

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The striking structural and anatomical parallels between the vasculature and the nervous system are reflected by the fact that these two organ systems appear to use related mechanisms during their development.1 Thus, it is not surprising that an increasing number of vascular biologists and researchers in the neuroscience field are fascinated by molecules that play an important role in both systems.
Vascular endothelial growth factor (VEGF) is a major regulator of new blood vessel growth and an important inducer of vascular permeability. However, during the last few years, it has become apparent that VEGF has additional non-vascular functions. In particular the identification of the neuropilins, receptors for the semaphorin family of proteins that mediate neuronal axon pathfinding, as coreceptors for various members of the VEGF family, as well as the detection of VEGF receptors on neurons and astrocytes, suggests that VEGF can act as a neurotrophic and neuroprotective factor in the central as well as in the peripheral nervous system. Thus, VEGF is a pleiotrophic factor with many important functions in the brain. This Chapter summarizes the current knowledge on VEGF expression, regulation and function in the adult nervous system.
The Members of the VEGF Family
VEGF (also designated as VEGF-A) is the founding member of a family of homodimeric glycoproteins that are structurally related to the platelet-derived growth factors (PDGF); this family also includes placenta growth factor (PlGF), VEGF-B, VEGF-C, VEGF-D and VEGF-E. This VEGF family of proteins binds selectively with different affinities to at least five distinct receptors (Fig. 1). Three of them belong to the superfamily of receptor tyrosine kinases and are termed VEGF receptor-1 (VEGFR-1), also called Flt-1 (fms-like tyrosine kinase 1), VEGFR-2, also called KDR (kinase insert-domain containing receptor) in humans, and Flk-1 (fetal liver kinase 1) in rodents, respectively, as well as VEGFR-3, also called Flt-4. The fourth and fifth receptors are neuropilin-1 and neuropilin-2.2 Most of the research on the VEGF family so far, especially with respect to the expression pattern and function in the nervous system, has focused on VEGF-A.
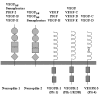
The secreted PlGF shows a strong structural homology to VEGF6 and exists as three isoforms, PlGF-1, PlGF-2 and PlGF-3, which result from alternative splicing from a single gene locus.5 PlGF is predominantly expressed in placenta and binds exclusively to VEGFR-1.7 Although no PlGF expression has been detected in human cortical brain tissue, expression was demonstrated in various tumors of the CNS.8,9 PlGF induces both proliferation and chemotaxis of endothelial cells in vitro and is angiogenic in vivo.6,10,11 These results support the proposal that PlGF is involved in tumor angiogenesis and that it is not required for vascular development in the brain. A recent report further demonstrates that embryonic angiogenesis in mice is not affected by PlGF deficiency, but that PlGF contributes to angiogenesis under pathological conditions.12
VEGF-B binds to both VEGFR-1 and neuropilin-1. VEGF-B is widely expressed, most prominently in heart and skeletal muscle, but also in mouse and human brain.13,14 The highest levels of VEGF-B in the brain are detected in neuronal-like cells of the hippocampus and the cerebral cortex.15 VEGF-B is implicated in angiogenesis by its role in the regulation of extracellular matrix degradation, cell adhesion and migration of endothelial cells.14
VEGF-C is a ligand for both VEGFR-2 and VEGFR-3.16 VEGF-C is synthesized as a prepropeptide and subsequently undergoes proteolytic maturation.17 VEGF-C mRNA is found in several tissues including heart, placenta, ovary, and small intestine,18 although it is undetectable in the normal brain.15 As VEGFR-3 is the main receptor for VEGF-C and is predominantly expressed on lymphatic endothelium, VEGF-C was therefore considered to be the prototypic lymphangiogenic factor.19 However, VEGF-C has also been shown to act on vascular endothelial cells both in vitro and in vivo.20–22 Indeed, mice which lacked a functional vegfr-3 gene showed defective blood vessel development in early stage mouse embryos.23 Thus, the VEGF-C/VEGFR-3 system has an essential role not only for lymphatic vessel formation but also for angiogenesis.
VEGF-D is structurally very similar to VEGF-C and it also binds to VEGFR-2 and VEGFR-3.24 VEGF-D is mitogenic for endothelial cells and thus may play a role in endothelial cell regulation. The expression of VEGF-D is prominent in heart and skeletal muscle, as well as in mesenchymal cells of the lung and skin, but shows only very weak expression in the brain.24,25
VEGF-E is the collective term for a group of proteins with homology to VEGF-A that are encoded by certain strains of the orf parapoxvirus, which affects goats, sheep and occasionally humans.5 It possesses about 25% amino acid identity to mammalian VEGF.26 VEGF-E binds with high affinity to VEGFR-2 and neuropilin-1, but neither to VEGFR-1 nor to VEGFR-3, inducing vascular permeability and potent angiogenic activity both in vitro and in vivo.27-29
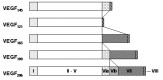
Finally, VEGF-A is ubiquitously expressed at low levels within the CNS by astrocytes and neurons.30 VEGF-A exists as several isoforms, derived from a single gene by alternative splicing (Fig. 2). The smallest form, VEGF121, is 121 amino acids long and does not bind to heparin, while the four larger forms, VEGF145, VEGF165, VEGF189 and VEGF206, all bind heparin with increasing affinity. VEGF189 and VEGF206 remain cell-attached while the smaller isoforms are secreted.5,10,31 VEGF was first isolated in 1983 as a factor leading to increased vascular permeability in tumors and was thus called vascular permeability factor (VPF).32 In 1989, VPF was independently isolated and cloned as an endothelial cell-specific mitogen.33 VEGF is expressed in virtually all cells in the body, however; expression in endothelial cells has mainly been found in vitro in cell culture, while it is undetectable in vivo.34 These findings indicate that VEGF acts as a paracrine factor on neighboring endothelial cells carrying the VEGF receptors rather than in an autocrine fashion. VEGF is such a potent regulator of vascular development that its dosage must be tightly regulated. Disruption of even a single allele of the VEGF gene in mice results in embryonic lethality due to severe vascular defects.35,36 VEGF-A binds to receptors, VEGFR-1 and VEGFR-2. Recently, the neuropilins have been identified as coreceptors for specific VEGF isoforms. While neuropilin-1 is a receptor only for the VEGF165 isoform,37 neuropilin-2 binds both VEGF165 and VEGF145.38 Targeted inactivation of VEGFR-1 and VEGFR-2 as well as neuropilin-1 in mice resulted in defects of blood vessel formation and embryonic lethality, demonstrating further the importance of VEGF-A for appropriate vascular development.39–41
In summary, VEGF-A and VEGF-B appear to be the only VEGF members expressed at significant levels in the CNS, whereas PlGF may be activated during pathological situations. While VEGF-A fulfills an important role in vascular development of the brain,42 it remains to be established whether VEGF-B has any physiological function in the CNS. I will, therefore, for the remaining part of this Chapter focus on the regulation and function of VEGF-A in the nervous system.
