for probing the mechanisms and kinetics of biochemical reactions. This would involve the use of redox or ion-transfer mediators specific for the reaction of interest.
Emerging Methodologies
Magnetic Resonance Force Microscopy
The development of proximal probe methods by which three-dimensional images of samples can be recorded with high spatial resolution in all three dimensions would represent a major breakthrough in chemical imaging technology. Such methods would allow scientists to “see below the surface.” One of the most promising emerging methods for achieving this goal is magnetic resonance force microscopy.269 This method takes advantage of the magnetic field gradient methods employed in conventional MRI as a means to resolve sample features. It also makes use of the sensitive probe-cantilever technologies developed for more conventional atomic force imaging experiments and the ability to generate extremely strong magnetic field gradients near the end of a sharp probe.
In this experiment, a magnetic proximal probe is scanned above the surface of a sample and the resonances of electron or nuclear spins are detected as a function of position (Figure 3.21). The magnetic probe is attached to a sensitive cantilever, whose motions are detected interferometrically. As the probe is passed above a region of the sample, a change in the spin state populations beneath the probe causes a deflection of the cantilever. The ultimate sensitivity of such experiments involves detection of single electron spins, requiring field gradients of 2 gauss/nm and sensitive cantilevers and cantilever deflection methods for detecting forces as small as 2 attonewtons (aN). In typical AFM imaging experiments, the forces detected are in the piconewton to nanonewton range. To achieve single nuclear spin sensitivity, field gradients of 200 gauss/nm will be required, along with force sensitivities down to 0.3 aN.

FIGURE 3.21
Magnetic resonance force microscope. SOURCE: Figure courtesy of Dan Rugar, IBM Almaden Research Center.
Magnetic resonance force microscopy would allow direct chemical imaging of samples to depths of about 100 nm for a system providing 1 nm lateral resolution. In such experiments, depth resolution is obtained by scanning the radio frequency employed to flip the electron or nuclear spins in the sample.
The Photonic Force Microscope
The scanning probe methods mentioned above are predominantly surface or near-surface methods. It is difficult to obtain images of the internal features or internal surfaces of a sample using these methods. One emerging proximal probe method that may allow these limitations to be overcome is photonic force microscopy. In this method, a micrometer- or submicrometer-sized particle (a dielectric or metallic bead) is optically trapped in the focus of a laser beam. The focal position of the laser is then changed as a means of moving the particle about for imaging purposes. As the particle moves it experiences different forces associated with internal variations in the sample composition and properties. The associated forces active on the probe particle can be detected and measured by optically monitoring the position of the bead within the laser focus. Subpiconewton forces can readily be detected by this method.
The photonic force microscope may yield a means for proximal probe imaging within fluid-containing voids and structures such as vesicles and living cells. Some current limitations of the photonic force microscope include the potential for incorporation of optical artifacts when internal structures of optically complex samples (such as cells) are to be studied. Coupling of optical features into the images of such a microscope arises via the dependence of the optical trapping potential on the optical properties of the medium through which the beam passes in reaching the focal point.
IMAGE PROCESSING
Chemical imaging is used to selectively detect, analyze, and identify chemical and biological samples, followed by visualization of the data in the dimension of interest. Some of the types of chemical image datasets that can be acquired are shown in Figure 3.22. The information of interest can range from composition, structure, and concentration to phase or conformational changes as a function of time or temperature. The expression “chemical image” describes a multidimensional dataset whose dimensions represent variables such as x, y, z spatial position, experimental wavelength, time, chemical species, and so forth. Image processing requires that the chemical images exist as digital images. A digital image is an image f(x,y) that has been digitized both in spatial coordinates and in brightness. The value of f at any point (x,y) is proportional to the brightness (or gray level) of the image at that point. The point at f(x,y) is an image pixel, and each pixel has an intensity value associated with it. This value may represent energy, radiation intensity, phase, height, et cetera, depending on the technique utilized. A digital image can be considered a matrix in which row and column indices identify a point in the image and the corresponding matrix element value identifies the gray level at that point. Image processing methods generally operate on the matrix representing the image.

FIGURE 3.22
Integration of spatial and spectral information. NOTE: RGB = red, green, blue. SOURCE: Julia E. Fulghum and Kateryna Artyushkova, University of New Mexico.
Many spectroscopic imaging techniques now utilize array detectors, which allow the rapid collection of both spectral and positional data.270 An entire spectra may be collected for each pixel on the array detector, creating a three dimensional cube consisting of both spatially resolved spectra and variable-dependent images (Figure 3.22). The complete integration of spatial and spectral information quite literally adds a new dimension to data analysis, providing the ability to examine the interdependence of spectral and spatial information, thereby improving understanding of the underlying chemical and physical attributes. The extraction of meaningful chemical information from spectroscopic imaging datasets (multivariate images) can require complex data analysis because more than 100 million data points can readily be acquired. Developing and implementing effective algorithms to obtain chemistry from spectrum imaging data is a critical and ongoing process.
Image processing is a subclass of signal processing concerned specifically with two- or three-dimensional datasets to improve image quality for human perception and/or additional mathematical analysis and interpretation.271 Image processing is required since chemical images acquired using analytical instrumentation can contain an overwhelming number of visual patterns generated by diverse processes. The images in an image dataset are assumed to be a composite of a chemical image and artifacts affecting image intensity that are generally unrelated to the chemical variable(s). One complicating factor in the development of image processing methods is that these artifacts are technique-specific, ranging from nonuniform illumination in optical techniques to tip artifacts in AFM.272 One of the primary goals of image processing, regardless of the technique used to generate the image, is to find a meaningful representation of the intensity distribution in a given image without introducing any artificial information by the imaging technique. That is, image processing should be carried out with caution in order to avoid excessive “beautifying” of data. The order in which image processing steps are carried out, however, and the importance of the methods mentioned here vary dramatically between imaging techniques.
A variety of methods can be used to visualize and process chemical image datasets, as listed in Table 3.1. In general, the goal is to facilitate interpretation of the dataset. In many cases the intended audience includes people that are not experts in the imaging technique, who are more interested in the chemical or spatial variation shown in the dataset. Image processing can thus have a variety of goals, including:
- enhancing contrast in images to highlight specific features;
- enhancing differences between images for correlation or comparison;
- correcting for noise, background (i.e., topography or other effects), or nonuniform illumination by removing or decreasing pixel intensities unrelated to chemical signals;
- decreasing image or dataset size to facilitate analysis (decrease analysis time) or to facilitate image correlation;
- identifying objects of interest (image segmentation, particle counting or identification);
- pre-processing (background correction, compression or binning, etc.) before utilizing multivariate analysis methods;
- quantifying image intensities;
- combining images to create (render) three-dimensional volumes (visualization).
TABLE 3.1
Various Methods to Visualize and Process Chemical Image Data Sets.
The ease with which these processes can be carried out using technique-specific software varies dramatically. Standard or add-in software on some X-ray and EM instruments can offer a range of processing options including particle counting,273 Fourier transform-based convolution methods,274 and tomographic image visualization,275 while software associated with some surface analysis techniques may include only basic image manipulations such as rescaling, addition, and subtraction. Although commercial software packages exist to implement most of the processing methods described here, in general, technique-specific requirements are barriers to the average user. Image processing methods that are routine in one user community may be state of the art or unavailable in a different user community.
It is also important to be aware of the way in which image processing methods are applied to an image, so that an appropriate choice of algorithm and operating conditions can be made. For example, filters operate on groups of pixels within an image, with the number of pixels affected depending on the kernel size chosen for the filter.276 Simple linear transformations, such as rotation or reflection, operate on single pixels, and results are independent of the value of neighboring pixels. Nonlinear transformations can dramatically alter image appearance, and Fourier transform-based methods convert images into the frequency domain for additional manipulation.
Initial Image Visualization
Frequently the first priority for the analyst is to generate an image or images that allow for visualization of heterogeneous chemical distributions in space or time. Image visualization methods vary from simply choosing a color scale for display of a single image to methods for displaying three-dimensional datasets. Simple gray-scale maps can be constructed from a single image. Different color scales can be utilized, and the contrast and brightness can be adjusted so that the information the analyst deems most important is emphasized. Multiple images from the same or different datasets can be viewed simultaneously for comparison. Scatter plots are frequently utilized for comparing two images. For more detailed comparisons among a small number of images, mapping individual images into red, green, and blue (RGB) channels creates composite color chemical images. For three-dimensional data, additional analysis tools are required, including the ability to extract spectra from a selected region of interest (ROI) for multispectral imaging datasets or rendering a three-dimensional volume or projection for depth arrays.277
Image Processing
A variety of factors can contribute to intensity in chemical images. Analytical microscope images can exhibit significant shading across the field of view. The shading might be caused by nonuniform illumination, nonuniform camera (detector) sensitivity, or even dirt and dust on lens surfaces if physical (rather than electrostatic or magnetic) lenses are present.278 In surface analysis methods such as XPS and atomic emission spectroscopy (AES) the analytical signal depends not only on surface composition but also on local topography.279 Subtraction of a background image is one of the simplest approaches to decreasing background effects, including nonuniform illumination, substrate reflectance, topography, and instrument response. This method is widely used in surface analysis methods as different as XPS and STM as well as in optically based imaging techniques.280
Data Extraction
A variety of tools are used for extracting image components useful in the representation or description of feature shapes. These include boundary extraction, skeletons, morphological filtering, thinning, and pruning. Image segmentation is used to separate objects of interest from the image background and is required by a variety of microscopy techniques. It is also one of the more difficult tasks in image processing.281 Segmentation algorithms generally are based on one of two properties of the image intensity values, either discontinuities in intensity values (such as edges) or similarity according to a set of predefined criteria. Edge detection is the most common approach for detecting meaningful discontinuities in gray level.282 An edge is defined as a set of connected pixels that lie on the boundary between two regions.
Morphological smoothing removes or attenuates both bright and dark artifacts or noise by performing an opening followed by a closing. Applying a morphological gradient highlights sharp gray-level transitions in the input image, while the top-hat transformation can be used to enhance details in the presence of shading. AFM images of biomolecules, and other structures similar in size, are enlarged because of the finite size of the probe tip.283 A method based on morphological image processing allows analysis and correction of the enlargement.
Image transformations are operations that alter the value of pixels in an image. Transformation results do not depend on the value of neighboring pixels. These include simple linear transformations such as image rotation, translation, and reflection that may be required for correlation of images acquired using different techniques, as well as nonlinear transformations such as shearing, which is used to skew objects.
Image Analysis
The next step in image processing involves using processed images for image analysis. The ultimate aim of image analysis is generally to extract quantitative information, which may be in the form of binary presence-absence categories or of measures of object location, length or area, shape statistics, and so forth. Shape characteristics, such as area, perimeter, compactness, topological descriptors, and internal characteristics such as color and texture are among the possible types of information that may be required. Texture is used to point to intrinsic properties of a surface or region, especially those that do not have a smoothly varying intensity.284 Texture is also used in the classification of images based on their appearance for segmentation of images into regions that have similar textural properties. Granulometry can be used to determine the number of particles, particle mean sizes, and nearest-neighbor distributions in images.285 The results can then be correlated with macroscopic properties, such as catalytic activity. Electron microscopy (TEM, SEM) is still the most commonly used technique for visualizing colloidal particles, although scanning probe microscopy (AFM), magnetic force microscopy [MFM]) is a serious alternative that provides more information about particles.286
Motion analysis and particle tracking methods enable users to follow the movement over time of tagged particles, such as fluorescently labeled cell surface molecules, microtubules, nucleic acids, lipids, and other objects with subpixel resolution.287 These methods allow scientists to measure x and y coordinates, velocity, mean displacement, mean vector length, and more.
Although, in theory, quantitative data obtained from chemical images should be useful in combination with information obtained using spectroscopic methods or bulk property measurements, image-to-property correlation is a problem poorly studied in chemical imaging. The most successful results have come from a combination of statistical image analysis, image processing, and multivariate analysis, as discussed below.
Multidimensional Image Processing
Image Compression
Image compression is an ongoing research topic in the field of image processing. A typical spectrum imaging experiment can result in the acquisition of more than 100 million data points, providing a strong impetus for automated data processing. Spectrum imaging datasets can, however, be too large to analyze efficiently, or at all, using most currently available software. For example, SIMS images are typically digitized at a resolution of 256 × 256 pixels and 16 bits of intensity information per pixel. A single image therefore requires at least 0.125 megabyte (MB) to store, and a typical image set with 64 images thus requires a minimum of 8 MB of storage. A three-dimensional SIMS analysis can involve acquisition of 4 to 8 three-dimensional datasets, resulting in 32 to 64 MB of data for only one analysis. With some types of X-ray and optical experiments, gigabyte-size datasets can result. Obviously data compression can be required to create manageable datasets.
Methods developed for traditional “lossy” image compression can be used in the compression of multispectral images. The discrete cosine transform (DCT), which is used to create standard joint photographic experts group (JPEG) images, has been widely used for lossy still image compression.288 Although it can be efficiently implemented and performs well for high-bit-rate compression, serious blocking artifacts are a well-known disadvantage for DCT-based coding. Binning is used routinely to decrease the size of SIMS and other spectral datasets by adding intensities over a given spectral or mass range.289 Discrete wavelet transforms (DWT) not only can overcome the blocking artifacts but also can achieve better performance overall. The wavelet transform (WT) is a new and very versatile technique that has been developed during the last decade.290 This approach has found applications in signal and image processing291 and, recently, also in chemistry.292
Developing a three-dimensional image based on an image stack can be a difficult task. In volume model visualization, information may be displayed as surfaces, interfaces, or intensity distributions through either surface or volume rendering. For automated visualization of volume data using isosurface extraction, segmentation is a necessary preprocessing step. Isosurface extraction applies a surface detector to the sample array, after which geometric primitives are fitted to the detected surfaces. These primitives are displayed using conventional surface-rendering algorithms. Isosurface extraction requires a variable contour value, and this value has a great effect on the appearance of the resulting volume.293
Multivariate Data Analysis Tools
Multidimensional datasets are frequently too large to analyze by visual inspection, and methods are required for reduction and analysis of these datasets. A variety of multivariate analysis (MVA) methods can be utilized to identify, extract, and correlate or classify information while reducing the overall dimensionality of the dataset.294 MVA methods include statistical, mathematical, and graphical methods that analyze multiple variables simultaneously. MVA soft modeling methods are used for deconvolution of the original data matrix, generally including only basic, if any, physical restrictions. Physical restrictions that are sometimes utilized include nonnegativity of concentrations or molar absorptivities or the Beer-Lambert law. Principal component analysis (PCA) and classification algorithms are among the most widely applied MVA methods.295 These methods have been successfully applied to TOF-SIMS,296, EDX-SEM,297 XPS,298 IR,299 and Raman300 data. These methods are not available in all instrument-specific software, however, and the image preprocessing required, as well as the image processing parameters, will be strongly technique-specific. It is best to consider the MVA methods that will be utilized in initial experimental design and data acquisition because the number and type of standards utilized and the type of data acquired will strongly affect the type of multivariate analyses that can be successfully applied.
Conventional chemometric techniques such as factor analysis, least-squares fitting, PCA, and principal components regression are powerful tools for determining the composition and concentration of samples with known constituents. Neural networks and data segmentation approaches have also been successful.301 PCA is currently one of the most commonly used methods; it transforms a number of possibly correlated variables into a smaller number of independent variables, called principal components. The first principal component accounts for as much of the variability in the data as possible, and each succeeding component accounts for a decreasing proportion of the remaining variability. The objective is to identify images that are globally correlated or anticorrelated. This information is then displayed as the loading of different images, and the pixels responsible for the correlations can be displayed in component images. The component images may be easier to interpret than pure variable images. The components can, in many cases, be connected to chemical components through a variety of methods ranging from visual inspection to additional processing such as classification or Simplisma. Principal component and cluster analysis are becoming more popular in analysis of SEM electron dispersive X-ray (EDX) data.302 The results from PCA can be difficult to interpret. Multivariate curve resolution (MCR) is a powerful technique for extracting chemical information from multivariate images (MIs). MCR is aimed at extracting the spectra and concentrations of individual components present in mixtures using a minimum set of initial assumptions.303 Purity-based methods show promise, and a simple, robust purity-based algorithm has been developed to initialize the MCR decomposition. Lack of selectivity, common in MI, generally results in a rotational ambiguity in factors extracted with MCR. Modifications of MCR methods are currently in development in order to reduce rotational ambiguities.304
In general, no software exists that is capable of visualizing multispectral imaging data, extracting those parts of the dataset that are significant, and deriving the required information. A wide range of multivariate methods have been utilized both singly and in combination, as illustrated by applications in SIMS, FTIR, SEM-EDX, Raman, NIR, and XPS.
