

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The abnormal accumulation of β-amyloid (Αβ) in the brain is an early and invariant feature of Alzheimer's disease and is believed to play a pivotal role in the etiology and pathogenesis of the disease. The concentration of Αβ is regulated by multiple enzymatic activities, including proteases responsible for its degradation. In this chapter we present evidence for endothelin-converting enzymes (ECEs) as Αβ-degrading enzymes. Overexpression of ECE1 in cultured cells reduces Αβ accumulation by up to 90%, and the enzyme is capable of directly cleaving Αβ at multiple sites. As ECEs are expressed in brain, reduced ECE activity by genetic mutation, altered transcriptional activity, or pharmacological inhibition, for example, may be a risk factor for Alzheimer's disease (AD). The risk of pharmacological reduction of ECE activity is of particular concern since ECE inhibitors are being developed for the treatment of hypertension and other disorders.
Introduction: The Endothelin-Converting Enzymes
The endothelin-converting enzymes (ECEs) are a class of type II integral membrane zinc metalloproteases (active site lumenal) named for their ability to hydrolyze a family of biologically inactive intermediates, big endothelins (big ETs), exclusively at a Trp21-Val/Ile22 bond to form the potent vasoconstrictors endothelins.1 This specific cleavage event appears to be determined in part by the secondary/tertiary structure of disulfide-bonded big ETs, as linear big ET-1 (in which the cysteines have been alkylated) is cleaved at multiple sites by ECE.2 Targeted disruption of the ECE-1 gene in mice revealed that this enzyme is in fact a physiologically relevant activating enzyme for both ET-1 and ET-3, and ECE-1 null mice die in utero or within minutes of birth due to severe craniofacial and cardiac defects.3 In addition to the specific cleavage of big ETs, ECE has been reported to hydrolyze several biologically active peptides in vitro, including bradykinin, neurotensin, substance P, and oxidized insulin B chain by cleaving on the amino side of hydrophobic residues.4,5
Two different endothelin-converting enzymes have been cloned. The first identified, ECE-1, is abundantly expressed in the vascular endothelial cells of all organs and is also widely expressed in nonvascular cells of tissues including brain, lung, pancreas, testis, ovary, and adrenal gland.6-8 Four isoforms of human ECE-1 differing only in the cytoplasmic tail are produced by a single gene located on chromosome 1 (1p36) through the use of alternate promoters.6,9-13 The four isoforms cleave big ETs with equal efficiency but differ primarily in their subcellular localization and tissue distribution.12,13 Human ECE1a is localized predominantly to the plasma membrane.12,13 Human ECE-1c and ECE-1d have also been reported to be localized predominantly to the plasma membrane with additional intracellular expression detected.12,13 In contrast, human ECE-1b appears to be localized exclusively intracellularly. Co-immunolocalization studies performed by Schweizer et al12 on human ECE-1b transfected CHO cells indicate the presence of this isoform in the trans-Golgi network (TGN). Azarani et al similarly demonstrated that human ECE-1b was located in an intracellular compartment when expressed in Madin-Darby canine kidney (MDCK) cells.14 In an endogenous ECE-1b and ECE-1c expressing cell line, ECV304, ECE-1 immunoreactivity was detected in intracellular Golgi-like structures as well as at the cell surface.12
ECE-2 is a homologous enzyme with catalytic activity similar to that of ECE-1. ECE-2 is localized intracellularly, has an acidic pH optimum, and is expressed most abundantly in the nervous system.15 Immunocytochemical analysis of endogenous ECE-2 in HUVEC cells revealed a punctate pattern of staining consistent with expression of ECE-2 in acidic intracellular vesicles of the constitutive secretory pathway.16 Like ECE-1, ECE-2 cleaves big ET-1 most efficiently among the three big ETs, and at least three isoforms of human ECE-2 are produced from a single gene on chromosome 3 (3q28q29).12,15,17 The catalytic activity, subcellular localization, and tissue distribution of the isoforms have not yet been compared.
ECE-2 null mice have been made and appear normal and healthy.18 In ECE-1/ECE-2 double knockout embryos the levels of two known products of ECE activity, ET-1 and ET-2, are remarkably similar to the ECE-1 nulls alone. The highly restricted tissue distribution of ECE-2 and modest effect of disruption of the ECE-2 gene on an ECE-1 null background suggests that ECE-2 may have physiological substrates distinct from the big ETs.18
Both ECE-1 and ECE-2 Are Expressed in Brain
While many classes of proteases may degrade Αβ in vitro, to be physiologically relevant Αβ-degrading enzymes must be expressed in the brain and their cellular localization must be consistent with having access to Αβ peptides. In the nervous system, ECE-1 immunoreactivity has been detected in fibers within the glial limitans, in neuronal processes and cell bodies of the cerebral cortex, in pyramidal cells of the neocortex and hippocampus, in astrocytes, and in Purkinje cells in the cerebellum.7,19-21 Northern blot analysis of bovine tissues revealed that ECE-2 is most abundantly expressed in neural tissues including cerebral cortex, cerebellum, and adrenal medulla.15 In mouse brain, ECE-2 is expressed in heterogeneous populations of neurons in the thalamus, hypothalamus, amygdala, dentate gyrus, and CA3.18 While detailed co-localization studies have not been performed, separate studies indicate that ECE and Αβ are present in the same cellular compartments. Human ECE-1b has been reported to be present in the TGN, a proposed site of Αβ40 generation in neuronal cells.12,22 ECE-2 is also likely to be present in the TGN and vesicles of the constitutive secretory pathway.15,16 The topology of the ECEs is such that the active site is within the lumen of organelles and vesicles, providing access to the Αβ peptide.
