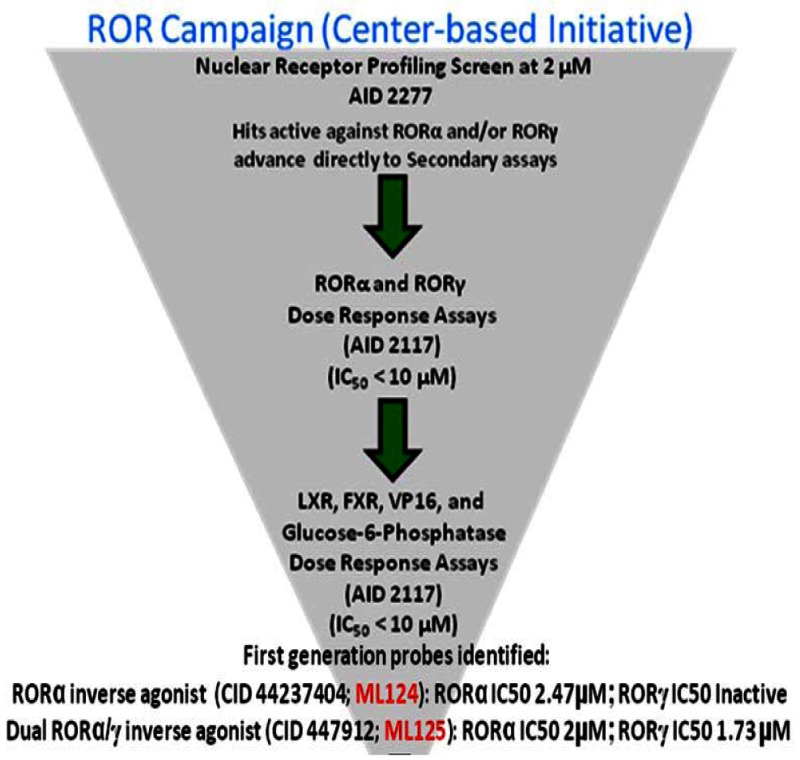
The Scripps Research Institute Molecular Screening Center (SRIMSC) implemented a Center-Based Component (CBC) that focuses on the use of a genomic screening platform to support selectivity profiling of high value compounds that emerge from the Molecular Libraries Probe Centers Network (MLPCN) program. One area of focus for the CBC was development of a nuclear receptor (NR) library containing expression vectors for all 48 human NRs in a GAL4 format. In an effort to validate the NR library (PubChem AID-2277), we screened it against a small chemical library containing mostly well characterized NR modulators. Analysis of the validation screen results revealed that the LXR agonist T0901317 (SID-85257301) was capable of repressing the activity of both GAL4-RORα and GAL4-RORγ, but not that of GAL4-RORβ. The activity of T0901317 was confirmed on wildtype receptor on native RORα/γ promoters and direct binding to these receptors was demonstrated. Compounds derived from these initial candidates were purchased as powders or synthesized at the SRIMSC and were tested for their ability to inhibit RORα and RORγ in luciferase-based reporter assays performed at a single concentration of 10 μM or in dose response assays starting at a nominal concentration of 20 μM. Compounds were subsequently counterscreened at a single concentration and/or dose response assays against the liver X receptor (LXR), the farnesoid X receptor (FXR), and glucose-6-phosphatase to determine selectivity. Finally, compounds of interest were tested at a single concentration of 10 μM against VP16 to determine whether they were non-selective or cytotoxic. The above Center-based probe development efforts resulted in the identification of two probes: The first probe, the benzenesulfonamide compound T0901317, ML125 (CID-447912; SID-85257301), previously identified as a selective agonist of LXR was identified here as a novel RORα/γ inverse agonist probe that decreases the transcriptional activity of both ROR receptors (IC50 values = 2.0 and 1.73 micromolar, respectively). The second probe, ML124 (CID-44237404; SID-85257298) synthesized at the SRIMSC, was found to decrease RORα transcriptional activity (IC50 value = 2.47 micromolar). ML124 represents an improvement over the prior art due to its lack of activity for LXR. ML124 does not have activity against RORγ (IC50 > 20 micromolar). These two probes are useful tools for examining ROR biology.
Assigned Assay Grant #: U54 MH084512
Screening Center Name & PI: Scripps Research Institute Molecular Screening Center (SRIMSC); Hugh Rosen
Chemistry Center Name & PI: SRIMSC; Hugh Rosen
Assay Submitter & Institution: Patrick Griffin, TSRI
PubChem Summary Bioassay Identifier (AID): 2139

Recommendations for scientific use of the probe (ML124 and ML125)
Limitations in state of the art. The retinoic acid receptor-related orphan receptors α and γ (RORα and RORγ) are two of these orphan receptors that have been demonstrated to play important roles in regulation of metabolism and immune function [1, 2]. Cholesterol and cholesterol sulfate have been suggested to be natural ligands for RORα [3, 4] and our recent work identified various oxysterols that bind to both RORα and RORγ with high affinity and regulate their activity [5, 6]. There is some controversy as to the nature of the constitutive activity of the RORs observed in cell-based assays. Although these molecules do bind tightly to the receptor, none alter the transcriptional output of the RORs in cell-based assays suggesting that while oxysterols bind to RORs, they do not induce a conformational change in the ligand binding domain that would be required to alter coactivator or corepressor interaction. Our data indicates that RORs display the constitutive activity in biochemical assays under conditions where the receptor would be expected to have no endogenous ligand present (denatured and refolded receptor) [5], but others have suggested that endogenous oxysterol ligands may copurify, leading to the observed constitutive activity [7]. Although the physiological significance of these natural ligands for the RORs is unclear, the potential utility of synthetic ligands that modulate the activity of these receptors is apparent. For example, loss of RORα in the staggerer mice results in mice resistant to weight gain and hepatic steatosis when placed on a high fat diet [8]. RORγ has been shown to be involved in development of Th17 cells that are implicated in autoimmune diseases, and loss of RORγ yields animals that are resistant to development of these diseases [9, 10]. RORα has been shown to be required for normal bone development and staggerer mice lacking functional RORα are osteopenic [11], suggesting that RORα agonists may have utility in the treatment of osteoporosis.
Probe Applications. These two probes are useful tools for examining ROR biology and represent an excellent starting point for development of ROR selective modulators. These probes may be useful for elucidating the role of RORs on lipid and glucose metabolism, and for therapeutic intervention in metabolic and immune disorders. More importantly, our results demonstrate for the first time that small molecules can be used to modulate the RORs and this finding has significant implications for the development of novel therapeutics for intervention in metabolic and immune disorders.
