

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Lo DC, Hughes RE, editors. Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton (FL): CRC Press/Taylor & Francis; 2011.

OVERVIEW
In 1872, physician George Huntington reported a familial form of chorea noted previously on Long Island by his father and grandfather, also physicians. More than a century later his comments about the disease now carrying his name, Huntington’s disease (HD), remain a clear description of its major clinical features (Huntington, 1872; reprinted in Huntington, 2003). Huntington described chorea in general as the “dancing propensities of those … affected,” in whom there “seems to exist some hidden power, something that is playing tricks, as it were, upon the will.” The familial form started “as an ordinary chorea might begin, by the irregular and spasmodic action of certain muscles as of the face, arms, etc. These movements gradually increase when muscles hitherto unaffected take on the spasmodic action….”
The disease, he further noted, “seems to obey certain fixed laws.” It is “confined to … a few families, is attended generally by all the symptoms of common chorea, hardly ever manifesting itself until adult or middle life, and then coming on gradually but surely, increasing by degrees, and often occupying years in its development, until the hapless sufferer is but a quivering wreck of his former self.” The “tendency to insanity,” Huntington observed, progresses so that the “mind becomes more or less impaired, in many amounting to insanity while in others, mind and body both gradually fail until death relieves them of their sufferings.”
The inheritance pattern was also clear to Huntington: “When either or both the parents have shown manifestations … one or more of the offspring almost invariably suffer from the disease … but if by any chance these children go through a life without it, the thread is broken….” Finally, he noted the relentless, fatal course: “I have never known a recovery … it seems at least to be one of the incurables.” Commenting later on Huntington’s description, Sir William Osler noted that “there are few instances in which a disease has been more accurately, more graphically, and more briefly described” (Stevenson, 1934).
The cardinal features of HD so aptly described by Huntington are even clearer to us now, nearly 140 years later. It is a dominantly inherited, neuropsychiatric disorder that affects successive generations of afflicted families. It progresses slowly over years with symptoms typically, but not always, beginning in adulthood. Although HD usually involves chorea and other abnormal movements, the progressive cognitive impairment and behavioral problems are perhaps even more disabling. The remarkable clinical variability and wide range in age of onset, even among affected members in the same family, are now recognized to stem directly from the type of mutation in HD, a dynamic repeat expansion in a polyglutamine-encoding CAG tract. Likewise, the clinical phenomenon of anticipation—the tendency for disease to begin earlier in successive generations—is the result of the propensity for further expansion of the disease-causing repeat on transmission to offspring. We now also recognize that not all HD is familial, as a small percentage of cases arises sporadically from newly expanded alleles. Naturally, such sporadic HD cases then constitute the first generation of new HD families.
The HD mutation, a CAG repeat expansion in the HTT gene (Huntington’s Disease Collaborative Research Group, 1993), is present in all affected persons. The high sensitivity and specificity of genetic testing for the mutation ensure that molecular confirmation of HD is straightforward in the clinic (Davis et al., 1994; Kremer et al., 1994). For persons in whom HD is clinically suspected, a gene test can quickly prove or disprove the diagnosis. For those who are known to be at risk for disease, presymptomatic testing is available after appropriate counseling (WFN Research Group on Huntington’s Chorea, 1994). Currently, in the absence of a preventive medication or disease-modifying therapy, only a small minority of at-risk persons choose to undergo presymptomatic testing (Quaid and Morris, 1993). But the ability to track presymptomatic HD gene carriers in long-term observational studies has greatly improved researchers’ and clinicians’ ability to chart the course of disease symptoms and signs, both before and after diagnosis. This knowledge will be crucial to the success of future preventive and symptomatic trials.
The discovery of the HD mutation has also shed light on possible disease mechanisms (Gatchel and Zoghbi, 2005; Nakamura and Aminoff, 2007; Todi et al., 2007). The CAG repeat expansion encodes a polymorphic stretch of the amino acid glutamine in the disease protein, a large polypeptide known as huntingtin (htt). This expanded polyglutamine tract is widely believed to be the key molecular culprit in HD (Mangiarini et al., 1996). Although the mechanism by which expanded polyglutamine in huntingtin causes brain dysfunction and neuronal cell death remains uncertain, growing knowledge about both the aberrant properties of the disease protein and the havoc it wreaks on cells has provided new insight into potential rational therapies (Di Prospero and Fischbeck, 2005), as discussed elsewhere in this book.
Although HD is a clinically distinctive neurodegenerative disease, polyglutamine expansions are not unique to HD. At least eight other neurodegenerative disorders are caused by polyglutamine expansions (Table 1.1) (Gatchel and Zoghbi, 2005; Riley and Orr, 2006; Todi et al., 2007). In many important ways, HD can be considered a flagship for other polyglutamine diseases. It is the most common among them: roughly 30,000 people are diagnosed with HD in the United States and Canada, with another ~150,000 at risk for disease. More than any other polyglutamine diseases, HD is studied by a vast array of clinicians and scientists who explore disease mechanisms, the natural course of disease, and potential therapies. In particular, the highly organized HD clinical research community is driving the push toward biomarkers and quantifiable clinical measures that will serve as the basis for successful human trials. Importantly, because all polyglutamine diseases may share common elements of pathogenesis, insights from the HD field could prove valuable for the thousands of people affected by other polyglutamine diseases.
TABLE 1.1
List of Known Polyglutamine Diseases, Their Disease Protein, Threshold Disease Repeat, and Major Clinical Features.
Although HD and other polyglutamine disorders are relatively uncommon, they have an impact disproportionate to their prevalence. In populations of European descent, the estimated prevalence of HD is approximately 4–9/100,000. Prevalence rates are much lower in populations of non-European ancestry. The aggregate prevalence of all other polyglutamine diseases, primarily spinocerebellar ataxias, may approach the lower bound of prevalence estimates for HD. Because HD and other polyglutamine diseases tend to strike in middle age and are slowly progressive, the economic impacts as a result of lost earnings and costs of care are enormous. The personal suffering and family disruption imposed by HD are devastating. The combination of usual midlife onset and dominant inheritance tends to pull HD families down the social scale, compromising the lifetime opportunities of both mutant allele carriers and their unaffected relatives.
Despite the many advances in our understanding of HD since discovery of the disease gene, HD remains, in Huntington’s words, “one of the incurables.” As the existence of this book attests, however, the knowledge gained since the gene discovery puts us in a position where this incurable status may soon change. Here we review HD, focusing on its major clinical features, neuropathological hallmarks, and current and possible future therapies. We emphasize literature from the past 15 years. Earlier studies are reviewed thoroughly in other books and chapters (Harper, 1991; Marshall, 2004). In keeping with this book’s purpose, we stress clinical and neuropathological aspects of HD that should be kept in mind as the field moves toward preventive therapy.
CLINICAL FEATURES
HD is, in every sense of the word, a “neuropsychiatric” disorder. No other disease so firmly bridges the disciplines of psychiatry and neurology. The classic clinical triad in HD is (1) progressive movement disorder, most commonly chorea; (2) progressive cognitive disturbance culminating in dementia; and (3) various behavioral disturbances that often precede diagnosis and can vary depending on the state of disease.
Learning from Woody
Physicians who run HD clinics are continually astonished by the diverse array of symptoms and signs that affected persons can develop. In one respect or another, each person with HD is clinically unique. But if there is a “typical” case of HD, it would be adult-onset disease manifested by chorea and behavioral changes that, over a 15–20-year period, slowly rob the affected person of his or her professional skills and social graces. Woody Guthrie, the famous activist and folksinger who died from HD in 1967, was by no means a typical person. Yet the course of his HD fits the description above (Arevalo et al., 2001). Reviewing his life story may help readers gain a personal perspective on this devastating disease.
Born in 1912, Guthrie proved to be a precocious boy and later a creative and prolific folksinger (among his thousands of songs are “This Land Is Your Land” and “So Long, It’s Been Good to Know You”). By the time Guthrie was a young adult, his mother had passed away from progressive dementia accompanied by disruptive behavior that led to her institutionalization. When Guthrie was 39, he was admitted to a detoxification center after an angry outburst toward his wife. This was the first of many institutions he would enter in the final 15 years of his life. At that time, Guthrie’s own notes described his various symptoms and his psychological state very well (Arevalo et al., 2001). He felt “terribly restless always. I get here and I want to be yonder. I get over yonder and I want to be back over here … I don’t trust anybody I see.” Later the same year he wrote:
Here’s my funny feeling over me again. That lost feeling. That gone feeling. That old empty whipped feeling. Shaky. Bad control. Out of control. Jumpy. Jerky. High tension. Least little thing knocks my ego down below zero mark. Everything cuts into me and hurts me several times more than it should … no bodily (physical) pains; just like my arms and legs and hands and feet and my whole body belongs to somebody else and not to me; so ashamed of myself … I spend every drop of my bodily strength trying to hide my trouble away so you can’t see it; trying to keep you from reading it in my face, or my eyes, or in any words I’d say … or in that stumbly way I walk around.
A short time later Guthrie wrote, “My chorea sure isn’t kidding these days. I feel it as a nervous fluttery heart condition along with a slight lack of control over my body at times.” As his disease progressed, the diagnosis was soon established as HD based on his progressive clinical signs (including chorea) and his mother’s illness. Eleven years later, Guthrie’s wife Marjorie described his now advanced symptoms at age 51:
Woody’s muscular condition continues to deteriorate. I find it more and more difficult to understand him when he speaks and this is when his fiery temper jumps out. His memory remains uncanny, but he just hasn’t got the muscular control to mouth the words. His balance is very poor … forever lighting up a new cigarette because the old one fell out of his mouth.
