Glycosyltransferases are the biosynthetic enzymes responsible for the construction of interglycosidic linkages and glycosidases catalyze the opposite reaction, hydrolysis of interglycosidic linkages. The diversity of natural glycans is reflected by the numerous glycosyltransferases and glycosidases encountered in nature, each exhibiting a defined substrate specificity. Natural glycans are often encountered in heterogeneous form and often produced in minute amounts making their isolation and characterization from natural sources often cumbersome. Therefore, glycobiology relies heavily on synthetic glycans, and synthetic methodology to produce glycans has witnessed tremendous progress (see Chapter 53). Glycosyltransferases and glycosidases offer advantages in the construction of glycans as these biocatalysts are very powerful under controlled conditions. This chapter summarizes recent developments in the use of (mutant) glycosidases and glycosyltransferases for the synthesis of tailored glycans, including combinations of both enzyme classes and in conjunction with chemically synthesized intermediates are presented.
MECHANISM OF GLYCOSYLTRANSFERASES AND GLYCOSIDASES
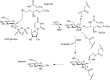
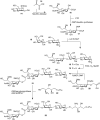
The mechanism of action of glycosyltransferases resembles the way an interglycosidic linkage is installed by chemical synthesis (Chapter 53). An activated donor saccharide, represented by UDP-glucose (Figure ) is condensed with an acceptor moiety (here, ceramide) to give, after expulsion of the leaving group (here, UDP), the glycoconjugate (glucosylceramide). In contrast to chemical synthesis, the regioselectivity is regulated by the enzyme active site that also selects for the donor and the acceptor. The glucosylceramide synthase (GCS)-catalyzed synthesis of glucosylceramide (Figure ) proceeds with “inversion” of the anomeric configuration, with the α-glucosidic linkage in UDP-glucose transformed into the β-glucosidic linkage in glucosylceramide. Nature employs a limited set of donor glycosides—most prominently the sugar nucleotides for Leloir-type glycosyltransferases, next to UDP-glucose for instance CMP-sialic acid, GDP-mannose—and the nature of the glycosyltransferase determines whether glycosylation proceeds with “retention” or “inversion” of configuration at the anomeric center. The mechanisms employed by most Leloir glycosyltransferases are now resolved.
Formation and hydrolysis of the glycosphingolipid, glucosylceramide. (GCS) Glucosylceramide synthase, (UDP) uridine diphosphate glucose, (GBA) acid glucosylceramidase.
Glucosylceramide is hydrolyzed by the lysosomal exo-glucosidase, acid glucosylceramidase (GBA). Hydrolysis takes place with “retention of configuration” and is the result of a double displacement mechanism. Upon protonation of the aglycon, ceramide is displaced in an SN2 substitution–like process to yield a covalent enzyme-glycoside adduct. Upon entry of water in the enzyme active site, the formed glycosyl linkage is hydrolyzed in another SN2-like process to release glucose from the enzyme active site. Next to retention of configuration, the hydrolysis of interglycosidic bonds can also take place with “inversion of configuration” and is normally the result of an SN2 displacement–like process of a protonated aglycon by water. Although not necessarily relevant for the product formation in nature (sugar hemiacetals being prone to mutarotation at physiological pH), the different mechanisms employed by “retaining glycosidases” (involvement of a covalent intermediate) and “inverting glycosidases” (no covalent intermediate involved) bears consequences for their use in glycan synthesis.
GLYCOSYLTRANSFERASE-MEDIATED SYNTHESIS OF GLYCANS
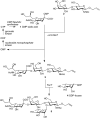
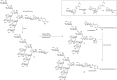
The use of Leloir-type glycosyltransferases in glycan synthesis requires access to the natural donor glycosides, which are sugar nucleotides. Thus, the intrinsic advantage of glycosyltransferase-mediated synthesis (excellent regio- and stereoselectivity) can be offset by the limitation on the access to the required donor glycosides. However, this challenge may be overcome by in situ biosynthesis/regeneration of the consumed sugar nucleotides. The power of glycosyltransferase-mediated glycan synthesis was demonstrated in 1992 by the synthesis of sialyl-Lewis x derivative 5 (Figure ). Allyl lactoside 2, derived by chemical synthesis (see Chapter 53), is reacted with CMP-sialic acid 1 using a recombinant α-2,3-sialyltransferase (α-2,3-SiaT) as the catalyst. The resulting trisaccharide 3 is further extended with GDP-fucose 4 as the donor and recombinant fucosyltransferase (FucT) as the catalyst to deliver allyl sialyl-Lewis x.
