Executive Summary
An overview of the submission details for the drug under review is provided in .
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disorder characterized by the degeneration of upper motor neurons (UMNs) and lower motor neurons (LMNs).1 Symptoms of ALS are typically first noticed when limb weakness occurs, though the first symptoms can also be bulbar and involve difficulty in speaking or swallowing. Over time, patients lose function in additional regions, such as other limbs and respiratory muscles. Progressive muscle weakness and eventual respiratory failure leads to death.2,3 ALS is a clinically heterogeneous disease in terms of presentation and rate of progression. There is no definitive test for diagnosing ALS, and there can be a long duration from symptom onset to diagnosis. The etiology of the disease is unknown.2 In a Canadian systematic review published in 2009,4 estimates of age-adjusted annual incidence of ALS ranged from 2.0 to 2.4 per 100,000 persons.
There is no cure for ALS. Health Canada–approved treatments for ALS include riluzole and edaravone. Riluzole is an oral medication that has been shown to extend tracheostomy-free survival by 2 months to 3 months in patients with ALS.5 Edaravone, a free radical scavenger thought to prevent oxidative damage to vascular endothelial cells and nerve cells, is currently available as an intravenously administered drug and has been found to slow the rate of decline in motor function.6 According to clinical expert opinion, IV edaravone is typically administered concomitantly with riluzole but had a low uptake at the time of this review in part because of the IV mode of administration and because it is a complex regimen.
This Reimbursement Review report evaluates orally administered edaravone, which is available as an oral suspension of 105 mg of edaravone per 5 mL of suspension. The Health Canada–approved indication is for the treatment of patients with ALS. According to the draft product monograph, the Health Canada–recommended dose of oral edaravone is 105 mg (5 mL) taken orally or via a feeding tube (percutaneous endoscopic gastrostomy or nasogastric tube). The recommended treatment regimen starts with an initial treatment cycle of daily dosing for 14 days followed by a 14-day drug-free period. Subsequent treatment cycles involve daily dosing for 10 days out of 14-day periods, followed by 14-day drug-free periods. Edaravone oral suspension should be taken in the morning after fasting overnight for at least 8 hours and waiting at least 1 hour before eating or drinking anything except water. For patients who are unable to fast overnight, the required fasting interval can be shortened depending on the type of meal. Patients treated with 60 mg of edaravone injection may be switched to 105 mg (5 mL) edaravone oral suspension using the same dosing frequency. Upon switching to edaravone oral suspension, patients should follow edaravone oral suspension dosing recommendations with regard to food consumption.7 Edaravone injection was recommended for reimbursement for the treatment of ALS by the CADTH Canadian Drug Expert Committee (CDEC) in March 2019, if CDEC-specified conditions were met.6
Stakeholder Perspectives
The information in this section is a summary of the input provided by the patient and clinician groups that responded to CADTH’s call for input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group, the ALS Society of Canada, submitted the patient input for this review. The submission was based on an online survey that collected input from 629 patients and caregivers from Ontario and Quebec and on telephone interviews with 7 patients who had experience with oral edaravone.
Respondents indicated that the most severe of ALS symptoms include difficulties with mobility (including walking and standing), gripping or holding things, muscle cramping or twitching, and fatigue caused by muscle exhaustion. These symptoms were also among the most important to control for people living with ALS, in addition to difficulties breathing, speaking, and swallowing. Patients indicated that their social life, travel and hobbies, and family life suffered the most. In addition, caregivers of patients with ALS highlighted a negative impact on emotional and psychological well-being, including pervasive feelings of overwhelming grief and struggles with mental health, including stress, anxiety, helplessness, and hopelessness. Loss of independence was mentioned as touching all aspects of patients’ lives and dramatically impacting caregivers, as patients eventually need help performing all daily tasks. Patients and caregivers reported treatment experience with riluzole and IV edaravone. Slowing disease progression, maintaining ability to participate in daily activities, and increasing survival were identified as the most important benefits from therapy. Access to riluzole and IV edaravone treatments was a problem for some patients. Other difficulties reported with edaravone were mostly related to the IV administration, including patients having to schedule activities of daily living around their infusion schedule and needing to have a port catheter implanted.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH for the purpose of the review indicated that there is currently no ideal treatment that prevents disease progression. At the time of this review, the only Health Canada–approved disease-modifying treatments for ALS were riluzole and IV edaravone, both showing modest benefits in slowing disease progression. The expert indicated that patients would usually be prescribed riluzole for its clinical benefits and because it is easily administered and well tolerated. Patients meeting the criteria for reimbursement would then be offered IV edaravone as an add-on therapy.
The mainstay of care for patients with ALS consists of symptom management and quality-of-life optimization. The clinical expert highlighted that patients should be diagnosed and followed by an ALS specialist as part of a multidisciplinary care team. The clinical expert noted that current standard of care involves following the patient at regular intervals and monitoring their physical, functional, emotional, and quality-of-life parameters. Medications are titrated appropriate to a patient’s condition and to their goals of care, in a palliative-focused approach.
According to the clinical expert, patients with the greatest need are those patients with preservation of the ability to complete at least 1 of their own activities of daily life. Based on the clinical expert’s experience, it would not be appropriate to recommend that patients would try to (and not) receive sufficient benefit from other treatments before initiating oral edaravone. Requiring the patient to demonstrate that a treatment has failed before introduction of another treatment would subject them to irreversible progression that would otherwise have been slowed had other therapies been given concurrently and would not be reflective of currently available evidence.
According to the clinical expert, the uptake of IV edaravone has so far been low, in part because the IV formulation is invasive and comes with a time-consuming administration schedule. The clinical expert considered that the oral formulation would be a well-received alternative, as many patients choose not to embark on the currently available IV formulation because of the caveats and excessive requirements and constraints related to IV infusion.
Clinician Group Input
One clinician group, the Canadian ALS Research Network (CALS), has provided input, which was in line with the input provided by the clinical expert consulted by CADTH. CALS acknowledged the need for ALS disease-modifying treatment options aiming at slowing disease progression, as well as the need for oral edaravone in clinical practice due to increased accessibility.
Drug Program Input
The drug program implementation questions were aimed at gaining insight from the clinical expert consulted by CADTH about whether the submitted trials would be sufficient to show bioequivalence between the oral and IV versions of edaravone. The clinical expert consulted by CADTH indicated that there is no reason to think that the efficacy profiles of the 2 versions would differ; however, whether they can be considered to display comparable bioavailability will be assessed by Health Canada during formal review. Both the drug plans and the clinical expert also noted that an oral version of edaravone would be a lot easier for patients to access than its IV formulation, reducing the risk of exposure to unnecessary infusion-associated adverse events (AEs) and decreasing health care system burden related to the IV administration. The clinical expert expects that both edaravone-naive and edaravone-experienced patients would be prescribed treatment with the oral version of the product, expanding the number of patients using the medication. The drug plans questioned the clinical expert regarding the existing prescribing criteria for IV edaravone. The clinical expert highlighted that ALS should be managed by a specialist and a multidisciplinary team. It is the clinical expert’s opinion that there is sufficient access to ALS specialists across the country and that there is no need to expand to family doctors.
Clinical Evidence
Pivotal Studies
Description of Studies
To inform on the use of oral edaravone compared to its IV formulation, 2 manufacturer-sponsored studies were included in this review. The single-dose, randomized, open-label study MT-1186-J03 (n = 42)8,9 evaluated the bioequivalence of the oral suspension and IV formulation of edaravone in healthy individuals who identified as Japanese. The study assessed drug concentration (in plasma and urine) of unchanged edaravone, sulphate conjugate, and glucuronide conjugate, as well as various pharmacokinetic parameters, including the area under the plasma concentration–time curve (AUC) and the maximum plasma concentration after administration (Cmax) with the bioequivalence limit (0.80 to 1.25). Oral edaravone was administered as an oral suspension at 105 mg for a single dose.
The multicentre, open-label, single-group study MT-1186-A01 (n = 185)10 evaluated the longer-term safety and tolerability of oral edaravone in patients with ALS living and functioning independently whose first symptom of ALS had occurred within the previous 3 years and who had a baseline forced vital capacity (FVC) greater than or equal to 70%. At the time of this review, the 24-week results were available. Patients received edaravone as a 105 mg oral suspension administered in accordance with the Health Canada–approved regimen. The concomitant use of riluzole was permitted throughout the study.
Efficacy Results
According to the sponsor’s conclusions, study MT-1186-J03 showed that oral suspension edaravone 105 mg was bioequivalent to an IV formulation of edaravone 60 mg in healthy volunteers who identified as Japanese. In this analysis, oral edaravone had equivalent AUC from zero up to the last quantifiable concentration time point (AUC0-t) and AUC from zero up to infinity with extrapolation of the terminal phase (AUC0-∞) of unchanged edaravone compared to the IV formulation, as both geometric mean ratio and 90% confidence interval (CI) were within the range of 0.80 to 1.25. As for Cmax, the geometric mean ratio and its lower limit at the 90% CI were also within the prespecified limits, while the upper limit of the 90% CI exceeded 1.25. Detailed results for each pharmacokinetic parameter are presented in .
Summary of Key Results From Pivotal Bioequivalence Study MT-1186-J03.
Harms Results
One patient in each treatment group reported an AE of mild intensity in the single-dose bioequivalence study MT-1186-J03; these AEs were not judged to be reasonably related to the investigational products by the investigator. No serious AEs (SAEs), no withdrawals due to AEs, and no AEs of special interest were reported in the study.
Results from the single-group safety study MT-1186-A01 in patients with ALS were reported for the 24-week interim analysis. Seventy-nine percent of patients experienced at least 1 AE; however, discontinuation due to AEs was low (6%), suggesting the harm profile might be considered acceptable. SAEs were reported by 11% of patients; the most frequently reported were likely related to the disease: ALS (n = 5), dyspnea (n = 3), and respiratory failure (n = 3). Six patients died over the 24-week study period; causes of death were respiratory failure (n = 3), pneumonia (n = 1), suicide (n = 1), and ALS (n = 1). Among AEs of special interest, 8 patients reported cardiac disorders. All cardiac events arose from electrocardiogram findings, with the exception of 1 patient with cardiac failure, so the sponsor considered that they did not reveal a signal of concern. Key harms results are presented in .
Summary of Key Results From Pivotal Single-Group Safety Study MT-1186-A01 — Interim Analysis at Week 24.
Critical Appraisal
The most significant limitation associated with the included trials is the study designs. The bioequivalence design in healthy participants and the open-label uncontrolled study are not sufficient to evaluate the comparative clinical value added for the drug in the target population for reimbursement. The key assumption of the submission is that as IV edaravone has been approved by Health Canada and recommended for reimbursement by CADTH, establishing bioequivalence is sufficient to establish the clinical value of oral edaravone. However, the 2 formulations (solution for injection and oral suspension) cannot be considered bioequivalent since they involve 2 different dosing forms. Whether they can be considered to display comparable bioavailability of edaravone upon administration is to be assessed by Health Canada during formal review. While there is merit and supporting precedent to the assumption of comparable bioavailability, there remains a degree of uncertainty as to the true treatment effects of oral edaravone given the bioequivalence study design (i.e., single administration, assessing pharmacokinetic parameters with estimates falling within a range of acceptable values to establish equivalence) and the lack of comparative evidence between the oral and IV formulations’ effects on clinical outcomes.
The single-dose bioequivalence study MT-1186-J03 does not inform on the efficacy of a Health Canada–approved dosing regimen of oral edaravone in patients with ALS in terms of outcomes relevant to patients living with the disease. That study MT-1186-A01 was an open-label, uncontrolled trial subjects the study to a high risk of bias and limits the conclusions that can be drawn from the findings. The lack of comparative data for the outcomes of motor function, mobility, muscle pain, and fatigue, as well as difficulty breathing and speaking, which were identified by patients with ALS as the most important symptoms to control according to the patient input received, is an important gap in the evidence.
Cost Information
At the submitted price of $9,200 per 1,050 mg of edaravone per 50 mL of suspension or $12,880 per package of two 735 mg (35 mL) bottles, the annual drug cost per patient of treatment with oral edaravone is $123,280 in the first year and $119,600 per subsequent year, which is equivalent to the drug acquisition cost of IV edaravone at publicly available prices. CADTH conducted a reanalysis of the sponsor-submitted cost comparison, considering that costs associated with IV administration and IV-related AEs differ in the first and subsequent years of therapy. In this analysis, where some of the IV administration costs were assumed to be borne by the sponsor’s patient support program (PSP), oral edaravone was associated with an average cost saving to the public health care payer of $1,649 per patient compared to IV edaravone in the first year of therapy, and $1,105 per patient in subsequent years of therapy.
The cost comparison assumes clinical similarity between the oral and IV formulations of edaravone, based on the sponsor’s submitted single-dose bioequivalence study and an uncontrolled, open-label safety study. CADTH was unable to account for uncertainties in the comparative clinical effectiveness and safety between edaravone products or for the confidential pricing and stipulations that may have been negotiated for IV edaravone.
Conclusions
Findings from the sponsor’s analysis of bioequivalence suggested that oral edaravone showed comparable bioavailability to its IV formulation in a population of healthy volunteers; however, this requires formal assessment by Health Canada. Since IV edaravone was found to slow the rate of decline in motor function in patients with ALS, comparable bioavailability would suggest that the same conclusion may apply to oral edaravone. Findings from a single-group safety study suggest that the harms profile of oral edaravone may be considered acceptable, and no major safety signal was identified. However, the level of confidence in the evidence is highly affected by several limitations, including the open-label, uncontrolled trial design of the study, which introduced a high risk of bias. The lack of comparative data with oral edaravone for the outcomes of motor function, mobility, muscle pain, and fatigue, as well as difficulty breathing and speaking, which were identified by patients with ALS as the most important symptoms to control according to the patient input received, remains a gap in the evidence. Input received from all sources, including patients with ALS, clinicians, and the clinical expert consulted by CADTH for this review, emphasized that an oral version of edaravone would be a lot easier for patients to access than its IV formulation, reducing the risk of exposure to unnecessary infusion-associated AEs and decreasing the burden related to the IV administration, both to the health care system and to patients with ALS themselves.
At the submitted price, the annual drug cost of oral edaravone is $123,280 per patient in the first year and $119,600 per patient in subsequent years, which is the same as the annual drug cost of IV edaravone. When costs associated with IV administration and IV-related AEs are considered, and taking into account the sponsor’s PSP, which funds some IV administration costs, oral edaravone is $1,649 less expensive per patient than IV edaravone in the first year and $1,105 less expensive in subsequent years. The results are based on publicly available list prices for IV edaravone and may not reflect actual prices paid by Canadian public drug plans.
