

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Forum on Drug Discovery, Development, and Translation; Shore C, Gee AW, Kahn B, et al., editors. Examining the Impact of Real-World Evidence on Medical Product Development: Proceedings of a Workshop Series. Washington (DC): National Academies Press (US); 2019 Feb 6.

Examining the Impact of Real-World Evidence on Medical Product Development: Proceedings of a Workshop Series.
Show detailsKey Messages Identified by Individual Workshop Participants
- Inclusion and exclusion criteria can be broadened in real-world trials so patients with comorbidities or concomitant treatments are included. These patients may require additional monitoring, and the trials will likely yield more generalizable results. (Califf, Horberg, Katz, London)
- When considering treatment restriction in real-world trials, researchers may consider a specific set of questions or categories for consideration in order to plan a trial that answers the research question and honors research participant safety and autonomy. (Alphs, Stein)
- Research driven by patients is iterative and considers patient needs and priorities, as well as patient experience. The focus of this type of research is to enable patients to make informed decisions about their health care. (Nowell)
- Researchers have two duties: maintaining the trial protocol and caring for the well-being of their patients. If these two duties conflict, caring for patients is the higher priority. (Alphs, Katz, Simon)
- Designing a study requires a decision about how to define the standard of care, which can be highly variable in different regions, for the control arm. This decision can affect the results of the study, and there is an ethical obligation to ensure that study participants are not receiving substandard care. (Califf, Hernandez, Stein)
In a real-world trial, there is a delicate balance between the needs of the trial and the needs of the patient, said Larry Alphs, deputy chief medical officer of Newron Pharmaceuticals. To make inferences from the study, researchers need to ensure a certain level of consistent treatment adherence. However, researchers must also ensure that patients receive adequate treatment for their condition, and that there is enough flexibility to handle unique patient needs and adverse events that arise. In this session, workshop participants explored how variability in treatment might affect results, and how to balance the needs of the study with patient autonomy and safety. Gregory Simon noted, “We're talking about real-world treatment, and by that we mean studying treatments with typical providers and typical patients, accepting that there will be highly variable quality of, and adherence to, treatment.”
ILLUSTRATIVE EXAMPLES
To explore the issues surrounding treatment quality in real-world trials, speakers at the second and third workshops presented case studies as illustrative examples of the considerations that go into treatment quality control and restriction.
Lithium for Suicide Prevention
For more than two decades, lithium has been proposed as a treatment to prevent suicide in patients with bipolar disorder and major depression, said Ira Katz, senior consultant for program evaluation at the U.S. Department of Veterans Affairs' (VA's) Office of Mental Health and Suicide Prevention. This idea has been supported by evidence from meta-analyses of observational studies and randomized controlled trials (RCTs) that were conducted for other purposes, although a propensity score-matched study of bipolar patients in the VA was equivocal. There had never been an adequately powered clinical trial on lithium, he said, and two previous trials had been terminated due to difficulties in enrollment. Seeing this need, Katz and his colleagues proposed the idea of carrying out a large RCT within the VA to test lithium as a treatment for preventing suicide. The VA, said Katz, was an ideal site for this research because there is a large patient population, approximately 140 medical centers, infrastructure for suicide prevention, and infrastructure for clinical trials.
The study, which is double blinded and placebo controlled, had enrolled around 360 people at the time of the workshop. These people had depression or bipolar disorder and had survived a suicide attempt. The projected sample size is around 1,600, and it is powered to detect about a 40 percent reduction rate of repeated suicide attempts, said Katz. There have been some problems with recruitment, said Katz, but the biggest issues have been questions about ecological validity (i.e., the extent to which the findings from the clinical trial can be generalized to treatments for patients in real-world settings). Simon noted that this type of study will become necessary with the development of a “new generation” of treatments for suicide prevention. “Our current way of evaluating treatments in mental health will be completely incapable of assessing that question,” he said.
Katz walked the workshop participants through a series of questions that the researchers needed to answer, and the considerations that went into answering the questions.
Does the Selection of Participants for RCTs Lower the Outcome Rate?