By age 53, Guthrie had stopped speaking entirely and could only communicate by pointing a wildly flailing arm at “yes” and “no” cards that Marjorie made for him. Eight months before he died in 1967 at 56 years of age, the folksinger Pete Seeger visited him. Woody was, Seeger noted,
in a wheelchair. He couldn’t walk anymore so the hospital attendant wheeled him out under a porch where it was warm…. You could see how much he wanted to be part of our little [group] … he tried to get his arms going but they were just flailing around like a windmill. He got to the point where it looked as though he might hurt himself so the attendant said, “You’d better quit playing.” (Arevalo et al., 2001)
Age of Onset, Natural History, and Diagnosis
Onset of manifest HD is defined conventionally by the presence of a motor disorder, usually the involuntary movements known as chorea (see “Movement Disorder” below). In some patients, other motor abnormalities may lead to a diagnosis of HD (Louis et al., 2000). Because the motor features of HD are often preceded by cognitive or behavioral features (see “Movement Disorder” below), the diagnostic criterion of a discernible motor disorder is a fairly conservative standard. In the earlier stages of HD, motor abnormalities may be subtle and may fluctuate depending on the state of arousal of the patient. Median age of diagnosis is approximately 40 years of age with a wide range in ages of onset. Onset before age 20 or after age 65 is relatively rare, but both very young and elderly new-onset HD patients are seen in tertiary referral clinics. Death generally occurs 15–20 years after diagnosis.
When the characteristic motor disorder is present in an individual from a well-characterized HD pedigree, diagnosis of manifest HD is straightforward (Kremer et al., 1994). Circumstances may arise, however, where information about the family history is limited or absent. In some individuals without an apparent family history of HD, covert adoption or nonpaternity, as well as a new mutation, may have occurred. Molecular testing is invaluable in these situations.
Movement Disorder
Although the range of movement disorders in HD extends beyond chorea, it remains the classic motor sign of HD. Derived from the Greek word for “dance,” chorea often begins as fleeting, suppressible movements that may appear as fidgetiness. Over the course of disease, chorea becomes more overt as it involves larger muscle groups. Patients often will incorporate chorea into purposeful movements, something known as parakinesia. Motor impersistence, the inability to sustain a voluntary muscular effort, is common in HD and often goes hand in hand with chorea. The “fly-catcher’s tongue” describes the difficulty many moderately advanced HD patients have in keeping their tongue from protruding beyond the lips.
By itself, chorea is usually not greatly disabling. For many HD clinicians chorea is not something that becomes a major target of treatment. However, flailing and continual chorea can be disabling or physically harmful, requiring treatment with dopamine-depleting agents such as tetrabenazine. In past decades, potent dopamine-blocking neuroleptic medications (e.g., haloperidol) were routinely used to control chorea in HD patients. More recently, however, clinicians have tended first to use newer atypical antipsychotic drugs in persons who experience severe chorea, especially when it is accompanied by psychiatric symptoms warranting antipsychotic use, such as delusions.
Dystonia, the involuntary contraction of muscles, is seen in most persons with HD but is especially prominent in younger-onset individuals. As “typical” adult-onset HD progresses, straightforward chorea can evolve into a more complicated constellation of movement disorders that increasingly include dystonia (Feigin et al., 1995). In addition, clumsiness and slowness of movements (bradykinesia) are common in those with HD, the latter especially in earlier-onset disease. Indeed, the juvenile-onset form of disease that manifests initially with bradykinesia, rigidity, and little or no chorea has been given its own name, the “Westphal variant.” Very early-onset cases also have a high incidence of epilepsy. The distinctive phenotype of very early-onset HD probably reflects the effects of the mutant allele on developing brains. Rather than single out one clinical subset of HD, it is perhaps better to think of motor signs in HD as spanning a spectrum—earlier-onset cases tending to have more bradykinesia and dystonia and later-onset cases tending to have more chorea (Louis et al., 2000). In many adult-onset HD patients, chorea will gradually progress and then begin to subside as dystonia and bradykinesia become more prominent motor features. The combination of chorea, bradykinesia, and dystonia sometimes leads to very unusual gait patterns that leave patients highly prone to falls. Ataxia and marked postural instability are common in those with more advanced HD.
Eye movement abnormalities occur early and persist throughout the course of disease. Persons with HD have difficulty maintaining fixation and develop slowed initiation and velocity of the rapid eye movements known as saccades (Lasker et al., 1987). As this worsens, they may need to thrust their head or blink to break fixation and move their eyes to gaze at a new target.
In summary, HD manifests with a remarkably wide range of movement disorders, partly because of the phenotypic heterogeneity stemming from differences in expanded repeat lengths and partly reflecting the fact that disease symptoms and signs change over time. The progressive movement disorder and increasingly widespread failure of the motor system contribute greatly to the physical disability and decreased life expectancy of individuals with HD. In particular, difficulties with swallowing (dysphagia), speech (dysarthria), and balance (frequent falls) become very debilitating. A common cause of death in those with advanced HD is aspiration pneumonia, reflecting severe dysphagia and general immobility (Sorensen and Fenger, 1992).
Cognitive Disorder
It was once suspected that cognitive decline only began when, or soon before, manifest HD was apparent. Recently, however, large-scale observational studies of HD gene carriers before diagnosis make it clear that subtle cognitive impairment is among the earlier manifestations in the disease process and is associated with progressive caudate atrophy (Aylward, 2007; Aylward et al., 2000). By the time of diagnosis, most subjects with HD have significant cognitive impairment readily measurable by neuropsychological testing. These early signs do not impede most activities of daily living, but individuals with demanding jobs requiring sustained concentration often find work increasingly stressful and difficult. The cognitive impairment progresses slowly over many years to frank dementia in most persons with HD.
In contrast to the dementia of Alzheimer’s disease (AD), HD results in a largely “subcortical” dementia characterized by slowness in initiating thought processes, difficulties with executive function, and problems with attentional and sequencing tasks (Paulsen et al., 1995; Rohrer et al., 1999). Although episodic memory is impaired, memory in general is relatively well preserved compared with, for example, AD. Because a battery of cognitive tests can detect abnormalities before diagnosis, elements of cognitive impairment likely will serve as important quantifiable measures in future clinical trials of candidate preventive agents in prediagnosis individuals.
One intriguing feature of the cognitive impairment in HD is the lack of insight subjects may have regarding their own symptoms (Hoth et al., 2007). Some individuals with overt signs of HD, evident to other family members, deny that they experience any motor or cognitive difficulties. Lack of insight is a typical sign of impaired frontal lobe function and may reflect early dysfunction of striatal neurons receiving prominent frontal lobe inputs.
Behavioral Disorder
The behavioral problems arising in HD can be the most vexing to the patient, family, and physician. These range from affective illness, most notably depression and apathy, to delusional behavior that can include, rarely, hallucinations (Caine and Shoulson, 1983; Paulsen et al., 2001). As is true of other progressive neurodegenerative diseases, the behavioral disorders of HD evolve during the course of illness. Most HD gene carriers will experience some behavioral symptoms before establishing the diagnosis (Close Kirkwood et al., 2002; Duff et al., 2007; Kirkwood et al., 2002). These may include depression, obsessive–compulsive behaviors (OCBs), irritability, and behavioral outbursts (Duff et al., 2007; Kirkwood et al., 2002). Family members often comment that patients’ personalities change in the years leading up to diagnosis, although this may be apparent only in retrospect.
Depression is particularly common in those with HD: between 30% and 50% of patients will develop depressive symptoms in the course of disease. The depression often responds very well to treatment, with selective serotonin reuptake inhibitor antidepressants commonly being the first agents tried. Often, however, there is superimposed apathy that is difficult to treat. Personality changes can occur that impair work performance and social interactions well before the diagnosis is established. This can lead to behavioral agitation, anxiety, alcohol abuse, marital problems, and antisocial behavior. Also common in HD patients are OCBs. These may overlap with features of rigidity and perseveration that also can be seen in individuals with frontal lobe dysfunction. OCBs, rigidity, and perseverative features probably all reflect striatal dysfunction.
The severity of psychiatric symptoms varies greatly in those with HD and does not correlate with dementia and chorea (Paulsen et al., 2001). Recent findings suggest that psychiatric problems are especially problematic in juvenile-onset cases (Ribai et al., 2007).
Some psychiatric symptoms tend to run in specific HD families. For example, overt psychotic behavior more likely will occur in someone from a family where one or more affected persons also suffer from psychosis (Tsuang et al., 2000).
An important cautionary note is the high rate of suicide in patients with HD (Almqvist et al., 1999; Lipe et al., 1993). The risk is significantly higher for those with HD gene-positive status, including both those with diagnosed HD and those who are prediagnosis. The risk may also be higher for family members who are not carriers of the gene, underscoring the tremendous stress and burden placed on families with HD (Lipe et al., 1993). Factors that increase the risk of suicide in gene-positive individuals include being single, lacking children, living alone, having depression, and already having manifest signs of HD. Degeneration of striatal neurons involved in limbic circuits is a possible substrate of depression in those with HD (Tippett et al., 2007).
Clinical Heterogeneity
In patients with HD, the remarkable range in symptoms and age of onset largely reflects differences in the length of the repeat expansion. As shown in Figure 1.1 (Langbehn et al., 2004) and documented in many publications, repeat length inversely correlates with age of onset (Andrew et al., 1993; Ashizawa et al., 1994; Duyao et al., 1993; Kieburtz et al., 1994; Stine et al., 1993). Because earlier- versus later-onset forms of disease typically manifest different motor symptoms, the differences in repeat length also explain, in part, the heterogeneous phenotype.
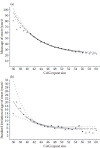
FIGURE 1.1
HD disease onset as a function of repeat length. Shown are population estimates based on an analysis of nearly 3,000 individuals seen at 40 HD centers. (a) Inverse correlation between mean age of onset and CAG repeat length. (b) Standard deviation of (more...)
Expanded repeat length accounts for approximately 50% of variability in age of onset. However, also note in Figure 1.1b that the variance in age of onset for a given repeat length is much greater with small expansions (e.g., 39) than with the rarer, large expansions (e.g., ~56). In other words, the range in age of disease onset for a repeat of 39 can span four decades or more, whereas a repeat of 56 has a narrow range. Clearly other genetic and environmental factors contribute to age of disease onset, particularly for the more common, smaller expansions (<45). Indeed, genetic modifiers of disease onset have already been identified. For example, Meyers and colleagues (Li et al., 2006) and Rubinsztein and colleagues (Rubinsztein et al., 1997; Zeng et al., 2006) separately have identified loci that influence the rate of onset. As mentioned earlier, familial aggregation of certain symptoms (e.g., psychosis) occurs in HD, and this too likely reflects genetic modifiers. It is hoped that further success in defining genetic risk factors/modifiers will shed light on biological pathways that contribute to disease.
