

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
The lysosomal storage disorders (LSD) are a group of about 50 diseases that are characterised by an accumulation of waste products in the lysosomes, resulting in the formation of large intracellular vacuoles (fig. 1).

Figure 1
Enlarged vacuoles in a lysosomal storage disease. The electron microscopy picture of a leukocyte cell from a patient affected with alpha-mannosidosis was kindly provided by Dr. Dag Malm.
Many of the diseases that we now know as lysosomal storage disorders were first described long before the discovery of the lysosome in 1955 by de Duve.1 As the structure and function of this organelle was defined and the different lysosomal proteins identified, the concept of lysosomal storage disorders evolved. The last decade has witnessed major advances in our understanding of the clinical, biochemical and genetical aspects of lysosomal storage diseases.
Although individually rare the lysosomal storage disorders as a group have a frequency of about 1/8000 live births,2,3 making this disease group a major challenge for the health care system. As an increasing number of patients with milder forms are being identified, the current figures may underestimate the actual frequencies of lysosomal storage disorders.
The group of lysosomal storage disorders is usually caused by the lack of a hydrolase, its activator or a transporter causing accumulation of specific substrates in the lysosomes for each disorder type. Recently defects of the vesicular transport in the endosomal/lysosomal system have been reported to cause phenotypes similar to lysosomal storage disorders, for example mucolipidosis type IV.4 This group of deficiencies includes the neuronal ceroid lipofuscinoses and a growing number of pigmentary disorders, and the elucidation of the function of these proteins will in a fundamental way increase our understanding on how the vesicular trafficking in the cell is regulated. The storage diseases are inherited in an autosomal-recessive fashion, except Fabry disease,5 Hunter disease (MPS II)6 and Danon disease7 that are all X-linked recessive. Certain disorders are more prevalent in certain geographic areas or among those of a particular ethnicity. For example the majority of patients with aspartylglucosaminuria8 and Salla disease9 are from Finland. In a similar fashion Gaucher disease10 and Tay-Sachs disease11 are almost 100 times more prevalent in Ashkenazi Jewish decent that in the general population.
The lysosomal storage diseases have a broad spectrum of clinical phenotypes. In addition the age of onset, severity of symptoms and central nervous system manifestation can vary markedly within a single disorder type. Several lysosomal storage disorders such as for example Gaucher disease,12 Tay Sachs disease,13 Pompe disease,14 beta-galactosidase deficiency15 have infantile, juvenile and adult forms.
The severity of a lysosomal storage disorder type will depend partially on the type of accumulating waste product. For example in beta-mannosidosis the accumulating product in affected goats and cows are the trisaccharide ManGlcNAcGlcNAc causing severe brain disease and early death,16,17 while in affected humans that instead accumulate the disaccharide ManGlcNAc the disease phenotype is very mild.18 The severity will also depend on which cells or tissues that accumulate the waste products. Other factors that affect the disease outcome are the genetic background and environmental influence. The cells and tissues have certain thresholds of enzymatic activities below which clinical manifestations occur. This is probably the reason how an infantile and a juvenile/adult form of the same disease can affect different tissues, as for example beta-galactosidase-deficiency, which is named GM1-gangliosidosis in its infantile form causing severe brain disease, while in the juvenile/adult form it is named MPS IV since the phenotype is reminiscent of mucopolysaccharidosis without brain involvement.15
Most of the patients with a lysosomal storage disorder are born apparently healthy and the symptoms develop progressively. The speed and severity of the evolving symptoms depend on many factors as discussed above. Important from a therapeutic point of view is that lysosomal proteins added to the extracellular space will reach the lysosomes, usually via lectins on the plasma membrane as the mannose 6-phosphate receptor and the mannose receptor.19,20 This concept is known as enzyme replacement therapy, and was first utilised successfully on patients with a mild form of Gaucher disease in the 1980s. The recombinant glucocerebrosidase was available for treatment in 1990.21,22 Since then enzyme replacement therapy is a reality for Fabry disease23 and Hurler disease (MPS 1).24 Clinical trials with recombinant human enzymes are ongoing in Pompe disease,25,26 MPS II27 and MPS VI.28 Enzyme replacement trials in several knock out mouse models, as for example acid sphingomyelinase deficiency (Niemann Pick disease)29 and lysosomal α-mannosidase deficiency (α-mannosidosis)30 have proven promising ensuring that clinical trials on new diseases are about to begin. Enzyme replacement by bone marrow transplantation continues to be effective in nonneuropathic Gaucher disease and in some forms of mucopolysaccharidosis31 but it has high morbidity and mortality that limits its use in lysosomal storage disorders. Gene therapy has not been sufficiently developed to merit clinical trials, but studies on animal models of lysosomal storage disorders32 have shown that it may become an alternative therapy as soon as the safety has been documented.33 Recently, however, the planning of phase 1 clinical trials of Batten disease using adeno associated vectors was reported.34 Another therapeutic approach is to decrease the access of substrate (substrate deprivation therapy). Recently an inhibitor of glucose transferase, N-butyldeoxynojirimycin, was shown effective in treatment of nonneuronopathic Gaucher disease.35 It is hope that within short future up to 50% of the lysosomal storage diseases may become treated by enzyme replacement. The targeting of enzyme drugs to all the cells that need them is still a major problem, especially through the blood-brain barrier. The success of therapy is dependent on an early intervention. The combination of new therapies and the combined frequency of lysosomal disorders of 1/8000 live births should make a newborn screening programme justifiable. Studies are continuing on finding markers that can be used in such a screening programme.36
The Lysosomal Storage Disorders
The lysosomal storage disorders were divided into the following 5 groups:
- Defects in glycan degradation
- Defects in lipid degradation
- Defects in protein degradation
- Defects in lysosomal transporters
- Defects in lysosomal trafficking
Table 1 lists 50 lysosomal storage disorders belonging to these groups.