Current Software
As discussed earlier, a significant limitation in the development of image processing is the technique-specific nature of the analysis. Several programs are available, or in development, for the application of multivariate methods. In general, these are still in the category of programs to be used by experts, because numerous decisions and assumptions must be made about data processing and interpretation. MATLAB,305 and packages associated with it, are widely used for the application of PCA and other commonly used multivariate methods.306 In addition to commercial add-ons, there is a user web site to which users post routines that may be of general interest. Although the posted routines can come and go, the current listing includes a graphical user interface (GUI) for visualizing three-dimensional volumetric data as well as a GUI for image segmentation and extraction.307 A software package developed by the remote sensing community, ENVI (Environment for Visualizing Images), is used by some research groups developing methods for the analysis of chemical images.308 ENVI combines both MVA and image visualization. A patented MCR automated software methodology has been developed for several instruments and is called expert spectral image analysis (AXSIA).309 Although not yet publicly available, initial publications indicate AXSIA is fast enough to efficiently extract meaningful chemical components from very large spectral image datasets. The MCR methodology in AXSIA works by fitting self-generated spectral shapes to the data using least-squares procedures. Physically realizable components are obtained by applying appropriate constraints (e.g., nonnegativity of concentrations and spectra) during the solution process. The number of chemical components is estimated through an eigenanalysis of the data cross-product matrix. As an example of technique-specific data processing requirements, for analysis of TOF-SIMS images, the data are optimally scaled to account for Poisson counting statistics. This provides maximum discrimination of chemical information from noise and allows detection of small features that would be otherwise overlooked. AXSIA was tested on SEM-EDX,310 TOF-SIMS,311 and XPS312 spectral imaging datasets, proving that algorithms as implemented in AXSIA are quick and efficient; they are able to process multigigabyte datasets in minutes using modern desktop computers.
Multitechnique Image Correlation
Complete characterization of a complex material requires information not only on the surface or bulk chemical components, but also on stereometric features such as size, distance, and homogeneity in three-dimensional space. It is frequently difficult to distinguish uniquely between alternative surface morphologies using a single analytical method and routine data acquisition and analysis. By combining imaging techniques that use different physical principles and, therefore, produce images representing different properties of the sample, complementary and redundant information becomes available.313 One important goal is data fusion, which refers to combining image data from multiple techniques to form a new image or volume that contains more interpretable chemical information than can be ascertained from a single technique. Successful data fusion can decrease ambiguities in the evaluation of materials chemistry and morphology; extend lateral and vertical spatial characterization of chemical phases; or enhance spatial resolution by utilizing techniques with nanometer spatial resolution (AFM or SEM) to enhance data from techniques with spatial resolutions of microns (XPS, chemical microscopy [CM], or FTIR). This approach can facilitate correlation of different physical properties; for example correlating phase information in AFM with chemical information in XPS images.
There are MVA techniques that can used in the combination of different imaging modalities by merging registered images from different techniques. This process can also be called intermodular imaging. For example, light microscopy can be used in a variety of different imaging modes that contain complementary information. Bright-field microscopy images result from optical attenuation by the sample, whereas phase contrast microscopy images show diffractive properties. The refractive properties of the sample are displayed in differential interference contrast (DIC) images.314 Leonardi and colleagues compared applications of polarized light, bright field, DIC, and SEM in the paper industry.315 Fluorescence microscopy can also be correlated with images acquired using light microscopy.316 Much effort has been expended in developing and modifying algorithms for matching images produced by different types of satellite remote sensing systems, such as optical sensors and synthetic aperture radar,317 some of which are transferable to laboratory image correlation.
Since Raman and NIR spectroscopies are complementary in nature, their combined usage offers the opportunity to describe heterogeneous mixtures in more detail. A novel sample referencing approach has been developed that allows data to be acquired from exactly the same area of the sample using both Raman and FT-NIR microscopies. The optimum images for the components are then overlaid, which gives rise to a combined chemical image that visually describes the entire formulation. Correlating imaging XPS and imaging FTIR data from polymer blends allowed for localized quantitative studies of surface segregation and phase separation phenomena.318 This approach is called chemical image fusion (CIF).319
Databasing and Data Mining
The acquisition of large datasets, followed by large numbers of such datasets, leads to issues of data cataloguing, analysis, and sharing—or databasing and data mining. Whether one is attempting to develop a method for sharing databases within a single research group or a specialized research community, numerous challenges remain in developing appropriate software for these tasks.
Databasing
Molecular databases and the associated data banks require the development of a conceptual structure for the information stored about the molecules, descriptive language representing the data, and methods for analysis enabling molecular modeling, similarity searches, classification, visualization, or other uses of the database.320 Currently, the Protein Data Bank (PDB; http:/www.rcsb.org/pdb/) is one of the best known examples of a molecular database. The PDB is a worldwide archive of three-dimensional structural data of biological macromolecules.321 The PDB is a common accentor to many structural databases.322 The success of the PDB in enabling the statistical analysis (bioinformatics) of protein structures suggests that a broader materials image and structural database would enable similar advances (using informatics) in the understanding of generic materials.
The development of imaging databases adds additional complexities compared to molecular databases. There, is, however, significant activity in the development of software for medical images. Picture archiving and communications systems (PACS) are utilized by an increasing number of laboratories and hospitals for the storage, retrieval, and sharing of images.323 The classical medical imaging technologies are advancing toward (1) higher resolution, (2) increased sensitivity, (3) standardized protocols, and (4) increasing application fields. The developments in items 1 to 4 will allow merging of data from different laboratories, exemplified in multimember screening studies conducted by Brown and colleagues.324
A database for functional magnetic resonance imaging (fMRI) provides a different example.325 A framework that will allow scientists access to raw data from published, peer-reviewed studies has already been established (fMRI Data Center). A more demanding goal is to compile the images in a database that will allow for data mining from image sets that are both highly heterogeneous and large in size. The fMRI Data Center has adopted several guiding tenets in the organization of its core database that highlight the complexity of this task:
- The database should be flexible enough to represent the broadest range of possible fMRI experimental paradigms.
- The database is organized hierarchically, with the study itself at the highest level.
- In addition to the high-level descriptive data of the study, meta-data characterization for, and pointers to, all neuroimaging data and time series are represented in the database in order to facilitate the broadest possible space over which accurate but efficient searches can be made.
- The database should be extensible and able to incorporate new studies, scans, or time course information as it becomes available.
Clearly, the data storage issue associated with archiving functional neuroimaging data is a serious one, not to mention the computational challenges of attempting to carry out analysis on such an archive. To address these issues, establishment of “near-line” and off-line data storage is being investigated. In addition, the search to provide more suitable computing resources for carrying out large-scale analysis research is also under way.326
There are a variety of other databases currently in development. The Global Image Database (GID) is a web-based structural central repository (http://www.gwer.ch/qv/gid/gid.htm) for scientifically annotated images. The GID was designed to manage images from a wide spectrum of imaging domains ranging from microscopy to automated screening. The development of the GID is aimed at facilitating the management and exchange of image data in the scientific community and the creation of new query tools for mining image data.327 Other databases include WebRacer, an image database that allows databasing and web serving of images; Soft Imaging System GmbH (http://www.soft-imaging.de/rd/english/420.htm); and Neuroinfo (http://www.neuroinformatica.com/faq.jsp), a software package designed to store and serve large arrays of microscopy data. Data acquired at various magnifications can be integrated to allow navigation of the data on a number of scales.
Image and Data Mining
Image mining involves the extraction of implicit knowledge, image-data relationships, or other patterns not explicitly stored in the images or between images and other alphanumeric data. Image mining is rapidly gaining attention among researchers in the field of data mining, information retrieval, and multimedia databases because of its potential in discovering useful image patterns that may push the various research fields to new frontiers. The fundamental challenge in image mining is to determine how low-level, pixel representation contained in a raw image or image sequence can be processed efficiently and effectively to identify high-level spatial objects and relationships.
Research in image mining can be classified broadly into two main directions. The first direction involves domain-specific applications where the focus is to extract the most relevant image features into a form suitable for data mining.328 The second direction involves general applications where the focus is to generate image patterns that maybe helpful in understanding the interaction between high-level human perceptions of images and low-level image features.329 The latter may lead to improvements in the accuracy of images retrieved from image databases.
Image mining is not simply an application of existing data mining techniques to the image domain because there are important differences between relational databases and image databases:
- Absolute versus relative values. In relational databases, the data values are often readily interpretable. For example, age is 35 is well understood. However, in image databases, the data values (e.g., pixel intensities) have a significance that will depend on the context. For example, a gray-scale value of 46 could appear darker than a gray-scale value of 87 if the surrounding context pixels values are all very bright.
- Spatial information. Another important difference between relational databases and image databases is that implicit spatial information is critical for interpretation of image contents, but there is no such requirement in relational databases. One approach to this problem is to extract position-independent features before searching for patterns between image datasets.
- Unique versus multiple interpretations. A third important difference is associated with the fact that multiple interpretations may apply to the same visual patterns observed in images. Traditional data mining algorithms, which associate a data pattern with a specific class or interpretation, are less useful for analysis of images. A new class of discovery algorithms is needed in response to the requirements for mining useful patterns from images.
The image database containing raw image data cannot be used directly for mining purposes. Raw image data have to be processed to generate information that is usable for high-level mining modules. An image mining system is often complicated because it employs various approaches and techniques ranging from image retrieval and indexing schemes to data mining and pattern recognition. A good image mining system is expected to provide users with effective access into the image repository and generation of knowledge and patterns underneath the images. Such a system typically encompasses the following functions: image storage, image processing, feature extraction, image indexing and retrieval, patterns, and knowledge discovery. Two different frameworks can be used to distinguish image mining systems: function-driven and information-driven image mining frameworks. The function-driven framework focuses on the functionalities of different component modules to organize image mining systems, while the latter is a hierarchical structure with an emphasis on the information needs at various levels in the hierarchy. Image mining techniques include object recognition, image indexing and retrieval, image classification and clustering, association rules mining, and neural network.330
Generating Images Through Theory and Simulation (Cyberinfrastructure)
Image databasing and data mining generally refer to the compilation and analysis of experimentally acquired images. Effective methods for exploring information obtained in chemical images acquired in the lab versus those developed through theory and simulation are also insufficiently developed. When one speaks of chemical imaging of samples, a subtle distinction can be drawn between (1) the creation of an image of a particular sample and (2) the creation of an image of a generic sample. In the former, there is no recourse but to make a literal measurement of the particular sample of the various types outlined in the other parts of this chapter. In this case, the role of cyberinfrastructure is primarily image manipulation as discussed above, although the attempts to address or understand a particular sample may be aided by whatever techniques have been used to understand generic samples. In the latter case, however, cyberinfrastructure and its underlying theoretical frameworks can provide computer-generated images that provide insight into the structure and function of generic samples. Moreover the simulations can also allow test cases for new paradigms for the cybertools used to manipulate data, process metrics, and render images using the experimental measurements.
Theoretical Models or Representations. The 1010 range of magnitude from the atomic scale to the bench top requires a significant amount of averaging (or compressing) to remove nonessential data. On the one hand, all the data cannot be stored easily, but on the other hand, it is a significant undertaking to search through all of the data. At present, this means pursuing multiscale techniques. One such hierarchical approach uses small-scale models to generate parameters for the next-scale model, and so forth. The lowest-scale system (at the molecular level) can be described by a combination of quantum mechanics and classical mechanics. It is the success of such models that has led to the utility and widespread use of molecular dynamics simulations. Yet what does one do at higher scales—perhaps use effective classical particles or integrative representations? Both are being done, although the methods are still in their infancy.
Yet another promising line of research lies in creating mesoscopic representations whose fundamental scale is somewhere within the 1010 range of distance scales and for which one defines closed (or fully consistent) equations of motion. At the macroscopic limit, hydrodynamic models are a very successful and standard example. More recent approaches include the Cahn-Hillard coarse-grained models and phase-field models. In some cases, one aims to ascertain the degree to which the systems exhibit self-similarity at various length scales; hence the lack of a specific parameterization—which would be necessary using reduced-dimensional models—is not of much importance.
Computer Simulation
To image either possible structures or trajectories of a system, one specifies the fundamental (smallest-length scale) representation, the interactions between objects in the system, and the appropriate equations of motion for this representation. When this fundamental representation is at the atomistic level, there is a wide array of choices for the interactions, ranging from the most computationally expensive, using high levels of ab initio calculations, to the least expensive, using parameterized force fields. At present, one can routinely run reasonable simulations consisting of about 100,000 atoms for up to a nanosecond using a few days of computational time. This is sufficient to obtain structural and dynamic information for many systems. However, this approach does not readily provide dynamic information about complex processes such as chemical reactions and full protein folding events. The latter have been treated, at present, with a variety of simplifying approximations or algorithms. For example, the solvent may be represented using mean-field forces, thereby removing the detailed descriptions of the solvent molecules. This is a common and elementary example of hierarchical models in which different regions of the system are treated with varying degrees of accuracy.
At another extreme, one can use effective mesoscopic models to characterize a given system and, in the best cases, obtain accurate structural information at much larger length scales than these found in atomic simulations. Such multiscaling techniques can be used to perform simulations for length scales as large a centimeters and for time scales as long as hours. However, they sacrifice detailed information about length scales shorter than micrometers.
Regardless of how the simulation is performed, the results must be analyzed. At present, only a limited number of cybertools are available for measuring the properties of simulations beyond merely recording the trajectories. Examples of such cybertools found routinely in many packages involve the calculation of correlation functions, triangulation of structure, Fourier transforms, clustering metrics, and informatics-based metrics.
Molecular Dynamics Simulations
At the atomic and molecular scale, typical simulation techniques use molecular dynamics (MD) to integrate the classical equations of motion.331 MD simulations are particularly desirable for current computational platforms because they are often highly parallelizable and limited only by the many-body terms in the force fields. Modern computers are sufficiently fast that many simulations can be run in real time to observe the molecular motions. Nonetheless asynchronous computing to obtain larger or longer MD simulations can readily be performed, often utilizing the same computing platforms. The necessity for such large-scale simulations lies in the fact that many processes cannot be isolated to a few interacting molecules, and they often require an explicit representation of the molecular environment. One extreme example using embarrassingly parallel computing is that of the Folding@Home project.332 Several packages are now available that simplify the process of implementing these algorithms to arbitrary systems, such as DL_POLY,333 and a module in NWCHEM.334 A larger number have been written specifically for biological systems such as CHARMM,335 TINKER,336 GROMACS,337 and NAMD.338 In the lowest order of accuracy, the force fields are generally pairwise, but they accommodate the largest systems for the longest trajectories. Nevertheless, as higher levels of accuracy are required, most modern potentials implemented in the cited codes also include higher-order corrections, including multipoles and polarization effects. In addition, so-called transferable force fields are increasingly being developed, allowing investigators a larger palette of molecules and larger portions of the phase diagram.339 For cases in which the force fields are unavailable or the underlying electron quantization is important, then Born-Oppenheimer Molecular Dynamics (BOMD), 340 Carr-Parrinello Molecular Dynamics (CPMD),341 or Atom-Centered Density Matrix Propagation (ADMP)342 have been integrated into commercial and free-ware computer codes.
In summary, at present there exist several cybertools for performing MD simulations of various systems by experts or near experts. These tools are fairly mature in capability, but the user interfaces have only recently started to make them available to scientists other than the experts in the computational chemistry community. The challenge will be to improve these cybertools to make their use completely transparent.
Multiscaling Simulations
While MD techniques are somewhat mature and have led to many successes in modeling the structure and dynamics of molecular systems, the use of multiscale methods connecting this microscopic level and the macroscopic world is growing now.343 Indeed the extensive set of contributions (more than 3500 pages) in the recent compendium344 describing multiscaling techniques serves to illustrate the importance of bridging this gap, as well as the breadth of techniques that are being aimed at it. Currently there exist many theories and a large effort in developing cybertools; however, the current state of the art does not contain mature computational packages at the level of the existing MD simulation packages. The development of such packages (and the associated advances in theoretical methods) in the context of addressing visualization of structures and dynamics is clearly an emerging area that would help advance chemical imaging.
Future Directions
Several promising avenues of research would greatly enhance the technological capacity of image processing, simulation, and modeling. To expedite the development of these various technologies, alterations in the field's current landscape are necessary. First, more accurate algorithms are needed for all imaging applications to ensure that the results generated from the algorithm are legitimate for varying implementations of the algorithm. One of the most critical needs associated with this task is a concerted effort to determine which algorithms are best suited for a particular image processing method. In addition, a more effective transfer of computer science advances in general image processing to scientific fields is urgently needed. To facilitate this transfer, rigorous collaborations between computer scientists and researchers using chemical imaging will be required. Furthermore, there is a serious gap in knowledge between the experts and routine users in the chemical imaging field. Often, researchers are unaware of or unfamiliar with which image processing methods, particularly multivariate methods, are most useful. If these rapid, target-specific routines could be incorporated into standard technique software it would accelerate widespread appropriate use in the field. There is also a need for investigators to incorporate the use of a priori information in order to optimize data analysis and image processing conditions. This requirement stems from the fact that many users apply image processing methods as though they know nothing about the sample(s) or system(s) when, in reality, users are rarely operating blindly. A related consideration is that most computer software is not optimized for speed or management of large datasets; researchers are therefore often discouraged from trying different approaches to the analysis of a single dataset. Increases in computational capacity are thus essential to generate novel image processing methods. Finally, to enhance our understanding of a variety of chemical processes, visualization methods must be developed and improved. Such methods are currently lagging behind image processing methods, and developed visualization methods have been implemented slowly in some chemical imaging communities. Developing visualization methods that correlate data across length and time scales and have the capacity to display and analyze such data will be crucial to the advancement of the field. Meanwhile, an ability to predict and describe structures at various levels using computer models is a powerful tool to help guide the visualization and interpretation of particular samples. For example, this ability could help bridge gaps in missing information or insufficient resolution (as long as it is used carefully.) Alternatively, it could help guide experimentalists to identify what regions of a structure would be particularly interesting. Thus, although such modeling would not image a particular structure, it is an extremely valuable tool that could help the overall effort.