A Potential Role for Endothelin-Converting Enzymes in Αβ Catabolism
The role that ECE may play in Alzheimer's disease (AD) is only beginning to be explored. While the data are controversial and factors other than ECE may contribute to endothelin levels, it is worth noting that endothelin levels have been reported to be decreased in the CSF of patients with AD when compared to nondemented control patients.23 Sib-pair analyses of genetic factors contributing to late onset AD have not excluded the region on chromosome 1 where the ECE-1 gene is located,24 and interestingly the human ECE2 gene is located on chromosome 3 in the vicinity of a possible AD locus.17,25,26 In this chapter we will present pharmacological and biochemical evidence for ECEs as Αβ degrading enzymes.
The Serendipitous Discovery of Metalloproteases as Important Modulators of Αβ Concentration in Vivo
In the mid-1990s a major focus of Alzheimer's disease research in our laboratory and others was the identification of the proteases responsible for the generation of Αβ peptides from a larger precursor, the β-amyloid precursor protein or βAPP. Two distinct proteolytic activities, termed β and γ secretase, are required for the release of Αβ from the membrane-bound βAPP, and the identification of specific inhibitors of these activities would aid greatly in the identification and characterization of the then-elusive proteases. As such, our laboratory screened an extensive collection of known protease inhibitors and other compounds for their ability to reduce Αβ secretion by a human CNS-derived cell line, H4. These screens revealed not only compounds that reduced Αβ concentration, but also some that dramatically increased the accumulation of the peptide. One of the most notable of these was the metalloprotease inhibitor phosphoramidon, which caused a 2–3-fold elevation of Αβ concentration in the medium of H4 cells (Fig. 7.1). This finding was particularly interesting to us as the effect on Αβ observed following phosphoramidon treatment was as great or greater than most of the AD-causing mutations that we and others had previously examined.27

To determine whether phosphoramidon could modulate Αβ concentration in vivo as well as in cell culture, we examined the effect of the compound following intravenous administration in guinea pigs, an animal model with an Αβ peptide sequence identical to that in humans. Phosphoramidon treatment caused a rapid, dose-dependent increase in the concentration of Αβ in the plasma of guinea pigs (Fig. 7.2), indicating that one or more phosphoramidon-sensitive proteases play an important role in regulating Αβ concentration in vivo.
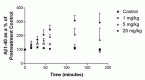
Phosphoramidon Increases Αβ Concentration in CNS-Derived Cell Lines Through the Inhibition of Intracellular Degradation
There are multiple mechanisms by which phosphoramidon could cause an increase in Αβ concentration. For example, inhibition of α-secretase, which cleaves within the Αβ peptide, could cause an increase in the production of Αβ by increasing the amount of substrate available for the Αβ-generating β- and γ-secretases. This mechanism, however, was unlikely to be responsible for the phosphoramidon-mediated effect on Αβ as the levels of other α-secretase derived APP products such as secreted APP-α (sAPPα), and C-terminal fragment α (CTFα) were unchanged in treated cells.28,29 An intriguing possibility was that phosphoramidon was somehow inhibiting the turnover of Αβ.
Proteolytic degradation of Αβ may occur at multiple sites, both intracellular and extracellular. To determine if the phosphoramidon-mediated increase in Αβ was due to inhibition of a secreted protease we first examined the amount of Αβ remaining in isolated H4 conditioned medium that was incubated at 37°C. While Αβ degradation was readily apparent, as assessed by loss of signal in our sandwich ELISAs with increasing incubation time, this degradation was not sensitive to phosphoramidon (data not shown). To examine whether the phosphoramidon induced increases in Αβ could be due to inhibition of a cell-surface protease we performed a spike experiment in which exogenous Αβ was added to the medium bathing H4 cells in the absence or presence of phosphoramidon. Exogenous Αβ was removed equally well in the presence or absence of phosphoramidon, indicating that the phosphoramidon-induced effect is not likely due to decreased internalization or to inhibition of a cell-surface or secreted protease.29 Collectively, these data suggested that phosphoramidon was exerting its effect on an intracellular event that eventually culminated in an increase in extracellular Αβ concentration. This is consistent with a report by Fuller et al showing an increase in cell-associated Αβ in phosphoramidon-treated SY5Y cells.28 In H4 cells, however, we have been unable to detect an elevation in intracellular Αβ levels, presumably due to rapid secretion of Αβ in this model system.
Endothelin-Converting Enzymes: Potential Targets for the Phosphoramidon-Mediated Increase in Αβ Concentration
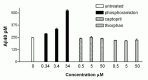
Phosphoramidon is known to inhibit several metalloproteases including ECE-1, ECE-2, neprilysin, and angiotensin-converting enzyme (ACE), but not insulin-degrading enzyme (IDE).30,31 Neprilysin and ACE, which are both capable of degrading Ab, have been reported to reside predominantly on the cell surface, although a soluble form of neprilysin is also present in serum and CSF.32-38 A recently identified phosphoramidon and thiorphan-sensitive neprilysin homologue SEP/NL1/NEPII is expressed both as a membrane-bound and secreted protease.39-41 While our spike experiments suggested that the phosphoramidon-induced increases in Αβ are not likely due to inhibition of a cell-surface or secreted enzyme, we decided to further evaluate a role for neprilysin and ACE in our cell culture system using more selective inhibitors of these enzymes, thiorphan and captopril, respectively. We have not yet been able to similarly analyze ECE, as a more selective inhibitor of ECE is not commercially available.
