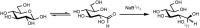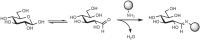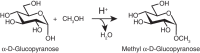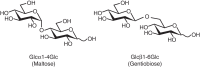This chapter covers the basic building blocks of glycans and fundamental considerations regarding glycan structure by introducing chemical concepts. Modes of linking glycans and structural depiction of the same are discussed to provide the groundwork for understanding longer glycans (Chapter 3).
INTRODUCTION TO GLYCAN TERMINOLOGY
In this book, as well as in the earlier editions, the term glycan is used. Still, a host of names are commonly used to refer to sugar polymers in other textbooks and the literature. In the 19th century, sugar-based substances were referred to as carbohydrates, or “hydrates of carbon,” that are based on the general formula C
x(H2O)n that also possess a carbonyl group, either an aldehyde or a ketone. Monosaccharides are the simplest of these polyhydroxylated carbonyl compounds (saccharide is derived from the Greek word for sugar or sweetness).
Monosaccharides are joined together to give rise to oligosaccharides or polysaccharides. Typically, the term “oligosaccharide” refers to any glycan that contains less than 20 monosaccharide residues connected by glycosidic linkages. The term “polysaccharide” is typically used to denote any linear or branched polymer consisting of monosaccharide residues, such as cellulose (Chapters 14 and 24). Thus, the relationship of monosaccharides to oligosaccharides or polysaccharides is analogous to that of amino acids and proteins or nucleotides and nucleic acids (polynucleotides).
The term “glycoconjugate” is often used to describe a macromolecule that contains monosaccharides covalently linked to proteins or lipids. The prefix “glyco” and the suffixes “saccharide” and “glycan” indicate the presence of carbohydrate constituents (e.g., glycoproteins, glycolipids, and proteoglycans). Just as is observed with proteins in nature, additional structural diversity can be imparted to glycans by modifying their hydroxyl groups with phosphate, sulfate, or acetyl esters, and/or their amino groups with acetyl or sulfate groups.
A carbohydrate may be termed “complex” if it contains more than one type of monosaccharide building unit. The glucose-based polymer cellulose is an example of a “simple” carbohydrate, whereas a galactomannan polysaccharide, composed of both galactose and mannose, is an example of a complex carbohydrate. However, even so-called simple glycans, such as cellulose and starch, often have very complex molecular structures in three dimensions. The term complex carbohydrates includes glycoconjugates, whereas the term carbohydrates per se would not. Additional nomenclature issues are covered in this chapter and Chapter 3. A more detailed and comprehensive listing of carbohydrate nomenclature rules has been published (see McNaught 1997 and Varki et al. 2015 in Further Reading at the end of this chapter, and Online Appendix 1B).
MONOSACCHARIDES: BASIC STRUCTURES AND STEREOISOMERISM
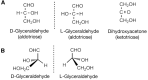
The classification of monosaccharide structures began in the late 19th century with the pioneering work of Emil Fischer. All simple monosaccharides have the general empirical formula Cx(H2O)n, where n is an integer ranging from 3 to 9. As mentioned briefly in Chapter 1, all monosaccharides consist of a chain of chiral hydroxymethylene units, which terminates at one end with a hydroxymethyl group and at the other with either an aldehyde group (aldoses) or an α-hydroxy ketone group (ketoses). Glyceraldehyde is the simplest aldose and dihydroxyacetone is the simplest ketose (Figure ). The structures of glyceraldehyde and dihydroxyacetone are distinct in that glyceraldehyde contains an asymmetric (chiral) carbon atom (Figure ), whereas dihydroxyacetone does not. With the exception of dihydroxyacetone, all monosaccharides have at least one asymmetric carbon atom, the total number being equal to the number of internal (CHOH) groups (n–2 for aldoses and n–3 for ketoses with n carbon atoms). The number of stereoisomers corresponds to 2k, where k equals the number of asymmetric carbon atoms. For example, an aldohexose with the general formula C6H12O6 and four asymmetric carbon atoms (i.e., four (CHOH) groups) can be described in 16 possible isomeric forms (Figure ).
Structures of glyceraldehyde and dihydroxyacetone. (A) Fischer projection. (B) D- and L-glyceraldehyde. The chiral central carbon in glyceraldehyde gives rise to two possible configurations of the molecule, termed D and L.
