In humans, smooth muscle cells (SMCs) are the main cell type in the artery medial layer, in pre-atherosclerotic diffuse thickening of the intima, and in all stages of atherosclerotic lesion development. SMCs secrete the proteoglycans responsible for the initial binding and retention of atherogenic lipoproteins in the artery intima, with this retention driving foam cell formation and subsequent stages of atherosclerosis. In this chapter we review current knowledge of the extracellular matrix generated by SMCs in medial and intimal arterial layers, their relationship to atherosclerotic lesion development and stabilization, how these findings correlate with mouse models of atherosclerosis, and potential therapies aimed at targeting the SMC matrix-lipoprotein interaction for atherosclerosis prevention.
Keywords:
Atherosclerosis, Biglycan, Diffuse intimal thickening, Extracellular matrix, Lipoprotein retention, Proteoglycans, Response to retention, Smooth muscle, Smooth muscle cells1. Introduction
Smooth muscle is the involuntary nonskeletal form of muscle cells, present in hollow organs such as the stomach, intestine, bladder and uterus, the respiratory tract, and arteries and veins of the circulatory system. Smooth muscle cells (SMCs) in the artery muscular or “medial” layer contract or relax to redistribute blood throughout the body and to regulate local blood pressure and blood vessel volume. In addition, in humans SMCs are the predominant cell type in the pre-atherosclerotic intima and all stages of atherosclerotic lesion development (Allahverdian et al. 2018). In the intimal layer, SMCs take on alternate phenotypes and roles including increased proliferation, increased production of matrix proteoglycans, uptake of lipoproteins and foam cell formation, loss of SMC markers and expression of macrophage markers, and stabilization of arteries against plaque rupture. In this chapter we review extracellular matrix (ECM) production by SMCs and its critical role in mediating the lipoprotein retention that drives atherosclerotic lesion development. We correlate these findings in humans with mouse models of atherosclerosis and how modification of the SMC-derived proteoglycan-lipoprotein interaction might be targeted as a means of preventing or treating atherosclerosis.
2. Smooth Muscle Cell Phenotype Switch and Extracellular Matrix Production
Vascular smooth muscle cells (SMCs), unlike skeletal and heart muscle cells, are highly plastic and show a spectrum of phenotypes in the medial and intimal artery layers, with contractile and synthetic being the extremes (Owens et al. 2004). SMCs switch their phenotypes depending on genetic preprogramming and local environmental signals including cytokines and mechanical forces. The majority of SMCs constituting the medial layer of healthy arteries exhibit a contractile phenotype with high levels of cytoplasmic contractile myofilaments, a low proliferation rate, and low protein synthesis. On the other end of the spectrum, intimal SMCs can exhibit either a contractile phenotype or a dedifferentiated, synthetic phenotype with reduced expression of contractile proteins and other differentiated SMC markers and high rates of proliferation and protein synthesis. The majority of SMCs in the intima are of synthetic phenotype. SMCs forming the fibrous cap over atheromas, however, retain SMC markers including α-SMA and show a more differentiated, contractile phenotype (Feil et al. 2014; Perisic Matic et al. 2016; Albarran-Juarez et al. 2016; Chappell et al. 2016). Although transdifferentiation of SMCs with partial or total loss of SMC markers and gain of some macrophage markers has been reported in both human and mouse atherosclerotic lesions, detailed studies have confirmed SMCs maintain their characteristics and do not function as macrophages (Feil et al. 2014; Allahverdian et al. 2014; Shankman et al. 2015; Vengrenyuk et al. 2015).
As the major cell type of the pre-atherosclerotic and atherosclerotic intima, the phenotypic characteristics and products generated by synthetic phenotype SMCs are key determinants of atherosclerotic lesion development (Nakashima et al. 2008). Some of these SMC products are components of the ECM (Wight 2018). ECM is a component of all tissues and organs including blood vessels that provides physical strength to the tissue and acts as a framework in which cells and other molecules are embedded and bound. In arteries, the composition of ECM varies in the intima, media, and adventitia, between healthy and atherosclerotic arteries, and with different stages of atherosclerosis. In particular, the marked increase in proteoglycan content of ECM in the pre-atherosclerotic diffuse intimal thickening (DIT) stage of intima development creates the conditions necessary for the initial retention of serum lipoproteins and initiation of foam cell and atherosclerotic lesion formation (Nakashima et al. 2008; Wight 2018).
3. Extracellular Matrix (ECM)
Extracellular matrix (ECM) is a non-cellular, well-organized, and highly dynamic three-dimensional network in which cells reside and function. ECM is comprised of both fibers and a non-fibrillar amorphous gelatinous material called ground substance. Fibrous protein components of ECM include collagens, elastin, fibronectin (FN), and laminins. The non-fibrillar substance consists of proteoglycans (including versican, perlecan, biglycan, and decorin), along with glycoproteins and hyaluronan. Proteoglycans consist of long chains of repeating disaccharides attached to a core protein, while glycoproteins are composed of short, highly branched chains of various monosaccharides with no repeating unit attached to a core protein (Fig. 1). Hyaluronan is a glycosaminoglycan molecule that has no protein core but forms a non-covalent complex with proteoglycans in the ECM.
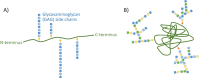
ECM components interact with resident cells through their cell surface receptors and regulate their morphology as well as functions such as growth, migration, differentiation, and survival (Theocharis et al. 2016). The composition and structure of ECM varies from tissue to tissue, and alteration of this composition changes the overall structure of the tissue and the function and properties of the embedded cells. Moreover, growth factors, cytokines, and chemokines that bind to ECM molecules are released in response to different stimuli and modify local cellular function (Theocharis et al. 2016). All cell types synthesize and secrete ECM, with alteration of cellular properties due to physiological and pathologic conditions modulating the composition of ECM produced. In the following sections, we focus primarily on ECM produced by, and affecting the function of, arterial SMCs.
3.1. Fibrillar Matrix
Collagens, the most abundant protein in the human body and the major component of the ECM, have widespread distribution among tissues from bone and cartilage to vessel walls. Collagens are categorized into fibril-forming varieties including types I, II and III, present mainly in dermis, tendon, bone, and cartilage, and non-fibrillar collagens such as types IV and VIII, present in the basement membrane – the layer between the endothelium/epithelium and underlying cells – and fibril-associated forms including collagen types IX and XII, which do not form fibers themselves but are associated with collagen fibers (Theocharis et al. 2019). Collagens I and III are the main collagen forms in the medial and intimal layers of medium and large arteries and are the predominant collagens providing tensile strength to the vessel wall (Shekhonin et al. 1985; Frantz et al. 2010; Yue 2014). Collagens make up approximately 40% of all ECM in normal arteries, 10% of early atherosclerotic lesions, and 50% in advanced lesions (Wight 2018). Among the different subtypes, type I collagen is more highly expressed in SMC-rich fibrous caps compared to SMC-poor plaque shoulders. Collagen fibers are thus believed to play an important role in plaque stability (Rekhter 1999).
Elastin fibers are another major fibrillar component of ECM that provide recoil and elasticity to tissues undergoing repeated stretching, such as arteries. Elastic fibers constitute about 50% of the ECM in normal artery walls but only 20% of early and 10% of late atherosclerotic lesions (Wight 2018). Elastin is produced by assembly and crosslinking of its soluble precursor tropoelastin (Wise and Weiss 2009). Normal arteries contain mature crosslinked elastin fibers and negligible amounts of tropoelastin. Using atherosclerotic models, Phinikaridou et al. have shown that accumulation of tropoelastin is associated with plaque progression and instability. They also found that human endarterectomy specimens with ruptured plaques have higher tropoelastin content than stable plaques, suggesting this marker can be used as an indicator of plaque instability (Phinikaridou et al. 2018).
