This chapter explores degradation and turnover of glycans in lysosomes, especially with respect to human genetic disorders, with representative glycans, illustrating features unique to different pathways. Degradation of oligomannosyl N-glycans removed from misfolded, newly synthesized glycoproteins is covered in Chapters 39 and 45.
LYSOSOMAL ENZYMES
Most glycans are degraded in lysosomes by highly ordered pathways using endo- and exoglycosidases, sometimes aided by noncatalytic proteins. Insights that unraveled these complex pathways emerged from studies of rare human genetic disorders called lysosomal storage diseases. In each disease, undigested molecules accumulate in lysosomes. Clever experiments combining enzymology with glycan structural analyses revealed the steps of the pathways and also unlocked the mechanisms of lysosomal enzyme targeting (Chapter 33).
Lysosomes contain approximately 50 to 60 soluble hydrolases that degrade various macromolecules. Most of the glycan-degrading enzymes (endo- and exoglycosidases and sulfatases) have pH optima between 4 and 5.5, but a few have higher pH optima, nearing neutral. Exoglycosidases cleave the glycosidic linkage of terminal sugars from the nonreducing end of glycans (the outermost left end of glycans for figures in this book, e.g., Figure ). Exoglycosidases recognize only one monosaccharide (rarely two) in a specific anomeric linkage and are much less particular about the structure of the molecule beyond that glycosidic linkage. This lack of specificity allows these enzymes to act on a broad range of substrates. However, exoglycosidases do not usually work unless all of the hydroxyl groups of the terminal sugar are unmodified. Acetate, sulfate, or phosphate groups usually have to be removed before action of the glycosidases. Esterases cleave acetyl groups and specific sulfatases remove the sulfate groups on glycosaminoglycans (GAGs) and N- or O-linked glycans. Endoglycosidases cleave internal glycosidic linkages of larger chains. These enzymes are often more tolerant of modifications of the glycan; in some cases, they require a modified sugar for optimal cleavage.
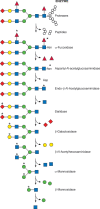
Degradation of complex-type N-glycans. The lysosomal degradation pathway of glycoproteins carrying complex-type glycans proceeds simultaneously on both the protein and glycan moieties. The N-glycans are sequentially degraded by the indicated exoglycosidases (more...)
Even though the lysosomal glycosidases perform similar reactions, their amino acid sequences are only ∼15%–20% identical to each other. Thus, there are no highly conserved glycosidase catalytic domains. Lysosomal enzymes are all N-glycosylated, and most are targeted to the lysosome by the mannose 6-phosphate pathway (Chapter 33), share aspects of the recognition marker for assembly of mannose 6-phosphate on N-linked glycans, and have affinity for mannose 6-phosphate receptors. The concentration of enzymes within the lysosome is difficult to determine. Proteinases such as cathepsins are estimated to be ∼1 mm; glycosidases are probably present at much lower concentrations.
GENETIC DEFECTS IN LYSOSOMAL DEGRADATION OF GLYCANS
About 50 known inherited diseases impair lysosomal degradation of macromolecules. Although each one is rare, together their occurrence is about one in every 5000 to 10,000 births. Loss of a single lysosomal hydrolase leads to the accumulation of its substrate as undegraded fragments in tissues and the appearance of related fragments in urine. Many of the human disorders have animal models. Tables , , and show some of the major clinical symptoms of diseases associated with the degradation of three classes of glycans. Many of the diseases share overlapping symptoms, and yet each disease has unique clinical features that allow it to be diagnosed. Many of the diseases also present with a range of severities. Usually, an infantile onset is the most severe, and the juvenile or adult onsets have milder symptoms. The later-onset forms may even affect organ systems distinct from those affected by early-onset forms. Hundreds of mutations have been mapped in the different disorders. The severity usually depends on the combination of mutated alleles. Predicting the disease severity (prognosis) from the specific mutation is generally difficult. Complete absence of a lysosomal hydrolase is uniformly severe. Hypomorphic alleles have variable residual glycosidase activity making their prognosis difficult. If there is a defect on mannose 6-phosphate–mediated targeting of lysosomal hydrolases, it will also lead to a lysosomal storage disease such as I-cell disease (mucolipidosis II) or pseudo-Hurler polydystrophy (mucolipidosis III) (Chapter 33).
