The use of chemical tools to inhibit glycosylation is a powerful approach for studying glycan functions and can serve as a starting point for drug discovery. This chapter discusses various types of inhibitors including those present in nature, those identified through rational design followed by synthesis, and those found by screening of chemical libraries.
ADVANTAGES OF INHIBITORS
Chapters 44, 45, and 49 describe natural and induced mutants with defects in glycosylation. These mutants have helped to define genes that encode various transferases and glycosidases, and in some cases alternate biosynthetic pathways have been uncovered. Mutants also provide insights into the function of glycosylation in cells and tissues and models for human inborn errors in metabolism and disease. However, one limitation of studying mutants is that the analyses are usually restricted to the cell or organism from which the mutant strain was isolated, and mutations in essential genes require conditional alleles.
Inhibitors of carbohydrate-processing enzymes—in particular, glycosyltransferases and glycosidases—provide another approach for studying glycosylation in cells, tissues, and whole organisms that avoids some of the problems associated with genetic models. Many of these compounds are small molecules that are taken up readily by cells and the effects can be reversed, enabling experimental designs that are difficult to achieve using genetic methods. Some compounds can also be absorbed through the gut, providing an opportunity for designing drugs to treat human diseases and disorders correlated with altered glycosylation (Chapter 57). Because the field is broad, only a selection of inhibitors that act on specific enzymes or metabolic pathways and that illustrate certain basic concepts are discussed here (Figure ).
Different classes of compounds for inhibiting glycosylation including those that prevent the formation of biosynthetic precursors, those that directly act on glycosidases and glycosyltransferases, and those that serve as primers/decoys and chain terminators. (more...)
INHIBITORS OF BIOSYNTHETIC PRECURSORS
A number of inhibitors have been described that block glycosylation by interfering with the metabolism of common precursors or intracellular transport activities. Some of these compounds act indirectly by impeding the transit of proteins between the endoplasmic reticulum (ER), Golgi, and trans-Golgi network. For example, brefeldin A (Figure ) causes retrograde transport of Golgi components located proximal to the trans-Golgi network back to the ER. Thus, treating cells with brefeldin A separates enzymes located in the trans-Golgi network from those found in the ER and Golgi and uncouples the assembly of the core structures of some glycans from later reactions, such as sialylation or sulfation. The drug can be used to examine if two pathways reside in the same compartment or share enzymes. Because the localization and array of the enzymes vary considerably in different cell types, extrapolating the effects of brefeldin A from one system to another is often difficult.
Some inhibitors act at key steps in intermediary metabolism in which glycosylation precursors are formed. For example, a glutamine analog, 6-diazo-5-oxo-L-norleucine (DON; Figure ) blocks many glutamine-dependent amidotransferases including glutamine:fructose-6-phosphate amidotransferase, the enzyme of the hexosamine biosynthetic pathway that forms glucosamine from fructose and glutamine (Chapter 5). Depressing glucosamine production in this way has a pleiotropic effect on glycan assembly because all of the major families contain N-acetylglucosamine or N-acetylgalactosamine. Given the nonspecific activity of DON, care should be taken to understand and limit nonspecific side effects.