Expected end-users of the probe in the research community. Probes ML124 and ML125 can be used by academic researchers studying cell biology, molecular biology, immunology, and lipid and glucose metabolism in particular. In addition, the identified probes will be useful for any researcher working in the field of drug discovery.
Relevant biology of the probes
RORα: The protein encoded by this gene is a member of the NR1 subfamily of nuclear hormone receptors. It can bind as a monomer or as a homodimer to hormone response elements upstream of several genes to enhance the expression of those genes. The specific functions of this protein are not known, but it has been shown to interact with NM23-2, a nucleoside diphosphate kinase involved in organogenesis and differentiation, as well as with NM23-1, the product of a tumor metastasis suppressor candidate gene. Four transcript variants encoding different isoforms have been described for this gene.
RORγ: The protein encoded by this gene is a DNA-binding transcription factor and is a member of the NR1 subfamily of nuclear hormone receptors. The specific functions of this protein are not known; however, studies of a similar gene in mice have shown that this gene may be essential for lymphoid organogenesis and may play an important regulatory role in thymopoiesis. In addition, studies in mice suggest that the protein encoded by this gene may inhibit the expression of Fas ligand and IL2. Two transcript variants encoding different isoforms have been found for this gene.
Probe SID-85257301 is a novel RORα/γ inverse agonist probe that binds to and decreases the transcriptional activity of both RORα and RORγ receptors. The second probe, SID-85257298 decreases RORα transcriptional activity and lacks activity for LXR and RORγ.
1. Introduction
The retinoic acid receptor-related receptors (RORs) are members of the nuclear receptor (NR) superfamily of transcription factors. Several NRs are still characterized as orphan receptors because ligands have not yet been identified for these proteins. Members of the nuclear receptor (NR) superfamily display a conserved domain structure with highly conserved DNA-binding and ligand-binding domains. Members of this family include the receptors for the steroid hormone, thyroid hormone, as well as for bile acids and oxysterols. Although many of the 48 NRs found in the human are characterized as ligand-activated transcription factors, a significant number of these proteins still have uncharacterized ligands. The retinoic acid receptor-related orphan receptors α and γ (RORA and RORC are the gene identifiers) are two of these orphan receptors that have been demonstrated to play important roles in regulation of metabolism and immune function [1, 2].
RORα has unusual potential as a therapeutic target for the “metabolic syndrome” which results in pathologies such as insulin resistance, dyslipidemia, hypertension, and a pro-inflammatory state, that greatly elevates the risk of diabetes and atherosclerosis[12]. The related RORγ demonstrates significant expression in metabolic tissues such as liver, adipose, and skeletal muscle [13]. These two receptors are implicated in several key aspects of this metabolic pathogenesis. For instance, the staggerer mouse, which carries a homozygous germline inactivation of RORα, shows low body weight, high food consumption [14–16], elevated angiogenesis in response to ischemia [17], susceptibility to atherosclerosis [16], and an abnormal serum lipid profile [18]. RORγ null mice exhibit normal plasma cholesterol levels, but when bred with the RORα staggerer mice, the resulting RORα/γ knockout exhibits hypoglycemia not found in the single mutant animals. These studies reveal the functional redundancy of RORα and RORγ in regulating blood glucose levels and highlight the need for RORα/γ ligands that can bind to these receptors and modulate their transcriptional activity[19, 20].
2. Materials and Methods
Descriptions of assays follow the summary tables.
Table
PubChem BioAssay Table.
Table
Table of Assay Rationale and Screening Statistics.
2.1. Assays
(Click on the hyperlinks to obtain itemized protocols directly from PubChem; see also Summary AID-2139)
RORα Inhibition Assays (PubChem AIDs 561, 610, and 2117)
This transcriptional cell-based assay (AID-561) utilizes a fusion of the DNA-binding domain of the yeast transcriptional factor Gal4 with the ligand-binding domain of target receptor RORα (encoded by the pFA-hROR α plasmid, Orphagen Pharmaceuticals) to regulate a luciferase reporter containing 5xGal4 response elements at its promoter region (pG5-luc, Stratagene). Both pFA-hRORα and pG5-luc plasmids are transiently co-transfected in CHO-K1 (Chinese Hamster Ovary) cells. The presence in this cell line of required coactivators allows the expression of luciferase driven by activated RORα nuclear receptors. Compounds that inhibit the basal transcription of luciferase are detected through the suppression of light emission using the SteadyLite luciferase detection kit (Perkin Elmer). Such compounds hence constitute potential inhibitors of the RORα nuclear receptor. The primary HTS assay was conducted in 1536-well format. All compounds were tested once at a 10 micromolar final concentration.
Dose response assay (AID-610): among 278 compounds selected during the primary screening, 273 compounds were assessed in dose-response experiments in 10 point, 1:3 serial dilutions starting at a nominal test concentration of 100 micromolar.
As with the primary screen, the dose-response assay (AID-610) utilizes a fusion of the DNA-binding domain of the yeast transcriptional factor Gal4 with the ligand-binding domain of target receptor RORα (encoded by the pFA-hRORα plasmid, Orphagen Pharmaceuticals) to regulate a luciferase reporter containing 5xGal4 response elements at its promoter region (pG5-luc, Stratagene). Both pFA-hRORα and pG5-luc plasmids are transiently co-transfected in CHO-K1 (Chinese Hamster Ovary) cells. The presence in this cell line of required co-activators allows the expression of luciferase driven by activated RORα nuclear receptors. MLSMR Compounds that inhibit the basal transcription of luciferase are detected through the suppression of light emission using the SteadyLite luciferase detection kit (Perkin Elmer). Such compounds hence constitute potential inhibitors of the RORα nuclear receptor. This assay was conducted in 1536-well format. In addition, compounds that were considered promising probe candidates were next tested in a 10-point dilution series starting at a nominal concentration of 20 micromolar. Six replicates were performed for each assay (AID-2117).
Human Nuclear Receptor Modulator Screen (AID-2277)
The purpose of this assay is to identify compounds that act as modulators of human nuclear receptors and to demonstrate the utility of the GAL4 nuclear receptor library. This assay screens endogenous and synthetic ligands against a GAL4 nuclear receptor library which was built by replacing the endogenous N-terminus and DNA-binding domain (DBD) of all 48 receptors with a GAL4 DBD. The fusion constructs consist of the GAL4 DBD, the hinge domain, ligand binding domain (LBD), and F domain if applicable, of the human receptors. Plasmids coding for full-length receptors were also included for some receptors. In this assay HEK293T cells are co-transfected with a single GAL4 receptor and a luciferase reporter containing an upstream activating sequence (UAS) recognized by the GAL4 DBD, followed by treatment with test compounds. As designed, compounds that modulate activity of a particular nuclear receptor will modulate the binding of the GAL4 DBD to the UAS, thereby modulating luciferase production, resulting in an increase or decrease in well luminescence. Each compound was evaluated using two plates of the GAL4 NR library for a total of six replicates, at a nominal test concentration of 2 micromolar.