Regulation of VEGF and VEGF Receptor Expression
A variety of physiological and pathological processes are associated with upregulation of components of the VEGF/VEGFR-system, including embryogenesis, the female reproductive cycle, pregnancy, wound healing, tumor growth, diabetic retinopathy and ischemic diseases (for a review see refs. 3,43). In the search for mechanisms and factors capable of influencing VEGF expression during these processes, many cytokines and growth factors have been shown to modulate VEGF gene expression.5 For example, TNFa and bFGF are able to induce VEGF gene expression in vitro;44,45 also glucose deficiency has been shown to increase VEGF expression.46 The most important and intensively studied inducer of VEGF gene expression, however, is hypoxia, which was demonstrated for the first time in the perinecrotic areas of glioblastomas.47,48 Under hypoxic conditions VEGF expression is mediated through the activation of specific hypoxia-inducible transcription factors, HIF-1 and HIF-2.49,50 In addition, VEGF upregulation during hypoxia is also achieved by an increase in the stability of its mRNA51 and by the efficient hypoxic translation of the VEGF mRNA which is mediated by an internal ribosomal entry site.52
HIF-1 is an ubiquitously expressed master regulator of oxygen homeostasis. It is a heterodimeric transcription factor that is composed of HIF-1α and HIF-1β (also known as the arylhydrocarbon receptor nuclear translocator ARNT) protein subunits. Whereas HIF-1β/ARNT is constitutively expressed, HIF-1α expression is induced in hypoxic cells. HIF-1 binds to the promoter/enhancer elements of hitherto more than 20 known hypoxia-inducible genes and stimulates their transcription. These genes play essential roles in the physiologic adaptation to hypoxia, including glycolysis, erythropoiesis, angiogenesis and vascular remodeling.53 Furthermore, it became evident that HIF-1 plays a central role in many human diseases, including myocardial ischemia, stroke and tumor growth.54 Recently, a close homologue of HIF-1α was identified and termed HIF-2α.50,55–57 In addition, it has been shown that ARNT2, a conserved ARNT homologue that is highly expressed in neurons, forms functional HIF complexes in vivo, leading to the hypothesis that HIF-1α/ARNT2 heterodimers may specifically mediate transcriptional responses in the nervous system.58
HIF-1α mRNA is constitutively expressed in virtually all organs and cells59 implicating a regulation of HIF-1 activity at a posttranscriptional level.60 HIF-1α protein levels are downregulated in normoxic cells by ubiquitination and rapid proteasomal degradation, a process that is mediated by the von Hippel-Lindau tumor suppressor protein (pVHL).53,61 The binding of pVHL to the α subunit requires a specific, oxygen-dependent, prolyl hydroxylation within the degradation domain of HIF-1α (Fig. 3).62,63 The involved prolyl-hydroxylase might therefore act as an oxygen sensor. As a result HIF-1α and also HIF-2α proteins are undetectable in normoxic tissues.64 However, expression of HIF-1α increases dramatically in hypoxic tissues, including the brain (e.g., during chronic hypoxia or ischemia).65–67

As a consequence of HIF-1 activation, VEGF transcription is increased during hypoxia.49 VEGF expression is upregulated mainly in astrocytes and neurons during hypoxic and ischemic events,34,67,68 but also in injured brain or spinal cord tissue.69–71 In addition, gene expression of both VEGFR-1 and VEGFR-2 is activated in hypoxic tissues.68,72,73 Whereas VEGFR-1 is a target gene for HIF-1 and is therefore directly upregulated by hypoxia,34,74 VEGFR-2 is not induced after short term hypoxic exposure34 but rather after prolonged periods of hypoxia, such as those occurring during ischemic events.68,75 VEGFR-2 has no HIF-1 consensus site in its promoter region74 but may be activated by HIF-2.76 Increased VEGF levels may activate VEGFR-2 expression through a positive feedback loop indicating a further regulatory pathway.77,78 The third receptor for VEGF, neuropilin-1, is upregulated in endothelial cells of cerebral blood vessels from hypoxic tissue after focal cerebral ischemia.79 For the other members of the VEGF family, as well as for VEGFR-3, upregulation by hypoxia has not been demonstrated conclusively, and it is so far unknown whether hypoxia-response elements allowing transcriptional regulation by either HIF-1 or HIF-2 are present in these genes.80,81
Pleiotropic Action of VEGF in the CNS
VEGF has long been considered a selective endothelial cell mitogen that promotes vasculogenesis and angiogenesis. In addition, it initiates endothelial procoagulant activity and induces vascular permeability. These effects are compatible with the expression of VEGF receptors specifically on vascular endothelial cells. However, other cell types, including hematopoietic progenitor cells, monocytes and trophoblasts also express functional VEGF receptors (for a review see ref. 2). More recently, additional cellular targets of VEGF were discovered in the CNS. Astrocytes and neurons have been reported to express either or both VEGFR-1 and VEGFR-2 during cerebral ischemia.83 VEGFR-2 expression has also been described in neural progenitor cells of the retina,84 and expression of VEGFR-1 was found on reactive astrocytes after VEGF stimulation,85 and in other models involving CNS trauma.86 These findings led to the hypothesis that VEGF might act as a direct neurotrophic or even neuroprotective factor. This concept is supported by the finding that other pro-angiogenic growth factors such as bFGF and PDGF have been shown to be neurotrophic as well.87 Furthermore, the identification of the neuropilins as coreceptors for different isoforms of VEGF37,38 implicates VEGF in the processes of neuronal growth and repair. Thus, VEGF may affect neuronal survival in a variety of pathological conditions indirectly by inducing angiogenesis and vascular permeability but also by a direct action on neurons and astrocytes.