An array of sugar analogs have been made with the hope that they might show selective inhibition of glycosylation. Some examples include 2-deoxy-D-glucose and fluorosugar analogs (3-deoxy-3-fluoro-D-glucosamine, 4-deoxy-4-fluoro-D-glucosamine, 6-deoxy-6-fluoro-D-N-acetylglucosamine, 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose, 2-deoxy-2-fluoro-L-fucose, and 3-fluorosialic acid), which all inhibit glycoprotein biosynthesis (Figure ). Early studies with 2-deoxyglucose showed that the analog was converted to UDP-2-deoxyglucose, as well as to GDP-2-deoxyglucose and dolichol-P-2-deoxyglucose. Inhibition of glycoprotein formation apparently occurs as a result of accumulation of various dolichol oligosaccharides containing 2-deoxyglucose, which cannot be elongated or transferred to glycoproteins normally. Although the mechanism of action of many of these molecules remains poorly understood, one better studied case is 2-deoxy-2-fluoro-L-fucose (Figure ). This compound acts as a biosynthetic precursor of the fucose salvage pathway (Chapter 5) to form GDP-2-deoxy-2-fluoro-fucose within cells, but the analog is a poor substrate for mammalian fucosyltransferases because the highly electronegative fluorine atom close to the anomeric center inductively stabilizes the oxocarbenium ion-like transition states used by the superfamily of glycosyltransferases (Chapter 6). This compound also causes feedback inhibition of the GDP-fucose biosynthetic enzyme GDP-mannose 4,6-dehydratase (GMD) leading to a loss of natural GDP-fucose within cells and a consequent decrease in the levels of fucosylation on all types of glycoconjugates. Notably, 2-deoxy-2-fluoro-L-fucose is orally available and is active both in vitro on cells as well as in vivo, leading to decreases in the levels of the carbohydrate epitope sialyl-Lewis x (sLex) (Chapter 14). Consistent with these effects, 2-deoxy-2-fluoro-L-fucose has shown potential, in preclinical models, at blocking tumor growth and metastasis as well as vaso-occlusive crisis caused by sLex-mediated adhesion of blood cells to selectin-expressing endothelial surfaces (Chapter 34) in transgenic sickle-cell mice. Similarly, 3-fluorosialic acid is converted by the NeuAc-salvage pathway (Chapter 5) within cells into CMP-3F-NeuAc and blocks formation of CMP-NeuAc, leading to profound decreases in sialylation of glycoconjugates both in cells and in vivo, suggesting potential use in cancers. Other sugar analogs, including 5-thio-N-acetylglucosamine and 4-deoxy-4-fluoro-N-acetylglucosamine, also lead to formation of unnatural donor sugars formed within cells that reduce pools of the natural nucleotide sugars and lead to consequent decreases in protein glycosylation. These compounds are proving useful research tools; however, care must be taken in interpreting the results of experiments using these compounds because they may have pleiotropic effects on glycan assembly caused by overlap of nucleotide precursors in different pathways.
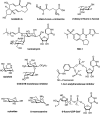
A number of natural products have been found to alter glycosylation. Tunicamycin belongs to a class of nucleoside antibiotics composed of uridine, an 11-carbon disaccharide called 2-amino-2,6,-dideoxyundecodialdose (tunicamine), and a fatty acid of variable length (13 to 17 carbons), branching, and unsaturation (Figure ). Tunicamycin derives its name from its antiviral activity, which occurs by inhibiting viral coat (or “tunica”) formation. The biosynthesis of tunicamycin has been defined, which may lead to analogs that are selective for different species.
Tunicamycin inhibits N-glycosylation in eukaryotes by blocking the transfer of N-acetylglucosamine-1-phosphate (GlcNAc-1-P) from UDP-GlcNAc to dolichol-P (catalyzed by GlcNAc phosphotransferase; GPT), thereby decreasing the formation of dolichol-PP-GlcNAc (Chapter 9). Other GlcNAc transferase reactions are not inhibited (e.g., GlcNAcTI–V), but the transfer of GlcNAc-1-P to undecaprenyl-P and the formation of undecaprenyl-PP-MurNAc pentapeptide (which is involved in bacterial peptidoglycan biosynthesis) are sensitive to tunicamycin (Chapter 21). The compound is a tight-binding competitive inhibitor, presumably because it resembles the donor nucleotide sugar. The Ki value for tunicamycin is ∼5×10−8
m, whereas the Km value for UDP-GlcNAc is ∼3×10−6
m. Given the key role of N-glycosylation in protein folding and quality control in the ER (Chapter 39), tunicamycin is cytotoxic to cells, and resistant mutants overproduce GPT. Similarly, transfection of cells with the cloned GPT confers resistance, suggesting that the variable dose of inhibitor required in different cells may reflect variation in enzyme levels. A more recently identified compound, NGI-1, offers an approach to blocking N-glycosylation that is complementary to use of tunicamycin. This compound acts as a direct inhibitor of oligosaccharyltransferase, the enzyme responsible for transferring the dolichol-PP-loaded glycan to asparagine.