RORγ Inhibition Assay (AID-2117)
The purpose of these assays is to identify compounds that inhibit RORγ activity. This assay employs the RORγ-expressing cell line from a GAL4 nuclear receptor library. In this assay, HEK293T cells co-transfected with a GAL4DBD-RORγ LBD construct (GAL4-RORγ) and a GAL4UAS-luciferase reporter construct are incubated for 18–24 hours with test compounds. The presence in this cell line of required co-activators allows the expression of luciferase driven by activated RORγ nuclear receptors. As designed, compounds that inhibit RORγ activity will prevent activation of the GAL4-RORγ construct, thereby preventing GAL4DBD-mediated activation of the GAL4UAS-luciferase reporter, leading to a decrease in well luminescence. Compounds were tested in a 10-point dilution series starting at a nominal concentration of 20 micromolar. Six replicates were performed for each assay.
LXR Activation Assay (AID-2117)
The purpose of these assays is to identify compounds that increase liver X receptor (LXR) activity. This assay employs the LXR-expressing cell line from a GAL4 nuclear receptor library. In this assay, HEK293T cells co-transfected with a GAL4DBD-LXRLBD construct (GAL4-LXR) and a GAL4UAS-luciferase reporter construct are incubated for 18–24 hours with test compounds. The presence in this cell line of required co-activators allows the expression of luciferase driven by activated LXR nuclear receptors. As designed, compounds that activate LXR activity will activate the GAL4-LXR construct, thereby increasing GAL4DBD-mediated activation of the GAL4UAS-luciferase reporter, leading to an increase in well luminescence. Compounds were tested in a 10-point dilution series starting at a nominal concentration of 20 micromolar. Six replicates were performed for each assay.
FXR Activation Assay (AID-2117)
The purpose of this assay is to identify compounds that increase farnesoid X receptor (FXR) activity. This assay employs the FXR-expressing cell line from a GAL4 nuclear receptor library. In this assay, HEK293T cells co-transfected with a GAL4DBD-FXRLBD construct (GAL4-FXR) and a GAL4UAS-luciferase reporter construct are incubated for 18–24 hours with test compounds. The presence in this cell line of required co-activators allows the expression of luciferase driven by activated FXR nuclear receptors. As designed, compounds that activate FXR activity will activate the GAL4-FXR construct, thereby increasing GAL4DBD-mediated activation of the GAL4UAS-luciferase reporter, leading to an increase in well luminescence. Compounds were tested in a 10-point dilution series starting at a nominal concentration of 20 micromolar. Six replicates were performed for each assay.
VP16 Inhibition Counterscreen Assay (AID-2117)
In this counterscreen assay the nuclear receptor plasmid was replaced by the GAL4DBD-VP16LBD plasmid, which expresses the strong transactivation domain of the herpes simplex virus Virion Protein 16 (VP16) fused to the GAL4 DBD. Cells are co-transfected with the 5xGAL4 response element (UAS) luciferase reporter to monitor GAL4DBD-VP16LBD activity, followed by incubation with test compounds for 18–24 hours. As designed, compounds that inhibit VP16 activity will decrease pGAL4DBD-VP16LBD activity, leading to reduced activation of the pG5-luc and decreased well luminescence. These compounds are likely to be nonselective inhibitors or cytotoxic. Compounds were tested in singlicate at a final nominal concentration of 10 micromolar. Six replicates were performed for each assay.
Glucose-6-Phosphatase (G6Pase) Promoter Assay (AID-2117)
The purpose of this assay is to determine whether probe candidates can modulate ROR target genes in cells. In these assays 293T cells were co-transfected with pS6 control plasmid or pS6 containing full length RORα along with G6Pase promoter. SRC-2 as a coactivator was also co-transfected with G6Pase promoter. Dose-response curve was determined by treating the transfected cells with varying concentrations of compound for 20 hours. Luciferase activity was measured and relative change was determined by normalizing to cells treated with vehicle only. Each data point was performed in eight replicates, n=8. Compounds were tested in a 10-point dilution series starting at a nominal concentration of 20 micromolar.
2.2. Probe Chemical Characterization
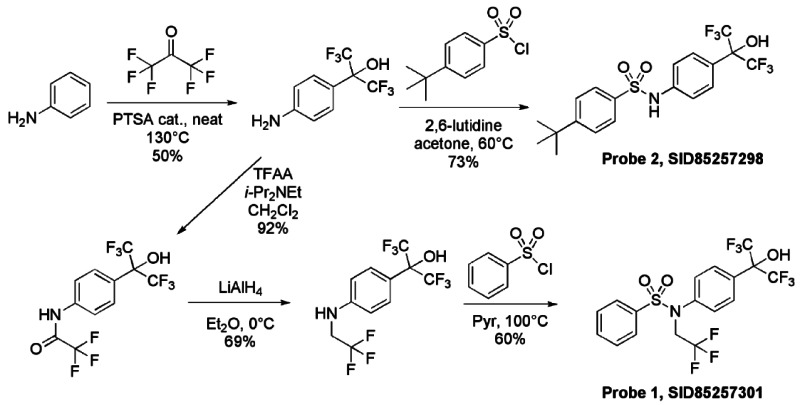
Synthetic route. The RORα and RORα/γ inverse agonist probes were synthesized as summarized below.
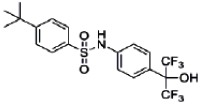
Probe 1 (ML125): N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]-N-(2,2,2- trifluoroethyl)benzenesulfonamide.
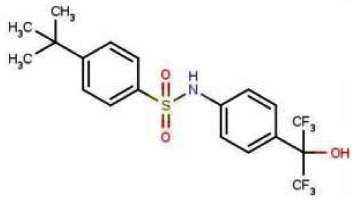
Probe 2: (ML124): 4-tert-butyl-N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]benzenesulfonamide.
