This chapter is focused on the methodology of the clinical component of the feasibility RCT. The methodology of the other elements of the wider study is described in the relevant later chapters.
Trial design
We performed a prospective feasibility RCT comparing appendicectomy with non-operative treatment in children with acute uncomplicated appendicitis. Recruitment lasted for 12 months and was open in three specialist paediatric surgery centres in England.
Participants
Participants were children aged 4–15 years inclusive with a clinical diagnosis of acute appendicitis, who would normally be treated with an appendicectomy as part of their standard care.
Inclusion criteria
Children aged 4–15 years.
Clinical diagnosis, either with or without radiological assessment, of acute appendicitis that, prior to study commencement, would be treated with appendicectomy.
Written informed parental consent, with child assent if appropriate.
Exclusion criteria
Clinical signs or radiological findings to suggest perforated appendicitis.
Presentation with appendix mass.
Previous episode of appendicitis or appendix mass treated non-operatively.
Major anaesthetic risk precluding allocation to the appendicectomy arm.
Known antibiotic allergy preventing allocation to the non-operative treatment arm.
Antibiotic treatment started at referring institution (defined as two or more doses administered).
Cystic fibrosis.
A positive pregnancy test.
Current treatment for malignancy.
Interventions
Non-operative treatment arm
Children randomised to non-operative treatment were treated according to a clinical pathway designed specifically for this trial. This treatment pathway comprised fluid resuscitation, a minimum of 24 hours of broad-spectrum intravenous (i.v.) antibiotics (per local antimicrobial policy), a minimum of 12 hours of nil by mouth (NBM) and regular clinical review to detect signs and symptoms of significant clinical deterioration, including, but not limited to, increasing fever, increasing tachycardia and increasing tenderness. After the initial 12-hour period of NBM, oral intake was advanced as tolerated. Children successfully treated without an operation were converted to oral antibiotics (per local policy) once they were afebrile for 24 hours and tolerating oral intake.
Clinical reviews were completed at approximately 24 and 48 hours post randomisation. Any children who showed signs of significant clinical deterioration by 24 hours, or at any point during the trial, were treated with appendicectomy. Children who were considered stable or improving continued with non-operative treatment. At 48 hours, any child who had not shown clinical improvement underwent an appendicectomy. The decision to continue non-operative treatment at these time points, or to recommend discontinuation of non-operative treatment and to recommend appendicectomy instead, was made by the treating consultant and based on clinical judgement rather than any specific features that are not evidence based. All reasons for a change in treatment were recorded in detail to guide a clinical pathway in a future trial.
Any child who received an appendicectomy for an incomplete response to non-operative treatment followed a standardised postoperative treatment regime already in use at each institution and identical to that used in the appendicectomy arm. The reason for having an appendicectomy was recorded.
Children treated non-operatively received a total of 10 days of antibiotics following randomisation, unless decided otherwise by the treating clinician. Children who received non-operative treatment were not routinely offered interval appendicectomy, but were counselled about the risk of recurrence.
Appendicectomy treatment arm
Children randomised to the appendicectomy arm underwent either open or laparoscopic appendicectomy at the surgeon’s discretion, performed by a suitably experienced trainee (as per routine current practice) or a consultant.
Participants received i.v. antibiotics from the time of diagnosis and were treated postoperatively with i.v. antibiotics according to existing institutional protocols; however, the following recommended regime was used to guide practice: children with acute uncomplicated appendicitis or a macroscopically normal appendix received no further antibiotics. Children with a perforated appendix (defined as a faecolith or faecal matter in the peritoneal cavity, or visualisation of a hole in the appendix) continued to receive i.v. antibiotics for a minimum of 3 days, and received a minimum total course of antibiotics of 5 days (i.v. and oral). The duration of antibiotic therapy was not standardised beyond this owing to anticipated variation in intraoperative findings and in response to treatment. The type of antibiotics used was identical to those used in the non-operative treatment arm in each centre. Any child failing to respond to first-line antibiotics was treated as was clinically appropriate with a longer course of antibiotics or a change in antibiotic therapy, with the choice of antibiotic determined by intraoperative swab or fluid culture.