Table 1
Lysosomal storage disorders.
Defects in Glycan Degradation
The most common group of lysosomal storage disorders, represented by about 30 diseases, results from defects of glycan degradation. The group can be divided into the following four subgroups:
- Defects in glycprotein degradation
- Defects in glycolipid degradation
- Defects in glycosaminoglycan degradation
- Defect in glycogen degradation
Defects in Glycoprotein Degradation (Glycoproteinoses)
Figure 2 depicts how the lysosomal exoglycosidases acts in sequence37 to cleave off monosaccharides from a complex type N-glycan. The hydrolases 5 (α1-6mannosidase) and C (chitobiase) are probably not required for the complete degradation as the glycosidic bonds cleaved by these also are cleaved by other lysosomal enzymes (fig. 2). Indeed, no lysosomal disorder involving these enzymes have been reported. The deficiencies of hydrolase 3 (β-hexosaminidase) and hydrolase 2 (β-galactosidase) cause defects in glycoprotein-, glycosaminoglycan- and glycolipid-degradation. As the defects of glycolipid degradation cause the most severe clinical phenotypes, both of these enzyme deficiencies are discussed under part b). The deficiencies of hydrolase 6 (β-mannosidase) and hydrolase 7 (α-N-acetylgalactosaminidase (α-galactosidase B)) are both extremely rare and exhibit large variations in clinical severities from no apparent clinical symptoms to severe brain disease.38,39 However, the combination of few patients, consanguineous parents and large variation in clinical symptoms makes it difficult to assess the consequences of these two diseases in humans. Possibly the severe clinical phenotypes reported in some patients are caused by other factors, and that in the absence of these the clinical symptoms are mild and the diseases thus possibly underdiagnosed.18,40
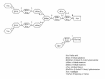
The deficiency of hydrolase 1 (sialidase) may be caused both by mutations in the sialidase gene 38 and in the gene encoding cathepsin A.41 Cathepsin A-deficiency causes combined sialidase and β-galactosidase deficiency due to its function in stabilising these two hydrolases.41 The early infantile forms cause mental retardation, dysostis multiplex, hepatosplenomegaly and early death. The clinical severities vary in continuum to the mild forms with normal life span and mild mental retardation.38,41 The deficiency of hydrolase A (α-fucosidase) does not normally cause hepatosplenomegaly and the typical symptoms are mental retardation, recurrent infections, growth retardation and dysostis multiplex. The clinical symptoms vary in continuum from the severe form with death before age of 10 and mild form with life span into adulthood.38 The deficiency of hydrolase 4 (lysosomal α-mannosidase) and hydrolase B (glycosylasparaginase (aspartylglucosaminidase)) cause uniquely accumulation of soluble oligosaccharides. Typical symptoms are mild mental retardation, recurrent infections, dysostis multiplex and hearing loss. Often these patients survive into adulthood.38,42
Defects in Glycolipid Degradation
This group includes defects in:
- Degradation of GM1 ganglioside
- Degradation of sulfatide
- Degradation of globotriaosylceramide
Gangliosides consist of a lipid moiety linked to a number of oligosaccharide structures differing in glycosidic linkage position, sugar configuration, neutral sugar and sialic acid content. The main gangliosides of the central nervous system (CNS) belong to the ganglio series, that is characterised by the tetrasaccharide Gal(β1-3)GalNAc(β1-4)Gal(β1-4)Glc to which residues of sialic acids are linked43 (fig. 3). The sulfatides are galactosylceramides to which sulphate is O-linked in 3-position.44 Galactosylceramide and sulfatide constitute a major part of the myelin layer in the CNS.45 As gangliosides and sulfatides serve important functions in brain membranes, the lack of enzymes involved in their degradation will change the lipid composition in brain and thus cause severe brain diseases. These diseases also exist as juvenile and adult forms associated with low residual enzyme activities. An exception is the hydrolysis of globotriaosylceramide by alpha-galactosidase A. Since this glycolipid is mainly represented in peripheral tissues, the lack of alpha-galactosidase A causes only mild symptoms without brain involvement.46

Figure 3
Degradation of GM1 ganglioside. (1) β-galactosidase, (2) β-hexosaminidase A, requiring GM2 activator protein (3) sialidase (4) glucocerebrosidase, requiring saposin C.