Introduction
Disease Background
ALS is a progressive neuromuscular disorder characterized by the degeneration of UMNs and LMNs.1 Symptoms of ALS are typically first noticed when limb weakness occurs, though the first symptoms can also be bulbar and involve difficulty in speaking or swallowing. Over time, patients lose function in additional regions, such as other limbs and respiratory muscles. Progressive muscle weakness occurs, and eventual respiratory failure leads to death. Patients present with symptoms in adulthood,2 and the median survival time from onset to death estimated from population-based studies ranges from 20 months to 36 months.3 ALS is a clinically heterogeneous disease in terms of presentation and rate of progression. For example, 5% to 10% of patients with ALS survive past 10 years from onset.3 Approximately 10% of ALS cases are familial ALS. The etiology of the disease is unknown, and at least 25 genes have been reproducibly shown to be associated with ALS.2
There is no definitive test for diagnosing ALS, and there can be a long duration from symptom onset to diagnosis. The diagnosis is based on clinical examination, electrophysiology tests, and exclusion of mimics. The lack of a useful biomarker of ALS contributes to delays in the diagnosis of ALS and difficulty in monitoring disease progression or activity in response to treatment.11 Expert consensus on diagnostic criteria was established in 1994 with the El Escorial criteria, and a subsequent version was established in 1999 as the El Escorial revised Airlie House criteria.12 The criteria have been mainly used for standardizing clinical trials as opposed to diagnosing patients in clinical practice.12 In the revised criteria, patients are categorized as having clinically “definite ALS,” “probable ALS,” “probable ALS — laboratory supported,” or “possible ALS.” The criteria are based on the presence of UMN or LMN signs in 4 regions of involvement — the brainstem, and the cervical, thoracic, and lumbosacral spinal cord — and are as follows:13
Definite ALS: Clinical UMN and LMN signs in 3 regions of involvement
Probable ALS: Clinical UMN and LMN signs in at least 2 regions, with some UMN signs rostral to the LMN signs
Probable ALS — laboratory supported: Clinical UMN and LMN signs in 1 region or UMN signs in 1 region accompanied by electrophysiological signs in at least 2 regions of the LMN
Possible ALS: Clinical UMN and LMN signs in 1 region, UMN signs in 2 or more regions alone, or LMN signs rostral to UMN signs (without proof of “probable ALS — laboratory supported”)
Disease Incidence
A systematic review published in 20094 summarized the results from 5 studies reporting incidence of ALS in 3 Canadian provinces, with 3 studies in Nova Scotia, 1 in Ontario, and 1 in Newfoundland and Labrador. Estimates of age-adjusted annual incidence per 100,000 persons ranged from 2.0 to 2.3 in 4 studies, with the fifth study estimating a crude annual incidence rate of 2.4 per 100,000 persons in Newfoundland and Labrador. Since the 2009 systematic review, 1 study of incident cases from 2010 to 2015 in British Columbia estimated a crude annual incidence rate of 3.29 per 100,000 persons14 and 1 study in the region of Saguenay-Lac-Saint-Jean in Quebec found an annual crude incidence of 3.01 per 100,000 persons during the period of 2005 to 2009.15
Standards of Therapy
There is no cure for ALS. Health Canada–approved treatments for ALS include riluzole and edaravone. Riluzole is an oral medication that has been shown to extend tracheostomy-free survival by 2 months to 3 months in patients with ALS.7 Riluzole is contraindicated for patients with hepatic disease or elevated liver enzymes, and AEs reported by patient respondents include cramps, diarrhea, heartburn, and feeling sick. Edaravone, a free radical scavenger thought to prevent oxidative damage to vascular endothelial cells and nerve cells, is currently available as an intravenously administered drug that has been found to slow the rate of decline in motor function.6 According to clinical expert opinion, it is typically administered concomitantly with riluzole but had a low uptake at the time of this review resulting from provincial drug coverage and patient choice due to the practicalities of the administration.
ALS symptoms may be managed (to varying degrees) by a range of pharmacologic therapies, including antidepressants, antianxiety and sleeping medications, medications to manage sialorrhea, and medications to address constipation.
Multidisciplinary nonpharmacologic care is important for managing symptoms and improving quality of life for patients with ALS. Multidisciplinary care optimally should involve a neurologist, a gastroenterologist, a respiratory physician, and a palliative care physician, as well as health care practitioners in the following areas: specialist nursing, physiotherapy, occupational therapy, nutrition, speech language pathology, and psychology.16-18 In Canadian clinics, the decision to introduce noninvasive ventilation is mostly based on patient symptoms (dyspnea, orthopnea, and morning headache), nocturnal oximetry, and FVC, and a survey published in 2010 found that 18.3% of patients with ALS living in Canada were using noninvasive ventilation.19 Patient intolerance and lack of access to a respirologist or ventilation technologist were identified as the most common barriers to utilization.19 As a second-line respiratory intervention, the use of invasive ventilation with a tracheostomy is associated with high cost and emotional and social impacts. The insertion of a gastrostomy feeding tube is recommended in Canadian,1 US,18 and European16 guidelines to supplement nutrition and stabilize weight loss. Decline in respiratory function, dysphagia, and weight loss factor in to the decision to place a feeding tube, though decision-making criteria vary between Canadian clinics.20
Key Characteristics of Edaravone IV and Riluzole.
Drug
This reimbursement review report evaluates orally administered edaravone, which is available as an oral suspension of 105 mg of edaravone per 5 mL of suspension. The Health Canada–approved indication is for the treatment of patients with ALS. According to the draft product monograph, the Health Canada–recommended dose of oral edaravone is 105 mg (5 mL) taken orally or via a feeding tube (percutaneous endoscopic gastrostomy or nasogastric tube). The recommended treatment regimen starts with an initial treatment cycle of daily dosing for 14 days followed by a 14-day drug-free period. Subsequent treatment cycles involve daily dosing for 10 days out of 14-day periods, followed by 14-day drug-free periods. Edaravone oral suspension should be taken in the morning after fasting overnight for at least 8 hours and waiting at least 1 hour before eating or drinking anything except water.7 For patients who are unable to fast overnight, the required fasting interval can be shortened depending on the type of meal. Patients treated with 60 mg of edaravone injection may be switched to 105 mg (5 mL) edaravone oral suspension using the same dosing frequency. Upon switching to edaravone oral suspension, patients should follow edaravone oral suspension dosing recommendations with regard to food consumption.7
Edaravone injection was recommended for reimbursement for the treatment of ALS by CDEC in March 2019,6 if CDEC-specified conditions were met:
Patient has a diagnosis of probable ALS or definite ALS.
Patient meets all of the following:
has scores of at least 2 points on each item of the ALS Functional Rating Scale — Revised (ALSFRS-R)
has an FVC greater than or equal to 80% of predicted FVC
has had ALS symptoms for 2 years or less
does not currently require permanent noninvasive or invasive ventilation.
Patient is under the care of a specialist with experience in the diagnosis and management of ALS.
Price is reduced.
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient advocacy group, the ALS Society of Canada, submitted the patient input for this review. The ALS Society of Canada is a registered charity working nationally to respond to the urgent unmet need for life-changing treatments through investments in research and engagement of various stakeholders to advocate for equitable, affordable, and timely access to proven therapies. The submission was based on an online survey disseminated in English and French in November 2021 and on telephone interviews with 7 patients who had experience with oral edaravone in February and March 2022. A total of 629 patients and caregivers responded to the online survey, primarily from Ontario and Quebec. Approximately 70% of respondents are, or were, caregivers to someone diagnosed with ALS, the remainder being patients living with the disease.
Respondents indicated that the most severe of ALS symptoms include difficulties with mobility (including walking and standing), gripping or holding things, muscle cramping or twitching, and fatigue caused by muscle exhaustion. These symptoms were also among the most important to control for people living with ALS, in addition to difficulties breathing, speaking, and swallowing. Patients indicated that their social life, travel and hobbies, and family life suffered the most. In addition, caregivers of patients with ALS highlighted a negative impact on emotional and psychological well-being, including pervasive feelings of overwhelming grief and struggles with mental health, including stress, anxiety, helplessness, and hopelessness. Loss of independence was mentioned as touching all aspects of patients’ lives and dramatically impacting caregivers, as patients eventually need help performing all daily tasks. Patients and caregivers reported treatment experience with riluzole and IV edaravone. Slowing disease progression, maintaining ability to participate in daily activities, and increasing survival were identified as the most important benefits from therapy. Access to riluzole and IV edaravone treatments was a problem for some patients. Other difficulties reported with edaravone were mostly related to the IV administration, including patients having to schedule activities of daily living around their infusion schedule and needing to have a port catheter implanted.
A copy of the patient input from the ALS Society of Canada is available with this report.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of ALS.
Unmet Needs
The clinical expert consulted by CADTH indicated that there is currently no treatment available to reverse the disease or to stop the progression of neurologic decline. Therefore, the mainstay of care for patients diagnosed with ALS consists of interventions and supports to manage symptoms. The 2 approved treatment options at the time of this review were riluzole and IV edaravone, both only showing modest benefits in slowing disease progression.
Place in Therapy
The clinical experts believed that an ideal treatment would delay or prevent disease progression (i.e., motor neuron degeneration), slow decline in lung capacity, reduce severity of symptoms, minimize AEs, reduce loss of cognition, improve health-related quality of life, and ultimately increase patients’ ability to continue to work, reduce burden on caregivers, and prolong life. Otherwise, the goals of treatment should include symptom management and quality-of-life optimization, which are priorities in patient care. An ideal treatment would also comprise a delivery mechanism that is safe, convenient, and pragmatic for patients and caregivers based on patient preference, stage of disease, and pathology.
At the time of this review, the only Health Canada–approved disease-modifying treatments for ALS were riluzole and IV edaravone, while AMX0035 is approved for use under the Special Access Program and is also under CADTH review. Riluzole, which acts by suppressing excessive motor neuron firing through inhibition of glutamate, prolongs survival by a median duration of 3 months. IV edaravone, which reduces oxidative stress, has been shown to slow the rate of clinical decline by 33% compared to IV placebo in a select group of patients with preserved respiratory function and disease duration of less than 2 years.
As the cause of ALS remains unknown, multiple pathogenic mechanisms are thought to be involved in the neuronal death process that starts even before the manifestation of clinical symptoms. Therefore, the clinical expert emphasized the importance of targeting all potential pathological pathways early. The expert indicated that patients would usually be prescribed riluzole for its clinical benefits and because it is easily administered and well tolerated. Patients meeting the criteria for reimbursement would then be offered IV edaravone. According to the clinical expert, the uptake of the medication has so far been low, in part because the IV formulation is invasive and comes with a time-consuming administration schedule. The coexistence of IV and oral edaravone formulations would provide options for drug administration based on patient preference, safety, and convenience, as well as progression and evolving pathology; however, the opinion of the clinical expert consulted by CADTH is that the more convenient and safer oral formulation of edaravone would be likely to replace the IV formulation.
Patient Population
The clinical expert indicated that patients must first be diagnosed with ALS by a specialist. Since no specific diagnostic biomarker exists, the diagnosis is made based on a patient’s history, physical examination, electrodiagnostic examination, and exclusion of alternative diagnoses. Suitability for treatment is therefore made clinically. The clinical expert anticipated that all patients with ALS could benefit from oral edaravone. However, the patients with the greatest need for a new treatment are those patients with a maintained physical motor function that could be preserved with the administration of edaravone. Patients whose disease is at a stage where the clinician believes there is no expected benefit from progression mitigation, who completely rely on the help of a caretaker, may be less suitable for treatment with oral edaravone. As with all treatment decisions, this would be made in consultation with the patient, informed by the clinical judgment of the treating physician.
Assessing Response to Treatment
The clinical expert mentioned that for patients with ALS, disease progression will continue despite best therapeutic efforts. Furthermore, the rate of disease progression varies between individuals, and as yet, there are no biomarkers to discern treatment response within a single patient. A specific treatment’s benefit in an individual is only apparent when compared against the average trajectory of natural disease history.
As such, the clinical expert indicated that current standard of care involves following the patient at regular intervals and monitoring their physical, functional, emotional, and quality-of-life parameters. Medications will be titrated appropriate to a patient’s condition and their goals of care, in a palliative-focused approach.
Discontinuing Treatment
According to the clinical expert, patients will have regular visits with their ALS care team to review their clinical status and goals of treatment. It is reasonable to expect that goals of treatment will change as the disease progresses. Therefore, medication could be continued until the philosophy of care shifts to a palliative focus and/or the clinician feels there is no expected benefit from progression mitigation.
Prescribing Conditions
The clinical expert highlighted the importance for patients to be appropriately diagnosed and followed by a multidisciplinary ALS clinic that delivers team-based care, including neurology, physiatry, respirology, and allied health professionals. The multidisciplinary care team will also address issues including communication, nutrition, swallowing, mobility, activities of daily living, respiratory care, cognition, psychosocial issues, medical management, and end-of-life care. Patients receiving edaravone should, at minimum, be followed by a neurologist or physiatrist experienced in the care of patients with ALS.
Based on the clinical expert’s experience, it would not be appropriate to recommend that patients try other treatments before initiating oral edaravone. There are no biomarkers to discern how well an individual patient would respond to a specific treatment. Though good clinical practice would allow time to confirm a patient’s tolerance on 1 medication, requiring the patient to demonstrate “failure” before introduction of another treatment would subject them to irreversible progression that would otherwise have been slowed had other therapies been given concurrently and would not be reflective of current evidence and practice.
Additional Considerations
The clinical expert consulted by CADTH indicated that the oral formulation of edaravone would provide clinicians and patients with a safe and pragmatic option for drug administration, given that it is expected to require less equipment (e.g., IV or central venous access) and personnel (e.g., medical staff to insert and maintain this equipment at the clinic or in the patient’s home) to administer. The clinical expert considered that the oral formulation would be a well-received alternative, as many patients choose not to embark on the currently available IV formulation because of the caveats and excessive requirements and constraints related to IV infusion.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
One clinician group provided input. CALS is a national network of clinicians working in academic health care centres across Canada and specializing in ALS research and clinical care. The network established in 2008 aims to connect Canadian ALS clinical research centres and to improve both patient and clinic participation in clinical research. All members of CALS were invited to participate in a virtual meeting in March 2022 to discuss key questions related to oral edaravone. Eleven CALS members from across Canada attended the meeting.
Overall, the clinician group input submission was consistent with the expertise provided by the clinical expert that was part of the CADTH review team.
A copy of the clinician group input is available with this report.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in .
Summary of Drug Plan Input and Clinical Expert Response.
Sponsor’s Summary of the Clinical Evidence
Note that the clinical evidence summarized in this section was prepared by the sponsor in accordance with the CADTH tailored review process and has not been modified by CADTH.
Pivotal Study MT-1186-J03
Details of Included Studies.