NOTES AND REFERENCES
- 1.
- The committee would like to thank the following individuals for contributions made to this chapter: Steven R Higgins, Wright State University; Takashi Ito, Kansas State University; Michael V Mirkin, Queens College, City University of New York (CUNY); Rachel K Smith, Pennsylvania State University; David O. Wipf, Mississippi State University; Kateryna Artyushkova, University of New Mexico; and Svitlana Pylypenko, University of New Mexico.
- 2.
- For the Ernst and Wuthrich Nobel lectures, see http://nobelprize
.org /chemistry/laureates /1991/ernst-lecture.html and http://nobelprize .org /chemistry/laureates /2002/wuthrich-lecture.html - 3.
- Rugar D, Budakian R, Mamin HJ, Chui BW. Single spin detection by magnetic resonance force microscopy. Nature. 2004;430:329–332. [PubMed: 15254532]
- 4.
- Greenberg YS. Application of superconducting quantum interference devices to nuclear magnetic resonance. Rev. Mod. Phys. 1998;70:175–222.
- 5.
- Hu KN, Yu HH, Swager TM, Griffin RG. Dynamic nuclear polarization with biradicals. J. Am. Chem. Soc. 2004;126:10844–10855. [PubMed: 15339160]
- 6.
- Han SI, Garcia S, Lowery TJ, Ruiz EJ, Seeley JA, Chavez L, King DS, Wemmer DE, Pines A. NMR-based biosensing with optimized delivery of polarized 129Xe to solutions. Anal. Chem. 2005;77:4008–4012. [PubMed: 15987104]
- 7.
- Godard C, Duckett SB, Polas S, Tooze R, Whitwood AC. Detection of intermediates in cobalt-catalyzed hydroformylation using para-hydrogen-induced polarization. J. Am. Chem. Soc. 2005;127:4994–4995. [PubMed: 15810814]
- 8.
- Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12:1–16. [PMC free article: PMC2312400] [PubMed: 12493823]
- 9.
- Tugarinov V, Ollerenshaw JE, Kay LE. Probing side-chain dynamics in high molecular weight proteins by deuterium. J. Am. Chem. Soc. 2005;127:8214–8225. [PubMed: 15926851]
- 10.
- Tycko R. Biomolecular solid state NMR: Advances in structural methodology and applications to peptide and protein fibrils. Annu. Rev. Phys. Chem. 2001;52:575–606. [PubMed: 11326075]
- 11.
- For the Lauterbur and Mansfield Nobel lectures, see http://nobelprize
.org /medicine/laureates /2003/lauterbur-lecture.html and http://nobelprize .org /medicine/laureates /2003/mansfield-lecture.html - 12.
- Moonen CTW, Bandettini PA, editors. Functional MRI. Berlin: Springer-Verlag; 1999.
- 13.
- Gillard J, Waldman A, Barer P. Clinical MR Neuroimaging: Diffusion, Perfusion, Spectroscopy. Cambridge, U.K.: Cambridge University Press; 2005.
- 14.
- 15.
- 16.
- Mcdermott R, Lee SK, Kahem B, Trabesinger AH, Pines A, Clarke J. Microtesla MRI with a superconducting quantum interference device. Proc. Natl. Acad. Sci. USA. 2004;101:7857–7861. [PMC free article: PMC419521] [PubMed: 15141077]
- 17.
- de Zwart JA, Ledden PJ, van Geldern P, Bodurka J, Chu R, Duyn JH. Signal-to-noise ratio and parallel imaging performance of a 16-channel receive-only brain coil array at 3.0 Tesla. Magn. Resonan. Med. 2004;51:22–26. [PubMed: 14705041]
- 18.
- Sodickson DK, McKenzie CA. A generalized approach to parallel magnetic resonance imaging. Med. Phys. 2001;28:1629–1643. [PubMed: 11548932]
- 19.
- Hu KN, Yu HH, Swager TM, Griffin RG. Dynamic nuclear polarization with biradicals. J. Am. Chem. Soc. 2004;126:10844–10855. [PubMed: 15339160]
- 20.
- Golman K, Olsson LE, Axelsson O, Mansson S, Karlson M, Petersson JS. Molecular imaging using hyperpolarized 13C. Br. J. Radiol. 2003;76:S118–127. [PubMed: 15572334]
- 21.
- Han SI, Garcia S, Lowery TJ, Ruiz EJ, Seeley JA, Chavez L, King DS, Wemmer DE, Pines A. NMR-based biosensing with optimized delivery of polarized 129Xe to solutions. Anal. Chem. 2005;77:4008–4012. [PubMed: 15987104]
- 22.
- Albert MS, Cates GD, Driehuys B, Happer W, Saam B, Springer CS, Wishnia A. Biological magnetic resonance imaging using laser-polarized 129Xe. Nature. 1994;370:199–201. [PubMed: 8028666]
- 23.
- Rugar D, Budakian R, Mamin HJ, Chui BW. Single spin detection by magnetic resonance force microscopy. Nature. 2004;430:329–332. [PubMed: 15254532]
- 24.
- Jaffer FA, Weissleder R. Molecular imaging in the clinical arena. JAMA. 2005;293:855–862. [PubMed: 15713776]
- 25.
- Michaels CA, Stranick SJ, Richter LJ, Cavanagh RR. Scanning near-field infrared microscopy and spectroscopy with a broadband laser source. J. Appl. Phys. 2000;88:4832.
- 26.
- a. Haka AS, Shafer-Peltier KE, Fitzmaurice M, Crowe J, Dasari RR, Feld MS. Diagnosing breast cancer using raman spectroscopy. Proc. Natl. Acad. Sci. 2005;102:12371–12376. [PMC free article: PMC1194905] [PubMed: 16116095]
b. Widjaja E, Chen TC, Morris MD, Ignelzi MA, McCreadie BR. Band-target entropy minimization (BTEM) applied to hyperspectral Raman image data. Appl. Spectrosc. 2003;57:1353–1362. [PubMed: 14658148]
c. Caspers PJ, Lucassen GW, Puppels GJ. Combined in vivo confocal Raman spectroscopy and confocal microscopy of human skin. Biophys. J. 2003;85:572–580. [PMC free article: PMC1303112] [PubMed: 12829511] - 27.
- a. Jeanmaire DL, Duyne RPV. Surface Raman spectroscoelectrochemistry. Part 1. Heterocyclic, aromatic, and aliphatic amines adsorbed to anodized silver electrodes. J. Electroanal. Chem. 1977;84:1–20.
b. Albrecht MG, Creighton JA. Anomalously intense Raman spectra of pyridine at a silver electrode. J. Am. Chem. Soc. 1977;99:5215–5217. - 28.
- Moskovits M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 1985;57:783–826.
- 29.
- Capadona LP, Zheng J, González JI, Lee TH, Patel SA, Dickson RM. Nanoparticle-free single molecule anti-Stokes Raman spectroscopy. Phys. l Rev. Lett. 2005;94:058301. [PubMed: 15783704]
- 30.
- Adams DM, Brus L, Chidsey CED, Creager S, Creutz C, Kagan CR, Kamat PV, Lieberman M, Lindsay S, Marcus RA, Metzger RM, Michel-Beyerle ME, Miller JR, Newton MD, Rolison DR, Sankey O, Schanze KS, Yardley J, Zhu XY. Charge transfer on the nanoscale: Current status. J. Physi. Chem. B. 2003;107:6668–6697.
- 31.
- Kneipp K, Yang W, Kneipp H, Itzkan I, Dasari RR, Feld MS. Population pumping of excited vibrational states by spontaneous surface-enhanced Raman scattering. Phys. Rev. Lett. 1996;76:2444–2447. [PubMed: 10060701]
- 32.
- Nie S, Emory SR. Probing single molecules and single nanoparticles by surface-enhanced Raman scattering. Science. 1997;275:1102–6. [PubMed: 9027306]
- 33.
- Shen YR. The Principles of Nonlinear Optics. New York: John Wiley & Sons.; 1984.
- 34.
- Duncan MD, Reintjes J, Manuccia TJ. Scanning coherent anti-Stokes Raman microscope. Opt. Lett. 1982;7:350. [PubMed: 19714017]
- 35.
- Zumbusch A, Holtom GR, Xie XS. Three-dimensional vibrational imaging by coherent anti-Stokes Raman scattering. Phys. Rev. Lett. 1999;82:4142.
- 36.
- Cheng JX, Xie XS. Coherent anti-Stokes Raman scattering microscopy: Instrumentation, theory, and applications. J. Phys. Chem. B. 2004;108:827.
- 37.
- Potma EO, Xie XS. CARS microscopy for biology and medicine. Opt. Photon. News. 2004;15:40.
- 38.
- Hartschuh A, Sanchez EJ, Xie XS, Novotny L. High-resolution near-field Raman microscopy of single-walled carbon nanotubes. Phys. Rev. Lett. 2003;90:095503. [PubMed: 12689234]
- 39.
- Weissleder R. A clearer vision for in vivo imaging. Nat. Biotech. 2001;19:316–317. [PubMed: 11283581]
- 40.
- a. Docherty FT, Clark M, McNay G, Graham D, Smith WE. Multiple labelled nanoparticles for bio detection. Faraday Dis. 2004;126:281–288. [PubMed: 14992413]
b. Cao YWC, Jin RC, Mirkin CA. Nanoparticles with Raman spectroscopic fingerprints for DNA and RNA detection. Science. 2002;297:1536–1540. [PubMed: 12202825] - 41.
- Kneipp J, Kneipp H, Rice WL, Kneipp K. Optical probes for biological applications based on surface-enhanced Raman scattering from indocyanine green on gold nanoparticles. Anal.l Chem. 2005;77:2381–2385. [PubMed: 15828770]
- 42.
- a. Bhargava R, Levin IW. Time-resolved Fourier transform infrared spectrosocpic imaging. Appl. Spectrosc. 2003;57:357–366. [PubMed: 14658631]
b. Chan KLA, Kazarian SG, Mavraki A, Williams DR. Fourier transform infrared imaging of human hair with a high spatial resolution without the use of a synchrotron. Appl. Spectrosc. 2005;59:149–155. [PubMed: 15720754]
c. OuYang H, Sherman PJ, Paschalis EP, Boskey A, Mendelsohn R. Fourier transform infrared microscopic imaging: Effects of estrogen and estrogen deficiency on fracture healing in rat femurs. Appl.Spectrosc. 2004;58:1–9. [PubMed: 14727714] - 43.
- For a comprehensive overview of recent developments in infrared imaging, note an upcoming book, Bhargava Rohit, Levin Ira, editors. Spectrochemical Analysis Using Infrared Multichannel Detectors. Blackwell Publishing; 2006. [Google Scholar].
- 44.
- Kneipp J, Miller LM, Joncic M, Kittel M, Lasch P, Beekes M, Naumann D. In situ identification of protein structural changes in prion-infected tissue. Bioch. Biophys. Acta. 2003;1639:152–158. [PubMed: 14636946]
- 45.
- Williams GP. High power THz synchrotron sources. Phil. Trans. R. Soc. Lond. A. 2004;362:403. [PubMed: 15306529]
- 46.
- Globus T, Woolard D, Bykovskaia M, Gelmont B, Werbos L, Samuels A. THz-frequency spectroscopic sensing of DNA and related biological materials. Int. J. High Speed Electron Syst. 2003;13:903–936.
- 47.
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotech. 2004;22:1567–1572. [PubMed: 15558047]
- 48.
- Haupts U, Maiti S, Schwille P, Webb WW. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Nat. Acad. Sci. USA. 1998;95:13573–13578. [PMC free article: PMC24860] [PubMed: 9811841]
- 49.
- Pologruto TA, Yasuda R, Svoboda K. Monitoring neural activity and Ca2+ with genetically encoded Ca2+ indicators. J. Neurosci. 2004;24:9572–9579. [PMC free article: PMC6730159] [PubMed: 15509744]
- 50.
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. [PMC free article: PMC1302065] [PubMed: 11964263]
- 51.
- Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307:538–544. [PMC free article: PMC1201471] [PubMed: 15681376]
- 52.
- Yao J, Larson DR, Zipfel WR, Webb WW. Dark fraction and blinking of water-soluble quantum dots in solution, Proc. SPIE: Nanobiophoton. Biomed. Appl. II. Cartwright AN, Osinski M, editors. Vol. 5705. Bellingham, WA: SPIE Press; 2005.
- 53.
- Ow H, Larson DR, Srivastava M, Baird BA, Webb WW, Wiesner U. Bright and stable core-shell fluorescent silica nanoparticles. Nano Lett. 2005;5:113–117. [PubMed: 15792423]
- 54.
- a. Magde D, Elson E, Webb WW. Thermodynamic fluctuations in a reacting system—Measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 1972;29:705–708.
b. Webb WW. Fluorescence correlation spectroscopy: Inception, biophysical experimentations, and prospectus. Appl. Opt. 2001;40:3969–3983. [PubMed: 18360431] - 55.
- Foquet M, Korlach J, Zipfel WR, Webb WW, Craighead HG. DNA fragment sizing by single molecule detection in submicrometer-sized closed fluidic channels. Anal. Chem. 2002;74:1415–1422. [PubMed: 11922312]
- 56.
- a. Xie XS, Trautman JK. Optical studies of single molecules at room temperature. Annu. Rev. Physl. Chem. 1998;49:441–480. [PubMed: 15012434]
b. (b) Moerner WE, Orrit M. Illuminating single molecules in condensed matter. Science. 1999;283:1670–1676. [PubMed: 10073924]
c. Ishijima A, Yanagida T. Single molecule nano-bioscience. Trends Biochem. Sci. 2001;26:438–444. [PubMed: 11440856] - 57.
- Lu HP, Xun L, Xie XS. Single-molecule enzymatic dynamics. Science. 1998;282:1877–1882. [PubMed: 9836635]
- 58.
- Stryer L, Haugland RP. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 1967;58:719–726. [PMC free article: PMC335693] [PubMed: 5233469]
- 59.
- Weiss S. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 2000;7:724–729. [PubMed: 10966638]
- 60.
- Zhuang X, Kim H, Pereira MJB, Babcock HP, Walter NG, Chu S. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–1476. [PubMed: 12029135]
- 61.
- Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. [PubMed: 12560545]
- 62.
- Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 1986;11:288. [PubMed: 19730608]
- 63.
- Strick T, Allemand JF, Croquette V, Bensimon D. Twisting and stretching single DNA molecules. Prog. Biophys. Mol. Bio. 2000;74:115. [PubMed: 11106809]
- 64.
- Visscher K, Schnitzer MJ, Block SM. Single kinesin molecules studied with a molecular force clamp. Nature. 1999;400:184–189. [PubMed: 10408448]
- 65.
- a. Bustamente C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu. Rev. Biochem. 2004;73:705–748. [PubMed: 15189157]
b. Allemand JF, Bensimon D, Croquette V. Stretching DNA and RNA to probe their interactions with proteins. Curr. Opin. Struct. Biol. 2003;13:266–274. [PubMed: 12831877] - 66.
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5 nm localization. Science. 2003;300(5628):2061–2065. [PubMed: 12791999]
- 67.
- Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA. 2000;97:8206–8210. [PMC free article: PMC26924] [PubMed: 10899992]
- 68.
- White JG, Amos WB, Fordham M. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light-microscopy. J. Cell. Biol. 1987;105:41–48. [PMC free article: PMC2114888] [PubMed: 3112165]
- 69.
- Pawley JB. Handbook of Biological Confocal Microscopy (The Language of Science). New York: Plenum; 1995.
- 70.
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence micrscopy. Science. 1990;248:73–76. [PubMed: 2321027]
- 71.
- Holtmaat A, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang XQ, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. [PubMed: 15664179]
- 72.
- Pologruto TA, Yasuda R, Svoboda K. Monitoring neural activity and Ca2+ with genetically encoded Ca2+ indicators. J. Neurosci. 2004;24:9572–9579. [PMC free article: PMC6730159] [PubMed: 15509744]
- 73.
- Maiti S, Shear JB, Williams RM, Zipfel WR, Webb WW. Measuring serotonin distribution in live cells with three-photon excitation. Science. 1997;275:530–532. [PubMed: 8999797]
- 74.
- a. Jung JC, Schnitzer MJ. Multiphoton endoscopy. Opt. Lett. 2003;28:902–904. [PubMed: 12816240]
b. Levene MJ, Dombeck DA, Molloy RP, Kasischke KA, Williams RM, Zipfel WR, Webb WW. In vivo multiphoton microscopy of deep brain tissue. J. Neurophysiol. 2004;91:1908–1912. [PubMed: 14668300]
c. Levene MJ, Dombeck DA, Williams RM, Skoch J, Hickey GA, Kasischke KA, Molloy RP, Ingelsson M, Stern EA, Klucken J, Backskai BJ, Zipfel WR, Hyman BT, Webb WW. In vivo multiphoton microscopy of deep tissue with gradient index lenses, Photon. West 2004. San Jose: International Society for Optical Engineering; 2004. - 75.
- Helmchen F, Fee MS, Tank DW, Denk W. A miniature head-mounted two-photon microscope: High-resolution brain imaging in freely moving animals. Neuron. 2001;31:903–912. [PubMed: 11580892]
- 76.
