4.1. Findings From the Literature
A total of two studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 3 and are described in section 4.2. There were no excluded studies, as all potentially relevant studies were included in the systematic review.

Figure 1
Flow Diagram for Inclusion and Exclusion of Studies.
Table 3
Details of Included Studies.
4.2. Included Studies
4.2.1. Description of Studies
The primary objective of PKU-016 was to evaluate the therapeutic effects of SAP compared with placebo on the symptoms of ADHD and global and executive function of patients with a confirmed diagnosis of PKU and symptoms of ADHD. Patients were required to have a mean decrease in Phe blood level of 20% or more from baseline to participate in the treatment periods of the trial. This was calculated as the difference between the mean of the baseline and screening values and the mean of the three lowest Phe blood levels during the first four weeks after initiating SAP.
PKU-016 (N = 206) was a double-blind, placebo-controlled, parallel-arm, phase 3b study consisting of two treatment periods of 13 weeks each: a double-blind randomized treatment period and an open-label treatment period, as illustrated in Figure 2. Eligible patients were stratified on the basis of presence of ADHD symptoms (yes or no), age (< 18 or ≥ 18 years), and use of ADHD medication (yes or no) and then randomized 1:1 by an interactive voice response system (IVRS) to receive SAP 20 mg/kg/day or placebo.
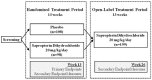
Figure 2
Study Design of Study PKU-016.
Patients who completed the 13-week randomized treatment period crossed over to the open-label treatment period where all patients were treated with SAP 20 mg/kg/day for an additional 13 weeks. A safety follow-up assessment was to be performed at 30 ± 7 days after completion of the open-label treatment period or after the early withdrawal visit.
For patients randomized to SAP in the randomized treatment period, assessments of Phe levels during the first four weeks of treatment (weeks 1 to 4) were used to determine the efficacy analysis population (Phe responders) based on the level of Phe reduction from baseline. For patients randomized to receive placebo in the randomized treatment period, assessments of Phe levels during the first four weeks of SAP treatment in the open-label treatment period (weeks 14 to 17) were used to determine the efficacy analysis population of Phe responders based on the level of Phe reduction from baseline.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬. Blood Phe levels were monitored throughout the study for safety and efficacy.
The primary objective of the SPARK trial was to evaluate the efficacy of SAP in increasing dietary Phe tolerance, as compared with dietary therapy alone, in infants and children with PKU who were aged four years or younger. In contrast to Study PKU-016, patients were required to be Phe responders (i.e., defined as a previous response to a BH4 test of 30% reduction from baseline in Phe levels) and to be in good control (i.e., Phe blood levels between 120 and 360 µmol/L) at study entry.
SPARK (n = 56) was an open-label, parallel-arm, phase 3b study with a duration of 26 weeks, as shown in Figure 3. Following completion of the 26-week treatment period, patients were able to enrol in an extension phase in which all patients were eligible to receive SAP plus a Phe-restricted diet. Patients were randomized 1:1 to either SAP plus a Phe-restrictive diet or a Phe-restricted diet alone. Randomization was stratified by age (i.e., < 12 months, 12 months to < 24 months, and 24 to < 48 months). According to the Clinical Study Report (CSR) for the SPARK study,11 the trial was open-label due to the inability to conceal Phe blood tests and results from the investigators, or parents or guardians of all enrolled patients, as the blood Phe levels and Phe response are essential in the care of infants and children with PKU.

Figure 3
Study Design of the Spark Study.
4.2.2. Populations
a) Inclusion and Exclusion Criteria
For PKU-016, the inclusion criteria stipulated that patients with a confirmed diagnosis of PKU were eligible to participate if they were at least eight years of age and willing to continue their current diet (i.e., typical diet for the three months prior to study entry) unchanged while participating in the trial. Key exclusion criteria were prior use of SAP within 16 weeks of randomization, initiation or adjustment of medication for the treatment of ADHD, depression, or anxiety within eight weeks of randomization, or medication that could interact with SAP (e.g., methotrexate, levodopa, or any phosphodiesterase type 5 [PDE-5] inhibitor) or a concurrent disease or condition that could interfere with study participation.