Treatment of H4 cells with phosphoramidon resulted in a greater than 2-fold elevation in Αβ40 accumulation, with a half-maximal effect occurring at a dose of approximately 7.5 μM (see Fig. 7.1). Treatment with thiorphan or captopril at concentrations greater than 1000 times the reported IC50 for the target enzymes in in vitro studies,42,43 but less than that required to inhibit ECE, failed to result in increases in extracellular Αβ (Fig. 7.3). These data indicated that the phosphoramidon-induced effect in H4 cells is not likely due to inhibition of neprilysin, SEP or ACE. Similar results were obtained for Αβ42 (data not shown). As endogenous ECE activity was readily detected in solubilized membranes of H4 cells using a big ET conversion assay6,44 (data not shown), our focus then shifted to evaluating a role for ECE in Αβ degradation.
Overexpression of Endothelin-Converting Enzyme-1 Results in a Significant Decrease in Extracellular Αβ Concentration That Is Completely Reversed by Treatment with Phosphoramidon
Evidence implicating a potential role for ECE in modulating Αβ concentration came further from the observation that CHO cells, which have no endogenous ECE activity,6 produce very high levels of Αβ when compared to most other cell types (personal observation). Conversely, HUVEC cells, which have high levels of endogenous ECE,45 accumulate very little Αβ unless treated with high concentrations of phosphoramidon (data not shown). To follow up on these casual observations and to further investigate the role of ECE in Αβ accumulation we cloned and stably transfected CHO cells with human ECE-1a and ECE-1b. Overexpression of either ECE-1a or ECE-1b in CHO cells, which lack endogenous ECE activity, resulted in a striking 75–90% reduction in Αβ40 and a 45–60% reduction in Αβ42 (Fig. 7.4). No significant changes were observed in the amount of sAPP accumulation in ECE-transfected cells compared to the vector controls, indicating that the cells were similarly viable and that general secretion is not affected by ECE overexpression. The reduction in Αβ concentration in ECE-transfected cells was completely reversed by treatment with phosphoramidon, indicating that the observed phenotype was likely due to the enzymatic activity of the overexpressed ECE.
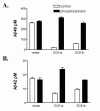
Increased Removal of Exogenous Αβ Is Apparent Only in ECE-1a Transfected Cells
In an endogenous ECE-expressing cell line, H4, extracellular Αβ removal does not appear to be affected by phosphoramidon treatment.29 To determine whether extracellular Αβ removal could account, at least in part, for the dramatic decrease in extracellular Αβ concentration in ECE-transfected cell lines, we spiked Αβ into the culture medium in the presence or absence of phosphoramidon and determined the percent removal by sandwich ELISA at 6 and 24 hours. Following a 6-hour incubation, removal of Αβ was similar in the culture medium of vector and ECE-transfected CHO cells, and was not affected by phosphoramidon treatment, although secretion of endogenous Αβ in phosphoramidon-treated ECE-transfected cells was increased 1.5 to 2-fold during the same time period.29 After a 24-hour incubation, we did observe a significant increase in the removal of the spiked-in Αβ in the medium of ECE-1a transfected cells compared to the vector controls (Fig. 7.5). No significant change in exogenous Αβ removal was observed in cells expressing ECE-1b. The ECE-1a induced increase in Αβ removal could be completely attenuated by phosphoramidon treatment, indicating that the effect was likely due to the enzymatic activity of ECE-1a (Fig. 7.5, inset).

These results suggest that ECE may contribute slightly to extracellular Αβ removal, at least in cells overexpressing human ECE-1a. However, the dramatic increase in Αβ concentration in ECE-1a expressing cells upon treatment with phosphoramidon (see Fig. 7.4) does not appear to be accounted for by the modest increase in exogenous Αβ degradation. While ECE-1a has been reported to be localized predominantly to the cell surface, this isoform has been shown to process big ET-1 intracellularly in CHO cells, most likely in secretory vesicles, as well as at the cell surface.46 Therefore, ECE-1a may similarly degrade Αβ intracellularly in CHO cells as it is being trafficked to the cell surface. Ultimately, the result of increased intracellular degradation is a decrease in extracellular Αβ accumulation.
