The health-care costs of Alzheimer’s disease (AD) have previously been estimated to be equivalent to the combined costs of cancer, stroke and heart disease. These costs are also predicted to increase further, with continued increases in life expectancy worldwide set against the current absence of long-term effective therapies.1–3 Thus, there remains an ongoing and urgent need for the discovery or development of better treatments for people with AD, particularly in the face of a number of failed intervention trials in recent years.4,5 The aim of these treatments could be to delay the onset or slow the progression of disease, which will extend participants’ quality of life and that of their families and carers and, as a result, reduce the associated health-care burden. There is a striking benefit to be had from a treatment that has even a modest improvement: any treatment that could delay the onset of AD by 5 years could halve its prevalence.6
Scientific background
There is scientific literature spanning several decades that suggests that hypertension in mid-life7,8 and later life9 and stroke10 increase the risk of dementia. Prior to the commencement of the Reducing pathology in Alzheimer’s Disease through Angiotensin taRgeting (RADAR) trial, we11 and others12 observed that angiotensin II (AngII)-targeting drugs [AngII type 1 receptor antagonists (AT1RAs) and angiotensin I converting enzyme 1 inhibitors (ACE1-Is)] resulted in a lower incidence of AD than other types of antihypertensive drugs. AT1RAs were also significantly more beneficial than ACE1-Is in slowing the rates of progression, hospitalisation and mortality.11,12 Since the RADAR trial commenced, there has been further observational evidence of the protective role of ACE1-Is and AT1RAs in clinical AD.13,14 Renin–angiotensin system (RAS)-acting drugs have been found to be associated with lower rates of conversion of mild cognitive impairment (MCI) to AD, as well as less AD-related tau neuropathology in both post-mortem brain tissue and cerebrospinal fluid (CSF) from living AD participants recruited to the Alzheimer’s Disease Neuroimaging Initiative (ADNI),15–17 whereas another analysis of the ADNI cohort showed that one of the earliest likely pathological events in the development of cognitive decline is cerebrovascular dysfunction.18 However, not all observational studies agree.19
Although in the vast majority of people with AD the underlying cause remains unclear, there is now acknowledgement of the likely role of vascular dysfunction in the development of AD and dementia;20 however, there remains a lack of understanding as to the possible mechanisms involved. In AD, the loss of acetylcholine and neurons due to the deposition in the brain of amyloid-β (Aβ) peptide and tau pathology is key.2 As mentioned, cerebrovascular dysfunction is likely to be the earliest pathological event in the development of AD.18 Such dysfunction [a likely contributor to reduced cerebral blood flow (CBF)], loss of cerebrovascular autoregulation, ischaemia and white matter hyperintensities (WMH) are all pathologies associated with, and are predictive of, loss of cognitive function21–25 and similarly common in AD.26–28 Previous studies have shown how hypertension is associated with plasma levels of Aβ29 and AD risk.30 Thus, molecular pathways that can connect the traditional pathological hallmarks of AD with mechanisms involved in blood pressure (BP) regulation and cerebrovascular dysfunction are likely to be strong candidates for intervention in AD, where they may act in addition to or independently of cerebrovascular-mediated pathology.
Angiotensin I converting enzyme 1 (ACE1) and neprilysin, which make AngII, are enzymes that are important to the function of the RAS, and the activity of both enzymes is elevated in the brain of AD patients.31,32 ACE1 activity is also elevated in the peripheral blood in patients with AD,33 whereas ACE1 and neprilysin degrade Aβ in vitro and in vivo;2 however, variation in the ACE1 gene (ACE1) is associated with lower plasma levels of ACE1 and also with AD risk.34 The multifunctional peptide AngII is likely to be an important factor in AD, particularly when in excess in the brain,35 because AngII signalling promotes the synthesis of the inflammatory mediator tumour necrosis factor alpha (TNFα),36,37 and has anticholinergic38,39 and antiglutamatergic effects,40 all of which are major sequelae of AD pathology.
Furthermore, AngII has been shown to promote the production of Aβ and phosphorylation of tau at the same amino acid residues that give rise to neurofibrillary tangles, which can be blocked by the AT1RA losartan.41,42 More recently, we have obtained further evidence of the detrimental effects of AngII in AD. We identified that there is reduced activity of the regulatory renin–angiotensin system (rRAS) pathway that, through the action of the enzyme angiotensin I converting enzyme 2 (ACE2), works to maintain levels of AngII [which is produced by ACE1 in what is now known as the classical renin–angiotensin system (cRAS) pathway], which has been previously reviewed.20 In these recent studies on post-mortem brain tissue, ACE2 activity was significantly reduced in brain tissue from elderly patients without dementia. In contrast, in AD patients ACE2 reductions were associated with increased Aβ and tau pathology and inversely correlated with ACE1 activity and AngII levels.35,43 These more recent rRAS findings show very positively that the balance between ACE1 and ACE2 activities, which has a significant bearing on levels of AngII, is highly relevant to the neuropathological basis of AD.