Direct Neurotrophic/Neuroprotective Actions of VEGF
The first evidence for a neurotrophic function for VEGF came from a series of experiments performed in the laboratory of Jeffrey Rosenstein, using murine brain explant cultures in vitro. The study was initially designed to investigate angiogenic reactions in the CNS after VEGF administration. This group found an increase in enlarged, dilated and branching vessels after VEGF stimulation in rat cortical slice explant cultures but also expression of VEGFR-1 on reactive astrocytes.85 Subsequently, in mesencephalic explant cultures they detected that VEGF administration led to the proliferation of astrocytes and also prevented dopaminergic neurons from cell death.88 The significance of VEGF-mediated astrocyte proliferation is unknown. Recent studies support the assertion that astrocytes are actively involved in the generation and maintenance of neuronal synapses.89 VEGF may sustain these effects on neurons by providing the necessary glial bed and thus lead to an indirect stabilization of neuronal function. This idea is supported by the finding that expression of both VEGFR-1 and VEGFR-2 in Müller cells of the developing retina is required for normal retinal development, from which it is surmised that the development of blood vessels and neural tissue takes place in a coordinated fashion, guided by the expression of VEGF receptors in glial cells.90 In addition, direct effects of VEGF on neuronal cell lines and primary neurons were also reported, including the role of VEGF in promoting the development and survival of rat photoreceptor cells.91 Also, addition of VEGF to the immortalized hippocampal neuronal cell line HN33 resulted in reduced cell death that was associated with an in vitro model of cerebral ischemia by doubling the number of surviving cells after 24 hours of combined oxygen and glucose deprivation.92 Furthermore, VEGF protects rat primary embryonic hippocampal neurons against glutamate-induced neurotoxicity.93 Six hours of pretreatment with VEGF before exposure to glutamate resulted in increased cell survival, inhibited upregulation of caspase-3-like activity and also attenuated DNA laddering. These results imply that VEGF is able to block neuronal apoptosis in specific cell lineages. Further evidence in support of this hypothesis comes from a recent in vivo study, where topical application of VEGF to the surface of an ischemic brain led to a significant reduction of infarct volume and neuronal damage some 24 hours after onset of experimentally induced stroke.94 The positive effect of VEGF administration after 24 hours is supportive of a direct protective effect of VEGF on the neurons itself. An indirect neuroprotective effect mediated by inducing angiogenesis (see below) would need more time, as new vessel growth during cerebral ischemia is initiated only 24 to 48 hours after occlusion of the artery.68 Alternatively, VEGF may improve survival of hypoxic endothelial cells in the penumbra surrounding the infarcted area, thus preserving a residual blood flow which then contributes to better neuronal survival.
In the peripheral nervous system, VEGF has also been implicated in neurotrophic and neuroprotective effects. VEGF is expressed by neurons of the superior cervical ganglia (SCG) and dorsal root ganglia (DRG), and VEGFR-2 is present on neurons within SCG and DRG, and also on Schwann cells.87 In these different cell types, VEGF and VEGFR-2 expression demonstrates temporal changes during early postnatal life, from which it may be concluded that there is developmental regulation of VEGF activity in peripheral ganglia.95 Addition of VEGF to explanted ganglia promoted cell survival, axonal outgrowth, and proliferation of Schwann cells.87 Topical application of VEGF to injured peripheral nerves, therefore, could have a beneficial effect on nerve regeneration by promoting the invasion of Schwann cells and new vessel growth.96,97 In addition, it has been shown that VEGF gene transfer in a rabbit ischemic hindlimb model, as well as in diabetic rats, had favorable effects on peripheral nerve function.98,99 The role of VEGF, and especially its inducibility by hypoxia, on neuronal survival was very recently underlined by a fascinating study in the laboratory of Peter Carmeliet. Transgenic mice with a deletion of the hypoxia-response element in the promoter of the VEGF gene demonstrated a reduction of baseline levels of VEGF specifically in neural tissue, and hypoxia-induced VEGF expression in the spinal cord and the brain was blunted. At the age of five months, the transgenic animals developed a progressive motor neuron degeneration, which was reminiscent of human amyotrophic lateral sclerosis.100
In the search for mechanisms that mediate the neuronal functions of VEGF, the interaction between VEGF and the neuropilins deserves attention. Neuropilins were initially identified according to their ability to bind secreted semaphorins of subclass 3.1 However, this semaphorin-neuropilin complex is unable to transmit a signal to the cell because neuropilins lack a signaling-competent cytoplasmic domain. Therefore, the binding of an additional class of receptors, the plexins, is required to form a fully functional semaphorin receptor complex. Alternatively, neuropilins can use VEGFR-2 as a coreceptor by interacting with specific isoforms of VEGF. The presence of neuropilin-1 on endothelial cells enhances the affinity of VEGF165 for its signaling receptor VEGFR-2, leading to increased endothelial cell chemotaxis and mitogenesis.37 Furthermore, neuropilin-1 may also use the tyrosine kinase activity of VEGFR-1 for signaling after semaphorin binding.101 In nervous tissue, secreted semaphorins control cell migration by forming tissue gradients that are sensed by receptor complexes consisting of plexins and neuropilins.4 Semaphorins act as repelling cues for the guidance of axons in developing nervous tissue. In the adult organism, semaphorins seem to inhibit axon growth after injury and maintain established neural pathways. Secreted semaphorins and VEGF compete with each other for binding to their natural receptors neuropilin-1 and VEGFR-2, respectively. To further complicate the situation, it has been demonstrated that neuropilin-1 binds with high affinity to VEGFR-1, thereby inhibiting the binding of neuropilin-1 to VEGF165.102 VEGFR-1 lacks a significant signaling activity and has therefore been considered to negatively regulate the VEGF-dependent activity by functioning as a sink receptor.2 Based on these results, a number of scenarios emerge, as to how VEGF action on neuronal cells could be mediated (Fig. 4). 1) VEGF can bind to VEGFR-2 present on neurons and signal via the kinase domain of this receptor. An additional complex formation with neuropilin-1 could modulate or enhance this signal. 2) As VEGFR-1 can act as a sink receptor, preventing VEGF binding to the signaling VEGFR-2, the binding of neuropilin-1 to VEGFR-1 could block this sink function and thereby increase the availability of VEGF for VEGFR-2 leading to an increased activity via this receptor. 