Defects in glycoprotein degradation—the glycoproteinoses
Defects in glycosaminoglycan (GAG) degradation—the mucopolysaccharidoses
Defects in glycolipid degradation
It is not clear whether accumulating different types of undegraded glycans leads to the different symptoms characteristic of each disease. There is no evidence that the stored material causes lysosomes to burst and spew their contents into the cytoplasm. However, increased lysosomal exocytosis or fusion of lysosomes with the plasma membrane has been noted for some disorders. Many lysosomal disorders have defects in autophagy. This can result in impaired clearance of damaged organelles like mitochondria. The pathology likely depends on the cell type and the cellular balance of synthesis and turnover rates. For instance, dermatan sulfate (DS), a GAG (Chapter 17), predominates in connective tissue, which might explain the bone, joint, and skin problems in mucopolysaccharidosis (MPS) I, II, VI, and VII. Keratan sulfate (KS), another GAG, is present in cartilage; therefore, MPS IV is largely a skeletal disease. Gangliosides (Chapter 11) are most abundant in neurons; therefore, gangliosidoses are predominantly brain disorders. The importance of glycogen for muscle explains the impact of Pompe disease, a lysosomal disease caused by a mutation of GAA gene encoding α-glucosidase important for glycogen degradation, on the heart and diaphragm, leading to rapid lethality in that disease. Secondary storage of glycans is a common finding in many of these disorders, highlighted by the accumulation of glycolipids in the cholesterol storage disease, Niemann–Pick type C and in several disorders with GAG storage. Given the balance between synthesis and degradation of multiple glycans, reducing synthesis somewhat by the use of an inhibitor may help to retard the accumulation of primary or secondary storage and reduce the pathology of some diseases.
GLYCOPROTEIN DEGRADATION
The great majority of N- and O-glycans reaching the lysosome contain only six sugars linked in one or two anomeric configurations: β-N-acetylglucosamine (βGlcNAc), α/β-N-acetylgalactosamine (α/βGalNAc), α/β-galactose (α/βGal), α/β-mannose (α/βMan), α-fucose (αFuc), and α-sialic acid (αSia). Each linkage should theoretically require only one anomer-specific glycosidase, assuming that each glycosidase ignores the underlying glycan. This number is close to the known number of enzymes in most glycan degradation pathways. However, some linkages require a specific enzyme outside of this group. For example, β-N-acetylhexosaminidase cleaves both βGlcNAc and βGalNAc residues. Degradation of the GlcNAcβAsn and GalNAcαSer/Thr linkages also requires specific enzymes.
Lysosomal Degradation of Complex-Type N-Glycans
Much of what we know about this pathway comes from analysis of products that accumulate in patients’ tissues or urine because of the absence of one of the degradative enzymes (Table ). Structural analyses of monosaccharide-labeled glycoproteins during degradation in perfused rat liver aided elucidation of the pathway. By conducting the latter studies in the presence of inhibitors of different lysosomal enzymes, a picture of simultaneous and independent bidirectional degradation of the protein and carbohydrate chains emerged (Figure ). The relative degradation rates vary depending on structural and steric factors of the protein and the sugar chains. The accumulation of GlcNAcβ1-4GlcNAcβAsn in cells that cannot cleave the GlcNAcβAsn linkage clearly shows that degradation of the sugar chain does not require advanced cleavage of the Asn. Much of the protein is probably degraded before N-glycan catabolism begins. Removal of core fucose (Fucα1-6GlcNAc) and probably any peripheral fucose residues linked to the outer branches of the chain (e.g., Fucα1-3GlcNAc) appears to be the first step in degradation, because patients lacking α-fucosidase still have intact N-glycans bound to asparagine. Glycosylasparaginase (aspartyl-N-acetyl-β-D-glucosaminidase) then cleaves the GlcNAcβAsn bond, producing a glycosylamine +Asp, not Asn. In rodents and primates, chitobiase (an endo-β-N-acetylglucosaminidase) removes the reducing N-acetylglucosamine, leaving the oligosaccharide with only one terminal GlcNAc. In many other species, splitting of the chitobiose linkage (GlcNAcβ1-4GlcNAc) uses the β-N-acetylhexosaminidase, mentioned below, as the last step in degradation. Either pathway appears to be effective, leaving the presence of chitobiase in some species unexplained. The oligosaccharide chain is then sequentially degraded by sialidases and/or α-galactosidase, followed by β-galactosidase, β-N-acetylhexosaminidase, and α-mannosidases. The remaining Manβ1-4GlcNAc (or Manβ1-4GlcNAcβ1-4GlcNAc in species lacking chitobiase) is cleaved by β-mannosidase to mannose and GlcNAc (or chitobiose, which is then cleaved to GlcNAc by β-N-acetylhexosaminidase).
Lysosomal sialidase (neuraminidase), β-galactosidase, and a serine carboxypeptidase called protective protein/cathepsin A form a complex in the lysosome that is required for efficient degradation of sialylated glycoconjugates. Cathepsin A protects β-galactosidase from rapid degradation and also activates the sialidase precursor, but the protection does not depend on the catalytic activity of cathepsin A. Mutations in this protective protein lead to galactosialidosis in which the simultaneous deficiencies in β-galactosidase and sialidase are secondary effects of defective cathepsin A.