Losartan, an AT1RA, is an effective antihypertensive drug over a wide range of ages. Losartan has been reported to cross the blood–brain barrier (BBB)44 and is the prototype drug developed for the class of AngII-blocking drugs that we, and others, have observed to be associated with reduced incidence of AD.11,12 Losartan also improves CBF,22 a surrogate marker of cognitive performance in humans,45–47 and limits neuronal damage following ischaemia in stroke rat models.23 Low-dose losartan (i.e. not reducing BP) has also been shown to reduce pathology and improve cognitive performance in transgenic mouse models of AD.48 Given its antihypertensive effect, it is also conceivable that it may have a beneficial effect in reducing ischaemia-mediated WMH.24
The timeliness of the RADAR trial
Since the commencement of the RADAR trial, to the best of our knowledge no clinical trials studying losartan or any related AT1RA drugs as an intervention in AD participants have been reported. The studies that preceded RADAR used losartan (50 mg) and reported modest and non-significant benefits on memory in non-demented hypertensive participants,49,50 which were thought to be independent of BP-lowering effects.51 Yet, as a hypertension trial, cognition was not the primary outcome and thus was probably underpowered for a more conventional study of cognition. There remain a limited number of systematic reviews that predate the initiation of RADAR and that have assessed the impact of BP-lowering treatments on cognitive decline.52 McGuinness et al.52 previously concluded, based on data from four randomised controlled trials (RCTs), that BP reduction was insufficient to prevent dementia and cognitive decline in hypertensive participants with no prior cerebrovascular disease. This was supported by secondary analysis of The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET) and Telmisartan Randomised AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND) trials, in which neither the ACE1-I (ramipril; Altace®, Aventis, Strasbourg, France) nor another AT1RA (telmisartan; Micardis®, Boerhinger Ingelheim, Ingelheim, Germany) reduced the risk of cognitive decline or the development of any type of dementia in participants with cardiovascular disease or diabetes.53 A small meta-analysis of hypertension treatment trials by Staessen54 also reported that BP-lowering did not reduce the dementia risk in populations with high cardiovascular morbidity. This finding was supported by a more recent systematic review of previous trials55 and two more recent meta-analyses of several observational longitudinal studies that explored whether or not different antihypertensive medications had any effect on cognitive decline or dementia.56,57 We previously thought that these studies had limited scope for translation to AD because the study participants were generally younger and were usually recruited exclusively because they had higher cardiovascular burden, and because cognitive outcome measures were not the primary objectives. In addition, the methods of cognitive assessment were less detailed or not as comprehensive as the typical battery of assessments normally used in clinical trials of AD and also rarely used objective outcome measures, such as imaging [e.g. change in various magnetic resonance imaging (MRI) measurements] or other biomarkers.58,59 We were of the opinion, prior to the commencement of RADAR that these negative findings made a larger-scale (e.g. Phase III) multicentre RCTs of an AT1RA in AD premature, and RADAR was designed to try to further provide supportive evidence.60
In the intervening years since the commencement of the RADAR trial, no formal clinical trials have tested as to whether or not losartan can change MRI-based measures of AD-related brain atrophy either alone or in combination with a standardised questionnaire-based assessment of cognition in AD participants. Indeed, the use of MRI-based approaches in AD trials has not been commonplace, although there is now growing recognition of their usefulness in trials.61,62 The protocol of a related, smaller (n = 100), Phase II three-arm US-based trial (NCT00605072), called the Antihypertensives and Vascular, Endothelial and Cognitive function (AVEC) trial, was announced around the time that the RADAR trial commenced. This smaller study recruited hypertensive participants with early (non-AD) cognitive impairment only, and it piloted the comparison of 1-year treatment of participants with candesartan (another AT1RA) with that of lisinopril (an ACE1-I) or hydrochlorothiazide (a diuretic) for their effect on memory and executive function, CBF (measured by transcranial Doppler) and central endothelial function (measured by changes in CBF in response to changes in end-tidal carbon dioxide).63 This pilot trial later reported encouragingly that the AT1RA candesartan outperformed the ACE1-I (lisinopril) and diuretic (hydrocholorthiazide) in preserving CBF and executive function in these hypertensive participants who had executive dysfunction.64 In addition, encouraging data, although again not specifically testing the impact that AngII targeting has in AD, were recently announced from the Systolic blood PRessure INtervention Trial Memory and cognition IN Decreased hypertension (SPRINT-MIND) substudy65 of the National Institutes of Health-funded Systolic blood Pressure INtervention Trial (SPRINT).66 The main SPRINT (NCT01206062) study compared the effectiveness of treating systolic BP, again in a population of hypertensive individuals (without diabetes or a history of stroke or AD) aged > 50 years, to lower than 120 mmHg (compared with the current normotensive threshold of 140 mmHg) on a primary outcome that was a composite of cardiovascular effects as a measure of all-cause mortality. The objective of the SPRINT-MIND substudy was to investigate the impact that the two BP-lowering approaches had on cognitive outcomes, which included the rates of probable dementia (the primary cognitive outcome), or on secondary cognitive outcomes of development of MCI or a composite outcome of the development of MCI and probable dementia. Another objective was to use MRI scanning to measure the impact on levels of small vessel disease in the brain, according to MRI-based measures.65 Although the main SPRINT trial was stopped early because it was becoming clear that according to its primary outcome measures clinical benefit was evident,66 there was evidence from SPRINT-MIND that lowering BP to below 120 mmHg could reduce rates of MCI and improve the composite secondary cognitive outcome measure. However, there was no significant evidence that the primary cognitive outcome, rate of probable dementia, was reduced. It is very plausible that stopping the main SPRINT trial early, and the impact that this had on follow-up periods in SPRINT-MIND, may have resulted in this aspect of the study being underpowered.65
It has been reassuring that these findings continue to show that the RADAR trial has been a timely and necessary formal investigation of the therapeutic effect of an available antihypertensive drug in a clinically defined population of AD participants. Meanwhile, the RADAR trial simultaneously allowed for a focused gold-standard way of testing the role of the RAS, particularly that of AngII, in the pathogenesis of AD. The results of RADAR will be the first to be announced in AD that test this hypothesis, to our knowledge. As such, they will provide the first clinical trial data to stand alongside what is still predominantly observational studies, testing the effects of various AT1RAs in reducing rates of MCI and/or rates of progression of dementia, or the effects of AT1RAs on other outcomes proposed to be indicative of age-related or dementia-associated cognitive decline (reviewed by Kehoe20).