Tunicamycin has been used extensively for studying the role of N-glycans in glycoprotein maturation, secretion, and function. The drug induces apoptosis preferentially in cancer cells, presumably because of alterations in glycosylation of various cell-surface receptors and signaling molecules and by inducing ER stress (Chapter 39). Thus, inhibition of N-glycan formation could be useful for treating cancer patients. Other potential applications include substrate reduction therapy for treatment of lysosomal storage disorders (Chapter 44), congenital disorders of glycosylation (Chapter 45), or naturally occurring mutations that create N-glycosylation sites in cell-surface receptors (gain-of-glycosylation mutants; Chapter 45). In a related vein, amphomycin, a lipopeptide, inhibits dolichol-P-mannose synthesis by apparently forming complexes with the carrier lipid dolichol-P. Other lipophilic compounds that bind lipid intermediates in bacterial cell wall synthesis also have been studied (Chapter 21).
INHIBITORS OF GLYCOSIDE FORMATION OR CLEAVAGE
Many different inhibitors of enzymes involved in glycoside formation or cleavage have been described, spanning a wide range of compound classes. These include natural products and synthetic derivatives, as well as acceptor and donor analogs obtained by chemical synthesis. The high-throughput screening of compound libraries and subsequent optimization of hits using medicinal chemistry approaches has also led to compounds that are both useful chemical tools and drugs. In this section, we highlight a few examples.
Example 1: Natural Product Inhibitors of Glycosidases
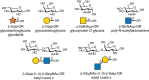
Plant alkaloids block N-linked glycosylation by inhibiting the processing glycosidases (α-glucosidases and α-mannosidases) involved in trimming nascent chains (Figure ). Unlike tunicamycin, which blocks glycosylation of glycoproteins entirely, these alkaloids inhibit the trimming reactions that occur after the Glc3Man9GlcNAc2 oligosaccharide is attached to a glycoprotein (Chapter 9). Treatment of cells with alkaloids results in the display of glycoproteins on the cell surface lacking the characteristic termini found on mature N-glycans (Chapter 14). α-Glucosidase inhibitors involved in the initial processing of N-glycans and in quality control of protein folding (Chapter 39) include castanospermine (from the seed of the Australian chestnut tree, Castanosperum australe), which inhibits α-glucosidases I and II, australine (also from C. australe), which preferentially inhibits α-glucosidase I, and deoxynojirimycin (from Streptomyces species), which preferentially inhibits α-glucosidase II (Figure ). Castanospermine and australine cause accumulation of fully glucosylated chains, whereas deoxynojirimycin results in chains containing one to two glucose residues. Unexpectedly, treating cells with these inhibitors revealed that some trimming of the mannose residues occurs independently of removal of the glucose residues through the action of the mechanistically intriguing GH99 endo-mannosidase (Chapter 9).
Examples of alkaloids that inhibit glycosidases involved in N-linked glycan biosynthesis.
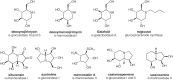
Swainsonine was first discovered in plants from the western United States (Astragalus species, also known as locoweed) and Australia (Swainsona canescens). Consumption of these plants by animals causes a severe abnormality called locoism and accumulation of glycoproteins in the lymph nodes. Swainsonine inhibits α-mannosidase II, causing accumulation of paucimannose oligosaccharides (Man4GlcNAc2 and Man5GlcNAc2) and hybrid-type chains at the expense of complex oligosaccharides. In addition, swainsonine inhibits the lysosomal α-mannosidase. Mannostatin A works in a similar way, but differs significantly in structure from swainsonine (Figure ). Other mannosidase inhibitors include deoxymannojirimycin and kifunensin, which selectively inhibit α-mannosidase I. These agents cause the accumulation of Man7–9GlcNAc2 oligosaccharides on glycoproteins.
All of the above listed inhibitors have in common polyhydroxylated ring systems that mimic the orientation of hydroxyl groups in the natural substrates, but a strict correlation between stereochemistry and enzyme target (α-glucosidase vs. α-mannosidase) does not exist. The compounds contain nitrogen, usually in place of the ring oxygen. To explain their activity, it is proposed that the nitrogen, which is protonated at physiological pH, mimics the transition state during the hydrolysis reaction, which has substantial positive character. Crystal structures for the α-mannosidase are available with a range of different bound inhibitors.