To conduct a study evaluating interventions to prevent suicidal behavior with reasonable sample sizes, Katz emphasized the need to enroll people who are at an increased risk for suicide. However, the process of enrolling in and consenting to an RCT makes it likely that people who are truly at risk will be “filtered out,” said Katz. Data from electronic medical records suggested that among those with depression or bipolar disorder who survive a suicide attempt, 15 percent reattempt during the year, he said. Researchers expected to see a far higher number during the course of the clinical trial because they assumed that many suicide attempts that were not documented in the medical record would be captured with the assessments included in the RCT protocol. However, experience in the study has been consistent with the 15 percent rate, said Katz, which was “really surprising and a puzzle.” After examination of other data, the researchers determined that what was likely happening was a counterbalancing effect: The suicide attempt rate was probably decreased due to filtering out from the RCT process, and at the same time, it was probably increased due to the RCT protocol that uncovered suicide attempts.
How Rigorous Should the Study Be About Diagnoses?
A second issue that the researchers faced, said Katz, was determining the inclusion and exclusion criteria for participants. Most RCTs want “clean patients,” that is, those with the diagnosis but without comorbidities. However, requiring “clean patients” could exclude those at highest risk for suicidal behavior, said Katz. The researchers decided to permit comorbidities, such as posttraumatic stress disorder (PTSD) or substance abuse, and there was no attempt to filter out primary versus secondary diagnoses of depression. Researchers also struggled with concomitant medications. Some medications, such as diuretics and angiotensin converting enzyme (ACE) inhibitors, make it more difficult to manage patients on lithium. The researchers proposed performing extra monitoring for these patients, but the Institutional Review Board (IRB) decided they could not be enrolled. After the research began, they discovered that 30 percent of otherwise eligible patients were excluded specifically because they were on ACE inhibitors. Katz and his colleagues successfully argued to the IRB that these patients had to be included if the trial participants were going to be a representative sample of the true patient population.
How Can Researchers Balance the Flexibility of Caring for Patients with the Need for Adherence to RCT Protocol?
Third, said Katz, there was the issue of finding a balance between providing appropriate care for patients and adhering closely to the RCT protocol. “Flexibility is essential but difficult,” said Katz. He relayed the story of a patient who had depression, PTSD, and a personality disorder, and was difficult to manage. The patient was frequently late for appointments or missed them entirely. In the sixth month of the study, the patient missed the required blood test and assessments. The investigators, however, continued to send the patient supplies of the study medication while encouraging the patient to come in for study assessments and care. The investigators believed this to be good clinical management for the patient, though they recognized that it may not have been consistent with good clinical practice for research. The study monitor criticized this decision and judged it to be a serious protocol violation. The question in designing clinical trials relevant to patients and complex issues, said Katz, is whether the protocol should fit the patient, or whether the patient should fit the protocol. This is a difficult dilemma to navigate, particularly in clinically difficult situations like suicide prevention, said Katz.
INTERSEPT and PRIDE: Real-World Mental Health Trials
Developing real-world evidence (RWE), Alphs said, is an iterative process that evolves over time. At the third workshop, Alphs explained that RWE is aimed at answering the basic questions, “Is this drug safe?” and “Is this drug effective?” As the body of information grows, researchers are able to focus their questions on specific populations that may be complex. The ultimate question is whether a drug is safe or effective for specific individuals within the broader population for which the drug has been shown to be safe and effective. However, it is unlikely there will ever be sufficient RWE to answer these questions at the individual patient level.
In real-world pragmatic clinical trials, said Alphs, patient treatment may need to be restricted to patients without many complicating medical problems for a period of time in order to generate answers about a drug's safety and efficacy. When considering treatment restriction, a number of general questions need to be asked:
- Which patients' treatments will be restricted from inclusion in the trial? What are their vulnerabilities that lead to these restrictions?
- What specific restrictions will be placed on treatment? What is the impact of these treatment restrictions?
- In which treatment settings will restrictions be applied? Are treatment practice and ethical considerations similar in all areas?
- How long are the restrictions to be in place? Will the treatment restrictions have enduring impact on morbidity and mortality?
- What is the value of the restrictions? What are the risk–benefit considerations of imposing these restrictions?
To answer these questions, Alphs and his colleagues developed a template that identifies the specific considerations when designing clinical trials, which are divided into six domains (see Table 7-1).
TABLE 7-1
Considerations for Study Design Restrictions.
Alphs presented two case studies to demonstrate how the issue of treatment restriction has been dealt with in real-world trials.
InterSePT: Clozapine Versus Olanzapine for Suicide Prevention
The first case study was an international trial that compared the use of clozapine and olanzapine for suicide prevention in patients with schizophrenia or schizoaffective disorder. Patients with these disorders, said Alphs, are at high risk of suicide behavior, with a lifetime risk of suicide attempts at 25 to 50 percent and a lifetime risk of death by suicide at 5 percent. This is an undertreated life-threatening mental health condition, and represents a major public health problem, said Alphs. There is stigma surrounding both schizophrenia and suicide, making them difficult problems to address.