Structure verification with 1H NMR and LCMS results
Probe 1 (ML125) was obtained as a white powder (m.p. 105°C) with 99% purity (HPLC analysis): 1H NMR (400 MHz, (CD3)2SO): 4.63 (q, J = 9.0 Hz, 2H), 7.29 (d, J = 8.8 Hz, 2H), 7.55–7.64 (m, 4H), 7.66 (d, J = 8.8 Hz, 2H), 7.73 (tt, J = 7.1, 1.6 Hz, 1H), 8.82 (s, 1H). 13C NMR (100 MHz, (CD3)2SO): 127.3 (2C), 127.7 (2C), 128.7 (2C), 129.4 (2C), 130.7, 133.8, 137.1, 140.4. Five carbon resonances are missing in the 13C spectrum of SID-85257301. These are the three carbons of the hexafluoropropanol moiety and the two carbons of the trifluoroethyl group. The fluorine coupling with these carbons gives multiplets which were difficult to detect in the 13C spectrum even with increased number of scans. FTIR: 3430, 1510, 1450, 1425, 1343, 1269, 1229, 1215, 1174, 1150, 1138, 1108, 1086, 976, 953, 928, 855, 769, 752, 735, 716, 705, 686, 673 cm−1. MS (ES-) m/z = 480 (found for C17H12F9NO3S-H+).
Probe 2 (ML124) was obtained as a white powder (m.p. 161°C) with >98% purity (HPLC analysis): 1H NMR (400 MHz, (CD3)2SO): 1.26 (s, 9H), 7.22 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.8 Hz, 2H), 7.59 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 8.58 (s, 1H), 10.66 (s, 1H). 13C NMR (100 MHz, (CD3)2SO): 30.7 (3C), 34.9, 118.3 (2C), 125.2, 126.2 (2C), 126.5 (2C), 127.9 (2C), 136.8, 139.6, 156.10. Three carbon resonances are missing in the 13C spectrum of SID-85257298. These are the carbons of the hexafluoropropanol moiety. The fluorine coupling with these carbons gives multiplets which were difficult to detect in the 13C spectrum even with increased number of scans. FTIR: 3430, 3245, 1517, 1471, 1403, 1332, 1308, 1267, 1228, 1192, 1162, 1150, 1112, 1088, 963, 926, 827, 752, 735, 702, 664 cm−1. MS (ES-) m/z = 454 (found for C19H19F6NO3S-H+).
Solubility. The solubility of the probes was measured in phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic and a pH of 7.4) at room temperature (23°C). The solubility of probe ML124 and ML125 was found to be 16 μM and 3 μM, respectively.
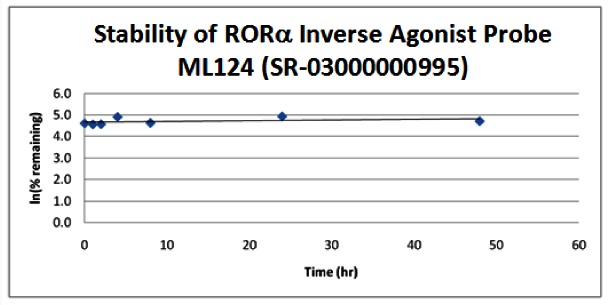
Stability. The stability of the probes was measured at room temperature (23ºC) in PBS (no antioxidants or other protectants; DMSO concentration below 0.1%). The stability, represented by the half-life, was found to be > 48 hours for both probes. Below is a graph showing loss of compound with time over a 48 hour period with a minimum of 6 time points. The table indicates the percent of compound remaining at the end of the 48 hours.

The probes were measured for their ability to form glutathione adducts. At concentrations of 100 μM reduced GSH, 10 μM of the probes do not appear to be Michael acceptors [21, 22].
CID, SID and ML# of the probe and five related analog samples submitted to the SMR collection
2.3. Probe Preparation
Detailed experimental procedures for synthesis of Probe 1 (ML125) and Probe 2 (ML124) follow.
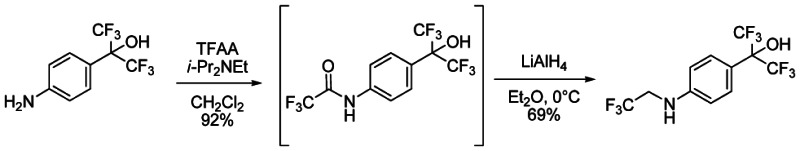
To a solution of 2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol1 (1.5M in THF, 1.29 mL, 1.93 mmol) in CH2Cl2 (2.7 mL) under argon were successively added at 0°C N, N-Diisopropylethylamine (370 μL, 2.12 mmol) and trifluoroacetic anhydride (268 μL, 1.93 mmol). The mixture was stirred for 10 min at 0°C, then 2 hours at room temperature. The solution was quenched by the addition of saturated NH4Cl solution and the organic phase was washed with saturated NH4Cl solution, brine, dried over Na2SO4, filtered and concentrated under reduce pressure to give 630 mg of 2,2,2-trifluoro-N-{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}acetamide (92%) as purple oil. The crude residue was used without purification for the next step. 1H NMR (400 MHz, (CD3)2SO): 7.72 (d, J = 8.8 Hz, 2H), 7.81 (d, J = 8.8 Hz, 2H), 8.75 (s, 1H), 11.46 (s, 1H).
To a solution of 2,2,2-trifluoro-N-{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}acetamide (630 mg, 1.77 mmol) in Et2O (2.7 mL) under argon was portion-wise added at 0°C lithium aluminum hydride (95%, 709 mg, 17.7 mmol). The mixture was stirred for 30 min at 0°C, then overnight at room temperature. The solution was cooled down at 0°C and quenched by H2O (800 μL), followed by NaOH (1N, 1.6 mL), then H2O (3*800 μL). The mixture was stirred 30 min at room temperature, filtered over Celite, washed by Et2O and the filtrate was concentrated under reduce pressure to give 414 mg of 1,1,1,3,3,3-hexafluoro-2-{4-[(2,2,2-trifluoroethyl)amino]phenyl}propan-2-ol (69%) as purple solid. The crude residue was used without purification for the next step. 1H NMR (400 MHz, (CD3)2SO): 3.52 (m, 2H), 6.60 (t, J = 6.8 Hz, 2H) 6.81 (d, J = 8.8, 2H), 7.38 (d, J = 8.8 Hz, 2H), 8.30 (s, 1H).
Synthesis of Probe 1 (ML125)

Probe 1, SID-85257301
To a solution of 1,1,1,3,3,3-hexafluoro-2-{4-[(2,2,2-trifluoroethyl)amino]phenyl}propan-2-ol (630 mg, 0.42 mmol) in pyridine (1.4 mL) under argon was added at room temperature benzenesulfonyl chloride (80 mg, 0.46 mmol). The mixture was heated overnight at 100°C, then diluted by toluene and concentrated under reduced pressure. The crude product was directly applied to a silica gel column and eluted with hexane-EtOAc (85/15) to obtain 121 mg of Probe 1 (SID-85257301) (60% yield, purity 99% by HPLC analysis) as a white powder (m.p. 105°C). Spectroscopic characterization data for Probe 1 (ML125) are provided in Section 2.2 above.