Diastereomers as well as alkylated and acylated analogs of the alkaloids have interesting and useful properties. Notably, the galactose-configured analog of deoxynojirimycin (Galafold; Figure ) has been approved as a pharmacological chaperone of the lysosomal α-galactosidase for the treatment of the lysosomal storage disease known as Fabry disease (Chapter 44). Mutations in the gene encoding the enzyme generally impair its ability to fold and traffic from the ER to lysosomes. Galafold can bind to the mutant α-galactosidase in the ER and help it fold and avoid degradation leading to its improved trafficking and activity within lysosomes. N-Butylation of deoxynojirimycin makes this glucosidase inhibitor an inhibitor of glycolipid biosynthesis, which is discussed in more detail further below. In other cases, alkylation of the amino group or acylation of the hydroxyl groups can improve the potency of the compound, presumably by facilitating uptake across the plasma and Golgi membranes. Some of these compounds have shown positive effects for treating diabetes, cancer, HIV infection, and lysosomal storage diseases (see Chapter 57), but some also induce male sterility. A major challenge with all of these enzymes is their limited specificity; thus while convenient and widely used, some caution is needed when interpreting resulting observations.
Example 2: Inhibitors of O-GlcNAc-Processing Enzymes
The importance of O-GlcNAc addition to many cytoplasmic and nuclear proteins (Chapter 19) has stimulated interest in developing agents to inhibit its addition by O-GlcNAc transferase (OGT) or its removal by O-GlcNAc-specific β-glucosaminidase (O-GlcNAcase [OGA]). These enzymes provide excellent examples of successful rational design and medicinal chemistry efforts directed to the development of glycoside hydrolase and glycosyltransferase inhibitors. Alloxan and streptozotocin affect O-GlcNAc addition, but these compounds lack specificity. The first potentially useful OGT inhibitor (OSMI-1; Figure ) was obtained by screening chemical libraries for compounds that displaced a fluorescent derivative of the donor sugar, UDP-GlcNAc. Structure-guided medicinal chemistry led to quinoline-containing ester (OSMI-4), which is a high-quality OGT inhibitor that is effective in cells. The active compounds do not block other N-acetylglucosamine addition reactions—for example, one involved in formation of the polysaccharide backbone of bacterial peptidoglycan (Chapter 21). In addition, 5SGlcNAc is another inhibitor of OGT that acts as a metabolic precursor leading to formation of UDP-5SGlcNAc. Its peracetylated form (Ac-5SGlcNAc) crosses cell membranes and undergoes deacetylation by nonspecific esterases to generate cell-active inhibitors. Modification of the N-acetyl group to give S5GlcNHex eliminated the need to have O-acetylation and provided a compound that could be used in vivo, although, like other metabolic precursor inhibitors, their use requires care because of possible off-target effects on other enzymes (e.g., GlcNAc transferases [Chapter 9]).
Inhibitors of O-GlcNAc-specific β-glucosaminidase (OGA) and O-GlcNAc transferase (OGT).
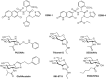
Several O-GlcNAcase inhibitors are based on N-acetylglucosamine. The first described, PUGNAc (Figure ) inhibits O-GlcNAcase at nanomolar concentrations, but also inhibits lysosomal β-hexosaminidases (HexA and HexB; Chapter 44). 1,2-Dideoxy-2′-propyl-α-D-glucopyranoso-[2,1-d]-Δ2′-thiazoline (NButGT) and the more potent aminothiazoline Thiamet-G are more specific and potent in cells than PUGNAc (Figure ). A rationally designed glucoimidazole, GlcNAcstatin, also potently inhibits O-GlcNAcase with good selectivity over HexA and HexB. These compounds inhibit the enzyme in cells and tissues, and Thiamet-G, in particular, has been used in animal models, providing new tools to study the function of O-GlcNAc. Use of these compounds has uncovered the potential of O-GlcNAcase inhibitors in various neurodegenerative diseases, which has spurred industrial pharmaceutical interest that has led to OGA inhibitors, such as the Thiamet-G analog MK-8719 (Figure ), advancing into Phase I clinical trials. Noncarbohydrate O-GlcNAcase inhibitors have also been uncovered, and highly potent brain permeable analogs have been used as positron emission tomography agents to examine the inhibition and distribution of O-GlcNAcase in human brain.