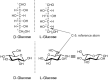
The numbering of carbon atoms follows the rules of organic chemistry nomenclature. The aldehyde carbon is referred to as C-1 and the carbonyl group in ketoses is referred to as C-2. The overall configuration (D or L) of each sugar is determined by the absolute configuration of the stereogenic center furthest from the carbonyl group (i.e., with the highest numbered asymmetric carbon atom; this is C-5 in hexoses and C-4 in pentoses). The configuration of a monosaccharide is most easily determined by representing the structure in a Fischer projection. If the OH (or other non-H group) is on the right in the Fischer projection, the overall configuration is D. If the OH (or other non-H group) is on the left, the overall configuration is L (Figure ). This figure also shows D- and L-glucose in the cyclic form (chair conformation) found in solution. Most vertebrate monosaccharides have the D configuration with the exception of fucose and iduronic acid (IdoA) L sugars. The Fischer projections shown in Figure illustrate the acyclic structures of all D-aldoses through the aldohexose group.
D- and L-glucopyranose in Fischer projection and chair conformation.
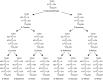
Fischer projections for the acyclic forms of the D series of aldoses, ranging from triose to hexose.
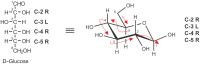
Any two sugars that differ only in the configuration around a single chiral carbon atom are called epimers. For example, D-mannose is the C-2 epimer of D-glucose, whereas D-galactose is the C-4 epimer of D-glucose (Figure ). Monosaccharide names are frequently abbreviated; most common are three-letter abbreviations for simple monosaccharides (e.g., Gal, Glc, Man, Xyl, Fuc). There are nine common monosaccharides found in vertebrate glycoconjugates (Figure ). Once incorporated into a glycan, these nine monosaccharide building blocks can be further modified to generate additional sugar structures. For example, glucuronic acid (GlcA) can be epimerized at C-5 to generate IdoA. Many more monosaccharides exist in glycoconjugates from other species and as intermediates in metabolism. We use a symbolic notation for the monosaccharides that are most abundant in vertebrate glycoconjugates (see Chapter 1).
Common monosaccharides found in vertebrates. N-Acetylneuraminic acid is the most common form of sialic acid.
MONOSACCHARIDES EXIST PRIMARILY IN CYCLIC FORM
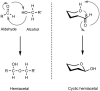
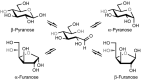
Monosaccharides exist in solution as an equilibrium mixture of acyclic and cyclic forms. The percentage of each form depends on the sugar structure. The cyclic form of a monosaccharide is characterized by a hemiacetal group formed by the reaction of one of the hydroxyl groups with the C-1 aldehyde or ketone. For reasons of chemical stability, five- and six-membered rings are most commonly formed from acyclic monosaccharides. Hexoses (six-carbon aldoses) and hexuloses (six-carbon ketoses) form six-membered rings via a C-1—O—C-5 ring closure; they form five-membered rings through a C-1—O—C-4 ring closure (Figure ). A five-membered cyclic hemiacetal is labeled a “furanose” and a six-membered cyclic hemiacetal is called a “pyranose.” Pentoses can form both pyranose and furanose forms.
Cyclization of acyclic D-glucose to form pyranose and furanose structures. The cyclization reaction produces both the α and β anomers (i.e., C-1 epimers).
Formation of Hemiacetals
Monosaccharides can also be represented as Haworth projections in which both five- and six-membered cyclic structures are depicted as planar ring systems, with the hydroxy groups oriented either above or below the plane of the ring (Figure ). Although not truly representative of the three-dimensional structure of a monosaccharide, the Haworth representation has been used since the late 1920s as an easy-to-draw formula that permits a quick evaluation of stereochemistry around the monosaccharide ring. The Haworth representations are preferably drawn with the ring oxygen atom at the top (for furanose) or the top right-hand corner (for pyranose) of the structure; the numbering of the ring carbons increases in a clockwise direction.
Conversion from Fischer to Haworth projection. Each hydroxyl group projected to the right in the Fischer projection points down in the Haworth formula.
For any D sugar, the conversion of a Fischer projection into a Haworth projection proceeds as follows: (1) any groups (atoms) that are directed to the right in the Fischer structure are given a downward orientation in the Haworth structure, (2) any groups (atoms) that are directed to the left in the Fischer structure are given an upward orientation in the Haworth structure, and (3) the terminal —CH2OH group is given an upward orientation in the Haworth structure. For an L sugar, (1) and (2) are the same, but the terminal —CH2OH group is projected downward.