Synthesis of Probe 2 (ML124)
Probe 2, SID-85257298
To a solution of 2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (1.5M in THF, 128 μL, 0.193 mmol) in acetone (643 μL) were successively added at room temperature 2,6-Lutidine (29 μL, 0.251 mmol) and 4-tert-butylbenzenesulfonyl chloride (46 mg, 0.193 mmol). The mixture was heated overnight at 80°C, then diluted by AcOEt and quenched at room temperature by the addition of saturated NaHCO3 solution. The aqueous phase was extracted two times by AcOEt and combined organic phases were dried over Na2SO4, filtrated and evaporated. The crude residue was purified by silica gel column and eluted with hexane-EtOAc (70/30) to obtain 64 mg of Probe 2 (SID-85257298) (73% yield, purity >98% by HPLC analysis) as a white powder (m.p. 161°C). Spectroscopic characterization data for Probe 2 (ML124) are provided in Section 2.2 above.
3. Results
3.1. Summary of Screening Results
This project was initiated as an outreach effort. During this outreach effort 64,925 substances from the MLPCN collection were tested in primary screening in singlicate to identify compounds that inhibit RORα activity (AID-561). A total of 278 substances were identified as active in the primary screen. The 273 available hits from this assay were then screened in titration assays in triplicate to determine RORα potency (AID-610). No RORα specific inverse agonists were identified and the project was pursued as a Center-based component (CBC). The SRIMSC CBC focuses on the use of a genomic screening platform to support selectivity profiling of high value compounds that emerge from the MLPCN program. One area of focus for the CBC was the development of a nuclear receptor (NR) library containing expression vectors for all 48 human NRs in a GAL4 format. In an effort to validate the NR library (AID-2277), we screened it against a small chemical library containing mostly well characterized NR modulators. Analysis of the results from this validation screen revealed that the LXR agonist T0901317 (SID-85257301) was capable of repressing the activity of both GAL4-RORα and GAL4-RORγ, but not that of GAL4-RORβ. The activity of T0901317 was confirmed on wildtype receptor on native RORα/γ promoters and direct binding to these receptors was demonstrated. This is an excellent example of the utility of the tools being developed within the SRIMSC CBC.
Compounds derived from these initial candidates were purchased as powders or synthesized at the SRIMSC and were tested for their ability to inhibit RORα and RORγ in luciferase-based reporter assays performed at a single concentration of 10 μM or in dose response assays starting at a nominal concentration of 20 μM. Compounds were subsequently counterscreened at a single concentration and/or dose response assays against the liver X receptor (LXR), the farnesoid X receptor (FXR), and glucose-6-phosphatase to determine selectivity. Finally, compounds of interest were tested at a single concentration of 10 μM against VP16 to determine whether they were non-selective or cytotoxic.
The Center-based probe development efforts resulted in the identification of two probes. The benzenesulfonamide compound T0901317 (SID-85257301) previously identified as a selective agonist of LXR (4) was identified here as a novel RORα/γ inverse agonist probe that decreases the transcriptional activity of both ROR receptors (IC50 values = 2.0 and 1.73 micromolar, respectively). The second probe, SID-85257298 synthesized at the SRIMSC, was found to decrease RORα transcriptional activity (IC50 value = 2.47 micromolar). SID-85257298 represents an improvement over the prior art due to its lack of activity for LXR. Probe compound SID-85257298 does not have activity against RORγ (IC50 > 20 micromolar). These two probes are useful tools for examining ROR biology.
Following the screening and hit validation campaign, a probe compound (SID-85257301) belonging to the benzenesulfonamide scaffold was identified. We searched the MLSMR and commercial sources for structurally related compounds. Analogs were purchased in powder form or re-ordered from the MLSMR in liquid form and tested in dose response assays against both RORα and RORγ. Additional compounds were synthesized, by using the synthesis scheme summarized in the following section, and were also tested in dose response assays against both RORα and RORγ, as well as other nuclear receptors such as LXR and FXR. Results of these assays are summarized in the SAR table below. We found that deletion of the N-trifluoroethyl substituent and replacement of the original N-benzenesulfonyl group of the original screening hit (SID-85257301) with a Np- tert-butylbenzenesulfonyl group gave a RORα selective inverse agonist (SID-85257298) that was inactive against both anti-targets, LXR and FXR. The assays have been summarized in Summary AID-2139.
3.2. Dose Response Curves for Probes
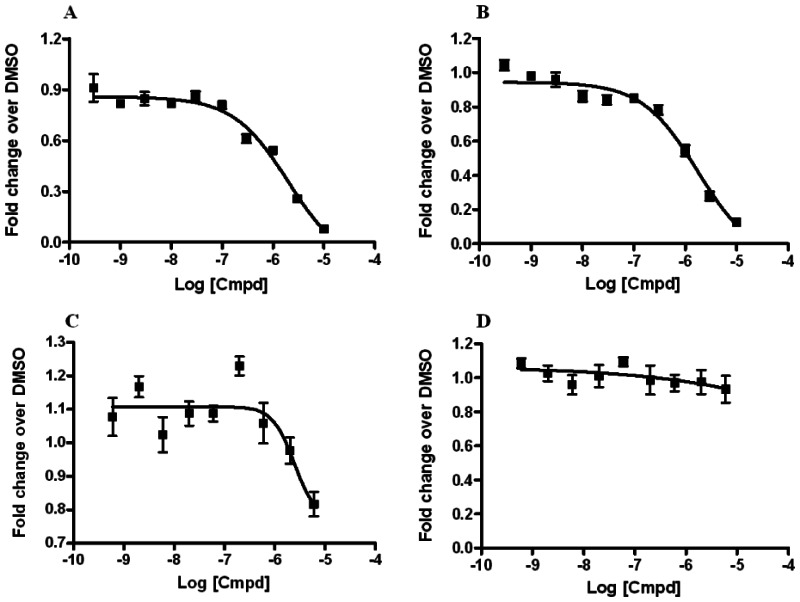
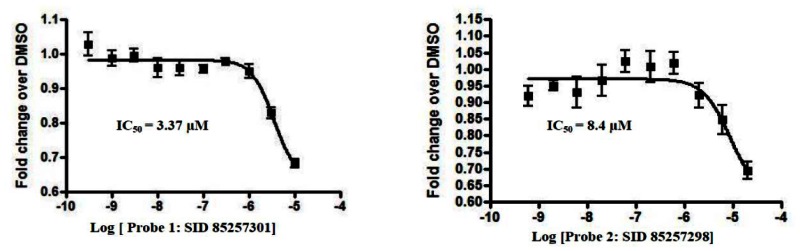
Dose-response curves of RORα and RORα/γ inhibitor probes ML125 and ML124 in the X cell-based assay
Ten-point, 1:3 serial dilution of probe compound ML125 (SID-85257301; A and B) and probe compound ML124 (SID-85257298; C and D) were tested in six replicate in both the RORα (A and C) and RORγ (B and D) inhibition assays the protocols described in the technical section of this report (see AID-2139 and AID-2117, respectively). Error bars represent the standard deviation of six replicates.