Given the relationship of repeat length to age of onset, one can generate “probability curves” that, for a given repeat length, plot the probability of developing manifest disease by a particular age (Langbehn et al., 2004). A series of such probability curves is shown in Figure 1.2. For larger expansions, the narrower range of disease onset is again clear (i.e., steeper slope). An important point illustrated by this figure is that rather modest expansions (≤40) may not lead to manifest disease until very late in life, and in some cases, HD symptoms may not surface before one dies from other age-related causes. In other words, shorter HD expansions are not fully penetrant alleles (Quarrell et al., 2007). There is also an underappreciation of the fact that HD manifesting after age 50 years is not as rare as once thought. Because a higher percentage of late-onset cases will be sporadic cases from newly expanded alleles, clinicians need to keep in mind that late-onset HD often does not have the typical positive family history (Falush et al., 2001).
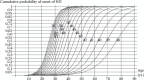
FIGURE 1.2
Cumulative probability curves of HD onset for various CAG repeat lengths. Numbers shown indicate repeat length for a given curve. (From Langbehn, D. R., Brinkman, R. R., Falush, D., Paulsen, J. S., and Hayden, M. R., Clin Genet 65, 267, 2004. With permission.) (more...)
Unlike age of onset, which strongly correlates with repeat length, rate of disease progression is not convincingly linked to repeat length. Some studies have found no correlation (Kieburtz et al., 1994), whereas others have, especially with late-stage outcomes (Marder et al., 2002). Ongoing, large observational studies of gene carriers may provide new data that resolve this uncertainty.
Huntington’s Disease Mimics
HD is a distinctive phenotype, but mimics of HD are encountered occasionally in clinical practice (Rosenblatt et al., 1998; Schneider et al., 2007). The list of choreiform disorders in the differential diagnosis is extensive. Many can be excluded based on the history or features of the physical examination. In doubtful cases, molecular testing is invaluable for identifying or excluding HD. More difficult to evaluate are a few inherited neurodegenerations that can mirror HD. These include other dominant disorders such as the polyglutamine diseases dentatorubral-pallidoluysian atrophy (DRPLA) and SCA17; Huntington’s disease-like 2, which is another expanded repeat disorder; and members of the neurodegeneration with brain iron accumulation spectrum such as neuroferritinopathy. Recessive and X-linked disorders in the neuroacanthocytosis family of diseases can also mimic HD. Molecular diagnoses are available for all of these diseases. Because among these disorders HD is by far the most common, a reasonable strategy is first to test for the presence or absence of the HD mutation. If absent, more extensive testing will be needed to establish a diagnosis. Establishing an accurate diagnosis is crucial for the family and the patient. The clinical implications are vastly different for a dominant versus a recessive disorder, and appropriate presymptomatic testing in other family members can be pursued only with an accurate diagnosis.
NEUROPATHOLOGICAL FEATURES
Non–Central Nervous System Changes
As with the other polyglutamine disorders, HD is primarily a disorder of the central nervous system (CNS). Huntingtin is expressed in many tissues, but the clinical features of HD reflect CNS dysfunction, and almost all histopathologic abnormalities are restricted to the brain. It is important to note that although clinical features and traditional histopathologic analyses point to HD as a CNS disease, investigations with more sensitive measures point to widespread, albeit subclinical, effects of expanded htt. Studies of peripheral blood and lymphoblasts derived from HD subjects have revealed aberrant gene regulation and mitochondrial abnormalities (Borovecki et al., 2005; Sawa et al., 1999). A recent study of urea cycle metabolites suggests a subclinical but detectable effect of expanded htt on hepatic function (Chiang et al., 2007). Several studies suggest abnormalities of another long-lived postmitotic tissue, muscle. Gene regulation studies, magnetic resonance spectroscopic studies, and biochemical studies suggest muscle mitochondrial abnormalities in HD, and muscle biopsy histology indicates the presence of significant but nonspecific abnormalities (Arenas et al., 1998; Lodi et al., 2000; Saft et al., 2005; Strand et al., 2005). An interesting recent observation is the description of testis abnormalities in HD. Testes are characterized by high-level expression of HTT mRNA. Van Raamsdonk et al. (2007) describe reduced numbers of germ cells and abnormal seminiferous tubule morphology.
Gross CNS Pathology
CNS changes, however, drive the mortality and morbidity associated with HD. A plausible explanation for the CNS selectivity of HD is that cell populations with limited lifespans do not live long enough to experience the damage caused by expanded polyglutamine htt. In this model, neurons are preferentially susceptible to the toxicity of expanded htt because of their long lifespan. Although this could explain the selective CNS pathology of HD, it explains neither the regional selectivity within the CNS nor the midlife onset of HD. Some unexplained interaction between expanded polyglutamine htt effects and normal aging phenomena must be invoked to explain the relatively late onset of HD.
Within the CNS, htt is expressed by neurons throughout the neuraxis without dramatic regional differences. Despite this, there is a definite regional pattern to HD pathology (Vonsattel and DiFiglia, 1998). Classic descriptions emphasize that HD is a striatal degeneration. This is true to a first approximation, but this traditional teaching point should not be exaggerated. Gross striatal atrophy is a prominent feature of HD. Careful pathologic analyses, performed usually on specimens of advanced HD, reveal a more complex picture (Figure 1.3). Thinning of the cortical mantle and decreased brain weights and volumes are common as well. Consistent with diffuse loss of neurons is the diffuse loss of cerebral white matter. Careful studies reveal neuronal loss in many regions, including the neocortex, cerebellum, hippocampal formation, substantia nigra, and brainstem nuclei. These findings reflect the widespread expression of htt and correlate well with the profound and diffuse clinical deficits found in patients with advanced HD, including pyramidal signs, ataxia, severe bulbar dysfunction, marked incoordination, and profound dementia.
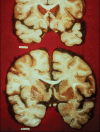
FIGURE 1.3
Gross neuropathology of HD. The top section is from an HD subject, whereas the lower section is from a control without neurologic disease. There is marked striatal atrophy with corresponding ventriculomegaly. There is also diffuse thinning of the cortical (more...)
An important question to consider is what structures are affected earlier in the course of HD. Identifying neuronal populations that are relatively susceptible to expanded polyglutamine htt effects might provide important clues to pathogenic mechanisms. The prevailing impression is that the striatal complex is affected early in HD, although there is also increasing evidence of early neocortical degeneration (Vonsattel and DiFiglia, 1998; see discussion of imaging results under “Striatal Pathology” below). An important development in the study of postmortem changes in HD was the introduction by Vonsattel et al. (1985) of a pathologic grading system based on the degree of striatal pathology. In earlier (lower) Vonsattel grades of pathology, the striatum does seem to be more affected than other brain regions. Vonsattel and colleagues also documented subregional differences in HD pathology in the striatal complex. In the extensive series of specimens examined by Vonsattel and colleagues, neurodegeneration within the striatal complex progressed in caudalto-rostral and dorsal-to-ventral gradients.
Striatal Pathology
Initial explorations of HD striatal pathology beyond classic histopathology involved biochemical assessments of neurotransmitter systems. These initial studies revealed loss of γ-aminobutyric acid (GABA)ergic and cholinergic markers with relative sparing of dopaminergic markers. These results suggested loss of intrinsic GABAergic and cholinergic neurons with relative sparing of extrinsic dopaminergic terminals. The similarity of these changes to the results of experimental excitotoxic lesions gave rise to the still popular excitotoxic hypothesis of neuronal death in HD.
The identification of striatal neuron subpopulations made it possible to pursue a finegrained analysis of striatal changes in HD. Striatal neuron populations are divided into two major groups: (1) aspiny interneurons whose projection arbors are restricted to the striatum; and (2) medium spiny projection neurons whose primary axons synapse in the downstream targets of the striatal complex. Striatal interneurons are further subdivided based on the morphology and neurochemical phenotype. Well-studied populations of striatal interneurons include the large aspiny cholinergic interneurons, which are probably synonymous with the tonically active neurons described in extracellular recording studies. These neurons are virtually spared in HD striatum (Ferrante et al., 1987). Interestingly, although there is solid evidence that cholinergic neuron perikarya persist even in those with advanced HD, striatal choline acetyltransferase (ChAT) levels decrease markedly. The discrepancy between decreasing ChAT activity and preserved perikarya suggests significant striatal cholinergic interneuron dysfunction, as opposed to neurodegeneration, in HD. Another spared subpopulation of striatal interneurons are those containing somatostatin, neuropeptide Y, and nitric oxide synthase, which persist even in advanced HD striatum (Ferrante et al., 1985).
Subpopulations of striatal projection neurons, all of which are GABAergic, are defined by their primary projection targets, coexpressed neuropeptides, and neurotransmitter receptors. Striatal neurons projecting to the different segments of the globus pallidus (external [GPe] and internal [GPi]) and the different components of the substantia nigra (dopaminergic pars compacta [SNc] and GABAergic pars reticulata [SNr]) form relatively segregated pools of neurons. Striato-GPe neurons are distinguished by the expression of enkephalins, dopamine D2 receptors, and adenosine A2a receptors, whereas the other striatal projection neuron pools tend to express tachykinins and dopamine D1 receptors. Some recent studies in nonhuman primates using sensitive tract tracing methods have cast doubt on the idea of segregated striatal projection neuron pools (Levesque and Parent, 2005). However, recent data from mice engineered to express green fluorescent protein under the control of the D1 or D2 promoters strongly support the existence of distinct pools of striatal projection neurons (Lobo et al., 2006). The existence of different pools of striatal projection neurons overlaps to some degree with another important aspect of striatal organization: the patch (striosome)–matrix differentiation. A variety of neurochemical markers differentiate the striatum into two compartments: (1) patches (striosomes), a geometrically complex interconnected tubular compartment; and (2) the surrounding matrix compartment. Striato-SNc neurons are disproportionately represented within the patch compartment.