Our selection of losartan as the intervention for the RADAR trial was based on several arguments.
First, we chose losartan because of the multifactorial functions described for AngII in the brain. These include being responsible for important physiological processes such as vasoconstriction, and numerous organ- and cell-based functions such as reduction in the release of the neurochemical acetylcholine, the promotion of inflammation and neuronal excitotoxicity,36–40,67 all of which are relevant to the pathogenesis of AD.
Second, selective antagonism of the AngII type 1 receptor by losartan does not inhibit ACE1 activity, which may be important because ACE1 is elevated in AD31,32 but has also been reported to degrade Aβ.31,68–70 For this reason, we did not wish to interfere with any potentially protective effects that ACE1 activity may have on Aβ pathology.2 Thus, AT1RAs may be better than ACE1-Is, at least in AD, which has been supported by various pharmacoepidemiological and pre-clinical studies,11–13,15,16,71–73 and they have been demonstrated to have beneficial effects on AD-related pathology and cognitive impairment in murine models of AD.48
Third, losartan, as the prototype AT1RA, had previously been found to be beneficial to cognitive function (in a 50-mg dose), compared with atenolol, in a small randomised study of their effects on cognitive function in very elderly hypertensive individuals (not in people with AD).49 It also had the longest use in a clinical setting, thereby providing the most expansive safety data at the time the study commenced. Losartan has been found to be well tolerated and has also been suggested, alongside ACE1-Is, to provide advantages over other antihypertensive medications with respect to quality of life in older people.74 This longevity in clinical use also offered additional economic advantages over most of the other AT1RAs in that it was already available in a generic form that would, in the event of a successful outcome, provide additional significant economic benefits in future large-scale studies and potential eventual use in a clinical setting.
Finally, given that there was evidence supporting the role of the RAS in AD that may be mediated through both cardiovascular and/or non-cardiovascular molecular mechanisms,49 we opted to test losartan in an AD population that included people with hypertension and those who were normotensive. This was intended to enhance the generalisability of any finding, while examining if any protective mechanisms may be operating independently of or in addition to the BP-lowering effects of these drugs. The decision was also a pragmatic one so that we could recruit from as typical an AD clinical population as possible and hence it would be easier to achieve our sample size. This is an important issue because a considerable proportion of AD participants would be otherwise excluded, depending on whether they had prior exposure to the intervention or related drugs that would make them ineligible, or were excluded because they had no prior hypertension. This intended approach, particularly the intention to recruit people who were normotensive, necessitated important design features in response to initial feedback on the study plan with key stakeholders, as is discussed in Patient and public involvement.
Hypothesis
The RADAR trial tested the hypothesis that blocking AngII signalling using losartan (100 mg) would reduce brain volume loss and, therefore, slow the clinical progression of mild-to-moderate AD.
Study design
This was a two-arm, double-blind, placebo-controlled, multicentre, individually randomised trial comparing the effects of 100 mg of losartan with placebo, preceded by a 1-month open-label phase. The main randomised phase allowed the testing of whether or not 12 months of treatment made any difference to structural brain changes, as measured using a comprehensive neuroimaging protocol involving MRI in both hypertensive and normotensive AD participants.
Duration of treatment and follow-up
We opted for a 12-month period of treatment and follow-up based on previous studies conducted by the ADNI,75–77 which found, using MRI, measurable brain atrophy among AD participants, equivalent to a mean 15.2 ml/year [standard deviation (SD) 8.6 ml/year]. We proposed that a relative between-group difference in atrophy rate of 25% could be considered a clinically meaningful difference that we would detect according to our sample size and power calculations. This relative difference is equivalent to an absolute difference in an annual atrophy rate of at least 3.8 ml/year in total brain volume (TBV).
Patient and public involvement
The study gained helpful input from patient and public involvement (PPI) at several stages of the study. The original design of the RADAR study was modified following consultation with some participants and carers whereby an open-label phase (described below) was introduced to address possible uncertainties that participants who were normotensive may have about taking a BP-lowering drug. Similarly, we sought feedback on the earliest versions of the study information sheets that were to be shared with participants and study partners and these were used at the outset of the study. However, as part of a nested qualitative study that was later conducted to explore other possible obstacles to recruitment,78 these information sheets were revisited and further revisions and simplifications made.
Open-label phase
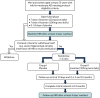
An open-label phase was introduced as a way of reassuring all participants, particularly normotensive participants, who may have been anxious about taking a BP-lowering drug, as highlighted by participants and carers initially consulted about the intended study. This phase allowed all participants to explore their ability to tolerate the intervention and suitability to be randomised, which would have involved a baseline MRI scan. Participants commenced the open-label phase if they met all of the appropriate criteria, as determined at the initial eligibility and consent visit, and subject to subsequent review of results on bloods taken at the same visit, according to protocol specified ranges. The open-label phase was therefore a 3- to 4-week pre-randomisation phase () that involved participants being on open-label active medication for a maximum of 14 days prior to randomisation.
Open-label phase of the trial.
The open-label drug was titrated upwards in participants over a period of 2 weeks, with blood testing carried out to ensure safety, consistent with clinical practice for anyone commencing AT1RAs. The participants were initially prescribed 25 mg of generic losartan (according to brands available locally) to be taken once per day for 7 days and asked to keep a diary of taking the medication and any symptoms that they experienced. They were seen after 7 days, by the research team, had further blood tests taken to ensure safety and asked questions regarding any contraindications up to that point. At this point they were prescribed 100 mg of losartan for a further 7 days and similarly asked to keep a diary of taking the medication and any symptoms that they experienced, which was again reviewed at the end of this period and blood tests taken once more. All participants were given a home use arm-cuff Omron blood pressure monitor (Omron Corporation, Kyoto, Japan) and asked to measure and record their BP daily while they were taking active medication during the 14-day open-label phase. This information was retrieved by the research team member at the visit to review the 7 days on the 100 mg of losartan dose (called the 14-day visit). Each participant was also given an information card with the details of their local research contact in case they needed these. Participants who did not experience or report any contraindications during the 7 days on 100 mg of losartan were prescribed the main study placebo to cover a further 4- to 14-day washout period.