Recombinant ECE-1 Degrades Αβ in vitro
Recombinant, soluble forms of ECE-1 (solECE-1) lacking the intracellular and transmembrane domains have been reported to hydrolyze big ET-1 with activity comparable to that of membrane-bound ECE-1a.5,44,47 To examine whether ECE-1 is capable of direct catabolism of Αβ we generated a soluble ECE-1 similar to those previously described. Incubation of synthetic Αβ40 and Αβ42 with this enzyme resulted in a nearly complete loss of the full-length peptides as detected by sandwich ELISA, and this reduction was completely blocked by incubation with ECE inhibitors.29
ECE-1 has been shown to cleave a number of biologically active peptides on the amino side of hydrophobic residues and appears not to cleave peptides smaller than 6 amino acids in length.5 Given this specificity, there are approximately 13 potential ECE cleavage sites in the Αβ40 peptide. Using HPLC, mass spectrometry, and NH2-sequence analysis29 we determined that soluble ECE-1 cleaves synthetic Αβ40 at at least three sites, resulting in the formation of Αβ fragments 1–16, 1–17, 1–19, and 20–40 (Fig. 7.6). Consistent with the known substrate specificity of ECE-1, each of these observed cleavages by solECE-1 occurred on the amino side of hydrophobic residues (Leu17, Val18, Phe20). Given that ECE-2 is highly homologous to ECE-1 and shares similar catalytic activity,15 ECE-2 is also likely capable of degrading Αβ.
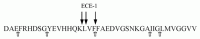
Kinetic Analysis of Αβ40 Cleavage by solECE-1
To determine the catalytic efficiency of Αβ cleavage by solECE-1 we calculated kcat/Km by measuring the rate of Αβ hydrolysis under second-order conditions at pH 6.5, the reported pH optimum for big ET-1 cleavage by the enzyme. Using this method, the kcat/Km for Αβ40 hydrolysis by solECE1 was determined to be (1.7 +/- 0.6) × 10-3 M-1sec-1 at pH 6.5, a value 15-fold lower than that for big ET-1 hydrolysis under the same conditions.29 Intrigued by a report indicating that solECE-1 cleaves bradykinin and substance P with an acidic pH optimum of approximately 5.6 compared to the optimum of pH 6.5 for big ET-1,48 we next compared the efficiency of solECE-1 hydrolysis of Αβ40 and big ET-1 at pH 5.6. Similar to bradykinin and substance P, solECE-1 cleaves Αβ40 greater than an order of magnitude more efficiently at pH 5.6 than at pH 6.5. The value of kcat/Km for big ET-1 hydrolysis at pH 5.6 is only 3-fold greater than that for Αβ40 at the same pH. This result may be particularly relevant as the TGN, where ECE-1b appears to be expressed, has an acidic pH. It may also help to explain why little exogenous Αβ is degraded by ECE-1a in transfected CHO cells.
Current Studies in Animal Models
A reasonable test for the physiological involvement of ECE or any enzyme in contributing to Αβ accumulation in the brain is to examine the effect of animals null for the enzyme. While several enzymes have been identified that can degrade Αβ in in vitro assays, only neprilysin has thus far been reported to influence Αβ accumulation in the brains of knock-out mice.33,49,50 Ongoing studies in our laboratory are focused on determining the effect of ECE deficiency on Αβ accumulation in the brains of knockout mice. As in the neprilysin knockout mice, elevated levels of Αβ in the brains of ECE knockout mice would indicate a physiological role for this enzyme family in regulating the accumulation of the peptide. Crossing the ECE knockout mice with a mouse model that develops AD-like pathology will further test the hypothesis that reduced ECE activity may critically alter the concentration of Αβ in the brain, accelerating and/or enhancing the development of senile plaques.
Summary: ECE Activity May Be One of Many Regulators of Αβ Accumulation in the Brain
It is likely that multiple proteases, both intracellular and extracellular, may play a role in determining Αβ concentration. Intracellular degradation of Αβ at the site of generation and/or within the secretory pathway may affect the extracellular concentration of the peptide by limiting the amount available for secretion. The concentration of secreted Αβ is further regulated by direct degradation by extracellular proteases and by receptor-mediated endocytosis or phagocytosis followed by lysosomal degradation. The relative contribution of Αβ degrading enzymes and other mechanisms of Αβ removal may vary in different regions of the brain and may also differ for Αβ40 and Αβ42. Decreases in any one of these mechanisms of Αβ removal, whether major or minor, may potentially result in increased Αβ accumulation and the development of AD pathology. Conversely, an increase in the activity of any enzyme capable of degrading Αβ may result in decreased accumulation of the peptide, potentially reducing the risk for AD.
We have presented extensive evidence that ECE is capable of degrading Αβ peptides, and studies in cell-based models suggest that the major site of ECE's effect on Αβ is intracellular, possibly in an acidic compartment. Even in cells overexpressing ECE-1a, an isoform expressed predominantly on the cell-surface, little ECE-mediated degradation of extracellular Αβ was observed. This may be due to the unfavorable kinetics of Αβ degradation by ECE at neutral pH and suggests that the dramatic reduction in Αβ concentration seen in these cells is due to degradation of Αβ within the biosynthetic, secretory, endocytic or other slightly acidic organelles and pathways. Interestingly, inhibition of this intracellular event(s) has the net result of a substantial increase in extracellular Ab.