Postoperatively, children with uncomplicated acute appendicitis or a normal appendix did not routinely have a nasogastric tube or a urinary catheter. They received oral intake as tolerated after surgery.
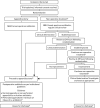
The participant flow through the two treatment arms is shown in .
Overview of the trial pathway. a, Non-operative treatment: NBM/sips for the initial 12 hours, minimum, then advance diet as tolerated; i.v. antibiotics for 24 hours, minimum, change to oral once afebrile for 24 hours, total course 10 days; and analgesia; (more...)
Discharge assessment
Criteria for discharge home were identical in both treatment arms and were as follows: vital signs within normal limits for age, afebrile for ≥ 24 hours, tolerating light diet orally, adequate oral pain relief and able to mobilise. We aimed to determine the feasibility of a blinded discharge assessment in a future RCT by attempting to complete a blinded discharge assessment for each participant as follows. Once a decision to discharge the child had been made, a member of the clinical team who had not been involved directly in the child’s treatment was asked to complete a discharge assessment. This assessor did not have prior knowledge of the randomisation or treatment received by the child. On completion of the discharge assessment, the assessor ‘guessed’ which treatment the child received. If the assessor became unblinded during the assessment, this was recorded.
Follow-up
All participants were given a diary card [see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/1419290/#/ (accessed February 2020)] to complete for the 14 days immediately following discharge from hospital and provided with a stamped addressed envelope to return this to the clinical trials unit on completion. This assessed whether or not the child had taken antibiotics and analgesia medication on each day following discharge, as well as an assessment of their recovery based on their ability to complete normal or full daily activities and attend school (if applicable). Finally, it also assessed whether or not parents had had to miss work as a result of their child’s illness. Follow-up appointments for all participants took place at 6 weeks and at 3 and 6 months following discharge, either in the outpatient clinic or in the clinical research facility at each centre. If a face-to-face appointment was not possible, the 3- and 6-month follow-ups were completed by telephone. Following an analysis of follow-up rates during the trial, we introduced an incentive in an attempt to improve follow-up rates. This was introduced in March 2018. All participants who attended all remaining follow-up visits from that point onwards were offered a £10 voucher.
Randomised controlled trial processes
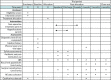
The schedule of enrolment, interventions and follow-up is shown in .
Overview of trial activity. AE, adverse event; HE, health economics.
Participant identification and recruitment
Participants were identified by the clinical team at the time of diagnosis and their eligibility was confirmed by the research team as soon as possible. Eligible patients were approached by the treating clinical teams with support from dedicated research nurses. Potential participants were provided with written information about the trial [see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/1419290/#/ (accessed February 2020) and shown a short video describing the study (see http://tinyurl.com/contract-f]. From the time of first discussing the trial with potential participants and their families, a maximum of 4 hours was permitted before a decision could be made regarding participation. This was to ensure that there was no delay in providing treatment as a result of considering trial participation. After receiving written informed consent (and assent from children aged ≥ 12 years who wished to give it), a member of the trial team randomised the participant to one of two treatment groups in a 1 : 1 ratio via an independent web-based system [TENALEA (Alea Clinical, Abcoube, the Netherlands)]. This online system allowed complete pre-randomisation concealment of treatment allocation and provided instant assignment to either the appendicectomy group or the non-operative treatment group. Minimisation was used to account for recruiting centre and to ensure balance between the groups in factors that may affect diagnostic accuracy and outcome of treatment. The factors taken into account were (1) sex, (2) age (4–8 years or 9–15 years), (3) duration of symptoms (onset of pain to recruitment to trial: < 48 hours or ≥ 48 hours) and (4) recruiting centre. In addition to the data required to complete randomisation, limited additional data were collected at baseline, including the use of any diagnostic imaging, and an Alvarado score21 (a scoring system used to help predict the severity of appendicitis) was calculated for each participant. This was used to provide an overview of the severity of illness of each child as a descriptive term; it was not used as a minimisation variable nor was a minimum Alvarado score used in the eligibility criteria.