The flexible washout period of 4–14 days was undertaken for several reasons. First, a minimum of 4 days was needed for drug washout. As, according to its original manufacturer, losartan has an estimated half-life of 2 hours and its metabolites have estimated half-lives of up to 8 hours (www.rxlist.com/cozaar-drug.htm), this allowed a generous period of at least five half-lives for elimination of the drug. Second, it allowed sufficient time to review the results of the blood tests taken to ensure safety after participants had taken the maximum dose for 7 days. Third, if the results of these safety bloods taken after the titration of losartan were as expected, there would be sufficient time to schedule a baseline MRI. Fourth, it allowed the assessment of participants’ ability to successfully take the overencapsulated intervention or placebo, which was larger than the normal tablet form, and ensure that there were no tolerability issues with the overencapsulated formulation. Finally, the provision of placebo during the washout period allowed participants to be inculcated into the trial dosage regimen that would continue in the randomised phase.
Throughout the open-label phase, consideration was given to any potential deviations from the BP levels and baseline blood biochemistry measurements taken at the eligibility visit [as assessed by the principal investigator and, if necessary, in further consultation with the RADAR clinical lead (Ian Wilkinson)]. Similarly, any concerns reported by participants or study companions to their local research team were reviewed and discussed with participants.
Randomised phase
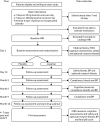
After successfully completing the open-label phase, participants were scheduled to undergo baseline MRI (see Primary outcome) and a cognitive assessment battery before being randomised (see Secondary outcomes). Following randomisation, participants underwent an identical upwards titration over 14 days, involving 7 days’ exposure to 25 mg of the intervention or placebo followed by 7 days at the 100-mg dose. Blood tests carried out to ensure safety were again taken on day 14 and more intervention was prescribed to last to the next visit at 3 months post randomisation. Subsequent visits that involved prescribing further allocations of intervention took place at approximately 3-month intervals, at which time drug adherence was also monitored by pill counts, collection of any adverse events (AEs) and conducting repeat assessments (). No dose modifications were made after the participant had entered the randomised phase. Participants who began to experience issues that they felt or that were deemed to be related to the intervention were told to stop taking the medication but remained in the study unless they expressed a desire to discontinue.
Participant procedures and data collection after confirmation of eligibility and willingness to participate. Safety bloods include measures of electrolytes, creatinine, and liver function tests according to protocol-defined ranges for inclusion/exclusion. (more...)
The randomised phase concluded 52 weeks after commencing the first dose of the intervention or placebo. A repeat 4-day washout was initiated and a final MRI was scheduled as close as possible to this as well as the final repeat assessments according to the assessment schedule (see ).
Throughout the trial period, the trial design was flexible to allow participants’ normal clinical care to continue under the supervision of primary or secondary care providers, as relevant. This included the use of other dementia-related treatments: we aimed for all participants to be on a stable dose of dementia treatments for approximately 3 months prior to study entry but also allowed any naturalistic dose adjustment that may be deemed necessary after entry to the trial.
Study setting
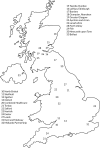
The RADAR trial was designed as a pragmatic multicentre RCT that recruited participants with mild-to-moderate AD from 23 NHS hospital trusts in the UK that routinely diagnose and treat participants with AD. lists all the recruitment sites and illustrates their geographical distribution. As some recruitment sites lacked their own MRI facilities, or could not meet the specifications needed for the facilities, patients from these sites underwent MRI at another location (see ). Sites’ eligibility to participate in RADAR was based on whether or not they had prior expertise in recruiting to clinical trials of AD and the capacity to provide MRI facilities to fulfil our neuroimaging protocol.
List of recruitment sites and scanner locations
Geographical location of RADAR sites.
Intervention
We used a maximum dose of overencapsulated losartan of 100 mg, which was titrated directly from 25 mg of overencapsulated losartan that was initially given for 7 days and reflected standard clinical practice. This titration approach was undertaken in both the randomised study and the preceding open-label phase already described. The placebo used in this study was similarly overencapsulated and made to match both doses and sourced from St Mary’s Pharmaceutical Unit (SMPU), Cardiff, UK. The maximum dose was chosen with the intention of achieving maximum exposure to the intervention and to ensure the strongest target (AngII type 1 receptor) engagement possible. There were no dose modifications after the participant had entered the randomised phase.
Given the age range of the participant population, we also made provision for the appropriate monitoring of BP. In the open-label phase, the participants themselves took daily measurements of their BP for the duration of the 14-day titration period on generic losartan. BP was also taken at almost every face-to-face meeting between participants and research teams (see ).
The intervention dose we used was higher than that reported in other studies. These studies used a dose of 50 mg of losartan and observed improvements to cognitive function in very elderly individuals with hypertension and in a secondary analysis in a hypertension study a modest benefit was reported. However, where non-significant benefits on memory were found in hypertensive participants who did not have AD49,50 were thought to be independent of BP-lowering effects.