Glycosyltransferase-mediated synthesis of sialyl-Lewis x. (CMP) Cytidine monophosphate, (CDP) cytidine diphosphate, (CTP) cytidine triphosphate.
In the first step of the assembly line, the expensive sugar nucleotide, CMP-sialic acid, is consumed, and on transfer of the sialic acid, cytosine monophosphate (CMP) is produced. With the aid of two consecutive kinases (nucleoside monophosphate kinase and pyruvate kinase), CMP can be transformed in situ into the corresponding triphosphate (CTP), which is then condensed by the enzyme CMP-sialic acid synthetase with sialic acid to regenerate CMP-Neu5Ac 2.
Glycosyltransferase-mediated synthesis of ganglio-oligosaccharides.
The synthesis of GalNAc-GD1a heptasaccharide 7 equipped with a biotin at the reducing end (replacing the sphingolipids present in the natural product) was accomplished by submitting synthetic lactoside 6 to the consecutive action of four glycosyltransferases, one of which (α-2,3-SiaT) was employed twice (Figure ). By using this method with various donor sugar nucleotides and glycosyltransferases, a comprehensive series of glycosphingolipid glycans and their analogs have been obtained. The methodology, especially with respect to enzymatic sialic acid introduction, is competitive when compared to chemical gangliosides synthesis.
Chemoenzymatic synthesis of a library of mammalian N-glycans.
The synthesis of complex, asymmetrically branched mammalian N-glycans was accomplished using combined chemical and glycosyltransferase-mediated enzymatic synthesis. As an example, decasaccharide 8 was prepared via contemporary solution phase chemical oligosaccharide synthesis (see Chapter 53). The asymmetrically branched decasaccharide features two nonreducing galactopyranose moieties, one of which is introduced as the tetra-acetate (in bold) whereas the other is unprotected. Decasaccharide 8 is thus designed to allow for specific enzymatic sialylation of the unprotected galactose residue (Figure ). After saponification, the intermediate undecasaccharide is expanded to well-defined oligosaccharide 9 making use of the substrate specificity of α-1,3-fucosyltransferase (α-1,3-FucT, introduction of two fucopyranoses), β-1,4-galactosyltransferase (β-1,4-GalT, twice), β-1,3-N-acetylglucosaminetransferase (β-1,3-GlcNAcT), and finally the sialyltransferase, ST6Gal-I. The chemoenzymatic strategy proved flexible and allowed for the generation of diverse N-glycans in which the reducing end is available for bioconjugation and for the preparation of glycan microarrays for protein binding studies.
FROM TRANSGLYCOSYLATION TO GLYCOSYNTHASES
In contrast to glycosyltransferase-mediated reactions, in which the equilibrium is shifted predominantly to (natural) product formation because of the intrinsic reactivity of donor glycosides, the equilibrium in a glycosidase-mediated reaction can be influenced such that the reaction proceeds in the opposite direction.
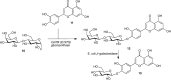
Under physiological conditions, with high water concentrations, glycosidases hydrolyze glycosidic linkages to produce the corresponding hemiacetal, either with retention (Figure ) or inversion of configuration at the anomeric center. Performing a glycosidase reaction in partly nonaqueous conditions, by addition of large excess of aglycon, by inducing kinetic conditions or by a combination of these allows for partial reversal of the reaction equilibrium. By this means and in a “transglycosylation” event, glycans can be constructed. Disadvantages of this method are that reaction conditions may be adverse to enzyme reactivity and/or stability and, moreover, that the formed product is in essence a substrate for glycosidase-catalyzed hydrolysis. This caveat can be circumvented by making use of mutant glycosidases (Figure ) in which the catalytic nucleophile in case of retaining glycosidases is mutated to an innocent bystander (depicted is an Asp to Ala substitution). Such a “glycosynthase” can be used to react a synthetic donor glycoside that bears the anomeric configuration corresponding to the intermediate enzyme glycosyl covalent adduct (see Figure ) with an appropriate nucleophile to construct a desired glycosidic linkage. The main advantage of this strategy is that, in principle, the mutant glycosidase has largely lost the ability to hydrolyze the formed product, because of the absence of the catalytic nucleophile. Many retaining glycosidases and, in recent years, inverting glycosidases have been mutated to glycosynthases, which have been used to produce a diverse array of glycans.
(A) Equilibrium in a retaining β-glucosidase. (B) Mutant-retaining β-glucosidase in which the catalytic nucleophile is substituted for a nonparticipating amino acid allows for the construction of β-glucosides.