The trial, called InterSePT (International Suicide Prevention Trial), was a 2-year, multicenter, randomized, open-label, rater-blinded study, said Alphs. The trial enrolled 980 patients at high risk for suicide. They were randomized to receive either clozapine or olanzapine and were followed for 2 years. At the time of the study, olanzapine was the leading state-of-the-art treatment for schizophrenia, although the standard of care has changed somewhat since the study was completed, noted Alphs. Several endpoints were assessed: significant worsening of suicidality, hospitalization to prevent suicide attempt, suicide attempt, and death by suicide. Blinded raters made these assessments and a blinded endpoint monitoring board judged whether or not an event had occurred. Alphs said that at the time of the study, scales to measure suicidality as a clinical trial endpoint did not exist, so the research team developed new scales to use for regularly monitoring suicidality.
The trial was not blinded, said Alphs, for several reasons. First, the side effect profiles of the two drugs are dramatically different, so it would have been very difficult to keep patients from knowing which treatment they were taking. Second, clozapine causes agranulocytosis in about 1 percent of the population, and as a consequence, requires regular blood draws that were not required for olanzapine treatment and, thus, unnecessary for patients randomized to that treatment arm. Finally, the study was not blinded because it would have been unethical to do so, said Alphs. The enrolled patients were at a high risk of suicide, having either been hospitalized for suicide ideation or having attempted suicide in the past year. Because the potential outcomes were so severe, and ethical considerations required that the research design minimize suicide attempts and deaths, the patients' clinicians needed the knowledge of treatment to flexibly manage their patients should they become suicidal.
Participants in the clozapine arm of the trial required regular visits for blood draws. Alphs noted these visits could have impacted the patients' suicidality, so all of the participants were required to come in for visits with the clinical staff on the same schedule, regardless of treatment arm. He said this requirement did not reflect the reality of olanzapine treatment in the real world, but it was “an absolute requirement for the safety of the study.” At each of these visits, the participants' suicidality was assessed. Alphs noted that this assessment is “good clinical practice that should probably be done in every case,” including real-world clinical practice. If the assessment found that the patient was highly suicidal, the patient was hospitalized to prevent a suicide attempt. If the patient's suicidality had worsened significantly, this was also considered an endpoint for the study.
The results of the study, said Alphs, indicated that patients treated with clozapine were less likely to exhibit suicidal behavior or be judged at imminent risk for suicide than patients treated with olanzapine (see Figure 7-1). Significantly fewer patients in the clozapine treatment arm attempted suicide, required hospitalization, required treatment with concomitant drugs to prevent suicide, or died by suicide. Alphs noted that in both treatment groups, the extensive study surveillance and regular clinical assessment likely prevented suicide in many of these high-risk patients. This study contributed to a U.S. Food and Drug Administration (FDA) decision to approve clozapine for reducing the risk of suicide in high-risk schizophrenic or schizoaffective patients, said Alphs.
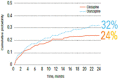
FIGURE 7-1
Cumulative probability of experiencing a significant suicide attempt or hospitalization to prevent suicide. SOURCE: Alphs presentation, July 17, 2018.
PRIDE: Oral Versus Injectable Antipsychotic for Treatment of Schizophrenia
The second case study that Alphs presented was a trial called PRIDE (Alphs et al., 2015). This study sought to determine if treatment with a long-acting injectable antipsychotic had advantages over oral antipsychotic treatments when provided to recently incarcerated persons with schizophrenia. Alphs said that “deinstitutionalization of the mentally ill over the past 50 years and changes in health policy have shifted the burden of care for mental illness [from mental hospitals] to jails and prisons.” Many mentally ill people in the United States are incarcerated, and the risk of reincarceration is high when people are not given access to treatment after leaving jail or prison. This study examined not only the potential clinical benefits of an injectable antipsychotic (see also Box 7-1), but also the potential economic benefits that could be gained if patients are adequately treated for mental illness.
BOX 7-1
Patient Compliance as Discussed by Individual Workshop Participants.
