This chapter focuses on less-easily categorized types of glycan linkages that occur on certain proteins or domains. O-linked sugars on epidermal growth factor (EGF)-like repeats (O-fucose, O-glucose, and O-GlcNAc) regulate Notch signaling and the functions of several other proteins. O-Fucosylation of thrombospondin type-1 repeats (TSRs) is required for folding of these domains in a number of secreted matricellular proteins. O-mannosylation of α-dystroglycan is essential for interactions with several extracellular matrix (ECM) proteins. Defects in the glycosyltransferases that add these glycans (O-fucose, O-glucose, O-GlcNAc, and O-mannose) result in human diseases. C-Mannosylation is a unique form of glycosylation in which mannose is linked through a carbon–carbon bond to tryptophan. O-Linked Glc-Gal disaccharides are added to hydroxylysine residues and play an important role in collagen fibril formation. Although the glycans described here are found on relatively few glycoproteins, they play specific and important roles in biology.
DISCOVERY OF NOVEL TYPES OF GLYCOSYLATION
In glycoproteins, the linkage between the first sugar of a glycan and the protein defines its glycosylation class (Chapter 1, Figure 1.7) including the common GlcNAc-N-Asn, GalNAc-O-Ser/Thr, and Xyl-O-Ser linkages present in glycoproteins and proteoglycans. The novel, nonclassical glycan Glcβ1–3Fucα-O-Thr was first discovered in human urine but garnered little interest at the time. However, finding O-fucose directly linked to various clotting proteins and signaling receptors, such as Notch, sparked considerable interest. Monoclonal antibodies that detect glycans on specific proteins such as α-dystroglycan provided tools to identify other novel glycans. In addition, mass spectrometry revealed unusual protein modifications, such as mannose linked to protein as a C-glycoside. Table describes many of the less common linkages synthesized in the endoplasmic reticulum (ER)–Golgi secretory pathway. Chapters 18 and 19 describe the very few known glycosylation linkages synthesized in the nucleus and cytoplasm.
Other classes of eukaryotic glycoprotein glycosylation
O-LINKED MODIFICATIONS IN EGF REPEATS
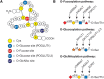
EGF repeats, also known as EGF domains, are small protein domains (∼40 amino acids) defined by six conserved Cys residues, which form three disulfide bonds (Figure ). They are found in a few hundred cell-surface and secreted proteins in metazoans and, depending on their sequence, may be modified with O-glycans as described in Table . Proteins with EGF repeats harboring these O-glycans include several involved in blood clot formation and dissolution, and the Notch family of receptors and canonical Notch ligands (Delta and Serrate/Jagged) involved in cell fate decisions. These glycan modifications are important because they regulate signal transduction during embryonic development and adult organ maintenance, cell differentiation, and the growth of several cancers. Moreover, mutations in several enzymes involved in the addition or elongation of these glycans have been found in human diseases (Chapter 45).
Modifications of epidermal growth factor (EGF) repeats. (A) EGF repeats can be modified by O-fucose, O-glucose, and O-GlcNAc. A schematic representation of an EGF repeat is shown: (yellow) conserved Cys residues; (gray lines between the cysteines) disulfide-bonding (more...)
The signaling pathway best known to be regulated by O-glycans on EGF repeats is the Notch signaling pathway. Notch was originally identified in Drosophila, and homologs have been found in all metazoans, with four Notch receptors in mammals. Activation of Notch signal transduction is controlled at numerous levels, and dysregulation of Notch signaling results in a number of human diseases, including several types of cancer and a variety of developmental disorders. Two classes of canonical ligands bind to and activate Notch signaling in Drosophila: Delta and Serrate. Mammals have three Delta-like homologs (DLL1, DLL3, and DLL4), and two Serrate homologs called Jagged 1 and 2 (JAG1 and JAG2). These ligands are single-pass transmembrane glycoproteins that bind to and trans-activate Notch receptors on an adjacent cell. Binding of Notch and ligands in the same cell often results in cis-inhibition of the pathway. Work in recent years has shown that the O-glycans on Notch receptors affect both trans-activatory and cis-inhibitory interactions between Notch receptors and their ligands. The extracellular domain of Notch contains up to 36 tandem EGF repeats (Figure ), many of which contain consensus sites for one or more types of O-glycan (Figure ). Notch EGF repeats can harbor O-fucose, O-GlcNAc and/or O-glucose (Figure ). As discussed below, these sugar modifications regulate various aspects of Notch signal transduction, sometimes in a partially redundant manner and sometimes via distinct mechanisms.