Degradation of GM1 Ganglioside
The degradation pathway of the GM1 ganglioside is illustrated in Figure 3 and the respective diseases caused by the lack of hydrolase 1 (beta-galactosidase), hydrolase 2 (beta-hexosaminidase) and hydrolase 4 (beta-glucocerebrosidase) are outlined in the table. The complete lack of these hydrolases results in early onset of symptoms usually about 4 months of age, severe neurological problems and death before the age of 4 years.12,13,15 The chronic (mild) forms of the defects can be quite complex showing a variety of clinical symptoms. The mild forms of beta-galactosidase deficiency (MPS IVB) and glucocerebrosidase deficiency (Gaucher disease type I) exhibit low residual activities and do not cause neurological deterioration. MPS IVB may be caused by the inability to degrade keratan sulfate. Gaucher disease type I results in engorged macrophages causing enlargement and dysfunction of the liver and spleen and damage to bone. The progression of the disease occurs slowly over decades.13 Gaucher disease type I was the first lysosomal storage disease that was successfully treated with enzyme replacement. 22
Degradation of Sulfatide
The inability to degrade sulfatides will cause accumulation of storage material in the brain and severe neurological symptoms, particularly by demyelination. Usually the disease begins at 3 months of age and soon progresses to severe mental and motor deterioration causing death before age of 2 years.44,47 Milder forms of the diseases causing a spectrum of clinical severities of brain disorders have been reported. The degradation of sulfatide occurs in two steps. In the first step the sulfate group is cleaved by 3-O-sulfogalactosyl cerebroside sulfatase (arylsulfatase A). The lack of this enzyme as well as the activator protein saposin B and Formyl-Glycin generating enzyme (FGE) which is required for the posttranslational modification of a cysteine in arylsulfatase A and other lysosomal sulfatases, cause symptoms typical for metachromatic leukodystrophy.44,48 In the second step the β-galactosyl group is cleaved from galactosylceramide. The lack of this cleavage causes Krabbe disease (globoid cell leukodystrophy).47
Degradation of Globotriaosylceramide
The globotriasylceramide is a glycolipid that is predominantly found in the vascular endothelium and not in the nervous tissue. Thus the defective degradation of this molecule does not affect the brain function to any large degree. Fabry disease is caused by the lack of lysosomal α-galactosidase. As the gene is X-linked the disease primarily affects males. Clinical manifestations appear in childhood or adolescense with pain in the extremities, angiokeratoma, hypohidrosis and retarded growth.46 The deposition of glycolipids occurs predominantly in lysosomes of endothelial, and smooth muscle cells of blood vessels. With increasing age severe renal impairment leads to renal failure, which contributes to the death. Enzyme replacement therapy is available for Fabry disease.23
Defects in Glycosaminoglycan Degradation (Mucopolysaccharidoses)
Defect in the degradation of glycosaminoglycans characterises the disease group mucopolysaccharidosis (MPS) (Table 1). Figure 4 illustrates the enzymes required in the degradation of heparan sulphate. The diseases in the MPS group share in a variable degree the clinical phenotypes. These include organomegaly, dysostis multiplex, decreased growth, recurrent infections and a chronic, progressive course of the disease.49 Most of the diseases do not affect the nervous system, and the disorders have thus been considered as potentially amenable to enzyme replacement therapy. Such therapy is available for MPS I,24 while clinical trials are ongoing in MPS II27 and MPS VI.28 As the degradation of different types of glycosaminoglycans may include similar glycosidases and sulfatases, the types of degradation products due to a single enzyme defect may originate from different types of partially degraded glycosaminoglycans.
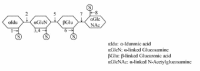
As shown in Figure 4, the first two step in the heparan sulfate degradation is catalysed by hydrolase 1 (iduronate sulfatase) and hydrolase 2 (alpha-iduronidase) which is required for the desulfatation of 2-sulfated iduronic acid residues and iduronic acids respectively, in dermatan sulfate and heparan sulfate. The lack of these enzyme activities causes Hunter syndrome (MPS II) and Hurler syndrome (MPS I) respectively.49 The gene encoding iduronate sulfatase is localised to chr. X and MPS I is thus restricted to males. The severe form of MPS I and II usually occurs between 2 and 4 years of age with progressive neurological and somatic involvement and death between 10 and 15 years. The mild forms are characterised by preservation of intelligence and survival into adulthood. The clinical variation is probably caused by residual enzyme activities.49 The deficiencies of hydrolase 3 (heparan N-sulfatase), enzyme 4 (Acetyl-CoA acetyltransferase), hydrolase 5 (alpha-N-acetylglucosaminidase) and hydrolase 8 (N-acetylglucosamine 6-sulfatase) cause Sanfilippo syndromes A-D (MPS IIIa-d),49 (Table 1). The syndromes are characterised by degeneration of the central nervous system (CNS) and mild somatic disease. The onset is usually between 2 and 4 years of age and severe CNS disorder is apparent by 6 to 10 years of age followed by early death.