Fibronectin (FN) is another fibril-forming glycoprotein of the ECM. Fibronectin monomers are produced and secreted by cells to form FN fibers (Theocharis et al. 2016). The fibrillar form of FN is a homodimer with monomers linked at the cell surface by disulfide bonds. A soluble monomeric form of fibronectin is also present in plasma, which cells can utilize to assemble into fibers. Fibronectin fibers have binding sites for each other to form multimers. They can also bind to cell surface receptors such as integrins, as well as to collagens, heparin, and fibrin, and act as a bridge to make a network (Theocharis et al. 2016, 2019; Yue 2014). Fibronectin along with fibrinogen and vitronectin is the major component of the provisional matrix present in the intimal space during early stages of atherosclerosis. Fibronectin therefore plays an important role in initiation of the plaque but also later in formation of the fibrous cap to stabilize the plaque (Rohwedder et al. 2012; Finney et al. 2017).
The laminin family comprises 16 heterotrimeric glycoproteins (Theocharis et al. 2016). Each laminin fiber contains one α-, one β-, and one γ-chain in five, four, and three genetic variants, respectively. Laminins are a major component of the basement membrane underlying endothelial cells and surrounding SMCs in the intima. Laminin fibers of the basement membrane bind to each other to make a network to which cells adhere and interact with collagen IV to stabilize the overall structure. Interaction of laminin with collagen is mediated through bridging molecules like the proteoglycan perlecan. Laminin fibers interact with resident cells through multiple cell surface receptors including integrins to regulate cellular differentiation, adhesion, and migration (Theocharis et al. 2016, 2019).
3.2. Proteoglycans: Non-fibrillar Components of the ECM
Proteoglycans are complex macromolecules consisting of a core protein (e.g., versican; the entire proteoglycan molecule is named according to its core protein) to which one or more glycosaminoglycan molecules (GAGs) are covalently attached (Wight 2018) (Fig. 1). GAGs are linear negatively charged polysaccharides of varying length made up of repeating disaccharide units. There are six types of GAGs: chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate (HS), heparin, keratan sulfate (KS), and hyaluronic acid (HA; also called hyaluronan). CS, DS, HS, and KS are linked to the different core proteins through O-glycosidic linkage to make distinct PG families: CSPGs, DSPGs, HSPGs, and KSPGs. Binding of more than one type of GAG to a single core protein creates various PG subtypes. Hyaluronan is the only GAG that is synthesized at the cell membrane and not in the Golgi apparatus, and is only present in a protein-free form. Proteoglycans are abundant in the ECM and are also present on the cell surface and intracellularly. They interact with growth factors, cytokines, cell surface receptors, and other ECM molecules, through which they participate in multiple cellular functions including signaling, proliferation, migration, adhesion, differentiation, and apoptosis (Wight 2018; Theocharis et al. 2016; Yue 2014). Almost all cell types in the vessel wall can synthesize PGs, with SMCs being the major contributor. Proteoglycan content of the normal vessel wall is low, only 4% of total ECM, but it increases dramatically up to 50% in early atherosclerosis and 20% in late lesions (Wight 2018). There are more than 20 different PGs in blood vessels. As discussed below, the negative charge of PGs plays a critical role in the binding and retention of positively charged lipoproteins in the artery intima, a pivotal step in atherosclerotic lesion initiation. Key features of PGs involved in atherosclerotic lesion development are discussed here and summarized in Table 1.
Table 1
Summary of the various proteoglycans found in the arterial wall, their respective GAG chains, location, and their key role in atherogenesis and vascular homeostasis
3.2.1. Versican
Versican is a large aggregating CSPG (Wight 2018) that exists in at least four different isoforms, created by alternative splicing of mRNA from a single gene (V0–V3). These variants differ by the length of their core proteins and the number of GAGs attached. Versican forms complexes with hyaluronan, a long-chain GAG, to make high molecular weight aggregates. These aggregates provide a swelling pressure that resists tissue shrinkage and also a viscoelastic pericellular matrix that allows SMCs to change their shape to prepare them for proliferation and migration (Wight and Merrilees 2004). Immunostaining of the ECM of human coronary arteries showed the presence of versican in all layers of the artery wall but increased in areas of atherosclerotic arteries with high SMC content such as diffuse intimal thickening (DIT), fibrous cap, and the edge of the lipid core in advanced lesions (Gutierrez et al. 1997; Kolodgie et al. 2002). SMCs are the main source of versican, and its expression is upregulated by factors including platelet-derived growth factor (PDGF) and transforming growth factor-β1 (TGF-β1). Both PDGF and TGF-β1 increase the number of versican transcripts and elongation of CS chains in cultured SMCs (Schonherr et al. 1991).
3.2.2. Biglycan
The small leucine-rich proteoglycan (SLRP) family includes small core proteins (biglycan, decorin) containing a number of leucine-rich repeats (LRRs) that can have CS, DS, or KS side chains (Wight 2018). Biglycan, together with decorin, belongs to the same class of SLRPs due to their homologous genes and proteins and the presence of ten LRRs sandwiched between cysteine-rich regions (Gutierrez et al. 1997). The N-terminal domain of biglycan typically contains two CS/DS side chains (Iozzo 1999) or two DSPG chains (O’Brien et al. 1998; Gutierrez et al. 1997). Biglycan is the primary proteoglycan found in the intima of normal and atherosclerotic human arteries (Little et al. 2008) and is enriched in areas such as DIT (discussed further in Sect. 4), the plaque core, and fibrous cap (Gutierrez et al. 1997). The presence of growth factors such as PDGF and TGF-β1 transforms biglycan into “hyperelongated biglycan,” which has more sulfate incorporated, conveying higher negative charge and longer GAG chains (Schonherr et al. 1993), and therefore increased binding capacity to low density lipoprotein (LDL) and other apolipoprotein B (apoB)-containing lipoproteins (Little et al. 2002). Biglycan concentrated in the deepest part of DIT colocalizes strongly with apoB in early and advancing fatty streaks and plays a critical role in mediating lipoprotein retention in the artery wall (outlined further in Sect. 4 below).
3.2.3. Decorin
Decorin is another member of the SLRP family (Iozzo 1999). Decorin’s N-terminus has a single CS/DS chain (Wight 2018). Decorin is expressed in the adventitia of normal and atherosclerotic arteries (Gutierrez et al. 1997). Decorin is also found, although to a somewhat lesser extent than biglycan, in the deep (outer) intima of coronary arteries in early stages of atherosclerosis, suggesting it also has a role in lipoprotein retention. It is also often found in association with collagen fibers in atherosclerotic plaque and may have a role in plaque stability by increasing collagen fibrillogenesis (Wight 2018).
3.2.4. Perlecan
Perlecan, the most abundant HSPG in the mouse artery wall (Nakashima et al. 2008), is located in the basement membrane between the endothelial cells and intimal SMCs (Tran-Lundmark et al. 2008). Perlecan functions as a permeability barrier but also facilitates lipoprotein retention (Tran-Lundmark et al. 2008). Mice that were deficient in both HS-perlecan and apoE were shown to have higher flux but lower arterial retention of lipoproteins compared to apoE-single null mice (Tran-Lundmark et al. 2008). Human atherosclerotic lesions have low expression of perlecan, and the role of perlecan in development of human atherosclerosis is not clear.
4. Interactions Between SMCs and the ECM
In addition to SMCs being the primary source of proteoglycans and other ECM components in arteries, the composition and organization of ECM itself plays an important role in the regulation of SMC phenotype, i.e., the interaction between SMCs and ECM is bidirectional. Integrins, transmembrane receptors linking the intracellular SMC cytoskeleton to the ECM, play a critical role in this bidirectional signaling across the plasma membrane and regulation of SMC phenotype by ECM components (Finney et al. 2017; Adiguzel et al. 2009).