Example 3: Rational Design of Acceptor and Donor Analogs
A number of specific glycosyltransferase inhibitors have been developed based on the concept that donor and acceptor substrate analogs might serve as inhibitors. For acceptor substrate analogs, the general strategy is to modify the hydroxyl group that acts as the nucleophile during formation of the glycosidic bond or groups in its immediate vicinity (Table ). Many designer compounds lack inhibitory activity, because modification of the targeted hydroxyl group prevents binding of the analog to the enzyme by interfering with hydrogen bonding networks that position the substrate; that is, these groups serve as “key polar groups.” In a smaller number of cases, the analogs show Ki values in the approximate range of the Km values for the unmodified substrate. As one might expect, the analogs usually act competitively with respect to the unmodified substrate, but in a few cases the inhibition pattern is more complex, suggesting possible binding outside the active site.
Synthetic substrate-based inhibitors of glycosyltransferases
Nucleotide sugar analogs provide opportunities for blocking classes of enzymes that use a common donor (e.g., all fucosyltransferases use GDP-fucose). Many nucleotide sugar derivatives have been synthesized (e.g., N- and O-substituted analogs of UDP-GalNAc), and several inhibit the enzymes in vitro. These have proven less useful in living cells because of poor uptake, but there are some notable exceptions, including the fluorescently tagged CMP-NeuAc analog in which the carbohydrate is replaced with an aryl group (1-G-m, Figure ), which has a nanomolar Ki values for a range of sialyltransferases. This compound has also been reported to block sialyltransferases within cells, although this compound has not been more widely adopted by the community. “Bisubstrate” transition-state analogs consist of the nucleoside sugar donor covalently linked to the acceptor substrate by way of a bridging group. In principle, this approach could lead to high-affinity inhibitors, in turn justifying the usually complex routes needed for their synthesis. However, the compounds reported to date have been only modest inhibitors with Ki values, at best, in the range of the Km values for the donors. Although such transition state mimics remain attractive targets, the results to date suggest new features need to be incorporated into their design.
Example 4: Inhibitors of Glycolipids and GPI Anchors
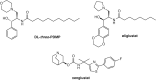
Reagents that alter the assembly of glycolipids in cells have been described. Xylosides (Figure ) have a mild effect on glycolipid formation, possibly because of the similarity between xylose and glucose and the assembly of a GM3-like compound (Neu5Acα2-3Galβ1-4Xylβ-O-R) on the primer. Because cells take up intermediates in glycolipid biosynthesis, they behave like synthetic glycoside primers. For example, glucosylceramide produces complex glycolipids when fed to cells. More direct competitive inhibitors of glucosylceramide synthase (GCS) have been generated with a view to their potential benefit in Gaucher disease (Chapter 44), which stems from accumulation of glucosylceramide caused by loss-of-function mutations in β-glucocerebrosidase (GCase). D-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP; Figure ) has been a widely used compound; however, it also inhibits the activity of purified lactosylceramide synthase. Medicinal chemistry efforts have led to the close analog eliglustat, which although it does not gain access to the central nervous system (CNS), is approved for treating type I Gaucher disease, a lysosomal storage disorder in which glucocerebrosidase is missing (Chapter 44). Its beneficial activity occurs through “substrate deprivation” by blocking synthesis of glycosphingolipids, thereby “depriving” the lysosome of substrate. Subsequent efforts have led to CNS permeable GCS inhibitors including venglustat, which is advancing through late stage clinical trials. The α-glucosidase inhibitor, N-butyldeoxynojirimycin, known commercially as miglustat, also inhibits GCS and is approved for treating Niemann–Pick disease type C and Gaucher disease.
Inhibitors of glycosphingolipid metabolism.