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. [PubMed: 15232110]
- 77.
- Fields RD. The other half of the brain. Sci. Am. 2004;290:54–61. [PubMed: 15045754]
- 78.
- Vishwasrao HD, Heikal AA, Kasischke KA, Webb WW. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J. Biol. Chem. 2005;280:25119–25126. [PubMed: 15863500]
- 79.
- Dombeck DA, Blanchard-Desce M, Webb WW. Optical recording of action potentials with second-harmonic generation microscopy. J. Neurosci. 2004;24:999–1003. [PMC free article: PMC6729824] [PubMed: 14749445]
- 80.
- a. Freund I, Deutsch M. 2nd-harmonic microscopy of biological tissue. Opt. Lett. 1986;11:94–96. [PubMed: 19730544]
b. Mertz J, Moreaux L. Second-harmonic generation by focused excitation of inhomogeneously distributed scatterers. Opt. Comm. 2001;196:325–330.
c. Williams RM, Zipfel WR, Webb WW. Interpreting second harmonic generation images of collagen I fibrils. Biophys. J. 2005;88:1377–1386. [PMC free article: PMC1305140] [PubMed: 15533922] - 81.
- Barad Y, Eisenber H, Horowitz M, Silberberg Y. Nonlinear scanning laser microscopy by third harmonic generation. Appl. Phys. Lett. 1997;70:922–924.
- 82.
- a. Moreaux L, Sandre O, Mertz J. Membrane imaging by second-harmonic generation microscopy. J. Opt. Soc. Am. B. 2000;17:1685–1694.
b. Cheng JX, Xie XS. Greens function formulation for third-harmonic generation microscopy. J. Opt. Soc. Am. B. 2002;19:1604–1610.
c. Cheng JX, Volkmer A, Xie XS. Theoretical and experimental characterization of coherent anti-Stokes Raman scattering microscopy. J. Opt. Soc. Am. B. 2002;19:1363–1375. - 83.
- Stubbs CD, Botchway SW, Slater SJ, Parker AW. The use of time-resolved fluorescence imaging in the study of protein kinase C localization in cells. BMC Cell Biol. 2005;6:22. [PMC free article: PMC1131895] [PubMed: 15854225]
- 84.
- Bowen BP, Enderlein J, Woodbury NW. Single-molecule fluorescence spectroscopy of TOTO on poly-AT and poly-GC DNA. Photochem.Photobiol. 2003;78(6):576–581. [PubMed: 14743865]
- 85.
- Uversky VN, Winter S, Lober G. Use of fluorescence decay times of 8-ANS-protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophys. Chem. 1996;60(3):79–88. [PubMed: 8679928]
- 86.
- a. Wan SK, Guo ZX, Kumar S, Aber J, Garetz BA. Noninvasive detection of inhomogeneities in turbid media with time-resolved log-slope analysis. J. Quant. Spectros. Radiat. Transfer. 2004;84:493–500.
b. Bordenave E, Abraham E, Jortusauskas G, Oberle J, Rulliere C. Single-shot correlation system for longitudinal imaging in biological tissues. Opt. Comm. 2002;208:275–283. [PubMed: 19424327] - 87.
- Inoue S. Video Microscopy. New York: Plenum Publishing Corporation; 1986.
- 88.
- Fujimoto JG, Pitris C, Boppart SA, Brezinski ME. Optical coherence tomography: An emerging technology for biomedical imaging and optical biopsy. Neoplasia. 2000;2:9–25. [PMC free article: PMC1531864] [PubMed: 10933065]
- 89.
- See for example: a. Dragsten PR, Webb WW, Paton JA, Capranic RR Auditory membrane vibrations—measurements at sub-angstrom levels by optical heterodyne spectroscopy. Science. 1974;185:55–57. [Google Scholar] b. Denk W, Webb WW Optical measurement of picometer displacements of transparent microscopic objects. Appl. Opt. 1990;29:2382–2391. [Google Scholar]
- 90.
- Gustafsson MGL. Extended resolution fluorescence microscopy. Curr. Opin. Struct. Biol. 1999;9:627–634. [PubMed: 10508771]
- 91.
- See for example: a. Sinclair MB, Timlin JA, Haaland DM, Werner-Washburne M Design, construction, characterization, and application of a hyperspectral microarray scanner. Appl. Opti. 2004;43:2079–2089. [Google Scholar] b. Schultz RA, Nielsen T, Zavaleta JR, Ruch R, Wyatt R, Garner HR Hyperspectral imaging: A novel approach for microscopic analysis. Cytometry. 2001;43:239–247. [Google Scholar] c. Zimmerman T, Rietdorf J, Pepperkok R Spectral imaging and its applications in live cell microscopy. Fed. Europ. Biochem.Soc. Lett. 2003;546:87–92. [Google Scholar]
- 92.
- Furuta T, Wang SSH, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY. Brominated 7-hydroxycoumarin-4-ylmethyls: Photolabile protecting groups with biologically useful cross sections for two photon photolysis. Proc. Natl. Aacd. Sci. USA. 1999;96:1193–1200. [PMC free article: PMC15439] [PubMed: 9990000]
- 93.
- Chklovskii DB, Mel BW, Svoboda K. Cortical rewiring and information storage. Nature. 2004;431:782–788. [PubMed: 15483599]
- 94.
- a. Zipfel WR, Williams RM, Christie RH, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton excited intrinsic fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. USA. 2003;100:7075–7080. [PMC free article: PMC165832] [PubMed: 12756303]
b. Zipfel WR, Williams RM, Webb WW. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotech. 2003;21:1369–1377. [PubMed: 14595365] - 95.
- Ouzounov DG, Moll KD, Foster MA, Zipfel WR, Webb WW, Gaeta AL. Delivery of nanojoule femtosecond pulses through large-core microstructured fibers. Opt. Lett. 2002;27:1513–1515. [PubMed: 18026490]
- 96.
- a. Leapman RD. Novel techniques in electron microscopy. Curr. Opin. Neurobiol. 2004;14:591–598. [PubMed: 15464893]
b. Frank J. Single-particle imaging of macromolecules by cryo-electron microscopy. Ann. Rev. Biophys. Biomol. Struct. 2002;31:303–319. [PubMed: 11988472] - 97.
- Rossmann MG, Morais MC, Leiman PG, Zhang W. Combining X-ray crystallography and electron microscopy. Structure. 2005;13:355–362. [PMC free article: PMC7173138] [PubMed: 15766536]
- 98.
- Rodgers DW. Cryocrystallography techniques and devices. In: Rossmann MG, Arnold E, editors. International Tables for Crystallography, Volume F: Crystallography of Biological Macromolecules. Dordrecht: Kluwer Academic Publishers; 2001. pp. 202–208.
- 99.
- a. Baker TS, Olson NH, Fuller SD. Adding the third dimension to virus life cycles: Three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 1999;63:862–922. [PMC free article: PMC98980] [PubMed: 10585969]
b. Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988;21:129–228. [PubMed: 3043536] - 100.
- a. Tao Y, Zhang W. Recent developments in cryoelectron microscopy reconstruction of single particles. Curr. Opin. Struct. Biol. 2000;10:127–136. [PubMed: 11042462]
b. van Heel M, Gowen B, Matadeen R, Orlova EV, Finn R, Pape T, Cohen D, Stark H, Schmidt R, Schatz M. Single particle electron cryo-microscopy: Towards atomic resolution. Q. Rev. Biophys. 2000;33:307–369. [PubMed: 11233408] - 101.
- a. Henderson R. Realizing the potential of electron cryomicroscopy. Q. Rev. Biophys. 2004;37:3–13. [PubMed: 17390603]
b. van Heel M, Gowen B, Matadeen R, Orlova EV, Finn R, Pape T, Cohen D, Stark H, Schmidt R, Schatz M. Single particle electron cryo-microscopy: Towards atomic resolution. Q. Rev. Biophys. 2000;33:307–369. [PubMed: 11233408] - 102.
- 103.
- Klie RF, Browning ND. Atomic scale characterization of a temperature dependence to oxygen vacancy segregation at SrTiO3 grain boundaries. Appl. Phys. Lett. 2000;77:3737–3739.
- 104.
- Klie RF, Zhu Y. Atomic resolution STEM analysis of defects and interfaces in ceramic materials. Micron. 2005;36(3):219–231. [PubMed: 15725591]
- 105.
- Williamson MJ, Tromp RM, Vereecken PM, Hull R, Ross FM. Dynamic microscopy of nanoscale cluster growth at the solid-liquid interface. Nat. Mat. 2003;2:532–536. [PubMed: 12872162]
- 106.
- Schmid AK, Bartelt NC, Hwang RQ. Alloying at surfaces by the migration of reactive two-dimensional islands. Science. 2000;290:1561–1564. [PubMed: 11090351]
- 107.
- Lobastov VA, Srinivasan R, Zewail AH. Four-dimensional ultrafast electron microscopy. Proc. Natl. Acad. Sci. 2005;102:7069–7073. [PMC free article: PMC1129142] [PubMed: 15883380]
- 108.
- a. Benninghoven A. Surface-analysis by secondary-ion mass-spectrometry (SIMS). Surf. Sci. 1994;300:246–260.
b. Chabala JM, Soni KK, Li J, Gavrilov KL, Levisetti R. High-resolution chemical imaging with scanning ion probe SIMS. Int. J. Mass Spectom. Ion Process. 1995;143:191–212. - 109.
- Wucher A, Sun S, Szakal C, Winograd N. Molecular depth profiling in ice matrices using C-60 projectiles. Appl. Surf. Sci. 2004;231-232:68–71.
- 110.
- McDonnell LA, Mize TH, Luxembourg SL, Koster S, Eijkel GB, Verpoorte E, de Rooij NF, Heeren ARM. Using matrix peaks to map topography: Increased mass resolution and enhanced sensitivity in chemical imaging. Anal. Chem. 2003;75:4373–4381. [PubMed: 14632039]
- 111.
- McDonnell LA, Piersma SR, Altelaar AFM, Mize TH, Luxembourg SL, Verhaert P, van Minnen J, Heeren RMA. Subcellular imaging mass spectrometry of brain tissue. J. Mass. Spectrom. 2005;40:160–168. [PubMed: 15706616]
- 112.
- a. Hercules DM, Novak FP, Viswanadham SK, Wilk ZA. Applications of laser microprobe mass-spectrometry in organic-analysis. Anal. Chim. Acta. 1987;195:61–71.
b. Seydel U, Haas M, Rietschel ET, Lindner B. Laser microprobe mass-spectrometry of individual bacterial organisms and of isolated bacterial compounds—A tool in microbiology. J. Microbiol. Methods. 1992;15:167–183.
c. Vaeck LV, Struyf H, Roy WV, Adams F. Organic and inorganic analysis with laser microprobe mass spectrometry. Part I: Instrumentation and methodology. Mass Spectrom. Rev. 1994;13:189–208. - 113.
- Bhattacharya SH, Raiford TJ, Murray KK. Infrared laser desorption/ionization on silicon. Anal. Chem. 2002;74:2228–2231. [PubMed: 12033331]
- 114.
- a. Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. [PubMed: 10866210]
b. McDonald WH, Yates JR. Proteomic tools for cell biology. Traffic. 2000;1:747–754. [PubMed: 11208064]
c. Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem. Rev. 2001;101:269–295. [PubMed: 11712248]
d. Godovac-Zimmermann J, Brown LR. Perspectives for mass spectrometry and functional proteomics. Mass Spectrom. Rev. 2001;20:1–57. [PubMed: 11223909] - 115.
- a. Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: A new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001;7:493–496. [PubMed: 11283679]
b. Gusev AI, Vasseur OJ, Proctor A, Sharkey AG, Hercules DM. Imaging of thin-layer chromatograms using matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 1995;67:4565–4570. - 116.
- Reyzer ML, Caldwell RL, Dugger TC, Forbes JT, Ritter CA, Guix M, Arteaga CL, Caprioli RM. Early changes in protein expression detected by mass spectrometry predict tumor response to molecular therapeutics. Cancer Res. 2004;64:9093–9100. [PubMed: 15604278]
- 117.
- Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. [PubMed: 15486296]
- 118.
- Chaurand P, Schwartz SA, Caprioli RM. Assessing protein patterns in disease using imaging mass spectrometry. J. Proteome Res. 2004;3:245–252. [PubMed: 15113100]
- 119.
- Chaurand P, Sanders ME, Jensen RA, Caprioli RM. Proteomics in diagnostic pathology: Profiling and imaging proteins directly in tissue sections. Am. J. Pathol. 2004;165:1057–1068. [PMC free article: PMC3118833] [PubMed: 15466373]
- 120.
- Binnig G, Rohrer H. Scanning tunneling microscopy. Helv. Phys. Acta. 1982;55:726–735.
- 121.
- Binnig G, Rohrer H, Gerber C, Weibel E. Surface studies by scanning tunneling microscopy. Phys. Rev. Lett. 1982;49:57–60.
- 122.
- Binnig G, Quate CF, Gerber C. Atomic force microscope. Phys. Rev. Lett. 56:930–933. [PubMed: 10033323]
- 123.
- a. Synge EH. A suggested method for extending microscopic resolution into the ultramicroscopic region. Phil. Mag. 1928;6:356–362.
b. Ash EA, Nicholls G. Super-resolution aperture scanning microscope. Nature. 1972;237:510–512. [PubMed: 12635200]
c. Lewis A, Isaacson M, Harootunian A, Muray A. Development of a 500 angstrom spatial resolution light microscope. Ultramicroscopy. 1984;13:227–232.
d. Pohl DW, Denk W, Lanz M. Optical stethoscopy: Image recording with resolution λ/20. Appl. Phys. Lett. 1984;44:651–653. - 124.
- Bard AJ, Fan FR, Kwak J, Lev O. Scanning electrochemical microscopy: Introduction and principles. Anal. Chem. 1989;61:132–138.
- 125.
- Engstrom RC, Weber M, Wunder DJ, Burgess R, Winquist S. Measurements within the diffusion layer using a microelectrode probe. Anal. Chem. 1986;58:844–848.
- 126.
- a. Chen CJ. Introduction to Scanning Tunneling Microscopy. New York: Oxford University Press; 1993.
b. Wiesendanger R. Scanning Probe Microscopy and Spectroscopy: Methods and Applications. New York: Cambridge University Press; 1994.
c. Bonnell DA. Scanning Probe Microscopy and Spectroscopy. New York: Wiley-VCH; 2001. - 127.
(a) Ibid.
b. Sarid D. Scanning Force Microscopy. New York: Oxford University Press; 1994.- 128.
- Riordan J. Physical Review Letters top ten. APS News. 2003;12(5):3–6.
- 129.
- Paesler MA, Moyer PJ. Near-Field Optics: Theory, Instrumentation, and Applications. New York: John Wiley & Sons; 1996.
- 130.
- Dunn RC. Luminescence in scanning tunneling microscopy on III-V nanostructures. Chem. Rev. 1999;99:2891–2927.
- 131.
- Mirkin MV, Bard AJ, editors. Scanning Electrochemical Microscopy. New York: Marcel Dekker; 2001.
- 132.
- a. Chen CJ. Introduction to Scanning Tunneling Microscopy. New York: Oxford University Press; 1993.
b. Wiesendanger R. Scanning Probe Microscopy and Spectroscopy: Methods and Applications. New York: Cambridge University Press; 1994.
c. Bonnell DA. Scanning Probe Microscopy and Spectroscopy. New York: Wiley-VCH; 2001.
d. Sarid D. Scanning Force Microscopy. New York: Oxford University Press; 1994.
e. Paesler MA, Moyer PJ. Near-Field Optics: Theory, Instrumentation, and Applications. New York: John Wiley & Sons; 1996.
f. Mirkin MV, Bard AJ, editors. Scanning Electrochemical Microscopy. New York: Marcel Dekker; 2001. - 133.
- Binh VT, Garcia N. Atomic metallic ion emission, field surface melting, and scanning tunneling microscopy tips. J. Phys. I. 1991;1:605–612.
- 134.
- Binh VT, Garcia N. On the electron and metallic ion emission from nanotips fabricated by field-surface-melting technique: Experiments on W and Au tips. Ultramicroscopy. 1992;42-44:80–90.
- 135.
- Hren JJ, Liu J. Field emission microscopy, The Handbook of Surface Imaging and Visualization. Hubbard AT, editor. New York: CRC Press; 1995.
- 136.
- Ehrlich G, Ernst N. Field ion microscopy and spectroscopy, The Handbook of Surface Imaging and Visualization. Hubbard AT, editor. New York: CRC Press; 1995.
- 137.
- Ruan L, Besenbacher F, Stensgaard I, Laegsgaard E. Atom resolved discrimination of chemically different elements on metal surfaces. Phys. Rev. Lett. 1993;70:4079–4082. [PubMed: 10054041]
See also note 115 (a).
- 138.
- Hamers RJ, Wang Y. Atomically-resolved studies of the chemistry and bonding at silicon surfaces. Chem. Rev. 1996;96:1261–1290. [PubMed: 11848789]
- 139.