Examination of postmortem HD material across a full spectrum of pathologic grades suggests a specific temporal order in degeneration of striatal projection neuron subpopulations. The early changes appear to be loss of striato-GPe neurons and perhaps striato-SNr neurons (Albin et al., 1992; Deng et al., 2004; Reiner et al., 1988). In contrast, striato-GPi neurons are relatively spared until later in the course of HD. This sequential pattern of neuronal loss correlates broadly with certain features of the natural history of HD. Oculomotor abnormalities, particularly abnormalities of saccadic eye movements, are early features of HD. As basal ganglia inputs to the superior colliculus come from SNr, the early loss of striato-SNr projection neurons correlates nicely with early saccadic abnormalities. The evolution of involuntary movements in HD correlates also with the evolution of striatal projection neuron changes. One predicted downstream consequence of initial degeneration of striato- GPe neurons is inhibition of the subthalamic nucleus. Decreased subthalamic activity is associated with choreoathetoid movements. In many patients, disease progression is associated with gradual worsening of choreoathetoid movements, which then peak in intensity and gradually decrease. The decrease in choreoathetosis is accompanied by worsening dystonia and bradykinesia. The apparent correlate in striatal pathology is generalized loss of striatal projection neurons and probably neurons within other nuclei of the basal ganglia (Albin et al., 1990). In a recent, interesting study, Tippett et al. (2006) looked specifically at a marker of striosomal striatal projection neurons in a broad spectrum of HD postmortem specimens. This work suggests a correlation between mood disorders in HD and striosomal pathology.
Neocortex is also affected significantly in HD. Recent neuroimaging data (see “Neuroimaging” below) suggest that this may be an early event in HD. Some data suggest that neocortical atrophy proceeds in tandem with striatal atrophy (Halliday et al., 1998; Macdonald and Halliday, 2002). As in the striatum, there is evidence for differential involvement of different cortical neuron populations in HD. Careful studies looking at changes in cortical laminae reveal differential loss of neurons in different layers. Pyramidal neurons of deeper cortical layers appear to be more affected in HD, although this may vary by cortical field (Hedreen et al., 1991; Sieradzan and Mann, 2001; Sotrel et al., 1991, 1993).
Unlike the neurofibrillary tangles of AD or the Lewy bodies of Parkinson’s disease, HD was believed historically not to exhibit characteristic abnormalities at the cellular level. This perception changed radically with the discovery in the R6/2 transgenic mouse model of neuronal intranuclear and cytoplasmic aggregates containing expanded polyglutamine htt, ubiquitin, and other proteins. These types of inclusions were also found in HD brain and in most other polyglutamine disorders. Although inclusions are found in the HD brain, their frequency is less than that seen in many of the murine genetic models (DiFiglia et al., 1997; Gutekunst et al., 1999). Perhaps because of the relatively short lifespan of mice, neuronal loss in murine genetic models is less severe than that seen in end-stage HD. It is possible that neuronal loss in HD is accompanied by a corresponding loss of neuronal inclusions.
Debate has arisen about the role, if any, of neuronal inclusions in disease pathogenesis. Suggestions for the role of inclusions include that they are (1) directly pathogenic structures; (2) markers of failed clearance of abnormal disease protein; (3) protective “sinks” for toxic polyglutamine proteins and associated factors; and (4) epiphenomena of the disease process (Ross and Poirier, 2005). A relevant fact from postmortem analysis is that neuronal inclusions are more readily found in some of the spared striatal interneuron populations than in the vulnerable striatal interneurons (Kuemmerle et al., 1999).
Neuroimaging
Whereas imaging methods were pursued originally to improve diagnosis of HD, recent studies have pursued imaging methods as part of efforts to develop biomarkers for HD. The hope is that novel imaging methods will provide sufficiently precise measures of disease state and progression to serve as state markers, as response markers, or even as surrogate endpoints in trials.
Computed tomography, magnetic resonance imaging (MRI), and positron emission tomography (PET) imaging methods have all been applied to HD. Most recent studies have concentrated on MRI morphometric analyses of regional brain volumes in the hope of establishing changes over time that will be useful as a marker of disease progression in symptomatic or presymptomatic HD subjects. Using observer-identified volumes of interest, Aylward and colleagues presented data suggesting progressive decrease in striatal volume well before the onset of manifest HD (Aylward, 2007). In their dataset, approximately 50% of the striatum had degenerated by onset of manifest HD. This approach seems promising for the development of a surrogate marker of disease progression that could be applied in both prediagnosis and manifest HD populations. On the other hand, the relatively small volume of the striatum and the apparent fact that considerable striatal atrophy occurs early in the course of HD may limit the dynamic range of striatal volume as a biomarker in studies of manifest HD. For presymptomatic subjects, the imaging arm of the PREDICT-HD observational study (Paulsen et al., 2008) will provide definitive data about the potential use of this approach as a biomarker.
Improved MRI morphometry analysis methods may provide additional or even superior biomarkers. Cortical mantle atrophy is a significant feature of HD pathology. The large extent of the cortex and the possibility of assessing many cortical regions offer the theoretical hope of a morphometric marker with very wide dynamic range. Improved morphometry methods make it possible to measure cortical thickness of many regions simultaneously. Rosas and colleagues pursued cortical morphometry in both symptomatic and presymptomatic HD subjects (Rosas et al., 2002, 2005). Their work suggests that cortical atrophy is an early event in HD, that it occurs in presymptomatic subjects, and that the magnitude of changes over relatively short intervals (e.g., 2–3 years) is sufficiently great to provide a biomarker of disease progression. In addition, this group’s work suggests that cortical degeneration is not uniform throughout the cortical mantle but occurs in a stereotyped sequence, with some cortical fields affected earlier than others (HD Rosas, personal communication). This exciting work awaits validation in suitable prospectively collected large datasets.
There has been less activity with PET imaging in HD research, but this remains an interesting potential imaging modality for HD. PET availability was limited historically to research centers with cyclotrons, but PET imaging has become a standard modality for many medical applications, and both scanner availability and the number of easily used tracers is expanding rapidly. The theoretical advantage of PET is its ability to image specific molecular targets. In HD, several PET studies have demonstrated decreases in striatal neurotransmitter markers, particularly striatal dopamine receptors. Changes in these markers appear to be an early feature of HD, consistent with the MRI morphometry data of Aylward and colleagues (Ginovart et al., 1997; van Oostrom et al., 2005; Weeks et al., 1996). Counterintuitively, some PET studies also demonstrate loss of striatal dopamine terminal markers in HD (Backman et al., 1997; Bohnen et al., 2000). This loss seems to be associated with more severe disease. It may reflect the loss of nigrostriatal dopaminergic neurons as a result of direct effects of perikaryal nigral polyglutamine toxicity or the loss of trophic support from striatal neurons.
Ongoing studies with MRI morphometry are likely to provide the critical data within the next 4–5 years. There is a very good chance that MRI morphometry will provide adequate biomarkers for evaluating progression and treatment response in HD.
THERAPY: PRESENT AND FUTURE
Symptomatic Therapy
Although HD may not yet be curable, some features are amenable to therapy (Bonelli and Hofmann, 2007). These include depression, which responds often to antidepressants, most commonly selective serotonin reuptake inhibitors or other, newer antidepressants. OCBs may also respond to antidepressants, as may agitation and irritability. Irritability and impulsive behavior are sometimes treated with anticonvulsants, such as valproic acid or carbamazepine. Anxiety can benefit from anxiolytic medications. When HD is accompanied by delusions or paranoid thoughts, atypical antipsychotic drugs are frequently used (less so the classic neuroleptic agents), and appropriate antipsychotic medications can improve behavioral outbursts. Thus far, limited trials of cognitive-enhancing agents used primarily in patients with AD, such as memantine (Beister et al., 2004), rivastigmine (de Tommaso et al., 2007), and donepezil, have shown only modest benefit. Bradykinesia and rigidity in younger-onset individuals can respond to dopaminergic agents used in parkinsonism (Jongen et al., 1980).
Chorea will respond to dopamine-blocking agents; for example, a recent well-controlled clinical trial proved that tetrabenazine reduces chorea in a dosedependent manner (Huntington Study Group, 2006). Many other medications have shown modest benefit for chorea (Bonelli et al., 2002; de Tommaso et al., 2005; Huntington Study Group, 2003; Kremer et al., 1999; O’Suilleabhain and Dewey, 2003; Verhagen Metman et al., 2002), but not all have been tested in well-controlled trials. Suppressing chorea per se is not an endpoint sought by many physicians who treat HD unless the chorea is disabling or so severe as to increase risk of injury. Although traditional neuroleptic agents or dopamine blockers such as tetrabenazine can be effective in reducing chorea, these medications are not without their side effects, including parkinsonism, gait disturbance, somnolence, and depression. Myoclonus, which is rare in HD and sometimes mistaken for chorea, can respond to valproic acid (Saft et al., 2006).
Assistive therapies such as physical, occupational, and speech therapy play an important role in the care of HD patients. In many cases, these are the primary therapeutic interventions and can be very useful for some patients. The efficacy of assistive interventions is often limited, however, by the cognitive and behavioral impairments of HD patients. As with many aspects of HD, the burden of maximizing benefits from assistive interventions falls on caregivers.
Although the focus of this book and the HD research community is on identifying a preventive medicine for HD, we must also continue efforts to seek out symptomatic medications and enhance practical measures (Nance, 2007) that can improve the lives of those currently affected by HD.
Toward Preventive Therapy
As our understanding of the molecular basis of HD becomes clearer, routes to preventive therapy are being considered (Di Prospero and Fischbeck, 2005; Feigin and Zgaljardic, 2002; Nakamura and Aminoff, 2007). Potential contributing factors to HD include perturbations in protein homeostasis, mitochondrial dysfunction, excessive or aberrant corticostriatal input, transcriptional dysregulation, loss of neurotrophic support, and impairments in axonal trafficking. Correspondingly, potential compounds or strategies considered for preventive treatment include histone deacetylase inhibitors (e.g., phenylbutyrate [Gardian et al., 2005]); antioxidants, mitochondrial enhancers, and energy substrates (e.g., coenzyme Q10 [Huntington Study Group, 2001] and creatine [Hersch et al., 2006]); neuroprotective compounds (e.g., lithium [Wei et al., 2001]); antiapoptotic compounds (e.g., minocycline [Huntington Study Group, 2004]); transglutaminase inhibitors (Dubinsky and Gray, 2006); reagents that block protein aggregation or assist protein folding; compounds to enhance autophagic clearance of mutant protein (Sarkar et al., 2007); inhibitors of the kynurenine 3-monooxygenase pathway (Giorgini et al., 2005); cell or gene replacement therapy (Bachoud-Levi et al., 2000; Bloch et al., 2004; Keene et al., 2007); and RNA interference reagents that “knock down” disease gene expression (Harper et al., 2005). The availability of numerous mouse models of HD in which disease progression can reliably be measured has allowed researchers to identify more than 20 compounds or reagents that slow disease in these models and therefore may warrant human trials (reviewed in Beal and Ferrante, 2004; Hersch and Ferrante, 2004). Two challenges at hand are (1) determining which molecules and compounds should be tried in human trials and (2) refining quantitative measures of HD in humans so that clinical trials can be performed faster and in fewer subjects without sacrificing statistical power.