The trial was a 15-month multicenter, randomized, open-label, rater-blinded study. Participants were randomized to either an injectable antipsychotic (paliperidone palmitate), or to oral antipsychotic treatment (one of seven frequently used oral antipsychotics). Participants could de-select the medications in this group if the medications were considered by the participant or his/her treating physician to be ineffective for them, said Alphs. After de-selection of unacceptable candidate oral treatments, patients randomized to oral treatment were further randomly assigned to one of the remaining acceptable oral antipsychotics. The trial was not blinded because it was known that all of the drugs were relatively safe and effective, and the difference between an injectable and an oral drug would have been impractical to blind. The endpoints, all of which were considered “treatment failure,” were time to hospitalization, time to suicide, time to arrest or reincarceration, and time to an intervention to prevent hospitalization or arrest.
The study found that treatment failure was 1.4 times more likely to occur during oral antipsychotic treatment than with injectable antipsychotic treatment, said Alphs. The mean days to treatment failure were nearly 6 months more for patients who received the injectable antipsychotic treatment. Using results from this study, the researchers applied economic modeling to stable schizophrenic Medicaid patients with similar clinical characteristics to predict health economic outcomes. This model estimated that, using the injectable treatment among all Medicaid patients with similar characteristics to those in the PRIDE study, more than $3 billion could be saved over an 18-month period, Alphs said.
DECISION AID
The general issues discussed by individual workshop participants in the first and second workshops were used to develop a decision aid for the third workshop (see Figure 7-2). As with the other decision aids, the intention was to outline some questions to consider in order to make thoughtful choices in RWE study design. Session moderator Jennifer Graff, vice president of comparative effectiveness research at the National Pharmaceutical Council, encouraged workshop participants to consider how much variation could be desired or accepted in terms of treatment, setting, or provider, and what elements of trial participant safety and autonomy could be most important. She asked, “What is our obligation to deliver safe care if we are watching what happens in the real world? What do we do and what are our obligations to deliver state-of-the-art care to patients enrolled in pragmatic trials?” Participants at the third workshop reflected on these questions and offered feedback on the decision aid throughout the course of their discussions.

FIGURE 7-2
Decision aid on questions to consider regarding participant safety and investigator control of treatments in a trial taking place in a community care setting. NOTE: This decision aid was drafted by some individual workshop participants based on the discussions (more...)
PATIENT-CENTERED RESEARCH
W. Benjamin Nowell, director of patient-centered research at the Global Healthy Living Foundation, spoke about his experiences developing a patient-powered research network (PPRN) for arthritis patients. The PPRN—called ArthritisPower—is a research registry with more than 16,000 patient participants who have rheumatic and musculoskeletal diseases. It was created in 2015 with support from the Patient-Centered Outcomes Research Institute, and is 1 of 33 networks within PCORnet, the National Patient-Centered Clinical Research Network. ArthritisPower has a smartphone app that is used to collect patient-reported outcomes (PROs), said Nowell. The app has a number of features: Patients can input and track their own symptoms and treatments, run analytics on their own data, send reports to providers, connect to other patients, and learn about research opportunities (see Box 7-2). Nowell noted that the app has evolved as patients have provided feedback. For example, the app initially only allowed patients to enter information about arthritis-specific drugs, but patients wanted to be able to enter information about all of their medications.
BOX 7-2
Features of ArthritisPower Research Registry, with Smartphone Application and User-Friendly Interface.
The fundamental assumption that drives ArthritisPower as a platform for patient-centered research, said Nowell, is that “it enables patients to make a decision about their health care.” In order to enable patients to make good decisions, facilitating access to relevant evidence and choosing study designs that are best suited to generating that evidence are needed, he said. Most important, he said, is determining which study designs and data sources will “permit us to answer the research question and engage our partners.” The patient-driven research process is iterative, said Nowell, and requires ongoing consideration of patient needs and priorities, the patient experience in the study, transparency, and consent.
Nowell gave an example of a study in which ArthritisPower is currently involved. The CHOICE (Comparative Health Outcomes in Immune-mediated diseases Collaborative) study, said Nowell, is a PCORnet demonstration project that involves multiple networks, including ArthritisPower and other PPRNs. The study aims to evaluate the comparative risks for infection, heart attack, and stroke, and to evaluate the comparative clinical effectiveness of various medications using PROs. Evaluating the effectiveness of medication, said Nowell, is of utmost importance to patients and providers—“Patients and doctors want to know what treatment works best for whom and under what circumstances.” Providers and patients need to make challenging decisions about treatment options, he said, which can be difficult to do when there are limited data. While most approved drugs work reasonably well for most patients most of the time, it is generally unknown exactly how well or how quickly the treatments work depending on the characteristics and preferences of a particular patient; this kind of evidence can be generated through PROs. Ultimately, he said, a patient is the only one who can determine how well a treatment is working to improve his or her quality of life. Data from multiple individual patients can be turned into information that physicians and other patients can use to make decisions about treatments, said Nowell.