Description of Study MT-1186-J03
Study MT-1186-J03 was designed as a 2-group, 2-period crossover study with the objective of investigating the bioequivalence between edaravone oral suspension and edaravone IV formulation in healthy subjects by assessing each pharmacokinetic (PK) parameter with the bioequivalence limit. The study was planned in accordance with the “Guideline for Bioequivalence Studies of Generic Products and the Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs - General Considerations.” A crossover design was selected for this study in order for PK parameters to be precisely compared in a small number of subjects.
The study took place at a single site in Japan, and 42 healthy Japanese adults were included. A randomization key code table was created and provided it for the investigator, and subjects were randomly allocated to one of two groups: advance administration of edaravone oral suspension group (PO - IV) and the advance administration of edaravone IV formulation group (IV - PO).
Inclusion and Exclusion Criteria
Inclusion Criteria
Subjects who met all of the following criteria and were capable of giving informed consent were included in the study:
Healthy adult male or female volunteers
Japanese
Subjects aged between 20 and 45 years at the time of informed consent
Subjects who have thoroughly understood the contents of the study and voluntarily provided written informed consent to participate in the study
Age-restricted healthy adult volunteers were selected as the study population in order for the subject backgrounds to be uniform as much as possible and for this study to conform with the “Guideline for Bioequivalence Studies of Generic Products”d and the Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs - General Considerationse, which specify that the subjects should be healthy adult volunteers in principle. In addition, the subjects were limited to Japanese in order for PK to be evaluated in Japanese.
Exclusion Criteria
Subjects who met any of the following criteria between screening and investigational product administration were excluded from the study:
Subjects with a current or previous history of cardiac, hepatic, renal, gastrointestinal, respiratory, psychiatric/nervous, hematopoietic, or endocrine diseases, and those whom the investigator (or sub-investigator) deems unsuitable for the study
History of drug or food allergies
History of alcohol or drug abuse or dependence
BMI of < 18.0 or > 30.0, or a body weight of < 50 kg [BMI formula: body weight (kg)/height (m)2, rounded to one decimal place]
Positive test for any of the following at screening: hepatitis B surface antigen, serological test for syphilis, hepatitis C virus antibody, or human immunodeficiency virus antigen/antibody
Any clinically significant 12-lead ECG abnormality or corrected QT interval using Fridericia’s formula (QTcF) interval ≥ 450 msec
Blood donation or sampling with a total volume of ≥ 400 mL within 12 weeks, ≥ 200 mL within 4 weeks, or ≥ 800 mL within one year before providing informed consent
Blood component donation or blood sampling within 2 weeks before providing informed consent
Subjects who have undergone any surgery known to affect the gastrointestinal absorption of drugs (except for appendectomy and herniotomy)
Female subjects who do not agree to use an effective method of contraception from screening or 2 weeks before the start of investigational product administration, whichever comes earlier, to 14 days after the completion (or discontinuation) of investigational product administration. Male subjects who do not agree to use an effective method of contraception from the start of investigational product administration to 14 days after the completion (or discontinuation) of investigational product administration
Subjects who have previously received edaravone
Subjects who have participated in another clinical study and received an investigational product within 12 weeks before providing informed consent
Subjects who have used any drugs other than the single use of acetylsalicylic acid within 7 days before the initiation of investigational product administration
Use of alcohol or any products containing xanthin or caffeine within 24 hours before screening and visit on Day −1
Use of any nutritional supplement(s) within 7 days before the initiation of investigational product administration
Use of grapefruit, grapefruit juice, or any processed food(s) containing these substances within 24 hours before screening and visit on Day −1
Use of any tobacco or nicotine-containing product(s) within 24 hours before screening and visit on Day −1
Female subjects who have a positive pregnancy test at screening and on Day −1, are pregnant or breast feeding, or plan to get pregnant during the study
Subjects judged by the investigator
The rationales for setting were to: ensure the safety of subjects and to exclude unhealthy subjects (exclusion criteria #1); to perform the study safely and ethically (#2, 3, 5, 6, 19); to reduce PK variability due to BMI differences (#4); to ensure the safety of subjects, volumes and intervals of blood sampling were set with reference to the “Enforcement Regulations for the Act on Securing a Stable Supply of Safe Blood Products” (#7, 8); to avoid a possible effect on PK (#9, 13, 15, 16, 17); To assure subject safety, even though there were no toxicity findings at the highest dose of 200 mg/kg in the reproductive and developmental toxicity studies (#10, 18); to avoid possible effects on assessment results of this study (#11, 14); and To perform the study ethically and to avoid any unpredictable effects of drugs whose efficacy and safety have not been established (#12).
Baseline Characteristics
No meaningful differences were found between the advance administration of edaravone oral suspension group (PO - IV) and the advance administration of edaravone IV formulation group (IV - PO) groups. There were no subjects who had medical history and complications at the start of the study.
Summary of Baseline Characteristics.
Interventions
A commercially available product of edaravone (RADICUT BAG for IV infusion 30 mg; Mitsubishi Tanabe Pharma Corporation, Osaka, Japan) was used for the IV formulation. A 105-mg dose of edaravone oral suspension was selected for assessment so it would provide PK parameters (AUCs) corresponding to those of the approved 60-minute IV infusion of edaravone 60 mg, as predicted in previous studiesf.
In order for bioequivalence between edaravone oral suspension and edaravone IV formulation to be examined, these drugs were to be administered in the fasting state for at least 10 hours and the fasting was to be continued until 4 hours after the administration.
Advance administration of edaravone oral suspension group (PO - IV)
Period I:
After fasting for at least 10 hours, the subjects drank 100 mL of water 1 hour before investigational product administration. After receiving administration of the investigational product, edaravone oral suspension 105 mg (105 mg/5 mL), the subjects drank 100 mL of water. They fasted until the completion of blood sampling performed 4 hours after the administration. Drinking water other than the water provided at the time of administration was prohibited from 1 hour before to 1 hour after investigational product administration.
Period II:
After fasting for at least 10 hours, the subjects received continuous IV infusion of edaravone IV formulation 60 mg (30 mg/100 mL formulation, 2 bags) over 1 hour. The subjects fasted until the completion of blood sampling performed 4 hours after the administration. Drinking water was prohibited from 1 hour before the investigational product administration to 1 hour after the completion of the administration.
Advance administration of edaravone IV formulation group (IV - PO)
Period I:
After fasting for at least 10 hours, the subjects received continuous IV infusion of edaravone IV formulation 60 mg (30 mg/100 mL formulation, 2 bags) over 1 hour. The subjects fasted until the completion of blood sampling performed 4 hours after the administration. Drinking water was prohibited from 1 hour before the investigational product administration to 1 hour after the completion of the administration.
Period II:
After fasting for at least 10 hours, the subjects drank 100 mL of water 1 hour before investigational product administration. After receiving administration of the investigational product, edaravone oral suspension 105 mg (105 mg/5 mL), the subjects drank 100 mL of water. They fasted until the completion of blood sampling performed 4 hours after the administration. Drinking water other than the water provided at the time of administration was prohibited from 1 hour before to 1 hour after investigational product administration.
Criteria for subject withdrawal
Subjects were to be withdrawn from the study if any of the following scenarios occurred:
The subject requests to withdraw from the study.
The subject is determined to be clearly ineligible as a study subject.
Study continuation becomes difficult for the subject due to the onset of an adverse event (AE).
Other cases where the investigator (or sub investigator) judges that the subject should be withdrawn from the study
Procedures for subject withdrawal
If a subject discontinued participation in the study between the end of investigational product administration in period I and the completion of safety assessment, the investigator (or sub investigator) was to take appropriate actions for the subject, and promptly report to the monitor regarding the subject’s withdrawal from the study. Within 3 days from the last dose, the investigator (or sub investigator) was to perform the tests and observations that were specified for the withdrawal assessment.
The investigator (or sub investigator) was to record the date, the reason for discontinuation along with detailed information, the course of events that had led to the discontinuation, and treatment that had been provided in the case report form (CRF). If the onset of an AE was the cause of the discontinuation of the subject, the investigator (or sub investigator) recorded the AE in the discontinuation section in the CRF. The date of discontinuation was the date when evaluation had been performed (the date of evaluation) at the time of discontinuation. However, when evaluation was impossible, the date of discontinuation was to be the date when it had been judged that the subject was withdrawn from the study.
If the subject missed the observations and tests that were to be performed within 3 days from the last dose, or if he/she did not return to visits after discontinuation, the investigator (or sub investigator) was to make attempts to follow him/her up in order to identify the reason and subsequent course, by letter or phone, and record the results in the discontinuation section in the CRF.
Outcomes
Plasma and urine concentrations of unchanged edaravone and its metabolites were assessed with validated methodologies. PK parameters evaluated for unchanged edaravone after both IV and oral administration were AUC from time 0 to the last quantifiable concentration time point (AUC0-t), AUC0-∞, Cmax, time to reach Cmax (tmax), terminal elimination half-life (t1/2), bioavailability, total clearance (CL) or apparent CL after oral administration, urinary excretion ratio of drug from time 0 to 48 hours, and renal clearance. Volume of distribution at steady state and volume of distribution during the terminal phase were evaluated for IV edaravone. For sulfate and glucuronide conjugates, assessments included AUC0-t, AUC0-∞, Cmax, tmax, and t1/2. PK analysis was conducted for all subjects who received ≥1 dose of edaravone oral suspension or edaravone IV and who had evaluable PK data.
Safety assessments included AEs, serious AEs (SAEs), adverse drug reactions (ADRs), and serious ADRs. The safety analysis set consisted of all subjects who received ≥1 dose of edaravone oral suspension or edaravone IV.
Statistical Analysisb,g
Descriptive statistics (number of subjects, mean value, standard deviation, minimum value, median value, and maximum value) were used to calculate for the numerical data, and frequency and percentage will be calculated for each category for the categorical and ordinal data. Demographic characteristics and other baseline characteristics, frequency and percentage were calculated for the discrete values, and descriptive statistics were calculated for the numerical data. The calculation was made for each group.
For assessment with the bioequivalence limit, the analysis of variance was conducted for the log-transformed AUC0-t, AUC0-∞, and Cmax of unchanged edaravone, which included factors accounting for the following sources of variation: sequence, subjects nested in sequences, period, and treatment. Estimates of the mean difference between formulations (oral suspension minus IV formulation) on the log scale and 90%CI for the difference were back transformed to present mean ratios and their 90%CIs for oral suspension to IV formulation. The estimated 90%CIs of the geometric mean ratios were examined to lay entirely within the standard bioequivalence limits of 0.80 and 1.25. For reference, the same analysis was also performed on other PK parameters of unchanged edaravone, such as tmax, AUC from time 0 up to the last sampling time point for all time points, and elimination rate constant from the central compartment (Kel). Values of tmax were not log-transformed prior to statistical analysis.
For the safety assessments, AEs were summarized by treatment and into multiple categories. Laboratory tests, vital signs,12-lead ECGs and physical examination data were summarized and presented.
Primary Outcome(s) of the Studies
Power Calculation
The necessary total number of subjects was based on the AUC0-∞ and Cmax data for unchanged drug obtained in previous studies f,h. The calculation was performed so that for AUC0-∞, the 2-sided 90% confidence interval (CI) of the mean ratio of edaravone oral suspension to edaravone IV would fall within the bioequivalence criterion of 0.80 to 1.25; and for Cmax, the lower limit of the 2-sided 90%CI would exceed 0.80. The intraindividual standard deviations of log-transformed AUC0-∞and Cmax were assumed to be 0.232 and 0.706, respectively, from the previous study. Assuming that the expected ratios of AUC0-∞ and Cmax of edaravone oral suspension to edaravone IV were 1.06 and 1.40 from the 4-parameter logistic model, the necessary total numbers of subjects calculated on the basis of 2 one-sided tests with a significance level of 5% and power ≥90% were 36 and 24, respectively. Accordingly, sample size was set at 42 with 21 subjects per group to allow 36 subjects to complete the 2 periods.
Parameters required for PK evaluation were selected with reference to the Guideline for Bioequivalence Studies of Generic Productsd and the Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs – General Considerationse, and Clinical Pharmacokinetic Studies of Pharmaceuticals i.
Statistical Test or Model
It should be stated if the analysis was based upon the intention-to-treat (ITT) or per-protocol (PP) population. All formal statistical tests of treatment effects were done at a two-sided significance level of 0.05. Point estimates were accompanied with two-sided 95% CIs where applicable.
Drug Concentrations and Confirmatory PK Assessments
The PK analysis set consisted of all subjects who received at least 1 dose of the investigational product and had evaluable PK data. The PK parameters were calculated using WinNonlin® software (version 6.3). Summaries of concentrations and PK parameters, and statistical analysis for unchanged edaravone, sulfate conjugate, and glucuronide conjugate were performed using data from subjects whose PK data were available in both formulations. For each formulation, individual plasma concentrations vs. actual time for unchanged edaravone, sulfate conjugate, and glucuronide conjugate were plotted on both linear/linear and log/linear scales. Mean plasma concentrations vs. nominal time curves were plotted on both linear/linear (+SD) and log/linear scales overlaid by both formulations.
The parameters AUC0-t, AUC0-∞ and Cmax of unchanged edaravone were log transformed prior to statistical analysis. The analysis was performed by analysis of variance (ANOVA), which included factors accounting for the following sources of variation: sequence, subjects nested in sequences, period, and treatment. Estimates of mean difference between formulations (oral suspension minus IV formulation) on the log scale and 90% CI for the difference (based on the residual mean square in the ANOVA) were back transformed to present means and 90%CI for the ratio of oral suspension to IV formulation.
90%CI for the ratio of AUC0-t, AUC0-∞ and Cmax of unchanged edaravone which lied entirely within the limits of 0.8000 to 1.2500 provides bioequivalence between IV formulation and oral suspension
Data Imputation Methods
For non-PK related AEs, if severity or relationship was found to be missing, the most severe occurrence would be imputed for the summary of interest. For safety summaries, only observed data was used and unless otherwise specified, missing safety data would not be imputed. For PK summaries, only observed data was used. Missing plasma concentration data would not be imputed. When calculating Ae and Ae%, missing urine concentration data would be imputed to 0.
Secondary Outcomes of the Studies
Reference PK Parameters
As reference, the same analysis as described in the primary outcomes section was also conducted on AUC0-t, AUC0-∞ and Cmax of sulfate conjugate and glucuronide conjugate and the other reference PK parameters of unchanged edaravone, sulfate conjugate, and glucuronide conjugate, such as tmax, AUC0-all, MRT0-∞, and Kel. Tmax was not log transformed prior to statistical analysis.
Safety Assessments
All AEs were coded according to MedDRA (version 22.0). Overall summary for the following category was conducted by treatment.
Subjects with at least one AE
Subjects with at least one ADR
Subjects with at least one SAE
Subjects with at least one serious ADR
Subjects with at least one AE leading to discontinuation of investigational product
Subjects with AE leading to death.