3) The neurotrophic action of VEGF could be an indirect one. As semaphorins inhibit axonal growth, VEGF could compete with semaphorins for neuropilin binding, thereby blocking the repellent function of semaphorins on neurons. This would result in an increased axonal outgrowth and better survival of neurons as previously demonstrated.87,97,101 4) Regulated expression of VEGFR-1 could modulate the level of available neuropilin-1 and thus influence the affinity of VEGFR-2 for VEGF as well as signaling via the semaphorin-plexin complexes. The balance between VEGF and semaphorin availability at their common receptors VEGFR-1, VEGFR-2 and neuropilin-1 may therefore modulate the migration, apoptosis, survival and proliferation of neural cells.101 The situation may be even more complex, as it is currently unknown whether neuropilin-1 and VEGFR-2 are able to directly interact. Furthermore, neuropilin-2 and the plexins may interfere with the other receptors as well. On the other hand, it is evident that semaphorins by competing with VEGF for binding to neuropilin-1 could influence the angiogenesis reaction mediated by VEGF.37

Angiogenesis
VEGF can also influence neuronal survival indirectly by inducing new blood vessel growth, thereby allowing increased transportation of oxygen and nutrients to hypoxic tissue. Formation and remodeling of new blood vessels is governed by three processes: vasculogenesis, angiogenesis and arteriogenesis. Vasculogenesis is defined as the differentiation of mesodermal progenitor cells (angioblasts) into endothelial cells in situ where they subsequently aggregate and form a primary vascular plexus.103 Vasculogenesis occurs primarily during embryonic development. However, recent studies suggest that endothelial precursor cells may exist in the bone marrow and contribute to new vessel formation or vascular remodeling by vasculogenesis also in the adult organism.104 Angiogenesis is defined as the formation of new blood vessels by sprouting of endothelial cells from pre-existing vessels or by intravascular subdivision (intussusception).105 Angiogenesis further refines the primitive embryonic vascular plexus and includes remodeling, a process that transforms the relatively uniformly sized vasculature into the network of small and large vessels, that finally undergoes maturation by recruiting perivascular cells, such as smooth muscle cells and pericytes. Angiogenesis is an important process during embryogenesis but also occurs in the adult in response to altered metabolic requirements, e.g., it can be triggered by hypoxia. The growth of new blood vessels is a complex process that requires the coordinated interaction of endothelial cells with the tissue environment.3 Transient phases of angiogenesis occur in various physiological processes in the adult organism, for example during the female reproductive cycle in the ovary and in the uterus, as well as during pregnancy and in wound healing. Furthermore, pathological neovascularization occurs in a variety of diseases, such as ophthalmic and rheumatic diseases, psoriasis, hemangioblastoma, solid tumor growth and also during ischemic diseases.106 Finally, arteriogenesis is the rapid proliferation of pre-existing collateral vessels that occurs in ischemic tissue (for a review see ref. 107).
The brain, with its high rate of oxidative metabolism, must rely on a steady supply of oxygen. A prolonged hypoxic period will lead to a reduction of tissue oxygenation with detrimental effects on proper brain function. In order to maintain oxygen delivery to the brain, the organism increases the vascular density in this organ, resulting in smaller intercapillary distances, which will finally restore tissue oxygenation.108,109 The brain is thus capable of structural and functional plasticity to balance energy supply and demand.110 Prolonged hypoxia increases VEGF expression in adult brain,34,111 thus implicating VEGF in the observed increase in vascular density. VEGF-mediated new vessel growth, can thus be considered as an physiologically adaptive response to tissue hypoxia. In addition, various pathophysiological events are associated with tissue hypoxia, such as brain injury, cerebral ischemia and also a variety of neurodegenerative diseases. Thus, although tissue hypoxia can lead to neuronal damage it is also the major stimulus capable of activating endogenous protective strategies. For example, activation of HIF-1 can promote cell survival in hypoxic and ischemic tissues via the upregulation of angiogenic and neuroprotective factors such as VEGF or erythropoietin.68,112 Indeed, increased expression of HIF-1 and HIF-2 has been reported during stroke.66,67 Induction of angiogenic processes through exogenous administration of VEGF (therapeutic angiogenesis) may therefore be important for the treatment of stroke, as has been shown for other ischemic disease such as hindlimb ischemia or myocardial infarction.3,43,113 However, a major concern precluding the use of VEGF in the CNS is the fact that this growth factor induces significant vascular permeability in vivo.
Vascular Permeability
Brain edema formation as a consequence of the disruption of the blood-brain barrier is a major problem in a variety of diseases of the CNS, including brain trauma, tumor growth and stroke. The upregulation of VEGF which occurs during these pathological processes might be responsible for the observed increase in vascular permeability and subsequent brain edema formation. Chronic hypoxia leads not only to an upregulation of VEGF mRNA in the brain, but also to an increased permeability in cortical vessels.114 Similarly, high altitude brain edema formation is associated with an increased VEGF production in the brain that can be blocked by a neutralizing anti-VEGF antibody (H.H.M., manuscript submitted). These results support the proposal that a strategy aimed at blunting brain edema formation by blocking VEGF action might be useful to enhance neuronal survival during brain trauma or stroke. Indeed, antagonism of VEGF action by using a VEGFR-1-IgG fusion protein, which sequesters VEGF, reduced edema formation and infarct size in a mouse model of cerebral ischemia.115 Thus, the usage of VEGF as therapeutic agent is context-dependent and therefore needs to be tightly controlled with respect to both time and dosage. This is evident from a recent study using a fibrin clot model of middle cerebral artery occlusion (MCAO) in rats. Late administration of VEGF (48 hours after MCAO) to the ischemic rats enhanced angiogenesis in the ischemic penumbra and significantly improved neurological recovery. However, early postischemic (1 hour after MCAO) administration of VEGF increased blood-brain barrier leakage and the size of the subsequent ischemic lesion.116 Thus, a therapeutic regimen for the treatment of stroke evolving from these results could potentially consist of the early inhibition of VEGF function thereby minimizing brain edema formation, followed by a later active VEGF treatment leading to angiogenesis and neuroprotection.