Ethics approval and research governance
Ethics approval for the study, and approval for all subsequent amendments to the study and study protocol, was given by South East Wales Research Ethics Committee C (MREC, later renamed Wales Research Ethics Committee 2 Cardiff) in February 2013 (reference number 12/WA/033). The clinical trial authorisation was given by the Medicines and Healthcare products Regulatory Agency (MHRA) in July 2013 (reference number 18524/0223/001/0001). The trial was registered with the International Standard Randomised Controlled Trial Number (ISRCTN) ISRCTN93682878 and with the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) under the reference number 2012-003641-15. Following the introduction of the new Health Regulation Authority the study was adopted on 16 June 2016. The trial was sponsored by North Bristol NHS Trust Research & Innovation (NBT R&I; reference 2625). There were no major changes to the original protocol or the statistical analysis plan during the study and a summary of changes made to the original protocol is given in .
Summary of changes to the original RADAR protocol approved by the MREC
The RADAR trial was overseen by a Trial Steering Committee (TSC) that comprised an independent chairperson and methodologist, external independent experts and lay members, as well as the study sponsor (NBT R&I). The TSC ordinarily met twice per year and more frequently for extraordinary meetings if deemed necessary. The safety and conduct of the study were overseen by an independently chaired Data Monitoring and Ethics Committee (DMEC) that convened every 6 months once data collection commenced and reported to the TSC ahead of each scheduled TSC meeting. The general operations of the trial were overseen by a Trial Management Group (TMG) that convened regularly during the study. The TMG was composed of all chief investigators and all co-applicants of the RADAR study and included the trial manager (TM), other co-opted members of the Dementia Research Centre University College London (DRC-UCL) team or members of the Bristol Randomised Trials Collaboration (BRTC) trials unit at the University of Bristol. The day-to-day administration of the study was conducted by the chief investigator, the TM and the trial administrator, with the support of the sponsor and funder as and when appropriate.
Participants
The trial sought to recruit people aged ≥ 55 years with mild-to-moderate AD diagnosed according to original National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria from NHS hospital trusts across England, Scotland and Northern Ireland where participants with AD are routinely diagnosed and treated. Participants could be male or female, hypertensive or normotensive and could already be taking licensed antidementia treatments.
Inclusion criteria
Women and men diagnosed with mild-to-moderate AD according to original NINCDS-ADRDA criteria80 and who met all of the following criteria were considered eligible:
aged ≥ 55 years (to maximise generalisability of the study and avoid exclusion of younger yet otherwise eligible potential participants)
had capacity to consent for themselves as judged by a member of the research team with appropriate training and experience
had a MMSE score of 15–28 at the consented eligibility assessment
scored ≤ 5 on a modified Hachinski Ischaemic Score
81had a previous computerised tomography (CT), single-photon emission computed tomography (SPECT) or MRI scan consistent with a diagnosis of AD
had a study companion who was willing to participate in the study.
Exclusion criteria
Participants were deemed ineligible if they met any of the following criteria:
were already receiving ACE1-Is, AT1RAs, the renin inhibitor aliskiren or potassium-sparing diuretics
had known intolerance to, or had experienced renal problems while taking, ACE1-Is or AT1RAs
were medically unsuitable for, or unwilling to undergo, MRI
had a primary neurodegenerative disease or potential cause of dementia other than AD
had a consistent BP (at eligibility visit) of < 115 mmHg systolic and < 70 mmHg diastolic or > 160 mmHg systolic and > 110 mmHg diastolic
experienced a fall in BP at the eligibility visit on standing from a seated position of > 20/10 mmHg associated with clinically significant symptoms or a fall > 30/15 mmHg
had previously had a cerebrovascular accident and significant residual impairment remained (note that transient ischaemic attack was not automatically a basis for exclusion)
had hypertrophic cardiomyopathy or clinically significant aortic valve stenosis
had an estimated glomerular filtration rate of < 30 ml/minute/1.73 m2
demonstrated evidence of liver disease or significant liver function test (LFT) derangement (aspartate transaminase/alkaline phosphatase/bilirubin more than two times the upper limit of normal) at the eligibility/consent visit
had blood levels of potassium > 6.0 mmol/l in a non-haemolysed blood sample
had primary neurodegenerative diseases or potential alternative cause of dementia other than AD
were female and had not yet reached the menopause (defined as having a period in the previous 12 months) and tested positive for pregnancy, were unwilling to take a pregnancy test prior to trial entry or were unwilling to undertake adequate precautions to prevent pregnancy for the duration of the trial
had any severe coincident medical disease, or other factor inhibiting compliance with the study medication or follow-up schedule (e.g. participant unlikely to survive the trial follow-up period because they had a terminal comorbid condition)
had participated in a previous Clinical Trial of an Investigational Medicinal Product (CTIMP) within 6 months of RADAR trial entry.
Recruitment procedure
Potential participants were identified at recruitment sites or associated patient identification centres (PICs) from pre-existing lists of patients interested in research or newly referred patients who were receptive to research participation. All sites had scope to recruit from primary care (if sites wanted to use primary care and appropriate arrangements were in place) and from the National Institute for Health Research (NIHR)-funded Join Dementia Research research register portal (see www.joindementiaresearch.nihr.ac.uk/), which was launched after RADAR commenced. Engagement with Join Dementia Research was either by research staff identifying registered potential participants or by registered potential participants contacting local RADAR recruitment centres. Some potential participants contacted the RADAR co-ordinating office or local sites after articles in the national or local press and media. We used social media channels [Twitter (Twitter, Inc., San Francisco, CA, USA) (www.twitter.com/@RadarTrialAD) and Facebook (Facebook, Inc., Menlo Park, CA, USA) (www.facebook.com/RADARTrialAD/)] in concert with various recruitment sites and patient interest channels to reinforce these messages and opportunities to participate, as well as providing information on the RADAR trial website (www.radar-trial.org.uk).
