Introduction
Cerebral autoregulation is the ability of the cerebral vasculature to maintain stable blood flow despite changes in blood pressure (or, more accurately, cerebral perfusion pressure).[1] Under normal circumstances, cerebral blood flow is regulated through changes in arteriolar diameter, which, in turn, drive changes in cerebrovascular resistance following the Hagen-Poiseuille equation. [2] Although decades of research have illuminated some underpinning mechanisms, the exact molecular means underlying autoregulation remain elusive. Various processes, including myogenic, neurogenic, endothelial, and metabolic responses, have been implicated in mediating cerebral vasomotor reactions. See Figure. Physiology of Cerebral Autoregulation.
Still, it is essential to differentiate carbon dioxide reactivity and flow-metabolism coupling from cerebral autoregulation.[3] Carbon dioxide reactivity describes vascular reactions in response to changes in the partial pressure of arterial carbon dioxide (PaCO2) but does not consider reactions to pressure changes. Flow-metabolism coupling, in comparison, involves regulating cerebral blood flow relative to local cellular demand, for example, as a consequence of neural activation during cognitive tasks. Similar to PaCO2 reactivity, flow-metabolism coupling, and the neurovascular unit function irrespective of fluctuations in cerebral perfusion pressure.[2]
With a working definition of autoregulation and an understanding of what it is not, researchers have developed technology that now boasts the ability to measure autoregulatory function in real-time, which may lead to the fine-tuning of long-established guidelines.[4][5] Updated guidelines may ameliorate clinical and functional outcomes after acute brain injury by individualizing cerebral perfusion pressure targets based on patients' unique hemodynamic physiology.[6]
Autoregulation is assessable by examining changes in cerebral blood flow, or its surrogates, in response to changes in cerebral perfusion pressure or mean arterial pressure as its surrogate.[2] Individualization of autoregulatory pressure ranges, together with the developing concept of an optimum mean arterial pressure landscape for the injured brain, represent a novel and innovative application of autoregulation neuromonitoring. This topic will be further discussed in the concluding section of this review.
Mechanism
Cerebral Blood Flow Regulation and Physiology
Cerebral oxygen delivery is a function of brain blood flow and blood oxygen content, whereby cerebral blood flow (CBF) is dependent on cerebral perfusion pressure (CPP) and inversely proportional to cerebrovascular resistance (CVR). Another way to conceptualize blood flow to the brain is via the pressure gradient between the supplying arteries (mean arterial pressure, MAP) and the cerebral venous system, the latter being approximately equivalent to intracranial pressure (ICP).
Vascular resistance within the brain reflects the smooth muscle tone of the vessels, which is partially influenced by mean arterial pressure (MAP). If CPP increases or decreases, the myogenic reflex will result in vasoconstriction or vasodilation, respectively. This assertion is the classical view of pressure-flow autoregulation. If intracranial pressure is stable, CPP is replaceable by MAP. In this manner, changes in brain blood flow can be measured for a range of blood pressures to determine autoregulation.
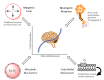
As mentioned in the introductory remarks, four mechanisms regulate cerebral blood flow: myogenic, neurogenic, endothelial, and metabolic processes. Each component appears in the figure below.[1] This section will review these classical mechanisms, with an important caveat that the interplay and relative contributions of each of these mechanisms are highly complex and poorly understood.
Myogenic Tone
Myogenic tone is produced when arteriole and small artery smooth muscle cells contract in response to increased pressure. In contrast, myogenic tone relaxes in response to decreased pressure. Transmural pressure changes, in turn, activate mechanically sensitive ion channels and proteins in the vessel wall, triggering various downstream cascades. For instance, membrane depolarization opens voltage-gated calcium channels, leading to an influx of calcium cations into the smooth muscle cell.[7] Calcium activates myosin light chain kinase (MLCK), which goes on to activate myosin by phosphorylation. Phosphorylated MLCK increases actin-myosin interaction, causing muscle cell contraction and vasoconstriction.
