

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Lo DC, Hughes RE, editors. Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton (FL): CRC Press/Taylor & Francis; 2011.

INTRODUCTION
The ultimate therapeutic goal for Huntington’s disease (HD) is to develop disease-modifying therapies able to (1) delay or prevent clinical illness in those who are at genetic risk; and (2) slow the progression and permit some recovery in those who have manifest clinical illness. Rapidly advancing basic and translational research has identified numerous potential targets for neuroprotection. Some targets may be generically neuroprotective and relevant for a variety of neurological insults, whereas others may be more selective for HD. None yet stand out sufficiently to enable concentrating efforts on just a few of these, with the exception of the huntingtin protein itself, which does not have a conventional pharmacology with which to work.
Each potential target for HD is approached by multiple strategies, primarily small molecules but also by RNA interference, antisense, gene therapy, or cellular therapy. These strategies start with families of compounds or biologicals. Medicinal chemistry, pharmacology, and biological assays winnow these families down by ordering them in terms of potency, favorable pharmacological properties, toxicity, teratogenicity, off-target effects, bioavailability, central nervous system (CNS) penetration, and so on. However, as helpful as in silico, in vitro, and in vivo models are, they provide only an incomplete understanding of target and treatment properties and disease modifying potential.
Indeed, for neurological disease, there is much more history with compounds working in cellular and in animal models and subsequently not working in human disease than there is of models successfully predicting effective therapies (e.g., in stroke or amyotrophic lateral sclerosis [ALS]). There may be more hope for HD because of its dominant genetic nature and the greater relevance of the models. In the end, target validation and prioritization, as well as discerning the potential risks and benefits of individual compounds, will have to come from clinical experiments in human subjects, particularly those with premanifest or manifest HD.
However, the capacity to conduct clinical trials is not close to keeping up with the numbers of compounds for which there are already some rationale and likelihood of safety and tolerability—and the gap is quickly widening. There are many reasons for this gap, and these mostly come down to limited resources of time, effort, money, investigators, and subjects. At-risk and affected individuals inexorably progressing toward clinical disease or through increasing disability provide an underlying urgency not only to do more testing of potential therapies but also to improve the process. In this context, the development and use of biomarkers in clinical trials for HD will have profound potential to increase the rate and accuracy with which treatments and by implication their targets, can be assessed.
Thus, there is a great need for the development of biomarkers for HD which are useful in early- and late-phase clinical trials. Although finding a dose range for a treatment in HD patients and testing for safety and tolerability are straightforward, it can be difficult to find signals that indicate that the desired pharmacological activity is occurring and is optimal, or that compare one agent with another. A further difficulty has been the lack of clinical or other outcome measures able to provide preliminary evidence of efficacy for neuroprotection that would help in the prioritization of compounds for large Phase III studies. Indeed, without such signals it is also very difficult to stop development of a compound short of its failure in a large-scale study.
Moreover, in premanifest HD, there may be no clinical measures to provide useful assessment of efficacy. In manifest HD, clinical symptoms progress slowly and are extremely variable, and their modulation does not intrinsically correspond to disease modification. Thus, although modulation of symptoms may point to a symptomatic benefit, improving symptoms does not necessarily predict slowing the disease process. For example, haloperidol can suppress chorea yet hasten death by worsening dysphagia.
Biomarkers that can indicate whether a potential disease-modifying therapy interacts with its target or affects disease processes (state) or progression in smaller, shorter early-phase trials are urgently needed to help decision making about therapeutic development, such that not every candidate has to be tested in large futility or Phase III studies. Biomarkers able to provide supportive or even primary evidence for efficacy would also facilitate late-phase trials. Indeed, the National Institutes of Health (NIH) Neuroscience Blueprint has made biomarker development for neurodegenerative diseases a high priority (http://neuroscienceblueprint.nih.gov).
However, although biomarker research is being embraced, there remains some confusion about biomarkers. This chapter will provide a framework for considering the development, assessment, and use of different types of biomarkers that could facilitate the development of neuroprotective therapies for HD.
Definitions
The field of biomarkers has been hampered by varying definitions of what a biomarker is and by the use of various adjectives to describe how thoroughly they have been assessed. A working group on biomarker definitions was convened by the NIH in 2000 to propose definitions for common use and to provide a framework for assessing their correspondence to clinical outcome measures.1 Biomarkers were defined as a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention. Biomarkers can also be considered to encompass technological improvements in looking at clinical signs, such as neuropsychological measures, eye movements, or quantitative movement measurements. Although these may also have value as outcome measures, for the purposes of this review these are better considered as refined clinical measures rather than biomarkers because they remain removed from the biology of the disease or of the treatments and may primarily measure symptoms, although with more sensitivity.
Surrogate endpoints were defined by the NIH Working Group on Biomarkers as a special subset of biomarkers intended to substitute for a clinical endpoint. A surrogate endpoint sufficiently correlated to a meaningful clinical outcome measure can be acceptable as a substitute for the clinical endpoint by the Food and Drug Administration (FDA) when considering whether an intervention is efficacious. Thus, surrogate endpoints can serve as a basis for regulatory approval, although this has been rare because establishing sufficient correspondence between a biomarker and a meaningful clinical endpoint is difficult and requires extensive validation in natural history and therapeutic studies. In other disease areas, established surrogate endpoints include blood pressure and cholesterol levels for cardiovascular drugs, blood glucose and glycohemoglobin for diabetes, viral RNA load and CD4 counts for HIV, and tumor size for antineoplastic agents. Although this standard is infrequently met, biomarkers need not become surrogate endpoints to be tremendously valuable. Likewise for HD, surrogate endpoints would be useful if they could improve the efficiency of efficacy studies. However, the main focus for biomarker research should be the development of useful purpose-driven biomarkers. These may ultimately include some able to serve also as surrogate endpoints.