People who made contact directly with recruitment sites, or were referred from the co-ordinating centre, or were identified as a potentially eligible and interested participant on Join Dementia Research were sent a letter of invitation, the initial participant information sheet, the initial companion information sheet, a reply slip and a prepaid envelope. Those with whom the study was discussed face to face were provided with the information packs to read and consider. The reply slip gave options for permission for the potential participant to be contacted by the research team with more information, permission for the research team to access medical records to confirm eligibility to the trial and permission to be contacted by the research team to take part in a telephone eligibility interview. Alternatively, the potential participant could indicate that they did not wish to participate in the trial.
Depending on the response given on the reply slips, further information was sent (for the participant and their companion with details about the MRI procedure) and general practitioners were contacted to find out which medications were currently being taken (to identify if the potential participant was being prescribed medications that would make them ineligible). Researchers telephoned all potential participants to introduce the study formally, to make initial eligibility checks and to arrange the first clinic appointment for consent and screening.
Enhancing site recruitment
During the course of RADAR recruitment, we embedded a qualitative component (before we had recruited 50% of the participants) to further explore the nature of trial site recruitment. This was intended to enhance recruitment and explore the impact of the design and conduct of the trial, and what organisation or training could possibly lead to improvements in recruitment in RADAR or in dementia trials in general. This would not have been possible without effective PPI.
Qualitative interviews were undertaken with a purposeful sample of research nurses and doctors responsible for screening and consent, and trial participants from a range of high- and low-recruiting trial sites to gain insights into barriers to and facilitators of recruitment. With informed consent, interviews were audio-recorded, transcribed and imported into NVivo 10 (QSR International, Warrington, UK) and analysed thematically.82 Data collection and analysis were conducted in parallel until data saturation was reached.83 Some of the findings of this nested study resulted in helpful revisions to our participant information packs that were considered to be overly complex and, therefore, were simplified.78 There were other, more generic, findings identified that were of note and will be discussed later in the overall discussion of findings as they are of relevance not only to the conduct of RADAR but also to clinical trial conduct in general.
Informed consent
Informed, written consent was obtained from every participant and their study companion during the screening visit to confirm their eligibility and before any other data collection took place. As per the study protocol, study participants had to have capacity to consent for themselves. Potential participants were also asked if they wished to consent to donating additional research bloods for future research that may further inform the results of the trial or AD-related research. At this time study participants were asked to identify a legal representative for the purposes of their involvement in the RADAR trial only. This representative was intended to be responsible for deciding whether or not it was in the participant’s best interests to continue in the trial should the participant lose capacity. The legal representative was usually the study companion who had consented to take part. All original signed and dated consent forms were held securely as part of the investigator site file at each of the recruitment sites, with a copy given to the participant, a copy put in the participant’s medical notes and a copy sent to the co-ordinating office for the purpose of central monitoring procedures by the sponsor. The eligibility criteria for the trial included consideration of the results of safety blood samples taken at the screening visit and subject to satisfying inclusion/exclusion criteria. Participants deemed to be ineligible for the study were informed of this and the reasons for it by site staff.
Randomisation
Participants who successfully completed the open-label phase were entered into the main part of the study. A baseline MRI (see Primary outcome and ) was then scheduled and undertaken approximately 4–14 days after completion of taking the active open-label generic losartan. Randomisation was contingent on successful baseline MRI for the primary outcome. Success was adjudicated by our pipeline quality assurance (QA) process provided by our collaborators at the DRC-UCL. Participants in whom a MRI scan was successfully undertaken were then randomised, via an online procedure or pin-access service by telephone hosted by Sealed Envelope (www.sealedenvelope.com), to receive either losartan or placebo in a ratio of 1 : 1 minimised by age and a baseline medial temporal lobe atrophy (MTA) score that encompasses the hippocampus and which was assessed for all sites by team members at the Clinical Research and Imaging Centre (CRIC), University of Bristol (www.bristol.ac.uk/cricbristol/). To expedite the randomisation process, so that it coincided as close as possible with the baseline MRI visit, and alongside the need to commence treatment as soon as possible, we used a subjective rating scale called the Scheltens scale84 of MTA score to gain some assessment of likely disease-associated atrophy and that has good inter-rater reliability.84 This allowed a rapid provision of some assessment of baseline atrophy, to serve as a minimisation variable to facilitate randomisation. Following randomisation, participants followed the same titration pattern as in the open-label phase, of 25 mg for 7 days and then 100 mg as a maintenance dosage with a 14-day monitoring and dispensing follow-up visit during the randomised phase. If the quality of the scan was insufficient, participants were asked to undergo another scan. If they declined, or the repeat scan(s) were still of insufficient quality (e.g. because the participant did not fully adhere to the MRI protocol), they were deemed to be ineligible for continued inclusion in the study and were informed of this outcome.
Summary of visits and assessments: after randomisation
Blinding
The study was double blinded, meaning that all participants and study companions, as well as all study personnel (except pharmacists at each site), were blinded to allocation at randomisation. There was scope within the protocol, in the interests of participant safety, for emergency unblinding to occur if a clinician believed that a treatment decision for a participant in certain circumstances could be influenced by the participant receiving losartan. With this in mind, a 24-hour emergency unblinding service was available to all research sites either through each local pharmacy service during working hours and out of hours or through a pharmacy nominated by the RADAR co-ordination team, in which case requests for emergency unblinding were documented by pharmacy staff and logged centrally by the TM. The procedure for any participant for whom emergency unblinding was undertaken was that they would discontinue taking the trial medication but remain part of the study unless they chose to withdraw. Furthermore, in the event of any emergency unblinding being required, the intention was that, where possible, all members of the research team (excluding trial pharmacists) would remain blinded, subject always to clinical need, and reasons for any instances of emergency unblinding would be determined by the TM. Any unblinding that occurred because of a serious adverse event (SAE) was appropriately documented in accordance with the sponsor’s procedures. All participants were given the relevant emergency contact details on their trial participation card when they consented to take part.