Fibrillar collagen I, a major component of ECM in the medial layer, induces a contractile and quiescent phenotype in human SMCs grown in culture, whereas monomeric (non-fibrillar) collagen type I, present in atherosclerotic plaque intima, enhances SMC proliferation, a characteristic feature of synthetic phenotype (Ichii et al. 2001; Koohestani et al. 2013; Yeh et al. 2012). This phenotypic shift was accompanied by alteration of gene expression in cultured human SMCs induced by polymerized compared to monomeric collagens (Ichii et al. 2001). Fibrillar collagen type I has also been shown to inhibit PDGF-induced proliferation and migration of SMCs (Raines et al. 2000). Collagen type IV and laminin, components of the basement membrane that surrounds arterial SMCs in the subendothelial space, induce a differentiated contractile phenotype in vascular SMCs grown in culture and diminish the synthetic phenotype induced by PDGF (Thyberg and Hultgardh-Nilsson 1994). In the same manner, integrins α1β1 and α7β1, which bind to collagen IV and laminin, respectively, are highly expressed in quiescent contractile SMCs and serve to maintain the spindle morphology and contractile phenotype of SMCs (Finney et al. 2017). Fibronectin, a major component of provisional matrix in the intima, induces both contractile and synthetic phenotype in vascular SMCs depending on the nature of integrins expressed by the local SMCs (Finney et al. 2017). Altogether various combinations of integrins expressed by SMCs and a variety of ECM components result in a complex SMC-ECM interaction that affects both SMC and ECM properties.
Smooth muscle cells also interact with non-fibrillar PG components of the ECM. Bingley et al. found that HSPGs extracted from rabbit aortae inhibit phenotypic change of the cultured SMCs and maintain them in a quiescent state (Bingley et al. 1998). Interestingly, they found that when applied in a periadventitial gel following injury of rabbit carotid artery, HSPGs inhibit neointima formation (Bingley et al. 1998). Different subtypes of versican have been shown to alter SMC phenotype differently (Wight et al. 2014). Hyaluronan, present in both early and advanced lesions (Krolikoski et al. 2019), enhances proliferation and migration of SMCs (Evanko et al. 1999) and when overexpressed in SMCs of apoE-deficient mice accelerates the progression of atherosclerosis (Chai et al. 2005).
5. SMC, ECM, and Lipoprotein Interactions in Diffuse Intimal Thickening (DIT)
Diffuse intimal thickening (DIT) is a layer of thickened intima composed of SMCs, elastin, and PGs. DIT is initiated in utero and is present in all humans in atherosclerosis-prone arteries such as the coronary arteries and abdominal aorta by the age of 2 years (Nakashima et al. 2008). DIT represents a pre-atherosclerotic stage that provides the “soil” for the initial deposition and retention of lipoproteins and subsequent stages of atherosclerosis (Nakashima et al. 2008; Dubland and Francis 2016). Versican and biglycan are the most abundant proteoglycans in this DIT layer of atherosclerosis-prone arteries, while the thin intimal layer of atherosclerosis-resistant arteries, such as the internal thoracic artery, is enriched in decorin (Merrilees et al. 2001).
Using immunohistochemical techniques, Nakashima et al. showed that biglycan is localized in the deeper region of DIT in human coronary arteries (Nakashima et al. 2007). Lipoproteins that diffuse in from the plasma are trapped and deposited in the deep intima in the early stages of atherosclerosis due to a charge-charge interaction between this proteoglycan and apolipoprotein B of the lipoproteins. In an in vitro model, overexpression of biglycan in rat vascular SMCs increased lipoprotein binding to matrix formed by the cultured cells, while expression of a GAG-deficient mutated biglycan blocked this binding and accumulation (O’Brien et al. 2004). This observation and colocalization of biglycan with apoB- and apoE-containing lipoproteins in both early and advanced human (Nakashima et al. 2007; O’Brien et al. 2004) and murine lesions (Kunjathoor et al. 2002) indicate the presence of biglycan is crucial for the initial and ongoing deposition of lipids in the artery wall.
Unlike biglycan, immunohistochemical studies show versican is predominantly localized in the superficial part of the thickened intima of coronary arteries (Merrilees et al. 2001). Although in vitro studies have shown versican is capable of binding to LDL and has a larger number of binding sites for LDL compared to biglycan (Camejo et al. 1988), it has not been detected in lipoprotein-rich areas of atherosclerotic lesions nor colocalized with apoE or apoB epitopes in either human or mouse atherosclerotic lesions (Wight and Merrilees 2004; Kunjathoor et al. 2002).
Decorin, like biglycan, is distributed in the deep intima although at lower levels and colocalizes with lipids with less intensity compared to biglycan (Nakashima et al. 2007; Otsuka et al. 2015). In vitro studies have shown decorin links LDL with collagen type I (Pentikainen et al. 1997), and in vivo studies have demonstrated colocalization of decorin with collagens and apoB in atherosclerotic lesions (Nakashima et al. 2007; Riessen et al. 1994). Moreover, overexpression of decorin in rat SMCs after balloon injury of the carotid arteries resulted in compact collagen-rich lesions with reduced intimal thickness (Fischer et al. 2000). These findings suggest that decorin has a primary role in collagen organization in atherogenesis and a secondary role in lipid retention.
Perlecan is a heparan sulfate proteoglycan and the most abundant PG in the mouse artery wall, present in basement membranes in the intima and media (Nakashima et al. 2008). Human atherosclerotic lesions express only low amounts of perlecan (Tran et al. 2007). Like biglycan in humans, perlecan is involved in lipid retention in mouse atherosclerosis (Kunjathoor et al. 2002). Vikramadithyan et al. have shown that heterozygous deficiency of perlecan in chow-fed apoE-deficient mice results in more than 70% reduction in early-stage atherosclerosis lesion size. However, these authors did not find any difference in lesion size in advanced atherosclerosis (Vikramadithyan et al. 2004). Tran-Lundmark et al. also showed more than 50% reduction in lesion size in HS-deficient perlecan apoE−/− mice compared to control apoE−/− mice (Tran-Lundmark et al. 2008).
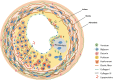
The expression of key artery wall ECM components and their roles including lipoprotein binding and fibrous cap formation are indicated in Fig. 2.
6. SMC-ECM Interaction in Lipid Retention
The ability of SMC-generated PGs in the human arterial intima to bind atherogenic apoB-containing lipoproteins mediates what is considered to be the critical and rate-limiting step in atherogenesis, from which subsequent steps of atheroma development ensue. This is the so-called “response-to-retention” hypothesis of atherosclerosis (Williams and Tabas 1995). The interaction between atherogenic lipoproteins and PGs involves an ionic or electrostatic interaction between positively charged amino acids of aopB on lipoproteins and negatively charged sulfate groups on the PGs generated by SMCs (Camejo et al. 1998; Boren et al. 1998). The role of deposition of lipoproteins in the intimal space and specific interactions of SMC-ECM molecules and apolipoproteins that mediate retention of lipoproteins are outlined below.
6.1. Deposition of Lipoproteins in the Artery Wall
Movement of lipoproteins from the plasma to the intimal space involves transport across the vascular endothelium via transcytosis, a process that is independent of LDL receptor function (Simionescu and Simionescu 1991). In contrast to endocytosis and lysosomal degradation of lipoproteins mediated by the LDL receptor, endothelial transcytosis delivers lipoproteins including LDL and other apoB-containing lipoproteins including very-low-density lipoproteins (VLDL), VLDL remnants (intermediate-density lipoproteins), chylomicron remnants (Yang et al. 2018), and presumably lipoprotein (a) via endocytic uptake, intracellular transport, and exocytosis into the ECM of the artery wall (Simionescu and Simionescu 1991; Fung et al. 2018). Although transcytosis is the rate-limiting step in the movement of LDL and other lipoproteins into the subendothelial layer, it does not equate to the amount of LDL retained in the intimal space (Vasile et al. 1983). This was supported by the in vivo observation that the rate of LDL entry into normal rabbit aorta exceeded the rate of LDL accumulation (Carew et al. 1984). The retention of LDL occurs more extensively in lesion-susceptible areas such as the aortic arch compared with lesion-resistant arteries such as the descending thoracic aorta (Schwenke and Carew 1988). The difference in LDL accumulation between atherosclerosis-susceptible and atherosclerosis-resistant sites is also apparently independent of LDL permeability, as an in vivo comparison of healthy and atherosclerosis-susceptible arterial segments in rabbits showed they have similar LDL permeability (Schwenke and Carew 1989). These findings indicate that retention of apoB-containing lipoproteins, rather than the rate of transcytosis or endothelial permeability, is the rate-limiting factor in the amount of deposition of these lipoproteins in the artery wall. These studies formed the basis for Williams and Tabas’ “response-to-retention” hypothesis, proposed in 1995, that highlighted the retention of apoB-containing lipoproteins as the major driver of atherogenesis (Williams and Tabas 1995).