- a. Poirier GE. Characterization of organosulfur molecular monolayers on Au(111) using scanning tunneling microscopy. Chem. Rev. 1997;97:1117–1127. [PubMed: 11851444]
b. Giancarlo LC, Flynn GW. Scanning tunneling and atomic force microscopy probes of self-assembled, physisorbed monolayers: Peeking at the peaks. Annu. Rev. Phys. Chem. 1998;49:297–336. [PubMed: 15012431]
c. Giancarlo LC, Flynn GW. Raising flags: Applications of chemical marker groups to study self-assembly, chirality, and orientation of interfacial films by scanning tunneling microscopy. Acc. Chem. Res. 2000;33:491–501. [PubMed: 10913238]
d. De Feyter S, Gesquiere A, Abdel-Mottaleb MM, Grim PCM, De Schryver FC, Meiners C, Sieffert M, Valiyaveettil S, Mullen K. Scanning tunneling microscopy: A unique tool in the study of chirality, dynamics, and reactivity in physisorbed organic monolayers. Acc. Chem. Res. 2000;33:520–531. [PubMed: 10955982] - 140.
- Stranick SJ, Parikh AN, Tao YT, Allara DL, Weiss PS. Phase separation of mixed-composition self-assembled monolayers into nanometer scale molecular domains. J. Phys. Chem. 1994;98:7636–7646.
- 141.
- a. Liu GY, Xu S, Qian Y. Nanofabrication of self-assembled monolayers using scanning probe lithography. Acc. Chem. Res. 2000;33:457–466. [PubMed: 10913234]
b. Kramer S, Fuierer RR, Gorman CB. Scanning probe lithography using self-assembled monolayers. Chem. Rev. 2003;103:4367–4418. [PubMed: 14611266]
c. Smith RK, Lewis PA, Weiss PS. Patterned self-assembled monolayers. Prog. Surf. Sci. 2004;75:1–68. - 142.
- Frenken JWM, Oosterkamp TH, Hendriksen BLM, Rost MJ. Pushing the limits of SPM. Mat. Today. 2005;8:20–25.
- 143.
- Rost MJ, Crama L, Schakel P, van Tol E, van Velzen-Williams GBEM, Overgauw CF, ter Horst H, Dekker H, Okhuijsen B, Seynen M, Vijftigschild A, Han P, Katan AJ, Schoots K, Schumm R, van Loo W, Oosterkamp TH, Frenken JWM. Scanning probe microscopes go video rate and beyond. Rev. Sci. Instrum. 2005;76:053710.
- 144.
- Pohl DW, Moller R. Tracking tunneling microscopy. Rev. Sci. Instrum. 1988;59:840–842.
- 145.
- Swartzentruber BS. Direct measurement of surface diffusion using atom-tracking scanning tunneling microscopy. Phys. Rev. Lett. 1996;76:459–462. [PubMed: 10061462]
- 146.
- a. Hamers RJ, Cahill DG. Ultrafast time resolution in scanned probe microscopies. Appl. Phys. Lett. 1990;57:2031–2033.
b. Hamers RJ, Cahill DG. Ultrafast time resolution in scanned probe microscopies: Surface photovoltage on Si(111)-(7×7). J. Vac. Sci. Technol. B. 1991;9:514–518.
c. Steeves GM, Freeman MR. Ultrafast scanning tunneling microscopy. Adv. Imag. Electron Phys. 2002;125:195–229.
d. Nunes G, Freeman MR. Picosecond resolution in scanning tunneling microscopy. Science. 1993;262:1029–1032. [PubMed: 17782049]
e. Kelly KF, Donhauser ZJ, Mantooth BA, Weiss PS. Expanding the capabilities of the scanning tunneling microscope, Scanning Probe Microscopy: Characterization, Nanofabrication and Device Application of Functional Materials. Vilarinho PM, Rosenwaks Y, Kingon A, editors. Vol. 186. Kluwer Academic; 2005. pp. 152–171. - 147.
- Hamers RJ. Atomic-resolution surface spectroscopy with the scanning tunneling microscopy. Annu. Rev. Phys. Chem. 1989;40:531–559.
- 148.
- Feenstra RM, Stroscio JA, Tersoff J, Fein AP. Atom-selective imaging of the GaAs(110) surface. Phys. Rev. Lett. 1987;58:1192–1197. [PubMed: 10034366]
- 149.
- Sykes ECH, Han P, Kandel SA, Kelly KF, McCarty GS, Weiss PS. Substrate-mediated interactions and intermolecular forces between molecules adsorbed on surfaces. Acc. Chem. Res. 2003;36:945–953. [PubMed: 14674785]
- 150.
- Berndt R, Gimzewski JK. Inelastic tunneling excitation of tip-induced plasmon modes on noble-metal surfaces. Phys. Rev. Lett. 1991;67:3796–3799. [PubMed: 10044828]
- 151.
- Alvarado SF, Renaud P, Abraham DL, Schonenberger CH, Arent DJ, Meier HP. Luminescence in scanning tunneling microscopy on III-V nanostructures. J. Vac. Sci. Technol. B. 1991;9:409–413.
- 152.
- Egusa S, Liau YH, Scherer NF. Imaging scanning tunneling microscope-induced electroluminescence in plasmonic corrals. Appl. Phys. Lett. 2004;84:1257–1259.
- 153.
- Haynes CL, Van Duyne RP. Nanosphere lithography: A versatile nanofabrication tool for studies of size-dependent nanoparticle optics. J. Phys. Chem. B. 2001;105:5599–5611.
- 154.
- Fromm DP, Sundaramurthy A, Schuck PJ, Kino GS, Moerner WE. Gap-dependent optical coupling of single “bowtie” nanoantennas resonant in the visible. Nano Lett. 2004;4:957–961.
- 155.
- Stipe BC, Rezaei MA, Ho W. Single-molecule vibrational spectroscopy and microscopy. Science. 1998;280:1732–1735. [PubMed: 9624046]
- 156.
- Jaklevic RC, Lambe J. Molecular vibration spectra by inelastic electron tunneling. Phys. Rev. Lett. 1966;165:1139–1142.
- 157.
- Albrecht TR, Akamine S, Zdeblick MJ, Quate CF. Microfabrication of integrated scanning tunneling microscope. J. Vac. Sci. Technol. A. 1990;8:317–318.
- 158.
- Bale M, Palmer RE. Microfabrication of silicon tip structures for multiple-probe scanning tunneling microscopy. J. Vac. Sci. Technol. B. 2002;20:364–369.
- 159.
- Kaiser WJ, Bell LD, Hecht MH, Davis LC. BEEM and the characterization of buried interfaces, Scanning Probe Microscopy and Spectroscopy: Theory, Techniques, and Applications. Bonnell DA, editor. New York: Wiley-VCH; 2001.
- 160.
- a. Chen CJ. Introduction to Scanning Tunneling Microscopy. New York: Oxford University Press; 1993.
b. Wiesendanger R. Scanning Probe Microscopy and Spectroscopy: Methods and Applications. New York: Cambridge University Press; 1994.
c. Bonnell DA. Scanning Probe Microscopy and Spectroscopy. New York: Wiley-VCH; 2001. - 161.
- Giessibl FJ. Atomic force microscopy's path to atomic resolution. Mat. Today. 2005;8:32–41.
- 162.
- Albrecht TR, Akamine S, Carver TE, Quate CF. Microfabrication of cantilever styli for the atomic force microscope. J. Vac. Sci. Technol. A. 1990;8:3386–3396.
- 163.
- Woolley AT, Cheung CL, Hafner JH, Lieber CM. Structural biology with carbon nanotube AFM probes. Chem. Biol. 2000;7:R193–R204. [PubMed: 11094343]
- 164.
- Nguyen CV, Stevens RMD, Barber J, Han J, Meyyappan M. Carbon nanotube scanning probe for profiling of deep-ultraviolet and 193 nm photoresist patterns. Appl. Phys. Lett. 2002;81:901–903.
- 165.
- a. Giessibl FJ. Atomic resolution of the silicon (111)-(7×7) surface by atomic force microscopy. Science. 1995;68:68–71. [PubMed: 17840059]
b. Fukui KL, Onishi H, Iwasawa Y. Atom-resolved image of the TiO2(110) surface by noncontact atomic force microscopy. Phys. Rev. Lett. 1997;79:4202–4205. - 166.
- Ohnesorge F, Binnig G. True atomic resolution by atomic force microscopy through repulsive and attractive forces. Science. 1993;260:1451–1456. [PubMed: 17739801]
- 167.
- a. Stolz M, Stoffler D, Aebi U, Goldsbury C. Monitoring biomolecular interactions by time-lapse atomic force microscopy. J. Struct. Biol. 2000;131:171–180. [PubMed: 11052889]
b. Lindsay SM. The scanning probe microscope in biology, Scanning Probe Microscopy and Spectroscopy: Theory, Techniques, and Applications. Bonnell DA, editor. New York: Wiley-VCH; 2001. - 168.
- Horber JKH, Miles MJ. Scanning probe evolution in biology. Science. 2003;302:1002–1005. [PubMed: 14605360]
- 169.
- Findeis MA. Approaches to discovery and characterization of inhibitors of amyloid beta-peptide polymerization. Biochim. Biophys. Acta. 2000;1502:76–84. [PubMed: 10899433]
- 170.
- Sulchek T, Yaralioglu GG, Quate CF, Minne SC. Characterization and optimization of scan speed for tapping-mode atomic force microscopy. Rev. Sci. Instrum. 2002;73:2928–2936.
- 171.
- Karrai K, Grober RD. Piezoelectric tip-sample distance control for near-field optical microscopes. Appl. Phys. Lett. 1995;66:1842–1844.
- 172.
- Sulchek T, Hsieh R, Adams JD, Yaralioglu GG, Minne SC, Quate CF, Cleveland JP, Atalar A, Adderton DM. High-speed tapping mode imaging with active Q control for atomic force microscopy. Appl. Phys. Lett. 2000;76:1473–1475.
See also note 159.
- 173.
- a. Bukofsky SJ, Grober RD. Video rate near-field scanning optical microscopy. Appl. Phys. Lett. 1997;71:2749–2751.
b. Magnussen OM, Polewska W, Zitzler L, Behm RJ. In situ video-STM studies of dynamic processes at electrochemical interfaces. Faraday Discuss. 2001;121:43–52. [PubMed: 12227584]
c. Frenken JWM, Oosterkamp TH, Hendriksen BLM, Rost MJ. Pushing the limits of SPM. Mat. Today. 2005;8:20–25.
d. Besenbacher F, Laegsgaard E, Stensgaard I. Fast scanning STM studies. Mat. Today. 2005;8:26–30.
e. Rost MJ, Crama L, Schakel P, van Tol E, van Velzen-Williams GBEM, Overgauw CF, ter Horst H, Dekker H, Okhuijsen B, Seynen M, Vijftigschild A, Han P, Katan AJ, Schoots K, Schumm R, van Loo W, Oosterkamp TH, Frenken JWM. Scanning probe microscopes go video rate and beyond. Rev. Sci. Instrum. 2005;76:053710. - 174.
- Humphris ADL, Hobbs JK, Miles MJ. Ultrahigh-speed scanning near-field optical microscopy capable of over 100 frames per second. Appl. Phys. Lett. 2003;83:6–8.
- 175.
- a. Takano H, Kenseth JR, Wong SS, O'Brien JC, Porter MD. Chemical and biochemical analysis using scanning force microscopy. Chem. Rev. 1999;99:2845–2890. [PubMed: 11749504]
b. Frisbie CD, Rozsnyai LF, Noy A, Wrighton MS, Lieber CM. Functional group imaging by chemical force microscopy. Science. 1994;265:2071–2073. [PubMed: 17811409]
c. Noy A, Vezenov DV, Lieber CM. Chemical force microscopy. Annu. Rev. Mater. Sci. 1997;27:381–421. - 176.
- Israelachvili J. Solvation forces and liquid structure, as probed by direct force measurements. Acc. Chem. Res. 1987;20:415–421.
- 177.
- a. Carpick RW, Salmeron M. Scratching the surface: Fundamental investigations of tribology with atomic force microscopy. Chem. Rev. 1997;97:1163–1194. [PubMed: 11851446]
b. Takano H, Kenseth JR, Wong SS, O'Brien JC, Porter MD. Chemical and biochemical analysis using scanning force microscopy. Chem. Rev. 1999;99:2845–2890. [PubMed: 11749504]
c. Frisbie CD, Rozsnyai LF, Noy A, Wrighton MS, Lieber CM. Functional group imaging by chemical force microscopy. Science. 1994;265:2071–2073. [PubMed: 17811409] - 178.
- Hoh JH, Cleveland JP, Prater CB, Revel JP, Hansma PK. Quantized adhesion detected with the atomic force microscope. J. Am. Chem. Soc. 1992;114:4917–4918.
- 179.
- Frisbie CD, Rozsnyai LF, Noy A, Wrighton MS, Lieber CM. Functional group imaging by chemical force microscopy. Science. 1994;265:2071–2073. [PubMed: 17811409]
- 180.
- a. Florin EL, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415–417. [PubMed: 8153628]
b. Lee GU, Chrisey LA, Colton RJ. Direct measurement of the forces between complementary strands of DNA. Science. 1994;266:771–773. [PubMed: 7973628]
c. Boland T, Ratner BD. Direct measurement of hydrogen bonding in DNA nucleotide bases by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1995;92:5297–5301. [PMC free article: PMC41681] [PubMed: 7777501] - 181.
- a. Noy A, Vezenov DV, Lieber CM. Chemical force microscopy. Annu. Rev. Mater. Sci. 1997;27:381–421.
b. Noy A, Sanders CH, Vezenov DV, Wond SS, Lieber CM. Chemically sensitive imaging in tapping mode by chemical force microscopy: Relationship between phase lag and adhesion. Langmuir. 1998;14:1508–1511. - 182.
- Hillborg H, Tomczak N, Olah A, Schonherr H, Vansco GJ. Nanoscale hydrophobic recovery: A chemical force microscopy study of UV/ozone-treated cross-linked poly(dimethylsiloxane). Langmuir. 2004;20:785–794. [PubMed: 15773106]
- 183.
- Ibid.
- 184.
- Takano H, Kenseth JR, Wong SS, O'Brien JC, Porter MD. Chemical and biochemical analysis using scanning force microscopy. Chem. Rev. 1999;99:2845–2890. [PubMed: 11749504]
- 185.
- Ibid.
- 186.
- Frisbie CD, Rozsnyai LF, Noy A, Wrighton MS, Lieber CM. Functional group imaging by chemical force microscopy. Science. 1994;265:2071–2073. [PubMed: 17811409]
- 187.
- Florin EL, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415–417. [PubMed: 8153628]
- 188.
- Boland T, Ratner BD. Direct measurement of hydrogen bonding in DNA nucleotide bases by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1995;92:5297–5301. [PMC free article: PMC41681] [PubMed: 7777501]
- 189.
- a. Lee GU, Chrisey LA, Colton RJ. Direct measurement of the forces between complementary strands of DNA. Science. 1994;266:771–773. [PubMed: 7973628]
b. Strunz T, Oroszlan K, Schafer R, Guntherodt HJ. Dynamic force spectroscopy of single DNA molecules. Proc. Natl. Acad. Sci. USA. 1999;96:11277–11282. [PMC free article: PMC18024] [PubMed: 10500167]
c. Noy A. Direct determination of the equilibrium unbinding potential profile for a short DNA duplex from force spectroscopy data. Appl. Phys. Lett. 2004;85:4792–4794.
d. Ling L, Butt HJ, Berger R. Rupture force between the third strand and the double strand within a triplex DNA. J. Am. Chem. Soc. 2004;126:13992–13997. [PubMed: 15506761] - 190.
- Williams JM, Han T, Beebe TP. Determination of single-bond forces from contact force variances in atomic force microscopy. Langmuir. 1996;12:1291–1295.
- 191.
- a. Fujihira M. Kelvin probe force microscopy of molecular surfaces. Ann. Rev. Mater. Sci. 1999;29:353–380.
b. Nonnenmacher M, O'Boyle MP, Wickramasinghe HK. Kelvin probe force microscopy. Appl. Phys. Lett. 1991;58:2921–2923. - 192.
- Fujihira M. Kelvin probe force microscopy of molecular surfaces. Ann. Rev. Mater. Sci. 1999;29:353–380.
- 193.
- Muller EM, Marohn JA. Microscopic evidence for spatially inhomogeneous charge trapping in pentacene. Adv. Mater. 2005;17:1410–1414. [PubMed: 34412424]
- 194.
- Johnson AS, Nehl CL, Mason MG, Hafner JH. Fluid electric force microscopy for charge density mapping in biological systems. Langmuir. 2003;19:10007–10010.
- 195.
- Wilson NR, MacPherson JV. Enhanced resolution electric force microscopy with single-wall carbon nanotube tips. J. Appl. Phys. 2004;96:3565–3567.
- 196.
- a. Kramer S, Fuierer RR, Gorman CB. Scanning probe lithography using self-assembled monolayers. Chem. Rev. 2003;103:4367–4418. [PubMed: 14611266]
b. Liu GY, Xu S, Qian Y. Nanofabrication of self-assembled monolayers using scanning probe lithography. Acc. Chem. Res. 2000;33:457–466. [PubMed: 10913234]
c. Nyffenegger RM, Penner RM. Nanometer-scale surface modification using the scanning probe microscope: Progress since 1991. Chem. Rev. 1997;97:1195–1230. [PubMed: 11851447] - 197.
- Piner RD, Zhu J, Xu F, Hong S, Mirkin CA. “Dip-pen” nanolithography. Science. 1999;283:661–663. [PubMed: 9924019]
- 198.