The importance of smooth muscle cell myogenic regulation is apparent in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Patients with CADASIL show a degree of smooth muscle cell degeneration in small cerebral arteries. Studies have demonstrated impaired myogenic autoregulatory functioning in both animal models and individuals with the genetic condition.[8][9][10] This disease results from a mutation in the NOTCH3 gene and is marked by recurrent ischemic strokes, cognitive impairment, subcortical dementia, mood disturbances like depression and apathy, as well as premature death.[11]
Lacombe et al. provided evidence that transgenic mice expressing a mutant NOTCH3 in vascular smooth muscle cells exhibited impaired cerebral vasoreactivity, including reduced responses to vasodilatory challenges and a shift of the lower limit of autoregulation toward higher pressures.[8] Interestingly, parenchymal arteries exhibit a greater basal tone than pial arteries. This difference may buffer the effects of rapid upstream changes in blood pressure on cerebral perfusion and attenuate the transmission of pulsatile mechanical stress into the brain's microcirculation. Disturbance of this basal tone may exacerbate the stroke burden in patients with CADASIL.
Neurogenic Response
Neurogenic mediation of cerebral vasoreactivity involves the control of small- and medium-sized vessel diameters. Neurons and other cell types like astrocytes and microglia secrete a variety of neurotransmitters with vasoactive properties. For instance, acetylcholine and nitric oxide are relatively potent vasodilators, while serotonin and neuropeptide Y stimulate vasoconstriction.[12] Through the creative use of infrared video-microscopy of interneurons and adjacent microvessels in rodents, Cauli et al. showed that increased depolarizing activity of single cortical interneurons results in precise vasomotor responses in neighboring microvessels.[13] They further showed that these neuronally induced vasomotor responses could be mimicked by the perivascular application of vasoactive neuropeptides directly on microvascular receptors.
On a larger scale, these changes in blood flow in response to neuronal activation are observable as the blood oxygen level-dependent (BOLD) signal employed in functional magnetic resonance imaging (fMRI). The BOLD response has been adapted in many fMRI studies investigating increased cerebral metabolic demand in cognitive tasks, spatial memory, visual processing, and across various disease states.
Regarding regional heterogeneity, the anterior circulatory system of the brain possesses denser sympathetic innervation than that of the posterior system. The anterior circulation is controlled mainly by adrenergic sympathetic relays from the superior cervical ganglion as they travel up the carotid arteries. The posterior vessels instead depend on the sympathetic chain via the vertebrobasilar arteries.
Autoregulation has also demonstrated greater effectiveness in the brainstem. For example, in severe hypertension in anesthetized cats, cerebral blood flow significantly increases in the anterior circulation, whereas the brainstem only requires modest increases in inflow.[14] This vascular resistance differential points to a likely regional incongruity in cerebral autoregulation.
This regional variability may play a key role in developing posterior reversible encephalopathy syndrome (PRES). This syndrome, which parenthetically is not always posterior or even reversible, is otherwise characterized radiologically by transient bilateral subcortical vasogenic edema in the posterior circulation.[15] Among several etiologic theories involving immunologic dysfunction, vasospasm, and blood-brain barrier breakdown, one interesting explanation for the edema's apparent posterior predilection is the relative dearth of sympathetic tone in that area, leading to poor autoregulation of blood flow in the setting of abrupt hypertensive episodes.
Metabolic Mechanism
The metabolic mechanism subserving autoregulation occurs in smaller vessels subject to changes in the local environment. Most notably, carbon dioxide overtly alters vasomotor responses; every 1 mmHg increase in PaCO2 corresponds to a roughly 4% increase in cerebral blood flow.[16] The concentration of cerebral carbon dioxide can accumulate and cause vasodilation in this fashion when, for example, hypotension below the lower autoregulatory limit results in tissue hypoperfusion and, thus, anaerobic respiration. The opposite physiology transpires in the setting of hyperperfusion with consequent decreases in PaCO2 and vasoconstriction.
There is a hypothesis that this vasomotor response is regulated by proton concentration in the smooth muscle of cerebral vessels. Proton gradients are regulated by carbonic anhydrase activity, the catalytic activity of which depends on the tight regulation of pH (normally hovering around 7.4 in the human body). Prolonged hypocapnia that generates tissue alkalosis may increase carbonic anhydrase activity.[2] Additionally, decreased oxygen partial pressures can increase cerebral blood flow; however, this effect does not occur unless severe hypoxemia is less than 50 mmHg. Similarly, severe hypoglycemia at levels of less than 2 mmol/L can lead to increases in cerebral blood flow.
Endothelial Mechanism
Lastly, endothelial tissue begets a gamut of signals that affect vascular tone. The endothelium secretes vasodilators like nitric oxide (NO) and vasoconstrictors like thromboxane A2 and endothelin-1 in a paracrine manner. Further, as an interesting bedside-to-bench endeavor, researchers have looked at the ability of statins to regulate autoregulation. In more detail, statins can upregulate nitric oxide synthase, causing cerebral artery dilation and increased cerebral blood flow.[17][18] This mechanism occurs by inhibiting small G-proteins known as Rho and Rac. Rho negatively regulates endothelial nitric oxide synthase. Statins inhibit Rho GTPase activity via inhibition of a process known as geranylgeranylation, a type of prenylation that ultimately confers nitric oxide synthase upregulation.