6.2. PG-Binding Sites in Lipoproteins
Lipoproteins delivered to the intima interact with the ECM via binding sites in their apolipoproteins (Usman et al. 2015). ApoB has been widely studied as the binding ligand of GAGs (Camejo et al. 1990; Srinivasan et al. 1988; Anber et al. 1997). Inhibition of the LDL-PG interaction by the presence of an anti-apoB antibody supports the observation that apoB is the binding site in lipoproteins (Sambandam et al. 1991). Modification of positively charged arginine and lysine residues in LDL abolished complex formation with CSPG, a GAG found in versican, biglycan, and decorin, indicating the importance of these amino acids in the apoB-PG interaction (Anber et al. 1997). The extent of apoB interaction with PGs is dependent on the degree of exposure of these positively charged residues in apoB (Camejo et al. 1990). The presence of hydropathic, hydroxyl-containing serine and threonine residues around positively charged lysine and arginine residues increases the hydrophilicity of these segments and enhances their ability to interact with PGs (Camejo et al. 1990). In addition, the hydrolysis of surface phospholipids on LDL by secretory phospholipase A2 results in smaller and denser LDL particles that have greater exposure of PG-binding apoB segments, accounting for the increased affinity of small dense LDL for biglycan and decorin when compared to native LDL (Sartipy et al. 1999).
Site B of apoB100, residues 3,359–3,369, has been proposed to be the principal site of binding with PGs (Boren et al. 1998). A single-point mutation in this region of apoB100 severely affects proteoglycan interactions, without affecting LDL receptor binding (Boren et al. 1998). Furthermore, Cardin and Weintraub identified the GAG-binding sequence of apoB100 to be either –X-B-B-X-B-X or –X-B-B-B-X-X-B-X where X is a hydropathic amino acid and B is either lysine or arginine (Cardin and Weintraub 1989). Chylomicron remnants, which are also atherogenic and deposit in the artery wall, contain the N-terminal 48% of apoB100 (apoB48) and therefore lack the GAG-binding Site B. GAG binding of apoB48 has been shown to be mediated by a lysine-rich cluster of amino acids in N-terminal residues 84–94, termed Site B-1b, exposed in this carboxyl-truncated form of apoB.
In addition to apoB, apoE and apoAI also colocalize with biglycan in human atherosclerotic plaques (O’Brien et al. 1998), but only apoE was found to have proteoglycan-binding domains (O’Brien et al. 1998; Olin et al. 2001). High-density lipoproteins (HDL) binding to GAGs appear to be limited to apoE-containing but not apoE-free HDL (Olin et al. 2001).
6.3. The Variability of Interactions Between Lipoproteins and Proteoglycans
Iverius initially demonstrated the interaction between GAGs and human plasma lipoproteins by equilibrating lipoproteins with a GAG-linked agarose gel, resulting in the interaction of LDL and VLDL, but not HDL, acetylated VLDL, or acetylated LDL, with heparin, DS, HS, and chondroitin-4-sulfate (Iverius 1972). Heparin and CSPGs have the same binding site in apoB (Sambandam et al. 1991). The position of sulfation of GAGs was not found to be important; however increased sulfation, which confers higher negative charge, correlates with increased lipoprotein binding (Sambandam et al. 1991), confirming the electrostatic nature of the interaction between negatively charged GAGs and positively charged apoB-binding sites. The strength of the interaction between VLDL and LDL with different GAGs was heparin > DS > HS ~ C4-S (Iverius 1972). Contrary to this observation, Vijayagopal et al. observed greater affinity between LDL and CS than HSPG (Vijayagopal et al. 1983). This was supported by another publication that found CS/DSPG from bovine aorta was more potent than HS-PG in inducing complex formation with LDL and that the strongest complex formed is between LDL and CS/DSPG (Radhakrishnamurthy et al. 1990). Parallel with Iverius’ observation, DSPG was determined to be more efficient in binding LDL than CSPG (Sambandam et al. 1991). The localization of lipoproteins in atherogenic plaques with biglycan and decorin (which may have DS side chains) as opposed to versican (a CSPG) aligns with this observation.
6.4. Effects of Binding and Retention of Lipoproteins
The retention of lipoproteins in the intima and their subsequent modification including oxidation and aggregation initiates a vicious cycle of further SMC proliferation and synthesis of elongated and more lipoprotein-attracting PGs that increase lipoprotein entrapment (Camejo et al. 1993; Chang et al. 2000). Macrophages infiltrating into the developing plaque secrete lipoprotein lipase, which can also act as a bridge between lipoproteins and PGs and thereby enhance lipoprotein retention. Macrophages also secrete inflammatory mediators that lead to further cell recruitment and plaque development (reviewed in Usman et al. 2015). The trapping of lipoproteins through interactions with PGs in the intima makes them a substrate for modifications including oxidation and aggregation. This converts the lipoproteins into ligands for scavenger receptors on macrophages and SMCs, leading to foam cell formation in both these cell types (Allahverdian et al. 2014; Wang et al. 2019). In this way, consistent with the response-to-retention theory of atherogenesis, SMC-dependent PG synthesis is the critical driver of subsequent stages of atherosclerotic lesion development.
7. Role of ECM Proteoglycans in Early Stages of Atherosclerosis Development in Animal Models
A key question, raised by the apparent pivotal importance of SMCs in producing the lipoprotein-binding PGs that drive lipoprotein retention and atherogenesis in humans, is how important this is in mice, the primary models used to study the determinants of atherosclerosis in man. Although small animals do not develop a pre-atherosclerotic SMC DIT layer like humans (Nakashima et al. 2008), several studies provide evidence that the initial binding of apoB-containing lipoproteins to subendothelial PGs in the artery wall is also a key step in atherogenesis in animal models of atherosclerosis. Perlecan and biglycan, but not versican, a major PG detected in human plaques, appear early in developing lesions in apoE−/− and LDLR−/− mice underneath the endothelium and in the absence of intimal thickening (Kunjathoor et al. 2002). The presence of these PGs around areas of extracellular lipid deposition suggests that they also contribute to lipoprotein retention in murine models of atherosclerosis. This supports the theory of PG-dependent lipoprotein retention, although these PGs are likely secreted by endothelial cells or activated macrophages rather than SMCs in the first stage of mouse atherosclerosis (Kinsella et al. 1997; Nugent et al. 2000; Chang et al. 2012). In intermediate and advanced lesions, PGs and lipoproteins are localized in SMC-rich areas of the mouse intima (Kunjathoor et al. 2002). In another study, Thompson et al. found that overexpression of biglycan by SMCs in LDLR−/− mice increases lipid retention and atherosclerosis development (Thompson et al. 2014). Moreover, apoE−/− mice heterozygous for deletion of perlecan have less atherosclerotic lesion area at early stages compared to apoE−/− mice with no deletion. This might be attributed partly to decreased lipoprotein retention in the perlecan+/− mice (Vikramadithyan et al. 2004). In another study, Skalen et al. used transgenic mice expressing recombinant LDL with proteoglycan binding site-defective apoB and showed those mice develop significantly less atherosclerosis compared to mice expressing wild-type control LDL (Skalen et al. 2002). These studies demonstrate a key role for PGs for initial subendothelial lipid retention at early stages of mouse atherosclerosis and a likely role for SMC-derived PGs in later stages of murine models of this disease.