To date, human clinical trials testing candidate preventive compounds have been discouraging. In the largest published study to date, the CARE-HD study, in which both coenzyme Q10 and remacemide were tested, coenzyme Q10 (600 mg) showed a statistically insignificant trend toward slowing disease (Huntington Study Group, 2001). A similar study of a higher dose of coenzyme Q10, the 2CARE trial (also sponsored by the Huntington Study Group), recently began. This study tests the effects of 2,400 mg/day on the progression of functional decline in HD subjects.
The field is now witnessing human trials that include potential biomarkers of disease as outcome measures. For example, a short-term study of creatine in a small number of HD subjects led to a reduction in a marker of DNA damage, 8-OH- deoxyguanosine (Hersch et al., 2006). We caution the reader that even though reducing levels of a peripheral marker of DNA damage sounds promising, we do not know that doing so would alter the disease process or disease progression in any way. As more potential biomarkers of disease surface, it will be vitally important to correlate changes in them with disease progression and with the clinical response to any preventive medication that does surface.
Search for Quantifiable Measures
This latter study points to the growing interest in identifying biomarkers for HD—for example, changes in brain imaging (reviewed earlier), metabolic/proteomic profiles (Dalrymple et al., 2007; Gomez-Anson et al., 2007), or gene expression (Borovecki et al., 2005) that might one day serve as reliable proxies for clinical benefit. To get there, however, we first need to know more about disease progression.
In the HD research community, efforts to define disease progression and response to medications have been aided greatly by a widely used standardized clinical rating scale, the Unified Huntington’s Disease Ratings Scale (UHDRS) (Huntington Study Group, 1996; Siesling et al., 1998). The UHDRS has four major components: motor function and cognitive, behavioral, and functional ability. It has been used by the large consortium of HD investigators known as the Huntington Study Group for 15 years and serves as the data collection base for two ongoing, large-scale observational trials: PREDICT-HD (Paulsen et al., 2006) and PHAROS-HD (Huntington Study Group PHAROS, 2006). The movement disorder of HD has classically been the barometer by which neurologists establish the diagnosis. Reliance on movement disorder as the sine qua non of HD diagnosis may change as these studies identify a nonredundant set of cognitive, psychiatric, and functional measures that are associated with motor scores.
With respect to developing therapeutics, it is important to recognize that clinical progression in HD and accompanying brain changes detectable by imaging can be quantified both before and after diagnosis. Studies have shown measurable decreases in total functional capacity, particularly in the early to middle years after diagnosis (Marder et al., 2000). Because the limited dynamic range of these measures in advanced disease makes it much more difficult to assess progression late in disease, effective preventive trials will almost certainly be carried out in early HD or prediagnosis subjects who are relatively close to the predicted age of onset. A subset of measures in the full UHDRS, perhaps supplemented with additional cognitive tests, likely will constitute a reliable, straightforward battery of tests with which clinicians in future trials can measure changes in rate of progression and more accurately pinpoint phenoconversion to manifest disease. As shown in Figure 1.4, an effective preventive medication would slow the rate to “phenoconversion.”
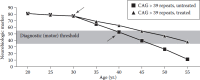
FIGURE 1.4
Intervention model for adult-onset HD. In this illustrated scenario, the downward-pointing arrow indicates administration of an effective neuroprotective agent in a treated individual, whereas the upward-pointing arrow indicates the point of clinical (more...)
Ongoing observational studies likely will generate the longitudinal data that allow the HD research community to establish associations between specific biomarkers and either conversion to manifest disease or progression of disease. Biochemical markers like 8-OH-deoxyguanosine or other metabolomic or genomic markers are being sought as state markers, progression markers, and response markers. Imaging markers look directly at brain changes and may prove to be particularly useful as progression/response markers and even prove to be suitable as surrogate endpoints. All putative biomarker methods will need to be validated against the well-developed clinical dataset and carefully evaluated to ensure that they have suitable statistical properties. Certain such markers may then become true surrogates for a clinical endpoint and thus permit trials of shorter duration in smaller numbers of subjects to identify preventive medications or experimental treatments. In short, “one of the incurables” might just become treatable after all.
SUMMARY
HD is a complex and clinically demanding disorder. Work over the past two decades has defined the clinical phenotype and neuropathology with increasing precision. This essential descriptive work provides the platform for an increasingly sophisticated body of clinical research aimed at developing tools for characterizing clinical, imaging, and other features of HD. These new tools will facilitate researchers’ ability to translate insights from the laboratory into rigorous clinical research and potential therapies for HD.
ACKNOWLEDGMENTS
HD and polyglutamine disease research in the Paulson and Albin laboratories is funded by RO1-NS38712 (HLP) and a VA Merit Review grant, the High Q Foundation, and RO3-NS054810 (RLA). We also thank Drs. Jane Paulsen, Doug Langbehn, and Andrew Lieberman for providing figures.
REFERENCES
- Albin R. L., Reiner A., Anderson K. D., Dure IV L. S., Handelin B., Balfour R., Whetsell W. O. Jr, Penney J. B., Young A. B. Preferential Loss of Striato-External Pallidal Projection Neurons in Presymptomatic Huntington’s Disease. Ann Neurol. 1992;31(4):425–30. [PubMed: 1375014]
- Albin R. L., Reiner A., Anderson K. D., Penney J. B., Young A. B. Striatal and Nigral Neuron Subpopulations in Rigid Huntington’s Disease: Implications for the Functional Anatomy of Chorea and Rigidity-Akinesia. Ann Neurol. 1990;27(4):357–65. [PubMed: 1972318]
- Almqvist E. W., Bloch M., Brinkman R., Craufurd D., Hayden M. R. A Worldwide Assessment of the Frequency of Suicide, Suicide Attempts, or Psychiatric Hospitalization after Predictive Testing for Huntington Disease. Am J Hum Genet. 1999;64(5):1293–304. [PMC free article: PMC1377865] [PubMed: 10205260]
- Andrew S. E., Goldberg Y. P., Kremer B., Telenius H., Theilmann J., Adam S., Starr E., Squitieri F., Lin B., Kalchman M. A., et al. The Relationship between Trinucleotide (Cag) Repeat Length and Clinical Features of Huntington’s Disease. Nat Genet. 1993;4(4):398–403. [PubMed: 8401589]
- Arenas J., Campos Y., Ribacoba R., Martin M. A., Rubio J. C., Ablanedo P., Cabello A. Complex I Defect in Muscle from Patients with Huntington’s Disease. Ann Neurol. 1998;43(3):397–400. [PubMed: 9506560]
- Arevalo J., Wojcieszek J., Conneally P. M. Tracing Woody Guthrie and Huntington’s Disease. Semin Neurol. 2001;21(2):209–23. [PubMed: 11442329]
- Ashizawa T., Wong L. J., Richards C. S., Caskey C. T., Jankovic J. Cag Repeat Size and Clinical Presentation in Huntington’s Disease. Neurology. 1994;44(6):1137–43. [PubMed: 8208412]
- Aylward E. H. Change in MRI Striatal Volumes as a Biomarker in Preclinical Huntington’s Disease. Brain Res Bull. 2007;72(2–3):152–58. [PubMed: 17352939]
- Aylward E. H., Codori A. M., Rosenblatt A., Sherr M., Brandt J., Stine O. C., Barta P. E., Pearlson G. D., Ross C. A. Rate of Caudate Atrophy in Presymptomatic and Symptomatic Stages of Huntington’s Disease. Mov Disord. 2000;15(3):552–60. [PubMed: 10830423]
- Bachoud-Levi A. C., Remy P., Nguyen J. P., Brugieres P., Lefaucheur J. P., Bourdet C., Baudic S., Gaura V., Maison P., Haddad B., et al. Motor and Cognitive Improvements in Patients with Huntington’s Disease after Neural Transplantation. Lancet. 2000;356(9246):1975–79. [PubMed: 11130527]
- Backman L. T., Robins-Wahlin B., Lundin A., Ginovart N., Farde L. Cognitive Deficits in Huntington’s Disease Are Predicted by Dopaminergic Pet Markers and Brain Volumes. Brain. 1997;120(12):2207–17. [PubMed: 9448576]