Related Testing
Methods to Measure Cerebral Autoregulation
Assessment of pressure autoregulation has traditionally been through calculating cerebral blood flow at two different equilibrium states of arterial blood pressure. These steady states correspond to particular cerebral blood flow values. One pressure measurement could be taken at baseline, and the second measured after manual or pharmacologic manipulation of blood pressure, at which point brain blood flow could get measured again. Because this approach involves stable pressures and flows, it is referred to as a static autoregulatory measurement.
The advent of transcranial Doppler (TCD) ultrasound allowed for the visualization of real-time blood-flow velocities (with a temporal resolution of approximately 5 msec), paving the road for dynamic assessments of autoregulation. Dynamic autoregulation refers to short-term, fast responses of blood flow to changes in systemic pressure. As TCD cannot measure flow directly, the blood flow velocity is useful as a surrogate. Pressure changes are inducible using stimuli such as body tilt, thigh-cuff release, or lower body negative pressure.[2]
While controlling the exact timing and magnitude of the hemodynamic stimulus provides a precision advantage, pressure manipulations in critically ill patients are potentially harmful. For example, a thigh-cuff inflation-deflation sequence may incite a precipitous pressure drop of up to 25 to 35 mmHg. In a patient with an ischemic stroke, this drop could cause secondary brain injury from significant hypoperfusion, particularly in a setting where there is autoregulatory physiology compromise in the first place.
Alternatively, one may isolate intracranial vessels without any particular blood pressure challenges and measure the CBF response to spontaneous arterial blood pressure fluctuations. This approach renders dynamic assessments of cerebral autoregulation safe and feasible for patients with acute brain injury. The dynamic response is likely to occur within 10 to 15 seconds, suggesting that arterioles can counter slower fluctuations in systemic blood pressure. Faster changes, such as those greater than 0.5 Hz, are not compensated – for example, those occurring with each cardiac systole. This selective compensation is referred to as the high-pass filter principle. The cerebrovascular system accordingly buffers against slow hemodynamic oscillations (0.01 to 0.4 Hz), while higher frequencies may pass unfiltered to the circulation of the brain.[6]
In addition to blood flow velocity, other intracranial signals are frequently helpful in dynamic vasoregulatory investigation. Examples include near-infrared spectroscopy (NIRS), local brain tissue oxygenation (PbtO2), and intracranial pressure (ICP) monitoring from cerebrospinal fluid (CSF) draining systems. The fundamental principle of these dynamic measurements is the same across methodologies – the input signal is blood pressure or volume change. The resulting change in the intracranial compartment acts as the output signal. Using spontaneous fluctuations of blood pressure and cerebral blood flow, researchers have devised several mathematical methods for modeling autoregulatory indices. This brief review will pay particular attention to transfer function analysis and the time-correlation approach, with subsequent nods to wavelet analysis and projection pursuit regression.
Transfer Function Analysis
Transfer function analysis (TFA) has its basis in linear, stationary modeling and a fast Fourier transform algorithm to compute spectral estimates of blood pressure and cerebral blood flow. Autoregulation, when properly functioning, attenuates the influence of blood pressure on brain blood flow velocity by preventing direct propagation of the pressure waveform at lower frequencies (usually under 0.2 Hz). Two key parameters – gain and phase shift – can be derived from TFA at each frequency. The gain reflects the compression of brain blood flow velocity amplitude changes in response to blood pressure. For example, a gain of 0.65 denotes that 65% of the relative amplitude of cerebral blood flow velocity gets attenuated with regard to a unit of change in arterial blood pressure. Phase shift quantifies the time lag between blood pressure and brain flow velocity at a given frequency and is represented in degrees or radians. Larger phase shifts between the two signals mean autoregulation properly buffers the cerebrovascular tree from blood pressure changes.