Classification of Biomarkers
Biomarkers can serve many purposes in the development of treatments for HD. Thus, it is useful to classify biomarkers to help convey the different purposes for which they can serve. To a large degree, these purposes are intrinsic to what is being measured. The more global the biomarker, the more able it may be to capture a large portion of the disease process and thus be more predictive of disease progression. More specific biomarkers linked to particular facets of disease biochemistry may have great appeal and measurement precision but also have a danger of capturing only a fraction of the disease process, and it can be difficult to understand what that fraction is.
Global biomarkers, such as tumor size for oncology or the size of the brain or of brain structures in neurodegeneration, are removed from the pathophysiology but can capture the global direction and severity of disease. Accordingly, global biomarkers are most likely to be useful in staging the presence, progression, or severity of disease, or as outcome measures able to reflect the response to a treatment in studies seeking evidence of possible clinical efficacy.
Process biomarkers are more specific laboratory measures such as levels of proteins, gene expression, compounds, or metabolites that capture a molecular/ biochemical aspect of the pathogenesis of the disease or of biological responses to the disease process. Process biomarkers can usefully demonstrate mechanisms of disease and pharmacological/pharmacodynamic responses but may only represent a component of the many contributors to disease. In a complex disorder like HD, in which a variety of pathogenic mechanisms have been implicated, a process biomarker may only capture a slice of the entire set of biochemical influences that make up the disease. Process biomarkers can be powerful for examining whether a compound appropriately affects its target (or other targets), for assessing dose, for comparing compounds for potency or toxicity, and for examining the biological bases of response variation between subjects (pharmacodynamic biomarkers).
However, the great specificity of a process biomarker often prevents it from reflecting the entire clinical disease process sufficiently for it to be informative about the impact a treatment might have on the clinical onset or progression of disease. For example, a process biomarker could show that a possible treatment successfully modulates its molecular target in an early-stage assessment, but the same biomarker may have an uncertain relationship to clinical efficacy, which might be better examined with other types of biomarkers. This discussion also illustrates the importance of matching biomarkers to the particular therapeutic development tasks at hand.
Another way of looking at this considers the known value of a biomarker rather than the biological modality from which it comes, in a more hierarchical manner. For HD, we can envision a hierarchical organization of biomarkers, as has been discussed for oncology.2 A diagnostic biomarker can identify processes that distinguish HD from non-HD. For example, the genetic test for HD can diagnose the presence or absence of risk for developing HD; other tests useful for clinical trials might diagnose the presence of the HD prodrome in premanifest individuals or the conversion from premanifest to manifest HD. Diagnostic biomarkers can serve classification purposes but may not be able to measure clinical aspects of HD such as severity, magnitude, and progression.
Pharmacodynamic biomarkers additionally have a connection to likely pathogenic, neuroprotective, or pharmacological mechanisms and thus can provide indications of a response to a therapy. This might include levels of compounds in blood or the brain, signs of toxicity or off-target interactions, or evidence that a compound is on target for its desired effects. For HD, pharmacodynamic biomarkers are especially useful in early-phase clinical trials to assess whether a treatment modulates a desired target, thus revealing pharmacological efficacy. However, even if a treatment were to have pharmacological efficacy, it may not translate to a clinical benefit if the biomarker measures an inconsequential process.
Progression biomarkers additionally have predictive power for a desired clinical outcome that has been confirmed in observational or therapeutic trials sufficiently to carry weight in decisions about late-phase development or to serve as potential surrogate endpoints. For HD, such biomarkers would correspond to clinical or biological progression of disease and thus could serve as primary or secondary endpoints for assessing clinical efficacy. Clearly, these biomarker types require increasing rigor of validation yet also provide increasing confidence about their ability to reflect a significant portion of the disease phenotype or even of treatment efficacy.
BIOMARKERS AND CLINICAL STAGES OF HD
The focus of this review is on individuals who are either premanifest or in the earlier stages of being symptomatic because they constitute the HD populations with the greatest potential for benefiting from neuroprotective treatments. Individuals are born at risk for HD by virtue of possessing the causative CAG expansion; therefore, CAG length is a diagnostic biomarker that classifies individuals as someday developing manifest HD symptoms. The actual length of the CAG expansion carries further information about age of onset and can be used to model predictions of symptom onset for clinical research3; however, there is still considerable spread in actual age of onset even for people close to expected onset,4 presumably as a result of other genetic and environmental influences.
Premanifest individuals are indistinguishable clinically from gene-negative individuals until they begin to approach the onset of unequivocal symptoms. There is a prodromal period lasting at least 10 years or more in which there may be cognitive, emotional, functional, and motor signs but little functional decline. Experimental neuropsychological, neuropathological, and neuroimaging studies are able to document changes in this period, and there is evidence that the brain is working harder as it compensates functionally for ongoing degenerative changes.5–15 Thus, the HD prodrome may ultimately be the most opportune time for neuroprotective therapy because it marks a period when individuals have active brain disease but remain completely functional. Because the HD prodrome has an underlying biology, diagnostic biomarkers that mark its onset or presence, pharmacodynamic biomarkers that indicate treatment responses, and progression biomarkers that correspond to its evolution toward manifest HD can be expected to provide the most useful outcome measures.
Currently, a therapeutic trial seeking to delay the clinical onset of HD (“phenoconversion”) would look for a differential rate of diagnoses of manifest HD between placebo and active treatment groups. To examine whether a treatment delays the onset of clinical symptoms in premanifest individuals with the HD genetic mutation, it has been estimated that 1,000–3,000 subjects and 3–6 years of follow-up evaluation are necessary to detect a large 30%–40% decrease in the frequency of symptom onset. Although the HD prodrome ends with the unequivocal presence of a movement disorder that permits a clinical diagnosis of manifest HD, the underlying biological processes that make up neurodegeneration are more continuous such that diagnosis likely marks some threshold being passed at which physiological compensation fails or observers gain sufficient sensitivity to detect symptomatic HD with confidence.