The following summaries were also conducted by treatment. These tables were ordered by International Agreed Order for System Organ Class (SOC) and then by alphabetical order for Preferred Term (PT).
Each of the summaries was done at the subject level - multiple occurrences of the same event within a subject was counted once in the summaries by SOC and PT; multiple occurrences of the same event within a subject was counted once in the maximum severity category (severe > moderate > mild) and/or maximum drug relationship category (reasonable possibility/no reasonable possibility) and/or the earliest duration. All AEs were listed.
For laboratory tests, absolute values and changes from baseline, except for urinalysis were summarized descriptively by group and scheduled visit. For urinalysis, number and percentage were presented. Shift tables presented the changes in clinically relevant categories from baseline to each scheduled post-baseline visit by group. All data including clinically relevant flagged were listed. Any data below the limit of quantification were treated as 0 in summary statistics.
Absolute values and changes from baseline for vital signs were summarized for the parameters (systolic blood pressure, diastolic blood pressure, pulse rate, body temperature) by treatment and scheduled time-point. All data were listed.
For 12-lead ECGs, Absolute values and changes from baseline were summarized for the parameters (heart rate, PR, RR, QRS, QT, QTcF) by treatment and scheduled time-point. The percentage of subjects with 12-lead ECG values outside pre-defined limit was summarized by treatment and scheduled time-point. All data (including overall evaluation) were listed.
For physical examinations, all data were listed.
Analysis Populations
All safety assessments were conducted on the safety analysis set (SAF) population. The consisted of all subjects who received at least 1 dose of the investigational product.
Sponsor’s Summary of the Results of Study MT-1186-J03
Patient Disposition
The study included 42 subjects (n = 21 in each group). The baseline demographic characteristics of the study population are summarized in . |||||||| subjects gave informed consent and were screened, however |||||| failed screening due to withdrawal of consent, were excluded based on the exclusion criteria, or withdrew for other reasons. While the eligible population included |||||| subjects, ||||| were kept as reserve subjects, with 42 subjects randomized to each of the advance administration of edaravone oral suspension group (PO - IV) and the advance administration of edaravone IV formulation group (IV - PO). summarizes the disposition of subjects in the study.
Exposure to Study Treatments
Study Treatments
The PK analysis set consisted of all subjects who received at least 1 dose of the investigational product and had evaluable PK data. The SAF also consisted of all subjects who received at least 1 dose of the investigational product. Investigational products were administered to a total of 42 subjects (21 subjects for each group) by both of a single IV infusion over 60 minutes and a single oral administration in fasted conditions. No subjects discontinued this study after the start of administration of the investigational product.
Concomitant Medications
One subject in the IV - PO group took magnesium oxide as medication due to AE (constipation) at 5 hours and 29 minutes after oral administration. However, taking into account the time of the administration of the concomitant drug, magnesium oxide was judged not to affect the PK of edaravone. No other concomitant medication use was noted in either treatment group.
Efficacy
Summary
In 42 healthy Japanese subjects, this study demonstrated that the 105 mg oral suspension of edaravone has equivalent AUC0-t and AUC0-∞ of unchanged edaravone to the approved 60 mg IV formulation [Geometric mean ratio (90% CI): 0.974 (0.914-1.038) for AUC0-t and 0.977 (0.917-1.041) for AUC0-∞]. Geometric mean ratio of Cmax of the oral suspension compared to the IV formulation and its lower limit of 90% CI were also within the range of 0.80 to 1.25, while the upper limit of 90% CI exceeded 1.25 as anticipated [Geometric mean ratio (90% CI): 1.217 (1.090-1.359)].
Overview of PK evaluation
Plasma Concentrations of Unchanged Edaravone and PK Parameters
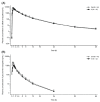
The results for the plasma concentration of unchanged edaravone for the oral suspension and IV formulation are summarized in and . Both oral and IV administration of edaravone showed a 3-phase elimination after reaching Cmax, and the plasma concentration-time profiles of unchanged edaravone were very similar regardless of the administration route. The median tmax values of unchanged edaravone in 105 mg oral suspension and 60 mg/60 min IV formulation were 0.50 and 1.00 hour, respectively. The mean AUC0-t, AUC0-∞ and t1/2 of unchanged edaravone were similar between two formulations. The mean absolute bioavailability (F) of the oral suspension was 57.3%.
Summary of Efficacy Outcomes.
Mean Plasma Concentration–Time Profiles of Unchanged Edaravone for the 105-mg Oral Suspension and the 60-mg IV Formulation (Log-Linear Plot).
Plasma Concentrations and PK Parameters of Sulfate and Glucuronide Conjugates
presents the plasma PK parameters and concentration-time profiles for the edaravone sulfate and glucuronide conjugates following administration of the 105 mg edaravone oral suspension and 60 mg edaravone IV. The mean plasma concentrations of both sulfate and glucuronide conjugates with the 105 mg oral suspension were higher than those with the 60 mg IV formulation, but the shape of the profiles and elimination patterns were similar between the two formulations after reaching Cmax.
Mean Plasma Concentration-Time Profiles of (A) Sulfate Conjugate and (B) Glucuronide Conjugate for Both Oral Suspension (105mg) and IV Formulation (60mg) of Edaravone.
Urine PK Parameters
Edaravone was eliminated into urine mainly as glucuronide conjugate and to a lesser extent as sulfate conjugate after administration of both the oral suspension and the IV formulation. The mean Ae% of sulfate conjugate for 48 hours after administration were 6.58% and 8.09% in 105 mg oral suspension and 60 mg/60 min IV formulation, respectively. The mean Ae% of glucuronide conjugate for 48 hours after administration were 59.8% and 78.4% in 105 mg oral suspension and 60 mg/60 min IV formulation, respectively. The mean Ae% of sum of unchanged edaravone, sulfate and glucuronide conjugates for 48 hours after administration were 67.0% and 87.3% in 105 mg oral suspension and 60 mg/60 min IV formulation, respectively. The urinary excretion of unchanged edaravone was low, and the composition ratios of unchanged edaravone and the metabolites in urine were similar for both administration routes. Urine PK parameters are presented in .
Urine PK Parameters of Unchanged Edaravone and of Sulfate and Glucuronide Conjugates.
Harms
Safety Evaluation Plan
The safety analysis set (SAF) consisted of all subjects who received at least 1 dose of the investigational products. Investigational products were administered to a total of 42 subjects (21 subjects for each group). No subjects discontinued this study after the start of administration of the investigational product.
Overview of Safety
A total of 2 AEs were reported in 2 subjects, one AE for each formulation (IV formulation: aspartate aminotransferase increased, oral suspension: constipation). All AEs were mild in severity and recovered. None of AEs were judged to be reasonably related to the investigational product by the investigator. No ADRs, SAEs, serious ADRs and AEs leading to discontinuation and/or to death occurred. A summary of AEs is presented in .
Hematology, biochemistry, coagulation, and urinalysis over time for patients were collected. No clear trends over time were observed for most safety laboratory parameters in any group during this study.
Absolute values and changes from baseline in vital sign parameters for patients were collected. Overall, all vital signs were stable, and there were no notable trends in any formulation during this study.
Absolute values and changes from baseline and overall evaluations in 12-lead ECG parameters were recorded for patients. No notable trends were observed in 12-lead ECG parameters in any formulation during this study.
Normal and abnormal status of physical examination by patient was recorded. No notable trends were observed in physical examination in any formulation during this study.
Overall, the two formulations for edaravone of 105 mg oral suspension and 60 mg IV formulation were well tolerated and there were no safety concerns in the results of this study.
Summary of Adverse Events.
Adverse Events
A total of 2 AEs were reported in 2 subjects, one AE for each formulation. Both AEs were mild in severity and recovered during the study. There were no AEs that were judged to be reasonably related to the investigational products by the investigator. One subject in the PO - IV group experienced aspartate aminotransferase increased on day 11, i.e., during the follow-up period after IV administration and her aspartate aminotransferase returned to normal range at the unscheduled visit on day 24 without treatment. Another subject in the IV - PO group experienced constipation after oral administration of the investigational product on day 4. The subject took the concomitant medication (magnesium oxide) for the AE on days 4 and 5 and recovered on day 6.
Serious Adverse Events
There were no serious adverse events (SAEs) reported in this study.
Withdrawals Due to Adverse Events
There were no withdrawals due to AEs in this study.
Adverse Events of Special Interest
There were no AEs of special interest in this study.
Bioequivalence
Assessment of Bioequivalence
The results of the statistical analysis of the AUC0-t and AUC0-∞ of the IV formulation of edaravone 60 mg compared with the oral suspension of edaravone 105 mg were determined to be equivalent (geometric mean ratio [90%CI], 0.97 [0.91-1.04] and 0.98 [0.92-1.04], respectively). The geometric mean ratio of Cmax of the 105-mg oral suspension compared to the 60-mg IV formulation was within prespecified limits, but the upper limit of 90%CI exceeded 1.25 (geometric mean ratio [90% CI], 1.22 [1.09-1.36]). The least squares mean difference and 90%CI for tmax (the 105-mg oral suspension minus the 60-mg IV formulation) was −0.56 (90%CI, −0.60 to −0.51). For the reference parameters, the geometric LS mean ratios and 90% CIs of unchanged edaravone between the 2 formulations were 0.976 (90% CI: 0.917-1.039) for AUC0-all, 1.064 (90% CI: 0.970-1.167) for MRT0-∞, 0.935 (90% CI: 0.758-1.153) for Kel, and LS mean ratio and 90% CI for tmax were −0.559 (90% CI: −0.604- −0.513).
The statistical analysis with bioequivalence limits for the confirmatory plasma PK parameters (Cmax, AUC0-t, and AUC0-∞ of unchanged edaravone, and the geometric mean ratios and 90%CIs of unchanged edaravone between the 2 formulations) and the bioequivalence of the reference plasma PK parameters are summarized in .
Statistical Analysis of Bioequivalence of Confirmatory and Reference Plasma PK Parameters of Unchanged Edaravone.
Pivotal Study MT-1186-A01
Details of Included Studies.
Description of Study MT-1186-A01
Study MT-1186-A01 was designed as a phase 3, multi-center, open-label, safety study of oral edaravone administered over 48 weeks in subjects with ALS. The primary objective of the study was to evaluate the safety and tolerability of oral edaravone in subjects with ALS over 24 and 48 weeks, and an exploratory objective to evaluate the efficacy of oral edaravone in subjects with ALS over 24 and 48 weeks was also established. The clinical study report included in the CADTH submission currently describes the analysis at 24 weeks, with the 48-week data expected in the second quarter of 2022. The duration of the study for individual subjects will be approximately 51 weeks, consisting of a screening period of up to 3 weeks, an open-label treatment period of up to 48 weeks, and a safety follow-up period of 2 weeks after the last dose.
The dose selected for this study delivers a similar exposure as the IV edaravone infusion delivered at a dose of 60 mg over 60 minutes. To establish the long-term safety of oral edaravone in subjects with ALS, an identical dosing schedule to the approved IV formulation was utilized in this study.
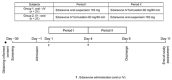
A total of 185 subjects were enrolled. 23 subjects from four Canadian sites (two in Alberta and two in Quebec) were included in this study. Subjects meeting the eligibility criteria were enrolled into the 48-week open-label treatment period and received 105 mg of oral edaravone, following an overnight fast, and subjects continued to fast for at least 1 to 2 hours post-dose before the next meal (e.g., breakfast). An initial treatment cycle with daily dosing for 14 days was followed by a 14-day drug-free period. Subsequent treatment cycles included daily dosing for 10 days out of a 14-day period, followed by a 14-day drug-free period. Treatment cycles were 4 weeks in duration. The study design is outlined in below.
Inclusion and Exclusion Criteria
Inclusion Criteria
Subjects who provided a signed and dated ICF to participate in the study. Subjects were able to (in the judgment of the Investigator) understand the nature of the study and all risks involved with participation in the study. Subjects were willing to cooperate and comply with all protocol restrictions and requirements.
Subjects were male or female, ≥18 to 75 years of age at the time the ICF was signed.
Subjects who were diagnosed with definite ALS, probable ALS, probable laboratory-supported ALS, or possible ALS according to the El Escorial revised criteria for the diagnosis of ALS.
Subjects who were living and functioning independently (e.g., able to eat, excrete, ambulate independently without assistance of others). The use of supportive tools and adaptive utensils was allowed.
Subjects who had a baseline %FVC ≥70%.
Subjects whose first symptom of ALS occurred within 3 years at the time of providing written informed consent.
Exclusion Criteria
Exclusions Related to General Health or Concomitant Conditions:
Subjects who underwent treatment for a malignancy or those with a pending biopsy result.
Subjects who had the presence or history of any clinically significant disease (except ALS) that could interfere with the objectives of the study (the assessment of safety and efficacy) or the safety of the subject, as judged by the Investigator.
Subjects of childbearing potential unwilling to use an acceptable method of contraception from the screening visit until 3 months after the last dose of study medication. Subjects who were sexually active and who would not agree to use contraception during the study period.
Subjects who were female and pregnant (a positive pregnancy test) or lactating at the Screening Visit (Visit 1).
Subjects who had a significant risk of suicide. Subjects with any suicidal behavior or suicidal ideation of type 4 (active suicidal ideation with some intent to act, without a specific plan) or type 5 (active suicidal ideation with specific plan and intent) based on the C-SSRS within the 3 months before the Screening Visit.
Subjects who had alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevations greater than 2 times the upper limit of normal (ULN) at screening.
Subjects with a glomerular filtration rate (GFR) <30 mL/min per 1.73 m2 at screening.
Exclusions Related to Medications:
Subjects who had a history of hypersensitivity to edaravone, any of the additives or inactive ingredients of edaravone, or sulfites.
Subjects who had hereditary fructose intolerance.
Subjects who participated in another study and were administered an investigational product within 1 month or 5 half-lives of the investigational agent, whichever was longer before providing informed consent for the present study.
Subjects who were unable to take their medications orally.
Baseline Characteristics
This study was conducted in 50 sites, located in Japan, the US, Canada, Germany, France, and Italy.
The enrolled population was mostly male (64%) and ranged in age from 22 to 75 years, with a median age of 61 years. Approximately 65% of the subjects were <65 years old, with 32% of subjects between 50 and 59 years old and 34% of subjects between 60 and 69 years old. Subjects who participated were located mostly in North America (51%) or Japan (35%). Demographic and baseline characteristics for the Enrolled Population are presented in .
Summary of Baseline Characteristics.
Interventions
Subjects received an oral dose of edaravone 105 mg suspension in the following regimen:
An initial treatment cycle with daily dosing for 14 days, followed by a 14-day drug-free period.
Subsequent treatment cycles with daily dosing for 10 days out of 14-day periods, followed by 14-day drug-free periods.
