N-Glycans affect glycoprotein folding because of their hydrophilic nature. In the endoplasmic reticulum (ER), the processing of N-glycans yields a series of truncated N-glycans that serve as checkpoints that dictate the life or death of many newly made membrane and secreted proteins. Other glycan modifications also may affect glycoprotein folding in the ER. This chapter describes glycan-mediated quality-control processes in the ER and Golgi apparatus and what happens to glycoproteins that fail their “final folding examination.”
CHAPERONES FACILITATE PROTEIN FOLDING
During protein synthesis, nascent polypeptides begin to assume their final three-dimensional conformation by passing through folding intermediates dictated, to a large extent, by their primary sequence. Proper folding involves the formation of secondary structures (α-helices and β-strands), burying of hydrophobic residues in the interior of the protein, the formation of disulfide bonds, and quaternary associations via oligomerization or multimerization. Together, these processes prevent unwanted protein aggregation that would interfere with protein functions. To make folding more efficient, cells use various chaperones. Two major cytoplasmic chaperone families consist of heat-shock proteins in the hsp60 and hsp70 gene families that bind to exposed hydrophobic domains on misfolded proteins and maintain their solubility as they acquire their final conformation. They also participate in the repair of damaged proteins and proteins that have not properly multimerized. Improperly folded proteins that fail to mature are eventually tagged by ubiquitination, which then leads to degradation by the proteasome.
Membrane proteins and proteins destined for secretion also undergo quality control during folding. Membrane and secretory proteins originate on membrane-bound ribosomes; translation and translocation into the lumen of the ER occur simultaneously in most cases. However, certain proteins are fully synthesized in the cytosol and translocated into the ER lumen posttranslationally. This latter process is more relevant in yeast than mammalian cells. The ER lumen is highly specialized for protein folding: Its oxidizing environment promotes disulfide bond formation and it is the main cellular reservoir of Ca++. To aid proper folding, a battery of molecular chaperones is present, including BiP/Grp78 (a glucose-regulated protein and member of the hsp70 family of chaperones), Grp94, and Grp170. In addition, the ER contains enzymes that promote proline cis–trans isomerization and protein disulfide bond formation, such as protein disulfide isomerases (PDI and ERp57, ERp59, and ERp72). The expression of many of these proteins increases during stress responses.
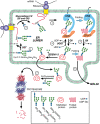
Unlike cytoplasmic proteins, most membrane and secreted proteins undergo modification with N-glycans (Figure ) as they enter the ER lumen. Oligosaccharyltransferase requires flexible domains of a polypeptide for glycosylation, and thus N-glycosylation evolved in eukaryotes to occur before protein folding. Accordingly, N-linked glycans affect the folding of glycoproteins directly by altering the biophysical properties of the protein. They can serve as signals to localize the folding machinery and to signal the folding status of the polypeptide. To achieve glycan-mediated folding, N-glycans are recognized by two lectin-like chaperones in the ER: calnexin (CNX) and calreticulin (CRT). These specialized chaperones require Ca++ for activity and bind to monoglucosylated forms of N-glycans, thus retaining the protein in the ER until proper folding occurs. The ER also contains three enzymes central to the process: α-glucosidases I and II (GI and GII), which trim glucose (Glc) from the Glc3Man9GlcNAc2 initially transferred to the protein and release the glycoprotein from CNX/CRT. The enzyme UDP-glucose glycoprotein glucosyltransferase (UGGT) reglucosylates the N-glycan if proper folding has not occurred. Glycoproteins that fail to fold, or to oligomerize properly, are marked with specific truncated N-glycans, and eventually retrotranslocated into the cytoplasm in which they are destroyed by N-deglycosylation and proteasomal degradation, a process called ER-associated degradation (ERAD). There are several ERAD pathways, which are described in the following sections.
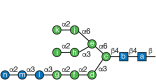
Mature N-glycan. Lettering (a–n) follows the order of addition of monosaccharides in the synthesis of Glc3Man9GlcNAc2-P-P-dolichol (Chapter 9). α-Glucosidase I (GI) removes residue n, and α-glucosidase II (GII) removes residues (more...)