Extracellular domain of a generic Notch receptor showing the numerous sites for POFUT1 and POGLUT1 modifications that are evolutionarily conserved in Drosophila Notch, mouse NOTCH1 and NOTCH2, and human NOTCH1 and NOTCH2. Epidermal growth factor (EGF) (more...)
O-Fucose Glycans
The α-linked O-fucose modification immediately precedes the third conserved Cys of certain EGF repeats (Figure ). The consensus motif for O-fucosylation is C2X4(S/T)C3 (C2 and C3 are the second and third conserved Cys of the EGF repeat). Nearly 100 proteins in mouse or human databases contain this sequence. O-Fucose can be elongated to a tetrasaccharide (Siaα2–3/6Galβ1-4GlcNAcβ1-3Fucα-O-Ser/Thr) in certain contexts (e.g., EGF1 from human clotting factor IX and EGF12 from mouse NOTCH1) (Table ), but not in others (e.g., EGF1 from human clotting factor VII and EGF5 from mouse NOTCH1).
Protein O-fucosyltransferase 1 (POFUT1) transfers fucose from GDP-Fuc to a properly folded EGF repeat containing the appropriate consensus sequence (Figure ). O-Fucose can be elongated by a β1-3 N-acetylglucosaminyltransferase (β3GlcNAcT) specific for O-fucose residues in a properly folded EGF repeat. The gene encoding the O-fucose-EGF β3GlcNAcT was originally identified in Drosophila as a modifier of Notch signaling called fringe. There are three mammalian homologs: Manic fringe (MFNG), Lunatic fringe (LFNG), and Radical fringe (RFNG). Each of the Fringe proteins catalyzes the transfer of GlcNAc from UDP-GlcNAc to O-fucose on an EGF repeat. The GlcNAc can be further elongated by a β4GalT (e.g., B4GALT1) and capped with a sialic acid, probably using the same sialyltransferases (α2-3/6SiaTs) as those that act on N- or other O-glycans (Chapter 14). Elongation past the disaccharide has not been observed to date on Drosophila Notch.
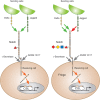
The O-fucose glycans play important roles in the Notch signal transduction pathway. Elimination of POFUT1 in Drosophila or mice results in embryonic lethality with developmental defects reminiscent of loss of Notch signaling. Moreover, cell-based assays and analysis of knock-in alleles of Drosophila Notch and mouse Notch1 lacking specific O-fucosylation sites indicate that POFUT1 and its downstream enzymes primarily, if not exclusively, regulate Notch signaling by adding O-fucose glycans to Notch receptors. Heterozygous mutations in the human POFUT1 gene cause a rare autosomal dominant skin discoloration disease called Dowling–Degos disease type 2 (DDD2; OMIM 615327). A homozygous POFUT1 variant causes a developmental disease termed POFUT1-CDG. POFUT1 is localized to the ER, in which it only modifies properly folded EGF repeats, suggesting that O-fucosylation may have some role in quality control. The Drosophila homolog of POFUT1 has chaperone activity and is proposed to play a role in the proper folding and trafficking of Notch. Loss of POFUT1 also affects the trafficking of Notch receptors to the surface of mammalian cells to varying extents. Structural studies reveal that the O-fucose on EGF12 directly participates in canonical Notch ligand binding. Of note, mutating this O-fucosylation site in Drosophila Notch and in mouse NOTCH1 results in embryonic lethality, with phenotypes milder than loss of Notch/NOTCH1 in each system, suggesting that other Notch O-fucosylation sites also contribute to the effects of POFUT1 on Notch signaling.