Intracellular Signaling Events
An important question in the field of neuroprotective aspects of VEGF is: through which intracellular signaling cascades are the various VEGF effects mediated? With regard to the possible therapeutic use of VEGF in the CNS, it would be helpful to know whether these several different VEGF-mediated effects use different signaling pathways which can be specifically activated (e.g., by low molecular compounds interfering with one pathway but not affecting the other). For example, it has been shown that distinct members of the Src kinase family (Src, Yes, Fyn) are required for different VEGF-mediated processes.117 Mice lacking Src or Yes, but not Fyn, showed no vascular permeability in response to VEGF administration, whereas induction of angiogenesis and endothelial cell survival required Src family kinase activity in general. In accordance with these results, blockage of Src, but not Fyn activity in mice provided cerebral protection following stroke, probably via a reduction in brain edema formation.118
In endothelial cells, VEGFR-2 is considered to be the major signaling receptor, while VEGFR-1 acts as a sink to trap an excess of VEGF. Endothelial proliferation is mediated via the Ras-Raf-MAP (mitogen-activated protein)-kinase pathway, while protein kinase C (PKC) activation is involved in endothelial migration and vascular permeability (for a review see ref. 2). The role of VEGF as a survival factor for endothelial cells is mediated by the phosphoinositol 3 (PI3) kinase-AKT signaling pathway.119 This situation may be similar in neuronal cells. A recent study, using antisense oligonucleotides for VEGFR-1 or VEGFR-2, supported the hypothesis that VEGFR-2, but not VEGFR-1, mediate neuroprotective effects. However, involvement of neuropilins was not investigated in this study. Neuroprotection was associated with the activation of PI3 kinase-AKT pathway as well as MEK/ERK signaling pathway.93 In the setting of cerebral ischemia in vivo and in various in vitro models of neuronal death, the neuroprotective function of VEGF is associated with an activation of VEGFR-2 expression and increased levels of phosphorylated AKT, implicating the PI3 kinase-AKT pathway.73,92,120 Figure 5 summarizes the major signal transduction pathways that are induced by activated VEGFR-2 in endothelial cells and neurons.

Conclusion
The VEGF/VEGFR receptor system is activated in a variety of pathological conditions in the CNS, most prominent during cerebral ischemia. But also in brain injury and neurodegenerative diseases, such as Alzheimer's disease, involvement of VEGF is discussed. VEGF expression is markedly enhanced in Alzheimer's disease implicating compensatory mechanisms to counter insufficient vascularity or reduced perfusion apparent in this condition.121 VEGF might therefore be an important factor in the pathophysiology of diseases that are associated with tissue hypoxia and may has also an important role for their treatment. VEGF can exert its neuroprotective function by at least two mechanisms. VEGF acts directly on neurons expressing various VEGF receptor types, thereby mediating its neuroprotective effect. Alternatively, VEGF induces angiogenesis, thereby allowing increased oxygen and nutrient transportation to ischemic or hypoxic tissues, which ultimately leads to protection of stressed neurons and possibly other cell types in the area. VEGF or VEGF-like agents may therefore be useful for the treatment of neurodegenerative as well as ischemic disorders of the CNS. A major risk of this form of therapy is that VEGF may also damage neurons by its effect of increasing permeability leading to brain edema formation. This precludes VEGF therapy as an efficient treatment for ischemic conditions such as stroke because serious adverse side effects may occur. As indicated above, a potential stroke therapy could therefore consist of early VEGF inhibition followed by a period of active VEGF treatment. Alternatively, one might search for additional factors capable of inducing angiogenesis and increasing neuronal survival, but devoid of the harmful effects associated with edema formation. The hematopoietic growth factor erythropoietin might be such a factor. Erythropoietin is expressed in the CNS, is induced by hypoxia like VEGF and has a very similar pattern of expression and functional role during cerebral ischemia. Indeed, erythropoietin has been shown to be neuroprotective as well as angiogenic in the brain,112 but does not seem to induce vascular permeability. Thus, therapeutic application of erythropoietin, which has been widely used as human therapeutic in the clinic, might be a more promising therapeutic approach. An alternative to the treatment of ischemic brain injury by application of VEGF alone, might be the combination of different angiogenic and neuroprotective growth factors. The combined application of VEGF with angiopoietins has been shown to protect vessels from VEGF-induced vascular leakage.122 However, the use of VEGF to treat patients with diseases of the CNS may lead to the induced growth of dormant tumors or facilitate atherosclerotic plaque progression.123 Thus, much more research is needed to establish the role of VEGF as neuroprotective agent, and whether the concept of therapeutic angiogenesis using VEGF is applicable as therapy for the treatment of neurodegenerative or ischemic diseases such as Alzheimer's disease or stroke. Furthermore, the potential damage to nervous tissue which may be caused by blocking an essential survival signal for neuronal cells mediated through the VEGF pathway should be avoided under any circumstances. The rapidly increasing number of reports demonstrating neurotrophic or neuroprotective functions of VEGF therefore warrants a careful re-evaluation of all antiangiogenic strategies (e.g., for treatment of solid tumor growth).
Acknowledgments
I gratefully acknowledge Sabine Raab and Marcel Groot for helpful discussion and Christopher Mitchell and Friedemann Kiefer for critically reading the manuscript. I was supported by a fellowship from the Max Planck Society.
References
- 1.
- Shima DT, Mailhos C. Vascular developmental biology: Getting nervous. Curr Opin Genet Dev. 2000;10:536–542. [PubMed: 10980432]
- 2.
- Clauss M. Molecular biology of the VEGF and the VEGF receptor family. Semin Thromb Hemost. 2000;26:561–569. [PubMed: 11129413]
- 3.
- Breier G. Functions of the VEGF/VEGF receptor system in the vascular system. Semin Thromb Hemost. 2000;26:553–559. [PubMed: 11129412]
- 4.
- Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: Cell guidance and beyond. Trends Cell Biol. 2000;10:377–383. [PubMed: 10932095]
- 5.
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. [PubMed: 11181169]
- 6.
- Maglione D, Guerriero V, Viglietto G. et al. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA. 1991;88:9267–9271. [PMC free article: PMC52695] [PubMed: 1924389]
- 7.
- Park JE, Chen HH, Winer J. et al. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646–25654. [PubMed: 7929268]
- 8.
- Hatva E, Kaipainen A, Mentula P. et al. Expression of endothelial cell-specific receptor tyrosine kinases and growth factors in human brain tumors. Am J Pathol. 1995;146:368–378. [PMC free article: PMC1869858] [PubMed: 7856749]
- 9.
- Nomura M, Yamagishi S, Harada S. et al. Placenta growth factor (PIGF) mRNA expression in brain tumors. J Neurooncol. 1998;40:123–130. [PubMed: 9892094]
- 10.
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. [PubMed: 9034784]
- 11.
- Clauss M. Functions of the VEGF receptor-1 (Flt-1) in the vasculature. Trends Cardiovasc Med. 1998;8:241–245. [PubMed: 14987558]
- 12.
- Carmeliet P, Moons L, Luttun A. et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nature Med. 2001;7:575–583. [PubMed: 11329059]
- 13.