Outcome measures and assessments
The full schedule of visits and assessment is summarised in . All main assessments (baseline, 6-month and 12-month MRI and face-to-face assessments) were completed by both the participant and their study companion. If consent had been originally given to donate additional research bloods, then these were collected at baseline, 14 days post randomisation and at the end of the study. Wherever possible the researcher arranged to meet the participant for face-to-face assessments and to have follow-up assessments where they felt most comfortable (e.g. at home or at the clinical research centre). Every attempt was made to achieve, as much as possible, consistency of location and time of assessments.
In the case of imaging-related primary and secondary outcome measures, each participant underwent the RADAR scanning protocol, which lasted approximately 30 minutes if all sequences were undertaken (see Primary outcome and Secondary outcomes). Scans and anonymised scan data from all sites were uploaded to a bespoke XNAT platform, and then transferred securely to the DRC-UCL team for QA. Where 12-month MRI differed from baseline in the protocol or positioning sufficiently to adversely affect primary outcomes, a rescan was requested. As previously mentioned, all scans were also independently awarded a visual MTA score, derived from what is more widely known as the Scheltens scale,84 by research staff at the CRIC. This score was intended to inform and expedite the randomisation process.
Primary outcome
The primary outcome was change in brain atrophy, determined by volumetric MRI (vMRI), between baseline and the 12-month follow-up after treatment with losartan. This outcome was measured at a pre-arranged NHS or clinical academic imaging centre. Brain atrophy as measured by vMRI is recognised as a surrogate marker of cognitive decline and AD pathology.75,76,85–88 Brain atrophy was defined as the absolute difference in TBV between baseline and after 12 months of treatment post randomisation measured using vMRI [T1-weighted magnetisation-prepared rapid-gradient echo (T1-MPRAGE)]. It was chosen as the primary outcome on the assumption that a difference between trial arms of at least 3.8 ml/year would be clinically meaningful.
All MRI was performed at clinical or clinical-academic imaging centres using either 1.5-T or 3-T MRI systems with high-resolution (1-mm isotropic) three-dimensional T1-MPRAGE. The analyses of the magnetisation-prepared rapid-gradient echo (MPRAGE) images were conducted in collaboration with the DRC-UCL. All trial sites piloted the validation of the RADAR MRI protocol under the guidance and support of our DRC-UCL colleagues. The UCL team have developed semiautomated computerised methods to derive brain structure volumes from single-time point MRI and rates of atrophy from serial MRI,89–92 similar to those previously reported for multicentre trials.93 QA of all scans, and the QA and editing of segmentations, was carried out using Medical Image Display and Analysis Software (MIDAS) version 6.6 (University College London, London, UK).90 Automated segmentations were performed using brain multi-atlas propagation and segmentation91 for whole brain areas and Similarity and Truth Estimation for Propagated Segmentations (STEPS)94 for hippocampal regions, prior to manual checks and edits if needed. All of the image analysts at DRC-UCL undergo training and regular validation on structure segmentation. Longitudinal change following registration was measured using a DRC-UCL implementation of K-means normalised boundary shift integral (KN-BSI)95 for brains or double-window KN-BSI for the hippocampus.92,95
Secondary outcomes
A number of secondary outcomes were included in this study (see ):
Rates of AD progression as assessed by changes in cognitive assessments, including the 11-item Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog),
96 the Neuropsychiatry Inventoric
97 and the MMSE,
98 as well as an examination of change to quality of life [measured using the Dementia Quality of Life Measure (DEMQOL) or the Dementia Quality of Life Measure by proxy (DEMQOL-Proxy)
99] and in ability to perform activities of daily living, assessed using the Bristol Activities of Daily Living Scale (BADLS).
100 These secondary outcomes were obtained during a face-to-face assessment by a researcher, who was blinded to the participant’s allocation. The assessments were conducted first at baseline, after successful baseline MRI, and subsequently at the 6-month follow-up visit, and again within 10 days of the 12-month MRI.
Change in WMH volume as determined by MRI using T2-weighted fluid-attenuated inversion recovery (T2-FLAIR), possible in only a subset of sites, to explore the effect of the intervention on white matter damage in participants, which exacerbates AD symptoms
101 and has been reported to predict 1-year cognitive decline.
24 The WMH volumes were automatically identified and quantified using the longitudinal extension
102 of a cross-sectional automated framework
103 that performs the analysis on T1- and T2-FLAIR images rigidly aligned to the T1 space, acquired during the same MRI session at baseline and at 1-year follow-up. This framework automatically derives the number of components required to appropriately model the data in a Gaussian mixture model
103 that simultaneously accounts for normal and unexpected observations. After convergence of the model, the same model structure is used across time points and candidate lesion voxels are identified based on intensity and anatomical location information. Clusters of selected voxels are then automatically classified as lesion or artefact.
In a small subset of sites where MRI facilities were suitable, and data of sufficient quality were available, we also explored the level of change in CBF by arterial spin labelling (ASL) methods. ASL data of sufficient quality were processed using MATLAB
® version 2019 (The MathWorks, Inc., Natick, MA, USA) and SPM12 [version 7219 (Statistical Parametric Mapping 12, The Wellcome Centre for Human Neuroimaging, University College London, London, UK)]-based pipeline ExploreASL software verion 1.0.0 (Amsterdam UMC, Amsterdam, the Netherlands)
104,105 to generate CBF maps as well as the grey matter spatial coefficient of variation (CoV). Although CBF is proposed to serve as a surrogate marker of cognitive performance,
45–47 ASL spatial CoV has been recently proposed as a surrogate global marker of cerebrovascular health.