Hydrolase 7 (β-glucuronidase) removes the glucuronic acid residues present in dermatan sulfate, chondroitin sulfate, dermatan sulfate and hyaluronan. The disease is characterised by dysostis multiplex, dysmorphic features, hepatosplenomegaly with a range of clinical severities. The infantile form resembles Hurler syndrome (MPS I). The mental retardation is modest. Hydrolase 6 (glucuronate-2-sulphatase) has not yet been associated with a lysosomal disease.49 Two lysosomal storage disorders in this group do not affect a hydrolase involved in the heparan sulfate degradation. These are N-acetylgalactosamine 4-sulfatase deficiency that cause Maroteaux-Lamy syndrome (MPS VI) and galactose 6-sulfatase deficiency that cause Morquio syndrome type A (MPS IVA), resulting from defects in dermatan and keratan sulfate degradation respectively. These diseases are characterised by short trunk dwarfism, skeletal dysplasia and mostly preservation of intelligense with an onset of symptoms at 2-4 years of age.49 The lack of lysosomal hyaluronidase causes the very rare disease MPS IX. The clinical symptoms of a single patient showed mild symptoms, including notable periarticular soft tissue masses, mild short stature, an absence of neurological or visceral involvement, and histological and ultrastructural evidence of a lysosomal storage disease.73
Defects in Glycogen Degradation
The defective degradation of glycogen in the lysosomes is caused by the lack of a single enzyme, lysosomal acid alpha-glucosidase resulting in glycogen storage disease type II (Pompe disease).14 The classic infantile form of the disease causes cardiomegaly, hypotonia, hepatomegaly and death before 2 years of age due to cardiorespiratory failure. There is an extreme variation of the clinical severities usually with skeletal muscle and cardiac involvement and a slower progressive course as compared to the infantile form. The milder forms appear to be associated with low residual activities. Clinical trials on enzyme replacement therapy are ongoing.26
Defects in Lipid Degradation
The defects in lipid degradation involve the two steps degradation of sphingomyelin to sphingosine and the ester hydrolysis of triglycerides and cholesteryl esters. The first step in the sphingomyelin degradation is a phosphodiester cleavage into ceramide and phosphocholine by acid sphingomyelinase. The lack of this enzyme results in Niemann Pick disease types A and B.50 Niemann Pick disease type A is an infantile form resulting in growth failure, hepatosplenomegaly and severe brain damage and leads to death before 2 years of age. Niemann pick disease type B is associated with low residual activity of acid sphingomyelinase and is phenotypically variable with typical symptoms as hepatosplenomegaly and pulmonary diffusion. Most of these patients have little neurological involvement and survival into adulthood. The second step in the sphingomyelin breakdown is the deamidation of ceramide into sphingosine and free fatty acid by acid ceramidase. The lack of this enzyme causes Farber disease.51 The classic infantile disease is characterised by painful swelling of joints, subcutaneous nodules and progressive hoarseness. In many cases there is a severe impairment of psychomotor development. Death usually occurs within 3 years of age. In the intermediate and mild forms of the disease the severities of the clinical symptoms vary considerably with life span ranging up to adulthood. The degrees of severities appear to correlate with the rate of sphingomyelin-derived ceramide degradation.51
The ester hydrolysis of triglycerides and cholesteryl esters is catalysed by acid lipase. Wolman disease is the infantile form of acid lipase deficiency causing hepatosplenomegaly, various gastrointestinal symptoms, adrenal calcification, failure to thrive. Death occurs before 1 year of age.52 Cholesteryl ester storage disease is a milder form caused by a low residual acid lipase activity. Typical clinical symptoms are atherosclerosis and hepatomegaly without mental retardation. In some patients hepatomegaly in adulthood may be the only symptom.
Defects in Protein Degradation
Diseases caused by the deficiency of lysosomal proteases are rare among the lysosomal storage disorders. Three disorders caused by the lack of cathepsin K, tripeptidyl-peptidase and palmitoyl-protein thioesterase are so far the only proteinase deficiencies reported. The lack of cathepsin K activity causes pycnodysostosis and the osteclasts in such patients display reduced capacity to degrade bone matrix during bone growth and remodelling. The disease is primarily a bone disease and common symptoms include short stature with a number of skeletal anomalies with typical dysmorphic features.53 Life span and intelligence are normal.
Lysosomal accumulation of autofluorescent, ceroid lipopigment material in various tissues and organs of which only brain tissue shows severe dysfunction and cell death are common features of the neuronal ceroid lipofuscinoses (NCLs). The deficiencies of palmitoyl-protein thioesterase (CLN1) and tripeptidyl-peptidase I (CLN2) result in the infantile (INCL) and late infantile (LINCL) forms of neuronal ceroid lipofuscinoses respectively.54 The symptoms are progressive neurogeneration, seizures, spacicity, dementia and blindness. The symptoms appear at about 1 year of age and death occurs around the age of 8 for INCL whereas the symptoms appear later in LINCL.54 Palmitoyl-protein thioesterase-deficiency (INCL) is prevalent in Finland with a carrier frequency of 1/50. The cause of the disease is not known, but may be related to a lysosomal accumulation of thioester-linked peptides in neurons. The physiological function of tripeptidyl peptidase is also not known. Functions in the degradation of neuropeptides55 and a role in antigen processing56 have been proposed.
Defects in Lysosomal Transporters
After lysosomal hydrolyses of macromolecules in the lysosomes the building blocks as monosaccharides and amino acids are transported through the lysosomal membrane into cytosol. Mutations in the sialic acid transporter (sialin) cause sialic acid storage disease.57 The infantile form of the disease (ISSD) is characterised by failure to thrive, hepatosplenomegaly, severe mental and motor retardation and dysostis multiplex. The children usually die before the age of 1 year. The juvenile/adult form of the disease is called Salla disease due to its prevalence in the Salla region of Finland. The children are born healthy, but develop psychomotoric delay and ataxia in infancy. Intelligence is moderately to severely reduced and they usually survive into adulthood.