The search for active compounds often benefits from serendipity and the synthesis of disaccharide (or larger) acceptor analogs with appropriate modifications is labor intensive, as is the preparation of complicated nucleotide sugar analogs. Nevertheless, the approach has yielded insights into the binding and reactivity of the glycan-processing enzymes, and substrate analogs with selectivity for particular enzymes have been developed in this way. Improvements in recombinant expression systems have led to increasing numbers of X-ray crystal structures, which provides clues for deriving mechanism-based inhibitors in the future (Chapter 6).
GLYCOSIDE PRIMERS AND CHAIN TERMINATORS
Glycoside Primers
The utility of any glycosyltransferase inhibitor ultimately depends on its ability to cross the plasma membrane and enter the Golgi where the glycosyltransferases reside. Unfortunately, many of the compounds described above lack activity in cells, presumably because their polarity and charge prevents their uptake. More than 40 years ago, Okayama and colleagues found that D-xylose in β-linkage to a hydrophobic aglycone (the noncarbohydrate portion of a glycoside) was taken up efficiently and inhibited the assembly of glycosaminoglycans on proteoglycans. Xylosides mimic the natural substrate, xylosylated serine residues in proteoglycan core proteins, and thus act as a substrate. “Priming” of chains occurs on the added xyloside, which diverts the assembly process from the endogenous core proteins and causes inhibition of proteoglycan formation. In general, cells incubated with xylosides secrete large amounts of individual glycosaminoglycan chains and accumulate proteoglycans containing truncated chains. The success of β-D-xylosides in altering proteoglycan biosynthesis suggested that other glycosides might function similarly (Figure ). Subsequent studies showed that β-N-acetylgalactosaminides prime oligosaccharides found on mucins and inhibit O-glycosylation of glycoproteins. Other active glycosides include β-glucosides, β-galactosides, β-N-acetylglucosaminides, and even disaccharides and trisaccharides. These latter compounds require conjugation to appropriate aglycones and acetylation to mask the polar carbohydrate hydroxyl groups. Cells contain several carboxyesterases that remove the acetyl groups and render the compounds available to the transferases in the Golgi.
Examples of glycoside primers. Structures shown are the compounds administered to cells; the depictions of the disaccharides do not show their acetylation. Below the names of the compounds are the glycan classes that are impacted.
Priming by glycosides occurs in a concentration-dependent manner, but the efficiency varies widely among different compounds and cell types. These variations may relate to the relative abundance of endogenous substrates, enzyme concentration and composition, the solubility of different glycosides, their susceptibility to hydrolysis, their uptake across the plasma membrane and into the Golgi, and their relative affinity for the glycosyltransferases. The type of chain made on a given primer also depends on concentration and aglycone structure, which may reflect selective partitioning of primers into different intracellular compartments or into different branches of biosynthetic pathways. Like priming, inhibition of glycoprotein, glycolipid, or proteoglycan formation occurs in a dose-dependent fashion, but the blockade is rarely complete, probably because of the inability of glycosides to mimic the entire endogenous substrates.
Primers represent starting points for tight-binding inhibitors with the properties described above. The compounds described in Figure could be converted to permeable acylated glycosides and tested in live cells for inhibitory activity. Active compounds could potentially become lead compounds for drugs to treat glycosylation-dependent diseases. Oligosaccharide priming may have beneficial effects as well. Xylosides, for example, can be absorbed through the gut, and when consumed at sufficient concentration, show antithrombotic activity. Many glycosides occur naturally because various organisms (especially plants) produce hydrophobic compounds as part of chemical defense and conjugate them to sugars to render them soluble. Thus, the human diet may contain various types of glycosides with interesting (and unknown) biological activities.
Care must be taken in interpreting the results of experiments using glycoside primers. For example, β-D-xylosides also prime glycans related in structure to glycosphingolipids and HNK-1. In some cases priming per se is not the mechanism responsible for inhibition of glycosylation, but rather inhibition occurs because of competitive binding of the primer to a target enzyme. Finally, primers could deplete cells of nucleotide sugars and have multiple effects on glycosylation. For example, 4-methylumbelliferone can be used to block hyaluronan biosynthesis. The precise mechanism of action is unknown but is thought to involve depletion of UDP-GlcA due to glucuronidation of the glycoside primer. Reduction in cellular UDP-GlcA levels in turn affects formation of sulfated glycosaminoglycans and other glucuronic acid–containing glycans and alter the pools of other nucleotide sugars, such as UDP-Xyl, which is produced in one step from UDP-GlcA (Chapter 5).