106,107 These data were acquired at the same time as the baseline and follow-up MRI at 12 months.
Change in BP over time is an important indicator (1) that the intervention was biologically active (i.e. observable reductions in BP in the intervention arm would be proof that the medication was active) and (2) of lowered BP, which may also alter the volume of WMH and improve CBF. As such, change to BP over time (visit-to-visit variation) may be a contributing factor to any observed change in cognitive function and thus could serve as an important confounding variable, even though previous studies have suggested that losartan may improve cognitive decline independent of lowered BP.
49–51,74 These data were collected at every visit in the study as described in
and
.
Measure of association between the primary MRI measures of atrophy and rate of cognitive decline. Given that atrophy, CBF and WMH volume are all considered proxies in various ways of cognitive decline, as summarised, it will be important to explore levels of association between the various assessment measures of cognitive function, activities of daily living and quality of life described. Examination of these will be important to explore which of the primary and secondary outcomes may be most relevant to use in any future large-scale clinical trials of AD involving losartan or related AT1RAs.
Examination of levels of drug compliance and tolerability across the main randomised phase and comparing rates across the active and placebo arms, as well as some examination of any potential differences between participants with hypertension and those who were normotensive. Data to inform this investigation, including AE data and safety bloods taken (with the exception of the 9-month visit, when no safety bloods were taken), were collected throughout the study at each follow-up visit and at the final 12-month face-to-face assessment.
Summary of visits and assessments: before randomisation
Sample size
Our proposed sample size for RADAR was based on previous studies conducted by the ADNI. These studies aim to optimise levels of recruitment to clinical trials of AD that involved MRI as an outcome measure.75–77 Previous studies have suggested that the 12-month atrophy that is measurable using MRI among AD patients is 15.2 ml/year (SD 8.6 ml/year) and that a relative between-group difference in atrophy rate of 25% is considered a clinically meaningful difference. This is equivalent to an absolute difference in TBV between the trial arms after 12 months of exposure to the intervention of 3.8 ml,108 equivalent to a standardised effect size of 0.44 SDs. We aimed to recruit and randomise a total of 228 participants, which would be sufficient to provide satisfactory primary outcome data on 182 participants for analysis, assuming 20% loss in primary outcome data. This was intended to provide us with 84% power to detect our target difference of at least 3.8 ml/year in 12-month atrophy (therapeutic benefit) with two-sided α = 0.05.
Adherence to intervention
We had defined compliance in the study to be based on participants having taken 80–120% of the intervention, as has been adopted in other studies.109 Instances of non-compliance by participants were reviewed at each follow-up visit and, where necessary, discussed with the principal investigator or delegated clinician to determine if it was appropriate for the participants in question to discontinue medication. In reality, compliance issues were noted when participants did not meet the lower threshold to be viewed as compliant, rather than the upper limit, because prudent prescribing and the frequency of follow-up visits ensured that participants were prescribed only up to the 100% amount needed between follow-up visits.
Statistical analysis
The analysis and reporting of this trial were undertaken in accordance with Consolidated Standards of Reporting in Trials (CONSORT) guidelines and followed a predefined statistical analysis plan that was agreed with the TSC prior to the completion of data collection. The primary comparative analyses between randomisation arms were conducted on an intention-to-treat basis, without the imputation of missing data, with due emphasis placed on confidence intervals (CIs) for the between-arm comparisons.
Preliminary analyses
Descriptive statistics of demographic and clinical measures were used to summarise the trial population and to compare the randomisation arms at baseline.
Primary analysis
The primary analysis employed linear regression to compare brain volume at 12-month follow-up between arms as randomised, adjusted for baseline brain volume, minimisation variables (age and Scheltens rating score) and recruitment site, to investigate differences in brain atrophy between the intervention and the control arms. The result of the linear regression model is presented as an adjusted difference in means between the intervention and control arms alongside the associated 95% CI and p-value for the comparison.
Secondary analyses
The effect of the intervention on secondary outcomes was investigated using appropriate regression models adjusted for baseline value of the outcome being examined, minimisation variables and recruitment site.
Sensitivity analyses
The sensitivity of the primary analysis to the impact of missing data was explored. Multiple imputation by chained equations (MICE) was used to impute missing primary outcome data. The imputation model included all variables that were part of the primary intention-to-treat analysis, secondary outcomes (from each time point) and baseline variables that were associated with missingness of the primary outcome. Twenty imputed data sets were generated and combined using Rubin’s rules and the primary analysis model was then repeated using the imputed data.
The impact that treatment compliance had on the data was investigated using the allocation-respecting method of complier average causal effects (CACE) via instrumental variable two-stage least squares regression, whereby outcomes of those who ‘complied’ with the intervention are compared with a group of ‘would-be compliers’ in the control arm. Treatment compliance was defined as (self-report of) taking 80–120% of study medication.
The impact that potential outliers had on the data was investigated by repeating the primary analysis model after excluding any participants whose brain volume measurement was > 3 SDs from the mean. The primary analysis was also repeated additionally adjusting for duration of follow-up.
Exploratory analyses
To investigate potential treatment effect modification, the primary analysis model was repeated with the inclusion of a treatment allocation by potential modifier interaction term. The potential treatment effect modifiers investigated were baseline hypertensive status, baseline MMSE, baseline age, duration of AD diagnosis, baseline brain volume and change in systolic BP. The trial was not designed to test interaction effects and, therefore, these analyses should be considered as exploratory in nature and the findings interpreted with due caution.