- Olofsson B, Pajusola K, Kaipainen A. et al. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci USA. 1996;93:2576–2581. [PMC free article: PMC39839] [PubMed: 8637916]
- 14.
- Olofsson B, Korpelainen E, Pepper MS. et al. Vascular endothelial growth-factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad Sci USA. 1998;95:11709–11714. [PMC free article: PMC21705] [PubMed: 9751730]
- 15.
- Lagercrantz J, Farnebo F, Larsson C. et al. A comparative study of the expression patterns for vegf, vegf-b/vrf and vegf-c in the developing and adult mouse. Biochim Biophys Acta. 1998;1398:157–163. [PubMed: 9689915]
- 16.
- Joukov V, Pajusola K, Kaipainen A. et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–298. [PMC free article: PMC449944] [PubMed: 8617204]
- 17.
- Joukov V, Sorsa T, Kumar V. et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16:3898–3911. [PMC free article: PMC1170014] [PubMed: 9233800]
- 18.
- Lee J, Gray A, Yuan J. et al. Vascular endothelial growth factor-related protein: A ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Natl Acad Sci USA. 1996;93:1988–1992. [PMC free article: PMC39896] [PubMed: 8700872]
- 19.
- Jeltsch M, Kaipainen A, Joukov V. et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997;276:1423–1425. [PubMed: 9162011]
- 20.
- Witzenbichler B, Asahara T, Murohara T. et al. Vascular endothelial growth factor-C (VEGF-C/VEGF-2) promotes angiogenesis in the setting of tissue ischemia. Am J Pathol. 1998;153:381–394. [PMC free article: PMC1852989] [PubMed: 9708799]
- 21.
- Enholm B, Jussila L, Karkkainen M. et al. Vascular endothelial growth factor-C: A growth factor for lymphatic and blood vascular endothelial cells. Trends Cardiovasc Med. 1998;8:292–297. [PubMed: 14987553]
- 22.
- Cao Y, Linden P, Farnebo J. et al. Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci USA. 1998;95:14389–14394. [PMC free article: PMC24383] [PubMed: 9826710]
- 23.
- Dumont DJ, Jussila L, Taipale J. et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282:946–949. [PubMed: 9794766]
- 24.
- Achen MG, Jeltsch M, Kukk E. et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. 1998;95:548–553. [PMC free article: PMC18457] [PubMed: 9435229]
- 25.
- Yamada Y, Nezu J, Shimane M. et al. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics. 1997;42:483–488. [PubMed: 9205122]
- 26.
- Lyttle DJ, Fraser KM, Fleming SB. et al. Homologs of vascular endothelial growth factor are encoded by the poxvirus orf virus. J Virol. 1994;68:84–92. [PMC free article: PMC236267] [PubMed: 8254780]
- 27.
- Ogawa S, Oku A, Sawano A. et al. A novel type of vascular endothelial growth factor, VEGF-E (NZ-7 VEGF), preferentially utilizes KDR/Flk-1 receptor and carries a potent mitotic activity without heparin-binding domain. J Biol Chem. 1998;273:31273–31282. [PubMed: 9813035]
- 28.
- Meyer M, Clauss M, Lepple-Wienhues A. et al. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999;18:363–374. [PMC free article: PMC1171131] [PubMed: 9889193]
- 29.
- Wise LM, Veikkola T, Mercer AA. et al. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc Natl Acad Sci USA. 1999;96:3071–3076. [PMC free article: PMC15896] [PubMed: 10077638]
- 30.
- Ogunshola OO, Stewart WB, Mihalcik V. et al. Neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain. Brain Res Dev Brain Res. 2000;119:139–153. [PubMed: 10648880]
- 31.
- Neufeld G, Cohen T, Gengrinovitch S. et al. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed: 9872925]
- 32.
- Senger DR, Galli SJ, Dvorak AM. et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. [PubMed: 6823562]
- 33.
- Keck PJ, Hauser SD, Krivi G. et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. [PubMed: 2479987]
- 34.
- Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA. 1998;95:15809–15814. [PMC free article: PMC28126] [PubMed: 9861052]
- 35.
- Carmeliet P, Ferreira V, Breier G. et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. [PubMed: 8602241]
- 36.
- Ferrara N, Carver-Moore K, Chen H. et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. [PubMed: 8602242]
- 37.
- Soker S, Takashima S, Miao HQ. et al. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. [PubMed: 9529250]
- 38.
- Gluzman-Poltorak Z, Cohen T, Herzog Y. et al. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165. J Biol Chem. 2000;275:18040–18045. [PubMed: 10748121]
- 39.
- Fong G -H, Rossant J, Gertsenstein M. et al. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. [PubMed: 7596436]
- 40.
- Shalaby F, Rossant J, Yamaguchi TP. et al. Failure of blood-island formation and vasculogenesis in flk-1- deficient mice. Nature. 1995;376:62–66. [PubMed: 7596435]
- 41.
- Kawasaki T, Kitsukawa T, Bekku Y. et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. [PubMed: 10518505]
- 42.
- Breier G, Albrecht U, Sterrer S. et al. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. [PubMed: 1592003]
- 43.
- Marti HH, Risau W. Angiogenesis in ischemic disease. Thromb Haemost. 1999;82 Suppl 1:44–52. [PubMed: 10695485]
- 44.
- Ryuto M, Ono M, Izumi H. et al. Induction of vascular endothelial growth factor by tumor necrosis factor a in human glioma cells. Possible roles of SP-1. J Biol Chem. 1996;271:28220–28228. [PubMed: 8910439]
- 45.
- Stavri GT, Zachary IC, Baskerville PA. et al. Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells. Synergistic action with hypoxia. Circulation. 1995;92:11–14. [PubMed: 7788904]
- 46.
- Shweiki D, Neeman M, Itin A. et al. Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: Implications for tumor angiogenesis. Proc Natl Acad Sci USA. 1995;92:768–772. [PMC free article: PMC42701] [PubMed: 7531342]
- 47.
- Shweiki D, Itin A, Soffer D. et al. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. [PubMed: 1279431]
- 48.
- Plate KH, Breier G, Weich HA. et al. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–848. [PubMed: 1279432]
- 49.
- Forsythe JA, Jiang B -H, Iyer NV. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. [PMC free article: PMC231459] [PubMed: 8756616]
- 50.
- Ema M, Taya S, Yokotani N. et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1a regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273–4278. [PMC free article: PMC20712] [PubMed: 9113979]
- 51.