The Regulation of Endogenous ECE Activity Is Complex, and Alterations May Influence Susceptibility to Alzheimer's Disease
The regulation of ECE expression is complex, as evidenced by the production of multiple isoforms of both ECE-1 and ECE-2 through the use of multiple promoters and alternative splicing. ECE-1 activity may be modulated at the level of transcription, mRNA stability, glycosylation, and zinc binding (see for example refs. 51-59). ECE-1 expression is increased under a number of pathophysiological conditions including but not limited to hypertension, congestive heart failure, subarachnoid hemorrhage, preeclampsia, and wound healing.55,60-62 In cultured endothelial cells, thrombin upregulates ECE-1 expression via the ERK pathway.63 It has been reported that ECE-1 activity is regulated transcriptionally and/or posttranscriptionally by endothelins (downregulated via ETB receptors), and angiotensin II (upregulated via ETA receptors).58,64-66 Glycosylation is required for the functional expression of ECE156,57 and altered glycosylation may be involved in the negative feedback regulation of ECE1 by ETB receptors.58 Superoxide, which may be generated during a variety of pathophysiological conditions, reversibly inhibits ECE by causing the release of zinc from the active site.59
Additional studies are required to determine the extent of the role of ECE activity in determining Αβ concentration in the brain, and the possible link between altered ECE activity and the development and/or progression of Alzheimer's disease. The complex regulation of ECE activity suggests that physiological conditions that cause a reduction in ECE activity in the brain may elevate Αβ levels and increase susceptibility to AD. Alterations in ECE activity in the brain (either in total activity or in the relative expression of ECE isoforms) may possibly occur in normal aging, the most common risk factor for AD. Genetic mutations in ECE that decrease its activity may also be identified that are causative of Alzheimer's disease in certain individuals. Equally important, however, is the possibility that there may be individuals with normally high levels of ECE activity who are at a reduced risk for the disease. A careful analysis of ECE activity in AD and control individuals is necessary to determine the extent of the involvement of this enzyme family in the development of AD.
Increased ECE Activity May Be Therapeutic for Alzheimer's Disease
Regardless of the extent of the influence of endogenous ECE activity on Αβ accumulation in vivo, increased activity of ECE or any other Αβ-degrading enzyme may be beneficial in AD. Up-regulation of ECE activity through gene therapy, transcriptional activation, or by reducing ECE turnover, for example, may provide a novel therapeutic approach for the treatment of AD. One obvious concern with this treatment method is that patients may become hypertensive. Up-regulation of ECE activity in the periphery of rats by intravenous injection of an adenoviral construct containing a secreted form of ECE does not, however, appear to result in increased circulating endothelin levels or in hypertension, indicating that ECE is not likely to be rate-limiting in the biosynthesis of ET under these conditions.67 Further, even if increased ECE activity does augment endothelin levels, endothelin receptor antagonists could be given in parallel to reduce or block any effect of increased endothelin levels. Approaches to increase Αβ catabolism by up-regulation of ECE or other Αβ-degrading enzymes such as neprilysin49 or IDE50 could be used alone or in conjunction with methods to prevent Αβ production, aggregation, and/or toxicity.
The Clinical Use of ECE Inhibitors Must Be Considered Carefully
ECE inhibitors have received a significant amount of pharmaceutical interest for their potential as anti-hypertensive drugs and for other ailments.60,68,69 It is likely that if these drugs enter the brain they will increase Αβ levels, potentially leading to the development and/or acceleration of AD in susceptible individuals. Our data argue for a careful evaluation of the effects of these compounds on Αβ accumulation prior to the initiation of human exposure. Based on the genetic mutations that cause AD, in which Αβ levels are elevated for decades prior to the development of the disease, this potential side effect of ECE inhibitors may not be observed for years and must be considered very carefully before clinical use of this class of compounds. The potential risks of neprilysin inhibition must also be carefully considered. Because most ECE and neprilysin inhibitors will inhibit both enzymes at certain concentrations, the use of these drugs may be particularly risky if ECE and neprilysin both play physiological roles in the degradation of Αβ in the brain. ET receptors may be a safer target for pharmacological interference with the endothelin system to reduce hypertension without the side effect of decreased Αβ catabolism.
Acknowledgements
This work was supported by a grant from the Alzheimer's Association to E.E.; Smith Fellowships to C.E. and E.E., a grant from the Bursak Foundation to support the efforts of E.E. and by the Mayo Foundation for Medical Education and Research. We thank Takeda Chemical Industries for their generous gifts of BNT77, BA27, and BC05 and Mona Watson and Cristian-Mihail Prada for their excellent technical assistance.
References
- 1.
- Turner AJ, Murphy LJ. Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol. 1996;51:91–102. [PubMed: 8615890]
- 2.
- Fahnoe DC, Johnson GD, Herman SB. et al. Disulfide bonds in big ET-1 are essential for the specific cleavage at the Trp(21)-Val(22) bond by soluble endothelin converting enzyme-1 from baculovirus/insect cells. Arch Biochem Biophys. 2000;373:385–93. [PubMed: 10620363]
- 3.
- Yanagisawa H, Yanagisawa M, Kapur RP. et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–36. [PubMed: 9449665]
- 4.
- Hoang MV, Turner AJ. Novel activity of endothelin-converting enzyme: hydrolysis of bradykinin. Biochem J. 1997;327(Pt 1):23–6. [PMC free article: PMC1218757] [PubMed: 9355729]
- 5.
- Johnson GD, Stevenson T, Ahn K. Hydrolysis of peptide hormones by endothelin-converting enzyme-1. A comparison with neprilysin. J Biol Chem. 1999;274:4053–8. [PubMed: 9933597]
- 6.
- Xu D, Emoto N, Giaid A. et al. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–85. [PubMed: 8062389]
- 7.
- Davenport AP, Kuc RE, Plumpton C. et al. Endothelin-converting enzyme in human tissues. Histochemical Journal. 1998;30:359–74.
- 8.