Glycans that have GalNAcβ1-4GlcNAc, GlcAβ1-3Gal, or Galα1-3Gal on the outer branches must first have these residues removed by β-N-acetylhexosaminidase, β-glucuronidase, and α-galactosidase, respectively, before any further digestion of the underlying oligosaccharide chains.
Lysosomal Degradation of Oligomannosyl N-Glycans
Oligomannosyl N-glycans that enter the lysosome are hydrolyzed by an α-mannosidase to yield Manα1-6Manβ1-4GlcNAc, a common intermediate of hybrid and complex-type N-glycan. A second α1-6-specific mannosidase can cleave this linkage in humans and rats, but only on molecules that have a single core region N-acetylglucosamine (i.e., those generated by chitobiase cleavage). Finally, β-mannosidase completes the degradation. Oligomannosyl N-glycans derived from dolichol-linked precursors or misfolded glycoproteins are handled differently (Chapter 39).
Degradation of O-Glycans
Degradation of typical αGalNAc-initiated O-glycans has not been systematically studied. Many of the outer structures of N-glycans are also found on O-glycans (Chapter 14); therefore, degradation of these glycans probably uses the same group of exoglycosidases as discussed above. An exception to this is the linkage region, GalNAcα-O-Ser/Thr. Patients with Schindler disease lack the α-N-acetylgalactosaminidase specific for αGalNAc and will not cleave αGlcNAc. This same enzyme probably removes terminal αGalNAc from blood group A–containing glycans (GalNAcα1-3Gal) and some glycolipids such as the Forssmann antigen (GalNAcα1-3GalNAcβ1-3Galα1-4Galβ1-4GlcβCer). Patients lacking α-N-acetylgalactosaminidase accumulate GalNAc-containing glycopeptides in their urine, but curiously they also accumulate more complex, extended glycopeptides containing N-acetylglucosamine, galactose, and sialic acid. The structures are the same as those found on some native glycoconjugates. Their production could result from a general slowdown of oligosaccharide degradation or may arise by reassembly of glycans on accumulated GalNAcα-O-Ser/Thr glycopeptides.
GLYCOSAMINOGLYCAN DEGRADATION
GAGs, including heparan sulfate (HS), chondroitin sulfate (CS), DS, KS, and hyaluronan, are degraded in a highly ordered fashion. The first three are O-xylose-linked to core proteins (Chapter 17), KS is N- or O-linked, and hyaluronan is a free glycan (Chapter 16). Some proteoglycans are internalized from the cell surface and the protein portion is degraded. The GAG chains are then partially cleaved by enzymes such as endo-β-glucuronidases or endohexosaminidases that clip at a few specific sites. Endoglycosidase cleavage creates multiple terminal residues that can be degraded by unique or overlapping sets of sulfatases and exoglycosidases. Structural analyses of partially degraded fragments in the lysosomes of cells from patients with MPS revealed both the degradation pathways and genetic defects responsible (Table ). The clinical severities and manifestations vary even with mutations in the same gene. For instance, MPS I is clinically subdivided into Hurler (most severe), Hurler–Scheie, and Scheie syndromes, although the three disorders represent a continuum of the same disease (Table ). Hurler–Scheie patients progress more slowly and die in early adulthood, whereas Scheie patients can survive to middle or old age. The milder forms of this disease do not cause intellectual disability.
Hyaluronan
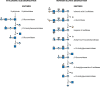
Hyaluronan (Chapter 16) is the largest and most abundant GAG: A 70-kg person degrades 5 g of hyaluronan (molecular mass 107 Da) per day. Degradation of hyaluronan involves a series of two endo-β-N-acetylhexosaminidases called hyaluronidases (Hyal-1 and Hyal-2), β-glucuronidase, and finally β-N-acetylhexosaminidase. Hyal-2 is active at low pH and is GPI-anchored on the cell surface (Chapter 12). This enzyme associates both with hyaluronan in lipid rafts and with an Na+/H+ exchanger to create an acidic microenvironment. Cleavage generates fragments of ∼20 kDa (approximately 50 disaccharides), which are internalized, delivered to endosomes, and then finally to lysosomes, in which the fragments are degraded by Hyal-1 into tetra- and disaccharides (Figure ). The exoglycosidases are thought to participate in the degradation of the larger fragments as well as that of the di-and tetrasaccharide units. Mutations in HYAL-1 cause MPS IX.
Degradation of hyaluronan and heparan sulfate (HS). (Left) Hyaluronidase (an endoglycosidase) cleaves large chains into smaller fragments, each of which is then sequentially degraded from the nonreducing end. (Right) Degradation of HS. An endo-β-glucuronidase (more...)