The O-fucose on Notch EGF repeats can be elongated by the activity of Fringe enzymes, which transfer a GlcNAc residue to O-fucose on Notch receptors (Figure ). Elimination of Fringe in Drosophila causes defects in wing, eye, and leg development, and ablation of Lfng in mice causes a severe defect in somite formation. Notably, Pofut1 loss-of-function phenotypes are more severe and broader than Fringe loss-of-function phenotypes in both mouse and fly embryos, indicating that elongation of O-fucose by Fringe proteins only regulates a subset of Notch-dependent developmental processes. Mutations in human LFNG result in malformed vertebrae and ribs (spondylocostal dysostosis type 3; OMIM 609813). Surprisingly, Fringe reduces Notch activation by Serrate/Jagged ligands, but potentiates Notch activation by Delta ligands (Figure ). For instance, Fringe-mediated elongation of O-fucose on EGF8 and EGF12 of the ligand-binding domain clearly enhances the affinity between Notch and Delta ligands, resulting in enhanced Notch activation. The fucose on NOTCH1 EGF12 functions like a surrogate amino acid as it directly participates in ligand binding. Regulation of Notch signaling by O-fucose glycans provides one of the clearest examples of how a signal transduction pathway may be regulated by altering the glycosylation of a receptor.
Notch signaling pathway. Notch exists on the cell surface as a heterodimer in which the extracellular domain is tethered to the transmembrane and intracellular domain by noncovalent, calcium-dependent interactions. Notch is activated by ligand (members (more...)
O-Glucose Glycans
The first β-linked O-glucose modification of EGF repeats described occurs between the first and second conserved Cys at the consensus sequence C1XSX(P/A)C2 (Figure ), which is found in approximately 50 proteins in mouse or human databases. This O-glucose glycan typically exists as the trisaccharide Xylα1–3Xylα1–3Glcβ-O-Ser, although mono- and disaccharide forms are also seen. The human gene encoding this protein O-glucosyltransferase is POGLUT1 (rumi in flies). Mutations in Poglut1/rumi cause developmental defects similar to Notch1/Notch mutations in mice and flies, although in both organisms, O-glucosylation is important in other glycoproteins as well. Similar to POFUT1, heterozygous mutations in human POGLUT1 cause an autosomal dominant skin pigmentation disease termed Dowling–Degos disease type 4 (DDD4; OMIM 615696). Moreover, recessive mutations in POGLUT1 have been identified in a new form of limb-girdle muscular dystrophy lacking any other phenotypes in the skin or other organ systems known to depend on Notch signaling (LGMDR21; OMIM 617232). Like POFUT1, POGLUT1 is localized to the ER and requires a properly folded EGF repeat as substrate. Although loss of Poglut1/rumi does not affect the surface expression of Notch in fly tissues and of NOTCH1 in several mammalian cell lines, it does affect NOTCH1 surface expression in some other mammalian cell lines. O-Glucosylation does not appear to affect Notch binding to canonical ligands and is likely required for Notch cleavage.
The genes encoding the xylosyltransferases are glucoside α1-3 xylosyltransferase (GXYLT1 and GXYLT2, shams in flies) and xyloside α1-3 xylosyltransferase (XXYLT1, Xxylt in flies) (Figure ). Genetic and cell-based studies on Notch xylosylation in flies have shown that (1) although addition of O-glucose to Notch promotes Notch signaling, addition of xylose residues to O-glucose on EGF16-20 of Drosophila Notch inhibits Notch signaling; (2) xylose residues on EGF16-20 selectively reduce the binding of Notch to trans-Delta, without affecting the binding of Notch to trans-Serrate or to cis-ligands; and (3) although loss of one copy of the fly GXYLT/shams does not have any phenotype by itself, it can affect Notch signaling when Notch is haploinsufficient or overexpressed.
Recently, a second β-linked O-glucose modification site has been identified between the third and the fourth Cys of the EGF repeats at the putative consensus sequence C3XNTXGSFXC4 (Figure ), which is found in more than 30 proteins in mouse and human databases. Homologs of POGLUT1, termed POGLUT2 and POGLUT3, are responsible for modifying this site. The O-glucose at this site has not been reported to be elongated past the monosaccharide.