CNX/CRT AND UGGT DETERMINE WHEN GLYCOPROTEINS ARE PROPERLY FOLDED
N-Glycosylation in the ER is considerably different from other types of glycosylation. It begins with the “en bloc” transfer of a 14-monosaccharide glycan to the polypeptide as it exits the translocon complex (Chapter 9). In vertebrates, addition of the N-glycan occurs on incompletely folded polypeptides. In bacteria, N-glycosylation can occur both on nascent polypeptides and on fully mature proteins (Chapter 21). N-Glycans modify the physical properties of glycoproteins by providing noncharged, bulky, hydrophilic groups that keep the protein in solution during folding. They also modulate protein conformation by forcing amino acids near the N-glycan into a hydrophilic environment.
N-Glycan processing starts immediately after transfer of the precursor sugar chain to the protein in the lumen of the ER. GI removes the terminal α1-2Glc and GII then removes both α1-3Glc units (Figure ). Although GII may remove the second Glc (designated m in Figure ) from glycoproteins with a single N-glycan, the trimming of the second Glc occurs more efficiently when there is a second N-glycan on the same protein. Depending on cell type, a variable number of Man residues may also be removed while glycoproteins remain in the ER. GII is an ER-soluble, heterodimeric protein. Its 100- to 110-kDa α-subunit contains the catalytic activity, whereas the 50- to 60-kDa β-subunit displays a canonical ER retention/retrieval sequence (XDEL), which is partially responsible for the subcellular location of the whole enzyme. The carboxy-terminal portion of the β-subunit contains a mannose receptor homologous (MRH) domain that is homologous to the domain in the Golgi mannose 6-phosphate receptor responsible for recognizing lysosomal enzymes and for their delivery to lysosomes (Chapters 9 and 33). The affinity of the GII MRH domain is higher for the Man9GlcNAc2 (M9) N-glycan and decreases for truncated N-glycans. Consequently, the GII activity toward N-glycans diminishes for truncated N-glycans created by ER mannosidases.
CNX and CRT bind to glycoproteins containing a single α1–3Glc residue (Figure ). CNX is a type I transmembrane protein and CRT is soluble, but both have ER retention/retrieval signals at their carboxyl terminus, retaining them in the ER. They have structurally similar lectin domains and comprise part of a large, weakly associated heterogeneous protein network that includes BiP/Grp78, ERp57, Grp94, and other ER-resident proteins that assist protein folding. Thereby, they act as interpreters of the N-glycan code to localize the folding machinery to a glycoprotein substrate. Both CNX and CRT are monovalent, low-affinity lectins for monoglucosylated, oligomannosyl N-glycans, but because of their different membrane-bound or soluble status within the ER lumen, their in vivo specificities are not identical. CNX mainly interacts with N-glycans close to the ER membrane, whereas CRT binds preferentially to glycoproteins in the ER lumen or to those that have large luminal domains. Binding of incompletely folded glycoproteins to the CNX/CRT complex prevents their exit from the ER and enhances folding efficiency by preventing aggregation and premature oligomerization/degradation and by facilitating the formation of native disulfide bonds. This latter task is performed by ERp57, a CNX/CRT-associated oxidoreductase. CNX and CRT are, therefore, unconventional chaperones that do not directly recognize protein moieties of folding intermediates like conventional chaperones, but rather they recognize particular N-glycans in the folding intermediates.
Model of quality control in glycoprotein folding. Proteins entering the endoplasmic reticulum (ER) are N-glycosylated by the oligosaccharyltransferase (OST) as they emerge from the translocon (SEC61) (1). Two glucose (Glc) residues are removed by the (more...)
Removal of the final Glc residue from N-glycans by GII prevents binding of the protein to CNX/CRT. If properly folded, these glycoproteins are packaged into COPII-coated vesicles and transferred to the Golgi (Figure ). However, if they remain unfolded, UGGT adds a single α1–3Glc to the glycan, thus recreating the identical N-glycan first recognized by CNX/CRT. Therefore, UGGT has the remarkable property of exclusively glucosylating glycoproteins that display a nonnative conformation. UGGT discriminates between different nonnative conformers, and preferentially glucosylates glycoproteins in their last stages of folding when they are displaying exposed hydrophobic amino acid patches (molten globules). In vitro, the enzyme also recognizes N-glycans linked to relatively short hydrophobic peptides or to hydrophobic aglycones, although less efficiently. Contrary to BiP/Grp78 that preferentially binds extended hydrophobic “sequences,” UGGT recognizes hydrophobic “surfaces.” This difference determines that glycoproteins are first recognized by chaperones like BiP/Grp78 and only later, at final folding stages, are epitopes created by UGGT recognized by CNX/CRT. UGGT modifies N-glycans located in the relative vicinity of amino acids that prevent rapid folding of a glycoprotein.