Heparan Sulfate
HS (Chapter 17) is first degraded by an endoglucuronidase, heparanase, followed by a well-ordered sequential degradation. An example is shown in Figure . A terminal iduronic acid-2-sulfate must be desulfated by iduronic acid-2-sulfatase to make the modified sugar a substrate for α-iduronidase. If glucuronic acid (GlcA)-2-sulfate is at this position, GlcA-2-sulfatase removes the sulfate before β-glucuronidase cleavage. The new terminal glucosamine sulfate (GlcNSO4) is the next sugar for cleavage, requiring three steps. Sulfate is first removed by N-sulfatase forming glucosamine, which cannot be cleaved by α-N-acetylglucosaminidase. The free amino group is N-acetylated by an N-acetyltransferase embedded in the lysosomal membrane, converting glucosamine to N-acetylglucosamine, which is a substrate of α-N-acetylglucosaminidase. In the second step, acetyl CoA donates the acetyl group to a histidine residue in the cytoplasmic domain of the N-acetyltransferase. The acetyl group then becomes available on the luminal side of the lysosomal membrane and is transferred at low pH to the amino group of glucosamine. If cleavage of glucuronic acid reveals a 6-O-sulfated αGlcNAc, the sulfate is removed by a specific GlcNAc-6-sulfatase before GlcNAc removal. Table provides a list of the enzymatic defects in HS degradation and the disorders that result.
Dermatan Sulfate and Chondroitin Sulfate
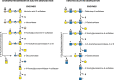
DS and CS are related GAG chains based on a repeating polymer of βGlcA and βGalNAc (Chapter 17). Figure shows that a combination of endoglycosidases, sulfatases, and exoglycosidases degrades DS in the lysosome. Iduronic acid-2-sulfatase is followed by α-iduronidase. The terminal GalNAc-4-SO4 can be removed by either of two pathways. In the first pathway, a GalNAc-4-SO4 sulfatase acts followed by β-N-acetylhexosaminidase A or B to remove N-acetylgalactosamine. In the second, β-N-acetylhexosaminidase A removes the entire GalNAc-4-SO4 unit followed by sulfatase cleavage. β-Glucuronidase cleaves the β-glucuronic acid residue and the process is repeated on the rest of the molecule.
Degradation of chondroitin/dermatan sulfates (CS/DS) and keratan sulfate (KS). (Left) Only DS degradation is shown. After removal of sulfated iduronic acid in two steps, further sequential degradation can proceed by two different routes. One route (straight (more...)
To degrade CS, GalNAc-6-SO4 sulfatase and GalNAc-4-SO4 sulfatase work in combination with β-N-acetylhexosaminidase A or B and β-glucuronidase. Hyaluronidases can also degrade CS, but no CS-specific endoglycosidases have been found.
Keratan Sulfate
KS is an N- or O-glycan with a heavily sulfated poly-N-acetyllactosamine chain. Mammalian cells do not have an endoglycosidase to break down KS (Figure ). The sequential action of sulfatases and exoglycosidases is needed. Galactose-6-SO4 sulfatase is the same enzyme that desulfates GalNAc-6-SO4 in CS degradation. This hydrolysis is followed by β-galactosidase digestion, leaving a terminal GlcNAc-6-SO4. Desulfation by the sulfatase followed by cleavage with β-N-acetylhexosaminidase A or B eliminates GlcNAc-6-SO4. Alternatively, β-N-acetylhexosaminidase A can directly release GlcNAc-6-SO4, followed by desulfation of the monosaccharide.
Linkage Region
Degradation of the core region of O-linked KS (skeletal type; type II) probably occurs by the same route as other O-glycans. The N-glycan corneal-type KS (type I) is probably degraded by the same set of enzymes used for N-glycan degradation. The O-xylose-linked GAG chains (DS, CS, and HS) all share a common core tetrasaccharide: GlcAβ1-3Galβ1-3Galβ1-4Xylβ-O-Ser. An endo-β-xylosidase has been detected in rabbit liver. How the linkage regions are degraded is not established.
Multiple Sulfatase Deficiency
A very rare human disorder is multiple sulfatase deficiency (MSD). All sulfatases are casualties of this defect because they all undergo a posttranslational conversion of an active site cysteine residue to a Cα-formylglycine (2-amino-3-oxopropionic acid) catalyzed by the Cα-formylglycine-generating enzyme (FGE), which is essential for activity. In essence, the SH group of cysteine is replaced by a double-bonded oxygen atom that probably acts as an acceptor for cleaved sulfate groups. Loss-of-function mutations in the FGE gene lead to inactive sulfatases. This deficit affects GAG degradation and any other sulfated glycan such as sulfatides.