- Liu GY, Xu S, Qian Y. Nanofabrication of self-assembled monolayers using scanning probe lithography. Acc. Chem. Res. 2000;33:457–466. [PubMed: 10913234]
- 199.
- Schonherr H, Chechik V, Stirling CJM, Vansco GJ. Monitoring surface reactions at an AFM tip: An approach to follow reaction kinetics in self-assembled monolayers on the nanometer scale. J. Am. Chem. Soc. 2000;122:3679–3687.
- 200.
- a. Minne SC, Yaralioglu G, Manalis SR, Adams JD, Zesch J, Atalar A, Quate CF. Automated parallel high-speed atomic force microscopy. Appl. Phys. Lett. 1998;72:2340–2342.
b. Saya D, Fukushima K, Toshiyoshi H, Hashiguchi G, Fujita H, Kawakatsu H. Fabrication of single-crystal Si cantilever array. Sens. Actuators A. 2002:281–287.
c. Chow EM, Yaralioglu GG, Quate CF, Kenny TW. Characterization of a two-dimensional cantilever array with through-wafer electrical interconnects. Appl. Phys. Lett. 2002;80:664–666. - 201.
- Piner RD, Zhu J, Xu F, Hong S, Mirkin CA. “Dip-pen” nanolithography. Science. 1999;283:661–663. [PubMed: 9924019]
- 202.
- Liu GY, Xu S, Qian Y. Nanofabrication of self-assembled monolayers using scanning probe lithography. Acc. Chem. Res. 2000;33:457–466. [PubMed: 10913234]
- 203.
- Ibid.
- 204.
- a. Higgins DA, Vanden Bout DA, Kerimo J, Barbara PF. Polarization-modulation near field scanning optical microscopy of mesostructured materials. J. Phys. Chem. 1996;100:13794–13803.
b. Higgins DA, Kerimo J, Vanden Bout DA, Barbara PF. A molecular yarn: Near field optical studies of self assembled, flexible, fluorescent fibers. J. Am. Chem. Soc. 1996;118:4049–4058.
c. Higgins DA, Liao X, Hall J, Mei E. Simultaneous near-field optical birefringence and fluorescence contrast applied to the study of dye-doped polymer-dispersed liquid crystals. J. Phys. Chem. B. 2001;105:5874–5882.
d. Vickery SA, Dunn RC. Direct observation of structural evolution in palmitic acid monolayers following Langmuir-Blodgett deposition. Langmuir. 2001;17:8204–8209.
e. Aoki H, Ito S. Two-dimensional polymer investigated by scanning near-field optical microscopy: Phase separation of polymer blend monolayer. J. Phys. Chem. B. 2001;105:4558–4564. - 205.
- a. Higgins DA, Liao X, Hall J, Mei E. Simultaneous near-field optical birefringence and fluorescence contrast applied to the study of dye-doped polymer-dispersed liquid crystals. J. Phys. Chem. B. 2001;105:5874–5882.
b. Aoki H, Ito S. Two-dimensional polymer investigated by scanning near-field optical microscopy: Phase separation of polymer blend monolayer. J. Phys. Chem. B. 2001;105:4558–4564. - 206.
- Hwang J, Tamm LK, Bohm C, Ramalingam TS, Betzig E, Edidin M. Nanoscale complexity of phospholipid monolayers investigated by near-field scanning optical microscopy. Science. 1995;270:610–614. [PubMed: 7570018]
- 207.
- Vickery SA, Dunn RC. Direct observation of structural evolution in palmitic acid monolayers following Langmuir-Blodgett deposition. Langmuir. 2001;17:8204–8209.
- 208.
- a. Garcia-Parajo MF, Veerman JA, Bouwhuis R, Vvallee R, van Hulst NF. Optical probing of single fluorescent molecules and proteins. ChemPhysChem. 2001;2:347–360. [PubMed: 23686956]
b. Höppener C, Siebrasse JP, Peters R, Kubitscheck U, Naber A. High-resolution near-field optical imaging of single nuclear pore complexes under physiological conditions. Biophys. J. 2005;88:3681–3688. [PMC free article: PMC1305514] [PubMed: 15695631]
c. Enderle T, Ha T, Ogletree DF, Chemla DS, Magowan C, Weiss S. Membrane specific mapping and colocalization of malarial and host skeletal proteins in the Plasmodium falciparum infected erythrocyte by dual-color near-field scanning optical microscopy. Proc. Natl. Acad. Sci. USA. 1997;94:520–525. [PMC free article: PMC19545] [PubMed: 9012816] - 209.
- a. Betzig E, Chichester RJ. Single molecules observed by near-field scanning optical microscopy. Science. 1993;262:1422–1425. [PubMed: 17736823]
b. Ruiter AGT, Veerman JA, Garcia-Parajo MF, van Hulst NF. Single molecule rotational and translational diffusion observed by near-field scanning optical microscopy. J. Phys. Chem. A. 1997;101:7318–7323. - 210.
- Higgins DA, Kerimo J, Vanden Bout DA, Barbara PF. A molecular yarn: Near field optical studies of self assembled, flexible, fluorescent fibers. J. Am. Chem. Soc. 1996;118:4049–4058.
- 211.
- Teetsov JA, Vanden Bout DA. Imaging molecular and nanoscale order in conjugated polymer thin films with near-field scanning optical microscopy. J. Am. Chem. Soc. 2001;123:3605–3606. [PubMed: 11472137]
- 212.
- Dragnea B, Preusser J, Schade W, Leone SR, Hinsberg WD. Transmission near-field scanning microscope for infrared chemical imaging. J. Appl. Phys. 1999;86:2795–2799.
- 213.
- a. Akhremitchev BB, Pollack S, Walker GC. Apertureless scanning near-field infrared microscopy of a rough polymeric surface. Langmuir. 2001;17:2774–2781.
b. Lahrech A, Bachelot R, Gleyzes P, Boccara AC. Infrared near-field imaging of implanted semiconductors: Evidence of a pure dielectric contrast. Appl. Phys. Lett. 1997;71:575–577.
c. Keilmann F. Vibrational-infrared near-field microscopy. Vibrat.Spectrosc. 2002;29:109–114. - 214.
- a. Jahncke CL, Paesler MA, Hallen HD. Raman imaging with near-field scanning optical microscopy. Appl. Phys. Lett. 1995;67:2483–2485.
b. Anderson N, Hartschuh A, Novotny L. Near-field Raman microscopy. Mat. Today. 2005;8:50–54. - 215.
- Zeisel D, Dutoit B, Deckert V, Roth T, Zenobi R. Optical spectroscopy and laser desorption on a nanometer scale. Anal. Chem. 1997;69:749–754.
- 216.
- a. Hartschuh A, Sanchez EJ, Xie XS, Novotny L. High-resolution near-field Raman microscopy of single-walled carbon nanotubes. Phys. Rev. Lett. 2003;90:095503. [PubMed: 12689234]
b. Pettinger B, Ren B, Picardi G, Schuster R, Ertl G. Nanoscale probing of adsorbed species by tip-enhanced Raman spectroscopy. Phys. Rev. Lett. 2004;92:096101. [PubMed: 15089490] - 217.
- Jahncke CL, Paesler MA, Hallen HD. Raman imaging with near-field scanning optical microscopy. Appl. Phys. Lett. 1995;67:2483–2485.
- 218.
- a. Veerman JA, Otter AM, Kuipers L, van Hulst NF. High definition aperture probes for near-field optical microscopy fabricated by focused ion beam milling. Appl. Phys. Lett. 1998;72:3115–3117.
b. Pilevar S, Edinger K, Atia W, Smolyaninov I, Davis C. Focused ion-beam fabrication of fiber probes with well-defined apertures for use in near-field scanning optical microscopy. Appl. Phys. Lett. 1998;72:3133–3135. - 219.
- Betzig E, Chichester RJ. Single molecules observed by near-field scanning optical microscopy. Science. 1993;262:1422–1425. [PubMed: 17736823]
- 220.
- Hollars CW, Dunn RC. Probing single molecule orientations in model lipid membranes with near-field scanning optical microscopy. J. Chem. Phys. 2000;112:7822–7830.
- 221.
- Higgins DA, Kerimo J, Vanden Bout DA, Barbara PF. A molecular yarn: Near field optical studies of self assembled, flexible, fluorescent fibers. J. Am. Chem. Soc. 1996;118:4049–4058.
- 222.
- a. Teetsov JA, Vanden Bout DA. Imaging molecular and nanoscale order in conjugated polymer thin films with near-field scanning optical microscopy. J. Am. Chem. Soc. 2001;123:3605–3606. [PubMed: 11472137]
b. DeAro JA, Weston KD, Buratto SK, Lemmer U. Mesoscale optical properties of conjugated polymers probed by near-field scanning optical microscopy. Chem. Phys. Lett. 1997;277:532–538.
c. Blatchford JW, Gustafson TL, Epstein AJ, Vanden Bout DA, Kerimo J, Higgins DA, Barbara PF, Fu DK, Swager TM, MacDiarmid AG. Spatially and temporally resolved emission from aggregates in conjugated polymers. Phys. Rev. B. 1996;54:R3683–R3686. [PubMed: 9986346] - 223.
- Paesler MA, Moyer PJ. Near-Field Optics: Theory, Instrumentation, and Applications. New York: John Wiley & Sons; 1996.
- 224.
- a. Higgins DA, Vanden Bout DA, Kerimo J, Barbara PF. Polarization-modulation near field scanning optical microscopy of mesostructured materials. J. Phys. Chem. 1996;100:13794–13803.
b. Vaez-Iravani M, Toledo-Crow R. Pure linear polarization imaging in near field scanning optical microscopy. Appl. Phys. Lett. 1993;63:138–140.
c. McDaniel EB, McClain SC, Hsu JWP. Nanometer scale polarimetry studies using a near-field scanning optical microscope. Appl. Opt. 1998;37:84–92. [PubMed: 18268563]
d. Goldner LS, Fasolka MJ, Noufler S, Nguyen HP, Bryant GW, Hwang J, Weston KD, Beers KL, Urbas A, Thomas EL. Fourier analysis near-field polarimetry for measurement of local optical properties of thin films. Appl. Opt. 2003;42:3864–3881. [PubMed: 12868825]
e. Lacoste T, Huser T, Prioli R, Heinzelmann H. Contrast enhancement using polarization-modulation scanning near-field optical microscopy. Ultramicroscopy. 1998;71:333–340. - 225.
- Humphris ADL, Hobbs JK, Miles MJ. Ultrahigh-speed scanning near-field optical microscopy capable of over 100 frames per second. Appl. Phys. Lett. 2003;83:6–8.
- 226.
- Bukofsky SJ, Grober RD. Video rate near-field scanning optical microscopy. Appl. Phys. Lett. 1997;71:2749–2751.
- 227.
- a. McNeill JD, Barbara PF. NSOM investigation of carrier generation, recombination, and drift in a conjugated polymer. J. Phys. Chem. B. 2002;106:4632–4639.
b. Higgins DA, Hall JE, Xie A. Optical microscopy studies of dynamic within individual polymer-dispersed liquid crystal droplets. Acc. Chem. Res. 2005;38:137–145. [PubMed: 15709733] - 228.
- a. Levy J, Nikitin V, Kikkawa JM, Awschalom DD, Samarth N. Femtosecond near-field spin microscopy in digital magnetic heterostructures. J. Appl. Phys. 1996;97:6095–6100. [PubMed: 10060561]
b. Guenther T, Lienau C, Elsaesser T, Glanemann M, Axt VM, Kuhn T, Eshlaghi S, Wieck AD. Coherent nonlinear optical response of single quantum dots studied by ultrafast near-field spectroscopy. Phys. Rev. Lett. 2002;89:057401. [PubMed: 12144462] - 229.
- a. McNeill JD, Barbara PF. NSOM investigation of carrier generation, recombination, and drift in a conjugated polymer. J. Phys. Chem. B. 2002;106:4632–4639.
b. Adams DM, Kerimo J, Liu CY, Bard AJ, Barbara PF. Electric field modulated near-field photo-luminescence of organic thin films. J. Phys. Chem. B. 2000;104:6728–6736. - 230.
- Higgins DA, Hall JE, Xie A. Optical microscopy studies of dynamic within individual polymer-dispersed liquid crystal droplets. Acc. Chem. Res. 2005;38:137–145. [PubMed: 15709733]
- 231.
- Davy S, Spajer M. Near field optics: Snapshot of the field emitted by a nanosource using a photosensitive polymer. Appl. Phys. Lett. 1996;69:3306–3308.
- 232.
- a. H'dhili F, Bachelot R, Lerondel G, Barchiesi D, Royer P. Near-field optics: Direct observation of the field enhancement below and apertureless probe using a photosensitive polymer. Appl. Phys. Lett. 2001;79:4019–4021.
b. Tarun A, Daza MRH, Hayazawa N, Inouye Y, Kawata S. Apertureless optical near-field fabrication using an atomic force microscope on photoresists. Appl. Phys. Lett. 2002;80:3400–3402.
c. Yin X, Fang N, Zhang X, Martini IB, Schwartz BJ. Near-field two-photon nanolithography using an apertureless optical probe. Appl. Phys. Lett. 2002;81:3663–3665. - 233.
- Yin X, Fang N, Zhang X, Martini IB, Schwartz BJ. Near-field two-photon nanolithography using an apertureless optical probe. Appl. Phys. Lett. 2002;81:3663–3665.
- 234.
- Davy S, Spajer M. Near field optics: Snapshot of the field emitted by a nanosource using a photosensitive polymer. Appl. Phys. Lett. 1996;69:3306–3308.
- 235.
- a. Tarun A, Daza MRH, Hayazawa N, Inouye Y, Kawata S. Apertureless optical near-field fabrication using an atomic force microscope on photoresists. Appl. Phys. Lett. 2002;80:3400–3402.
b. Yin X, Fang N, Zhang X, Martini IB, Schwartz BJ. Near-field two-photon nanolithography using an apertureless optical probe. Appl. Phys. Lett. 2002;81:3663–3665. - 236.
- Sun S, Leggett GJ. Matching the resolution of electron beam lithography by scanning near-field photolithography. Nano Lett. 2004;4:1381–1384.
- 237.
- Lee MB, Atoda N, Tsutsui K, Ohtsu M. Nanometric aperture arrays fabricated by wet and dry etching of silicon for near-field optical storage application. J. Vac. Sci. Technol. B. 1999;17:2462–2466.
- 238.
- Bukofsky SJ, Grober RD. Video rate near-field scanning optical microscopy. Appl. Phys. Lett. 1997;71:2749–2751.
- 239.
- Sanchez EJ, Novotny L, Xie XS. Near-field fluorescence microscopy based on two-photon excitation with metal tips. Phys. Rev. Lett. 1999;82:4014–4017.
- 240.
- a. Jahncke CL, Paesler MA, Hallen HD. Raman imaging with near-field scanning optical microscopy. Appl. Phys. Lett. 1995;67:2483–2485.
b. Anderson N, Hartschuh A, Novotny L. Near-field Raman microscopy. Mat. Today. 2005;8:50–54.
c. Hartschuh A, Sanchez EJ, Xie XS, Novotny L. High-resolution near-field Raman microscopy of single-walled carbon nanotubes. Phys. Rev. Lett. 2003;90:095503. [PubMed: 12689234]
d. Pettinger B, Ren B, Picardi G, Schuster R, Ertl G. Nanoscale probing of adsorbed species by tip-enhanced Raman spectroscopy. Phys. Rev. Lett. 2004;92:096101. [PubMed: 15089490] - 241.
- Krug JT, Sanchez EJ, Xie XS. Design of near-field optical probes with optimal field enhancement by finite difference time domain electromagnetic simulation. J. Chem. Phys. 2002;116:10895–10901.
- 242.
- a. Novotny L, Sanchez EJ, Xie XS. Near-field optical imaging using metal tips illuminated by higher-order Hermite-Gaussian beams. Ultramicroscopy. 1998;71:21–29.
b. Sanchez EJ, Novotny L, Xie XS. Near-field fluorescence microscopy based on two-photon excitation with metal tips. Phys. Rev. Lett. 1999;82:4014–4017. - 243.
- a. Adams DM, Kerimo J, Liu CY, Bard AJ, Barbara PF. Electric field modulated near-field photo-luminescence of organic thin films. J. Phys. Chem. B. 2000;104:6728–6736.
b. Mei E, Higgins DA. Near-field optical microscopy studies of electric-field-induced molecular reorientation dynamics. J. Phys. Chem. A. 1998;102:7558–7563. - 244.
- a. Adams DM, Kerimo J, Liu CY, Bard AJ, Barbara PF. Electric field modulated near-field photo-luminescence of organic thin films. J. Phys. Chem. B. 2000;104:6728–6736.
b. Mei E, Higgins DA. Near-field optical microscopy studies of electric-field-induced molecular reorientation dynamics. J. Phys. Chem. A. 1998;102:7558–7563.
c. Moerner WE, Plakhotnik T, Irngartinger T, Wild UP, Pohl DW, Hecht B. Near-field optical spectroscopy of individual molecules in solids. Phys. Rev. Lett. 1994;73:2764–2767. [PubMed: 10057186] - 245.