UGGT also glucosylates incompletely assembled multimeric complexes of fully mature monomers because it recognizes hydrophobic surfaces exposed in the absence of the appropriate subunits. UGGT is a relatively large, soluble, ER protein (150- to 170-kDa) that uses UDP-Glc as sugar donor and requires millimolar Ca++ concentrations, typically found within the ER lumen. UGGT has at least two domains. The amino-terminal one, comprising ∼80% of the molecule, recognizes abnormal protein conformers. This domain contains three tandemly repeated, thioredoxin-like domains that are frequently found in ER proteins involved in protein folding and quality-control processes. A crystal structure of the third domain showed that it contains a large hydrophobic patch hidden by a flexible carboxy-terminal helix. The carboxy-terminal domain has homology with other glycosyltransferases and is responsible for the catalytic activity.
Together, UGGT and the CNX/CRT complex ensure that only properly folded and multimerized glycoproteins move from the ER to the Golgi. Unlike GII, the activity of UGGT does not decrease on reduction of N-glycan Man content. Therefore, ER α-mannosidase catalyzed removal of Man from N-glycans results in longer half-lives of UGGT reaction products that may be recognized by CNX/CRT. It is possible that the differential activities of UGGT and GII toward N-glycans with reduced Man content provides glycoproteins with a last opportunity to achieve proper folding before proteasomal degradation. The ER folding and quality-control process is not foolproof, however. Some misfolded proteins escape the ER and others may misfold after they exit. Thus, there are additional checkpoints to correct mistakes that occur later in the secretory pathway.
ER–Golgi intermediate compartment 53 (ERGIC53) is a type I membrane protein that binds to oligomannosyl N-glycans in a Ca++-dependent manner. Its luminal, carbohydrate-binding domain is similar to those of soluble lectins from leguminous plants. ERGIC53 cycles between the ER and the ERGIC. The mammalian protein contains both ER-targeting (dilysine) and ER-exit (diphenylalanine) determinants at its carboxyl terminus, which bind, respectively, to COPI and COPII coatomers involved in trafficking between the ER and Golgi. ERGIC53 loads a subset of glycoproteins (coagulation factors V and VIII and cathepsins C and Z) into COPII-coated vesicles leaving the ER for the Golgi. Mutation of ERGIC53 causes combined factor V and VIII deficiency, indicating an important role in secretion of these, and probably other glycoproteins. Vesicular integral protein 36 (VIP36) is another Golgi lectin that also binds glycoproteins containing oligomannosyl N-glycans. VIP36 may facilitate transport of glycoproteins from the ERGIC to cis-Golgi cisternae, or retrieve glycoproteins bearing N-glycans that did not undergo conversion to Man5GlcNAc2 by cis-Golgi α-mannosidases IA, IB, and/or IC. Conceivably, this would provide an opportunity for new rounds of trimming and eventual formation of complex N-glycans. Both ERGIC53 and VIP36 are up-regulated as part of the unfolded protein response. Whether other lectins recognizing specific glycans are involved in post-ER quality control is still an open question.
REMOVING MISFOLDED GLYCOPROTEINS FROM THE CNX/CRT/UGGT CYCLE
Despite the presence of a battery of classical chaperones and the CNX/CRT/UGGT cycle in the ER, the process of glycoprotein folding is relatively inefficient, with as much as 80% of some newly made proteins never maturing. How do cells distinguish between misfolded glycoproteins and folding intermediates if both entities have almost identical structural features such as exposed hydrophobic patches? How do cells pull glycoproteins destined for degradation out from futile reglucosylation–deglucosylation cycles if those glycoproteins are indeed very efficiently glucosylated by UGGT? How are terminally misfolded glycoproteins driven to the proteasome for degradation? The key discriminating factor appears to be the structure of the truncated N-glycans that result from the relatively long stay of a misfolded glycoprotein in the ER lumen.