GLYCOSPHINGOLIPID DEGRADATION
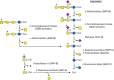
Glycosphingolipids (Chapter 11) are degraded from the nonreducing end by exoglycosidases while they are still bound to the lipid moiety ceramide. Because glycosphingolipids share some of the same outer sugar sequences found in N- and O-glycans (Chapter 14), many of the same glycosidases are used for their degradation (Figure ). However, specific hydrolases cleave the glucose–ceramide and galactose–ceramide bonds. Besides specific enzymes, noncatalytic sphingolipid activator proteins (SAPs or saposins) help to present the lipid substrates to enzymes for cleavage. Ceramide glycanase (endoglycoceramidases) from leeches and earthworms release the entire glycan chain from the lipid, much like PNGase releases N-glycans from proteins.
Degradation of glycosphingolipids. Required activator proteins are shown in parentheses and individual enzymes are as shown in previous figures in this chapter. (SAP) Saposin.
Specialized Issues for Glycolipid Degradation
Some exoglycosidases are unique to glycolipid degradation, and their absence causes glycolipid storage diseases (Table ). Glucocerebrosidase, also called β-glucoceramidase, is specific for the degradation of the GlcβCer bond. The loss of this enzyme causes Gaucher's disease. Heterozygous mutations in this enzyme have been shown to be among the most prevalent and robust genetic risk factors for Parkinson's disease, highlighting a role for glycolipid degradation in the context of more common neurological disorders. A specialized β-galactosidase, β-galactoceramidase, hydrolyzes the bond between galactose and ceramide and can also cleave the terminal galactose from lactosylceramide. Loss of this enzyme produces Krabbe disease. Galactosylceramide is often found with a 3-sulfate ester (sulfatide) and a specific sulfatase (arylsulfatase A) is needed for its removal before β-galactosylceramidase action. Loss of this sulfatase causes metachromatic leukodystrophy and the accumulation of sulfatide. Glycolipids terminated with α-galactose residues (globo-series; Chapter 11) are degraded by a specific α-galactosidase, the loss of which causes Fabry disease.
Activator Proteins
Sugars that lie too close to the lipid bilayer apparently give limited access to soluble lysosomal enzymes, and additional proteins called saposins are required to present the substrates to the enzymes. Saposins (SAPs), also called “liftases,” form complexes with multiple degradative enzymes for more efficient hydrolysis of short glycolipids close to the membrane. SAPs are derived from a 524-amino-acid precursor called prosaposin, which is processed into four homologous activator proteins each comprising ∼80 amino acids: saposins A, B, C, and D. Despite their homology, each saposin has different properties. SAP-A and SAP-C help β-galactosyl- and β-glucosylceramidase degradation, respectively. SAP-B assists arylsulfatase A, α-galactosidase, sialidase, and β-galactosidase. The activation mechanism of SAPs is thought to be similar to that of the GM2 activator described below. SAP-D and SAP-B also assist in sphingomyelin degradation by sphingomyelinase. Complete absence of prosaposin is lethal in humans, and deficiencies in SAP-B and SAP-C lead to defects that clinically resemble arylsulfatase A deficiency (metachromatic leukodystrophy) and Gaucher's disease. These symptoms might be predicted based on the enzymes that these saposins assist.
GM2 activator protein forms a complex with either GM2 or GA2 and presents them to β-N-acetylhexosaminidase A for cleavage of β-GalNAc. The activator protein binds a molecule of glycolipid, forming a soluble complex, which can be cleaved by the hexosaminidase. The resulting product is then inserted back into the membrane, and the activator presents the next GM2 molecule, and so on. Genetic loss of this activator protein causes the accumulation of GM2 and GA2, resulting in the AB variant of GM2 gangliosidosis.
Topology for Degradation and Roles of Lipids
Endocytic vesicles deliver membrane components to the lysosome for degradation and are often seen as intralysosomal multivesicular bodies (MVBs). They appear like vesicles within the lysosome and are especially prominent in patients with glycolipid storage disorders. How do “vesicles within vesicles” form and what is their function? The internal surface of the lysosomal membrane has a thick, degradation-resistant glycocalyx of integral and peripheral membrane proteins decorated with poly-N-acetyllactosamines, which protect the lysosomal membrane against destruction. However, these proteins would also shield incoming membranes from degradation if vesicle fusion simply occurred. By creating multiple internal membranes seen in typical MVBs, the target molecules are exposed to the soluble lysosomal enzymes, and digestion proceeds efficiently on the membrane surfaces of these internal vesicles.