- Beal M. F., Ferrante R. J. Experimental Therapeutics in Transgenic Mouse Models of Huntington’s Disease. Nat Rev Neurosci. 2004;5(5):373–84. [PubMed: 15100720]
- Beister A., Kraus P., Kuhn W., Dose M., Weindl A., Gerlach M. The N-Methyl-D- Aspartate Antagonist Memantine Retards Progression of Huntington’s Disease. J Neural Transm Suppl. 2004;(68):117–22. [PubMed: 15354397]
- Bloch J., Bachoud-Levi A. C., Deglon N., Lefaucheur J. P., Winkel L., Palfi S., Nguyen J. P., Bourdet C., Gaura V., Remy P., et al. Neuroprotective Gene Therapy for Huntington’s Disease, Using Polymer-Encapsulated Cells Engineered to Secrete Human Ciliary Neurotrophic Factor: Results of a Phase I Study. Hum Gene Ther. 2004;15(10):968–75. [PubMed: 15585112]
- Bohnen N. I., Koeppe R. A., Meyer P., Ficaro E., Wernette K., Kilbourn M. R., Kuhl D. E., Frey K. A., Albin R. L. Decreased Striatal Monoaminergic Terminals in Huntington Disease. Neurology. 2000;54(9):1753–59. [PubMed: 10802780]
- Bonelli R. M., Hofmann P. A Systematic Review of the Treatment Studies in Huntington’s Disease since 1990. Expert Opin Pharmacother. 2007;8(2):141–53. [PubMed: 17257085]
- Bonelli R. M., Mahnert F. A., Niederwieser G. Olanzapine for Huntington’s Disease: An Open Label Study. Clin Neuropharmacol. 2002;25(5):263–65. [PubMed: 12410058]
- Borovecki F., Lovrecic L., Zhou J., Jeong H., Then F., Rosas H. D., Hersch S. M., Hogarth P., Bouzou B., Jensen R. V., et al. Genome-Wide Expression Profiling of Human Blood Reveals Biomarkers for Huntington’s Disease. Proc Natl Acad Sci U S A. 2005;102(31):11023–28. [PMC free article: PMC1182457] [PubMed: 16043692]
- Caine E. D., Shoulson I. Psychiatric Syndromes in Huntington’s Disease. Am J Psychiatry. 1983;140(6):728–33. [PubMed: 6221669]
- Chiang M. C., Chen H. M., Lee Y. H., Chang H. H., Wu Y. C., Soong B. W., Chen C. M., Wu Y. R., Liu C. S., Niu D. M., et al. Dysregulation of C/Ebpalpha by Mutant Huntingtin Causes the Urea Cycle Deficiency in Huntington’s Disease. Hum Mol Genet. 2007;16(5):483–98. [PubMed: 17213233]
- Close Kirkwood S., Siemers E., Viken R. J., Hodes M. E., Conneally P. M., Christian J. C., Foroud T. Evaluation of Psychological Symptoms among Presymptomatic HD Gene Carriers as Measured by Selected Mmpi Scales. J Psychiatr Res. 2002;36(6):377–82. [PubMed: 12393306]
- Dalrymple A., Wild E. J., Joubert R., Sathasivam K., Bjorkqvist M., Petersen A., Jackson G. S., Isaacs J. D., Kristiansen M., Bates G. P., et al. Proteomic Profiling of Plasma in Huntington’s Disease Reveals Neuroinflammatory Activation and Biomarker Candidates. J Proteome Res. 2007;6(7):2833–40. [PubMed: 17552550]
- Davis M. B., Bateman D., Quinn N. P., Marsden C. D., Harding A. E. Mutation Analysis in Patients with Possible but Apparently Sporadic Huntington’s Disease. Lancet. 1994;344(8924):714–17. [PubMed: 7915776]
- de Tommaso M., Di Fruscolo O., Sciruicchio V., Specchio N., Cormio C., De Caro M. F., Livrea P. Efficacy of Levetiracetam in Huntington Disease. Clin Neuropharmacol. 2005;28(6):280–84. [PubMed: 16340384]
- de Tommaso M., Di Fruscolo O., Sciruicchio V., Specchio N., Livrea P. Two Years’ Follow-up of Rivastigmine Treatment in Huntington Disease. Clin Neuropharmacol. 2007;30(1):43–46. [PubMed: 17272969]
- Deng Y. P., Albin R. L., Penney J. B., Young A. B., Anderson K. D., Reiner A. Differential Loss of Striatal Projection Systems in Huntington’s Disease: A Quantitative Immunohistochemical Study. J Chem Neuroanat. 2004;27(3):143–64. [PubMed: 15183201]
- Di Prospero N. A., Fischbeck K. H. Therapeutics Development for Triplet Repeat Expansion Diseases. Nat Rev Genet. 2005;6(10):756–65. [PubMed: 16205715]
- DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science. 1997;277(5334):1990–93. [PubMed: 9302293]
- Dubinsky R., Gray C. Cyte-I-Hd: Phase I Dose Finding and Tolerability Study of Cysteamine (Cystagon) in Huntington’s Disease. Mov Disord. 2006;21(4):530–33. [PubMed: 16258942]
- Duff K., Paulsen J. S., Beglinger L. J., Langbehn D. R., Stout J. C. Psychiatric Symptoms in Huntington’s Disease before Diagnosis: The Predict-HD Study. Biol Psychiatry. 2007;62(12):1341–46. [PubMed: 17481592]
- Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M., Abbott M., et al. Trinucleotide Repeat Length Instability and Age of Onset in Huntington’s Disease. Nat Genet. 1993;4(4):387–92. [PubMed: 8401587]
- Falush D., Almqvist E. W., Brinkmann R. R., Iwasa Y., Hayden M. R. Measurement of Mutational Flow Implies Both a High New-Mutation Rate for Huntington Disease and Substantial Underascertainment of Late-Onset Cases. Am J Hum Genet. 2001;68(2):373–85. [PMC free article: PMC1235271] [PubMed: 11225602]
- Feigin A., Kieburtz K., Bordwell K., Como P., Steinberg K., Sotack J., Zimmerman C., Hickey C., Orme C., Shoulson I. Functional Decline in Huntington’s Disease. Mov Disord. 1995;10(2):211–14. [PubMed: 7753064]
- Feigin A., Zgaljardic D. Recent Advances in Huntington’s Disease: Implications for Experimental Therapeutics. Curr Opin Neurol. 2002;15(4):483–89. [PubMed: 12151847]
- Ferrante R. J., Beal M. F., Kowall N. W., Richardson E. P. Jr, Martin J. B. Sparing of Acetylcholinesterase-Containing Striatal Neurons in Huntington’s Disease. Brain Res. 1987;411(1):162–66. [PubMed: 2955849]
- Ferrante R. J., Kowall N. W., Beal M. F., Richardson E. P. Jr, Bird E. D., Martin J. B. Selective Sparing of a Class of Striatal Neurons in Huntington’s Disease. Science. 1985;230(4725):561–63. [PubMed: 2931802]
- Gardian G., Browne S. E., Choi D. K., Klivenyi P., Gregorio J., Kubilus J. K., Ryu H., Langley B., Ratan R. R., Ferrante R. J., et al. Neuroprotective Effects of Phenylbutyrate in the N171-82q Transgenic Mouse Model of Huntington’s Disease. J Biol Chem. 2005;280(1):556–63. [PubMed: 15494404]
- Gatchel J. R., Zoghbi H. Y. Diseases of Unstable Repeat Expansion: Mechanisms and Common Principles. Nat Rev Genet. 2005;6(10):743–55. [PubMed: 16205714]
- Ginovart N., Lundin A., Farde L., Halldin C., Backman L., Swahn C. G., Pauli S., Sedvall G. Pet Study of the Preand Post-Synaptic Dopaminergic Markers for the Neurodegenerative Process in Huntington’s Disease. Brain. 1997;120(3):503–14. [PubMed: 9126061]
- Giorgini F., Guidetti P., Nguyen Q., Bennett S. C., Muchowski P. J. A Genomic Screen in Yeast Implicates Kynurenine 3-Monooxygenase as a Therapeutic Target for Huntington Disease. Nat Genet. 2005;37(5):526–31. [PMC free article: PMC1449881] [PubMed: 15806102]
- Gomez-Anson B., Alegret M., Munoz E., Sainz A., Monte G. C., Tolosa E. Decreased Frontal Choline and Neuropsychological Performance in Preclinical Huntington Disease. Neurology. 2007;68(12):906–10. [PubMed: 17372125]
- Gutekunst C. A., Li S. H., Yi H., Mulroy J. S., Kuemmerle S., Jones R., Rye D., Ferrante R. J., Hersch S. M., Li X. J. Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J Neurosci. 1999;19(7):2522–34. [PMC free article: PMC6786077] [PubMed: 10087066]
- Halliday G. M., McRitchie D. A., Macdonald V., Double K. L., Trent R. J., McCusker E. Regional Specificity of Brain Atrophy in Huntington’s Disease. Exp Neurol. 1998;154(2):663–72. [PubMed: 9878201]
- Harper Peter S., editor. Huntington’s Disease, Major Problems in Neurology. Philadelphia: W.B. Saunders Company, Ltd; 1991.
- Harper S. Q., Staber P. D., He X., Eliason S. L., Martins I. H., Mao Q., Yang L., Kotin R. M., Paulson H. L., Davidson B. L. RNA Interference Improves Motor and Neuropathological Abnormalities in a Huntington’s Disease Mouse Model. Proc Natl Acad Sci U S A. 2005;102(16):5820–25. [PMC free article: PMC556303] [PubMed: 15811941]
- Hedreen J. C., Peyser C. E., Folstein S. E., Ross C. A. Neuronal Loss in Layers V and VI of Cerebral Cortex in Huntington’s Disease. Neurosci Lett. 1991;133(2):257–61. [PubMed: 1840078]
- Hersch S. M., Ferrante R. J. Translating Therapies for Huntington’s Disease from Genetic Animal Models to Clinical Trials. NeuroRx. 2004;1(3):298–306. [PMC free article: PMC534928] [PubMed: 15717031]
- Hersch S. M., Gevorkian S., Marder K., Moskowitz C., Feigin A., Cox M., Como P., Zimmerman C., Lin M., Zhang L., et al. Creatine in Huntington Disease Is Safe, Tolerable, Bioavailable in Brain and Reduces Serum 8oh2’dg. Neurology. 2006;66(2):250–52. [PubMed: 16434666]
- Hoth K. F., Paulsen J. S., Moser D. J., Tranel D., Clark L. A., Bechara A. Patients with Huntington’s Disease Have Impaired Awareness of Cognitive, Emotional, and Functional Abilities. J Clin Exp Neuropsychol. 2007;29(4):365–76. [PubMed: 17497560]
- Huntington G. On Chorea. J Neuropsychiatry Clin Neurosci. 2003;15(1):109–12. [PubMed: 12556582]
- Huntington G. On Chorea. Medical and Surgical Reporter: A Weekly Journal. 1872;26(15):317–21.