- a. DeAro JA, Moses D, Buratto SK. Near-field photoconductivity of stretch-oriented poly(para-phenylene vinylene). Appl. Phys. Lett. 1999;75:3814–3816.
b. Gray MH, Hsu JWP, Giovane L, Bulsara MT. Effect of anisotropic strain on the crosshatch electrical activity in relaxed GeSi films. Phys. Rev. Lett. 2001;86:3598–3601. [PubMed: 11328032]
c. McNeill CR, Frohne H, Holdworth JL, Dastoor PC. Near-field scanning photocurrent measurements of polyfluorene blend devices: Directly correlating morphology with current generation. Nano Lett. 2004;4:2503–2507. - 246.
- Kwak J, Bard AJ. Scanning electrochemical microscopy. Theory of the feedback mode. Anal. Chem. 1989;61:1221–1227.
- 247.
- a. James PJ, Garfias-Mesias LF, Moyer PJ, Smyrl WH. Scanning electrochemical microscopy with simultaneous independent topography. J. Electrochem. Soc. 1998;145:L64–L66.
b. Lee Y, Ding Z, Bard AJ. Combined scanning electrochemical/optical microscopy with shear force and current feedback. Anal. Chem. 2002;74:3634–3643. [PubMed: 12175147] - 248.
- Ibid.
- 249.
- Fan FRF, Bard AJ. Imaging of biological macromolecules on mica in humid air by scanning electrochemical microscopy. Proc. Nat. Acad. Sci. USA. 1999;96:14222–14227. [PMC free article: PMC24418] [PubMed: 10588687]
- 250.
- Basame SB, White HS. Scanning electrochemical microscopy of metal/metal oxide electrodes. Analysis of spatially localized electron-transfer reactions during oxide growth. Anal. Chem. 1999;71:3166–3170. [PubMed: 21662907]
- 251.
- Fernandez JL, Walsh DA, Bard AJ. Thermodynamic guidelines for the design of bimetallic catalysts for oxygen electroreduction and rapid screening by scanning electrochemical microscopy. M-Co (M: Pd, Ag, Au). J. Am. Chem. Soc. 2005;127:357–365. [PubMed: 15631486]
- 252.
- Liu B, Rotenberg SA, Mirkin MV. Scanning electrochemical microscopy of living cells: Different redox activities of nonmetastatic and metastatic human breast cells. Proc. Nat. Acad. Sci. USA. 2000;97:9855–9860. [PMC free article: PMC27604] [PubMed: 10963658]
- 253.
- Liu B, Bard AJ, Mirkin MV, Creager SE. Electron transfer at self-assembled monolayers measured by scanning electrochemical microscopy. J. Am. Chem. Soc. 2004;126:1485–1492. [PubMed: 14759206]
- 254.
- Zhang J, Slevin CJ, Morton C, Scott P, Walton DJ, Unwin PJ. New approach for measuring lateral diffusion in Langmuir monolayers by scanning electrochemical microscopy (SECM): Theory and application. J. Phys. Chem. B. 2001;105:11120–11130.
- 255.
- a. Zhang J, Slevin CJ, Morton C, Scott P, Walton DJ, Unwin PJ. New approach for measuring lateral diffusion in Langmuir monolayers by scanning electrochemical microscopy (SECM): Theory and application. J. Phys. Chem. B. 2001;105:11120–11130.
b. O'Mullane AP, Macpherson JV, Unwin PR, Cervera-Montesinos J, Manzanares JA, Frehill F, Vos JG. Measurement of lateral charge propagation in [Os(Bpy)2(Pvp)Ncl]Cl thin films: A scanning electrochemical microscopy approach. J. Phys. Chem. B. 2004;108:7219–7227. - 256.
- a. Bath BD, White HS, Scott ER. Electrically facilitated molecular transport. Analysis of the relative contributions of diffusion, migration, and electroosmosis to solute transport in an ion-exchange membrane. Anal. Chem. 2000;72:433–442. [PubMed: 10695125]
b. Uitto OD, White HS. Scanning electrochemical microscopy of membrane transport in the reverse imaging mode. Anal. Chem. 2001;72:533–539. [PubMed: 11217758] - 257.
- a. Liu B, Rotenberg SA, Mirkin MV. Scanning electrochemical microscopy of living cells: Different redox activities of nonmetastatic and metastatic human breast cells. Proc. Nat. Acad. Sci. USA. 2000;97:9855–9860. [PMC free article: PMC27604] [PubMed: 10963658]
b. Yasykawa T, Kaya T, Matsue T. Dual imaging of topography and photosynthetic activity of a single protoplast by scanning electrochemical microscopy. Anal. Chem. 1999;71:4637–4641. - 258.
- Liu B, Rotenberg SA, Mirkin MV. Scanning electrochemical microscopy of living cells: Different redox activities of nonmetastatic and metastatic human breast cells. Proc. Nat. Acad. Sci. USA. 2000;97:9855–9860. [PMC free article: PMC27604] [PubMed: 10963658]
- 259.
- Wittstock G, Schuhmann W. Formation and imaging of microscopic enzymatically active spots on an alkanethiolate-covered gold electrode by scanning electrochemical microscopy. Anal. Chem. 1999;69:5059–5066.
- 260.
- Turyan I, Matsue T, Mandler D. Patterning and characterization of surfaces with organic and biological molecules by the scanning electrochemical microscope. Anal. Chem. 2000;72:3431–3435. [PubMed: 10952523]
- 261.
- Frenken JWM, Oosterkamp TH, Hendriksen BLM, Rost MJ. Pushing the limits of SPM. Mat. Today. 2005;8:20–25.
- 262.
- a. James PJ, Garfias-Mesias LF, Moyer PJ, Smyrl WH. Scanning electrochemical microscopy with simultaneous independent topography. J. Electrochem. Soc. 1998;145:L64–L66.
b. Lee Y, Ding Z, Bard AJ. Combined scanning electrochemical/optical microscopy with shear force and current feedback. Anal. Chem. 2002;74:3634–3643. [PubMed: 12175147] - 263.
- Gray MH, Hsu JWP, Giovane L, Bulsara MT. Effect of anisotropic strain on the crosshatch electrical activity in relaxed GeSi films. Phys. Rev. Lett. 2001;86:3598–3601. [PubMed: 11328032]
- 264.
- Treutler TH, Wittstock G. Combination of an electrochemical tunneling microscope (ECSTM) and a scanning electrochemical microscope (SECM): Application for tip-induced modification of self-assembled monolayers. Electrochim. Acta. 2003;48:2923–2932.
- 265.
- a. Macpherson JV, Unwin PJ. Combined scanning electrochemical-atomic force microscopy. Anal. Chem. 2000;72:276–285. [PubMed: 10658320]
b. Kranz C, Friedbacher G, Mizaikoff B. Integrating an ultramicroelectrode in an AFM cantilever—Combined technology for enhanced information. Anal. Chem. 2001;73:2491–2500. [PubMed: 11403290] - 266.
- a. James PJ, Garfias-Mesias LF, Moyer PJ, Smyrl WH. Scanning electrochemical microscopy with simultaneous independent topography. J. Electrochem. Soc. 1998;145:L64–L66.
b. Lee Y, Ding Z, Bard AJ. Combined scanning electrochemical/optical microscopy with shear force and current feedback. Anal. Chem. 2002;74:3634–3643. [PubMed: 12175147] - 267.
- Lee Y, Ding Z, Bard AJ. Combined scanning electrochemical/optical microscopy with shear force and current feedback. Anal. Chem. 2002;74:3634–3643. [PubMed: 12175147]
- 268.
- Alpuche-Aviles MA, Wipf DO. Impedance feedback control for scanning electrochemical microscopy. Anal. Chem. 2001;73:4873–4881. [PubMed: 11681463]
- 269.
- a. Sidles JA. Folded Stern-Gerlach experiment as a means for detecting nuclear magnetic resonance in individual nuclei. Phys. Rev. Lett. 1992;68:1124–1127. [PubMed: 10046086]
b. Rugar D, Yannoni CS, Sidles JA. Mechanical detection of"5agnetic resonance. Nature. 1992;360:563–566. - 270.
- a. Gremlich HU, Yan B. Infrared and Raman Spectroscopy of Biological Materials. New York: Marcel Dekker; 2001.
b. Lewis EN, Treado PJ, Reeder RC, Story GM, Dowres AE, Marcott C, Levin IW. FTIR spectroscopic imaging using an infrared focal-plane array detector. Anal. Chem. 1995;67:3377–3381. [PubMed: 8686889]
c. Kotula PG, Keenan MR, Michael JR. Automated analysis of SEM X-ray spectral images: A powerful new microanalysis tool. Microsc. Microanal. 2003;9:1–17. [PubMed: 12597783]
d. Artyushkova K, Fulghum J. XPS imaging, Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. Briggs D, Grant JT, editors. Chichester, Manchester, UK: IM publications and SurfaceSpectra Limited; 2003.
e. Winograd N. Prospects or imaging TOF-SIMS: From fundamentals to biotechnology. Appl. Surf. Sci. 2003;203:13–19.
f. Treado PJ, Nelson MP, Myers TH. Current state of the art in Raman microscopy and chemical imaging. Microbeam Anal. 2000, Proc. 2000;165:37–38. - 271.
- a. Oberholzer M, Ostreicher M, Christen H, Bruhlmann M. Methods in quantitative image analysis. Histochem. Cell Biol. 1996;105:333–355. [PubMed: 8781988]
b. Bovik A. Handbook of Image and Video Processing. San Diego, CA: Academic Press; 2000. - 272.
- Wilson DL, Kump KS, Eppell SJ, Marchant RE. Morphological restoration of atomic-force microscopy images. Langmuir. 1995;11:265–272.
- 273.
- Jak MJJ, Konstapel C, van Kreuningen A, Verhoeven J, van Gastel R, Frenken JWM. Automated detection of particles, clusters and islands in scanning probe microscopy images. Surf. Sci. 2001;494:43–52.
- 274.
- Meijering EHW, Niessen WJ, Viergever MA. Quantitative evaluation of convolution-based methods for medical image interpolation. Med. Image Anal. 2001;5:111–126. [PubMed: 11516706]
- 275.
- a. Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 1995;28(2):171–193. [PubMed: 7568675]
b. Bajaj C, Yu ZY, Auer M. Volumetric feature extraction and visualization of tomographic molecular imaging. J. Struct. Biol. 2003;144:132–143. [PubMed: 14643216]
c. Baldock B, Gilpin CJ, Koster AJ, Ziese U, Kadler KE, Kielty CM, Holmes DF. Three-dimensional reconstructions of extracellular matrix polymers using automated electron tomography. J. Struct. Biol. 2002;138:130–136. [PubMed: 12160709] - 276.
- Al-Kofahi KA, Can A, Lasek S, Szarowski DH, Dowell-Mesfin N, Shain W, Turner JT, Roysam B. Median-based robust algorithms for tracing neurons from noisy confocal microscope images. IEEE Trans. Info. Tech. Biomed. 2003;7:302–317. [PubMed: 15000357]
- 277.
- Udupa JK. Three-dimensional visualization and analysis methodologies: A current perspective. Radiographics. 1999;19:783–806. [PubMed: 10336203]
- 278.
- a. Leong FJWM, Brady M, McGee JO. Correction of uneven illumination (vignetting) in digital microscopy images. J.Clin. Pathol.y. 2003;56:619–621. [PMC free article: PMC1770032] [PubMed: 12890815]
b. Tomazevic D, Likar B, Pernus F. Comparative evaluation of retrospective shading correction methods. J. Microsc. 2002;208:212–223. [PubMed: 12460452] - 279.
- a. Lippert T, Ortelli E, Panitz JC, Raimondli F, Wambach J, Wei J, Wokuan A. Imaging—XPS/Raman investigation on the carbonization of polyimide after irradiation at 308 nm. Appl. Phys. A. 1999;69:S651–S654.
b. Artyushkova K, Fulghum JE. Multivariate analysis applied to topographical corrections in X-ray photoelectron images. Surf. Inter. Anal. 2004;36:1304–1313.
c. Allen GC, Hallam KR, Eastman JR, Graveling GJ, Ragnarsdottir VK, Skuse DR. XPS analysis of polyacrylamide adsorption to kaolinite, quartz and feldspar. Surf. Interface Anal. 1998;26:518–523. - 280.
- a. Jak MJJ, Konstapel C, van Kreuningen A, Verhoeven J, van Gastel R, Frenken JWM. Automated detection of particles, clusters and islands in scanning probe microscopy images. Surf. Sci. 2001;494:43–52.
b. Lippert T, Ortelli E, Panitz JC, Raimondli F, Wambach J, Wei J, Wokuan A. Imaging—XPS /Raman investigation on the carbonization of polyimide after irradiation at 308 nm. Appl. Phys. A. 1999;69:S651–S654. - 281.
- a. Oberholzer M, Ostreicher M, Christen H, Bruhlmann M. Methods in quantitative image analysis. Histochem. Cell Biol. 1996;105:333–355. [PubMed: 8781988]
b. Tyler B. Interpretation of TOF-SIMS images: Multivariate and univariate approaches to image de-noising, image segmentation and compound identification. Appl. Surf. Sci. 2003;203-204:825–831.
c. Newsam S, Bhagavathy S, Kenney C, Manjunath BS, Fonseca L. Object-based representations of spatial images. Acta Astronaut. 2001;48:567–577. - 282.
- Oberholzer M, Ostreicher M, Christen H, Bruhlmann M. Methods in quantitative image analysis. Histochem.Cell Biol. 1996;105:333–355. [PubMed: 8781988]
- 283.
- Wilson DL, Kump KS, Eppell SJ, Marchant RE. Morphological restoration of atomic-force microscopy images. Langmuir. 1995;11:265–272.
- 284.
- a. Newsam S, Bhagavathy S, Kenney C, Manjunath BS, Fonseca L. Object-based representations of spatial images. Acta Astronaut. 2001;48:567–577.
b. Bernasconi A, Antel SB, Collins DL, Bernasconi N, Olivier A, Dubeau F, Pike GB, Andermann F, Arnold DL. Texture analysis and morphological processing of magnetic resonance imaging assist detection of focal cortical dysplasia in extra-temporal partial epilepsy. Ann. Neurol. 2001;49:770–775. [PubMed: 11409429] - 285.
- a. Dur JC, Elsass F, Chaplain V, Tessier D. The relationship between particle-size distribution by laser granulometry and image analysis by transmission electron microscopy in a soil clay fraction. European J. Soil Sci. 2004;55:265–270.
b. Costa LD, Rodrigues CA, de Souza NC, Oliveira ON. Statistical characterization of morphological features of layer-by-layer polymer films by image analysis. J. Nanosci. Nanotech. 2003;3:257–261. [PubMed: 14509969] - 286.
- a. Rasa M, Philipse AP. Scanning probe microscopy on magnetic colloidal particles. J. Magn. Magn. Mat. 2002;252:101–103.
b. Plaschke M, Schafer T, Bundschuh T, Manh TN, Knopp R, Geckeis H, Kim JI. Size characterization of bentonite colloids by different methods. Anal. Chem. 2001;73:4338–4347. [PubMed: 11569829] - 287.
- a. Gerlich D, Mattes J, Eils R. Quantitative motion analysis and visualization of cellular structures. Methods. 2003;29:3–13. [PubMed: 12543067]
b. Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 2005;151:182–195. [PubMed: 16043363] - 288.
- a. Ashin R, Morimoto A, Nagase M, Vaillancourt R. Image compression with multiresolution singular value decomposition and other methods. Math. Comp. Modeling. 2005;41:773–790.
b. Schelkens P, Munteanu A, Barbarien J, Galca M, Giro-Nieto X, Cornelis J. Wavelet coding of volumetric medical datasets. IEEE Trans. Medi. Imag. 2003;22:441–458. [PubMed: 12760560] - 289.
- Wickes BT, Kim Y, Castner DG. Denoising and multivariate analysis of time-of-flight SIMS images. Surface and Interface Analysis. 2003;35:640–648.
- 290.
- a. Daubechies I. Orthnormal bases of compactly supported wavelets. Commu. Pure Appl.Math. 1988;41:909–996.
b. Mallat S. A theory for multiresolution signal decomposition. IEEE Tran. Pattern Anal. Machine Intell. 1989;11:674–693. - 291.
- Starck JL, Bijaoui A. A signal processing filtering and deconvolution by the wavelet transform. Signal Proc. 1994;35:195–211.
- 292.
- Wolkenstein M, Hutter H, Nikolov SG, Grasserbauer M. Improvement of SIMS image classification by means of wavelet de-noising. Fresenius J. Anal.l Chem. 1997;357:783–788.
- 293.
- a. Barequet G, Sharir M. Piecewise-linear interpolation between polygonal slices. Comput. Vis. Image Und. 1996;63:251–272.
b. Goshtasby A, Turner DA, Ackerman LV. Matching of tomographic image slices. IEEE Trans. Med. Imaging. 1992;11:507–516. [PubMed: 18222892]
c. Chen E, Williams L. View interpolation for image synthesis. SIGGRAPH '93. 1993. http://citeseer.ist.psu .edu/chen93view.html. - 294.