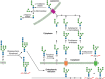
Mammalian cells have, in addition to Man1B1, three additional homologs called EDEMs (ER degradation-enhancing α-mannosidase-like) proteins, which initially were thought to be devoid of activity. However, when either Man1B1 or the EDEMs were individually knocked out in human or chicken cells, conversion of M9 to M8B (Figure ) was mainly performed by EDEM2, and the contribution of Man1B1 to that particular demannosylation step was minimal. On the other hand, conversion of M8B to M7 lacking residues i and k (M7BC; Figure ) was performed by EDEMs 1 and 3. Recent evidence suggests that Man1B1, initially considered as ER mannosidase I, localizes to Golgi, and appears to work as a backup enzyme to cleave off residue i in the Golgi as this residue is rather resistant to the action of Golgi α-mannosidase I. The M7BC isomer exposes an α1–6Man (residue j; Figure ), which is specifically recognized by an ER lectin (OS9 in mammals and Yos9p in Saccharomyces cerevisiae). Like GII, this lectin also contains an MRH domain that binds N-glycans smaller than M9, including M7BC. OS9/Yos9p forms part of the complex that translocates misfolded glycoproteins from the ER lumen to the cytosol (ERAD-L; see below). Both the conversion of M9 to M8B and the conversion of M8B to M7BC are slow processes in mammalian cells when compared with deglucosylation reactions. This affords two checkpoints ensuring that only misfolded glycoproteins, and not folding intermediates, are driven toward proteasomal degradation after a relatively long ER residence. In yeast, only an ER α-mannosidase and one EDEM-like protein called Htm1p are present. The former transforms M9 to M8B very rapidly in practically all glycoproteins, whereas the latter converts M8B to M7BC slowly, thus affording only one checkpoint for the exclusive degradation of terminally misfolded glycoproteins. Htm1p forms a complex with PDI. More recent evidence further suggests that mammalian EDEMs act as active α-mannosidases forming complexes with distinct oxidoreductases. In the case of yeast the oxidoreductase-Htm1p association enhances the α-mannosidase activity of the latter and the complex participates in the recognition of ERAD substrates. However, not all glycoproteins with N-glycans terminating in α1–6Man are driven to degradation. For instance, 3-hydroxy-3-methylglutaryl acetyl-coenzyme-A reductase, a key ER enzyme in sterol biosynthesis, carries Man5GlcNAc2 and Man6GlcNAc2 N-glycans that do not target it for degradation (see below).
RETROTRANSLOCATION OF MISFOLDED GLYCOPROTEINS TO THE CYTOSOL
Retrotranslocation of misfolded glycoproteins from the ER lumen to the cytosol for proteasomal degradation ultimately depends on various protein complexes, some of which contain integral ER membrane proteins. ER membrane proteins with folding defects in the cytosolic domain are extracted from the ER membrane by the Doa10 complex via ERAD-C, whereas those in which the folding defect is present in the luminal (ERAD-L) or membrane (ERAD-M) domains use the Hrd1 complex. Both Doa10p and Hrd1p are integral membrane proteins with E3 ligase activity in their cytosolic portion. Other proteins in these complexes include chaperones, proteins with E2 ubiquitin-conjugating activity or proteins that recognize misfolded proteins. For instance, glycoproteins with N-glycans containing a terminal α1–6Man are bound by Yos9p and undergo ERAD only if the N-glycans are in unstructured protein, a feature recognized by Hrd3p in the Hrd1 complex. Finally, proteins common to both complexes such as yeast Cdc48 (p97 in mammals) are responsible for membrane extraction of misfolded proteins in an ATP-dependent manner. Although the pore by which ERAD-L substrates are actually transported to the cytosol has not been identified, it is known that the substrates must be unfolded in the ER lumen. Although studies on ERAD complexes have been mainly performed in S. cerevisiae, mammalian cells have homologs of nearly all proteins described in the yeast complexes.