O-GlcNAc Glycans
Although β-linked O-GlcNAc is very common on proteins in the nuclear and cytosolic compartments (Chapter 19), the same modification can be found in comparatively few membrane and secreted proteins. Addition of O-GlcNAc occurs between the fifth and sixth conserved Cys of an EGF repeat at the putative consensus C5XXXX(S/T)GX2-3C6 (Figure ) in mouse and Drosophila Notch. However, sites have been mapped on only a few proteins. The O-GlcNAc can be elongated by β1,4Gal followed by a sialic acid to form Siaα2-3/6Galβ1-4GlcNAc. The EGF-specific O-GlcNAc transferase, EOGT, is distinct from OGT, the GlcNAc-transferase that modifies nuclear and cytoplasmic proteins (Chapter 19). Inactivation of Drosophila Eogt is pupal lethal and causes a wing-blistering defect if deleted in the wing. The Drosophila phenotype can be explained by an EOGT substrate Dumpy and no obvious Notch-deficient phenotypes have been observed. However, genetic interactions between eogt and Notch pathway gene mutations occur in the fly and mutations in human EOGT result in skin and skeletal problems in AOS (Adams–Oliver syndrome type 4; OMIM 615297), a disorder with links to the Notch pathway. Consistently, EOGT in mice regulates vascular development and integrity by potentiating Delta-like ligand-mediated Notch signaling.
O-FUCOSE MODIFICATION OF THROMBOSPONDIN TYPE 1 REPEATS (TSRs)
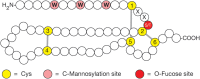
O-Fucose is also found on TSRs. Similar to EGF repeats, TSRs are small protein domains (50–60 amino acids) with six conserved Cys that form three disulfide bonds (Figure ). Like EGF repeats, TSRs are found in a subset of cell-surface and secreted proteins in metazoans, and they appear to function in protein–protein interactions. The O-fucose site occurs between the first and second conserved Cys residues at the putative consensus sequence C1X2(S/T)C2 in TSRs in about 50 proteins (Table ). The O-fucose on TSRs can be elongated to form the disaccharide Glcβ1-3Fucα-O-Ser/Thr, first observed in 1975 in glycosides from human urine. The enzyme that fucosylates TSRs is POFUT2. It is expressed in many cells and tissues and is distinct from POFUT1. Elimination of Pofut2 in mice results in early embryonic lethality with defects in gastrulation. The gene encoding the β3-glucosyltransferase that elongates O-fucose on TSRs is B3GLCT. Mutations in B3GLCT in humans result in a multifaceted severe developmental disorder known as Peters-plus syndrome (OMIM 261540). Elimination of B3glct in mice phenocopies many of the defects seen in Peters-plus syndrome. TSRs are also often frequently modified with C-mannose (Figure ), although the relationship between the two modifications is not yet clear. A number of biologically interesting proteins are known or predicted to contain this modification, including thrombospondin-1 and -2, all ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs; ADAMTS 1–20) and ADAMTS-like proteins, properdin, and F-spondin (Table ). O-Fucosylation of TSRs in several proteins including ADAMTS 13 and ADAMTS-like 1 is needed for their secretion, suggesting that O-fucosylation of TSRs may be involved in quality control or folding, similar to the predicted role of O-fucose on EGF repeats in Drosophila. Like POFUT1, POFUT2 is an ER-localized enzyme that only fucosylates properly folded TSRs. POFUT2 and B3GLCT appear to function in a novel ER quality control pathway required for proper folding of TSRs. In thrombospondin-1, the region of the TSR modified with O-fucose mediates interactions between TSRs and several cell-surface receptors. Thus, the presence of O-fucose in this region could influence interactions between TSRs and adjacent cells, much as O-fucose on EGF repeats regulates Notch ligand binding. TSRs allow proteins like the thrombospondins to bind to cells and/or to other components in the ECM, an essential step in the antiangiogenic functions of these proteins.
Modifications of thrombospondin type-1 repeats (TSRs). These repeats have six conserved Cys (yellow), three disulfide bonds (gray lines), and consensus sites for O-fucose (red) and C-mannose (pink) addition. W, tryptophan; S, serine; T, threonine; X, (more...)
O-MANNOSE GLYCANS
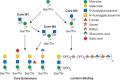
O-Mannose α-linked to protein was first identified in yeast in the 1950s (Chapter 23). Fungi elongate this mannose with a battery of mannosyltransferases to form extensions similar to those on their N-glycans. O-Mannose was first identified in mammals in 1979, linked to a rat brain proteoglycan. O-Mannose glycans account for up to one-third of all O-glycans in some mammalian tissues, including brain. O-Mannose glycans in mammals are quite varied and belong to three core classes (Figure ). Defects in O-mannosylation of α-dystroglycan, which is the major extracellular glycoprotein component of the dystrophin–glycoprotein complex, result in multiple forms of congenital muscular dystrophy (CMD). These diseases are known as the secondary dystroglycanopathies and include Walker–Warburg syndrome, muscle–eye–brain disease, Fukuyama congenital muscular dystrophy, and various forms of limb-girdle muscular dystrophy (Chapter 45). Some of the O-mannose glycans on α-dystroglycan serve as binding sites for ECM proteins that contain laminin globular domains. These same O-mannose glycans function as receptors for certain arenaviruses, and the loss of these glycans is also associated with metastasis in multiple cancers.