Chain Terminators
Chain terminators are compounds that are introduced into a growing glycan by a glycosyltransferase, but in so doing introduces a functionality that prevents further elongation. Mannosamine acts as a metabolic inhibitor that inhibits GPI anchor formation both in Trypanosoma brucei and in mammalian cells by the formation of ManNH2-Man-GlcNH2-PI. Apparently, mannosamine in its activated form (GDP-ManNH2) is used as a substrate in the second mannosyltransferase reaction, but the ManNH2-Man-GlcNH2-PI intermediate will not act as a substrate for the next α2-mannosyl-transferase (Chapter 12). GlcNR-phosphatidylinositols with different substituents (R) act as substrate analogs and some act as suicide inhibitors in vitro. Another class of inhibitors is based on fatty acid analogs that only trypanosomes incorporate into GPI anchors. Trypanosomes, unlike their mammalian hosts, incorporate myristic acid into GPI anchors by exchanging myristic acid for other fatty acids in the phosphatidylinositol moiety. By making a series of analogs, an inhibitor was found that is highly toxic to trypanosomes in culture and nontoxic to mammalian cells (10-(propoxy)decanoic acid). Such reagents are drug candidates for treating trypanosomiasis, which is endemic in sub-Saharan regions of Africa. Additional examples of chain terminators have been reported. For example, fluorinated sugar nucleotides (Figure ) are incorporated into growing oligosaccharides by carbohydrate polymerases, but the resulting products lack the hydroxyl group to enable further polymerization.
STRUCTURE-BASED RATIONAL DESIGN
The increasing number of X-ray crystallographic and cryo-EM investigations of carbohydrate processing enzymes has provided structural information that has facilitated the rational design of new inhibitors, often with very high potency. These compounds have been used as research tools and some have also advanced to clinically used drugs.
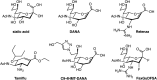
Studies of influenza neuraminidase exemplify the power of rationally designed drugs that have successfully been marketed as drugs. The crystal structure for influenza neuraminidase was obtained in 1983, and many other enzymes have since been characterized from other sources. Even before the crystal structure had been obtained, a neuraminidase inhibitor was designed by assuming that the hydrolysis reaction involved an oxocarbenium ion–like transition state with significant positive charge accumulation at the anomeric center. This would result in C-2 and C-3 adopting a planar configuration, and therefore compounds that mimicked this geometry were hoped to have inhibitory activity. Indeed, Neu5Ac-2-ene (DANA; Figure ) has a micromolar Ki value. Interestingly, this compound inhibits most sialidases, but does not inhibit the trypanosome trans-sialidase and only weakly bacterial sialidases.
Structure of neuraminidase inhibitors. Chemical structure of sialic acid (Neu5Ac, 2-deoxy-2,3-dehydro-N-acetyl neuraminic acid), DANA; 4-amino-DANA; 4-guanidino-DANA (Relenza, zanamivir); (3R, 4R, 5S)-4-acetamido-5-amino-3-(1-ethylpropoxyl)-1-cyclohexane-1-carboxylic (more...)
A visual inspection of the X-ray structure of influenza neuraminidase with DANA bound showed that two glutamate residues lined a pocket near carbon 4 of the sialic acid analog. The pocket is fairly open, suggesting that a bulkier substituent at this position might be tolerated. A substrate analog containing a positively charged guanidinium group instead of the hydroxyl at carbon 4 (4-guanidino-DANA; Figure ) is a remarkably potent influenza neuraminidase inhibitor (Ki=10−11
m). The higher affinity is presumably due to a salt bridge formed between the charged guanidinium group and the carboxylates lining the pocket. The analog is nearly a million times less potent on human sialidases, leading to its approval and use as the anti-influenza drug Relenza (Chapter 57). It does not work on bacterial sialidases, however, because the equivalent pocket is filled with an arginine group and is only a modest (low-micromolar) inhibitor of the human neuraminidases. Next-generation analogs of Relenza including the close derivative laninamivir as well as the guanidinium-containing peramivir have also been developed.