N-Glycans are removed from unfolded glycoproteins during proteasomal degradation. A cytoplasmic peptide:N-glycanase (PNGase, N-glycanase, NGLY1) plays an important role in both removing the glycan and constructing an efficient predegradation complex (Figure ). NGLY1 recognizes only misfolded or denatured glycoproteins. The enzyme is bound to the proteasome by subunits S4 and HR23B as a complex with Cdc48, a component of ERAD complexes. Degradation of the released N-glycan occurs in two stages. First, cleavage occurs between the two GlcNAcs (chitobiose) in the core of the N-glycan via a cytoplasmic endo-β-N-acetylglucosaminidase. A cytoplasmic α-mannosidase cleaves up to four Man residues to generate Man5GlcNAc (Man c, d, e, f, and g in Figure ). This glycan is then taken into the lysosome via an ATP-dependent lysosomal membrane transporter for final degradation to monosaccharides. Whether these free glycans have any function in the cytosolic/nuclear compartment before they are degraded has not been investigated.
Degradation of oligomannosyl N-glycans in the endoplasmic reticulum (ER), cytoplasm, and lysosomes. Specific pathways exist for the degradation of free glycans released from misfolded glycoproteins and glycopeptides generated within the ER. Dolichol-linked (more...)
O-GLYCOSYLATION REACTIONS IN ER QUALITY CONTROL
Protein O-fucosyltransferases (Ofut1 in Drosophila; POFUT1 and POFUT2 in mammals) are soluble ER enzymes that O-fucosylate Ser/Thr residues. Ofut1 and POFUT1 add fucose to epidermal growth factor (EGF)–like repeats, whereas POFUT2 adds fucose to thrombospondin (TSR) type 1–like repeats (Chapter 13). EGF and TSR domains are present as tandem repeats in numerous proteins of multicellular organisms. Ofut1/POFUT1 and POFUT2 selectively fucosylate only properly folded protein domains, suggesting a role of this glycosylation in folding quality control. Indeed, several POFUT2 targets including ADAMTSL1 and ADAMTSL2 (scaffolding proteins in the extracellular matrix), ADAMTS9 (a matrix remodeling protease), and ADAMTS13 (the von Willebrand factor cleaving protease) strictly require O-fucosylation for efficient secretion. Indeed, O-fucosylation appears to stabilize folded TSRs. The case of Ofut1/POFUT1 is a bit more ambiguous. Studies performed in Drosophila on the efficiency of cell-surface expression of the Ofut1 substrate Notch showed that Ofut1 is required as a chaperone for Notch folding and cell-surface expression, and that chaperone activity is largely independent of the O-fucosyltransferase activity. In contrast, POFUT1 does not appear to be absolutely required for cell-surface expression of Notch receptors in all mammalian cells, suggesting that cell-specific chaperones may be able to compensate for loss of POFUT1 in certain contexts. It has been reported recently that O-mannosylation in S. cerevisiae ER might be responsible for terminating futile cycles of association–dissociation between proteins unable to attain their native conformation and Kar2p (the budding yeast homolog of mammalian BiP/Grp78 chaperone).
THE ER QUALITY-CONTROL MACHINERY AND VIABILITY
The loss of CNX and CRT in most multicellular organisms causes a severe phenotype. Mice deficient in CNX reach full term, but half die within 2 days of birth, and very few survive beyond 3 months. The runts that survive have obvious motor disorders with loss of large myelinated nerve fibers. CRT-deficient mice and ERp57-null mice also show embryonic lethality. In contrast, CNX and CRT null Caenorhabditis elegans mutants are viable. Yeast has CNX or CNX-like lectins (but not CRT), whereas trypanosomatid protozoa have only CRT. Another important conserved mammalian ER lectin resident is calmegin, expressed only in the testes during spermatogenesis. Calmegin shares ∼60% homology with CNX in mice. Mice deficient in calmegin are nearly sterile despite producing normal-looking sperm. The sperm are defective in migration into the oviducts and they do not adhere to the zona pellucida. UGGT is absent from S. cerevisiae but not from Schizosaccharomyces pombe cells. The enzyme is not required for the viability of the fission yeast or single mammalian cells grown under normal conditions. The enzyme is also dispensable for growth of some multicellular organisms, such as plants. However, loss of UGGT is embryonically lethal in mice. This fact, as well of the strict requirement of UGGT for the viability of S. pombe cells only when grown under severe ER stress conditions, point to a restricted set of glycoproteins absolutely requiring UGGT for proper folding.