Of note, TFA can only rationalize linear relationships between arterial blood pressure and mean flow velocity, which is why coherence usually accompanies TFA to test the linearity between the two waveforms. Generally, a coherence above 0.5 is considered acceptable for TFA. Regarding frequency bands, values for gain, phase-shift, and coherence get reported in three bins: very low (0.02 to 0.07 Hz), low (0.07 to 0.2 Hz), and high (0.2 to 0.5 Hz) ranges.[19] The high-pass filter principle of autoregulation translates to reductions of coherence and gain with increases in phase shift. These modulations result in the relative desynchronization between blood pressure and cerebral blood flow oscillations. Additionally, because vasomotor adaptation is slow and requires roughly 10 to 15 seconds, autoregulation is most likely to function at lower frequencies.[6]
Time Domain Analysis
This method measures the degree of correlation between blood pressure and various cerebral output signals. A rolling Pearson correlation coefficient gets calculated between 30 consecutive, time-averaged (10 sec) values of arterial blood pressure and cerebral blood flow (or its surrogates). The resulting coefficient provides an estimate of autoregulatory function respective to each variable. The mean cerebral blood flow velocity coefficient is Mx, while the tissue oxygenation index (TOx) derives from NIRS.
There are over 20 cerebral autoregulation indices, which have apparent pros and cons for autoregulation research. Perhaps the most rigorously studied index is the pressure reactivity index (PRx), which derives from ICP instead of cerebral blood flow velocity or tissue oxygenation.[2] Cerebral perfusion pressure (CPP = MAP – ICP) may also be substituted for arterial blood pressure. Each index enlists a unique threshold for impaired autoregulation, with a range of 0.069 to 0.46, depending on devices used to measure cerebral blood flow or its surrogate.[3]
In all cases, a positive correlation coefficient reflects synchrony between the two signals, suggesting impaired cerebral autoregulation, whereby systemic pressures passively propagate to the cerebral vasculature. Meanwhile, a negative or near-zero coefficient implies active buffering of the cerebral vasculature against blood pressure changes and, thus, intact autoregulatory physiology.
Wavelet Analysis
This approach, also known as multimodal pressure-flow analysis, represents an alternative to classical spectral analyses, such as fast Fourier transform, and considers both the time and frequency content of the signal. The wavelet analysis produces maps of phase shift and coherence between blood pressure and cerebral blood flow velocity over a range of frequencies and time points. Enforcing a minimal coherence threshold and focusing the analysis on areas in the time-frequency map with a high degree of correlation increases the reliability of the phase shift estimation. Signal decomposition with wavelet analysis has also been applied to tissue oxygenation using NIRS.[20]
Projection Pursuit Regression
Projection pursuit regression (PPR) is a non-parametric method wherein a model is not specified a priori but derives directly from the variables of interest (i.e., arterial pressure and cerebral blood flow). The analysis modifies a linear transfer function between input (blood pressure) and output (brain blood flow). A linear autoregressive transfer function gets passed through kernel functions, also known as ridge functions, determined by minimizing the mean squared error.[21] The method characterizes the non-linear relationship between pressure and flow and identifies regions wherein this relation changes. The gain (i.e., the slope) of the pressure-flow relation within each region provides a measure of the effectiveness of autoregulation within that region.
A 2016 study by Santos et al. used PPR to show that patients suffering from delayed cerebral ischemia (DCI) after subarachnoid hemorrhage had distinctive hemodynamic profiles concerning those not suffering from DCI.[22] The authors then invoked previously found pharmacologic effects on PPR-derived autoregulation parameters. After combining their results with those parameters, the research team argued that myogenic dysfunction leads to vasospasm, sympathetic overaction, and cholinergic dysfunction leads to DCI, while deficits in all three pathophysiologic mechanisms beget both vasospasm and DCI.
In the last two decades, these autoregulatory indices have also served to generate optimum cerebral perfusion pressures and ideal pressure ranges based on the lower and upper limits of autoregulation. Steiner et al. published a landmark study in 2002 using continuous autoregulation monitoring to identify optimal cerebral perfusion pressure in patients with traumatic brain injury.[23] This optimal pressure is calculated by plotting cerebral autoregulation indices against a range of blood pressures over 4-hour monitoring periods and fitting a U-shaped curve to the data to identify the blood pressure range at which autoregulation is most preserved. The hypothesis surrounding this window of cerebral perfusion pressures is that brain arterioles can maintain a constant cerebral blood flow with the largest possible autoregulatory reserve at those pressures. At an individual level in the critical care setting, a continuous estimation of an ideal cerebral perfusion pressure presents an attractive target for hemodynamic management.