Thus, phenoconversion is essentially a binary variable of very low power but very high clinical relevance. It is of such low power that early-phase clinical trials in premanifest subjects would have little possibility of providing preliminary evidence for efficacy. In contrast, biomarkers could be used to detect disease and pharmacodynamic responses to treatment during the prodrome, without awaiting phenoconversion. Furthermore, biomarkers could provide continuous variables with slopes of change during the HD prodrome and early symptomatic period that could be monitored, and comparisons could be made between treatment groups. Biological responses to treatments or changes of slope in biomarker measures as a result of treatment could be detected much more sensitively than phenoconversion and provide evidence of an intervention being disease modifying in Phase II-sized studies. Such data would be essential to help justify the effort and expense that an efficacy study in premanifest HD would entail. It may also provide evidence that a treatment shown to slow clinical progression in manifest HD is behaving similarly in premanifest HD. Thus, clinical and biological markers able to detect the HD prodrome and measure its progression would enable the identification of subjects in this period and facilitate performing informative therapeutic trials.
An important issue in designing a clinical trial in the premanifest population is that the vast majority of individuals (>95%) at risk for carrying the HD genetic mutation have not desired genetic testing.16 Some of the reasons for this include fear of genetic discrimination, the lack of effective treatment, and concern about the negative consequences of testing. Focus groups with at-risk individuals have revealed that many would be averse to taking part in clinical trials if informative genetic testing is required. In fact, many would willingly take an experimental medication and risk side effects in a clinical trial without genetic testing, understanding that there would be a 50% chance of not having the gene mutation. Performing a clinical trial only in subjects who have had genetic testing thereby raises a concern about creating an incentive for genetic testing along with its negative consequences in subjects wishing to participate. Currently, because relatively few individuals have pursued genetic testing, it is necessary to include individuals who do not know their genetic status in clinical research. Because a biomarker can be a surrogate (albeit a less sensitive one) for the HD genetic test, it will be important to treat biomarker data with the same concerns about confidentiality and blinding.
Once diagnosed clinically, manifest HD has a highly variable phenotype. Some of this variation is inherent in the disease. For example, different affected individuals can have predominant motor, predominant cognitive, or predominant psychiatric presentations and different rates of progression. Moreover, the severity of symptoms can be modulated by many temporary factors such as mood, nutrition, medications, and sleep disturbances. Despite great day-to-day variability in symptoms, progression is slow when assessed by an integrated measure of functional capacity (e.g., the Total Functional Capacity [TFC] scale).17 For manifest HD, significant reduction of functional decline over time is the sine qua non of slowing progression and has been acceptable to the FDA as meeting their legal mandate that a potential treatment must have clinical significance.
Currently, neuroprotection efficacy studies in symptomatic patients with HD rest on a primary clinical outcome measure (TFC scale) requiring, for example, about 600 subjects and 5 years of follow-up evaluation to detect a 20% slowing of functional decline (1:1 randomized placebo controlled trial). Thus, the expense, time, and great magnitude of effort needed to test efficacy means that few interventions can be tested. More sensitive clinical measures have been explored and may help, but none has demonstrated clear promise for increasing clinical trial power dramatically, and none has been validated as improving on the TFC scale. Furthermore, having large numbers of subjects on placebo treatment for years in these trials is an unfortunate necessity.
In this context, biomarkers corresponding to disease progression could help assess efficacy, could supplement the TFC scale and other clinical endpoints in symptomatic subjects, and may ultimately serve as surrogate endpoints enabling the testing of disease modification in fewer subjects more quickly. Biomarkers of disease biological activity can be used in these studies to help answer whether treatments have the desired pharmacodynamic effects, whether dosing is optimal, and whether there are potential biological explanations for response heterogeneity. Finally, pharmacodynamic and other biomarker responses in early-phase studies suggestive of disease modification could help greatly in deciding whether to proceed to large efficacy studies and in enabling refinement of study design.
BIOMARKER DEVELOPMENT AND CHARACTERIZATION
The attributes of an ideal biomarker include pathophysiological relevance, sensitivity and specificity for HD and for treatment effects, reliability (accuracy, precision, robustness, reproducibility), practicality (must be minimally invasive, affordable), and simplicity (does not require unusual skills or equipment, scalable to multicenter trials). For HD, regional brain atrophy has very high pathophysiological relevance as it likely reflects neurodegeneration directly. However, the relevance of process biomarkers, such as levels of small molecules in blood or cerebrospinal fluid (CSF), enzyme activities, or gene expression may be uncertain because the exact manner in which mutant huntingtin causes neurodegeneration has not yet been established. Nevertheless, connections can be made to processes known to play roles in pathogenesis, such as energy compromise, oxidative stress, and transcriptional disruption, and such levels can serve as diagnostic, progression, and pharmacodynamic biomarkers. Reliability, practicality, sensitivity, and specificity of biomarkers must be assessed experimentally or statistically. There are no hard criteria for these, but their positive and negative attributes must be balanced against the applications for which they are needed and any available alternatives.
Criteria for biomarker assessment and validation are defined by the nature of the questions that the biomarker is intended to address, the degree of certainty required, and assumptions about relationships to clinical endpoints. Validation is a continuous process that evolves as new information is accumulated from preclinical, early-phase clinical, and late-phase clinical studies. Brooks et al.18 presented criteria for the assessment of potential biomarkers in studies of progression in Parkinson’s disease (PD), which can be adapted to the characterization of biomarkers for HD as follows:
- The biomarker should correlate with the likelihood or progression of premanifest disease activity, with the development of clinical HD in premanifest individuals, or with clinical deterioration in symptomatic individuals.
- The biomarker should be objective (amenable to blinded or centralized assessment).
- The biomarker should be reproducible (reliable).