Defiency of the cystine transporter (cystinosin) results in the storage disorder cystinoses. The affected children are usually born healthy and develop signs of kidney disease before age of 1 year.58 Symptoms include dehydration, acidosis, vomiting, failure to grow. The renal glomerular damage progresses with age, requiring dialysis or transplantation at 6 to 12 years of age. Oral cysteamine therapy has proven to be efficient in the systemic depletion of cystine, and patients receiving this drug before the age of 2 years display a delayed clinical onset of the disease.
Defects in Trafficking
Deficiencies in trafficking have recently been recognised to cause several lysosomal disorders. The deficient proteins may not be directly linked to a lysosomal location, but may be present in the trafficking route of lysosomal proteins from endoplasmic reticulum (ER) to the lysosomes. Thus, both cytosolic proteins as well as ER/Golgi/endosome/lysosome localised proteins involved in trafficking may cause lysosomal storage disorders.
Given the large number of proteins involved in trafficking one may expect the discovery of new protein deficiencies of this group in the near future.
The mucolipidosis diseases are due to the simultaneous lysosomal storage of lipids together with water-soluble substances. The classical trafficking defects are the mucolipidosis type II and III (I-cell disease). These diseases are caused by the defective activity of UDP-Nacetylglucosamine- phosphotransferase, a cis-Golgi localised enzyme that recognises and attaches GlcNAc-1-P onto terminal mannose-residues on N-glycans in lysosomal hydrolases.59 Defects in this transferase cause mislocalisation of lysosomal hydrolases into the extracellular milieu. The transferase is composed of two gene products, the α/β-subunit and the gamma-subunit. Mutations in the γ-subunit appear to be the major cause of mucolipidosis type III. This is a mild disorder with onset of symptoms about 2 to 4 years of age and a slow progression of symptoms with survival into adulthood.60 The symptoms share many features with the mild form of MPS I. Mucolipidosis type II is a more severe disease with death before the age of 8 years, but so far no mutations causing this disease has been reported.
Mucolipidosis type IV (MLIV) is characterized by psychomotor retardation and ophthalmological abnormalities. Severely affected as well as milder patients have been described. Over 80% of the MLIV patients are Ashkenazi Jews; the estimated heterozygote frequency in this population is 1/100.61 A broad spectrum of storage material stems from an abnormal endocytosis process in cells from MLIV patients. The missing protein, mucolipin-1 is a cation channel that seems to be involved in the acidification and normal endosomal function.4
Deficiency of LAMP-2 (Danon disease) belongs to the hereditary myopathies characterized by the development of autophagic vacuoles.62 LAMP-2 deficiency is a rare X-linked disorder.63 It is characterized clinically by cardiomyopathy, myopathy and variable mental retardation. The pathological hallmark of the disease is intracytoplasmic vacuoles containing autophagic material and glycogen in skeletal and cardiac muscle cells. The role of LAMP 2 is not yet unravelled, but it appears to be linked to the trafficking of lysosomal hydrolases.64
The defect in trafficking of cholesterol causing lysosomal/endosomal accumulation of unesterified cholesterol results in Niemann Pick disease type C (NP-C). This accumulation is particularly pronounced in liver and spleen but more deleteriously in brain, where it correlates with severe neuronal dysfunction.65 About 95% of the patients have mutations in the NPC1-gene, and the remainder in the NPC2-gene.66 Although the functions of these gene products are not known, it is believed that NPC1 is involved in regulation of the membrane trafficking in the endosomal/lysosomal system. The clinical manifestations are heterogeneous. Most patients have progressive neurological disease and hepatosplenomegaly. The symptoms appear most often in childhood and death occurs in the teenage years or early adulthood. The adult forms of NP-C cause psychiatric illness and dementia.
The neuronal ceroid lipofuscinoses (NCL) are a group of diseases that are characterized by progressive accumulation of autofluorescing waxy lipopigments (ceroid-lipofuscin) within the brain, accumulation of hydrophobic proteins in lysosomes, progressive neuroretinal symptomatology and cerebral atrophy. NCLs are caused by at least 8 mutant genes (CLN1-CLN8).67 Several lines of evidence have suggested that the CLN-gene products have roles in the protein trafficking in the endosomal/lysosomal system. Whereas the soluble lysosomal hydrolases CLN1 and CLN2 appears to be involved in lysosomal/endosomal proteolysis, CLN3, CLN6 and CLN8 are membrane proteins that may be involved in vesicular trafficking. CLN3-deficiency causing juvenile neuronal ceroid lipofuscinosis (Batten disease) is a form of NCL that is characterized by onset of neuroretinal symptoms between 4 and 10 years.67 It is the most common type of NCL in the United States and Europe. CLN3 has a proposed orthologue in Drosophilia, BTN1 that appears to be linked to the regulation of endocytosis.68 CLN6 and CLN8 are both transmembrane protein residing in the endoplasmic reticulum.69,70 CLN8-deficiency causes progressive epilepsy with mental retardation (Northern epilepsy). It is characterized by the onset of generalized seizures at between five and ten years of age, with progressive deterioration of mental development thereafter.67 The symptoms of CLN6-deficiency are similar. The functions of CLN6 and 8 remain to be found. Based on sequence homology analysis CLN8 has been suggested to be involved in lipid synthesis in the endoplasmic reticulum,70 but the relation to lysosomal dysfunction remains obscure. Possibly these proteins are involved in the ER to Golgi transport of lysosomal proteins.