The strategy is exemplified by the synthesis of flavonoid glucoside 13 (Figure ). Synthetic α-fluorolactoside 10 is treated with phenol 11 in the presence of the E197S mutant of the Humicola insolens Cel7B endoglucosidase. This glycosynthase proved highly flexible with respect to the acceptor phenol, allowing for the construction of a small library of flavonoid glycosides represented by phenolic disaccharide 12. Subsequent enzymatic removal of the nonreducing galactospyranoside provided flavonoid glucoside 13.
Glycosynthase-mediated synthesis of flavonoid glycosides.
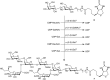
By combining the strengths of glycosyltransferases, glycosynthases, and chemical synthesis, the total synthesis of ganglioside LLG-3 from the neurogenic starfish was accomplished (Figure ). N-Carboxybenzyl(Cbz)-protected CMP-sialic acid 17 was prepared by first performing a Neu5Ac aldolase-catalyzed aldol reaction of mannosamine derivative 14 and pyruvate 15. The resulting sialic acid derivative 16 was reacted with CTP under the agency of CMP-Neu5Ac synthetase to give donor sialoside 17. In a sialyltransferase-catalyzed reaction, compound 17 was condensed with α-fluorolactoside 10 to give trisaccharide 18, in which the amine was unmasked by palladium-catalyzed hydrogenolysis to give trisaccharide, equipped with an anomeric fluoride for glycosynthesis and an amine for chemical amide bond formation. The sequence commenced by condensation of synthetic sialic acid derivative 20 with the free amine in 19 under amide bond-forming conditions, to give 21. Condensation of tetrasaccharidyl fluoride 21 with the double (E351S/D341Y) mutant of the bacterial endoglycosidase EGCase II gave lysolipid 23 in good yield. The free amine in 23 can be condensed with a fatty acid or alternatively with a fluorescent reporter group.
A combined glycosyltransferase/glycosynthase/chemical synthesis of a lysosphingolipid.
Glycosynthases derived from endoglycosidases have been used for the construction of structurally well-defined N-glycoproteins (Figure ). Hexosaminidases, both exo- and endo-types, hydrolyze N-acetylglucosamine-containing glycosidic linkages with retention of the anomeric configuration. In contrast to most other retaining glycosidases, some retaining hexosaminidases do not employ an enzyme active site nucleophile in the nucleophilic displacement of the aglycon, but rather utilize the N-acetyl group in the substrate for this purpose (Figure , insert). As a result, an intermediate oxazolinium ion intermediate is produced that after nucleophilic attack of water yields the hemiacetal with retention of configuration. Endo-hexosaminidases can be employed in transglycosylation reactions and, in mutant form, as glycosynthases.
Glycosynthase mediated synthesis of homogeneous peptide N-glycans.
A demonstration of an endo-hexosaminidase-derived glycosynthase in action is given by the synthesis of Man9GlcNAc2-glycopeptide 28 from a glycoprotein from HIV-1. The sequence of steps commences with homogeneous Man9GlcNAc2Asn 24 that can be prepared by exhaustive proteolytic digestion of soybean glycoproteins by pronase. Wild-type endohexosaminidase, Endo-A, recognizes this high-mannose N-glycan and hydrolyses the GlcNAc-GlcNAc glycosidic linkage, to yield Man9GlcNAc 25. After peracetylation, treatment with trimethylsilyl bromide and borontrifluoride etherate, and global deprotection provided oxazolidinium ion 27. This compound is a good substrate for mutant (N175A) endohexosaminidase, endo-M, which was identified by site-directed mutagenesis as a putative glycosynthase activity. In the final step, the efficacy of the endo-M glycosynthase was demonstrated by the construction of high-mannose N-glycan 28 from 26 and 27.
OUTLOOK
Enzymatic synthesis has proven to be a powerful methodology for the construction of complex glycans. Chemoenzymatic synthesis can be a suitable alternative for chemical synthesis, as demonstrated by the many applications of sialyltransferases for the construction of complicated or intractable sialic acid-containing glycans. The marriage of enzymatic synthesis and chemical synthesis is very promising. The elaboration by means of glycosyltransferases and/or glycosynthases has proven to be a highly effective strategy to prepare large and complex glycans and glycoconjugates. Advances in chemical and enzymatic syntheses provide the means to construct most natural or designed glycans.
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Nathaniel Finney and David Rabuka and appreciate helpful comments and suggestions from Xi Chen.