3.3. Scaffold/Moiety Chemical Liabilities
There are no known chemical liabilities known for Probe 1 (ML125) and Probe 2 (ML124). While both compounds have 6 and 9 fluorine atoms, respectively, both have acceptable solubility in PBS (3 and 16 mM, respectively—see Section 2.2).
Analogs were synthesized or purchased in powder form or re-ordered from the MLSMR in liquid form and tested in dose response assays against RORα, RORγ, VP16, LXR, and FXR. Results for these compounds are summarized in the SAR table below and in AID-2117.
Analogs of T0901317 (SID-85257301) were purchased as powders or synthesized at the SRIMSC and were tested for their ability to inhibit RORα and RORγ in luciferase-based reporter assays performed at a single concentration of 10 μM or in dose response assays starting at a nominal concentration of 20 μM. Compounds were subsequently counterscreened at a single concentration and/or dose response assays against the liver X receptor (LXR), the farnesoid X receptor (FXR), and glucose-6-phosphatase to determine selectivity. Finally, compounds of interest were tested at a single concentration of 10 μM against VP16 to determine whether they were non-selective or cytotoxic. This led to the identification of CID-44237404 that was declared as a first generation selective RORα inverse agonist probe ML124 (SR-03000000995; SID-85257298: MLS-002554296).
Efforts to identify a more potent RORα-selective inverse agonist have continued. We have explored changes in the arylsulfonamide unit, the aniline unit, and the linker connecting the two. The SAR Table below provides structures of more than new 30 analogs synthesized or purchased to explore these points; data for CID- 44237404 and the analogs declared in the first –generation RORα probe report are also included. From these results it is apparent that the sulfonamide unit cannot be replaced by a carboxamide, and thus far all substituents introduced to replace the bis(trifluoromethyl)carbinol unit of the original probe have eliminated or substantially reduced activity. The most productive change has been the discovery that replacement of the arylsulfonamide unit of probe ML124 (CID-44237404) with a thiophenyl sulfonamide group, as in SR- 06000113335 (“SR-3335”) has led to a ca. 5-fold increase in potency as an RORα inverse agonist, while maintaining selectivity vs. RORγ (inactive). [Note that this SR-3335 compound is a next generation selective RORα probe discussed in a separate probe report]. However, introduction of additional substituents on the thiophene ring have led to decrease in activity as RORα inverse agonists, and has led to re-emergence of activity against RORγ. The SAR table summarizing these results is attached below. In future work, we will continue to explore structural modifications of this inverse agonist series, with the objective of further increasing potency while maintaining selectivity for RORα vs. RORγ.
Subsequent SAR efforts were performed after the present ML124 and ML125 probe report was submitted, Those efforts led to the identification of an improved, selective second-generation RORα inverse agonist probe, ML176 [also known as SR-06000113335 (“SR-3335”)]. ML176 is ca. 5-fold more potent than ML124 as a RORα inverse agonist, while maintaining electivity vs. RORγ (inactive). More than 30 analogs of ML124 and ML176 are reported in the second generation ML176 probe report.

3.4. SAR Table
3.5. Cellular Activity
The probes were tested in a variety of cell-based assays performed by the SRIMSC and assay provider. These assays were performed to determine the probe’s selectivity and potency. The results of these studies demonstrated that probe ML125 is a selective RORα inverse agonist, while probe ML124 is a dual RORα/γ inverse agonist. Further, probe ML124 was were tested in a variety of cell-based assays performed by the SRIMSC and assay provider. These assays were performed to determine the probe’s selectivity and potency. The results of these studies demonstrated that probe ML125 is a selective RORα inverse agonist, while probe ML124 is a dual RORα/γ inverse agonist.

ML124 Suppresses RORα and RORγ mediated IL-17 transcription
HEK293 cells were transiently transfected with the IL-17-dependent reporter construct, renilla luciferase, and vectors containing full-length RORα (+RORα), full-length RORγ (+RORγ), or empty vector alone (endogenous, “no receptor”). Twenty-four hours later cells were treated with DMSO or increasing concentrations of ML124. Twenty-four hours post-treatment, IL-17 activity was determined by luciferase assay. The data are normalized to the vehicle (DMSO) treated cells.
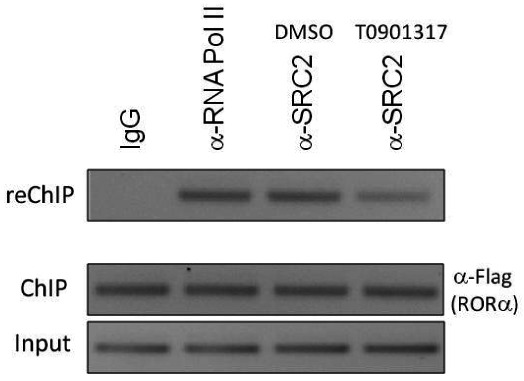
Modulation of Glucose-6-Phosphatase (G6Pase) promoter activity by ML124 in HepG2 cells
Sequential chromatin immunoprecipitation (ChIP/reChIP) assay illustrating that probe ML124 (10 μM) treatment reduces the ability of RORα to recruit SRC-2 to a G6Pase gene promoter. HepG2 cells overexpressing Flag-tagged RORα were treated with vehicle (DMSO) or 10 μM ML124 for 24 h followed by sequential ChIP. The first immunoprecipitation was performed using α-Flag antibody and the second immunoprecipitation was performed using α-SRC-2 antibody. Mouse IgG was used as a negative control and anti-RNA pol II antibody was used as a positive control.