The concept of lysosomes not only having an endpoint function in the degradation, but also function in the regulated secretion in immune cells and melanocytes, has resulted in the finding of a number of lysosomal diseases where the trafficking of the lysosomes to the plasma membrane is deficient.71 A number of human autosomal diseases give rise to both pigmentation and immune dysfunction, which is the hallmark for this group of lysosomal storage diseases. The first gene to be mapped was from patients affected with Chediak-Higashi syndrome. The patients are characterised by hypopigmentartion and enlarged lysosomes in all cell types. The defective gene LYST (lysosomal trafficking regulator) is a cytosolic protein that plays a role in regulating membrane fusion. Patients affected with Griscelli syndrome also exhibit hypopigmentation but the lysosomes are normal in size. The defective genes are RAB27A (type 2) and MYOV (type 1) and melanophilin (type 3) which are involved in the fusion of melanosomes with the plasma membrane. Patients affected with Chediak-Higashi and Griscelli syndromes have severe neurological problems due to infiltration of activated T-cells and macrophages in the brain. The disorders known as Hermansky-Pudlak syndrome (HPS) are a group of genetic diseases resulting from abnormal formation of intracellular vesicles. The respective proteins contribute to the formation of organella such as melanosomes and lysosomes. The syndrome has evolved into a group of genetically distinct disorders characterized by oculocutaneous albinism, a storage pool deficiency, and impaired formation or trafficking of intracellular vesicles.71,72 There are now seven disorders in this group, but taking into account that there are 16 mouse models of this syndrome, it is likely that new genetic types of Hermansky-Pudlak syndrome will appear.74 The best characterised protein defect is the one that causes Hermansky-Pudliak type 2 (HPS-2).75 This disorder is caused by mutations in ADTB3A, which codes for the cytosolic protein beta3A subunit of the adaptor protein-3 complex, AP3. This coat protein complex has been localized to the clathrin coat of trans Golgi network as well as to a peripheral endosomal compartment. AP3 is known to play a role assisting in vesicle formation from the trans-Golgi network or late endosomes.
References
- 1.
- de Duve C. Lysosomes, a new group of cytoplasmic particles. In: Hayashi T, ed. Subcellular Particles. Ronald Press. 1959:128–159.
- 2.
- Meikle PJ, Hopwood JJ, Clague AE. et al. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–54. [PubMed: 9918480]
- 3.
- Poorthuis BJ, Wevers RA, Kleijer WJ. et al. The frequency of lysosomal storage diseases in the Netherlands. Hum Gen. 1999;105:151–156. [PubMed: 10480370]
- 4.
- Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N. et al. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum Mol Genet. 2004;13(6):617–27. [PubMed: 14749347]
- 5.
- Masson C, Cisse I, Simon V. et al. Fabry disease: A review. Joint Bone Spine. 2004;71(5):381–3. [PubMed: 15474388]
- 6.
- Hopwood JJ, Bunge S, Morris CP. et al. Molecular basis of mucopolysaccharidosis type II: Mutations in the iduronate-2-sulphatase gene. Hum Mutat. 1993;2(6):435–42. [PubMed: 8111411]
- 7.
- Nishino I, Fu J, Tanji K. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–10. [PubMed: 10972294]
- 8.
- Ikonen E, Peltonen L. Mutations causing aspartylglucosaminuria (AGU): a lysosomal accumulation disease. Hum Mutat. 1992;1(5):361–5. [PubMed: 1301945]
- 9.
- Varho TT, Alajoki LE, Posti KM. et al. Phenotypic spectrum of Salla disease, a free sialic acid storage disorder. Pediatr Neurol. 2002;26(4):267–73. [PubMed: 11992753]
- 10.
- Horowitz M, Pasmanik-Chor M, Borochowitz Z. et al. Prevalence of glucocerebrosidase mutations in the israeli ashkenazi jewish population. Hum Mutat. 1998;12(4):240–4. [PubMed: 9744474]
- 11.
- Myerowitz R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum Mutat. 1997;9(3):195–208. [PubMed: 9090523]
- 12.
- Beutler E, Grabowski GA. Gaucher disease. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3635–3668.
- 13.
- Gravel RA, Kaback MM, Proia RL. et al. The GM2 Ganglisiodosis. In: Schriver CR, Beaudet AL, Sly WS, Valle D,eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3827–3876.
- 14.
- Hirshhorn R, Reuser A. Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3389–3420.
- 15.
- Beta-galactosidase deficiency (beta-galactosidosis). In: Suzuki Y, Oshima A, Nanba E, Schriver CR, Beaudet AL, Sly WS, Valle D, eds. GM1 gangliosidosis and Morquio B disease. 2001:3775–3809.
- 16.
- Patterson JS, Jones MZ, Lovell KL. et al. Neuropathology of bovine beta-mannosidosis. J Neuropathol Exp Neurol. 1991;50(5):538–46. [PubMed: 1895144]
- 17.
- Boyer PJ, Jones MZ, Rathke EJ. et al. Regional central nervous system oligosaccharide storage in caprine beta-mannosidosis. J Neurochem. 1990;55(2):660–4. [PubMed: 2370553]
- 18.
- Bedilu R, Nummy KA, Cooper A. et al. Variable clinical presentation of lysosomal beta-mannosidosis in patients with null mutations. Mol Genet Metab. 2002;77(4):282–90. [PubMed: 12468273]
- 19.