- Korth P, Bohle RM, Corvol P. et al. Cellular distribution of endothelin-converting enzyme-1 in human tissues. J Histochem Cytochem. 1999;47:447–62. [PubMed: 10082746]
- 9.
- Valdenaire O, Rohrbacher E, Mattei MG. Organization of the gene encoding the human endothelin-converting enzyme (ECE-1). J Biol Chem. 1995;270:29794–8. [PubMed: 8530372]
- 10.
- Shimada K, Takahashi M, Ikeda M. et al. Identification and characterization of two isoforms of an endothelin-converting enzyme-1. FEBS Lett. 1995;371:140–4. [PubMed: 7672114]
- 11.
- Schmidt M, Kroger B, Jacob E. et al. Molecular characterization of human and bovine endothelin converting enzyme (ECE-1). FEBS Lett. 1994;356:238–43. [PubMed: 7805846]
- 12.
- Schweizer A, Valdenaire O, Nelbock P. et al. Human endothelin-converting enzyme (ECE-1): three isoforms with distinct subcellular localizations. Biochem J. 1997;328(Pt 3):871–7. [PMC free article: PMC1218999] [PubMed: 9396733]
- 13.
- Valdenaire O, Lepailleur-Enouf D, Egidy G. et al. A fourth isoform of endothelin-converting enzyme (ECE-1) is generated from an additional promoter molecular cloning and characterization. Eur J Biochem. 1999;264:341–9. [PubMed: 10491078]
- 14.
- Azarani A, Boileau G, Crine P. Recombinant human endothelin-converting enzyme ECE-1b is located in an intracellular compartment when expressed in polarized Madin-Darby canine kidney cells. Biochem J. 1998;333(Pt 2):439–48. [PMC free article: PMC1219603] [PubMed: 9657986]
- 15.
- Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;270:15262–8. [PubMed: 7797512]
- 16.
- Russell FD, Davenport AP. Evidence for intracellular endothelin-converting enzyme-2 expression in cultured human vascular endothelial cells. Circ Res. 1999;84:891–6. [PubMed: 10222335]
- 17.
- Lorenzo MN, Khan RY, Wang Y. et al. Human endothelin converting enzyme-2 (ECE2): characterization of mRNA species and chromosomal localization. Biochim Biophys Acta. 2001;1522:46–52. [PubMed: 11718899]
- 18.
- Yanagisawa H, Hammer RE, Richardson JA. et al. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J Clin Invest. 2000;105:1373–82. [PMC free article: PMC315458] [PubMed: 10811845]
- 19.
- Barnes K, Walkden BJ, Wilkinson TC, Turner AJ. Expression of endothelin-converting enzyme in both neuroblastoma and glial cell lines and its localization in rat hippocampus. J Neurochem. 1997;68:570–7. [PubMed: 9003042]
- 20.
- Sluck JM, Lin RC, Katolik LI. et al. Endothelin converting enzyme-1-, endothelin-1-, and endothelin-3-like immunoreactivity in the rat brain. Neuroscience. 1999;91:1483–97. [PubMed: 10391453]
- 21.
- Nakagomi S, Kiryu-Seo S, Kiyama H. Endothelin-converting enzymes and endothelin receptor B messenger RNAs are expressed in different neural cell species and these messenger RNAs are coordinately induced in neurons and astrocytes respectively following nerve injury. Neuroscience. 2000;101:441–9. [PubMed: 11074166]
- 22.
- Hartmann T, Bieger SC, Bruhl B. et al. Distinct sites of intracellular production for Alzheimer's disease Αβ40/42 amyloid peptides. Nat Med. 1997;3:1016–20. [PubMed: 9288729]
- 23.
- Yoshizawa T, Iwamoto H, Mizusawa H. et al. Cerebrospinal fluid endothelin-1 in Alzheimer's disease and senile dementia of Alzheimer type. Neuropeptides. 1992;22:85–8. [PubMed: 1407412]
- 24.
- Kehoe P, Wavrant-De Vrieze F. et al. A full genome scan for late onset Alzheimer's disease. Hum Mol Genet. 1999;8:237–45. [PubMed: 9931331]
- 25.
- Poduslo SE, Yin X, Hargis J. et al. A familial case of Alzheimer's disease without tau pathology may be linked with chromosome 3 markers. Hum Genet. 1999;105:32–7. [PubMed: 10480352]
- 26.
- Tanzi RE, Kovacs DM, Kim TW. et al. The gene defects responsible for familial Alzheimer's disease. Neurobiol Dis. 1996;3:159–68. [PubMed: 8980016]
- 27.
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Αβ isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:172–87. [PubMed: 10899442]
- 28.
- Fuller SJ, Storey E, Li QX. et al. Intracellular production of beta A4 amyloid of Alzheimer's disease: modulation by phosporamidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry. 1995;34:8091–98. [PubMed: 7794922]
- 29.
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–8. [PubMed: 11337485]
- 30.
- Kukkola PJ, Savage P, Sakane Y. Differential structure-activity relationships of phosphoramidon analogues for inhibition of three metalloproteases: endothelin- converting enzyme, neutral endopeptidase, and angiotensin-converting enzyme. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S65–8. [PubMed: 8587470]
- 31.