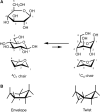
The planar Haworth structures are distorted representations of the actual molecules. The preferred conformation of a pyranose ring is the “chair” conformation, similar to the structure of cyclohexane. The conversion from Haworth projection to chair conformation leaves the downward or upward orientation of ring substituents unaltered. Two chair conformations can be distinguished and designated as 4C1 and 1C4, respectively (Figure ), and these conformers can interconvert by a process called the “ring flip.” The first numeral in the chair conformer designation (superscript) indicates the number of the ring carbon atom above the “seat of the chair (C)” and the second numeral (subscript) indicates the number of the ring carbon atom below the plane of the seat (spanned by C-2, C-3, C-5, and the ring O). Chair conformations are designated from structures with the ring oxygen atom in the top right-hand corner of the ring “seat,” resulting in the clockwise appearance of the ring numbering. To determine the stereochemistry in the chair form as it corresponds to the Fischer projection, one can locate C-6 and then trace along the carbon skeleton of the sugar, bisecting the C—O and C—H bonds formed from each atom. The OH (or OR) and H groups are found on the right (R) or left (L) sides, just as in the Fischer projection (Figure ).
Chair conformations. (A) β-D-Glucose in Haworth projection and in its 4C1 and 1C4 chair conformations; (B) envelope and twist conformations for a five-membered ring structure.
Conversion from Fischer to chair projection formula; (R) right; (L) left. Red arrows illustrate the path to follow along the sugar backbone when correlating the stereochemistry of the Fischer projection with the chair conformation.
The more structurally accurate chair representations are preferred to Haworth projections for depicting pyranoses. However, Haworth projections are convenient and are commonly used to depict furanoses. The furanose ring is rather flexible and not entirely flat in any of its energetically favored conformations; for example, it has a slight pucker when viewed from the side, as seen in the representations of the so-called envelope and twist (or skew) conformations (Figure ). Because furanoses can adopt many low-energy conformations, researchers have adopted the Haworth projection as a simple means to avoid this complexity.
CHEMISTRY AT THE ANOMERIC CENTER
Mutarotation
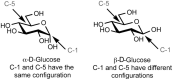
When cyclized into rings, monosaccharides acquire an additional asymmetric center derived from the carbonyl carbon atom (Figure ). The new asymmetric center is termed the “anomeric carbon” (i.e., C-1 in the ring form of glucose). Two stereoisomers are formed by the cyclization reaction because the anomeric hydroxy group can assume two possible orientations. When the configurations are the same at the anomeric carbon and the stereogenic center furthest from the anomeric carbon, the monosaccharide is defined as the α anomer. When the configurations are different, the monosaccharide is defined as the β anomer (Figure ). Unlike the other stereocenters on the monosaccharide ring, which are configurationally stable, the anomeric center can undergo an interconversion of stereoisomers via the process of mutarotation. Catalyzed by dilute acid or base, the reaction proceeds by the reverse of the cyclization reaction. The monosaccharide ring opens up and then recloses to form a ring with the other anomeric configuration (Figure ). The term mutarotation derives from the rapid change in optical rotation (denoted [α] D) that is observed when an anomerically pure form of a monosaccharide is dissolved in water. For example, β-D-glucopyranose shows an initial rotation of +19°, whereas the α anomer shows an initial rotation of +112°. When either anomer is allowed to undergo the mutarotation reaction, an equilibrium mixture containing both anomers is obtained, producing a rotation of +52.5°.
Determination of configuration at the anomeric center.
Oxidation and Reduction
Generally, the acyclic (aldehyde or ketone) form of a monosaccharide is only present in minor amounts in an equilibrium mixture (<0.01%). Nevertheless, the open-chain aldehydes or ketones can participate in chemical reactions that drive the equilibrium and eventually consume the sugar.
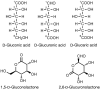
Aldoses and ketoses were historically referred to as “reducing sugars” because they responded positively in a chemical test that effected oxidation of their aldehyde and hydroxyketone functionalities, respectively. The carboxylic acid formed by oxidation of the aldehyde in an aldose is referred to as a glyconic acid (e.g., gluconic acid is the oxidation product of glucose). It is also possible to oxidize the hydroxyl groups of monosaccharides, most notably the terminal OH group (i.e., C-6 of glucose). In this reaction, a glycuronic acid is produced, and if both terminal groups are oxidized, the product is a glycaric acid. The three acids derived from D-glucose are illustrated in Figure . These compounds have a tendency to undergo intramolecular cyclization reactions, preferably yielding six-membered lactones. Two examples of lactonized forms are shown in Figure . Oxidized forms of monosaccharides can be found in nature. For example, GlcA is an abundant component of many glycosaminoglycans (see Chapter 17).
Oxidized forms of D-glucose.
Conversion of a monosaccharide to a tritium-labeled alditol by reduction with NaB3H4.