Treatment cycles were every 4 weeks. The dose of edaravone was taken after an overnight fast and subjects continued to fast for at least 1 to 2 hours post-dose before the next meal (e.g., breakfast).
This was an uncontrolled safety study. With the previous establishment of efficacy for IV edaravone in the treatment of ALS, placebo treatment was not considered appropriate. Concomitant use of riluzole was permitted throughout the study.
Withdrawal of Subjects
A subject was withdrawn from the study if the subject met any of the following criteria:
Subject requested to be withdrawn from the study;
Had been found ineligible for participation in the study;
The investigator (or subinvestigator) judged that continuation of the study would be difficult for subject due to AEs (e.g., hypersensitivity reactions);
Was pregnant;
Required tracheotomy;
Required permanent assisted mechanical ventilation (≥23 hours/day);
The investigator (or subinvestigator) judged that continuation of the study would be inappropriate for subject due to exacerbation of the primary disease;
ALT or AST of greater than 5 times the ULN;
Noncompliance with study medication (after consultation with sponsor or designee).
If a subject was withdrawn prematurely from the study, the date and the reason for withdrawal was recorded in the electronic Case Report Form (eCRF).
In the event that a subject dropped out of the study at any time, the reason for discontinuation was fully documented in the source documents and the eCRF. The Investigator site personnel documented the AEs and any other assessments in the source documents and made every effort to complete all required early termination (ET) assessments. Study sites followed up with subjects via phone calls at Weeks 24, 36, and 48 for event assessments. Subjects who withdrew from the study following enrollment were not allowed to re-enter the study.
Outcomes
The safety and tolerability of edaravone was evaluated and included the following safety assessments:
Adverse events (AEs), adverse drug reactions (ADRs), and treatment-emergent adverse events (TEAEs; e.g., grade, incidence, severity);
Physical examination;
Body weight;
12-lead electrocardiogram (ECG) parameters;
Vital signs (heart rate, sitting systolic and diastolic blood pressure, and axillary, oral, or tympanic body temperature);
Laboratory safety assessments (e.g., hematology, chemistry, and urinalysis);
Unsteadiness and sensory evaluation (e.g., assessment of unsteadiness and peripheral sensation will be evaluated by assessment of vibratory sensation with a tuning fork applied to the lateral side of the right and left ankles);
Columbia-Suicide Severity Rating Scale (C-SSRS);
Percentage of predicted forced vital capacity (%FVC).
The functional and survival assessments of oral edaravone efficacy was also evaluated using the following:
Change in ALSFRS-R from baseline to each visit;
Time (days) to death, tracheostomy, or permanent assisted mechanical ventilation.
Statistical Analysis
The long-term safety and tolerability of oral edaravone was evaluated using descriptive statistics. For the exploratory efficacy analysis, point estimates and their associated 95% confidence intervals (CI) were presented. All data from all subjects enrolled into the study were included in subject data listings.
Continuous data were summarized descriptively using the number in the analysis population set (N), the number of observations (n), mean, standard deviation (SD), median, minimum, and maximum. Categorical data were summarized using frequency counts and percentages. The denominator for the percentages was the total number of subjects in the treatment group and analysis population being presented, unless otherwise specified. For visit-specific data, the number of subjects with non-missing observations at the visit in question were used as the denominator for percent calculations. Unknown, Not Done, Not Applicable and other classifications of missing data were not considered. Unscheduled or repeated assessments were not included in summary tables but were included in listings.
Primary Outcome(s) of the Studies
Power Calculation
During the initial protocol development stage, a 30% dropout rate (based on the results of a previous Phase III study for the IV formulation of edaravone, Study MCI-186-19) was assumed over the course of the study, and thus approximately 150 subjects were planned to be enrolled to receive treatment with oral edaravone (105 mg) to obtain 1-year long-term safety data from approximately 100 subjects, However, while the study was ongoing and based upon the potential for a higher than expected premature termination rate due to the coronavirus disease (COVID-19) pandemic, the sample size was revised to enroll approximately 185 subjects to receive treatment with oral edaravone (105 mg) to obtain 1-year long-term safety data from approximately 100 subjects, meeting the requirement of ICH E1 guideline for long-term safety.
Statistical Test or Model
The long-term safety and tolerability of oral edaravone was evaluated using descriptive statistics.
Adverse Events: Adverse events were coded according to the MedDRA version 23.0. The frequency and incidence of TEAEs were summarized by SOC and PT. The SOC was sorted by International order; then within SOC, PT was sorted by PT code.
The following summaries were provided:
A Summary table of the overall incidence (number and percentage) and the number of events were provided for TEAE, TEAE related to study drug, severe TEAEs, treatment-emergent serious adverse event (TESAE), TEAEs leading to study treatment discontinuation, and TEAEs leading to death.
A Summary table of the overall incidence (number and percentage) will be provided for Peripheral Neuropathy Standardized MedDRA query (SMQ) TEAEs
The numbers and proportions of subjects were calculated for the following:
TEAEs by SOC and PT
TEAEs by SOC, PT and severity
Most Common (≥5% of subjects) TEAEs by SOC and PT
TEAEs related to study drug by SOC and PT
TEAEs related to study drug by SOC, PT, and severity
TESAEs by SOC and PT
TESAEs related to study drug by SOC and PT
Severe TEAEs by SOC and PT
Severe TEAEs related to study drugs by SOC and PT
TEAEs leading to study treatment discontinuation by SOC and PT
TEAEs by SOC, PT, and relationship to study drug
TESAEs by SOC, PT, and relationship to study drug
TEAEs leading to death by SOC and PT
TEAEs of Peripheral Neuropathy SMQ by SOC and PT
Serious TEAEs of Peripheral Neuropathy SMQ by SOC and PT
Physical Examinations: Physical examination findings including reason not done were listed for the safety analysis population.
12-Lead Electrocardiogram: All ECG parameters were listed and analyzed for the safety analysis population. The ECGs were assessed by the investigator and deemed ‘Normal’, ‘Abnormal, not clinically significant’ (Abnormal, NCS), and ‘Abnormal, clinically significant’ (Abnormal, CS) and tabulated by visit up to Week 24 using frequency counts and percentages. In addition, the numerical ECG parameters and their change from baseline generated by the central ECG laboratory were summarized by descriptive statistics for each parameter by visit. A shift table describing the number and percentage of subjects shifting from non- potentially clinically significant values (PCSV) at baseline to PCSV at any time post- baseline values during treatment period was produced. The percentages were calculated from the number of subjects with available baseline values and any time post-baseline value.
Vital Signs Including Body Weight: Vital sign measurements (heart rate, supine and standing blood pressure [both systolic and diastolic], body temperature, and weight) and their change from baseline were listed and summarized for the safety analysis population using descriptive statistics by visit up to Week 24. Furthermore, supine minus standing blood pressure (both systolic and diastolic) and their change from baseline were summarized with descriptive statistics by visit. The body weight values and change from baseline to each post-baseline visit up to Week 24 were plotted by visit. A shift table describing the number and percentage of subjects shifting from non-PCSV at baseline to PCSV at any time post-baseline during treatment period was produced. The percentages were calculated from the number of subjects with a baseline value and any time post-baseline value. The number and percentage of subjects with orthostatic hypotension were tabulated by visit.
Laboratory Tests: All laboratory data were listed and analyzed for the safety analysis population. Laboratory data and change from baseline (hematology, biochemistry, or urinalysis) were summarized with descriptive statistics (continuous variables) or as distributions (categorical variables) by visit up to Week 24 except for pregnancy test parameter. A shift table from baseline to each visit up to Week 24 was produced for selected variables as described below. The shift categories for out of reference range were “Low, Normal, and High” for hematology, biochemistry, and urinalysis, and “Normal and Abnormal” for urinalysis (qualitative value). A shift table describing the number and percentage of subjects shifting from non-PCSV at baseline to PCSV post-baseline at any time during treatment period was produced. The percentages were calculated from the number of subjects with available baseline values and any time post-baseline value.
Unsteadiness and Sensory Evaluation: The unsteadiness and sensory evaluations were listed and analyzed for the safety analysis population. For numbness and unsteadiness, the number and percentages of subjects with ‘present’ or ‘absent’ were summarized by each visit up to Week 24. In addition, severity was summarized for each visit with the number and percentage of subjects in each category: ‘Normal/ Mild/ Moderate /Severe.’ For this summary, the subjects with ‘Absent’ were classified and counted as ‘Normal.’ A shift table from each baseline category up to Week 24 was summarized using number and percentages. Vibratory sensation values and change from baseline to each analysis visit window were summarized descriptively for right and left side of the ankle.
Columbia Suicide Severity Rating Scale (C-SSRS): The C-SSRS was analyzed and listed for the safety analysis population. The frequency and percentage of subjects with each response for suicidal ideation, intensity of ideation, and suicidal behavior items were summarized for subjects’ lifetime history (Screening to past 3 months, screening to lifetime) and during the treatment period (Weeks 12 to since last visit, Week 24 to since last visit). The distribution of responses for most severe suicidal ideation and suicidal behavior were also presented for lifetime history and the treatment period.
Percent Predicted Forced Vital Capacity (%FVC): The %FVC values and change from baseline to each post-baseline visit up to Week 24 was listed, plotted, and analyzed. The changes from baseline and their associated 95% CI were estimated separately for each visit, from the same MMRM analysis using LSMEANS estimates. In addition, frequency counts and percent for categorical %FVC (70% ≤%FVC, 50% <%FVC<70%, and %FVC ≤50%) were displayed at each visit.
Data Imputation Methods
For safety summaries, only observed data was used. Missing safety data was not imputed.
Subgroup Analyses
Subgroup analysis was performed for the following parameters:
TEAEs by SOC and PT stratified by region
TEAEs by SOC and PT stratified by previous exposure to edaravone
Unsteadiness and sensory evaluation stratified by region
%FVC stratified by region
%FVC stratified by previous exposure to edaravone
Laboratory test stratified by region
Laboratory test stratified by previous exposure to edaravone
Vital signs stratified by region
12-Lead ECGs stratified by region
Secondary Outcomes of the Studies
For exploratory efficacy analysis, continuous data were summarized at each analysis visit using summary statistics. Absolute values and changes from baseline were presented. All categorical endpoints were summarized at each analysis visit, using frequency tabulations.
As the primary purpose of this study was to explore the safety of edaravone and not to perform confirmatory analyses, there was no formal hypothesis testing performed and adjustments for multiplicity were not required.
ALSFRS-R Change from Baseline
The ALSFRS-R score of each item, domain score, and total score were listed. The ALSFRS-R total score and change from baseline to each post-baseline visit up to Week 24 were plotted by visit.
The changes from baseline to all post-baseline visits until Week 24 in the ALSFRS-R score were estimated using a mixed model for repeated measures (MMRM) analysis. The model included response data from all post-baseline visits with no imputation for missing data. The ALSFRS-R score at baseline, previous exposure to edaravone, concomitant riluzole, and visit at Weeks 4, 12, and 24 were included as fixed factors in the model. An unstructured covariance structure was assumed, and the denominator degrees of freedom was computed using the Kenward-Roger method.
In case the model did not converge with the unstructured covariance structure, the heterogeneous compound symmetry (CSH) and the heterogeneous Toeplitz structure (TOEPH) were to be used instead (in that order). The changes from baseline and their associated 95% Cis were estimated, separately for each visit, from the same MMRM analysis using LSMEANS estimates.
Time to Death, Tracheostomy, or Permanent Assisted Mechanical Ventilation
The time to first occurrence of death, tracheostomy, or permanent assisted mechanical ventilation were derived as follows:
In case the event mentioned above was observed any time up to the last observed visit date until Week 24, then the time variable for each subject was calculated as: The date of the event - First date of study drug + 1
In case the event mentioned above was not observed any time up to the last observed visit date until Week 24, a right censoring was performed for each subject at the last observed date of treatment. The time variable for each subject was calculated as: Last observed Date - First date of study drug + 1
Indicator (censoring) variable was created to indicate an event (0) if the event was observed or censoring (1) if the event was not observed and the subject was either discontinued or was ongoing at the Week 24 database lock.
The following parameters were listed, plotted using Kaplan-Meier (KM) methods, and summarized by KM methods with 95% CI, the number of events and percentage.
The time to first onset of death, tracheostomy, or permanent assisted mechanical ventilation.
Data Imputation Methods
Data for the efficacy analysis was not imputed.
Subgroup Analysis
Subgroup analysis was performed for the following parameters:
Change from baseline to Week 24 in ALSFRS-R stratified by region
Change from baseline to Week 24 in ALSFRS-R stratified by previous exposure to edaravone
Time to death, tracheostomy, or permanent assisted mechanical ventilation stratified by region
Time to death, tracheostomy, or permanent assisted mechanical ventilation stratified by previous exposure to edaravone.
Analysis Populations
The Enrolled Population included 185 subjects (85.6% of all screened subjects) and the Safety Analysis Population included 185 subjects (100.0% of the Enrolled Population). The PK Analysis Population included 39 subjects (21.1% of the Enrolled Population).
Sponsor’s Summary of the Results of Study MT-1186-A01
Subject Disposition
A total of 216 subjects were screened, of which 185 (85.6%) subjects were enrolled and 31 (14.4%) subjects were screen failures. Reasons for screen failure included study entry criteria not met (25 [11.6%] subjects), COVID-19 (4 [1.9%] subjects), and withdrawal by the subject (2 [0.9%] subjects).
A total of 160 (86.5%) subjects in the Enrolled Population completed the 24-week study period. Of the 24 (13.0%) subjects who discontinued, the most frequent reasons for discontinuation were withdrawal by the subject (8 [4.3%] subjects), adverse event (7 [3.8%] subjects), and death (6 [3.2%] subjects). One additional subject completed the 24-week study period but did not return to the site for Week 24 procedures. The subject disposition is outlined in .
Exposure to Study Treatments
Study Treatments
The Safety Analysis Population consisted of 185 subjects who received at least 1 dose of study drug. Overall mean treatment compliance was 99.55% (SD 5.34%), ranging from 28.6% to 103.1%. One subject (0.5%) had compliance <80%, and no subjects had compliance >120%. The mean exposure to edaravone was 61.3 days and the total exposure was 31.1 person years.
Concomitant Medications
3 subjects took 4 medications during the 24-week study period that were not reported until after database lock for the 24-week analysis. One subject took Movicol for an AE of constipation, 1 subject took ketoconazole and terbinafine hydrochloride for an AE of eczema, and 1 subject took esomeprazole magnesium hydrate for gastritis prophylaxis. The majority of subjects reported concomitant use of riluzole (161 [87.0%] subjects).
Harms
Study MT-1186-A01 was designed to evaluate the safety and tolerability of oral edaravone in subjects with ALS over 24 and 48 weeks, and an exploratory objective to evaluate the efficacy of oral edaravone in subjects with ALS over 24 and 48 weeks was also established. The clinical study report currently describes the analysis at 24 weeks, with the 48-week data expected in the second quarter of 2022. The duration of the study for individual subjects will be approximately 51 weeks, consisting of a screening period of up to 3 weeks, an open-label treatment period of up to 48 weeks, and a safety follow-up period of 2 weeks after the last dose.