MVB formation starts with inward budding of the limiting endosomal membrane. Lipids and proteins are sorted to either the internal or the limiting membrane. Ubiquitination of cargo proteins targets them to internal vesicles of MVBs, but ubiquitin-independent routes also exist. Intra-endosomal membranes and limiting endosomal membrane have different lipid and protein compositions. Membrane segregation and lipid sorting prepare the internal membranes for lysosomal degradation. During maturation of the internal membranes, cholesterol is continually stripped away (to <1%) and a negatively charged lipid bis(monoacylglycero)phosphate (BMP) increases up to 45%. This molecule is highly resistant to phospholipases. BMP also stimulates sphingolipid degradation on the inner membranes of acidic compartments. Because BMP is not present in the lysosomal outer membrane, the unique lipid profile ensures that degradation by hydrolases and membrane-disrupting SAPs occurs without digesting the lysosomal outer membrane.
DEGRADATION AND RESYNTHESIS
Degradation of glycans is not always complete. Partially degraded or incomplete glycans on glycoproteins, glycopeptides, and glycosphingolipids can be internalized within a functional Golgi compartment containing sugar nucleotides and glycosyltransferases and then elongated. In the case of glycosphingolipids, this pathway makes a substantial contribution to total cellular synthesis, but in glycoproteins it probably makes a relatively small contribution. These processes can be salvage and repair mechanisms or may play an integral part in an unidentified physiological pathway.
Reglycosylation and Recycling of Glycoproteins
The half-life of certain membrane proteins is longer than the half-life of their sugar chains. Terminal monosaccharides turn over faster than those near the reducing end of the glycan, suggesting that terminal sugars are removed by exoglycosidases. Cleavage may occur at the cell surface or when proteins are endocytosed in the course of normal membrane recycling. Because the mildly acidic late endosomes contain lysosomal enzymes with a fairly broad pH range, terminal sugars such as sialic acid can be cleaved. If the endocytosed proteins are not degraded in lysosomes, the proteins may reencounter sialyltransferases in the Golgi, become resialylated, and appear again on the cell surface. A similar situation may occur if a protein has lost both sialic acid and galactose residues from its glycans. Some membrane proteins synthesized in the presence of oligosaccharide processing inhibitors can still reach the cell surface in an unprocessed form. Subsequent incubation in the absence of inhibitors leads to normal processing over time. The extent of processing and the kinetics depend on the protein and the cell type. Because Golgi enzymes are not distributed identically in all cells, the extent of reprocessing is variable. Most studies have monitored N-glycans, but membrane proteins with O-glycans probably behave similarly.
Glycosphingolipid Recycling
The majority of the newly synthesized glucosylceramide arrives at the cell surface by a Golgi-independent cytoplasmic pathway. Some of this material, as well as glucosylceramide generated by partial degradation of complex glycosphingolipids, can recycle to the Golgi. There, like glycoproteins, simple glycosphingolipids can serve as acceptors for the synthesis of longer sugar chains. In the lysosome, sphingosine is produced by complete degradation of glycosphingolipids and reused. Together, these pathways, especially the latter, may account for the majority of complex glycolipid synthesis in many cells. Depending on the physiological state of the cell and synthetic demands, the de novo pathway starting from serine and palmitoyl CoA may account for only 20%–30% of the total synthesis. Shuttling the recycled components through and among the various organelles appears to involve vimentin intermediate filaments. Mechanisms and details of this process are presently lacking.
SALVAGE OF MONOSACCHARIDES
Monosaccharide salvage is discussed in Chapter 5. Monosaccharides derived from glycan degradation are transported back into the cytoplasm using transporters specific for neutral sugars, N-acetylated hexoses, anionic sugars, sialic acid, and glucuronic acid. Turnover of glycans is high and the salvage of sugars can be quite substantial. There have been few studies comparing the actual contributions of exogenous monosaccharides, those salvaged from glycan turnover, and those generated from de novo synthesis. Cells probably differ in their preference for each source of monosaccharides. These differences may explain why monosaccharide therapy used to treat some glycosylation disorders is effective in certain cells and not others.
BLOCKING DEGRADATION
Lysosomal degradation of glycans can be blocked by raising the intralysosomal pH, by inactivating a particular glycosidase, by mutation, or by protein-specific inhibitors.
Lysosomal enzymes usually have acidic pH optima, and adding ammonium chloride or chloroquine slows degradation by increasing the intralysosomal pH. However, some lysosomal enzymes have relatively broad pH optima, higher than the lysosome. Some active “lysosomal” enzymes can be found in early and late endosomes, which have a higher pH than lysosomes.