8. The Role of the ECM-SMC Interaction in Plaque Stability and Rupture
Stable plaque is defined by the presence of a thick fibrous cap lying between the necrotic core and luminal surface of the plaque. It consists of SMCs embedded in a collagen-proteoglycan matrix with varying degrees of macrophage and lymphocyte infiltration and has a critical role in maintaining integrity of the plaque. Unstable plaques are prone to erosion and rupture that leads to platelet adhesion and luminal thrombus formation, the most common cause of acute coronary syndromes and sudden cardiac death (Allahverdian et al. 2018). Plaque rupture involves breakage of a large lipid/necrotic core with large luminal thrombus, thin fibrous cap, and a dense infiltration of inflammatory cells (Kolodgie et al. 2004).
Extracellular matrix components of atherosclerotic lesions have a key role in maintaining stability of plaques and accumulate in topographically distinct patterns in different plaque types. Kolodgie et al. found that the fibrous caps of stable lesions show increased amounts of versican, biglycan, and collagen type I staining and significantly less decorin and hyaluronan (Kolodgie et al. 2002). With SMCs being the main producer of collagen and PGs in the fibrous matrix (Pietila and Nikkari 1983), it is not surprising that the number of SMCs in fibrous caps is directly correlated with plaque stability (Allahverdian et al. 2018). One study found collagen VIII deficiency in apoE−/− mice resulted in thinning of the fibrous cap, which suggests that this collagen plays an important role in protecting the plaque from rupture (Lopes et al. 2013).
Eroded plaques show an intense staining for hyaluronan, versican, and collagen type III at the plaque/thrombus interface and weak staining for biglycan and decorin. Cell surface receptor for hyaluronan, CD44, has been shown to be expressed by a subset of SMCs at the plaque/thrombus interface in erosions (Kolodgie et al. 2002) and mediates adhesion of platelets to hyaluronan (Day 1999). Exposure of versican-hyaluronan to flowing blood at the de-endothelialized surface of the eroded plaque promotes platelet attachment and may play an important role in the development of thrombosis via a CD44-dependent mechanism (Koshiishi et al. 1994).
Fibrous caps owe their strength and resistance to rupture to their collagen fiber content and therefore the content and synthetic capacity of collagen-producing SMCs (Doran et al. 2008; Libby 2008). Thinned fibrous cap areas contain very few SMCs with little to absent staining for PGs or hyaluronan and scattered strands of collagen type I (Kolodgie et al. 2002). Factors that alter the abundance or function of SMCs thereby alter ECM production by SMCs and plaque stability.
8.1. Factors Involved in Plaque Instability
Pro-inflammatory cytokines have been shown to reduce the number of intimal SMCs, thereby compromising ECM production (Geng et al. 1997; Kockx and Knaapen 2000). Various cytokines and growth factors, normally present in atherosclerotic plaques, also either stimulate or suppress collagen synthesis by vascular SMCs (Amento et al. 1991). Moreover, reduction of SMC density in plaque and the fibrous cap as a result of apoptosis results in diminished ECM production and may lead to plaque instability. von der Thusen et al. found that induction of vascular SMC apoptosis by overexpression of p53 in the fibrous cap of carotid plaques of apoE−/− mice results in reduction of cell density, cap thinning, and plaque destabilization (von der Thusen et al. 2002). Low endothelial shear stress (ESS) has been shown to induce SMC apoptosis (Qi et al. 2008) and therefore indirectly affect collagen content of atherosclerotic plaques. Frontini et al. found that lipid loading of human vascular SMCs impairs their ability to assemble fibrillar collagen and fibronectin (Frontini et al. 2009), which may contribute to reducing plaque stability. Others found that enhancement of cholesterol efflux preserves the assembly of fibrillar collagen and fibronectin by SMCs (Beyea et al. 2012).
An imbalance between synthesis and degradation of ECM leads to thinning of the fibrous cap and plaque rupture. Members of the matrix metalloproteinases (MMP) and tissue inhibitor of metalloproteinase (TIMP) families can alter the quantity and composition of ECM and affect plaque stability (Newby et al. 2009). Although macrophages are the main source of MMPs in atherosclerosis (Shah et al. 1995), secretion of different MMPs by SMCs also plays an important role in cap thinning and plaque destabilization. Members of the MMP family with collagenase activity, including MMP-1, MMP-8, MMP-13, and the activator of MMP collagenases, MMP-14, weaken the plaque by degrading collagen fibers (Libby 2008; Deguchi et al. 2005). The concentration, production, and expression of MMP-9 (Loftus et al. 2000), MMP-1, and MMP-13 (Sukhova et al. 1999) are significantly higher in unstable compare to stable carotid plaques.
Low endothelial shear stress (ESS) has also been shown to play an important role in plaque instability. In an ex vivo model of porcine carotid atherosclerosis, low ESS resulted in an increase in MMP expression (Gambillara et al. 2005). Low ESS is also associated with a diminished SMC and collagen content, a high MMP expression and activity, and a marked thinning of the fibrous cap in mouse carotid and porcine coronary plaques (Cheng et al. 2006; Koskinas et al. 2013).
9. Therapeutic Targets and Interventions
The critical role of SMC-dependent PG synthesis and lipoprotein retention in subsequent atherosclerosis development makes the PG-lipoprotein interaction a potentially profound and novel point of therapeutic intervention to prevent ischemic vascular disease. Vazquez and colleagues introduced the chP3R99 monoclonal antibody (mAb) against ECM GAGs as a potential therapeutic for atherosclerosis in 2012 (Soto et al. 2012). The mAb binds to sulfated GAGs such as heparin, HS, and DS and had the strongest affinity for CS in aorta (Soto et al. 2012). It prevented 70% of LDL-CS complex formation and abolished 80% of LDL oxidation in vitro (Soto et al. 2012). The mAb also demonstrated the capacity to (1) prevent foam cell formation by the inhibition of LDL retention and oxidation in the arteries of rats; (2) decrease the intima-media ratio, a measure of DIT formation; and (3) decrease macrophage infiltration in the aorta of New Zealand white rabbits (Soto et al. 2012). Thereafter, they delivered the mAb chP3R99 via subcutaneous injections to apoE−/− mice fed a high-fat and high-cholesterol diet and determined that the vaccination stimulated an immune response against various sulfated GAGs via the induction of a secondary anti-idiotype response (Ab2) that mirrors GAG antigenic determinants (Brito et al. 2012). The Ab2 gave rise to synthesis of anti-idiotype antibodies (Ab3) that have a specificity homologous to that of Ab1 (Brito et al. 2012). Further research using chP3R99 in the same mouse model established the chP3R99-LALA variant restricted the progression of atherosclerosis in apoE−/− mice by preventing initial CS-LDL complex formation (Delgado-Roche et al. 2015). Both the efficiency of the chP3R99-LALA mAb in initiating the anti-idiotype cascade and anti-atherogenic benefits were dose-dependent in apoE−/− mice but were independent of the sex and age of the mice (Sarduy et al. 2017). The mAb was also shown to decrease inflammation and halt lesion development of advanced stages of atherosclerosis in male apoE−/− mice (Brito et al. 2017). These findings strongly confirm the relevance of the “response-to-retention” hypothesis of atherogenesis and provide the exciting possibility of developing an immunization that could block the PG-lipoprotein interaction lifelong as a novel means of preventing atherosclerosis.
10. Conclusion
Smooth muscle cells (SMCs) are the most abundant cell type in the healthy human artery wall as well as the pre-atherosclerotic and atherosclerotic intimal layer. The ECM components secreted by SMCs are critical factors in maintaining normal arterial architecture and stability but also in predicting propensity to atherosclerosis. In particular, the secretion of PGs by intimal SMCs promotes the initial and ongoing binding and retention of atherogenic lipoproteins in the artery wall. This retention of lipoproteins is necessary for further development of atherosclerosis, apparently in mice as well as in humans, making the proteoglycan-lipoprotein interaction a novel and exciting potential point of intervention for the future treatment and prevention of atherosclerosis.
Acknowledgments
This work is supported by a Heart and Stroke Foundation Grant-In-Aid to GAF.