- Biomarker changes should be specific to changes in disease status (e.g., they should not be distorted by pharmacological treatments or coexisting medical conditions).
- Biomarker assessment itself should be safe and well tolerated.
- Ideally, the biomarker should be inexpensive and easy to use.
- The biomarker should be able to be assessed repeatedly in the same patient so as to provide measures of change and progression.
Discovery and validation of biomarkers proceed in phases.19 First, promising directions are identified and prioritized in preclinical and retrospective or cross-sectional clinical material; these could be nonbiased screening (“omic”) approaches or candidate-based approaches. Examples include metabolomic and gene expression screening analyses, brain-wide regional morphometry, and testing of mechanistic assays such as oxidative stress markers or indole levels. Evaluation of sensitivity, specificity, and dynamic range helps prioritize candidates, and replication in additional sample or subject sets is important to validate the potential biomarkers and their prioritization. Validation at this level can be sufficient to identify diagnostic and pharmacodynamic biomarkers.
Next, identified biomarkers are tested in relevant prospective clinical cohorts for their assessment as continuous variables and to examine their correspondence to desired clinical indicators, such as time, specific clinical features, clinical progression, response to therapy, and response to covariates (e.g., gender, age, smoking, medications, and concomitant illnesses). These studies enable comparing markers, developing algorithms for combining them, and determining slopes and variability, which help define useful sample intervals and sizes.
Finally, prospective biomarkers are tested in definitive large-scale observational or therapeutic studies to assess their feasibility and power to predict the onset of disease, disease progression, or the response to a disease-modifying therapy. These progressive analyses enable true estimates of the power of the biomarkers to make diagnostic and therapeutic predictions and lead to an accumulation of information about their feasibility, generalizability, and cost.
It is well known that population-level associations between biomarkers and clinical outcomes, even when strong and highly statistically significant, do not necessarily translate into precise predictions at an individual level. For example, the mean age at HD onset for a population of at-risk subjects can be estimated precisely as a function of CAG repeat length, but the variability around this prediction for any single individual remains high. Predictive values of tests for disease status based on a continuous biomarker can be described by receiver operating characteristic (ROC) curves,20 which plot sensitivity against specificity. The area under an ROC curve represents the probability that an HD subject will have a higher (or lower) value of the biomarker than a healthy subject and is a useful summary measure of predictive ability that can be used for rank ordering biomarker effectiveness. Using clinically meaningful criteria, the predictive ability of biomarkers can be calculated for distinguishing controls from premanifest or manifest subjects, premanifest subjects with detectable disease activity from those without, premanifest subjects who develop manifest disease from those who do not, and “fast progressors” from “slow progressors” among premanifest and manifest subjects (intrinsically or in response to treatment). Importantly, ROC curves do not depend on the absolute scale of raw data measurements, which enables comparisons among different types of biomarkers. They also display true- and false-positive rates, which are particularly relevant.
Biomarker Modalities—Neuroimaging
Modern neuroimaging, including structural (mainly computed tomography and magnetic resonance imaging [MRI]), biochemical (magnetic resonance spectroscopy), and functional neuroimaging (mainly positron emission tomography [PET], single-photon emission computed tomography, and functional MRI [fMRI]), may enable the visualization of early brain changes in vivo. These methods are largely noninvasive, are increasingly accessible in clinical practice, and, together with the clear relevance to the neuropathology, make neuroimaging an appealing potential biomarker for HD. Until recently, most efforts to use morphometric neuroimaging in HD have been focused on the striatum because it is so severely affected and because it is composed of structures that can be easily delineated. However, if HD is truly a “polytypic process,” any imaging analysis limited to the study of a small number of structures may fail to fully characterize the distribution and temporal course of pathology or to explain the clinical symptoms of HD accurately enough to be of value in clinical trials. Thus, while the relative contributions of the cortex and striatum to clinical symptoms of HD remain to be elucidated, treatments aimed solely at preserving striatal function or striatal anatomy may fail to ameliorate clinical symptoms (Figure 11.1).
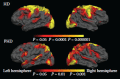
FIGURE 11.1
Cortical changes occur early and are extensive in HD. Top, maps of statistically significant thinning in 33 patients with early HD compared with ageand sex-matched subjects. Bottom, maps of statistically significant thinning during the premanifest period, (more...)
Development of neuroimaging biomarkers for HD has been strongly supported by recent technological advancements in structural neuroimaging methods, which have led to improvements in the acquisition of images with better spatial resolution and gray/white contrast, as well as in the availability of automated methods for detailed morphometric analyses, with improved sensitivity and reliability compared with traditional manual methods and which enable evaluation of the entire brain. Diffusion tensor imaging is another emerging technology that enables examination of the brain at a microstructural level. MRI measures of cortical, white matter and subcortical neurodegeneration are detectable years before symptom onset15,21–23 and provide a sensitive measure of progression in premanifest and symptomatic individuals. These changes correlate with disabling symptoms and functional decline as measured using the TFC scale24 and may be responsive to neuroprotective treatments providing potentially far greater experimental power than clinical outcome measures. These readily available, noninvasive methods allow for multiple repeated evaluations over the course of longitudinal studies and may lend themselves well as biomarkers in clinical drug trials.
Several studies in both premanifest and early symptomatic studies using fMRI (which uses changes in regional blood flow during neuronal activity to identify regions of brain activated during performance of a cognitive task) have shown alterations in patterns of activation in HD.25–27 However, although potentially appealing as a biomarker of early neuronal dysfunction, there are several limitations to its use in clinical trials: fMRI activation patterns can vary significantly across subjects28; fMRI is highly susceptible to trivial differences in conditions and study design; there are inherent limitations to its use in longitudinal studies; and, finally, it is unclear how changes in activation may actually reflect neuropathology directly.29,30
PET imaging has the potential to provide information about neurochemical, hemodynamic, or metabolic processes that may be disrupted in HD. Several studies have shown abnormalities in glucose utilization, striatal raclopride binding, and microglial activation.31–34 Several PET tracers have been used in PD to study pathophysiological mechanisms; however, no existing tracer has yet been identified that could serve as a surrogate endpoint in clinical trials in PD.35 Finally, PET imaging has been proposed as a potential biomarker for disease-modifying trials in Alzheimer’s disease (AD)36,37; however, the generalizability of these methods for multicenter studies and for HD is uncertain.