3.6. Profiling Assays
Human Nuclear Receptor Profiling Assay. In addition to the above cellular assays, we have also obtained profiling results for the probe at 2 μM using a 48 human nuclear receptor library. This assay has been published in PubChem as AID-2277. The nuclear receptor library was plated into 384-well plates. HEK293T cells were reverse transfected with the well-specific construct and the UAS luciferase reporter pGL4.31 using Fugene6 transfection reagent in a final volume of 40 microliters. Control wells containing constructs encoding for the GAL4 DBD alone (pBind) or GAL4 fused to VP16 were also analyzed. After 24 hours, optimized compounds (2 micromolar final concentration) or DMSO was added to the plates and allowed to incubate for 20 hours. Next, 40 microliters of BriteLite was added to all wells and luciferase activity was measured on the PerkinElmer Envision 2104. Compounds that attenuate the GAL4-VP16-dependent luciferase activity are considered promiscuous or cytotoxic. To correct for plate-to-plate variance, sample data was normalized to wells containing vector only (69 wells) to determine Normalized Data Values (NDV). For each nuclear receptor, the average of NDV treated with a particular test compound was divided by the average NDV of wells treated with DMSO. This resulted in a fold-activation or fold-inhibition value for each receptor-compound pair. Because the activity of VP16 should not be impacted by these compounds and a change in this value is indicative of cell toxicity or general disruptions in transcription/translation, this fold-change value was then normalized to the VP16 fold-change. Compounds that induced in any receptor an average fold change that was three standard deviations from the VP16 value were considered active for that particular receptor.
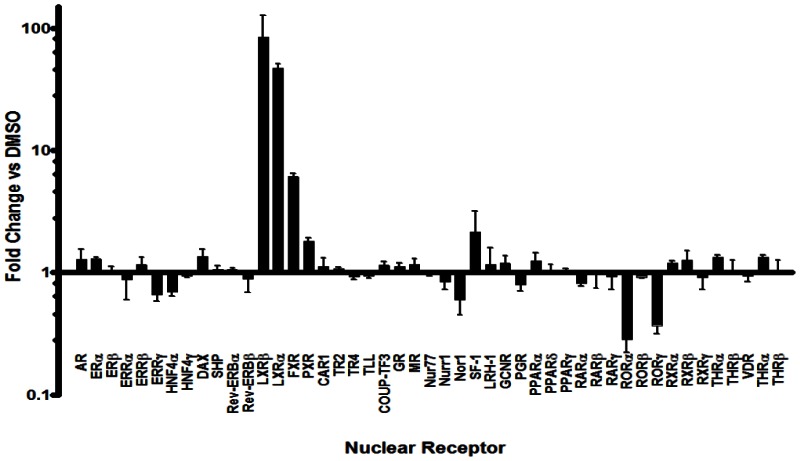
Gal4 Nuclear Receptor Profiling of Ml125. Gal4 NR clones were reverse transfected with a UAS reporter construct into HEK293T cells. After 24 hours, the LXRα agonist T0901317 (2 μM final concentration) or DMSO was added and incubated for 20 hours. The luciferase activity of each construct was measured and normalized to the mock (vector alone), then the fold change in signal compared to DMSO is calculated (n=6).
4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
Cholesterol, cholesterol sulfate, and 25-hydroxycholesterol have been shown to be ligands of the RORs. Although these molecules bind tightly to the receptor, none alters the transcriptional output of the RORs in cell-based assays suggesting that while some oxysterols bind to RORs, they do not induce a conformational change in the ligand binding domain that would be required to alter coactivator or corepressor interaction. Therefore, prior to this probe report there were no published functional modulators of the RORs. Regardless, sterols would represent a limited chemical starting point for optimization of potency and isoform selectivity.
Approach taken in the absence of an ROR modulator
The promiscuity of ML125 as well as other nuclear receptor ligands indicates that there are privileged structures (chemotypes) that bind to a range of these receptors. Since it is possible to utilize these promiscuous ligands as points to initiate development of receptor selective ligands, we set out to profile the activity of a collection of well characterized NR ligands against all human nuclear receptors. Recently, we developed a GAL4 nuclear receptor library containing all 48 human receptors to facilitate selectivity profiling of putative NR modulators. In an effort to demonstrate the utility of the NR library, a collection of 65 well characterized NR modulators including the LXR agonist T0901317 (ML125) was assembled. Interestingly, when this chemical set was tested against the GAL4 NR library, it was discovered that in addition to its expected activity, ML125 was a potent inhibitor of the nuclear receptors RORα and RORγ (retinoid-related orphan receptor-alpha and -gamma; [NR1F1] and [NR1F3]) yet afforded little or no activity on RORβ (retinoid-related orphan receptor-beta; [NR1F2]).
The RORs have emerged as attractive drug targets for the treatment of metabolic disorders and inflammatory disease. So with ML125 we demonstrate, for the first time, that a synthetic ligand can bind directly to and modulate the transcriptional activity of RORα and RORγ. ML125 was found to directly bind to RORα and RORγ with high affinity resulting in modulation of the receptor’s ability to interact with transcriptional cofactor proteins. ML125 repressed RORα/γ-dependent transactivation of ROR responsive reporter genes and in HepG2 cells reduced recruitment of the coactivator SRC2 by RORα at an endogenous ROR target gene. Using siRNA, we demonstrate that repression of the gluconeogenic enzyme glucose-6-phosphatase in HepG2 cells by ML125 is ROR-dependent and not due to the compounds LXR activity.
In summary, ML125 represents a novel chemical probe to examine RORα/γ function. Also, this compound, with a chemically tractable scaffold, represents an excellent starting point for medicinal chemistry towards the development of ROR selective modulators. More importantly, our results demonstrate for the first time that small molecules can be used to target the RORs for potential therapeutic intervention in metabolic and immune disorders. While ML125 does activate LXRα and FXR, it is presented as a probe based on the wealth of published pharmacological studies with this compound. In these published studies the action of ML125 is attributed exclusively to LXRα activation including observations that are not easily attributable to LXRα function.
Chemical optimization of ML125
A recent survey of the patent literature does not reveal any specific patent applications on synthetic ROR modulators. While there are patents filed on ML125, they are all specific to LXR activity. Additionally, the modifications made to obtain selectivity over LXR as demonstrated with ML124, render these compounds inactive on LXR thus they do not appear as examples in the ML125 patent material. We have filed a provisional application which is currently under conversion to non-provisional on the general scaffold covering ML124 as selective modulators of RORα and RORγ.
4.2. Mechanism of Action Studies
Modulation of RORα mediated Glucose-6-Phosphatase (G6Pase) promoter activity. In addition to the cellular and profiling assays above, it was important to determine whether the probes ML125 and ML124 could block expression of relevant ROR target genes in cells. In these assays 293T cells were co-transfected with pS6 control plasmid or pS6 containing full length RORα along with G6Pase promoter. SRC-2 as a coactivator was also co-transfected with G6Pase promoter. Dose-response curve was determined by treating the transfected cells with varying concentrations of compound for 20 hours. Luciferase activity was measured and relative change was determined by normalizing to cells treated with vehicle only. Each data point was performed in eight replicates and represented as mean ±SEM, n=8. The maximum concentration tested was 10 μM.