FURTHER READING
Ichikawa Y, Lin YC, Dumas DP, Shen GJ, Garcia-Junceda E, Williams MA, Bayer R, Ketcgam C, Walker LE, Paulson JC, Wong CH. 1992. Chemical-enzymatic synthesis and conformational analysis of sialyl Lewis X and derivatives. J Am Chem Soc
114:
9283–9298. doi:10.1021/ja00050a007 [
CrossRef]
Henrissat B, Davies G. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol
7:
637–644. doi:10.1016/s0959-440x(97)80072-3 [
PubMed: 9345621] [
CrossRef]
Williams SJ, Withers SG. 2002. Glycosynthases: mutant glycosidases for glycoside synthesis. Aust J Chem
55:
3–12. doi:10.1071/ch02005 [
CrossRef]
Coutinho PM, Deleury E, Davies GJ, Henrissat B. 2003. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol
328:
307–317. doi:10.1016/s0022-2836(03)00307-3 [
PubMed: 12691742] [
CrossRef]
Blixt O, Vasiliu D, Allin K, Jacobsen N, Warnock D, Razi N, Paulson JC, Bernatchez, Gilbert M, Wakarchuk W. 2005. Chemoenzymatic synthesis of 2-azidoethyl-ganglio-oligosaccharides GD3, GT3, GM2, GD2, GT2, GM1, and GD1a. Carbohydr Res
340:
1963–1972. doi:10.1016/j.carres.2005.06.008 [
PubMed: 16005859] [
CrossRef]
Rising TWDF, Claridge TDW, Moir JWB, Fairbanks AJ. 2006. Endohexosaminidase M: exploring and exploiting enzyme substrate specificity. ChemBioChem
7:
1177–1180. doi:10.1002/cbic.200600183 [
PubMed: 16800015] [
CrossRef]
Bennet CS, Wong CH. 2007. Chemoenzymatic approaches to glycoprotein synthesis. Chem Soc Rev
36:
1227–1238. doi:10.1039/B617709C [
PubMed: 17619683] [
CrossRef]
Yang M, Davies GJ, Davis BG. 2007. A glycosynthase catalyst for the synthesis of flavonoid glycosides. Angew Chem Int Ed
46:
3885–3888. doi:10.1002/anie.200604177 [
PubMed: 17304599] [
CrossRef]
Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. 2008. Mutants of
Mucor hiemalis endo-β-
N-acetylglucosaminidase show enhanced transglycosylation and glycosynthase-like activities. J Biol Chem
283:
4469–4479. doi:10.1074/jbc.m707137200 [
PubMed: 18096701] [
CrossRef]
Pukin AV, Florack DEA, Brochu D, van Lagen B, Visser GM, Wennekes T, Gilbert M, Zuilhof H. 2011. Chemoenzymatic synthesis of biotin-appended analogues of gangliosides GM2, GM1, GD1a and GalNAc-GD1a for solid-phase applications and improved ELISA tests. Org Biomol Chem
9:
5809–5815. doi:10.1039/c1ob00009h [
PubMed: 21727969] [
CrossRef]
Schmaltz RM, Hanson SR, Wong CH. 2011. Enzymes in the synthesis of glycoconjugates. Chem Rev
111:
4259–4307. doi:10.1021/cr200113w [
PubMed: 21749134] [
CrossRef]
Rich JR, Withers SG. 2012. A chemoenzymatic total synthesis of the neurogenic starfish ganglioside LLG-3 using an engineered and evolved synthase. Angew Chem Int Ed
51:
8640–8643. doi:10.1002/anie.201204578 [
PubMed: 22821741] [
CrossRef]
Armstrong A, Withers SG. 2013. Synthesis of glycans and glycopolymers through engineered enzymes. Biopolymers
99:
666–674. doi:10.1002/bip.22335 [
PubMed: 23821499] [
CrossRef]
Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons GJ. 2013. A general strategy for the chemoenzymatic synthesis of asymmetrically branched
N-glycans. Science
341:
379–383. doi:10.1126/science.1236231 [
PMC free article: PMC3826785] [
PubMed: 23888036] [
CrossRef]
Zhang J, Chen C, Gadi MR, Gibbons C, Guo Y, Cao X, Edmunds G, Wang S, Liu D, Yu J, Wen L, Wang PG. 2018. Machine-driven enzymatic oligosaccharide synthesis by using a peptide synthesizer. Angew Chem Int Ed Engl
57:
16638–16642. doi:10.1002/anie.201810661 [
PMC free article: PMC6402783] [
PubMed: 30375138] [
CrossRef]