References
- Adiguzel E, Ahmad PJ, Franco C, Bendeck MP (2009) Collagens in the progression and complications of atherosclerosis. Vasc Med 14(1):73–89. [PubMed: 19144782]
- Albarran-Juarez J, Kaur H, Grimm M, Offermanns S, Wettschureck N (2016) Lineage tracing of cells involved in atherosclerosis. Atherosclerosis 251:445–453. [PubMed: 27320174]
- Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA (2014) Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 129(15):1551–1559. [PubMed: 24481950]
- Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML (2018) Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res 114(4):540–550. [PMC free article: PMC5852505] [PubMed: 29385543]
- Amento EP, Ehsani N, Palmer H, Libby P (1991) Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb 11(5):1223–1230. [PubMed: 1911708]
- Anber V, Millar JS, McConnell M, Shepherd J, Packard CJ (1997) Interaction of very-low-density, intermediate-density, and low-density lipoproteins with human arterial wall proteoglycans. Arterioscler Thromb Vasc Biol 17(11):2507–2514. [PubMed: 9409221]
- Beyea MM, Reaume S, Sawyez CG, Edwards JY, O’Neil C, Hegele RA et al (2012) The oxysterol 24(s),25-epoxycholesterol attenuates human smooth muscle-derived foam cell formation via reduced low-density lipoprotein uptake and enhanced cholesterol efflux. J Am Heart Assoc 1(3):e000810. [PMC free article: PMC3487330] [PubMed: 23130136]
- Bingley JA, Hayward IP, Campbell JH, Campbell GR (1998) Arterial heparan sulfate proteoglycans inhibit vascular smooth muscle cell proliferation and phenotype change in vitro and neointimal formation in vivo. J Vasc Surg 28(2):308–318. [PubMed: 9719326]
- Boren J, Olin K, Lee I, Chait A, Wight TN, Innerarity TL (1998) Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest 101(12):2658–2664. [PMC free article: PMC508856] [PubMed: 9637699]
- Brito V, Mellal K, Portelance SG, Perez A, Soto Y (2012) deBlois D, et al. induction of anti-anti-idiotype antibodies against sulfated glycosaminoglycans reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 32(12):2847–2854. [PubMed: 23087361]
- Brito V, Mellal K, Zoccal KF, Soto Y, Menard L, Sarduy R et al (2017) Atheroregressive potential of the treatment with a chimeric monoclonal antibody against sulfated glycosaminoglycans on pre-existing lesions in apolipoprotein E-deficient mice. Front Pharmacol 8:782. [PMC free article: PMC5672559] [PubMed: 29163168]
- Camejo G, Olofsson SO, Lopez F, Carlsson P, Bondjers G (1988) Identification of Apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycans. Arteriosclerosis 8(4):368–377. [PubMed: 3395272]
- Camejo G, Rosengren B, Olson U, Lopez F, Olofson SO, Westerlund C et al (1990) Molecular basis of the association of arterial proteoglycans with low density lipoproteins: its effect on the structure of the lipoprotein particle. Eur Heart J 11(Suppl E):164–173. [PubMed: 2226525]
- Camejo G, Fager G, Rosengren B, Hurt-Camejo E, Bondjers G (1993) Binding of low density lipoproteins by proteoglycans synthesized by proliferating and quiescent human arterial smooth muscle cells. J Biol Chem 268(19):14131–14137. [PubMed: 8314779]
- Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G (1998) Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis 139(2):205–222. [PubMed: 9712326]
- Cardin AD, Weintraub HJ (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9(1):21–32. [PubMed: 2463827]
- Carew TE, Pittman RC, Marchand ER, Steinberg D (1984) Measurement in vivo of irreversible degradation of low density lipoprotein in the rabbit aorta. Predominance of intimal degradation. Arteriosclerosis 4(3):214–224. [PubMed: 6324731]
- Chai S, Chai Q, Danielsen CC, Hjorth P, Nyengaard JR, Ledet T et al (2005) Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res 96(5):583–591. [PubMed: 15705963]
- Chang MY, Potter-Perigo S, Tsoi C, Chait A, Wight TN (2000) Oxidized low density lipoproteins regulate synthesis of monkey aortic smooth muscle cell proteoglycans that have enhanced native low density lipoprotein binding properties. J Biol Chem 275(7):4766–4773. [PubMed: 10671509]
- Chang MY, Chan CK, Braun KR, Green PS, O’Brien KD, Chait A et al (2012) Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J Biol Chem 287(17):14122–14135. [PMC free article: PMC3340194] [PubMed: 22351750]
- Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD et al (2016) Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res 119(12):1313–1323. [PMC free article: PMC5149073] [PubMed: 27682618]
- Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ et al (2006) Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113(23):2744–2753. [PubMed: 16754802]
- Day AJ (1999) The structure and regulation of hyaluronan-binding proteins. Biochem Soc Trans 27(2):115–121. [PubMed: 10093718]
- Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM et al (2005) Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation 112(17):2708–2715. [PubMed: 16230484]
- Delgado-Roche L, Brito V, Acosta E, Perez A, Fernandez JR, Hernandez-Matos Y et al (2015) Arresting progressive atherosclerosis by immunization with an anti-glycosaminoglycan monoclonal antibody in apolipoprotein E-deficient mice. Free Radic Biol Med 89:557–566. [PubMed: 26454078]
- Doran AC, Meller N, McNamara CA (2008) Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol 28(5):812–819. [PMC free article: PMC2734458] [PubMed: 18276911]
- Dubland JA, Francis GA (2016) So much cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol 27(2):155–161. [PubMed: 26836481]
- Evanko SP, Angello JC, Wight TN (1999) Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 19(4):1004–1013. [PubMed: 10195929]
- Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M et al (2014) Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 115(7):662–667. [PubMed: 25070003]
- Finney AC, Stokes KY, Pattillo CB, Orr AW (2017) Integrin signaling in atherosclerosis. Cell Mol Life Sci 74(12):2263–2282. [PMC free article: PMC5427000] [PubMed: 28246700]
- Fischer JW, Kinsella MG, Clowes MM, Lara S, Clowes AW, Wight TN (2000) Local expression of bovine decorin by cell-mediated gene transfer reduces neointimal formation after balloon injury in rats. Circ Res 86(6):676–683. [PubMed: 10747004]
- Frantz C, Stewart KM, Weaver VM (2010) The extracellular matrix at a glance. J Cell Sci 123(Pt 24):4195–4200. [PMC free article: PMC2995612] [PubMed: 21123617]
- Frontini MJ, O’Neil C, Sawyez C, Chan BM, Huff MW, Pickering JG (2009) Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res 104(7):832–841. [PubMed: 19229059]
- Fung KYY, Fairn GD, Lee WL (2018) Transcellular vesicular transport in epithelial and endothelial cells: challenges and opportunities. Traffic 19(1):5–18. [PubMed: 28985008]
- Gambillara V, Montorzi G, Haziza-Pigeon C, Stergiopulos N, Silacci P (2005) Arterial wall response to ex vivo exposure to oscillatory shear stress. J Vasc Res 42(6):535–544. [PubMed: 16179795]
- Geng YJ, Henderson LE, Levesque EB, Muszynski M, Libby P (1997) Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 17(10):2200–2208. [PubMed: 9351390]
- Gutierrez P, O’Brien KD, Ferguson M, Nikkari ST, Alpers CE, Wight TN (1997) Differences in the distribution of versican, decorin, and biglycan in atherosclerotic human coronary arteries. Cardiovasc Pathol 6(5):271–278. [PubMed: 25989722]
- Ichii T, Koyama H, Tanaka S, Kim S, Shioi A, Okuno Y et al (2001) Fibrillar collagen specifically regulates human vascular smooth muscle cell genes involved in cellular responses and the pericellular matrix environment. Circ Res 88(5):460–467. [PubMed: 11249868]