Biological Fluids and Candidate Molecular Biomarkers
CSF is in contact with the brain and thus is likely to pick up biochemical signals from brain disease. It is relatively acellular, so its potential lies primarily in the measurement of small molecules and proteins. CSF biomarkers have been developed for AD that are diagnostic, may correspond to progression, and are potentially pharmacodynamic. There are also possible biomarkers in CSF for ALS and PD. CSF has some limitations in that its collection is a significantly inconvenient and time-consuming procedure with some morbidity and expense, and so may be best suited for studies without many repeated measurements. Some changes have been noted in the CSF of HD patients, including monoamine metabolites38; tryptophan pathway metabolites39; F2-isoprostanes, a marker of lipid peroxidation and oxidative stress40; and measures of transglutaminase activity,41 which is involved in protein cross-linking and antioxidant responses. However, none has yet been studied extensively enough to determine whether it has potential as a clinically useful biomarker.
Blood and urine are peripheral fluids with the obvious advantage of being easily obtained with a minimum of effort and risk. However, it may be counterintuitive that useful biomarkers of HD could be found in blood or urine because we are most interested in detecting and treating neurodegeneration. For a peripheral marker to represent the “state” of HD, it would either have to leak from the brain or represent pathology occurring in the periphery. Leakage can be considered broadly to include molecules escaping the brain, the effects of escaped molecules on peripheral biochemistry, and disturbances of the means by which the brain customarily acts on the periphery (e.g., neuroendocrine, metabolic, autonomic, and behavior). Any of these paths have the potential to indicate the presence and progression of HD and its response to treatment.
Effects of mutant huntingtin in the periphery independent from the brain could also readily indicate disease activity and responses to treatments. Progressive processes could also occur in the periphery, although they may or may not mirror progression in the brain sufficiently to serve as biomarkers corresponding to neurodegeneration. In the end, the correspondence of a biomarker to disease features is determined experimentally by correlation with clinical endpoints; therefore, fully understanding their source is not a requirement. Nevertheless, the closer such a biomarker is to a relevant disease mechanism, the more likely it is to be useful.
Blood consists of cellular (red blood cells, leukocytes, or platelets) and liquid components (serum or plasma) that are typically separated for analyses. Most studies have either examined small molecules or proteins free in plasma or contained in leukocytes (buffy coat preparation separated from red blood cells by centrifugation) or platelets. Because leukocytes are cellular, they also provide an opportunity to examine markers associated with organelles, cell membranes, and cellular and nuclear processes (e.g., proteolysis, gene expression). Platelets can also be isolated and express an interesting subset of proteins and receptors that are in common with neurons. Lymphocytes survive from weeks to lifelong (as is the case for memory lymphocytes); thus, biochemical and molecular changes may accumulate as the disease progresses over time. Moreover, in HD, accumulating oxidative damage to nuclear and mitochondrial DNA and accumulating transcriptional alterations in longer-lived erythrocytes and erythropoietic progenitor cells may further contribute to progressive biochemical and gene expression changes in blood cells. Thus, the turnover of blood cells may not hamper their potential for providing biomarkers of disease progression.
Blood liquids can pick up molecules from the entire body, as well as the brain. Although the blood–brain barrier (BBB) may limit CNS-to-blood transfer, other tissues and organs in the body have their own diverse leakiness into the blood stream. Because the mutant huntingtin protein is expressed ubiquitously throughout the body and because it is so promiscuously interactive,42 it would be surprising if it does not affect biochemistry and gene expression much more widely than just in the brain, even if these effects are mostly clinically silent. In fact, effects of HD on peripheral cells such as lymphocytes and peripheral tissues, such as muscle, have been detected.43–45 Although studies in blood in HD are in their infancy, a number of promising candidate biomarkers have already been identified in survey studies using various “omic” platforms and candidate-based studies measuring specific molecules. Not surprisingly, these potential HD biomarker candidates mostly cluster around pathways involved in the pathogenesis or expression of HD as understood in neurons. These include oxidative stress, energy compromise, transcriptional alterations, neurotrophin alterations, cell death pathways, inflammation, and proteolysis.
OXIDATIVE STRESS AND INFLAMMATION
Evidence for energy compromise and oxidative stress being significant contributors to the pathogenesis of HD, and not just fallout from neurodegeneration, has progressively accumulated. Peripheral markers of these processes have included measurement of oxidized molecules (DNA, lipids) in blood and urine,46 –50 endogenous antioxidants,48,51 branch chain amino acids as products of metabolism,52 and levels of the hormones ghrelin and leptin, which are involved in energy balance and weight maintainance.53 Plasma levels of 8-hydroxy-2-deoxy guanosine, a marker of oxidized DNA, have been used as a pharmacodynamic biomarker in a study of creatine monohydrate as a potential neuroprotective therapy (Figure 11.1).46 Several studies have identified proinflammatory molecules as being elevated in HD, although it is uncertain whether there is a primary connection between inflammatory responses in the brain or the periphery to pathogenesis, or whether there may be inflammatory responses to oxidative or other stresses. These molecules include C-reactive protein in plasma,47 and a proteomic analysis54 identified interrelated cytokine and complement pathway activations (clusterin, α2-macroglobulin, interleukin 6, C7, C9). These latter molecules have had some correlation with HD stage and so are candidate progression biomarkers awaiting further validation.