Biosynthetic pathway of O-mannose glycans. The O-mannose glycans belong to three core classes depending on extension from protein-linked mannose by a sole β1-2GlcNAc (core M1), by two GlcNAcs in β1-2 and β1-6 linkages (core M2), (more...)
The initiating O-mannose for α-dystroglycan is transferred by O-mannosyltransferases POMT1 and POMT2 that function together (Figure ). However, there are multiple protein O-mannosyltransferases that catalyze the addition of O-mannose to Ser/Thr residues on a host of proteins in the ER using dolichol-P-mannose as the donor. Core M1 O-mannose glycans (Figure ) are then generated by the action of a Golgi-localized N-acetylglucosaminyltransferase (POMGNT1) that adds β1-2GlcNAc to O-mannose on glycoproteins. Core M1 glycans can be further extended by enzymes that also act in other pathways, including galactosyltransferases, fucosyltransferases, glucuronyltransferases, and sialyltransferases. Core M1 glycans can also be converted to Core M2 O-mannose glycans by the addition of a β1-6GlcNAc to the O-mannose, catalyzed by the N-acetylglucosaminyltransferase MGAT5B. This new branch on core M2 glycans can also be extended by the glycosyltransferases that act on core M1 (Figure core extensions). Core M1 and M2 O-mannose glycans have been identified on multiple mammalian proteins besides α-dystroglycan.
The core M3 glycans (Figure ) possess key functional properties and have to date been identified only on the mammalian protein α-dystroglycan. Core 3 O-mannose is extended in the ER by an N-acetylglucosaminyltransferase (POMGNT2) that adds a β1-4GlcNAc. This disaccharide is a substrate for an N-acetylgalactosaminyltransferase (B3GALNT2) that adds β1-3GalNAc to the GlcNAc. This trisaccharide is a substrate for a secretory pathway kinase (POMK) that adds a 6-linked phosphate to the core mannose. The immediate next steps are the addition of a ribitol-5-P in a phosphodiester linkage to the GalNAc catalyzed by fukutin (FKTN), followed by the addition of another ribitol-5-P in a phosphodiester linkage to the first ribitol by fukutin-related protein (FKRP). A D-ribitol-5-phosphate cytidylytransferase (CRPPA) generates the sugar nucleotide CDP-ribitol that FKTN and FKRP use. RXYLT1 and B4GAT1 glycosyltransferases add a xylose and a glucuronic acid, respectively, to the outermost ribitol. This action serves to prime the core M3 glycan for the addition of a repeating disaccharide (Xyl-α3-GlcA-β3-)n that is catalyzed by the dual activity LARGE1 (or LARGE2) glycosyltransferase. This repeating disaccharide, termed matriglycan, serves as the binding site for ECM proteins containing laminin globular domains, including laminins, agrin, pikachurin, and Eyes shut homolog, as well as the spike protein of certain arenaviruses. Mutations in the genes encoding all of the enzymes involved in the synthesis of the M3 glycan have been identified in CMD patients (Chapter 45).