Overview of Safety
At 24 weeks, TEAEs were experienced by 146 (78.9%) subjects. Of these, 60 TEAEs related to study treatment (none were severe) were reported by 36 (19.5%) subjects, 30 severe TEAEs were reported by 17 (9.2%) subjects, 17 TEAEs leading to study treatment discontinuation were reported by 11 (5.9%) subjects, and 6 TEAEs leading to death were reported in 6 (3.2%) subjects. A total of 24 TESAEs were reported by 21 (11.4%) subjects. After the 24-week database lock, 7 subjects reported TEAEs that occurred during the 24-week study period. None of these TEAEs were severe, serious, related to study drug, or led to discontinuation or death. An overall summary of TEAEs is provided in .
The most commonly reported TEAEs by system organ class (SOC) were musculoskeletal and connective tissue disorders (66 [35.7%] subjects); injury, poisoning, and procedural complications (49 [26.5%] subjects); and gastrointestinal disorders (48 [25.9%] subjects). The most commonly reported TEAEs by preferred term (PT) were muscular weakness (30 [16.2%] subjects), fall (29 [15.7%] subjects), and fatigue (14 [7.6%] subjects). A summary of common TEAEs is provided in .
Overall Summary of Treatment-Emergent Adverse Events.
Common (Reported by ≥ 5% Subjects) Treatment-Emergent Adverse Events.
Adverse Events
A total of 36 (19.5%) subjects reported a TEAE related to study drug. The most commonly reported TEAEs related to study drug by SOC were nervous system disorders (11 [5.9%] subjects), gastrointestinal disorders (9 [4.9%] subjects), musculoskeletal and connective tissue disorders (6 [3.2%] subjects), and general disorders and administration site conditions (6 [3.2%] subjects). The most commonly reported TEAEs related to study drug by PT were fatigue (6 [3.2%] subjects), dizziness (5 [2.7%] subjects), and headache (4 [2.2%] subjects).
Deaths: Six (3.2%) subjects died during the study period: 3 (1.6%) subjects died from respiratory failure and 1 (0.5%) subject died each from pneumonia, completed suicide, and ALS.
Serious Adverse Events
A total of 21 (11.4%) subjects reported a treatment-emergent serious adverse events (TESAE). The most frequently reported TESAEs by SOC were respiratory, thoracic, and mediastinal disorders (9 [4.9%] subjects); nervous system disorders (5 [2.7%] subjects); and infections and infestations (3 [1.6%] subjects). The most frequently reported TESAEs by PT were ALS (5 [2.7%] subjects), dyspnea (3 [1.6%] subjects), and respiratory failure (3 [1.6%] subjects). No TESAEs were related to study drug, as determined by the Investigator.
Withdrawals Due to Adverse Events
A total of 11 (5.9%) subjects reported a TEAE leading to study treatment discontinuation. Three (1.6%) subjects reported respiratory, thoracic, and mediastinal disorders leading to discontinuation. No other TEAE by SOC of PT was reported by >2 subjects.
Adverse Events of Special Interest
Cardiac disorders were reported in eight subjects. Of the cardiac TEAEs reported, 2 subjects with atrial fibrillation and 1 subject each with cardiac failure, sinus arrhythmia, sinus tachycardia, supraventricular tachycardia, tachycardia, ventricular extrasystoles, tachyarrhythmia, ECG signs of ventricular hypertrophy, and ECG signs of myocardial infarction were included. Other than 1 TEAE of cardiac failure, these cardiac events arose from ECG findings. The cardiac TEAEs did not reveal a signal of concern. Most of the cardiac TEAEs were asymptomatic ECG findings or were confounded by medical history or concurrent AEs.
Other Safety Assessments
Clinical Laboratory Evaluation: No clear trends over time were observed for safety laboratory parameters, and There were no clear trends in terms of the number of subjects with shifts from normal to abnormal values for most safety laboratory parameters. In the Safety Analysis Population, the following AEs related to laboratory parameters were reported:
ALT increased: 1 (0.5%) subject
AST increased: 3 (1.6%) subjects
Blood cholesterol increased: 1 (0.5%) subject
Blood creatine phosphokinase increased: 3 (1.6%) subjects
Blood glucose increased: 1 (0.5%) subject
Blood lactate dehydrogenase increased: 1 (0.5%) subject
Blood potassium increased: 1 (0.5%) subject
Hepatic enzyme increased: 1 (0.5%) subject
C-reactive protein increased: 1 (0.5%) subject
Pulmonary function test decreased: 1 (0.5%) subject
Liver function test increased: 1 (0.5%) subject
Total neuropathy score: 2 (1.1%) subjects
Vital Signs: Summaries of vital signs by visit and change from baseline by visit and by visit and region were collected. Overall, all vital signs were stable, and there were no notable trends during the study.
Electrocardiogram: Data on 12-lead ECG parameters and change from baseline by visit and by visit and region were collected. Distribution of out-of-normal range values of 12-lead ECG by visit and by visit and region is provided in and shifts of potentially clinically significant 12-Lead ECGs by region were recorded. There were no notable trends in 12-lead ECG parameters.
Physical Examination: There were no notable trends seen during physical examination in any group during the study. However, in the Safety Analysis Population, 6 (3.2%) subjects reported a TEAE of weight decreased.
Unsteadiness and Sensory Evaluation: Data for numbness and unsteadiness with severity was collected at each visit. Shifts from baseline in numbness and unsteadiness at each visit was also recorded, as was vibratory sensation information. No notable trends were observed during the study.
Columbia-Suicide Severity Rating Scale (C-SSRS): Summary information from C-SSRS administered to subjects was gathered, and no notable trends in suicidal ideation or behavior were observed during study treatment.
Forced Vital Capacity Percentage: At baseline, mean %FVC was ||||% (SD ||||%). The LS mean change from baseline was ||||% (95% CI |||||||||) at Week 4, ||||% (95% CI ||||||||||) at Week 12, and |||||% (95% CI -||||||||||) at Week 24.
Safety Conclusions
Study drug was generally well tolerated. TEAEs were experienced by 146 (78.9%) subjects. Of these, 60 TEAEs related to study treatment were reported by 36 (19.5%) subjects, 30 severe TEAEs were reported by 17 (9.2%) subjects, 17 TEAEs leading to study treatment discontinuation were reported by 11 (5.9%) subjects, and 6 TEAEs leading to death were reported in 6 (3.2%) subjects. A total of 24 TESAEs were reported by 21 (11.4%) subjects.
The most commonly reported TEAEs by SOC were musculoskeletal and connective tissue disorders (66 [35.7%] subjects); injury, poisoning, and procedural complications (49 [26.5%] subjects); and gastrointestinal disorders (48 [25.9%] subjects). The most commonly reported TEAEs by PT were muscular weakness (30 [16.2%] subjects), fall (29 [15.7%] subjects), and fatigue (14 [7.6%] subjects).
Six (3.2%) subjects died during the study period: 3 (1.6%) subjects died from respiratory failure and 1 (0.5%) subject died each from pneumonia, completed suicide, and ALS. The most frequently reported TESAEs by SOC were respiratory, thoracic, and mediastinal disorders (9 [4.9%] subjects); nervous system disorders (5 [2.7%] subjects); and infections and infestations (3 [1.6%] subjects). The most frequently reported TESAEs by PT were ALS (5 [2.7%] subjects), dyspnea (3 [1.6%] subjects), and respiratory failure (3 [1.6%] subjects). No TESAEs or severe TEAEs were related to study drug, as determined by the Investigator. ALS is known to be associated with respiratory complications.
No trends were observed in safety laboratory parameters, vital signs, 12-lead ECGs, or physical examinations. Percentage of the predicted FVC decreased over the study period.
The most frequently reported TEAEs, TESAEs, and TEAEs leading to death were associated with ALS disease progression or were non-specific symptoms, and no unique safety concerns were identified in the results of the MT-1186-A01 study.
Efficacy
The exploratory efficacy analysis evaluated the functional and survival assessments of oral edaravone efficacy. This evaluation was secondary to the main safety analysis.
ALS Functional Rating Scale
At baseline, the mean ALSFRS-R total score was 40.0 (SD 4.5). The change from baseline in LS mean ALSFRS-R score at Week 4, Week 12, and Week 24 was −0.8 (95% CI −1.3 to −0.4), −3.0 (95% CI −3.7 to −2.4), and −5.6 (95% CI −6.5 to −4.8), respectively. The LS mean score of each domain decreased over the 24 weeks. ALSFRS-R results and changes from baseline by visit for the Safety Analysis Population are provided in .
Time to Death, Tracheostomy, or Permanent Assisted Mechanical Ventilation: A summary of number (%) and time to death, tracheostomy, or permanent assisted mechanical ventilation up to the last observed visit for the Safety Analysis Population is provided in . Six (3.2%) subjects died during the study period. No subjects received a tracheostomy or permanent assisted mechanical ventilation during the study period. Time to death, tracheostomy, or permanent assisted mechanical ventilation was not calculated for these subjects.
Baseline and Changes From Baseline in ALSFRS-R Results by Visit (Safety Analysis Population)j.
Time to Death, Tracheostomy, or Permanent Assisted Mechanical Ventilation Up to Last Observed Visit (Safety Analysis Population).
CADTH’s Critical Appraisal of the Clinical Evidence
CADTH conducted a critical appraisal of the clinical studies for edaravone oral suspension based on the summary of the evidence provided by the sponsor.
Internal Validity
Study Design, Intervention, and Comparator
The most significant limitation associated with the included trials is the study designs. The bioequivalence design in healthy participants and the open-label uncontrolled study are not sufficient to evaluate the comparative clinical value added for the drug in the target population for reimbursement. The key assumption of the submission is that as IV edaravone has been approved by Health Canada and recommended for reimbursement by CADTH, establishing bioequivalence is sufficient to establish the clinical value of oral edaravone. However, the 2 formulations (solution for injection and oral suspension) cannot be considered bioequivalent since they involve 2 different dosing forms. Whether they can be considered to display comparable bioavailability of edaravone upon administration is to be assessed by Health Canada during formal review. While there is merit and supporting precedent to the assumption of comparable bioavailability, there remains a degree of uncertainty as to the true treatment effects of oral edaravone given the bioequivalence study design (i.e., single administration, assessing pharmacokinetic parameters with estimates falling within a range of acceptable values to establish equivalence) and the lack of comparative evidence between the oral and IV formulations’ effects on clinical outcomes.
Since safety study MT-1186-A01 was an uncontrolled trial, it was not designed to inform on the comparative efficacy of oral edaravone, even if this was a secondary outcome of the trial. With the previous establishment of efficacy for IV edaravone in the treatment of ALS, the sponsor did not consider it appropriate to include a comparative placebo control group.
Selection, Allocation, and Disposition of Patients
Patients were randomly allocated to 1 of the 2 treatment groups of the open-label, single-dose study MT-1186-J03 using a randomization key code table. No patient withdrew from the trial. Baseline characteristics were balanced between treatment groups.
Study MT-1186-A01 was an open-label, uncontrolled trial. Therefore, randomization and allocation concealment — 2 important strategies to minimize biases in clinical trials — do not apply here, subjecting the study to a high risk of bias and limiting the conclusions that can be drawn from the study. After a follow-up period of 24 weeks, 13% of patients had withdrawn from the study, which did not lead to any particularly significant concerns.
Outcome Measures
The bioequivalence and pharmacokinetic parameters measured in study MT-1186-J03 are considered appropriate, objective, and reliable outcome measures; however, 2 different drug formulations (solution for injection and oral suspension) with 2 different dosing forms cannot be considered bioequivalent, although they may display comparable bioavailability after administration. This is not for CADTH to evaluate, but for Health Canada to assess during formal review. The patient-reported outcomes such as AEs in study MT-1186-A01 are considered more subjectively measured outcomes, especially in the context of an open-label trial.
In study MT-1186-A01, efficacy was a secondary exploratory outcome assessed using the ALSFRS-R. The ALSFRS-R is a well-studied tool with demonstrated construct validity and internal consistency reliability, its use being supported by the FDA as a measure of treatment effect on function in daily living.51 However, the respiratory subscale does not correlate strongly with percent FVC, and the minimal clinically important difference for the slope of the ALSFRS-R score over time is based on expert opinion, leading to a degree of subjectivity in the items. More importantly, the trial did not include a control group, preventing adequate assessment of the real effect of the drug on the natural history of the disease over time.
Statistical Analysis
For assessment with the bioequivalence limit in study MT-1186-J03, analysis of variance was conducted on the following pharmacokinetic parameters: AUC0-t, AUC0-∞, and Cmax. The estimated 90% CIs of the geometric mean ratios were examined, and if they lay entirely within the limits of 0.80 and 1.25, then the bioequivalence between IV formulation and oral suspension was concluded. The analysis was performed by analysis of variance, which included factors accounting for the following sources of variation: sequence, subjects nested in sequences, period, and treatment.
Study MT-1186-A01 evaluated the safety and tolerability of edaravone using descriptive statistics in the absence of a control group.
External Validity
Patient Selection
Study MT-1186-J03 was performed with healthy individuals who identified as Japanese and had no medical history or complications at baseline. Selection of healthy volunteers is typical of bioequivalence studies, but the population is not representative of the real-life patients for whom edaravone is intended. This would not, however, significantly impact the confidence in the results.
Inclusion and exclusion criteria appeared relevant and reasonable in study MT-1186-A01. The trial included adult patients with ALS living and functioning independently whose first symptom of ALS had occurred within the previous 3 years and who had a baseline FVC greater than or equal to 70%. Some categories of patients were excluded, such as patients with an FVC less than 70%, patients who require the assistance of others for daily function, and patients with ALS symptom onset more than 3 years ago. Therefore, the findings are not generalizable to these categories of patients.
Treatment Regimen and Length of Follow-up
In both studies, the dosage of edaravone was aligned with the Health Canada–approved dosing. Study MT-1186-J03 was a single-dose study to assess bioequivalence, thus justifying the short follow-up duration. In study MT-1186-A01, the administration schedule of edaravone was aligned with the Health Canada product monograph. The study planned to follow patients for 48 weeks; however, results submitted to CADTH described the analysis at 24 weeks, since the 48-week data were not yet available at the time of the Reimbursement Review. There was no information reported as to how issues related to interim analyses were handled, such as the adequacy of type I error rate control across multiple time points and the appropriateness of methods used to maintain trial integrity with interim analyses.
That 87% of patients took riluzole in study MT-1186-A01, which was allowed throughout the trial duration, is representative of clinical practice. However, its use also acts as a confoundant, with the result being that AEs reported in the study may also be attributable to the concomitant medication.