Clinical Significance
Applications of Autoregulation Neuromonitoring
Numerous studies in recent years have demonstrated that large differences between actual mean arterial pressure and an optimal, calculated mean arterial pressure (based on autoregulatory status) are associated with poor outcomes. These papers encompass traumatic brain injury, intracerebral hemorrhage, subarachnoid hemorrhage, ischemic stroke, adults undergoing cardiac bypass surgery, children with moyamoya vasculopathy, and neonates with hypoxic-ischemic encephalopathy.[3][4]
Nevertheless, blood pressure management guidelines persistently recommend a single, fixed target value for many critically ill patients. For example, the American Heart Association endorses a systolic blood pressure of less than 140 mmHg after intracerebral hemorrhage; they also suggest systolic pressures under 160 mmHg before aneurysm obliteration and less than 140 mmHg after clipping or coiling of the aneurysm following subarachnoid hemorrhage. The same society recommends systolic readings of less than 180 mmHg after intravenous recombinant tissue plasminogen activator for large-vessel occlusion ischemic stroke. These guidelines, however, do not currently consider autoregulation-guided hemodynamic management of critically ill patients. In this omission, many questions in the field of neuromonitoring are left unanswered.[3][6] For instance, is it feasible to effectively personalize MAP targets based on an individual’s dynamic autoregulatory composition? Might this method be clinically beneficial?
Notwithstanding such unanswered questions, the science of autoregulation has come a long way since 1959, when Dr. Niels Lassen published the first consolidated review on cerebral autoregulation.[24] Speaking perhaps to the incremental yet potentially groundbreaking nature of the scientific investigation, Lassen concludes his review with the following remarks: “These major findings and the wealth of additional observations have substantially increased our understanding of this important area of human physiology. Undoubtedly our knowledge is still incomplete at various points. However, a solid foundation for relevant physiological thinking and future studies has been established.” It is over 60 years later, and autoregulation research is at the precipice of tangibly translatable use at the bedside, as clinical trials of autoregulation-guided therapy are underway across Europe (NCT02982122).
With the idea of an individualized, optimal cerebral perfusion pressure range in mind, researchers have carried out many observational studies in both adult and pediatric settings to personalize hemodynamic care. These studies now stand on the shoulders of Lassen’s autoregulatory curve, just now seeing eye-to-eye with society-endorsed guidelines that recommend impersonal, imprecise hemodynamic management of patients with cerebrovascular disease. Nonetheless, randomized controlled trials are lacking. Furthermore, the specialized equipment to monitor and determine optimal pressures is expensive and demanding, not universally available, and requires a moderate degree of training and interpretation abilities. ICM+ software licenses (University of Cambridge, UK) are required to perform this work, so data regarding effectiveness in randomized trials are needed before the widespread distribution of licenses becomes a reality.
Physiology of Cerebral Autoregulation. This illustration shows the four classical mechanisms contributing to cerebral autoregulation. Through myogenic tone, transmural pressure influences arterial diameter through direct smooth muscle contraction or relaxation. (more...)
References
- 1.
Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation.
Cerebrovasc Brain Metab Rev. 1990 Summer;2(2):161-92. [
PubMed: 2201348]
- 2.
Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage.
Nat Rev Neurol. 2013 Mar;9(3):152-63. [
PubMed: 23419369]
- 3.
Rivera-Lara L, Zorrilla-Vaca A, Geocadin RG, Healy RJ, Ziai W, Mirski MA. Cerebral Autoregulation-oriented Therapy at the Bedside: A Comprehensive Review.
Anesthesiology. 2017 Jun;126(6):1187-1199. [
PubMed: 28383324]
- 4.
Petersen NH, Silverman A, Wang A, Strander S, Kodali S, Matouk C, Sheth KN. Association of Personalized Blood Pressure Targets With Hemorrhagic Transformation and Functional Outcome After Endovascular Stroke Therapy.
JAMA Neurol. 2019 Oct 01;76(10):1256-1258. [
PMC free article: PMC6664391] [
PubMed: 31355872]
- 5.
Silverman A, Kodali S, Strander S, Gilmore EJ, Kimmel A, Wang A, Cord B, Falcone G, Hebert R, Matouk C, Sheth KN, Petersen NH. Deviation From Personalized Blood Pressure Targets Is Associated With Worse Outcome After Subarachnoid Hemorrhage.
Stroke. 2019 Oct;50(10):2729-2737. [
PMC free article: PMC6756936] [
PubMed: 31495332]
- 6.
Wang A, Ortega-Gutierrez S, Petersen NH. Autoregulation in the Neuro ICU.
Curr Treat Options Neurol. 2018 May 17;20(6):20. [
PubMed: 29770889]
- 7.
Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure.