Nowell emphasized that engaging patients is more than a “one and done” conversation, and that patients need to be engaged in different ways at different times throughout the process. In addition, different types of patients with different perspectives need to be engaged, he said. Although some patients are very familiar with the terms and concepts of clinical research, other patients need help understanding how research works and how it can impact them. In response to Nowell's presentation, Graff suggested that the patient's perspective be somehow captured in the decision aid (see Figure 7-2).
CONTEXT OF THE DECISION
As with any decision about how to design a study, said Peter Stein, deputy director of the Office of New Drugs at FDA's Center for Drug Evaluation and Research, the first consideration is the context of the decision to be made: What is the research question? What is the intended use of the evidence that is generated? What is the level of evidence that is needed to make the decision? Stein said the answers to these questions can help guide decisions about patient treatment within a real-world study. For example, if the study is aimed at establishing efficacy for a new intervention, the researchers may want to tightly control patient treatment to find the most precise estimate of efficacy. However, if the study is about an intervention that is already known to be safe and efficacious, and the research question is broader, a study design that allows for flexibility in patient treatment may be more appropriate. For example, if the research question is how an intervention works in an expanded population in the real world, a trial in which patient treatment was tightly controlled might not generate generalizable evidence about real-world usage. Stein pointed to the study on oral versus injectable antipsychotics presented by Alphs, and said that the decisions about patient treatment in this case were based on what the study sought to discover. That is, the study was designed to look at patient outcomes on these two drugs in a real-world setting. Closely monitoring the group that was taking the oral antipsychotic—or ensuring compliance through directly observed therapy—would have defeated the purposes of the research. If research is being conducted for the purpose of a regulatory decision, said Stein, there are specific parameters of how the treatment and the comparative treatment were administered in order to make decisions regarding relative efficacy (see Box 7-3 about regulatory decision making).
BOX 7-3
Regulatory Decision Making Regarding Clinical Strategy as Discussed by Schneeweiss.
Another consideration when making decisions about patient treatment, said Stein, is the clinical context and the patients to be studied: What is the nature of the disease? Is it progressive or non-progressive? Are vulnerable populations involved and what is their susceptibility to harm? What are the current available treatments?
There is a trade-off, said Stein, between internal validity and generalizability. Depending on the specific research question, the intended use of the evidence, and safety concerns, a researcher might choose to emphasize one of these factors over the other.
OBLIGATION TO PATIENTS
Investigators, said Simon, have “dual interests.” Investigators have a duty to uphold the protocol of a trial to ensure that the research question is answered; meanwhile, they also have a duty to the safety and well-being of participants. These dual interests can sometimes conflict, and as Simon noted, “We want to make sure the duty to the participant always trumps the duty to the protocol.”
Safety Monitoring
Researchers have an obligation to ensure that participants in research are receiving treatment that is appropriate for their condition, and that the treatments are safe, said Alphs. When a relatively new treatment is under investigation, said Alphs, the obligation to patients is greater; there are going to be more restrictions on treatment and there is a need for substantial safety monitoring. By contrast, when a treatment has been used safely and effectively for many years, some of the requirements and safety monitoring can be relaxed. Simon explained two reasons to monitor for safety:
- 1.
For a new treatment, safety monitoring is essential to learn about the unknown adverse effects and to make inferences about how the treatment may or may not work in the real world; and
- 2.
For a treatment for which the adverse effects are already known, safety monitoring is about “doing the right thing” for the participants.
These two uses of safety monitoring, said Simon, require different procedures and study designs. Alphs noted that in the trials on treatment for suicidality, the side effects of agranulocytosis and weight gain were known. In this case, the research was not aimed primarily at discovering more about clozapine's (or olanzapine's) adverse effects, but there was still an obligation to the patients to monitor for potential safety events.
Inclusion of Patients and Real-World Experiences
Researchers' responsibility to patients, said Simon, includes an obligation to include in trials patients who “do not behave as we would always hope.” Patients who behave in understandably human ways need to be involved in trials so that results are generalizable to the real-world population, he said. Robert Califf added that patients with comorbidities or concomitant drugs are more likely to experience adverse events, but in traditional RCTs, these patients are excluded. In addition, the issue of how a treatment works for a real-life patient—with comorbidities and concomitant drugs—is what providers and patients “really want to know,” said Michael Horberg. If treatments are only tested on patients without these complexities, providers are left grasping for answers on how to treat their real-world patients, such as an older person with hypertension, diabetes, and advanced HIV. “There are real consequences” of limiting trials in this way, concluded Califf.