The carbonyl groups of aldoses and ketoses also can be reduced with sodium borohydride (NaBH4) to form polyhydroxy alcohols, referred to as alditols. This reaction is widely used to introduce a radiolabel at C-1 of the monosaccharide by reduction with NaB3H4 (Figure ).
Schiff Base Formation
The aldehyde and ketone groups of monosaccharides can also undergo Schiff base formation with amines or hydrazides, forming imines and hydrazones, respectively (Figure ). This reaction is often used to conjugate the monosaccharide to proteins (via their lysine residues) or to biochemical probes such as biotin hydrazide. It should be noted that the imines formed with amino groups are not stable to water and are typically reduced with sodium cyanoborohydride (NaCNBH3) in a process termed reductive amination.
Conjugation of a monosaccharide to an amino group by formation of an imine. The filled circle represents any small molecule or macromolecule containing an amine.
As aldehydes, reducing sugars can also form Schiff bases with amino groups of the lysine residues in proteins. This nonenzymatic process that links glycans to proteins is termed “glycation” and is distinct from “glycosylation,” which involves the formation of a glycosidic bond between the sugar and protein. Glycation products can undergo further reactions that lead to the formation of protein cross-links, and these can have pathogenic consequences (i.e., they are immunogenic and change the properties of the protein). Glycation products of glucose accumulate at higher levels in diabetics than in healthy individuals because of elevated blood glucose levels. These modified proteins are thought to underlie some of the pathologies associated with diabetes.
Glycosidic Bond Formation
Two monosaccharide units can be joined together by a glycosidic bond—this is the fundamental linkage among the monosaccharide building blocks found in all oligosaccharides. The glycosidic bond is formed between the anomeric carbon of one monosaccharide and a hydroxyl group of another. In chemical terms, a hemiacetal group reacts with an alcohol group to form an acetal. Glycosidic bonds can be formed with virtually any hydroxylated compound, including simple alcohols such as methanol (Figure ) or hydroxy amino acids such as serine, threonine, and tyrosine. Indeed, glycosidic linkages are formed between sugars and these amino acids within proteins to form glycoproteins (see Chapters 9 and 10). Like the hemiacetal, the acetal or glycosidic linkage can exist in two stereoisomeric forms: α and β. But unlike the hemiacetal, the acetal is configurationally stable under most conditions. Thus, once a glycosidic bond is formed, its configuration is maintained. Like acetals in general, glycosidic bonds can be hydrolyzed in dilute acid, generating the constituent monosaccharides from oligosaccharides.
Glycoside formation. Conversion of a hemiacetal into an acetal.
Glycosidic bond construction is the central challenge of glycan synthesis and immense efforts have been devoted to high-yielding and stereoselective glycosylation reactions. An overview of glycan synthesis strategies is provided in Chapters 53 and 54.
CHEMISTRY OF MONOSACCHARIDE FUNCTIONAL GROUPS
Methylation of Hydroxyl Groups
The hydroxyl groups present in both monosaccharides and oligosaccharides can be chemically modified without affecting the glycosidic linkages. Methylation is used in the structural analysis of glycans (see Chapter 50). Natural products containing partially methylated glycans are known and a number of methyltransferases have been identified.
Esterification of Hydroxyl Groups
A variety of different enzymes can esterify the hydroxyl groups of glycans to transiently vary glycan structure. Esterification is sometimes required for interactions with other biomolecules. The most important types of sugar esters in nature are phosphate esters (including diphosphate esters), acyl esters (with acetic acid or fatty acids), and sulfate esters. Acyl esters can sometime migrate to other hydroxyl groups on the same monosaccharide.
Deoxygenation of Hydroxyl Groups
The replacement of monosaccharide hydroxyl groups with hydrogen atoms forms deoxysugars. Nature has evolved enzymes to perform this reaction in a minimum number of steps, whereas chemically multistep procedures are required. Deoxygenation of ribose within a ribonucleotide to form the 2-deoxyribonucleotide is a critical reaction in DNA biosynthesis. Fucose (Fuc), one of the common vertebrate monosaccharides, is deoxygenated at C-6 during its biosynthesis from mannose (Chapter 5).
Amino Groups
Many monosaccharides contain N-acetamido groups, such as GlcNAc, GalNAc, and NeuNAc. Free amino groups, formed by de-N-acetylation of the N-acetamido group, are rare and found in heparan sulfate (HS) (Chapter 17), glycosylphosphatidylinositol (GPI) anchors (Chapter 12), neuraminic acid (Chapter 15), and in many bacterial glycan structures (Chapter 20). Amino groups can be modified with sulfates, similar to hydroxyl groups, as found in HS.