- Ikeda E, Achen MG, Breier G. et al. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor (VEGF) in C6 glioma cells. J Biol Chem. 1995;270:19761–19766. [PubMed: 7544346]
- 52.
- Stein I, Itin A, Einat P. et al. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: Implications for translation under hypoxia. Mol Cell Biol. 1998;18:3112–3119. [PMC free article: PMC108893] [PubMed: 9584152]
- 53.
- Wenger RH. Mammalian oxygen sensing, signalling and gene regulation. J Exp Biol. 2000;203:1253–1263. [PubMed: 10729275]
- 54.
- Semenza GL. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000;14:1983–1991. [PubMed: 10950862]
- 55.
- Tian H, McKnight SL, Russell DW. Endothelial pas domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. [PubMed: 9000051]
- 56.
- Flamme I, Fröhlich T, von Reutern M. et al. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. [PubMed: 9178256]
- 57.
- Hogenesch JB, Chan WK, Jackiw VH. et al. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–8593. [PubMed: 9079689]
- 58.
- Maltepe E, Keith B, Arsham AM. et al. The role of ARNT2 in tumor angiogenesis and the neural response to hypoxia. Biochem Biophys Res Commun. 2000;273:231–238. [PubMed: 10873592]
- 59.
- Wenger RH, Rolfs A, Marti HH. et al. Nucleotide sequence, chromosomal assignment and mRNA expression of mouse hypoxia-inducible factor-1α Biochem Biophys Res Commun. 1996;223:54–59. [PubMed: 8660378]
- 60.
- Wenger RH, Kvietikova I, Rolfs A. et al. Hypoxia-inducible factor-1a is regulated at the post-mRNA level. Kidney Int. 1997;51:560–563. [PubMed: 9027739]
- 61.
- Maxwell PH, Wiesener MS, Chang GW. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. [PubMed: 10353251]
- 62.
- Ivan M, Kondo K, Yang HF. et al. HIFa targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–468. [PubMed: 11292862]
- 63.
- Jaakkola P, Mole DR, Tian YM. et al. Targeting of HIF a to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. [PubMed: 11292861]
- 64.
- Talks KL, Turley H, Gatter KC. et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. [PMC free article: PMC1850121] [PubMed: 10934146]
- 65.
- Chavez JC, Agani F, Pichiule P. et al. Expression of hypoxia-inducible factor-1a in the brain of rats during chronic hypoxia. J Appl Physiol. 2000;89:1937–1942. [PubMed: 11053346]
- 66.
- Bergeron M, Yu AY, Solway KE. et al. Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur J Neurosci. 1999;11:4159–4170. [PubMed: 10594641]
- 67.
- Jin KL, Mao XO, Nagayama T. et al. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1α by global ischemia in rat brain. Neuroscience. 2000;99:577–585. [PubMed: 11029549]
- 68.
- Marti H J H, Bernaudin M, Bellail A. et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. [PMC free article: PMC1876841] [PubMed: 10702412]
- 69.
- Papavassiliou E, Gogate N, Proescholdt M. et al. Vascular endothelial growth factor (vascular permeability factor) expression in injured rat brain. J Neurosci Res. 1997;49:451–460. [PubMed: 9285521]
- 70.
- Bartholdi D, Rubin BP, Schwab ME. VEGF mRNA induction correlates with changes in the vascular architecture upon spinal cord damage in the rat. Eur J Neurosci. 1997;9:2549–2560. [PubMed: 9517460]
- 71.
- Sköld M, Cullheim S, Hammarberg H. et al. Induction of VEGF and VEGF receptors in the spinal cord after mechanical spinal injury and prostaglandin administration. Eur J Neurosci. 2000;12:3675–3686. [PubMed: 11029637]
- 72.
- Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/flk and flt in lungs exposed to acute or chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. [PMC free article: PMC295709] [PubMed: 7706486]
- 73.
- Jin KL, Mao XO, Nagayama T. et al. Induction of vascular endothelial growth factor receptors and phosphatidylinositol 3'-kinase/Akt signaling by global cerebral ischemia in the rat. Neuroscience. 2000;100:713–717. [PubMed: 11036205]
- 74.
- Gerber H -P, Condorelli F, Park J. et al. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes; flt-1, but not flk- 1/KDR, is up-regulated by hypoxia. J Biol Chem. 1997;272:23659–23667. [PubMed: 9295307]
- 75.
- Li J, Brown LF, Hibberd MG. et al. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–H1811. [PubMed: 8928889]
- 76.
- Kappel A, Rönicke V, Damert A. et al. Identification of vascular endothelial growth factor (VEGF) receptor-2 (Flk-1) promoter/enhancer sequences sufficient for angioblast and endothelial cell-specific transcription in transgenic mice. Blood. 1999;93:4284–4292. [PubMed: 10361126]
- 77.
- Kremer C, Breier G, Risau W. et al. Up-regulation of flk-1/vascular endothelial growth factor receptor 2 by its ligand in a cerebral slice culture system. Cancer Res. 1997;57:3852–3859. [PubMed: 9288799]
- 78.
- Shen B -Q, Lee DY, Gerber H -P. et al. Homologous up-regulation of KDR/Flk-1 receptor expression by vascular endothelial growth factor in vitro. J Biol Chem. 1998;273:29979–29985. [PubMed: 9792718]
- 79.
- Zhang ZG, Tsang W, Zhang L. et al. Up-regulation of neuropilin-1 in neovasculature after focal cerebral ischemia in the adult rat. J Cereb Blood Flow Metab. 2001;21:541–549. [PubMed: 11333364]
- 80.
- Gleadle JM, Ebert BL, Firth JD. et al. Regulation of angiogenic growth factor expression by hypoxia, transition metals, and chelating agents. Am J Physiol. 1995;268:C1362–C1368. [PubMed: 7541940]
- 81.
- Enholm B, Paavonen K, Ristimaki A. et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 m-RNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475–2483. [PubMed: 9188862]
- 82.
- Zhu H, Bunn HF. How do cells sense oxygen? Science. 2001;292:449–451. [PMC free article: PMC3040953] [PubMed: 11292863]
- 83.
- Lennmyr F, Ata KA, Funa K. et al. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. [PubMed: 9737551]
- 84.
- Yang XJ, Cepko CL. Flk-1, a receptor for vascular endothelial growth-factor (VEGF), is expressed by retinal progenitor cells. J Neurosci. 1996;16:6089–6099. [PMC free article: PMC6579183] [PubMed: 8815891]
- 85.