Glycosylation inhibitors are covered in Chapter 55 but some of these agents also inhibit specific lysosomal enzymes. For example, swainsonine blocks lysosomal α-mannosidase as well as the α-mannosidase II involved in glycoprotein processing. Sheep and cattle become neurologically deranged by eating food rich in swainsonine, which is also called locoweed. These temporary symptoms are likely induced because lysosomal α-mannosidase is inhibited. Undegraded oligosaccharides probably accumulate in affected animals. Although many inhibitors block various enzymes in vitro, they may not be effective in cells or whole animals because they may not enter the lysosome at sufficient concentrations.
THERAPY FOR LYSOSOMAL ENZYME DEFICIENCIES
Study of lysosomal enzyme defects provided major insights into catabolic pathways and showed their importance to human health. Mannose 6-phosphate targeting of lysosomal enzymes was discovered by showing that cocultivation of fibroblasts from patients with different storage diseases led to the disappearance of stored material in both types of cells. Each supplied the corrective factors (i.e., lysosomal enzymes) that the other cell lacked by delivery of secreted enzymes through cell-surface mannose 6-phosphate receptors (Chapter 33). Cross-correction provides the mechanistic basis for an important mode of therapy and highlights the fact that only a relatively small amount of enzyme may be needed to prevent accumulation of storage products and the resulting pathology. Some estimates suggest that <5% of normal β-N-acetylhexosaminidase activity may be sufficient to prevent pathological symptoms of Tay–Sachs disease. In fact, the index Scheie patient with α-L-iduronidase deficiency had <1% normal activity in fibroblasts and lived 77 years.
Current therapeutic approaches include enzyme replacement therapy (ERT), substrate reduction therapy (SRT), enzyme enhancement therapy (EET), and hematopoietic stem cell transplantation (HSCT). Gene replacement therapy is a promising future approach because it would avoid the sustained delivery necessary for ERT. Accumulation of stored undegraded material occurs because the rate of glycan synthesis is greater than its degradation at steady state. SRT reduces the glycan's synthetic rate to counter low glycosidase activity. This hypothesis was tested in a mouse model of Sandhoff disease by using N-butyldeoxynojirimycin, an inhibitor of glucosylceramide synthase, which catalyzes the first step in glycosphingolipid biosynthesis. When wild-type mice were treated with this compound, the amount of glycosphingolipids fell 50%–70% in all tissues without obvious pathological effects. When this compound was given to Sandhoff disease mice, the accumulation of GM2 in the brain was blocked and the amount of stored ganglioside reduced. Thus, reducing the synthesis of the primary precursor reduced the load of GM2 to levels that were degradable by the glycosidase-deficient mice. Because this compound inhibits the first biosynthetic step, theoretically this drug could reduce the accumulation of storage products in any glycolipid storage disorder.
In clinical trials, N-butyldeoxynojirimycin (miglustat, Zavesca) was effective for many patients with mild-to-moderate Gaucher's disease, although improvement varied, as did side effects. Small trials in infantile Tay–Sachs disease patients did not arrest neurological deterioration but prevented macrocephaly. Further clinical studies are likely to reveal the potential and limitations of SRT.
EET or pharmacological chaperone therapy (PCT) is based on the concept that inhibitors of an enzyme can act as molecular chaperones to stabilize the mutated enzymes in the endoplasmic reticulum (ER). When the enzyme reaches the lysosome, the inhibitor dissociates at low pH and the abundant stored material is gradually digested. The overall increase in activity is small, but it can have significant clinical benefits. Clinical trials are in progress for treating Fabry disease, Gaucher type I, and Pompe disease, whereas preclinical trials are being performed for GM1 and GM2 gangliosidosis. A potential problem is that some inhibitors may not cross the blood–brain barrier, limiting their effectiveness in the central nervous system (CNS). The advent of high-throughput screening methods and large chemical libraries increase the potential of this approach. In addition, allosteric site binders can be selected that remain bound to the enzyme, and any successful compound could be used in combination with ERT to further enhance activity.
ERT has been quite successful for treating Gaucher's disease. Injection of glucocerebrosidase carrying mannose-terminated N-glycans targets the enzyme to the macrophage/monocytes, which are the primary sites of substrate accumulation. In use now for many years, the added enzyme consistently improves patients’ clinical features with minimal side effects. The cost of treatment is high, and high doses are no more effective than lower doses for improving visceral and hematological pathologies. The proven effectiveness makes glucocerebrosidase the favorite therapy for these patients, but terminal mannose residues cannot be used to target the enzyme to other cells or organs or to enable treatment of other lysosomal storage disorders. Insufficient infused enzyme crosses the blood–brain barrier thus, the enzyme (Cerezyme) works very well for type I disease without neurological involvement but not for the less common Gaucher's with neurological involvement. In addition, patients sometimes develop antibodies against the injected human protein.