GLYCOSIDIC LINKAGES
A variety of linkages can be formed between two monosaccharides. The glycosidic linkage can give rise to two possible stereoisomers at the anomeric carbon of one sugar (α or β). Also, the many hydroxyl groups of the other sugar permit several possible regioisomers. Two glucose residues, for example, can be joined together in numerous ways, as illustrated by maltose (Glcα4Glc) and gentiobiose (Glcβ6Glc) (Figure ). These isomers have very different three-dimensional structures and biological activities. A monosaccharide can engage in more than two glycosidic linkages, thus serving as a branchpoint. The common occurrence of branched sequences (as opposed to the linear sequences that are found in almost all peptides and oligonucleotides) is unique to glycans and contributes to their structural diversity.
Two isomeric disaccharides.
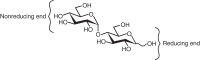
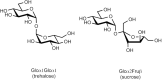
The relationship of the glycosidic bond to oligosaccharides is analogous to the relationship of the amide bond to polypeptides and the phosphodiester bond to polynucleotides. However, amino acids and nucleotides are linked in only one fashion during the formation of polypeptides and nucleic acids, respectively; there is no stereochemical or regiochemical diversity in these biopolymers. The number of monomeric residues contained in an oligosaccharide is designated in the nomenclature—disaccharide, trisaccharide, and so on. Just as polypeptides have amino and carboxyl termini and polynucleotides have 5′ and 3′ termini, oligosaccharides have a polarity that is defined by their reducing and nonreducing termini (Figure ). The reducing end of the oligosaccharide bears a free anomeric center that is not engaged in a glycosidic bond and thus retains the chemical reactivity of the aldehyde. However, it continues to be referred to as reducing end even when it is engaged in a linkage (e.g., to the hydroxyl of serine or threonine in glycoproteins). Structures are commonly written from the nonreducing end on the left toward the reducing end on the right. For some structures, there is no reducing end. For example, the common disaccharides sucrose and trehalose have glycosidic linkages between the anomeric centers of two monosaccharide constituents (Figure ).
Reducing and nonreducing ends of a disaccharide.
Nonreducing disaccharides.
The glycosidic linkage is the most flexible part of a disaccharide structure. Whereas the chair conformation of the constituent monosaccharides is relatively rigid, the torsion angles around the glycosidic bond (φ, ψ, and ω; Figure ) can vary. Thus, a disaccharide of well-defined primary structure can adopt multiple conformations in solution that differ in the relative orientation of the two monosaccharides. The combination of structural rigidity and flexibility is typical of complex carbohydrates and essential to their biological functions.
Torsion angles that define the conformation of the glycosidic linkages φ, ψ, and ω. (A) Newman projection along the C1—O1 bond illustrating φ for the 1-6 glycosidic bond. (B) Newman projection along the C6′—O1 (more...)
Glycans are linked to other biomolecules, such as lipids or amino acids within polypeptides, through glycosidic linkages to form glycoconjugates (see Chapters 9, 10, 11, and 12). Glycans are often referred to as the glycone of a glycoconjugate and the noncarbohydrate component is named the aglycone. The glycan may be a mono- or an oligosaccharide.
In conclusion, monosaccharide building blocks can be linked to various regio- and stereochemistries, and the resulting oligosaccharides can be assembled on protein or lipid scaffolds (see Chapter 3).
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Carolyn R. Bertozzi and David Rabuka and appreciate helpful comments and suggestions from Dirk Lefeber, Todd Lowary, and Sriram Neelamegham.
FURTHER READING
El Khadem HS. 1988. Carbohydrate chemistry: monosaccharides and their oligomers. Academic, San Diego.
Allen HJ, Kisailus EC. 1992. Glycoconjugates: composition, structure, and function. Marcel Dekker, New York.
McNaught AD. 1997. Nomenclature of carbohydrates. Carbohydr Res
297: 1–92. doi:10.1016/S0008-6215(97)83449-0 [
PubMed: 9042704] [
CrossRef]
Bill MR, Revers L, Wilson IBH. 1998. Protein glycosylation. Kluwer Academic, Boston.
Boons G-J. 1998. Carbohydrate chemistry. Blackie Academic and Professional, London.
Stick RV. 2001. Carbohydrates: the sweet molecules of life. Academic, New York.
Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T,, et al.
2015. Symbol nomenclature for graphical representations of glycans. Glycobiology
25: 1323–1324. doi:10.1093/glycob/cwv091 [
PMC free article: PMC4643639] [
PubMed: 26543186] [
CrossRef]