Outcome Measures
The choice of outcome measures for assessing bioequivalence in study MT-1186-J03 was considered adequate. Harms outcomes were also assessed appropriately in study MT-1186-A01 with the use of AEs as the primary outcome measure.
However, no comparative data were reported for the outcomes of motor function, mobility, muscle pain, and fatigue, as well as difficulty breathing and speaking, which were identified by patients with ALS as the most important symptoms to control according to the patient input received. This is an important gap in the evidence.
Sponsor-Submitted Cost Information
The sponsor submitted a cost comparison of oral edaravone to IV edaravone for the treatment of ALS. At the submitted price of $9,200 per 1,050 mg of edaravone per 50 mL of suspension or $12,880 per package of two 735 mg (35 mL) bottles, the annual cost per patient of treatment with oral edaravone is $123,280 in the first year and $119,600 per subsequent year (), excluding markups and dispensing fees. When comparing drug costs alone, the annual cost of therapy with oral edaravone is the same as that of IV edaravone at Ontario Drug Benefit Exceptional Access Program list prices.
When also considering IV administration and AE costs, the sponsor estimated that IV edaravone would be associated with an average of |||||| per patient per year in AE-related hospitalization costs as well as an average of ||||||||| per patient per year in IV infusion costs (e.g., catheter insertion, maintenance, and removal), for a total of ||||||||| per patient per year in health care costs (). CADTH notes that the sponsor has accounted for the proportion of these costs covered by its PSP. Further detail on these costs can be found in . As oral edaravone would not be associated with these costs, the sponsor concluded that the use of oral edaravone would result in a saving of ||||||||| per patient per year.
Sponsor’s Drug Acquisition Cost Comparison.
Sponsor’s Associated Health Care Costs.
CADTH’s Cost Comparison Analysis.
Critical Appraisal of Cost Information
Lack of comparative safety and efficacy evidence: Findings from the sponsor’s bioequivalence study showed that oral edaravone was equivalent to its IV formulation in a population of healthy volunteers.
8 Since IV edaravone was found to slow the rate of decline in motor function in patients with ALS, bioequivalence suggests the same conclusion may apply to oral edaravone. A single-group safety study suggests the harms profile of oral edaravone may be considered acceptable, with no major safety signals identified.
10 However, the lack of comparative data between oral and IV edaravone for efficacy outcomes identified as important by patients with ALS in stakeholder input (e.g., motor function, mobility, muscle pain, fatigue, difficulty breathing and speaking) remains a gap in evidence and increases uncertainty.
Subsequent years of therapy not considered: The sponsor reported that the use of oral edaravone would be associated with a savings of ||||||||| per patient per year compared to IV edaravone due to savings related to IV administration and IV-related AEs, taking into account the sponsor’s PSP, which funds some of these costs. However, the sponsor appears to consider only the first year of therapy in this assessment. According to the inputs provided by the sponsor, subsequent years of therapy with IV edaravone are associated with reduced administration costs due to a slight reduction in the frequency of peripherally inserted central catheter line insertion and the 1-time cost associated with an implantable port.
CADTH Reanalyses
CADTH revised the sponsor’s analysis to consider costs in subsequent years in addition to those in the first year of therapy. CADTH used the sponsor’s provided inputs (, ) to recalculate the incremental savings associated with oral edaravone compared to IV edaravone arising from IV administration and IV-related AEs. For the first year of therapy, CADTH reanalysis was aligned with the sponsor’s in that oral edaravone was associated with a weighted average savings of $1,561 in administration costs and $89 in IV-related AE costs, for a total average incremental savings of $1,649 per patient compared to IV edaravone. For subsequent years, the CADTH reanalysis suggests that oral edaravone was associated with an average saving of $1,016 in administration costs and $89 in IV-related AE costs, for a total average incremental saving of $1,105 per patient per year.
CADTH was unable to account for uncertainties in the comparative effectiveness and safety between edaravone products or for the confidential pricing and stipulations that may have been negotiated for IV edaravone.
Issues for Consideration
Confidential prices available for comparator: IV edaravone underwent pan-Canadian Pharmaceutical Alliance pricing negotiations, concluding with a letter of intent.
24 It is therefore likely that IV edaravone is reimbursed by jurisdictional drug plans at a confidential price that is less than publicly available list prices.
Hidden costs within the price of IV edaravone may not be accounted for: For IV edaravone, the sponsor runs a PSP that funds administration costs for many patients in either PSP-funded infusion clinics or in the patient’s home by PSP-funded nurses (see for details). Some IV administration costs are therefore borne by the sponsor and not the health care system; this was considered within the submitted cost comparison. However, the cost of providing such a program may have been factored into the negotiation and subsequent price of IV edaravone. The need for the sponsor to fund this PSP will be substantially lessened as patients switch to or are initiated on the oral product rather than the IV product, and thus this reduction in PSP expenses may require additional consideration during price negotiations for the oral product.
Oral edaravone may allow for greater flexibility to accommodate patients’ lifestyles: Some patients may find oral edaravone more conducive to maintaining their lifestyle, while possible, than IV edaravone due to the avoidance of regular hour-long infusions required by the IV formulation,
7 potentially easier travel as long as the storage requirements of the oral formulation are met,
7 and — according to the clinical expert consulted by CADTH — the increased potential for activities such as swimming that are made difficult by the need for peripherally inserted central catheter lines for some patients using the IV formulation. Patient input provided for this review indicated patients experienced difficulties related to IV edaravone including scheduling activities of daily living around their infusions and difficulties regarding port catheter insertion; patients with experience using oral edaravone reported no such issues.
Another treatment for ALS may become available: At the time of this review, Health Canada
25 and CADTH
26 were also reviewing sodium phenylbutyrate and ursodoxicoltaurine (Albrioza), also known as AMX0035,
27 for the treatment of ALS. While the clinical expert consulted by CADTH for this review did not believe AMX0035 would be a direct comparator to oral or IV edaravone, and instead may be used sequentially in combination with riluzole and edaravone for some patients, the arrival of an additional therapy may alter prescribing patterns. A publicly accessible cost of AMX0035 therapy relative to edaravone was not available at the time of this review.
Discussion
Summary of Available Evidence
To inform on the use of oral edaravone compared to its IV formulation, 2 manufacturer-sponsored studies were included in this review.
The single-dose, randomized, open-label bioequivalence study MT-1186-J03 (n = 42)8,9 evaluated the bioequivalence of an oral suspension and an IV formulation of edaravone in healthy individuals who identified as Japanese. The key outcomes were the pharmacokinetic parameters of AUC and Cmax.
The second study was a multicentre, open-label, single-group study, MT-1186-A01 (n = 185),10 that evaluated the longer-term safety and tolerability of oral edaravone in patients with ALS living and functioning independently whose first symptom of ALS had occurred within the previous 3 years and who had a baseline FVC greater than or equal to 70%. At the time of the review, the 24-week results were available. Patients received edaravone as a 105 mg oral suspension administered in accordance with the Health Canada–approved regimen. The concomitant use of riluzole was permitted throughout the study.
Interpretation of Results
Efficacy
Based on the sponsor’s analysis, results from the single-dose, randomized, open-label bioequivalence study MT-1186-J03 show that an oral suspension of edaravone 105 mg was equivalent to an IV formulation of edaravone 60 mg in healthy volunteers who identified as Japanese. In this analysis, oral edaravone had equivalent AUC0-t and AUC0-∞ of unchanged edaravone compared to the IV formulation, as both geometric mean ratio and 90% CI were within the range of 0.80 to 1.25. As for Cmax, the geometric mean ratio and its lower limit of the 90% CI were also within the prespecified limits, while the upper limit of the 90% CI exceeded 1.25.
An exploratory efficacy analysis was reported in the open-label, single-group study MT-1186-A01, evaluating the functional assessment of oral edaravone efficacy using the ALSFRS-R. The least squares mean score of each domain decreased over the 24 weeks. The change from baseline in least squares mean in the ALSFRS-R score at week 4, week 12, and week 24 was –0.8 (95% CI, –1.3 to –0.4), –3.0 (95% CI, –3.7 to –2.4), and –5.6 (95% CI, –6.5 to –4.8), respectively. The ALSFRS-R is a well-studied tool with demonstrated construct validity and internal consistency reliability and is supported by the FDA as a measure of treatment effect on function in daily living. However, the level of confidence in the evidence is highly affected by several limitations, most importantly the open-label uncontrolled trial design of the study that introduces a high risk of bias. Therefore, no efficacy conclusions could be drawn from these findings.
The key assumption of the submission hinges on the previous recommendation that IV edaravone be reimbursed for the treatment of ALS (with conditions) and on oral edaravone being bioequivalent to the IV formulation. The finding of bioequivalence would establish the clinical effectiveness of the oral formulation relative to the IV 1. However, the 2 formulations (solution for injection and oral suspension) cannot be considered bioequivalent since they involve 2 different dosing forms. Whether they can be considered to display comparable bioavailability of edaravone upon administration is to be assessed by Health Canada during formal review. While bioavailability is an accepted approach for market access, it makes it difficult to determine the clinical value of the product in the absence of comparative assessments with clinically important outcomes in ALS. This is in part because the population in which the comparable bioavailability was established was in healthy participants, not in those with ALS. In addition, comparable bioavailability may be claimed based on an established range in which pharmacokinetic values may fall, and therefore there will be a range of treatment effects in practice settings, which is particularly notable in a disease that has a heterogeneous natural history. Although the interventional study, MT-1186-A01, examined clinical outcomes, no concrete conclusions could be drawn because of the aforementioned limitations of the study. Therefore, while it is accepted that oral edaravone appears to display comparable bioavailability to the IV formulation, there remains uncertainty as to what the true treatment effect will be in Canadian patients with ALS.
Additionally, the submission highlighted the difficult administration of IV edaravone and that the oral formulation provides an option for patients to avoid the issues surrounding IV administration. Indeed, both patient and clinician input to CADTH specified many issues with the IV administration. According to the clinical expert consulted by CADTH, the uptake of IV edaravone has so far been low, in part because the IV formulation is invasive and comes with a time-consuming administration schedule. The clinical expert believed that the oral formulation would be a well-received alternative, as many patients choose not to embark on the currently available IV formulation because of the caveats and excessive requirements and constraints related to IV infusion. Indeed, the patient input received highlighted the difficulties related to the IV administration of edaravone, including patients having to schedule activities of daily living around their infusion schedule and needing to have a port catheter implanted. Both the drug plans and the clinical expert noted in their inputs that an oral version of edaravone would be a lot easier for patients to access than its IV formulation, reducing the risk of exposure to unnecessary infusion-associated AEs and decreasing health care system burden related to the IV administration. No comparative data were provided regarding outcomes like patient preference or improved treatment adherence between the 2 modes of administration.
Harms
One patient in each treatment group reported an AE of mild intensity in the single-dose bioequivalence study MT-1186-J03; these AEs were not judged to be reasonably related to the investigational products by the investigator. No SAEs, no withdrawals due to AEs, and no AEs of special interest were reported in the study.
Results from the single-group safety study MT-1186-A01 in patients with ALS were reported for the 24-week interim analysis. Seventy-nine percent of patients experienced at least 1 AE; however, discontinuation due to AEs was low (6%), suggesting the harm profile might be considered acceptable. SAEs were reported by ||| of patients; the most frequently reported were ||||||||||||||||||||||||||||||||||||||||||||||||||| patients died over the 24-week study period; causes of death were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. There is no evidence to assess the comparative AE profiles of IV edaravone and oral edaravone. Information reported in the Health Canada draft product monograph7 and in the CADTH Reimbursement Review for IV edaravone28 suggests that the oral version is not associated with an increased incidence of AEs.
Cost
At the submitted price of $9,200 per 1,050 mg of edaravone per 50 mL of suspension or $12,880 per package of two 735 mg (35 mL) bottles, the annual drug cost per patient of treatment with oral edaravone is $123,280 in the first year and $119,600 per subsequent year, which is equivalent to the drug acquisition cost of IV edaravone at publicly available prices. CADTH conducted a reanalysis of the sponsor-submitted cost comparison, considering that costs associated with IV administration and IV-related AEs differ in the first and subsequent years of therapy. In this analysis, where some of the IV administration costs were assumed to be borne by the sponsor’s PSP, oral edaravone was associated with an average cost saving to the public health care payer of $1,649 per patient compared to IV edaravone in the first year of therapy, and $1,105 per patient in subsequent years of therapy.
The cost comparison assumes clinical similarity between the oral and IV formulations of edaravone, based on the sponsor’s submitted single-dose bioequivalence study and an uncontrolled, open-label safety study. CADTH was unable to account for uncertainties in the comparative clinical effectiveness and safety between edaravone products or for the confidential pricing and stipulations that may have been negotiated for IV edaravone.
Conclusions
Findings from the sponsor’s analysis of bioequivalence suggested that oral edaravone showed comparable bioavailability to its IV formulation in a population of healthy volunteers; however, this requires formal assessment by Health Canada. Since IV edaravone was found to slow the rate of decline in motor function in patients with ALS, comparable bioavailability would suggest that the same conclusion may apply to oral edaravone. Findings from a single-group safety study suggest that the harms profile of oral edaravone may be considered acceptable, and no major safety signal was identified. However, the level of confidence in the evidence is highly affected by several limitations including the open-label uncontrolled trial design of the study, which introduced a high risk of bias. The lack of comparative data with oral edaravone for the outcomes of motor function, mobility, muscle pain, and fatigue, as well as difficulty breathing and speaking, which were identified by patients with ALS as the most important symptoms to control according to the patient input received, remains a gap in the evidence. Input received from all sources, including patients with ALS, clinicians, and the clinical expert consulted by CADTH for this review, emphasized that an oral version of edaravone would be a lot easier for patients to access than its IV formulation, reducing the risk of exposure to unnecessary infusion-associated AEs and decreasing the burden related to IV administration both to the health care system and to patients with ALS themselves.
At the submitted price, the annual drug cost of oral edaravone is $123,280 per patient in the first year and $119,600 per patient in subsequent years, which is the same as the annual drug cost of IV edaravone. When costs associated with IV administration and IV-related AEs are considered, and the sponsor’s PSP which funds some IV administration costs is taken into account, oral edaravone is $1,649 less expensive per patient than IV edaravone in the first year, and $1,105 less expensive in subsequent years. The results are based on publicly available list prices for IV edaravone and may not reflect actual prices paid by Canadian public drug plans.