Data collection and analysis
Data were recorded by dedicated research nurses at each site directly into an electronic, secure, web-based case report form (Medidata Rave® database; Medidata Solutions, Inc., New York, NY, USA). Data analysis was performed by the study statistician, who was blinded to treatment allocation by the use of coded data. As this is a feasibility study, all analyses were treated as preliminary and exploratory and data are reported descriptively. Feasibility outcomes (number of eligible patients, recruitment/retention rates, reasons for non-participation, success of blinding of the discharge assessor), treatment outcomes and complications are presented as simple summary statistics with 95% confidence intervals (CIs). Clinical outcomes were compared between treatment groups in an exploratory analysis.
Trial oversight
A Study Management Group (SMG) was responsible for overseeing the day-to-day management of the trial. An independent Trial Steering Committee and Data Monitoring and Safety Committee were convened to provide oversight of the trial. Their roles and responsibilities, which included adverse event (AE) monitoring, were agreed at the beginning of the trial and documented in specific charters. Specific processes to report AEs in a timely manner to the relevant committee were agreed.
Protocol, registration and ethics approval
The trial was carried out in accordance with a published protocol22 that was developed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials – Children (SPIRIT-C) guidance,23 and was registered prior to recruitment of the first participant (International Standard Randomised Controlled Trial Number: ISRCTN15830435). The overall study was given ethics approval by the Hampshire A Research Ethics Committee (reference number 16/SC/0596). The study is reported in accordance with the Consolidated Standards of Reporting Trials 2010 statement.24
Outcomes
Primary outcome
The primary outcome was to assess the feasibility of conducting a multicentre RCT testing the clinical effectiveness and cost-effectiveness of a non-operative treatment pathway for the treatment of acute uncomplicated appendicitis in children. This was evaluated as the proportion of eligible patients who were approached and recruited to the study over 12 months.
Secondary outcomes
The secondary outcomes were predominantly centred on the qualitative and COS substudies, contributing to the development of a future RCT. Those specifically relating to the RCT are marked with an ‘*’:
*Determine the willingness of parents, children and surgeons to take part in a randomised trial comparing operative treatment with non-operative treatment, and identify the anticipated recruitment rate. This was assessed from audio-recorded family–surgeon recruitment consultations; interviews with patients, parents, surgeons and nurses; surgeon surveys; and focus groups.
Identify strategies to optimise surgeon–family communication using the consultation and interview data.
Design a future RCT from the perspectives of stakeholders at participating sites (children, parents, surgeons, nurses, etc.), informed by the consultation and interview data, surgeon surveys and focus groups.
Assess the equipoise and willingness of UK paediatric surgeons to participate in a future RCT through surgeon surveys and focus groups.
*Assess the clinical outcomes of trial treatment pathways, including (1) overall success of initial non-operative treatment (measured as the number of participants who were randomised to non-operative treatment and discharged from hospital without appendicectomy), (2) complications of disease and treatment (measured during hospital stay and during the 6-month follow-up period) and (3) the rate of recurrent appendicitis during the 6-month follow-up period.
*Assess the performance of the study procedures, including retention of participants for the duration of the trial and the feasibility of outcome-recording and data collection systems.
Sample size
Participants were recruited from three centres for 12 months. It was expected that each centre would treat 80–100 children with acute appendicitis per year, with an estimate that at least 130 would be eligible out of the 240–300 potential patients. As this was a feasibility study, we did not specify a specific sample size, but aimed to define our recruitment rate within an approximate 10% margin of error. Based on an anticipated study population available for recruitment of approximately 130 participants, we would be able to estimate a true 40% recruitment rate with a 95% CI of 31% to 49% and a true 50% recruitment rate with a 95% CI of 41% to 59%. These numbers of participants in the feasibility RCT would be adequate to test treatment pathway procedures, data collection methods and loss to follow-up.
Changes to the original protocol
Version 3, 4 July 2017
Change to co-investigator at St George’s Hospital.
Reference to online patient video access.
Consent process oversight by Southampton Clinical Trials Unit (SCTU).
Telephone consent process for qualitative substudy.
Specification of office hours for randomisation backup.
Timeline for questionnaire completion.
Time frame for AE reporting.
Update to COS protocol.