J Physiol. 1998 Apr 01;508 ( Pt 1)(Pt 1):199-209. [
PMC free article: PMC2230857] [
PubMed: 9490839]
- 8.
Lacombe P, Oligo C, Domenga V, Tournier-Lasserve E, Joutel A. Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy.
Stroke. 2005 May;36(5):1053-8. [
PubMed: 15817893]
- 9.
Joutel A, Monet-Leprêtre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, Lemaire-Carrette B, Domenga V, Schedl A, Lacombe P, Hubner N. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease.
J Clin Invest. 2010 Feb;120(2):433-45. [
PMC free article: PMC2810078] [
PubMed: 20071773]
- 10.
Pfefferkorn T, von Stuckrad-Barre S, Herzog J, Gasser T, Hamann GF, Dichgans M. Reduced cerebrovascular CO(2) reactivity in CADASIL: A transcranial Doppler sonography study.
Stroke. 2001 Jan;32(1):17-21. [
PubMed: 11136908]
- 11.
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil.
Lancet Neurol. 2009 Jul;8(7):643-53. [
PubMed: 19539236]
- 12.
Hamel E. Perivascular nerves and the regulation of cerebrovascular tone.
J Appl Physiol (1985). 2006 Mar;100(3):1059-64. [
PubMed: 16467392]
- 13.
Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways.
J Neurosci. 2004 Oct 13;24(41):8940-9. [
PMC free article: PMC6730057] [
PubMed: 15483113]
- 14.
Faraci FM, Mayhan WG, Heistad DD. Segmental vascular responses to acute hypertension in cerebrum and brain stem.
Am J Physiol. 1987 Apr;252(4 Pt 2):H738-42. [
PubMed: 3565591]
- 15.
Tetsuka S, Ogawa T. Posterior reversible encephalopathy syndrome: A review with emphasis on neuroimaging characteristics.
J Neurol Sci. 2019 Sep 15;404:72-79. [
PubMed: 31349066]
- 16.
Yoshihara M, Bandoh K, Marmarou A. Cerebrovascular carbon dioxide reactivity assessed by intracranial pressure dynamics in severely head injured patients.
J Neurosurg. 1995 Mar;82(3):386-93. [
PubMed: 7861215]
- 17.
Endres M, Laufs U. Effects of statins on endothelium and signaling mechanisms.
Stroke. 2004 Nov;35(11 Suppl 1):2708-11. [
PubMed: 15375300]
- 18.
Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: facts, myths, and missing links.
Neurocrit Care. 2009;10(3):373-86. [
PubMed: 19127448]
- 19.
Claassen JA, Meel-van den Abeelen AS, Simpson DM, Panerai RB., international Cerebral Autoregulation Research Network (CARNet). Transfer function analysis of dynamic cerebral autoregulation: A white paper from the International Cerebral Autoregulation Research Network.
J Cereb Blood Flow Metab. 2016 Apr;36(4):665-80. [
PMC free article: PMC4821028] [
PubMed: 26782760]
- 20.
Tian F, Tarumi T, Liu H, Zhang R, Chalak L. Wavelet coherence analysis of dynamic cerebral autoregulation in neonatal hypoxic-ischemic encephalopathy.
Neuroimage Clin. 2016;11:124-132. [
PMC free article: PMC4753811] [
PubMed: 26937380]
- 21.
Tan CO. Defining the characteristic relationship between arterial pressure and cerebral flow.
J Appl Physiol (1985). 2012 Oct 15;113(8):1194-200. [
PMC free article: PMC3472492] [
PubMed: 22961266]
- 22.
Santos GA, Petersen N, Zamani AA, Du R, LaRose S, Monk A, Sorond FA, Tan CO. Pathophysiologic differences in cerebral autoregulation after subarachnoid hemorrhage.
Neurology. 2016 May 24;86(21):1950-6. [
PMC free article: PMC4887123] [
PubMed: 27164675]
- 23.
Steiner LA, Czosnyka M, Piechnik SK, Smielewski P, Chatfield D, Menon DK, Pickard JD. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury.
Crit Care Med. 2002 Apr;30(4):733-8. [
PubMed: 11940737]
- 24.
LASSEN NA. Cerebral blood flow and oxygen consumption in man.
Physiol Rev. 1959 Apr;39(2):183-238. [
PubMed: 13645234]
Disclosure: Andrew Silverman declares no relevant financial relationships with ineligible companies.
Disclosure: Nils Petersen declares no relevant financial relationships with ineligible companies.