O-GLYCANS IN COLLAGENS
Proteins with collagen domains are modified by a disaccharide, Glcα1-2Galβ-, which is assembled on hydroxylysine (Hyl) residues. First recognized on collagen in the 1960s, this O-glycan has been observed in other proteins that have collagen-like modules, including adiponectin and pulmonary surfactant proteins. Primitive forms of collagen in sponges and sea anemones also have disaccharide-modified Hyl. The first step in the pathway is the hydroxylation of lysine to Hyl via three lysyl hydroxylases. The two-step glycosylation rapidly follows on most of the available acceptor sites. Galactosytransferases COLGALT1 and COLGALT2 initiate collagen glycosylation in the ER. Autosomal recessive mutations in COLGALT1 have been identified in two unrelated patients with a brain vessel disease associated with fragility of cerebral vessels (OMIM 618360). Importantly, similar phenotypes are observed in some patients with collagen type IV mutations, highlighting the critical role of collagen glycosylation in vascular integrity. Interestingly, although the hydroxylation and addition of galactose are conserved in all animal species examined and occur by the action of two separate enzymes, giant viruses have one bifunctional enzyme capable of hydroxylating lysine and adding glucose instead of galactose to collagen. It is worth noting that the enzyme that adds glucose to the galactose on collagens remains to be identified, although human lysyl hydroxylase 3 (LH3) has been suggested to have this activity besides hydroxylation of lysine. Mice lacking LH3 fail to glycosylate collagen IV properly, which causes embryonic lethality and deposition of misfolded collagen in the ER. Glycosylation is thought to proceed until the protein folds and assembles into the well-known collagen triple helix. Some studies suggest that the extent of glycosylation controls or influences the rate of triple-helix formation and, in turn, the size of collagen fibrils. Other studies suggest that excessive glycosylation of hydroxylysines makes them poor substrates for extracellular enzymatic deamination, the first step in cross-linking collagen.
C-MANNOSYLATION
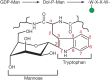
Another type of glycosylation, first detected in human urine, is called C-mannosylation. The C-1 atom of a single mannose residue is added in α-linkage to the C-2 atom of the indole moiety of tryptophan (Trp) in target proteins (Figure ). Note that this is a rare glycosidic linkage, as it is a C–C bond rather than a C–O or C–N bond. Structural analysis, biosynthetic studies in mammalian cell lines, and a specific antibody against the modification suggest that C-mannosylation is widespread, but it appears to be absent in bacteria and yeast. Conserved C-mannosylation sites are mainly found in proteins with TSRs and in type I cytokine receptors. The critical glycosylation sequence is WXXW/C. In TSRs, the C-mannosylation motif can have up to three Trp residues (WXXWXXWXXC), all three of which can be C-mannosylated (Figure ). Synthetic peptides with this motif can be C-mannosylated in vitro, and more than 300 mammalian proteins have this sequence. Several complement proteins contain TSRs with C-mannose. For instance, in properdin (which is a positive regulator of complement), 14 of 17 consensus sites are fully modified. The biosynthetic donor for C-mannosylation in the ER is Dol-P-Man, which is produced from GDP-Man (Chapter 5). A gene encoding a C-mannosyltransferase has been identified in Caenorhabditis elegans (DPY-19) and mutants in DPY-19 cause a neuroblast migration defect. Mammalian genomes encode four DPY-19 homologs, two of which have been demonstrated to have C-mannosyltransferase activity. Protein C-mannosylation is thought to occur before or during protein folding in the ER. Moreover, loss of C-mannosylation results in the ER accumulation of some DPY-19 target proteins. Therefore, C-mannosylation is likely to aid in protein folding in the ER. Given the widespread presence of C-mannosylation on proteins and recent identification of the enzymes responsible for this posttranslational modification, biological functions should be revealed soon.
Biosynthetic pathway for C-mannosylation and structural details of a C-mannosylated tryptophan. The α-linked mannose is shown in the 1C4 conformation.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Thierry Hennet, Tadashi Suzuki, and Jon Agirre.
FURTHER READING
Hennet T. 2019. Collagen glycosylation. Curr Opin Struct Biol
56: 131–138. doi:10.1016/j.sbi.2019.01.015 [
PubMed: 30822656] [
CrossRef]
Shcherbakova A, Preller M, Taft MH, Pujols J, Ventura S, Tiemann B, Buettner FF, Bakker H. 2019. C-mannosylation supports folding and enhances stability of thrombospondin repeats. eLife
8: e52978. doi:10.7554/elife.52978 [
PMC free article: PMC6954052] [
PubMed: 31868591] [
CrossRef]
Yu H, Takeuchi H. 2019. Protein
O-glucosylation: another essential role of glucose in biology. Curr Opin Struct Biol
56: 64–71. doi:10.1016/j.sbi.2018.12.001 [
PubMed: 30665188] [
CrossRef]
John A, Järvå MA, Shah S, Mao R, Chappaz S, Birkinshaw RW, Czabotar PE, Lo AW, Scott NE, Goddard-Borger ED. 2021. Yeast- and antibody-based tools for studying tryptophan C-mannosylation. Nat Chem Biol
17: 428–437. doi:10.1038/s41589-020-00727-w [
PubMed: 33542533] [
CrossRef]