- Bijsterbosch MK, Donker W, van de Bilt H. et al. Quantitative analysis of the targeting of mannose-terminal glucocerebrosidase. Predominant uptake by liver endothelial cells. Eur J Biochem. 1996;237(2):344–9. [PubMed: 8647071]
- 20.
- Zhu Y, Li X, Kyazike J. et al. Conjugation of mannose 6-phosphate-containing oligosaccharides to acid alpha -glucosidase improves the clearance of glycogen in Pompe mice J Biol Chem 2004, [Epub ahead of print]. [PubMed: 15383547]
- 21.
- Whittington R, Goa KL. Alglucerase. A review of its therapeutic use in Gaucher's disease. Drugs. 1992;44(1):72–93. [PubMed: 1379912]
- 22.
- Grabowski GA, Barton NW, Pastores G. et al. Enzyme therapy in type 1 Gaucher disease: Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122:33–39. [PubMed: 7985893]
- 23.
- Mignani R, Cagnoli L. Enzyme replacement therapy in Fabry's disease: Recent advances and clinical applications. J Nephrol. 2004;17(3):354–63. [PubMed: 15365954]
- 24.
- Wraith JE, Clarke LA, Beck M. et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr. 2004;144(5):581–8. [PubMed: 15126990]
- 25.
- Van den Hout JM, Kamphoven JH, Winkel LP. et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113(5):e448–57. [PubMed: 15121988]
- 26.
- Lachmann RH. alpha-glucosidase (CHO) (Genzyme). Curr Opin Investig Drugs. 2004;5(10):1101–10. [PubMed: 15535432]
- 27.
- Muenzer J, Lamsa JC, Garcia A. et al. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): A preliminary report. Acta Paediatr Suppl. 2002;91(439):98–9. [PubMed: 12572850]
- 28.
- Harmatz P, Whitley CB, Waber L. et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr. 2004;144(5):574–80. [PubMed: 15126989]
- 29.
- Bae JS, Jang KH, Schuchman TR. et al. Comparative effects of recombinant acid sphingomyelinase administration by different routes in niemann-pick disease mice. Exp Anim. 2004;53(5):417–21. [PubMed: 15516789]
- 30.
- Roces DP, Lullmann-Rauch R, Peng J. et al. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: A preclinical animal study. Hum Mol Genet. 2004;13(18):1979–88. [PubMed: 15269179]
- 31.
- Hoogerbrugge PM, Brouwer OF, Bordigoni P. et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet. 1995;345(8962):1398–402. [PubMed: 7760610]
- 32.
- Zheng Y, Rozengurt N, Ryazantsev S. et al. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol Genet Metab. 2003;79(4):233–44. [PubMed: 12948739]
- 33.
- Barranger JM, Novelli EA. Gene therapy for lysosomal storage disorders. Expert Opin Biol Ther. 2001;1(5):857–67. [PubMed: 11728220]
- 34.
- Mandel RJ, Burger C. Clinical trials in neurological disorders using AAV vectors: Promises and challenges. Curr Opin Mol Ther. 2004;6(5):482–90. [PubMed: 15537049]
- 35.
- Elstein D, Hollak C, Aerts JM. et al. Sustained therapeutic effects of oral miglustat (Zavesca, N -butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J Inherit Metab Dis. 2004;27(6):757–66. [PubMed: 15505381]
- 36.
- Meikle PJ, Ranieri E, Simonsen H. et al. Newborn screening for lysosomal storage disorders: Clinical evaluation of a two-tier strategy. Pediatrics. 2004;114(4):909–16. [PubMed: 15466084]
- 37.
- Aronson NN, Kuranda MJ. Lysosomal degradation of Asn-linked glycoproteins. FASEB J. 1989;3:2615–2622. [PubMed: 2531691]
- 38.
- Thomas G. Disorders of glycoprotein degradation: alpha-mannosidosis, beta-Mannosidosis, Fucosidosis and Sialidosis. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3507–3534.
- 39.
- Desnick RJ, Schindler L. Alpha-N-acetylgalactosaminidase deficiency: Schindler disease. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3483–3506.
- 40.
- Sakuraba H, Matsuzawa F, Aikawa S. et al. Structural and immunocytochemical studies on alpha-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki disease). J Hum Genet. 2004;49:1–8. [PubMed: 14685826]
- 41.
- d'Azzo A, Andria G, Strisiugli P. et al. Galactosialidosis. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3811–3826.
- 42.
- Aula P, Jalanko A, Peltonen L. Aspartylglucosaminuria. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3535–3550.
- 43.
- von FiguraK, Gieselman V, Jaeken J. Metachromatic leukodystrophy. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3695–3724.
- 44.
- Sonnino S, Chigorno V. Ganglioside molecular species containing C18- and C20-sphingosine in mammalian nervous tissues and neuronal cell cultures. Biochim Biophys Acta. 2000;1469:63–77. [PubMed: 10998569]
- 45.
- Coetzee T, Suzuki K, Popko B. New perspectives on the function of myelin galactolipids. Trends Neurosci. 1998;21:126–130. [PubMed: 9530920]
- 46.
- Desnick RJ, Ioannou YA, Eng CM. alpha-Galactosidase A deficiency; Fabry Disease. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3507–3534.
- 47.