- Huntington’s Disease Collaborative Research Group. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–83. [PubMed: 8458085]
- Huntington Study Group, govt. Tetrabenazine as Antichorea Therapy in Huntington Disease: A Randomized Controlled Trial. Neurology. 2006;66(3):366–72. [PubMed: 16476934]
- Huntington Study Group. Minocycline Safety and Tolerability in Huntington Disease. Neurology. 2004;63(3):547–49. [PubMed: 15304592]
- Huntington Study Group. Dosage Effects of Riluzole in Huntington’s Disease: A Multicenter Placebo-Controlled Study. Neurology. 2003;61(11):1551–56. [PubMed: 14663041]
- Huntington Study Group. A Randomized, Placebo-Controlled Trial of Coenzyme Q10 and Remacemide in Huntington’s Disease. Neurology. 2001;57(3):397–404. [PubMed: 11502903]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and Consistency. Mov Disord. 1996;11(2):136–42. [PubMed: 8684382]
- Huntington Study Group PHAROS. At Risk for Huntington Disease: The Pharos (Prospective Huntington at Risk Observational Study) Cohort Enrolled. Arch Neurol. 2006;63(7):991–96. [PubMed: 16831969]
- Jongen P. J., Renier W. O., Gabreels F. J. Seven Cases of Huntington’s Disease in Childhood and Levodopa Induced Improvement in the Hypokinetic–Rigid Form. Clin Neurol Neurosurg. 1980;82(4):251–61. [PubMed: 6165509]
- Keene C. D., Sonnen J. A., Swanson P. D., Kopyov O., Leverenz J. B., Bird T. D., Montine T. J. Neural Transplantation in Huntington Disease: Long-Term Grafts in Two Patients. Neurology. 2007;68(24):2093–98. [PubMed: 17562830]
- Kieburtz K., MacDonald M., Shih C., Feigin A., Steinberg K., Bordwell K., Zimmerman C., Srinidhi J., Sotack J., Gusella J., et al. Trinucleotide Repeat Length and Progression of Illness in Huntington’s Disease. J Med Genet. 1994;31(11):872–74. [PMC free article: PMC1016662] [PubMed: 7853373]
- Kirkwood S. C., Siemers E., Viken R., Hodes M. E., Conneally P. M., Christian J. C., Foroud T. Longitudinal Personality Changes among Presymptomatic Huntington Disease Gene Carriers. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15(3):192–97. [PubMed: 12218712]
- Kremer B., Goldberg P., Andrew S. E., Theilmann J., Telenius H., Zeisler J., Squitieri F., Lin B., Bassett A., Almqvist E., et al. A Worldwide Study of the Huntington’s Disease Mutation. The Sensitivity and Specificity of Measuring Cag Repeats. N Engl J Med. 1994;330(20):1401–06. [PubMed: 8159192]
- Kremer B., Clark C. M., Almqvist E. W., Raymond L. A., Graf P., Jacova C., Mezei M., Hardy M. A., Snow B., Martin W., et al. Influence of Lamotrigine on Progression of Early Huntington Disease: A Randomized Clinical Trial. Neurology. 1999;53(5):1000–11. [PubMed: 10496259]
- Kuemmerle S., Gutekunst C. A., Klein A. M., Li X. J., Li S. H., Beal M. F., Hersch S. M., Ferrante R. J. Huntington Aggregates May Not Predict Neuronal Death in Huntington’s Disease. Ann Neurol. 1999;46(6):842–49. [PubMed: 10589536]
- Langbehn D. R., Brinkman R. R., Falush D., Paulsen J. S., Hayden M. R. A New Model for Prediction of the Age of Onset and Penetrance for Huntington’s Disease Based on Cag Length. Clin Genet. 2004;65(4):267–77. [PubMed: 15025718]
- Lasker A. G., Zee D. S., Hain T. C., Folstein S. E., Singer H. S. Saccades in Huntington’s Disease: Initiation Defects and Distractibility. Neurology. 1987;37(3):364–70. [PubMed: 2950337]
- Levesque M., Parent A. The Striatofugal Fiber System in Primates: A Reevaluation of Its Organization Based on Single-Axon Tracing Studies. Proc Natl Acad Sci U S A. 2005;102(33):11888–93. [PMC free article: PMC1187973] [PubMed: 16087877]
- Li J. L., Hayden M. R., Warby S. C., Durr A., Morrison P. J., Nance M., Ross C. A., Margolis R. L., Rosenblatt A., Squitieri F., et al. Genome-Wide Significance for a Modifier of Age at Neurological Onset in Huntington’s Disease at 6q23-24: The HD Maps Study. BMC Med Genet. 2006;7 [PMC free article: PMC1586197] [PubMed: 16914060]
- Lipe H., Schultz A., Bird T. D. Risk Factors for Suicide in Huntington’s Disease: A Retrospective Case Controlled Study. Am J Med Genet. 1993;48(4):231–33. [PubMed: 8135306]
- Lobo M. K., Karsten S. L., Gray M., Geschwind D. H., Yang X. W. Facs-Array Profiling of Striatal Projection Neuron Subtypes in Juvenile and Adult Mouse Brains. Nat Neurosci. 2006;9(3):443–52. [PubMed: 16491081]
- Lodi R., Schapira A. H., Manners D., Styles P., Wood N. W., Taylor D. J., Warner T. T. Abnormal in Vivo Skeletal Muscle Energy Metabolism in Huntington’s Disease and Dentatorubropallidoluysian Atrophy. Ann Neurol. 2000;48(1):72–76. [PubMed: 10894218]
- Louis E. D., Anderson K. E., Moskowitz C., Thorne D. Z., Marder K. Dystonia- Predominant Adult-Onset Huntington Disease: Association between Motor Phenotype and Age of Onset in Adults. Arch Neurol. 2000;57(9):1326–30. [PubMed: 10987900]
- Macdonald V., Halliday G. Pyramidal Cell Loss in Motor Cortices in Huntington’s Disease. Neurobiol Dis. 2002;10(3):378–86. [PubMed: 12270698]
- Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., et al. Exon 1 of the HD Gene with an Expanded Cag Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell. 1996;87(3):493–506. [PubMed: 8898202]
- Marder K., Sandler S., Lechich A., Klager J., Albert S. M. Relationship between Cag Repeat Length and Late-Stage Outcomes in Huntington’s Disease. Neurology. 2002;59(10):1622–24. [PubMed: 12451208]
- Marder K., Zhao H., Myers R. H., Cudkowicz M., Kayson E., Kieburtz K., Orme C., Paulsen J., Penney J. B. Jr, Siemers E., et al. Rate of Functional Decline in Huntington’s Disease. Huntington Study Group. Neurology. 2000;54(2):452–58. [PubMed: 10668713]
- Marshall F. J. Clinical Features and Treatment of Huntington’s Disease. In: Watts R. L., Koller W. C., editors. In Movement Disorders: Neurologic Principles & Practice. New York: McGraw-Hill; 2004. pp. 589–601.
- Nakamura K., Aminoff M. J. Huntington’s Disease: Clinical Characteristics, Pathogenesis and Therapies. Drugs Today (Barc). 2007;43(2):97–116. [PubMed: 17353947]
- Nance M. A. Comprehensive Care in Huntington’s Disease: A Physician’s Perspective. Brain Res Bull. 2007;72(2–3):175–78. [PubMed: 17352943]
- O’Suilleabhain P., Dewey R. B. Jr. A Randomized Trial of Amantadine in Huntington Disease. Arch Neurol. 2003;60(7):996–98. [PubMed: 12873857]
- Paulsen J. S., Butters N., Sadek J. R., Johnson S. A., Salmon D. P., Swerdlow N. R., Swenson M. R. Distinct Cognitive Profiles of Cortical and Subcortical Dementia in Advanced Illness. Neurology. 1995;45(5):951–56. [PubMed: 7746413]
- Paulsen J. S., Hayden M., Stout J. C., Langbehn D. R., Aylward E., Ross C. A., Guttman M., Nance M., Kieburtz K., Oakes D., et al. Preparing for Preventive Clinical Trials: The PREDICT-HD Study. Arch Neurol. 2006;63(6):883–90. [PubMed: 16769871]
- Paulsen J. S., Langbehn D. R., Stout J. C., Aylward E., Ross C. A., Nance M., Guttman M., Johnson S., MacDonald M., Beglinger L. J., et al. Detection of Huntington’s Disease Decades before Diagnosis: The Predict-HD Study. J Neurol Neurosurg Psychiatry. 2008;79(8):874–80. [PMC free article: PMC2569211] [PubMed: 18096682]
- Paulsen J. S., Ready R. E., Hamilton J. M., Mega M. S., Cummings J. L. Neuropsychiatric Aspects of Huntington’s Disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310–14. [PMC free article: PMC1737562] [PubMed: 11511702]
- Quaid K. A., Morris M. Reluctance to Undergo Predictive Testing: The Case of Huntington Disease. Am J Med Genet. 1993;45:41–45. [PubMed: 11657397]
- Quarrell O. W., Rigby A. S., Barron L., Crow Y., Dalton A., Dennis N., Fryer A. E., Heydon F., Kinning E., Lashwood A., et al. Reduced Penetrance Alleles for Huntington’s Disease: A Multi-Centre Direct Observational Study. J Med Genet. 2007;44(3) [PMC free article: PMC2598018] [PubMed: 17361007]
- Reiner A., Albin R. L., Anderson K. D., D’Amato C. J., Penney J. B., Young A. B. Differential Loss of Striatal Projection Neurons in Huntington Disease. Proc Natl Acad Sci U S A. 1988;85(15):5733–37. [PMC free article: PMC281835] [PubMed: 2456581]
- Ribai P., Nguyen K., Hahn-Barma V., Gourfinkel-An I., Vidailhet M., Legout A., Dode C., Brice A., Durr A. Psychiatric and Cognitive Difficulties as Indicators of Juvenile Huntington Disease Onset in 29 Patients. Arch Neurol. 2007;64(6):813–19. [PubMed: 17562929]
- Riley B. E., Orr H. T. Polyglutamine Neurodegenerative Diseases and Regulation of Transcription: Assembling the Puzzle. Genes Dev. 2006;20(16):2183–92. [PubMed: 16912271]