The presence of the guanidinium group requires these drugs to be either inhaled or injected. Subsequent studies focused on dispensing this functionality. Replacement of the pyranose ring with a cyclohexene to mimic the planar ring of the proposed intermediate in hydrolysis, protecting the carboxylate as an ester that is hydrolyzed after ingestion, and replacing the guanidinium with an amine led to an orally active, widely used analog, the anti-influenza drug Tamiflu (Figure ; Chapter 57). Crystal structures for other sialidases have allowed the design of species-specific analogs. This rational approach to inhibitor design holds great potential, not only for neuraminidase inhibitors, but also, as described above, for O-GlcNAcase and OGT inhibitors. These examples illustrate how structure can guide design of carbohydrate-based inhibitors as well as heterocyclic inhibitors that have been more commonly advanced as drugs.
These advances have stimulated greater interest into influenza neuraminidase as well as the four human neuraminidases, which have poorly understood physiological roles. A major challenge associated with viral neuraminidase inhibitors—most strikingly, Tamiflu—is the rapid development of mutations that confer resistance. An alternative approach to circumvent this problem has been to develop mechanism-based inhibitors that covalently inhibit the enzyme. One example, FaxGuDFSA, has a fluoride leaving group, the displacement of which by the neuraminidase forms a glycosyl–enzyme intermediate. This intermediate has an appreciable half-life because the electronegative fluorine destabilizes the oxocarbenium ion-like transition state that leads to its breakdown. Accordingly, this compound can protect mice against infection. Moreover, the emergence of resistant influenza strains is slower than against Tamiflu.
In addition to driving increased effort against influenza neuraminidase, efforts to create inhibitors of the human neuraminidases have benefited from the fact that DANA and Relenza are modest and nonselective inhibitors of these enzymes. Medicinal chemistry efforts using DANA as a starting point has yielded useful inhibitors for these enzymes by exploiting structural differences in their active sites. One example, C9-4HMT-DANA (Figure ), is the most potent and selective inhibitor of any of these enzymes having an ∼100 nm Ki value and exhibiting 500-fold selectivity over any of the other family members. Doubtless, structures of these human enzymes are likely to accelerate the development of new selective inhibitors.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Manfred Wulhrer.
FURTHER READING
Brown JR, Crawford BE, Esko JD. 2007. Glycan antagonists and inhibitors: a fount for drug discovery. Crit Rev Biochem Mol Biol
42: 481–515. doi:10.1080/10409230701751611 [
PubMed: 18066955] [
CrossRef]
Gloster TM, Vocadlo DJ. 2012. Developing glycan processing enzyme inhibitors as enabling tools for glycobiology. Nat Chem Biol
8: 683–694. doi:10.1038/nchembio.1029 [
PubMed: 22810773] [
CrossRef]
Tu Z, Lin YN, Lin CH. 2013. Development of fucosyltransferase and fucosidase inhibitors. Chem Soc Rev
42: 4459–4475. doi:10.1039/c3cs60056d [
PubMed: 23588106] [
CrossRef]
Kallemeijn WW, Witte MD, Wennekes T, Aerts JM. 2014. Mechanism-based inhibitors of glycosidases: design and applications. Adv Carbohydr Chem Biochem
71: 297–338. doi:10.1016/b978-0-12-800128-8.00004-2 [
PubMed: 25480507] [
CrossRef]
Selnick HG, Hess JF, Tang C, Liu K, Schachter JB, Ballard JE, Marcus J, Klein DJ, Wang X, Pearson M,, et al.
2019. Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. J Med Chem
62: 10062–10097. doi:10.1021/acs.jmedchem.9b01090 [
PubMed: 31487175] [
CrossRef]
Howlader MA, Guo T, Chakraberty R, Cairo CW. 2020. Isoenzyme-selective inhibitors of human neuraminidases reveal distinct effects on cell migration. ACS Chem Biol
15: 1328–1339. doi:10.1021/acschembio.9b00975 [
PubMed: 32310634] [
CrossRef]