ERT trials with other recombinant lysosomal enzymes have progressed. Enzyme replacement therapy is available for Fabry disease, Pompe disease, and mucopolysaccharidosis types I, II, IVA, and VI and is under development for others. Combination therapy using ERT and a pharmacological chaperone to stabilize the therapeutic enzyme in the bloodstream is being considered. Recombinant α-galactosidase for Fabry disease works well and recombinant α-L-iduronidase for MPS I works very well with the intermediate severity form (Hurler–Scheie), but its effect is unclear for patients with the more common neurological form (Hurler syndrome). HSCT, presumably providing α-L-iduronidase in CNS from the donor-derived microglia, is effective to MPS I. Recombinant N-acetylgalactosamine-4-sulfatase (arylsulfatase B; Naglazyme) was approved for MPS VI, as was α-glucosidase (Myozyme) for treating Pompe disease patients. Iduronic acid-2-sulfatase (Elaprase) is approved for treating Hunter syndrome. These results clearly validate this approach. The efficacy of most of these enzymes relies on the engineering of Man-6-P-containing glycans. This is one of the foremost examples of how the clinically motivated exploration of a disorder (I-cell disease) led to the discovery of Man-6-P-based targeting of lysosomal enzymes (Chapter 33) and the development of an entire industry targeting therapy for multiple diseases. The availability of animals deficient in specific lysosomal enzymes offers appropriate systems to test the efficacy of corrective genes, compounds, and enzymes.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Junko Matsuda and Nathan Lewis.
FURTHER READING
Neufeld EF, Lim TW, Shapiro LJ. 1975. Inherited disorders of lysosomal metabolism. Annu Rev Biochem
44: 357–376. doi:10.1146/annurev.bi.44.070175.002041 [
PubMed: 806251] [
CrossRef]
Winchester B. 2005. Lysosomal metabolism of glycoproteins. Glycobiology
15: 1R–15R. doi:10.1093/glycob/cwi041 [
PubMed: 15647514] [
CrossRef]
Winchester B. 2014. Lysosomal diseases: diagnostic update. J Inherit Metab Dis
37: 599–608. doi:10.1007/s10545-014-9710-y [
PubMed: 24711203] [
CrossRef]
Clarke LA, Hollak CE. 2015. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract Res Clin Endocrinol Metab
29: 219–235. doi:10.1016/j.beem.2014.08.010 [
PubMed: 25987175] [
CrossRef]
Coutinho MF, Matos L, Alves S. 2015. From bedside to cell biology: a century of history on lysosomal dysfunction. Gene
555: 50–58. doi:10.1016/j.gene.2014.09.054 [
PubMed: 25275857] [
CrossRef]
Deng H, Xiu X, Jankovic J. 2015. Genetic convergence of Parkinson's disease and lysosomal storage disorders. Mol Neurobiol
51: 1554–1568. doi:10.1007/s12035-014-8832-4 [
PubMed: 25099932] [
CrossRef]
Espejo-Mojica ÁJ, Alméciga-Díaz CJ, Rodríguez A, Mosquera Á, Díaz D, Beltrán L, Díaz S, Pimentel N, Moreno J, Sánchez J, et al.
2015. Human recombinant lysosomal enzymes produced in microorganisms. Mol Genet Metab
116: 13–23. doi:10.1016/j.ymgme.2015.06.001 [
PubMed: 26071627] [
CrossRef]
Oh DB. 2015. Glyco-engineering strategies for development of therapeutic enzymes with improved efficacy for the treatment of lysosomal storage diseases. BMB Rep
48: 438–444. doi:10.5483/bmbrep.2015.48.8.101 [
PMC free article: PMC4576951] [
PubMed: 25999178] [
CrossRef]
Parenti G, Andria G, Valenzano KJ. 2015. Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther
23: 1138–1148. doi:10.1038/mt.2015.62 [
PMC free article: PMC4817787] [
PubMed: 25881001] [
CrossRef]
Breiden B, Sandhoff K. 2019. Lysosomal glycosphingolipid storage diseases. Annu Rev Biochem
88: 461–485. doi:10.1146/annurev-biochem-013118-111518 [
PubMed: 31220974] [
CrossRef]
Ryan E, Seehra G, Sharma P, Sidransky E. 2019. GBA1-associated parkinsonism: new insights and therapeutic opportunities. Curr Opin Neurol
32: 589–596. doi:10.1097/wco.0000000000000715 [
PubMed: 31188151] [
CrossRef]
Tancini B, Buratta S, Delo F, Sagini K, Chiaradia E, Pellegrino RM, Emiliani C, Urbanelli L. 2020. Lysosomal exocytosis: the extracellular role of an intracellular organelle. Membranes (Basel)
10: 406. doi:10.3390/membranes10120406 [
PMC free article: PMC7764620] [
PubMed: 33316913] [
CrossRef]