- Rohrer D., Salmon D. P., Wixted J. T., Paulsen J. S. The Disparate Effects of Alzheimer’s Disease and Huntington’s Disease on Semantic Memory. Neuropsychology. 1999;13(3):381–88. [PubMed: 10447299]
- Rosas H. D., Hevelone N. D., Zaleta A. K., Greve D. N., Salat D. H., Fischl B. Regional Cortical Thinning in Preclinical Huntington Disease and Its Relationship to Cognition. Neurology. 2005;65(5):745–47. [PubMed: 16157910]
- Rosas H. D., Liu A. K., Hersch S., Glessner M., Ferrante R. J., Salat D. H., van der Kouwe A., Jenkins B. G., Dale A. M., Fischl B. Regional and Progressive Thinning of the Cortical Ribbon in Huntington’s Disease. Neurology. 2002;58(5):695–701. [PubMed: 11889230]
- Rosenblatt A., Ranen N. G., Rubinsztein D. C., Stine O. C., Margolis R. L., Wagster M. V., Becher M. W., Rosser A. E., Leggo J., Hodges J. R., et al. Patients with Features Similar to Huntington’s Disease, without Cag Expansion in Huntingtin. Neurology. 1998;51(1):215–20. [PubMed: 9674805]
- Ross C. A., Poirier M. Opinion: What is the Role of Protein Aggregation in Neurodegeneration? Nat Rev Mol Cell Biol. 2005;6(11):891–98. [PubMed: 16167052]
- Rubinsztein D. C., Leggo J., Chiano M., Dodge A., Norbury G., Rosser E., Craufurd D. Genotypes at the Glur6 Kainate Receptor Locus Are Associated with Variation in the Age of Onset of Huntington Disease. Proc Natl Acad Sci U S A. 1997;94(8):3872–76. [PMC free article: PMC20534] [PubMed: 9108071]
- Saft C., Lauter T., Kraus P. H., Przuntek H., Andrich J. E. Dose-Dependent Improvement of Myoclonic Hyperkinesia Due to Valproic Acid in Eight Huntington’s Disease Patients: A Case Series. BMC Neurol. 2006;6 [PMC free article: PMC1413552] [PubMed: 16507108]
- Saft C., Zange J., Andrich J., Muller K., Lindenberg K., Landwehrmeyer B., Vorgerd M., Kraus P. H., Przuntek H., Schols L. Mitochondrial Impairment in Patients and Asymptomatic Mutation Carriers of Huntington’s Disease. Mov Disord. 2005;20(6):674–79. [PubMed: 15704211]
- Sarkar S., Perlstein E. O., Imarisio S., Pineau S., Cordenier A., Maglathlin R. L., Webster J. A., Lewis T. A., O’Kane C. J., Schreiber S. L., et al. Small Molecules Enhance Autophagy and Reduce Toxicity in Huntington’s Disease Models. Nat Chem Biol. 2007;3(6):331–38. [PMC free article: PMC2635561] [PubMed: 17486044]
- Sawa A., Wiegand G. W., Cooper J., Margolis R. L., Sharp A. H., Lawler J. F. Jr, Greenamyre J. T., Snyder S. H., Ross C. A. Increased Apoptosis of Huntington Disease Lymphoblasts Associated with Repeat Length-Dependent Mitochondrial Depolarization. Nat Med. 1999;5(10):1194–98. [PubMed: 10502825]
- Schneider S. A., Walker R. H., Bhatia K. P. The Huntington’s Disease-Like Syndromes: What to Consider in Patients with a Negative Huntington’s Disease Gene Test. Nat Clin Prac Neurol. 2007;3(9):517–25. [PubMed: 17805246]
- Sieradzan K. A., Mann D. M. The Selective Vulnerability of Nerve Cells in Huntington’s Disease. Neuropathol Appl Neurobiol. 2001;27(1):1–21. [PubMed: 11298997]
- Siesling S., van Vugt J. P., Zwinderman K. A., Kieburtz K., Roos R. A. Unified Huntington’s Disease Rating Scale: A Follow Up. Mov Disord. 1998;13(6):915–19. [PubMed: 9827615]
- Sorensen S. A., Fenger K. Causes of Death in Patients with Huntington’s Disease and in Unaffected First Degree Relatives. J Med Genet. 1992;29(12):911–14. [PMC free article: PMC1016212] [PubMed: 1479606]
- Sotrel A., Paskevich P. A., Kiely D. K., Bird E. D., Williams R. S., Myers R. H. Morphometric Analysis of the Prefrontal Cortex in Huntington’s Disease. Neurology. 1991;41(7):1117–23. [PubMed: 1829794]
- Sotrel A., Williams R. S., Kaufmann W. E., Myers R. H. Evidence for Neuronal Degeneration and Dendritic Plasticity in Cortical Pyramidal Neurons of Huntington’s Disease: A Quantitative Golgi Study. Neurology. 1993;43(10):2088–96. [PubMed: 8413971]
- Stevenson Charles S. A Biography of George Huntington, M.D. In: Henry E Sigerist., editor. In Bulletin of the Institute of the History of Medicine. Baltimore: The Johns Hopkins University; 1934. pp. 53–76.
- Stine O. C., Pleasant N., Franz M. L., Abbott M. H., Folstein S. E., Ross C. A. Correlation between the Onset Age of Huntington’s Disease and Length of the Trinucleotide Repeat in It-15. Hum Mol Genet. 1993;2(10):1547–49. [PubMed: 8268907]
- Strand A. D., Aragaki A. K., Shaw D., Bird T., Holton J., Turner C., Tapscott S. J., Tabrizi S. J., Schapira A. H., Kooperberg C., et al. Gene Expression in Huntington’s Disease Skeletal Muscle: A Potential Biomarker. Hum Mol Genet. 2005;14(13):1863–76. [PubMed: 15888475]
- Tippett L. J., Waldvogel H. J., Thomas S. J., Hogg V. M., van Roon-Mom W., Synek B. J., Graybiel A. M., Faull R. L. Striosomes and Mood Dysfunction in Huntington’s Disease. Brain. 2007;130 (Pt 1):206–21. [PubMed: 17040921]
- Todi S. V., Williams A. J., Paulson H. L. Polyglutamine Disorders Including Huntington’s Disease. In: Waxman Stephen G., editor. In Molecular Neurology. Burlington, VT: Elsevier Academic Press; 2007. pp. 257–75.
- Tsuang D., Almqvist E. W., Lipe H., Strgar F., DiGiacomo L., Hoff D., Eugenio C., Hayden M. R., Bird T. D. Familial Aggregation of Psychotic Symptoms in Huntington’s Disease. Am J Psychiatry. 2000;157(12):1955–59. [PubMed: 11097960]
- van Oostrom J. C., Maguire R. P., Verschuuren-Bemelmans C. C., Veenma-van der Duin L., Pruim J., Roos R. A., Leenders K. L. Striatal Dopamine D2 Receptors, Metabolism, and Volume in Preclinical Huntington Disease. Neurology. 2005;65(6):941–43. [PubMed: 16186542]
- Van Raamsdonk J. M., Murphy Z., Selva D. M., Hamidizadeh R., Pearson J., Petersen A., Bjorkqvist M., Muir C., Mackenzie I. R., Hammond G. L., et al. Testicular Degeneration in Huntington Disease. Neurobiol Dis. 2007;26(3):512–20. [PubMed: 17433700]
- Verhagen Metman L., Morris M. J., Farmer C., Gillespie M., Mosby K., Wuu J., Chase T. N. Huntington’s Disease: A Randomized, Controlled Trial Using the NMDA- Antagonist Amantadine. Neurology. 2002;59(5):694–99. [PubMed: 12221159]
- Vonsattel J. P., DiFiglia M. Huntington Disease. J Neuropathol Exp Neurol. 1998;57(5):369–84. [PubMed: 9596408]
- Vonsattel J. P., Myers R. H., Stevens T. J., Ferrante R. J., Bird E. D., Richardson E. P. Jr. Neuropathological Classification of Huntington’s Disease. J Neuropathol Exp Neurol. 1985;44(6):559–77. [PubMed: 2932539]
- Weeks R. A., Piccini P., Harding A. E., Brooks D. J. Striatal D1 and D2 Dopamine Receptor Loss in Asymptomatic Mutation Carriers of Huntington’s Disease. Ann Neurol. 1996;40(1):49–54. [PubMed: 8687191]
- Wei H., Qin Z. H., Senatorov V. V., Wei W., Wang Y., Qian Y., Chuang D. M. Lithium Suppresses Excitotoxicity-Induced Striatal Lesions in a Rat Model of Huntington’s Disease. Neuroscience. 2001;106(3):603–12. [PubMed: 11591460]
- WFN Research Group on Huntington’s Chorea. Guidelines for the Molecular Genetics Predictive Test in Huntington’s Disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Neurology. 1994;44(8):1533–36. [PubMed: 8058167]
- Zeng W., Gillis T., Hakky M., Djousse L., Myers R. H., MacDonald M. E., Gusella J. F. Genetic Analysis of the Grik2 Modifier Effect in Huntington’s Disease. BMC Neurosci. 2006;7 [PMC free article: PMC1618398] [PubMed: 16959037]
- Review Chapter 33: the history of movement disorders.[Handb Clin Neurol. 2010]Review Chapter 33: the history of movement disorders.Lanska DJ. Handb Clin Neurol. 2010; 95:501-46.
- Review Chorea: A Journey through History.[Tremor Other Hyperkinet Mov (N...]Review Chorea: A Journey through History.Vale TC, Cardoso F. Tremor Other Hyperkinet Mov (N Y). 2015; 5. Epub 2015 May 28.
- [Huntington chorea in children].[Arch Fr Pediatr. 1993][Huntington chorea in children].Turpin JC. Arch Fr Pediatr. 1993 Feb; 50(2):119-22.
- Huntington's disease: what we learned from the original essay.[Neurologist. 2003]Huntington's disease: what we learned from the original essay.Okun MS. Neurologist. 2003 Jul; 9(4):175-9.
- Review Mouse Models for Validating Preclinical Candidates for Huntington’s Disease.[Neurobiology of Huntington's D...]Review Mouse Models for Validating Preclinical Candidates for Huntington’s Disease.William Yang X, Gray M. Neurobiology of Huntington's Disease: Applications to Drug Discovery. 2011
- Huntington’s Disease - Neurobiology of Huntington's DiseaseHuntington’s Disease - Neurobiology of Huntington's Disease
- Cytochrome b, partial (mitochondrion) [Tropheus kasabae]Cytochrome b, partial (mitochondrion) [Tropheus kasabae]gi|13776|emb|CAA78077.1|Protein
Your browsing activity is empty.
Activity recording is turned off.
See more...