Acknowledging and welcoming human behavior is sometimes the only way to answer certain types of questions, said Horberg. For example, a study on HIV preexposure prophylaxis consistently advised sero-discordant couples to practice safer sex and to use condoms. However, during the course of the trial, there were more than 100 pregnancies, and the fact that these pregnancies happened without the transmission of HIV was one way that researchers could show that the therapy was effective, he said. Another example of necessary inclusion, said Simon, is seen in the case of the lithium trial for prevention of suicide. Katz had reported that patients who were taking diuretics or ACE inhibitors were initially excluded from the study. However, said Califf, the people most at risk for suicide may be exactly this population of older men with chronic health problems who are taking such medications. Excluding them from the trial not only reduces generalizability, but leaves this at-risk population without knowledge of what treatments might work.
At the third workshop, Alex John London, Clara L. West Professor of Ethics and Philosophy at Carnegie Mellon University, added a comment about how the view on vulnerable populations in research has changed over the years, through the lens of the Council for International Organizations of Medical Sciences (CIOMS) guidelines. The initial view was “protectionist,” he said; there were concerns about involving vulnerable populations in research. Now, CIOMS has revised its guidelines to a “justice-based approach,” which encourages the participation of vulnerable patients so that research can generate the information necessary to treat these populations. Simon concluded, “Unless we welcome in the way the real world works, we are never going to answer these questions.”
Standard of Care
Califf said that when researchers are designing trials, they have to decide on one of three options for the treatment for the control group: the best available care, usual care, or “whatever care is available where you are doing it.” If the research question is specifically about how a treatment works in a real-world setting, using a standard of care that is above the usual care for the control group may not provide an accurate answer, said Stein. However, he noted, there is always an obligation to ensure that the care delivered to the control arm is not substandard care. Simon said that although the effect of a treatment might be magnified if the control arm received substandard care, doing so would be unethical. Adrian Hernandez noted that research often seeks to understand whether one treatment is better than the standard of care, but that the standard of care can vary significantly by region, due to clinical practice and access issues. This variation can make it challenging to detect whether and to what extent a new treatment is better than the standard.
Simon asked Katz about balancing the needs of both the research question and the extremely vulnerable patients in the study on lithium for suicide prevention. Simon noted that the researchers had an obligation to stay engaged with the patients in the placebo arm, and to closely monitor for suicide and to hospitalize the patients if they suspected the possibility of a suicide attempt. However, at some point, the researchers might control and augment the care of participants so much that it would prevent the outcome from ever occurring and guarantee a null result, he said. Katz responded that there was extensive debate about this issue in defining outcomes for the study. The final approach to addressing this problem, at least in part, was to include hospital admissions with documentation that the reason for admission was specifically for prevention of suicidal behavior. This addresses the issue, but it “softens” the outcome. To minimize bias in deciding what events should be considered outcomes, the study uses a process of outcome adjudication, based on reviews of study documents and medical records by independent clinician-investigators blinded to treatment assignment. In general, the VA may be a unique site for this type of research because the baseline of care for suicide prevention at the VA is probably above the community standard. There is significant infrastructure in place, and there are requirements for flagging patients at risk of suicide and facilitating their access to care.
Marc Berger added that another option is to use observational data; these could allow researchers to study the outcomes of usual care without making fraught decisions about obligations to research subjects. Observational research can provide great insight into real-world effectiveness, he said. He gave the example of an oral drug and an inhaled corticosteroid used for childhood asthma. The oral drug was not as efficacious, but was equally effective in the real world because children did not want to use their inhalers at school, he said. Gaining this insight from an RCT would not be possible, but it is important information that can be used to make patient treatment decisions, he said. Evidence from observational data can be used, said Berger, along with evidence from RCTs or pragmatic clinical trials to build a “corpus of evidence” about a treatment. (See Chapter 9 for further discussion of observational trials.)
- How Tightly Should Investigators Attempt to Control or Restrict Treatment Qualit...How Tightly Should Investigators Attempt to Control or Restrict Treatment Quality in a Pragmatic or Real-World Trial? - Examining the Impact of Real-World Evidence on Medical Product Development
Your browsing activity is empty.
Activity recording is turned off.
See more...