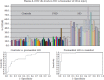
FIGURE 11.2
Individual 8-hydroxy-2- deoxyguanosine (8-OHDG) levels in plasma from ageand gender-matched controls (mean = 13.5), premanifest HD (PHD) subjects (mean = 18.1), and early manifest HD subjects (mean = 45.3). Levels are higher in PHD subjects than in controls (more...)
TRANSCRIPTIONAL MARKERS
Many studies have demonstrated direct and indirect effects on gene transcription by mutant huntingtin, and there are growing connections between these effects and the pathogenesis of HD. The greatest potential for transcriptional biomarkers lies in measuring gene transcription in cellular blood, particularly leukocytes, or in measuring gene products more generally in blood cells and fluids. Skeletal muscle obtained by muscle biopsy also has potential,45 although variability and changes in nutrition, weight, and muscle use in the course of HD are potential complicating factors. Survey and targeted approaches to transcription have used microarrays for discovery and quantitative polymerase chain reaction for confirmation.45,55,56 Borovecki et al.55 developed a 12-gene marker set that differentiated control and HD subjects, correlated with progression, and responded pharmacodynamically to treatment with an experimental histone deacetylase inhibitor. However, this same marker set was not confirmed by Runne et al.,56 who failed to observe any strong signals in blood.
Such studies suggest great potential in transcriptomic markers but also caution about their sensitivity. The genes in these studies arose mostly from microarray screens. Other genes already known to be affected in the brain in HD have been examined as possible biomarkers by assessing their gene products in blood. Brain-derived neurotrophic factor (BDNF) is a neurotrophin that could play a role in neurodegeneration; it is down-regulated in the HD brain57 and can cross the BBB. Ciammola et al.58 have studied serum levels of BDNF in HD subjects and demonstrated that it is reduced in HD and that this reduction could be related to CAG length and to disease severity. Another gene that is profoundly repressed in the brain in HD is the adenosine A2a receptor. Maglioni et al.59 and Varani et al.60 studied A2a receptor function in platelets from HD subjects and found an increase in receptor binding and a potential correspondence with proximity to clinical onset of HD.
SMALL MOLECULES
Biological tissues and fluids contain thousands of small molecules. Metabolomic profiling enables the comparison of small molecules between different groups or conditions, as well as determination of which specific molecules might differentiate among them. Metabolomic profiling has been applied to serum61 and plasma from HD subjects, and it is clear that there are rich changes in small molecule populations that can differentiate between HD and controls and could potentially serve as biomarkers. The promise in metabolomics is to identify candidate small molecules or metabolic pathways for further assessment as task-specific biomarkers in clinical studies. One such promising pathway is kynurenine metabolism,47 in which multiple alterations have been identified in HD blood.
HUNTINGTIN
Finally, worth special mention as a potential biomarker is the mutant huntingtin protein itself, although this has been technically difficult to measure so far because of its insolubility. Measurement of huntingtin would be extremely important as a variety of potential therapies are targeted against either huntingtin itself or against some of its functional properties. Measuring huntingtin levels, its physical state, its proteolytic products, or its interactions with other molecules would all have distinct values as biomarkers, especially as pharmacodynamic indicators.62 Thus, developing reliable and sensitive methods for measuring huntingtin is a high priority in the HD biomarker field.
REFERENCES
- 1.
- De Gruttola V.G, Clax P., De Mets D.L., et al. Considerations in the evaluation of surrogate endpoints in clinical trials: summary of a National Institutes of Health workshop. Controlled Clinical Trials. 2001;22:485–502. [PubMed: 11578783]
- 2.
- Floyd E., Floyd E., McShane T.M. Development and use of biomarkers in oncology drug development. Toxicol Pathol. 2004;32 Suppl 1:106–15. [PubMed: 15209410]
- 3.
- Langbehn D.R., Brinkman R.R., Falush D., et al. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. 2004;65:267–77. [PubMed: 15025718]
- 4.
- Andrew S., Theilmann J., Almqvist E., et al. DNA analysis of distinct populations suggests multiple origins for the mutation causing Huntington disease. Clin Genet. 1993;43:286–94. [PubMed: 8370147]
- 5.
- Aylward E.H., Codori A.M., Rosenblatt A., et al. Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington’s disease. Mov Disord. 2000;15:552–60. [PubMed: 10830423]
- 6.
- Blekher T., Johnson S.A., Marshall J., et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology. 2006;67:394–99. [PubMed: 16855205]
- 7.
- Blekher T.M., Yee R.D., Kirkwood S.C., et al. Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington’s disease. Vision Res. 2004;44:2729–36. [PubMed: 15358067]
- 8.
- Brandt J., Shpritz B., Codori A.M., et al. Neuropsychological manifestations of the genetic mutation for Huntington’s disease in presymptomatic individuals. J Int Neuropsychol Soc. 2002;8:918–24. [PubMed: 12405543]
- 9.
- Feigin A., Ghilardi M.F., Huang C., et al. Preclinical Huntington’s disease: compensatory brain responses during learning. Ann Neurol. 2006;59:53–59. [PMC free article: PMC2519955] [PubMed: 16261565]
- 10.
- Gutekunst C.A., Li S.H., Yi H., et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–34. [PMC free article: PMC6786077] [PubMed: 10087066]
- 11.
- Ho A.K., Sahakian B.J., Robbins T.W., et al. Random number generation in patients with symptomatic and presymptomatic Huntington’s disease. Cogn Behav Neurol. 2004;17:208–12. [PubMed: 15622016]
- 12.
- O’Donnell B.F., Wilt M.A., Hake A.M., et al. Visual function in Huntington’s disease patients and presymptomatic gene carriers. Mov Disord. 2003;18:1027–34. [PubMed: 14502670]
- 13.