4.3. Planned Future Studies
In future work, we will continue to explore structural modifications of this inverse agonist series, with the objective of further increasing potency while maintaining selectivity for RORα vs. RORγ. ML125 represents a chemical tool that has been studied extensively and is useful to scientists that are interested in pharmacological observations made that are not easily attributable to LXR and FXR. ML124 represents a probe that is significantly less well studied, but is devoid of LXR and FXR activity. We have ongoing studies showing efficacy of ML124 and close analogs in animal models of diabetes 20 and autoimmune disorders. Subsequent to this probe report, SAR studies have uncovered RORα selective analogs. While these compounds have modest potency in cellular and biochemical assays, they show efficacy in animal models. Regardless, future effects are focused on structural studies and additional SAR to drive potency. More recent, we have uncovered a unique scaffold that offers RORγ isoform selectivity. Thus, the two probes presented here have provided an excellent foundation for the development of chemical probes to explore the biology of the RORs.
5. References
- 1.
- Jetten AM, Kurebayashi S, Ueda E. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog Nucleic Acid Res Mol Biol. 2001;69:205–47. [PubMed: 11550795]
- 2.
- Solt LA, Griffin PR, Burris TP. Ligand regulation of retinoic acid receptor-related orphan receptors: implications for development of novel therapeutics. Curr Opin Lipidol. 2010;21(3):204–11. [PMC free article: PMC5024716] [PubMed: 20463469]
- 3.
- Kallen JA, Schlaeppi JM, Bitsch F, Geisse S, Geiser M, Delhon I, Fournier B. X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha. Structure. 2002;10(12):1697–707. [PubMed: 12467577]
- 4.
- Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem. 2004;279(14):14033–8. [PubMed: 14722075]
- 5.
- Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, Garcia-Ordonez RD, Stayrook KR, Zhang X, Novick S, Chalmers MJ, Griffin PR, Burris TP. Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands. J Biol Chem. 2010;285(7):5013–25. [PMC free article: PMC2836105] [PubMed: 19965867]
- 6.
- Wang Y, Kumar N, Crumbley C, Griffin PR, Burris TP. A second class of nuclear receptors for oxysterols: Regulation of RORalpha and RORgamma activity by 24S-hydroxycholesterol (cerebrosterol). Biochim Biophys Acta. 2010;1801(8):917–23. [PMC free article: PMC2886165] [PubMed: 20211758]
- 7.
- Jin L, Martynowski D, Zheng S, Wada T, Xie W, Li Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol Endocrinol. 2010;24(5):923–9. [PMC free article: PMC2870936] [PubMed: 20203100]
- 8.
- Lau P, Fitzsimmons RL, Raichur S, Wang SC, Lechtken A, Muscat GE. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet-induced obesity. J Biol Chem. 2008;283(26):18411–21. [PubMed: 18441015]
- 9.
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28(1):29–39. [PMC free article: PMC2587175] [PubMed: 18164222]
- 10.
- Ivanov BS II, McKenzie L, Zhou CE, Tadokoro A, Lepelley JJ, Lafaille DJ, Cua, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–33. [PubMed: 16990136]
- 11.
- Meyer T, Kneissel M, Mariani J, Fournier B. In vitro and in vivo evidence for orphan nuclear receptor RORalpha function in bone metabolism. Proc Natl Acad Sci U S A. 2000;97(16):9197–202. [PMC free article: PMC16845] [PubMed: 10900268]
- 12.
- Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Arterioscler Thromb Vasc Biol. 2004;24(2):13–8. [PubMed: 14766739]
- 13.
- Medvedev A, Yan ZH, Hirose T, Giguere V, Jetten AM. Cloning of a cDNA encoding the murine orphan receptor RZR/ROR gamma and characterization of its response element. Gene. 1996;181(1-2):199–206. [PubMed: 8973331]
- 14.
- Bertin R, Guastavino JM, Portet R. Effects of cold acclimation on the energetic metabolism of the staggerer mutant mouse. Physiol Behav. 1990;47(2):377–80. [PubMed: 2333349]
- 15.
- Guastavino JM, Bertin R, Portet R. Effects of the rearing temperature on the temporal feeding pattern of the staggerer mutant mouse. Physiol Behav. 1991;49(2):405–9. [PubMed: 2062915]
- 16.
- Mamontova A, Seguret-Mace S, Esposito B, Chaniale C, Bouly M, Delhaye-Bouchaud N, Luc G, Staels B, Duverger N, Mariani J, Tedgui A. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circulation. 1998;98(24):2738–43. [PubMed: 9851961]
- 17.
- Besnard S, Silvestre JS, Duriez M, Bakouche J, Lemaigre-Dubreuil Y, Mariani J, Levy BI, Tedgui A. Increased ischemia-induced angiogenesis in the staggerer mouse, a mutant of the nuclear receptor Roralpha. Circ Res. 2001;89(12):1209–15. [PubMed: 11739287]
- 18.
- Raspe E, Duez H, Gervois P, Fievet C, Fruchart JC, Besnard S, Mariani J, Tedgui A, Staels B. Transcriptional regulation of apolipoprotein C-III gene expression by the orphan nuclear receptor RORalpha. J Biol Chem. 2001;276(4):2865–71. [PubMed: 11053433]
- 19.
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14(22):2831–8. [PMC free article: PMC317060] [PubMed: 11090131]
- 20.
- Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Burris TP, Griffin PR. The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2- trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Mol Pharmacol. 2010;77(2):228–36. [PMC free article: PMC2812071] [PubMed: 19887649]
- 21.
- Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37(6):1242–50. [PMC free article: PMC3202349] [PubMed: 19282395]
- 22.
- Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem Res Toxicol. 2009;22(10):1736–42. [PubMed: 19803472]
Footnotes
- 1
Farah, B. S.; Gilbert, E. E.; Sibilia, J. P. J. Org. Chem. 1965, 30, 1001.
Publication Details
Author Information and Affiliations
Authors
N Kumar,1 LA Solt,1 J Conkright,1 Y Wang,1 MA Istrate,1 SA Busby,1 RD Garcia-Ordonez,1 P Nuhant,2 T Burris,1 BA Mercer,3 P Hodder,1,3 WR Roush,2 H Rosen,4 and PR Griffin1,*.Affiliations
Publication History
Received: January 3, 2009; Last Update: March 11, 2011.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Kumar N, Solt LA, Conkright J, et al. Campaign to identify novel modulators of the Retinoic acid receptor-related Orphan Receptors (ROR) 2009 Jan 3 [Updated 2011 Mar 11]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.