- Ansorge S, Bohley P, Kirschke H. et al. The insulin and glucagon degrading proteinase of rat liver: a metal-dependent enzyme. Biomed Biochim Acta. 1984;43:39–46. [PubMed: 6372797]
- 32.
- Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze beta-amyloid(1–40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides. 1995;16:647–52. [PubMed: 7479298]
- 33.
- Iwata N, Tsubuki S, Takaki Y. et al. Identification of the major Αβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. [PubMed: 10655101]
- 34.
- Hu J, Igarashi A, Kamata M. et al. Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide Αβ, retards Αβ aggregation, deposition, fibril formation and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–47868. [PubMed: 11604391]
- 35.
- Turner AJ, Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. Faseb J. 1997;11:355–64. [PubMed: 9141502]
- 36.
- Corvol P, Williams TA, Soubrier F. Peptidyl dipeptidase A: angiotensin I-converting enzyme. Methods Enzymol. 1995;248:283–305. [PubMed: 7674927]
- 37.
- Spillantini MG, Sicuteri F, Salmon S. et al. Characterization of endopeptidase 3.4.24.11 “enkephalinase” activity in human plasma and cerebrospinal fluid. Biochem Pharmacol. 1990;39:1353–6. [PubMed: 2322317]
- 38.
- Soleilhac JM, Lafuma C, Porcher JM. et al. Characterization of a soluble form of neutral endopeptidase-24.11 (EC 3.4.24.11) in human serum: enhancement of its activity in serum of underground miners exposed to coal dust particles. Eur J Clin Invest. 1996;26:1011–7. [PubMed: 8957208]
- 39.
- Ikeda K, Emoto N, Raharjo SB. et al. Molecular identification and characterization of novel membrane-bound metalloprotease, the soluble secreted form of which hydrolyzes a variety of vasoactive peptides. J Biol Chem. 1999;274:32469–77. [PubMed: 10542292]
- 40.
- Ghaddar G, Ruchon AF, Carpentier M. et al. Molecular cloning and biochemical characterization of a new mouse testis soluble-zinc-metallopeptidase of the neprilysin family. Biochem J. 2000;347(Pt 2):419–29. [PMC free article: PMC1220974] [PubMed: 10749671]
- 41.
- Ouimet T, Facchinetti P, Rose C. et al. Neprilysin II: A putative novel metalloprotease and its isoforms in CNS and testis. Biochem Biophys Res Commun. 2000;271:565–70. [PubMed: 10814502]
- 42.
- Roques BP, Beaumont A. Neutral endopeptidase-24.11 inhibitors: from analgesics to antihypertensives? Trends Pharmacol Sci. 1990;11:245–9. [PubMed: 2166369]
- 43.
- Gronhagen-Riska C, Fyhrquist F. Purification of human lung angiotensin-converting enzyme. Scand J Clin Lab Invest. 1980;40:711–9. [PubMed: 6269175]
- 44.
- Ahn K, Herman SB, Fahnoe DC. Soluble human endothelin-converting enzyme-1: expression, purification, and demonstration of pronounced pH sensitivity. Arch Biochem Biophys. 1998;359:258–68. [PubMed: 9808768]
- 45.
- Ahn K, Pan S, Beningo K. et al. A permanent human cell line (EA.hy926) preserves the characteristics of endothelin converting enzyme from primary human umbilical vein endothelial cells. Life Sci. 1995;56:2331–41. [PubMed: 7791520]
- 46.
- Parnot C, Le Moullec JM, Cousin MA. et al. A live-cell assay for studying extracellular and intracellular endothelin-converting enzyme activity. Hypertension. 1997;30:837–44. [PubMed: 9336381]
- 47.
- Korth P, Egidy G, Parnot C. et al. Construction, expression and characterization of a soluble form of human endothelin-converting-enzyme-1. FEBS Lett. 1997;417:365–70. [PubMed: 9409753]
- 48.
- Fahnoe DC, Knapp J, Johnson GD. et al. Inhibitor potencies and substrate preference for endothelin-converting enzyme-1 are dramatically affected by pH. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S22–5. [PubMed: 11078325]
- 49.
- Iwata N, Tsubuki S, Takaki Y. et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. [PubMed: 11375493]
- 50.
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–80. [PubMed: 11683988]
- 51.
- Orzechowski HD, Gunther A, Menzel S. et al. Endothelial expression of endothelin-converting enzyme-1 beta mRNA is regulated by the transcription factor Ets-1. J Cardiovasc Pharmacol. 1998;31(Suppl 1):S55–7. [PubMed: 9595399]
- 52.
- Orzechowski HD, Richter CM, Funke-Kaiser H. et al. Evidence of alternative promoters directing isoform-specific expression of human endothelin-converting enzyme-1 mRNA in cultured endothelial cells. J Mol Med. 1997;75:512–21. [PubMed: 9253714]
- 53.
- Funke-Kaiser H, Orzechowski HD, Richter M. et al. Human endothelin-converting enzyme-1 β mRNA expression is regulated by an alternative promoter. J Cardiovasc Pharmacol. 1998;31(Suppl 1):S7–9. [PubMed: 9595385]
- 54.
- Barker S, Khan NQ, Wood EG. et al. Effect of an antisense oligodeoxynucleotide to endothelin-converting enzyme-1c (ECE-1c) on ECE-1c mRNA, ECE-1 protein and endothelin-1 synthesis in bovine pulmonary artery smooth muscle cells. Mol Pharmacol. 2001;59:163–9. [PubMed: 11160849]
- 55.