- Paulsen J.S., Zimbelman J.L., Hinton S.C., et al. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington’s disease. AJNR Am J Neuroradiol. 2004;25:1715–21. [PMC free article: PMC8148746] [PubMed: 15569736]
- 14.
- Reading S.A., Dziorny A.C., Peroutka L.A., et al. Functional brain changes in presymptomatic Huntington’s disease. Ann Neurol. 2004;55:879–83. [PubMed: 15174024]
- 15.
- Rosas H.D., Tuch D.S., Hevelone N.D., et al. Diffusion tensor imaging in presymptomatic and early Huntington’s disease: selective white matter pathology and its relationship to clinical measures. Mov Disord. 2006;21:1317–25. [PubMed: 16755582]
- 16.
- Tibben A., Niermeijer M.F., Roos R.A., et al. Understanding the low uptake of presymptomatic DNA testing for Huntington’s disease. Lancet. 1992;340:1416. [PubMed: 1360126]
- 17.
- Marder K., Zhao H., Myers R.H., et al. Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology. 2000;54:452–58. [PubMed: 10668713]
- 18.
- Brooks D.J., Frey K.A., Marek K.L., et al. Assessment of neuroimaging techniques as biomarkers of the progression of Parkinson’s disease. Exp Neurol. 2003;184 Suppl 1:S68–79. [PubMed: 14597329]
- 19.
- Pepe M.S., Etzioni R., Feng J.D.Z., et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93:1054–61. [PubMed: 11459866]
- 20.
- Pepe M. The Statistical Evaluation of Medical Tests for Classification and Prediction. Oxford: Oxford University Press; 2003.
- 21.
- Douaud G., Gaura V., Ribeiro M.J., et al. Distribution of grey matter atrophy in Huntington’s disease patients: a combined ROI-based and voxel-based morphometric study. Neuroimage. 2006;32:1562–75. [PubMed: 16875847]
- 22.
- Reading S.A., Yassa M.A., Bakker A., et al. Regional white matter change in pre-symptomatic Huntington’s disease: a diffusion tensor imaging study. Psychiatry Res. 2005;140:55–62. [PubMed: 16199141]
- 23.
- Rosas H.D., Hevelone N.D., Zaleta A.K., et al. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745–47. [PubMed: 16157910]
- 24.
- Rosas H.D., Salat D.H., Lee S.Y., et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131:1057–68. [PMC free article: PMC2657201] [PubMed: 18337273]
- 25.
- Georgiou-Karistianis N., Sritharan A., Farrow M., et al. Increased cortical recruitment in Huntington’s disease using a Simon task. Neuropsychologia. 2007;45:1791–800. [PubMed: 17321554]
- 26.
- Thiruvady D.R., Georgiou-Karistianis N., Egan G.F., et al. Functional connectivity of the prefrontal cortex in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:127–33. [PMC free article: PMC2077648] [PubMed: 17028117]
- 27.
- Zimbelman J.L., Paulsen J.S., Mikos A., et al. fMRI detection of early neural dysfunction in preclinical Huntington’s disease. J Int Neuropsychol Soc. 2007;13:758–69. [PubMed: 17697407]
- 28.
- Kim J.S., Reading S.A., Brashers-Krug T., et al. Functional MRI study of a serial reaction time task in Huntington’s disease. Psychiatry Res. 2004;131:23–30. [PubMed: 15246452]
- 29.
- Gavazzi C., Nave R.D., Petralli R., et al. Combining functional and structural brain magnetic resonance imaging in Huntington disease. J Comput Assist Tomogr. 2007;31:574–80. [PubMed: 17882035]
- 30.
- Wolf R.C., Vasic N., Schonfeldt-Lecuona C., et al. Dorsolateral prefrontal cortex dysfunction in presymptomatic Huntington’s disease: evidence from event-related fMRI. Brain. 2007;130:2845–57. [PubMed: 17855375]
- 31.
- Feigin A., Tang C., Ma Y., et al. Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain. 2007;130:2858–67. [PMC free article: PMC4455546] [PubMed: 17893097]
- 32.
- Pavese N., Andrews T.C., Brooks D.J., et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: a PET study. Brain. 2003;126:1127–35. [PubMed: 12690052]
- 33.
- van Oostrom J.C., Maguire R.P., Verschuuren-Bemelmans C.C., et al. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology. 2005;65:941–3. [PubMed: 16186542]
- 34.
- Tai Y.F., Pavese N., Gerhard A., et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66. [PubMed: 17400599]
- 35.
- Ravina B., Eidelberg D., Ahlskog J.E., et al. The role of radiotracer imaging in Parkinson disease. Neurology. 2005;64:208–15. [PubMed: 15668415]
- 36.
- Cummings J.L., Doody R., Clark C. Disease-modifying therapies for Alzheimer disease: challenges to early intervention. Neurology. 2007;69:1622–34. [PubMed: 17938373]
- 37.
- Pike K.E., Savage G., Villemagne V.L., et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–44. [PubMed: 17928318]
- 38.
- Kurlan R., Caine E., Rubin A., et al. Cerebrospinal fluid correlates of depression in Huntington’s disease. Arch Neurol. 1988;45:881–83. [PubMed: 2456053]
- 39.
- Schwarcz R., Tamminga C.A., Kurlan R., et al. Cerebrospinal fluid levels of quinolinic acid in Huntington’s disease and schizophrenia. Ann Neurol. 1988;24:580–82. [PubMed: 2977086]
- 40.
- Montine T.J., Beal M.F., Robertson D., et al. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurology. 1999;52:1104–05. [PubMed: 10102447]
- 41.
- Jeitner T.M., Bogdanov M.B., Matson W.R., et al. N(epsilon)-(gamma-L-glutamyl)-L- lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington’s disease. J Neurochem. 2001;79:1109–12. [PubMed: 11739625]
- 42.
- Kaltenbach L.S., Romero E., Becklin R.R., et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. [PMC free article: PMC1866352] [PubMed: 17500595]
- 43.