- Iozzo RV (1999) The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem 274(27):18843–18846. [PubMed: 10383378]
- Iverius PH (1972) The interaction between human plasma lipoproteins and connective tissue glycosaminoglycans. J Biol Chem 247(8):2607–2613. [PubMed: 4336380]
- Kinsella MG, Tsoi CK, Jarvelainen HT, Wight TN (1997) Selective expression and processing of biglycan during migration of bovine aortic endothelial cells. The role of endogenous basic fibroblast growth factor. J Biol Chem 272(1):318–325. [PubMed: 8995264]
- Kockx MM, Knaapen MW (2000) The role of apoptosis in vascular disease. J Pathol 190(3):267–280. [PubMed: 10685061]
- Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN et al (2002) Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler Thromb Vasc Biol 22(10):1642–1648. [PubMed: 12377743]
- Kolodgie FD, Burke AP, Wight TN, Virmani R (2004) The accumulation of specific types of proteoglycans in eroded plaques: a role in coronary thrombosis in the absence of rupture. Curr Opin Lipidol 15(5):575–582. [PubMed: 15361794]
- Koohestani F, Braundmeier AG, Mahdian A, Seo J, Bi J, Nowak RA (2013) Extracellular matrix collagen alters cell proliferation and cell cycle progression of human uterine leiomyoma smooth muscle cells. PLoS One 8(9):e75844. [PMC free article: PMC3770620] [PubMed: 24040420]
- Koshiishi I, Shizari M, Underhill CB (1994) CD44 can mediate the adhesion of platelets to hyaluronan. Blood 84(2):390–396. [PubMed: 8025267]
- Koskinas KC, Sukhova GK, Baker AB, Papafaklis MI, Chatzizisis YS, Coskun AU et al (2013) Thin-capped atheromata with reduced collagen content in pigs develop in coronary arterial regions exposed to persistently low endothelial shear stress. Arterioscler Thromb Vasc Biol 33(7):1494–1504. [PMC free article: PMC3954496] [PubMed: 23640495]
- Krolikoski M, Monslow J, Pure E et al (2019) Matrix Biol 78-79:201–218. [PMC free article: PMC6249119] [PubMed: 29792915]
- Kunjathoor VV, Chiu DS, O’Brien KD, LeBoeuf RC (2002) Accumulation of biglycan and perlecan, but not versican, in lesions of murine models of atherosclerosis. Arterioscler Thromb Vasc Biol 22(3):462–468. [PubMed: 11884291]
- Libby P (2008) The molecular mechanisms of the thrombotic complications of atherosclerosis. J Intern Med 263(5):517–527. [PMC free article: PMC2562742] [PubMed: 18410595]
- Little PJ, Tannock L, Olin KL, Chait A, Wight TN (2002) Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol 22(1):55–60. [PubMed: 11788461]
- Little PJ, Osman N, O’Brien KD (2008) Hyperelongated biglycan: the surreptitious initiator of atherosclerosis. Curr Opin Lipidol 19(5):448–454. [PubMed: 18769225]
- Loftus IM, Naylor AR, Goodall S, Crowther M, Jones L, Bell PR et al (2000) Increased matrix metalloproteinase-9 activity in unstable carotid plaques. A potential role in acute plaque disruption. Stroke 31(1):40–47. [PubMed: 10625713]
- Lopes J, Adiguzel E, Gu S, Liu SL, Hou G, Heximer S et al (2013) Type VIII collagen mediates vessel wall remodeling after arterial injury and fibrous cap formation in atherosclerosis. Am J Pathol 182(6):2241–2253. [PMC free article: PMC3668035] [PubMed: 23567639]
- Merrilees MJ, Beaumont B, Scott LJ (2001) Comparison of deposits of versican, biglycan and decorin in saphenous vein and internal thoracic, radial and coronary arteries: correlation to patency. Coron Artery Dis 12(1):7–16. [PubMed: 11211169]
- Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K (2007) Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol 27(5):1159–1165. [PubMed: 17303781]
- Nakashima Y, Wight TN, Sueishi K (2008) Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res 79(1):14–23. [PubMed: 18430750]
- Newby AC, George SJ, Ismail Y, Johnson JL, Sala-Newby GB, Thomas AC (2009) Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb Haemost 101(6):1006–1011. [PMC free article: PMC2850555] [PubMed: 19492140]
- Nugent MA, Nugent HM, Iozzo RV, Sanchack K, Edelman ER (2000) Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc Natl Acad Sci U S A 97(12):6722–6727. [PMC free article: PMC18716] [PubMed: 10841569]
- O’Brien KD, Olin KL, Alpers CE, Chiu W, Ferguson M, Hudkins K et al (1998) Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation 98(6):519–527. [PubMed: 9714108]
- O’Brien KD, Lewis K, Fischer JW, Johnson P, Hwang JY, Knopp EA et al (2004) Smooth muscle cell biglycan overexpression results in increased lipoprotein retention on extracellular matrix: implications for the retention of lipoproteins in atherosclerosis. Atherosclerosis 177(1):29–35. [PubMed: 15488862]
- Olin KL, Potter-Perigo S, Barrett PH, Wight TN, Chait A (2001) Biglycan, a vascular proteoglycan, binds differently to HDL2 and HDL3: role of apoE. Arterioscler Thromb Vasc Biol 21(1):129–135. [PubMed: 11145944]
- Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV et al (2015) Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: a pathology study. Atherosclerosis 241(2):772–782. [PMC free article: PMC4510015] [PubMed: 26058741]
- Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84(3):767–801. [PubMed: 15269336]
- Pentikainen MO, Oorni K, Lassila R, Kovanen PT (1997) The proteoglycan decorin links low density lipoproteins with collagen type I. J Biol Chem 272(12):7633–7638. [PubMed: 9065418]
- Perisic Matic L, Rykaczewska U, Razuvaev A, Sabater-Lleal M, Lengquist M, Miller CL et al (2016) Phenotypic modulation of smooth muscle cells in atherosclerosis is associated with downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler Thromb Vasc Biol 36(9):1947–1961. [PMC free article: PMC8991816] [PubMed: 27470516]
- Phinikaridou A, Lacerda S, Lavin B, Andia ME, Smith A, Saha P et al (2018) Tropoelastin: a novel marker for plaque progression and instability. Circ Cardiovasc Imaging 11(8):e007303. [PMC free article: PMC6130848] [PubMed: 30214669]
- Pietila K, Nikkari T (1983) Role of the arterial smooth muscle cell in the pathogenesis of atherosclerosis. Med Biol 61(1):31–44. [PubMed: 6341723]
- Qi YX, Qu MJ, Long DK, Liu B, Yao QP, Chien S et al (2008) Rho-GDP dissociation inhibitor alpha downregulated by low shear stress promotes vascular smooth muscle cell migration and apoptosis: a proteomic analysis. Cardiovasc Res 80(1):114–122. [PubMed: 18540023]
- Radhakrishnamurthy B, Srinivasan SR, Vijayagopal P, Berenson GS (1990) Arterial wall proteoglycans--biological properties related to pathogenesis of atherosclerosis. Eur Heart J 11(Suppl E):148–157. [PubMed: 2226523]
- Raines EW, Koyama H, Carragher NO (2000) The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann N Y Acad Sci 902:39–51; discussion-2. [PubMed: 10865824]
- Rekhter MD (1999) Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res 41(2):376–384. [PubMed: 10341837]
- Riessen R, Isner JM, Blessing E, Loushin C, Nikol S, Wight TN (1994) Regional differences in the distribution of the proteoglycans biglycan and decorin in the extracellular matrix of atherosclerotic and restenotic human coronary arteries. Am J Pathol 144(5):962–974. [PMC free article: PMC1887362] [PubMed: 8178945]
- Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Duner P, Nilsson J et al (2012) Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med 4(7):564–576. [PMC free article: PMC3407945] [PubMed: 22514136]
- Sambandam T, Baker JR, Christner JE, Ekborg SL (1991) Specificity of the low density lipoprotein-glycosaminoglycan interaction. Arterioscler Thromb 11(3):561–568. [PubMed: 2029496]
- Sarduy R, Brito V, Castillo A, Soto Y, Grinan T, Marleau S et al (2017) Dose-dependent induction of an idiotypic cascade by anti-glycosaminoglycan monoclonal antibody in apoE(−/−) mice: association with atheroprotection. Front Immunol 8:232. [PMC free article: PMC5334371] [PubMed: 28316603]