- Shao R, Yan W, Rockey DC. Regulation of endothelin-1 synthesis by endothelin-converting enzyme-1 during wound healing. J Biol Chem. 1999;274:3228–34. [PubMed: 9915864]
- 56.
- Schweizer A, Loffler BM, Rohrer J. Palmitoylation of the three isoforms of human endothelin-converting enzyme-1. Biochem J. 1999;340(Pt 3):649–56. [PMC free article: PMC1220295] [PubMed: 10359648]
- 57.
- Nelboeck P, Fuchs M, Bur D. et al. Glycosylation of Asn-632 and Asn-651 is important for functional expression of endothelin-converting enzyme-1. J Cardiovasc Pharmacol. 1998;31(Suppl 1):S4–6. [PubMed: 9595384]
- 58.
- Ehrenreich H, Loffler BM, Hasselblatt M. et al. Endothelin converting enzyme activity in primary rat astrocytes is modulated by endothelin B receptors. Biochem Biophys Res Commun. 1999;261:149–55. [PubMed: 10405338]
- 59.
- Lopez-Ongil S, Senchak V, Saura M. et al. Superoxide regulation of endothelin-converting enzyme. J Biol Chem. 2000;275:26423–7. [PubMed: 10833511]
- 60.
- Loffler BM. Endothelin-converting enzyme inhibitors: current status and perspectives. J Cardiovasc Pharmacol. 2000;35:S79–82. [PubMed: 10976788]
- 61.
- Nishikawa S, Miyamoto A, Yamamoto H. et al. Preeclamptic serum enhances endothelin-converting enzyme expression in cultured endothelial cells. Am J Hypertens. 2001;14:77–83. [PubMed: 11206686]
- 62.
- Kwan AL, Bavbek M, Jeng AY. et al. Prevention and reversal of cerebral vasospasm by an endothelin-converting enzyme inhibitor, CGS 26303, in an experimental model of subarachnoid hemorrhage. J Neurosurg. 1997;87:281–6. [PubMed: 9254094]
- 63.
- Eto M, Barandier C, Rathgeb L. et al. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ Res. 2001;89:583–90. [PubMed: 11577023]
- 64.
- Naomi S, Iwaoka T, Disashi T. et al. Endothelin-1 inhibits endothelin-converting enzyme-1 expression in cultured rat pulmonary endothelial cells. Circulation. 1998;97:234–6. [PubMed: 9462522]
- 65.
- Barton M, Shaw S, d'Uscio LV. et al. Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: role of ETA receptors for endothelin regulation. Biochem Biophys Res Commun. 1997;238:861–5. [PubMed: 9325182]
- 66.
- Morawietz H, Goettsch W, Szibor M. et al. Angiotensin-converting enzyme inhibitor therapy prevents upregulation of endothelin-converting enzyme-1 in failing human myocardium. Biochem Biophys Res Commun. 2002;295:1057–61. [PubMed: 12135601]
- 67.
- Telemaque S, Emoto N, deWit D. et al. In vivo role of endothelin-converting enzyme-1 as examined by adenovirus-mediated overexpression in rats. J Cardiovasc Pharmacol. 1998;31 Suppl 1:S548–50. [PubMed: 9595539]
- 68.
- Gray GA, Webb DJ. The endothelin system and its potential as a therapeutic target in cardiovascular disease. Pharmacol Ther. 1996;72:109–48. [PubMed: 8981573]
- 69.
- Kitas EA, Loffler BM, Daetwyler S. et al. Synthesis of triazole-Tethered pyrrolidine libraries: novel ECE inhibitors. Bioorg Med Chem Lett. 2002;12:1727–30. [PubMed: 12067547]
- Introduction: The Endothelin-Converting Enzymes
- Both ECE-1 and ECE-2 Are Expressed in Brain
- A Potential Role for Endothelin-Converting Enzymes in Αβ Catabolism
- The Serendipitous Discovery of Metalloproteases as Important Modulators of Αβ Concentration in Vivo
- Phosphoramidon Increases Αβ Concentration in CNS-Derived Cell Lines Through the Inhibition of Intracellular Degradation
- Endothelin-Converting Enzymes: Potential Targets for the Phosphoramidon-Mediated Increase in Αβ Concentration
- Overexpression of Endothelin-Converting Enzyme-1 Results in a Significant Decrease in Extracellular Αβ Concentration That Is Completely Reversed by Treatment with Phosphoramidon
- Increased Removal of Exogenous Αβ Is Apparent Only in ECE-1a Transfected Cells
- Recombinant ECE-1 Degrades Αβ in vitro
- Kinetic Analysis of Αβ40 Cleavage by solECE-1
- Current Studies in Animal Models
- Summary: ECE Activity May Be One of Many Regulators of Αβ Accumulation in the Brain
- The Regulation of Endogenous ECE Activity Is Complex, and Alterations May Influence Susceptibility to Alzheimer's Disease
- Increased ECE Activity May Be Therapeutic for Alzheimer's Disease
- The Clinical Use of ECE Inhibitors Must Be Considered Carefully
- Acknowledgements
- References
- Αβ Degradation by Endothelin-Converting Enzymes - Madame Curie Bioscience Databa...Αβ Degradation by Endothelin-Converting Enzymes - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...