- Gizatullina Z.Z., Lindenberg K.S., Harjes P., et al. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol. 2006;59:407–11. [PubMed: 16437579]
- 44.
- Saft C., Zange J., Andrich J., et al. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov Disord. 2005;20:674–79. [PubMed: 15704211]
- 45.
- Strand A.D., Aragaki A.K., Shaw D., et al. Gene expression in Huntington’s disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863–76. [PubMed: 15888475]
- 46.
- Hersch S.M., Gevorkian S., Marder K., et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology. 2006;66:250–52. [PubMed: 16434666]
- 47.
- Stoy N., Mackay G.M., Forrest C.M., et al. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J Neurochem. 2005;93:611–23. [PubMed: 15836620]
- 48.
- Chen C.M., Wu Y.R., Cheng M.L., et al. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun. 2007;359:335–40. [PubMed: 17543886]
- 49.
- Christofides J., Christofides M., Bridel M., Egerton M., et al. Blood 5-hydroxytryptamine, 5-hydroxyindoleacetic acid and melatonin levels in patients with either Huntington’s disease or chronic brain injury. J Neurochem. 2006;97:1078–88. [PubMed: 16573644]
- 50.
- Liu C.S., Cheng W.L., Kuo S.J., et al. Depletion of mitochondrial DNA in leukocytes of patients with poly-Q diseases. J Neurol Sci. 2008;264:18–21. [PubMed: 17720200]
- 51.
- Klepac N., Relja M., Klepac R., et al. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: a cross-sectional study. J Neurol. 2007;254:1676–83. [PubMed: 17990062]
- 52.
- Mochel F., Charles P., Seguin F., et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS ONE. 2007;2:e647. [PMC free article: PMC1919424] [PubMed: 17653274]
- 53.
- Popovic V., Svetel M., Djurovic M., et al. Circulating and cerebrospinal fluid ghrelin and leptin: potential role in altered body weight in Huntington’s disease. Eur J Endocrinol. 2004;151:451–55. [PubMed: 15476444]
- 54.
- Dalrymple A., Wild E.J., Joubert R., et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–40. [PubMed: 17552550]
- 55.
- Borovecki F., Lovrecic L., Zhou J., et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl Acad Sci U S A. 2005;102:11023–28. [PMC free article: PMC1182457] [PubMed: 16043692]
- 56.
- Runne H., Kuhn A., Wild E.J., et al. Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proc Natl Acad Sci U S A. 2007;104:14424–29. [PMC free article: PMC1964868] [PubMed: 17724341]
- 57.
- Zuccato C., Liber D., Ramos C., et al. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharm Res. 2005;52:133–39. [PubMed: 15967378]
- 58.
- Ciammola A., Sassone J., Cannella M., et al. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am J Med Genet B Neuropsychiatr Genet. 2007;144:574–77. [PubMed: 17427191]
- 59.
- Maglione V., Giallonardo P., Cannella M., et al. Adenosine A2A receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005;139:101–5. [PubMed: 16184606]
- 60.
- Varani K., Bachoud-Levi A.C., Mariotti C., et al. Biological abnormalities of peripheral A(2A) receptors in a large representation of polyglutamine disorders and Huntington’s disease stages. Neurobiol Dis. 2007;27:36–43. [PubMed: 17512749]
- 61.
- Underwood B.R., Broadhurst D., Dunn W.B., et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain. 2006;129:877–86. [PubMed: 16464959]
- 62.
- Weiss A., Abramowski D., Bibel M., et al. Single-step detection of mutant huntingtin in animal and human tissues: A bioassay for Huntington’s disease. Anal Biochem. 2009;395:8–15. [PubMed: 19664996]
- Review Value of Invertebrate Genetics and Biology to Develop Neuroprotective and Preventive Medicine in Huntington’s Disease.[Neurobiology of Huntington's D...]Review Value of Invertebrate Genetics and Biology to Develop Neuroprotective and Preventive Medicine in Huntington’s Disease.Neri C. Neurobiology of Huntington's Disease: Applications to Drug Discovery. 2011
- Review Neuroprotection for Huntington's disease: ready, set, slow.[Neurotherapeutics. 2008]Review Neuroprotection for Huntington's disease: ready, set, slow.Hersch SM, Rosas HD. Neurotherapeutics. 2008 Apr; 5(2):226-36.
- Review High-Throughput and High-Content Screening for Huntington’s Disease Therapeutics.[Neurobiology of Huntington's D...]Review High-Throughput and High-Content Screening for Huntington’s Disease Therapeutics.Varma H, Lo DC, Stockwell BR. Neurobiology of Huntington's Disease: Applications to Drug Discovery. 2011
- Measuring hot flashes: summary of a National Institutes of Health workshop.[Mayo Clin Proc. 2004]Measuring hot flashes: summary of a National Institutes of Health workshop.Miller HG, Li RM. Mayo Clin Proc. 2004 Jun; 79(6):777-81.
- Review Therapeutic Update on Huntington's Disease: Symptomatic Treatments and Emerging Disease-Modifying Therapies.[Neurotherapeutics. 2020]Review Therapeutic Update on Huntington's Disease: Symptomatic Treatments and Emerging Disease-Modifying Therapies.Dash D, Mestre TA. Neurotherapeutics. 2020 Oct; 17(4):1645-1659.
- Biomarkers to Enable the Development of Neuroprotective Therapies for Huntington...Biomarkers to Enable the Development of Neuroprotective Therapies for Huntington’s Disease - Neurobiology of Huntington's Disease
- ACACB [Myotis davidii]ACACB [Myotis davidii]Gene ID:102759463Gene
- C770_RS28270 [Sinorhizobium meliloti GR4]C770_RS28270 [Sinorhizobium meliloti GR4]Gene ID:25013735Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...