- Rosenstein JM, Mani N, Silverman WF. et al. Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:7086–7091. [PMC free article: PMC22748] [PubMed: 9618543]
- 86.
- Krum JM, Rosenstein JM. VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp Neurol. 1998;154:57–65. [PubMed: 9875268]
- 87.
- Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. [PMC free article: PMC6783109] [PubMed: 10407014]
- 88.
- Silverman WF, Krum JM, Mani N. et al. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience. 1999;90:1529–1541. [PubMed: 10338318]
- 89.
- Temburni MK, Jacob MH. New functions for glia in the brain. Proc Natl Acad Sci USA. 2001;98:3631–3632. [PMC free article: PMC33328] [PubMed: 11274377]
- 90.
- Robinson GS, Ju M, Shih S -C, Xu X, McMahon G, Caldwell R, Smith L E H. Nonvascular role for VEGF: VEGFR-1, 2 activity is critical for neural retinal development. FASEB J. 2001;15:1215–1217. [PubMed: 11344092]
- 91.
- Yourey PA, Gohari S, Su JL. et al. Vascular endothelial cell growth factors promote the in vitro development of rat photoreceptor cells. J Neurosci. 2000;20:6781–6788. [PMC free article: PMC6772847] [PubMed: 10995821]
- 92.
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. [PMC free article: PMC27841] [PubMed: 10963684]
- 93.
- Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP. et al. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: Signal transduction cascades. FASEB J. 2001;15:1218–1220. [PubMed: 11344093]
- 94.
- Hayashi T, Abe K, Itoyama Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab. 1998;18:887–895. [PubMed: 9701350]
- 95.
- Sondell M, Kanje M. Postnatal expression of VEGF and its receptor flk-1 in peripheral ganglia. Neuroreport. 2001;12:105–108. [PubMed: 11201066]
- 96.
- Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor stimulates Schwann cell invasion and neovascularization of acellular nerve grafts. Brain Res. 1999;846:219–228. [PubMed: 10556639]
- 97.
- Sondell M, Sundler F, Kanje M. Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci. 2000;12:4243–4254. [PubMed: 11122336]
- 98.
- Schratzberger P, Schratzberger G, Silver M. et al. Favorable effect of VEGF gene transfer on ischemic peripheral neuropathy. Nat Med. 2000;6:405–413. [PubMed: 10742147]
- 99.
- Schratzberger P, Walter DH, Rittig K. et al. Reversal of experimental diabetic neuropathy by VEGF gene transfer. J Clin Invest. 2001;107:1083–1092. [PMC free article: PMC209283] [PubMed: 11342572]
- 100.
- Oosthuyse B, Moons L, Storkebaum E. et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. [PubMed: 11381259]
- 101.
- Bagnard D, Vaillant C, Khuth S -T. et al. Semaphorin 3A-vascular endothelial growth factor-165 balance mediates migration and apoptosis of neural progenitor cells by the recruitment of shared receptor. J Neurosci. 2001;21:3332–3341. [PMC free article: PMC6762465] [PubMed: 11331362]
- 102.
- Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor Flt-1. J Biol Chem. 2000;275:26690–26695. [PubMed: 10842181]
- 103.
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. [PubMed: 8689573]
- 104.
- Asahara T, Takahashi T, Masuda H. et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–3972. [PMC free article: PMC1171472] [PubMed: 10406801]
- 105.
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. [PubMed: 9109485]
- 106.
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995; 1:27–31. [PubMed: 7584949]
- 107.
- Buschmann I, Schaper W. Arteriogenesis versus angiogenesis: Two mechanisms of vessel growth. News Physiol Sci. 1999;14:121–125. [PubMed: 11390835]
- 108.
- Harik SI, Hritz MA, LaManna JC. Hypoxia-induced brain angiogenesis in the adult rat. J Physiol (London). 1995;485:525–530. [PMC free article: PMC1158011] [PubMed: 7545234]
- 109.
- Boero JA, Ascher J, Arregui A. et al. Increased brain capillaries in chronic hypoxia. J Appl Physiol. 1999;86:1211–1219. [PubMed: 10194205]
- 110.
- LaManna JC, Harik SI. Brain metabolic and vascular adaptations to hypoxia in the rat. Adv Exp Med Biol. 1997;428:163–167. [PubMed: 9500043]
- 111.
- Kuo N -T, Benhayon D, Przybylski RJ. et al. Prolonged hypoxia increases vascular endothelial growth factor mRNA and protein in adult mouse brain. J Appl Physiol. 1999;86:260–264. [PubMed: 9887138]
- 112.
- Marti HH, Bernaudin M, Petit E. et al. Neuroprotection and angiogenesis: A dual role of erythropoietin in brain ischemia. News Physiol Sci. 2000;15:225–229. [PubMed: 11390915]
- 113.
- Greenberg DA. Angiogenesis and stroke. Drug News Perspect. 1998;11:265–270. [PubMed: 15616645]
- 114.
- Ment LR, Stewart WB, Fronc R. et al. Vascular endothelial growth factor mediates reactive angiogenesis in the postnatal developing brain. Brain Res Dev Brain Res. 1997;100:52–61. [PubMed: 9174246]
- 115.
- van Bruggen N, Thibodeaux H, Palmer JT. et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. [PMC free article: PMC409867] [PubMed: 10587525]
- 116.
- Zhang ZG, Zhang L, Jiang Q. et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. [PMC free article: PMC517814] [PubMed: 11018070]
- 117.
- Eliceiri BP, Paul R, Schwartzberg PL. et al. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. [PubMed: 10635317]
- 118.
- Paul R, Zhang ZG, Eliceiri BP. et al. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. [PubMed: 11175854]
- 119.
- Gerber HP, McMurtrey A, Kowalski J. et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3'-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. [PubMed: 9804796]
- 120.
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor rescues HN33 neural cells from death induced by serum withdrawal. J Mol Neurosci. 2000;14:197–203. [PubMed: 10984196]
- 121.
- Kalaria RN, Cohen DL, Premkumar D R D. et al. Vascular endothelial growth factor in Alzheimer's disease and experimental cerebral ischemia. Mol Brain Res. 1998;62:101–105. [PubMed: 9795165]
- 122.
- Thurston G, Rudge JS, Ioffe E. et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. [PubMed: 10742156]
- 123.
- Celletti FL, Waugh JM, Amabile PG. et al. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–429. [PubMed: 11283668]
- Vascular Endothelial Growth Factor - Madame Curie Bioscience DatabaseVascular Endothelial Growth Factor - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...