- Wenger DA, Suzuki K, Suzuki Y. et al. Galactosylceramide lipidosis: Globoid cell leukodystrophy (Krabbe disease). In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3669–3694.
- 48.
- Dierks T, Schmidt B, Borissenko LV. et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 2003;113(4):435–44. [PubMed: 12757705]
- 49.
- Neufeld E, Muenzer J. The Mucopolysaccharidoses.In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds Metabolic and Molecular Bases of Inherited Disease 2001:3669–3694.
- 50.
- Schuchman EH, Desnick RJ. Niemann-Pick Disease Types A and B: Acid sphingomyelinase Deficiencies. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3589–3610.
- 51.
- Moser HW, Linke T, Fensom AH. et al. Acid ceramidase deficiency: Farber lipogranulomatosis. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3573–3588.
- 52.
- Assmann G, Seedorf U. Acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. In: Schriver CR, Beaudet AL, Sly WS, Valle D,eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3551–3572.
- 53.
- Gelb BD, Bromme D, Desnick RJ. Pycnodysostosis: Cathepsin K deficiency In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3453–3468.
- 54.
- Hofmann SJ, Peltonen L. The neuronal ceroid lipofuscinoses. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3877–3896.
- 55.
- Kopan S, Sivasubramaniam U, Warburton MJ. The lysosomal degradation of neuromedin B is dependent on tripeptidyl peptidase-I: Evidence for the impairment of neuropeptide degradation in late-infantile neuronal ceroid lipofuscinosis. Biochem Biophys Res Commun. 2004;319(1):58–65. [PubMed: 15158442]
- 56.
- Kloetzel PM, Ossendorp F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr Opin Immunol. 2004;16(1):76–81. [PubMed: 14734113]
- 57.
- Aula P, Gahl WA. Disorders of free sialic acid storage. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:5109–5120.
- 58.
- Gahl WA, Thoene JG, Schneider JA. Cystinosis: A disorder of lysosomal memmbrane transport. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:5085–5108.
- 59.
- Raas-Rothschild A, Cormier-Daire V, Bao M. et al. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J Clin Invest. 2000;105(5):673–81. [PMC free article: PMC289169] [PubMed: 10712439]
- 60.
- Kornfeld S, Sly WS. I-Cell Disease and pseudo-hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3469–3482.
- 61.
- Bach G. Mucolipidosis type IV. Mol Genet Metab. 2001;73(3):197–203. [PubMed: 11461186]
- 62.
- Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep. 2003;3(1):64–9. [PubMed: 12507414]
- 63.
- Nishino I, Fu J, Tanji K. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–10. [PubMed: 10972294]
- 64.
- Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13(3):137–45. [PubMed: 12628346]
- 65.
- Patterson MC, Vanier MT, Suzuki K. et al. Niemann-pick disease type c: A lipid trafficking disorder. In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3611–3634.
- 66.
- Sturley SL, Patterson MC, Balch W. et al. The pathophysiology and mechanisms of NP-C disease. Biochim Biophys Acta. 2004;1685(1-3):83–7. [PubMed: 15465429]
- 67.
- Hofman SL, Peltonen L. The neuronal ceroid lipofuscinoses In: Schriver CR, Beaudet AL, Sly WS, Valle D, eds. Metabolic and Molecular Bases of Inherited Disease. 2001:3877–3896.
- 68.
- Luiro K, Yliannala K, Ahtiainen L. et al. Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum Mol Genet. 2004;13(23):3017–27. [PubMed: 15471887]
- 69.
- Mole SE, Michaux G, Codlin S. et al. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res. 2004;298(2):399–406. [PubMed: 15265688]
- 70.
- Winter E, Ponting CP. TRAM, LAG1 and CLN8: Members of a novel family of lipid-sensing domains? Trends Biochem Sci. 2004;27(8):381–3. [PubMed: 12151215]
- 71.
- Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: The secrets of secretory lysosomes. Science. 2004;305(5680):55–9. [PubMed: 15232098]
- 72.
- Huizing M, Gahl WA. Disorders of vesicles of lysosomal lineage: The Hermansky-Pudlak syndromes. Curr Mol Med. 2002;2(5):451–67. [PubMed: 12125811]
- 73.
- Triggs-Raine B, Salo TJ, Zhang H. et al. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc Natl Acad Sci. 1999;96:6296–300. [PMC free article: PMC26875] [PubMed: 10339581]
- 74.
- Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet. 2004;131C(1):75–81. [PubMed: 15452859]
- 75.
- Clark RH, Stinchcombe JC, Day A. et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse Nat Immunol 20034(11):1111–20. Epub 2003. [PubMed: 14566336]
- Introduction
- The Lysosomal Storage Disorders
- Defects in Glycan Degradation
- Defects in Glycoprotein Degradation (Glycoproteinoses)
- Defects in Glycolipid Degradation
- Degradation of GM1 Ganglioside
- Degradation of Sulfatide
- Degradation of Globotriaosylceramide
- Defects in Glycosaminoglycan Degradation (Mucopolysaccharidoses)
- Defects in Glycogen Degradation
- Defects in Lipid Degradation
- Defects in Protein Degradation
- Defects in Lysosomal Transporters
- Defects in Trafficking
- References
- Lysosomal Storage Disorders - Madame Curie Bioscience DatabaseLysosomal Storage Disorders - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...