- a. Geladi P, Hans G. Multivariate Image Analysis. New York: John Wiley & Sons; 1996.
b. Geladi P, Isaksson H, Lindquist L, Wold S, Esbensen K. Principal component analysis of multivariate images. Chemomet. Intell. Lab. Sys. 1989;5:209–220.
c. de Juan A, Tauler R, Dyson R, Marcolli C, Rault M, Maeder M. Spectroscopic imaging and chemometrics: A powerful combination for global and local sample analysis. Trac-Trends Anal. Chem. 2004;23:70–79.
d. Richards JA. Remote Sensing Digital Image Analysis: An Introduction. New York: Springer; 1999.
e. Stubbings TC, Wolkenstein MG, Hutter H. Comparison of different approaches to analytical images classification. J. Trace Microprobe Tech. 1999;17:1–16.
f. Prutton M, Wilkinson DK, Kenny PG, Mountain DL. Data processing for spectrum-images: Extracting information from the data mountain. Appl. Surf. Sci. 1999;145:1–10. - 295.
- a. Geladi P, Isaksson H, Lindquist L, Wold S, Esbensen K. Principal component analysis of multivariate images. Chemomet. Intell. Lab. Sys. 1989;5:209–220.
b. Prutton M, Wilkinson DK, Kenny PG, Mountain DL. Data processing for spectrum-images: Extracting information from the data mountain. Appl. Surf. Sci. 1999;145:1–10. - 296.
- a. Wickes BT, Kim Y, Castner DG. Denoising and multivariate analysis of time-of-flight SIMS images. Surf.Interface Anal. 2003;35:640–648.
b. Coullerez G, Lundmark S, Malmstrom E, Hult A, Mathieu HJ. ToF-SIMS for the characterization of hyperbranched aliphatic polyesters: Probing their molecular weight on surfaces based on principal component analysis (PCA). Surf Interface Anal. 2003;35:693–708.
c. Peterson RE, Tyler BJ. Surface composition of atmospheric aerosol: Individual particle characterization by TOF-SIMS. Appl. Surf. Sci. 2003;203:751–756.
d. Tyler B. Interpretation of TOF-SIMS images: Multivariate and univariate approaches to image de-noising, image segmentation and compound identification. Appl. Surf. Sci. 2003;203:825–831. - 297.
- a. Kotula PG, Keenan MR, Michael JR. Automated analysis of SEM X-ray spectral images: A powerful new microanalysis tool. Microsc. Microanal. 2003;9:1–17. [PubMed: 12597783]
b. Roseman AM. FindEM—A fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 2004;145:91–99. [PubMed: 15065677]
c. Jembrih D, Schreiner M, Peev M, Krejsa P, Clausen C. Identification and classification of iridescent glass artifacts with XRF and SEM/EDX. Mikrochim Acta. 2000;133:151–157.
d. Flesche H, Nielsen AA, Larsen R. Supervised mineral classification with semiautomatic training and validation set generation in scanning electron microscope energy dispersive spectroscopy images of thin sections. Math. Geol. 2000;32:337–366.
e. Egelandsdal B, Christiansen KF, Host V, Lundby F, Wold JP, Kvaal K. Evaluation of scanning electron microscopy images of a model dressing using image feature extraction techniques and principal component analysis. Scanning. 1999;21:316–325. - 298.
- a. Artyushkova K, Fulghum JE. Multivariate image analysis methods applied to XPS imaging data sets. Surf Interface Anal. 2002;33:185–195.
b. Peebles DE, Ohlhausen JA, Kotula PG, Hutton S, Blomfield C. Multivariate statistical analysis for x-ray photoelectron spectroscopy spectral imaging: Effect of image acquisition time. J. Vacuum Sci. Techn. A. 2004;22:1579–1586.
c. Walton J, Fairley N. Quantitative surface chemical-state microscopy by X-ray photoelectron spectroscopy. Surf Inter Anal. 2004;36:89–91. - 299.
- a. Duponchel L, Elmi-Rayaleh W, Ruckebusch C, Huvenne JP. Multivariate curve resolution methods in imaging spectroscopy: Influence of extraction methods and instrumental perturbations. J.Chem. Info. Com. Sci. 2003;43:2057–2067. [PubMed: 14632458]
b. Budevska BO, Sum ST, Jones TJ. Application of multivariate curve resolution for analysis of FT-IR microspectroscopic images of in situ plant tissue. Appl. Spectrosc. 2003;57:124–131. [PubMed: 14610947]
c. Budevska BO. Minimization of optical non-linearities in Fourier transform-infrared microspectroscopic imaging. Vibrational Spectrosc. 2000;24:37–45. - 300.
- a. Zhang DM, Ben-Amotz D. Enhanced chemical classification of Raman images in the presence of strong fluorescence interference. Appl. Spectrosc. 2000;54:1379–1383.
b. Sasic S, Clark DA, Mitchell JC, Snowden MJ. A comparison of Raman chemical images produced by univariate and multivariate data processing–A simulation with an example from pharmaceutical practice. Analyst. 2004;129:1001–1007.
c. Drumm CA, Morris MD. Microscopic Raman line-imaging with principal component analysis. Appl.Spectrosc. 1995;49:1331–1337.
d. Gallagher NB, Shaver JM, Martin EB, Morris J, Wise BM, Windig W. Curve resolution for multivariate images with applications to TOF-SIMS and Raman. Chemomet. Intell. Lab. Sys. 2004;73:105–117. - 301.
- a. Bonnet N. Multivariate statistical methods for the analysis of microscope image series: Applications in materials science. J.Microsc.—Oxford. 1998;190:2–18.
b. Hadjiiski L, Munster S, Oesterschulze E, Kassing R. Neural network correction of nonlinearities in scanning probe microscope images. J. Vacuum Sci. Tech. B. 1996;14:1563–1568.
c. Velthuizen RP, Hall LO, Clarke LP. Feature extraction for MRI segmentation. J. Neuro. 1999;9:85–90. [PubMed: 10208105] - 302.
- a. Roseman AM. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 2004;145:91–99. [PubMed: 15065677]
b. Jembrih D, Schreiner M, Peev M, Krejsa P, Clausen C. Identification and classification of iridescent glass artifacts with XRF and SEM/EDX. Mikrochim. Acta. 2000;133:151–157.
c. Flesche H, Nielsen AA, Larsen R. Supervised mineral classification with semiautomatic training and validation set generation in scanning electron microscope energy dispersive spectroscopy images of thin sections. Math. Geol. 2000;32:337–366.
d. Egelandsdal B, Christiansen KF, Host V, Lundby F, Wold JP, Kvaal K. Evaluation of scanning electron microscopy images of a model dressing using image feature extraction techniques and principal component analysis. Scanning. 1999;21:316–325. - 303.
- a. Tauler R, Smilde A, Kowalski BJ. Selectivity, local rank, 3-way data analysis and ambiguity in multivariate curve resolution. J. Chemom. 1995;9:31–58.
b. Duponchel L, Elmi-Rayaleh W, Ruckebusch C, Huvenne JP. Multivariate curve resolution methods in imaging spectroscopy: Influence of extraction methods and instrumental perturbations. J. Chem. Infor. Com. Sci. 2003;43:2057–2067. [PubMed: 14632458] - 304.
- a. Hadjiiski L, Munster S, Oesterschulze E, Kassing R. Neural network correction of nonlinearities in scanning probe microscope images. J. Vacuum Sci. Tech. B. 1996;14:1563–1568.
b. Duponchel L, Elmi-Rayaleh W, Ruckebusch C, Huvenne JP. Multivariate curve resolution methods in imaging spectroscopy: Influence of extraction methods and instrumental perturbations. J. Chem. Info. Com. Sci. 2003;43:2057–2067. [PubMed: 14632458] - 305.
- MATLAB: The Language of Technical Computing. The Mathworks, Inc.; Natick, Massachusetts:
- 306.
- PLS_toolbox. Eigenvector Research Inc.; Wenatchee, Washington:
- 307.
- 308.
- ENVI: The Environment for Visualizing Images. The Research Systems, Inc.; Boulder, Colorado:
- 309.
- Kotula PG, Keenan MR, Michael JR. Automated analysis of SEM X-ray spectral images: A powerful new microanalysis tool. Microsc. Microanal. 2003;9:1–17. [PubMed: 12597783]
- 310.
- Ibid.
- 311.
- a. Smentkowski VS, Ohlhausen JA, Kotula PG, Keenan MR. Multivariate statistical analysis of time-of-flight secondary ion mass spectrometry images-looking beyond the obvious. Appl. Surf. Sci. 2004;231-232:245–249.
b. Ohlhausen JAT, Keenan MR, Kotula PG, Peebles DE. Multivariate statistical analysis of time-of-flight secondary ion mass spectrometry images using AXSIA. Appl. Surf. Sci. 2004;231-232:230–234. - 312.
- Peebles DE, Ohlhausen JA, Kotula PG, Hutton S, Blomfield C. Multivariate statistical analysis for x-ray photoelectron spectroscopy spectral imaging: Effect of image acquisition time. J. Vacuum Sci. Tech. A. 2004;22:1579–1586.
- 313.
- a. Pohl C. Tools and methods for fusion of images of different spatial resolution. Int.Arch.Photogram. Remote Sens. 1999;32:W6.
b. Hall DL, McMullen SAH. Mathematical Techniques in Multisensor Data Fusion. Norwood, Massachusetts: Artech House; 2004. - 314.
- Glasbey CA, Martin NJ. Multimodal microscopy by digital image processing. J. Microsc.—Oxford. 1996;181:225–237.
- 315.
- Leonardi AJ, Blakistone BA, Kyryk SW. Application of microscopy in the paper industry: Case histories of the Mead corporation. Food Struc. 1990;9:203–213.
- 316.
- Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science. 2003;300:82–86. [PubMed: 12677057]
- 317.
- Richards JA. Remote Sensing Digital Image Analysis: An Introduction. New York: Springer; 1999.
- 318.
- a. Artyushkova K, Fulghum J. XPS imaging, Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. Briggs D, Grant JT, editors. Chichester, Manchester, UK: IM publications and SurfaceSpectra Limited; 2003.
b. Artyushkova K, Wall B, Koenig J, Fulghum JE. Correlative spectroscopic imaging: XPS and FT-IR studies of PVC/PMMA polymer blends. Appl. Spectrosc. 2000;54:1549–1558. - 319.
- Clarke FC, Jamieson MJ, Clark DA, Hammond SV, Jee RD, Moffat AC. Chemical image fusion. The synergy of FT-NIR and Raman mapping microscopy to enable a more complete visualization of pharmaceutical formulations. Anal. Chem. 2001;73:2213–2220. [PubMed: 11393843]
- 320.
- Carugo O, Pongor S. The evolution of structural databases. Trends Biotechnol. 2002;20:498–501. [PubMed: 12443870]
- 321.
- a. Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng ZK, Green RK, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE. The RCSB Protein Data Bank: A redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. [PMC free article: PMC540011] [PubMed: 15608185]
b. Venclovas C, Ginalski K, Kang C. Sequence-structure mapping errors in the PDB: OB-fold domains. Protein Sci. 2004;13:1594–1602. [PMC free article: PMC2279972] [PubMed: 15133161]
c. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. [PMC free article: PMC102472] [PubMed: 10592235] - 322.
- a. Fischer D, Pas J, Rychlewski L. The PDB-Preview database: A repository of in-silico models of “on-hold” PDB entries. Bioinformatics (Oxford). 2004;20:2482–2484. [PubMed: 15073011]
b. Fitzkee NC, Fleming PJ, Rose GD. The protein coil library: A structural database of nonhelix, nonstrand fragments derived from the PDB. Proteins—Struc. Function Bioinform. 2005;58:852–854. [PubMed: 15657933] - 323.
- Nattkemper TW. Multivariate image analysis in biomedicine. J. Biomed. Inform. 2004;37:380–391. [PubMed: 15488751]
- 324.
- Brown J, Buckley D, Coulthard A, Dixon AK, Dixon JM, Easton DF, Eeles RA, Evans DG, Gilbert FG, Graves M, Hayes C, Jenkins JP, Jones AP, Keevil SF, Leach MO, Liney GP, Moss SM, Padhani AR, Parker GJ, Pointon LJ, Ponder BA, Redpath TW, Sloane JP, Turnbull LW, Walker LG, Warren RM. Magnetic resonance imaging screening in women at genetic risk of breast cancer: Imaging and analysis protocol for the UK multicentre study. Magn. Reson. Imaging. 2000;18(7):765–776. [PubMed: 11027869]
- 325.
- a. Van Horn JD, Grethe JS, Kostelec P, Woodward JB, Aslam JA, Rus D, Rockmore D, Gazzaniga MS. The Functional Magnetic Resonance Imaging Data Center (fMRIDC): The challenges and rewards of large-scale databasing of neuroimaging studies. Philosoph. Trans. R Soc. Lond. B Biol. Sci. 2001;356:1323–1339. [PMC free article: PMC1088517] [PubMed: 11545705]
b. Van Horn JD, Gazzaniga MS. Databasing fMRI studies—Towards a “discovery science” of brain function. Nat. Rev. Neurosci. 2002;3:314–318. [PubMed: 11967562] - 326.
- Van Horn JD, Gazzaniga MS. Databasing fMRI studies—Towards a “discovery science” of brain function. Nat. Rev. Neurosci. 2002;3:314–318. [PubMed: 11967562]
- 327.
- Gonzalez-Couto E, Hayes B, Danckaert A. The life sciences Global Image Database (GID). Nucleic Acids Res. 2001;29:336–339. [PMC free article: PMC29843] [PubMed: 11125130]
- 328.
- a. Fayyad U, Haussler D, Stolorz P. Mining scientific data. Commun. ACM. 1996;39:51–57.
b. Hsu W, Lee ML, Goh KG. Image mining in IRIS: Integrated retinal information system. SIGMOD Record. 2000;29:593–593. - 329.
- Cooke CD, Ordonez C, Garcia EV, Omiecinski E, Krawczynska EG, Folks RD, Santana CA, DeBraal L, Ezquerra NF. Data mining of large myocardial perfusion SPECT (MPS) databases to improve diagnostic decision making. J. Nucl. Med. 1999;40(suppl.S):293P–293P.
- 330.
- Andrienko G, Andrienko N. Knowledge-based visualization to support spatial data mining. Lect. Notes Comp. Sci. 1999;1642:149–160.
- 331.
- a. Allen MP, Tildesley DJ. Computer Simulation of Liquids. Oxford: Clarendon Press; 1989.
b. Frenkel D, Smit B. Understanding Molecular Simulation. San Diego, CA: Academic Press; 2002. - 332.
- Larson SM, Snow CD, Shirts M, Pande VS. Computational Genomics. Grant R, editor. Norwich, UK: Horizon Press; 2002.
- 333.
- Todorov IT, Smith W. DL_POLY_3: The CCP5 national UK code for molecular-dynamics simulations. One contribution of 12 to a theme “Discrete element modelling: Methods and applications in the environmental sciences.” Phil. Trans. R. Soc. Lond. A. 2004;362:1835–1852. [PubMed: 15306418]
- 334.
- Kendall RA, Apra E, Bernholdt DE, Bylaska EJ, Dupuis M, Fann GI, Harrison RJ, Ju J, Nichols JA, Nieplocha J, Straatsma TP, Windus TL, Wong AT. High performance computational chemistry: An overview of NWChem a distributed parallel application. Computer Phys. Comm. 2000;128:260–283.
- 335.
- a. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983;4:187–217.
b. MacKerell AD Jr, Brooks B, Brooks CL III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The energy function and its parameterization with an overview of the program, The Encyclopedia of Computational Chemistry. Schleyer PvR, et al., editors. Chichester, U.K.: John Wiley & Sons; 1998. - 336.
- a. Ponder JW, Richards FM. An efficient newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 1987;8:1016–1024.
b. Ren P, Ponder JW. Consistent treatment of inter- and intramolecular polarization in molecular mechanics calculations. J. Comput. Chem. 2002;23:1497–1506. [PubMed: 12395419] - 337.
- van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meulenhoff PJ, Tieleman DP, Sijbers ALTM, Feenstra KA, van Drunen R, Berendsen HJC. Gromacs User Manual Version 3.2. 2004. wwwgromacsorg.
- 338.
- Laxmikant K, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comp. Phys. 1999;151:283–312.
- 339.
- Siepmann JI. Challenges in the development of transferable force fields for phase equilibrium calculations, Forum 2000: Fluid Properties for New Technologies, Connecting Virtual Design with Physical Reality. Rainwater JC, et al., editors. Boulder, CO: NIST Special Publication; 2001. pp. 110–112.
- 340.
- Bolton K, Hase WL, Peshlherbe GH. Modern Methods for Multidimensional Dynamics Computation in Chemistry. Thompson DL, editor. Singapore: World Scientific; 1998.
- 341.
- Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985;55:2471. [PubMed: 10032153]
- 342.
- a. Schlegel HB, Millam JM, Iyengar SS, Voth GA, Daniels AD, Scuseria GE, Frisch MJ. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001;114:9758–9763.
b. Iyengar SS, Schlegel HB, Millam JM, Voth GA, Scuseria GE, Frisch MJ. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001;115:10291–10302.
c. Schlegel HB, Iyengar SS, Li X, Millam JM, Voth GA, Scuseria GE, Frisch MJ. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002;117:8694–8704. - 343.
- a. Yip S. Synergistic materials science. Nat. Mat. 2003;2:3. [PubMed: 12652660]
b. Yip S, editor. Handbook of Materials Modeling. New York: Springer; 2005. - 344.
- Ibid.
- Imaging Techniques: State of the Art and Future Potential - Visualizing Chemistr...Imaging Techniques: State of the Art and Future Potential - Visualizing Chemistry
Your browsing activity is empty.
Activity recording is turned off.
See more...