Abbreviations
-
AE
adverse event
-
ALS
amyotrophic lateral sclerosis
-
ALSFRS-R
ALS Functional Rating Scale — Revised
-
AUC
area under the plasma concentration–time curve
-
AUC0-t
area under the plasma concentration–time curve from zero up to the last quantifiable concentration time point
-
AUC0-∞
area under the plasma concentration–time curve from zero up to infinity with extrapolation of the terminal phase
-
Cmax
maximum plasma concentration after administration
-
CALS
Canadian ALS Research Network
-
CDEC
CADTH Canadian Drug Expert Committee
-
CI
confidence interval
-
FVC
forced vital capacity
-
LMN
lower motor neuron
-
PSP
patient support program
-
SAE
serious adverse event
-
SD
standard deviation
-
UMN
upper motor neuron
Appendix 1. Additional Economic Information
Note that this appendix has not been copy-edited.
Additional Details on the Sponsor’s Submission
Sponsor’s Health Resource Use.
Appendix 2. Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Summary of Key Take-Aways.
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budgetary impact of reimbursing oral edaravone for the treatment of ALS compared to the reference scenario in which considered IV edaravone only. The BIA was conducted from a Canadian drug plan payer perspective over a 3-year time horizon (2023 to 2025) using an epidemiological approach and included drug acquisition costs. The sponsor also submitted a scenario analysis from a health care payer perspective which considered drug acquisition costs as well as administration costs associated with peripherally inserted central catheter line or implantable port insertion and maintenance for IV edaravone, and costs associated with IV-site infections and infusion site thrombosis. Data for the model was obtained from: Statistics Canada,32 the NIHB annual report,33 real-world data from the sponsor’s PSP (not provided), the sponsor’s internal projections, the Ontario Case Costing Initiative,29 and formulary-specific costs.30,34 Key inputs to the BIA are documented in .
Key assumptions included:
No patients discontinue edaravone treatment once initiated.
Patients already using IV edaravone will switch to oral edaravone in the same proportion as new patients who will start oral edaravone rather than IV edaravone.
Patients already using IV edaravone do not need an introductory treatment cycle upon switching to oral edaravone.
Patients using oral edaravone will not experience AEs (scenario).
Summary of Key Model Parameters.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of oral edaravone for the treatment of ALS would be associated with an incremental cost of $2,597,291 in Year 1, $4,521,761 in Year 2, and $6,651,801 in Year 3, for a 3-year total incremental budget impact of $13,770,852. When other health care costs were considered, i.e., IV administration and IV-related AEs, the reimbursement of oral edaravone would be associated with a 3-year total incremental budget impact of $13,494,762.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Many inputs depend on internal sponsor’s data and forecasting: The sponsor’s estimates of the current and anticipated proportion of ALS patients using edaravone, the proportion of patients reimbursed for edaravone through a public plan, the expected uptake of oral edaravone, and the proportion of IV edaravone patients using each method of IV administration were all based on the sponsor’s internal PSP data, claims data, and forecast data.
22,35,36 As such, validation of these inputs by CADTH was not possible.
NIHB population was inappropriately calculated: The sponsor incorporated the number of NIHB clients reported in March 2020
33 as the size of the NIHB population in the base year, 2022. A more current NIHB 2020/2021 Annual Report has been published with updated population numbers.
37 Additionally, the sponsor’s model added the NIHB population to that of the provincial populations estimated by Statistics Canada.
32 NIHB clients living within the borders of a province are counted within provincial population data, thus the NIHB population was double counted in the sponsor’s analysis. Finally, NIHB clients residing within Ontario who are under 25 or over 65 years of age are eligible for reimbursement by Ontario Drug Benefit and thus should be counted as Ontario Drug Benefit clients rather than NIHB clients for the purposes of modelling the budgetary impact of reimbursing oral edaravone.
CADTH used the number of NIHB clients reported in March 2021
37 within each province and inflated these numbers by 6 months using the growth rates of the NIHB population within each province as reported for 2020 to 2021 to estimate the population in Q4 2021, equivalent to the time point used to inform the baseline year for the other jurisdictions.
35 The population of NIHB clients living within each province was then subtracted from the populations of those provinces.
32,37 Finally, NIHB clients residing within Ontario who are under 25 or over 65 years of age were considered to be part of Ontario’s eligible population for the purpose of the BIA.
37
Uptake of oral edaravone may be underestimated: The sponsor estimated that the reimbursement of oral edaravone would increase the total proportion of ALS patients using edaravone; from approximately 10% (all IV edaravone), to approximately 12% in Year 1, 14% in Year 2, and 15% in Year 3, split between oral and IV edaravone (method used to derive the estimate not provided). While the clinical expert consulted by CADTH agreed that 10% was a reasonable estimate for the proportion of ALS patients currently using IV edaravone, they believed that the reimbursement of the oral formulation would lead to a greater uptake of oral edaravone than estimated by the sponsor, estimating that 25% of ALS patients would use oral edaravone by Year 3 of its availability.
CADTH estimated that for the new drug scenario, 15% of ALS patients would use edaravone in Year 1, 20% in Year 2, and 25% in Year 3 in its base case reanalysis. A scenario analysis was conducted where this was reduced to 12%, 16%, and 20%.
Proportion of edaravone patients using the oral formulation may be underestimated: The sponsor estimated that of patients using edaravone, 72.2% would be using the oral formulation in Year 1 of its reimbursement, 78.6% in Year 2, and 79.4% in Year 3, regardless of whether the patient was previously using IV edaravone or was edaravone naive. The clinical expert consulted by CADTH estimated that once the oral formulation was available, due to patient preference in avoiding infusions, as well as potentially reduced health system costs, and increased safety due to the absence of IV-related AEs, 100% of patients who were previously edaravone naive would begin on the oral form, while approximately 95% of IV patients would switch to the oral formulation within the first year of its reimbursement.
CADTH assumed 100% of new edaravone patients would use the oral formulation in the new drug scenario, and 80%, 95%, and 95% of patients previously using the IV formulation would instead use the oral formulation in years 1, 2, and 3, respectively. As it is likely to take some months for all patients who will switch from the IV to the oral formulation to do so, the proportion of edaravone-experienced patients using the oral formulation in Year 1 was estimated as 80% rather than 95%.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analyses by ensuring the NIHB population was not double counted, increasing the market uptake of edaravone, and increasing the proportion of edaravone patients using the oral formulation. The changes applied to derive the CADTH base case are described in .
CADTH Revisions to the Submitted Budget Impact Analysis.
The results of the CADTH step-wise reanalysis are presented in summary format in and a more detailed breakdown is presented in . When considering the drug plan payer perspective, CADTH reanalyses suggest that reimbursement of oral edaravone will be associated with a 3-year budgetary incremental cost of $38,710,109. When a health care payer perspective is taken, CADTH reanalyses suggest the resulting 3-year budgetary incremental cost of reimbursing oral edaravone would be slightly reduced to $38,359,198.
Results of the BIA are highly dependent on the estimated increase in the proportion of ALS patients using edaravone due to the reimbursement of the oral formulation. CADTH therefore conducted a scenario analysis assuming a reduced overall uptake of edaravone of 12%, 16%, and 20% in years 1, 2, and 3, respectively. CADTH also conducted a scenario assuming that 65% of edaravone patients would be publicly reimbursed to explore uncertainty in the proportion of patients covered by public drug plans. See .
Summary of the CADTH Reanalyses of the BIA.
Detailed Breakdown of the CADTH Reanalyses of the BIA.
Appendix 3. Sponsor References
a. Mitsubishi Tanabe Pharma Corporation. Clinical Study Report, Study MT-1186-J03. 2021.
b. Mitsubishi Tanabe Pharma Corporation. Clinical Study Protocol, Study MT-1186-J03. 2019.
c. Shimizu
H, Nishimura
Y, Shiide
Y, Yoshida
K, Hirai
M, Matsuda
M, et al.
Bioequivalence Study of Oral Suspension and Intravenous Formulation of Edaravone in Healthy Adult Subjects. Clin Pharmacol Drug Dev. 2021;10(10):1188-97.
[
PMC free article: PMC8518908] [
PubMed: 33955162]
d. Guideline for Bioequivalence Studies of Generic Products (PFSB/ELD Notification No. 0229-10 dated February 29, 2012).
e. Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs - General Considerations (March 2014).
f. Shimizu H, Nishimura Y, Shiide Y, et al. Evaluation of pharmacokinetics, safety, and drug-drug interactions of an oral suspension of edaravone in healthy adults.
Clin Pharmacol Drug Dev. 2021. 10.1002/cpdd.925. [
PMC free article: PMC8518673] [
PubMed: 33704925] [
CrossRef]
g. Mitsubishi Tanabe Pharma Corporation. Statistical Analysis Plan, Study MT-1186-J03. 2019.
h. Shimizu
H, Inoue
S, Endo
M, et al.
A randomized, single-blind, placebo-controlled, 3-way crossover study to evaluate the effect of therapeutic and supratherapeutic doses of edaravone on QT/QTc interval in healthy subjects. Clin Pharmacol Drug Dev.
2021;10(1):46–56.
[
PMC free article: PMC7818234] [
PubMed: 32543120]
i. Clinical Pharmacokinetic Studies of Pharmaceuticals” (PMSB/ELD Notification No.796 dated June 1, 2001).
j. Mitsubishi Tanabe Pharma Corporation. Clinical Study Report, Study MT-1186-A01. 2021.
l. RADICAVA and RADICAVA (edaravone) Product Monograph (Draft). Jersey City, New Jersey: Mitsubishi Tanabe Pharma America, Inc., a US subsidiary of Mitsubishi Tanabe Pharma Corporation; 2021.
m. Mitsubishi Tanabe Pharma Canada. MTP Patient Support Real-World Evidence interim analysis. Data on file.
s. Ontario Case Costing Initiative. Procedure 1IS53LALF: Implant int dev v cava OA implant CVC w injection port. Ambulatory procedure, 2017-18.
https://hsim.health.gov.on.ca/hdbportal/ Accessed: 2022 Feb 7.
References
- 1.
Shoesmith
C, Abrahao
A, Benstead
T, et al.
Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. Can Med Assoc J.
2020;192(46):E1453-E1468.
[
PMC free article: PMC7683000] [
PubMed: 33199452]
- 2.
Brown
RH, Al-Chalabi
A. Amyotrophic lateral sclerosis. N Engl J Med.
2017;377(2):162-172.
[
PubMed: 28700839]
- 3.
- 4.
Wolfson
C, Kilborn
S, Oskoui
M, Genge
A. Incidence and prevalence of amyotrophic lateral sclerosis in Canada: a systematic review of the literature. Neuroepidemiology.
2009;33(2):79-88.
[
PubMed: 19494548]
- 5.
- 6.
- 7.
Radicava (edaravone oral suspension) 105mg/5mL and (edaravone injection) Solution, 30mg/100mL (0.3mg/mL), intravenous administration [product monograph]. Jersey City (NJ): Mitsubishi Tanabe Pharma America, Inc.; 2022 Nov 8:
https://pdf.hres.ca/dpd_pm/00068094.PDF. Accessed 2022 Nov 11.
- 8.
Clinical Study Report: MT-1186-J03. Bioequivalence study of oral suspension and intravenous formulation of edaravone in healthy adult subjects [internal sponsor's report]. Osaka (Japan): Mitsubishi Tanabe Pharma Group; 2019 Dec 13.
- 9.
Shimizu
H, Nishimura
Y, Shiide
Y, et al.
Bioequivalence study of oral suspension and intravenous formulation of edaravone in healthy adult subjects. Clinical pharmacology in drug development.
2021;10(10):1188-1197.
[
PMC free article: PMC8518908] [
PubMed: 33955162]
- 10.
Clinical Study Report: MT-1186-A01. A phase 3, multi-center, open-label, safety study of oral edaravone administered over 48 weeks in subjects with amyotrophic lateral sclerosis (ALS) [internal sponsor's report]. Jersey City (NJ): Mitsubishi Tanabe Pharma Development America; 2021 Sep 10.
- 11.
Turner MR, Bowser R, Bruijn L, et al. Mechanisms, models and biomarkers in amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degeneration. 2013;14 Suppl 1(0 1):19-32. [
PMC free article: PMC4284067] [
PubMed: 23678877]
- 12.
Agosta
F, Al-Chalabi
A, Filippi
M, et al.
The El Escorial criteria: strengths and weaknesses. Amyotrophic lateral sclerosis & frontotemporal degeneration.
2015;16(1-2):1-7.
[
PubMed: 25482030]
- 13.
Brooks
BR, Miller
RG, Swash
M, Munsat
TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord.
2000;1(5):293-299.
[
PubMed: 11464847]
- 14.
Golby
R, Poirier
B, Fabros
M, Cragg
JJ, Yousefi
M, Cashman
N. Five-year incidence of amyotrophic lateral sclerosis in British Columbia (2010-2015). Can J Neurol Sci.
2016;43(6):791-795.
[
PubMed: 27476760]
- 15.
Lareau-Trudel
E, Fortin
E, Gauthier
M, Lavoie
S, Morissette
E, Mathieu
J. Epidemiological surveillance of amyotrophic lateral sclerosis in Saguenay region. Can J Neurol Sci.
2013;40(5):705-709.
[
PubMed: 23968945]
- 16.
Andersen
PM, Abrahams
S, Borasio
GD, et al.
EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol.
2012;19(3):360-375.
[
PubMed: 21914052]
- 17.
Hogden
A, Foley
G, Henderson
RD, James
N, Aoun
SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. Journal of multidisciplinary healthcare.
2017;10:205-215.
[
PMC free article: PMC5446964] [
PubMed: 28579792]
- 18.
Miller
RG, Jackson
CE, Kasarskis
EJ, et al.
Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology.
2009;73(15):1218-1226.
[
PMC free article: PMC2764727] [
PubMed: 19822872]
- 19.
Ritsma
BR, Berger
MJ, Charland
DA, et al.
NIPPV: prevalence, approach and barriers to use at Canadian ALS centres. Can J Neurol Sci.
2010;37(1):54-60.
[
PubMed: 20169774]
- 20.
Benstead
T, Jackson-Tarlton
C, Leddin
D. Nutrition with gastrostomy feeding tubes for amyotrophic lateral sclerosis in Canada. Can J Neurol Sci.
2016;43(6):796-800.
[
PubMed: 27039940]
- 21.
- 22.
Drug Reimbursement Review sponsor submission: Radicava (edaravone), 105mg/5mL oral suspension
[internal sponsor's package]. Toronto: Mitsubishi Tanabe Pharma Canada, Inc; 2022.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
Budget Impact Analysis
[internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Radicava (edaravone), 105mg/5mL oral suspension.
Toronto: Mitsubishi Tanabe Pharma Canada, Inc; 2022.
- 36.
Mitsubisi Tanabe Pharma Canada Inc. response to April 28, 2022 request for additional information regarding Radicava review: Information regarding the budget impact analysis
[internal additional sponsor's information]. Toronto: Mitsubishi Tanabe Pharma Canada, Inc.; 2022.
- 37.
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
Sponsor: Mitsubishi Tanabe Pharma Canada, Inc.
Therapeutic area: Amyotrophic lateral sclerosis