- Sartipy P, Camejo G, Svensson L, Hurt-Camejo E (1999) Phospholipase A(2) modification of low density lipoproteins forms small high density particles with increased affinity for proteoglycans and glycosaminoglycans. J Biol Chem 274(36):25913–25920. [PubMed: 10464335]
- Schonherr E, Jarvelainen HT, Sandell LJ, Wight TN (1991) Effects of platelet-derived growth factor and transforming growth factor-beta 1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J Biol Chem 266(26):17640–17647. [PubMed: 1894644]
- Schonherr E, Jarvelainen HT, Kinsella MG, Sandell LJ, Wight TN (1993) Platelet-derived growth factor and transforming growth factor-beta 1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler Thromb 13(7):1026–1036. [PubMed: 8318504]
- Schwenke DC, Carew TE (1988) Quantification in vivo of increased LDL content and rate of LDL degradation in normal rabbit aorta occurring at sites susceptible to early atherosclerotic lesions. Circ Res 62(4):699–710. [PubMed: 3349573]
- Schwenke DC, Carew TE (1989) Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis 9(6):908–918. [PubMed: 2590068]
- Shah PK, Falk E, Badimon JJ, Fernandez-Ortiz A, Mailhac A, Villareal-Levy G et al (1995) Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 92(6):1565–1569. [PubMed: 7664441]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM et al (2015) KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21(6):628–637. [PMC free article: PMC4552085] [PubMed: 25985364]
- Shekhonin BV, Domogatsky SP, Muzykantov VR, Idelson GL, Rukosuev VS (1985) Distribution of type I, III, IV and V collagen in normal and atherosclerotic human arterial wall: immunomorphological characteristics. Coll Relat Res 5(4):355–368. [PubMed: 3902343]
- Simionescu N, Simionescu M (1991) Cellular interactions of lipoproteins with the vascular endothelium: endocytosis and transcytosis. Target Diagn Ther 5:45–95. [PubMed: 1797171]
- Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL et al (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417(6890):750–754. [PubMed: 12066187]
- Soto Y, Acosta E, Delgado L, Perez A, Falcon V, Becquer MA et al (2012) Antiatherosclerotic effect of an antibody that binds to extracellular matrix glycosaminoglycans. Arterioscler Thromb Vasc Biol 32(3):595–604. [PubMed: 22267481]
- Srinivasan SR, Vijayagopal P, Eberle K, Dalferes ER Jr, Radhakrishnamurthy B, Berenson GS (1988) Low density lipoprotein binding affinity of arterial wall isomeric chondroitin sulfate proteoglycans. Atherosclerosis 72(1):1–9. [PubMed: 3214455]
- Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC et al (1999) Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 99(19):2503–2509. [PubMed: 10330380]
- Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK (2016) Extracellular matrix structure. Adv Drug Deliv Rev 97:4–27. [PubMed: 26562801]
- Theocharis AD, Manou D, Karamanos NK (2019) The extracellular matrix as a multitasking player in disease. FEBS J 286(15):2830–2869. [PubMed: 30908868]
- Thompson JC, Tang T, Wilson PG, Yoder MH, Tannock LR (2014) Increased atherosclerosis in mice with increased vascular biglycan content. Atherosclerosis 235(1):71–75. [PMC free article: PMC4054690] [PubMed: 24816040]
- Thyberg J, Hultgardh-Nilsson A (1994) Fibronectin and the basement membrane components laminin and collagen type IV influence the phenotypic properties of subcultured rat aortic smooth muscle cells differently. Cell Tissue Res 276(2):263–271. [PubMed: 8020062]
- Tran PK, Agardh HE, Tran-Lundmark K, Ekstrand J, Roy J, Henderson B et al (2007) Reduced perlecan expression and accumulation in human carotid atherosclerotic lesions. Atherosclerosis 190(2):264–270. [PubMed: 16620836]
- Tran-Lundmark K, Tran PK, Paulsson-Berne G, Friden V, Soininen R, Tryggvason K et al (2008) Heparan sulfate in perlecan promotes mouse atherosclerosis: roles in lipid permeability, lipid retention, and smooth muscle cell proliferation. Circ Res 103(1):43–52. [PMC free article: PMC2765377] [PubMed: 18596265]
- Usman A, Ribatti D, Sadat U, Gillard JH (2015) From lipid retention to immune-mediate inflammation and associated angiogenesis in the pathogenesis of atherosclerosis. J Atheroscler Thromb 22(8):739–749. [PubMed: 26156748]
- Vasile E, Simionescu M, Simionescu N (1983) Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol 96(6):1677–1689. [PMC free article: PMC2112465] [PubMed: 6853599]
- Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO et al (2015) Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol 35(3):535–546. [PMC free article: PMC4344402] [PubMed: 25573853]
- Vijayagopal P, Srinivasan SR, Radhakrishnamurthy B, Berenson GS (1983) Hemostatic properties and serum lipoprotein binding of a heparan sulfate proteoglycan from bovine aorta. Biochim Biophys Acta 758(1):70–83. [PubMed: 6222769]
- Vikramadithyan RK, Kako Y, Chen G, Hu Y, Arikawa-Hirasawa E, Yamada Y et al (2004) Atherosclerosis in perlecan heterozygous mice. J Lipid Res 45(10):1806–1812. [PubMed: 15258195]
- von der Thusen JH, van Vlijmen BJ, Hoeben RC, Kockx MM, Havekes LM, van Berkel TJ et al (2002) Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation 105(17):2064–2070. [PubMed: 11980686]
- Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE et al (2019) Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 39(5):876–887. [PMC free article: PMC6482082] [PubMed: 30786740]
- Wight TN (2018) A role for proteoglycans in vascular disease. Matrix Biol 71-72:396–420. [PMC free article: PMC6110991] [PubMed: 29499356]
- Wight TN, Merrilees MJ (2004) Proteoglycans in atherosclerosis and restenosis: key roles for versican. Circ Res 94(9):1158–1167. [PubMed: 15142969]
- Wight TN, Kinsella MG, Evanko SP, Potter-Perigo S, Merrilees MJ (2014) Versican and the regulation of cell phenotype in disease. Biochim Biophys Acta 1840(8):2441–2451. [PMC free article: PMC4074575] [PubMed: 24401530]
- Williams KJ, Tabas I (1995) The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 15(5):551–561. [PMC free article: PMC2924812] [PubMed: 7749869]
- Wise SG, Weiss AS (2009) Tropoelastin. Int J Biochem Cell Biol 41(3):494–497. [PubMed: 18468477]
- Yang H, Zhang N, Okoro EU, Guo Z (2018) Transport of apolipoprotein B-containing lipoproteins through endothelial cells is associated with apolipoprotein e-carrying HDL-like particle formation. Int J Mol Sci 19(11):3593. [PMC free article: PMC6274886] [PubMed: 30441770]
- Yeh YT, Lee CI, Lim SH, Chen LJ, Wang WL, Chuang YJ et al (2012) Convergence of physical and chemical signaling in the modulation of vascular smooth muscle cell cycle and proliferation by fibrillar collagen-regulated P66Shc. Biomaterials 33(28):6728–6738. [PubMed: 22770800]
- Yue B (2014) Biology of the extracellular matrix: an overview. J Glaucoma 23(8 Suppl 1):S20–S23. [PMC free article: PMC4185430] [PubMed: 25275899]
Publication Details
Author Information and Affiliations
Authors
Sima Allahverdian,1 Carleena Ortega,1 and Gordon A. Francis 1.
1.Affiliations
Corresponding author.Publication History
Published online: December 19, 2020.
Copyright
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Publisher
Springer, Cham (CH)
NLM Citation
Allahverdian S, Ortega C, Francis GA. Smooth Muscle Cell-Proteoglycan-Lipoprotein Interactions as Drivers of Atherosclerosis. 2020 Dec 19. In: von Eckardstein A, Binder CJ, editors. Prevention and Treatment of Atherosclerosis: Improving State-of-the-Art Management and Search for Novel Targets [Internet]. Cham (CH): Springer; 2022. doi: 10.1007/164_2020_364