In addition to combined factor V and VIII deficiency caused by ERGIC53 mutations (see above), other congenital diseases result in retention of defective proteins in the ER and their eventual proteasomal degradation. For instance, the most common mutation in cystic fibrosis patients, CFTR-ΔF508, leads to improper folding of this chloride channel and its retention in the ER. Importantly, the mutation affects CFTR transit to the plasma membrane but not its transport activity. Similarly, UGGT recognition of a brassinosteroid receptor mutant triggers retention in the ER of Arabidopsis thaliana cells. Thus, certain diseases caused by ER retention of defective glycoproteins might be ameliorated by inhibitors of UGGT activity. One beautiful example of how the fine sensitivity of UGGT allows modulation of basic physiological processes is its role during antigen presentation by the major histocompatibility complex I (MHC I). Here, MHC I complexes loaded with suboptimal peptides are recognized by UGGT, thus triggering their retention in the ER. On the contrary, complexes formed with high-affinity peptides are poor UGGT substrates and are able to be presented on the cell surface. Many congenital diseases such as lysosomal storage diseases also result in the accumulation of misfolded glycoproteins in the ER, thus overloading the folding machinery. An estimated 20%–30% of normal proteins misfold, so it is not surprising that small differences in the kinetics of conformational maturation and degradation can tip the balance to ERAD for lack of sufficient chaperones and other factors that facilitate folding.
Although most glycoproteins transiently associate with CNX/CRT during their maturation in the ER in vivo, other ER folding and quality-control mechanisms do the job quite efficiently in cultured cells. However, viral glycoproteins such as vesicular stomatitis virus G protein, HIV glycoprotein gp120, and M protein from hepatitis B virus, absolutely require CNX/CRT for proper folding and exit from the ER. Several viruses use components of the ERAD machinery to evade immune surveillance in the host. For instance, cytomegalovirus proteins bind to MHC I complexes and deliver them to E3 ligase complexes that result in their eventual proteasomal degradation. Similarly, expression of HIV ER protein Vpu results in the proteasomal degradation of CD4. SV40 and proteins such as cholera toxin are retrotranslocated to the ER through the secretory pathway where they use some ERAD components to reach the cytosol where they produce deleterious effects.
ACKNOWLEDGMENTS
The authors acknowledge contributions to the previous version of this chapter from Jeffrey D. Esko and Hudson H. Freeze and helpful comments and suggestions from Taroh Kinoshita and Kelley Moremen.
FURTHER READING
Helenius A, Aebi M. 2004. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem
73:
1019–1049. doi:10.1146/annurev.biochem.73.011303.073752 [
PubMed: 15189166] [
CrossRef]
Lehrman MA. 2006. Stimulation of N-linked glycosylation and lipid-linked oligosaccharide synthesis by stress responses in metazoan cells. Crit Rev Biochem Mol Biol
41:
51–75. doi:10.1080/10409230500542575 [
PubMed: 16595294] [
CrossRef]
Benyair R, Ogen-Shtern N, Lederkremer GZ. 2015. Glycan regulation of ER-associated degradation through compartmentalization. Semin Cell Dev Biol
41:
99–109. doi:10.1016/j.semcdb.2014.11.006 [
PubMed: 25460542] [
CrossRef]
Caramelo JJ, Parodi AJ. 2015. A sweet code for glycoprotein folding. FEBS Lett
589:
3379–3387. doi:10.1016/j.febslet.2015.07.021 [
PubMed: 26226420] [
CrossRef]
Harada Y, Hirayama H, Suzuki T. 2015. Generation and degradation of free asparagine-linked glycans. Cell Mol Life Sci
72:
2509–2533. doi:10.1007/s00018-015-1881-7 [
PubMed: 25772500] [
CrossRef]
Satoh T, Yamaguchi T, Kato K. 2015. Emerging structural insights into glycoprotein quality control coupled with N-glycan processing in the endoplasmic reticulum. Molecules
20:
2475–2491. doi:10.3390/molecules20022475 [
PMC free article: PMC6272264] [
PubMed: 25647580] [
CrossRef]