For the SPARK study, the inclusion criteria stated that infants and children younger than four years of age with confirmed clinical and biochemical PKU (including at least two previous Phe blood levels ≥ 400 µmol/L obtained on two separate occasions) and previous response to a BH4 test could be enrolled in the trial. Previous response to a BH4 test required that the BH4 dose was 20 mg/kg/day, the duration of the test was at least 24 hours, and that a 30% decrease in Phe blood levels occurred. Additional inclusion criteria were that the child have a defined level of dietary Phe tolerance, good adherence to dietary treatment, and maintenance of Phe blood levels within the therapeutic target range (120 to 360 µmol/L) over a four-month period prior to screening. Key exclusion criteria were previous use or exposure to SAP or any registered or unregistered preparation of BH4, previous diagnosis of BH4 deficiency, or medication or concurrent disease or condition that could affect the trial outcomes.
b) Baseline Characteristics
In PKU-016, the mean age of included patients was 23.1 ± 11.5 years, whereas in the SPARK study, the mean age of enrolled patients was 21.2 ± 12.1 months, as shown in Table 4. In both trials, general baseline demographic and disease characteristics were similar between treatment groups and the majority of patients (~94% to 98%) were white, reflective of the geographic location of the trials.
Table 4
Summary of Baseline Demographic and Disease Characteristics (Intention-to-Treat Population).
In PKU-016, more males (53.9%) enrolled in the study than females (46.1%), with the difference most apparent in the SAP + diet group (i.e., 58.2% males and 41.8% females), as shown in Table 4. There was also a higher proportion of patients in the age range of ≥ 18 years (n = 120) compared with < 18 years (n = 86); however, the relative proportions within the treatment groups were similar. Although the mean (SD) Phe blood levels at baseline were higher in the placebo + diet group compared with the SAP + diet group, this difference was not statistically significant or clinically relevant. Approximately one-third of patients in both treatment arms had previous ADHD symptoms and ▬% ▬% of patients were on ADHD medications at the time of study enrolment.
In PKU-016, the primary efficacy population consisted of 118 patients (49 females and 69 males) who had a Phe blood level reduction ≥ 20% from baseline within the first four weeks of SAP treatment (i.e., Phe responders). Of these, 57 (52.8%) patients were in the placebo + diet arm and 61 (62.2%) patients were in the SAP + diet arm. A total of 38 (32.2%) Phe responders (n = 19 in each treatment arm) had ADHD symptoms present at baseline. In these patients, the mean ADHD-RS/ASRS total score at baseline was 31.2 (placebo + diet) and 28.9 (SAP + diet). The contribution of the Inattention subscale score was greater than the Hyperactivity-Impulsivity subscale score at baseline. The mean Inattention subscale scores were 19.2 (placebo + diet) and 18.0 (SAP + diet), whereas the mean HyperactivityImpulsivity subscale scores were 12.0 (placebo + diet) and 10.9 (SAP + diet). Seven (12.3%) of the 57 Phe responders in the placebo arm and five (8.2%) of the 61 Phe responders in the SAP arm were on ADHD medications at baseline. The mean ± SD Phe blood level at baseline in the Phe responders was 789.5 ± 464.97 µmol/L (placebo + diet) and 680.2 ± 435.44 µmol/L (SAP + diet).
In the SPARK study, there were also more males than females enrolled, with the difference most apparent in the SAP + diet arm (i.e., 59.3% males and 40.7% females). Other demographic and disease characteristics were balanced and the majority of patients had either mild (▬%) or moderate (▬%) PKU disease severity.
4.2.3. Interventions
In PKU-016, patients received either SAP 100 mg tablets (▬ to the commercially available Kuvan 100 mg tablets except for the addition of a blue coating to ensure blinding) or matched placebo. All doses (20 mg/kg) were administered orally once daily after a meal. The IVRS calculated the daily dose of SAP or placebo by multiplying each patient’s total body weight (in kg) at screening by 20 mg/kg and then rounding up to the nearest 100 mg to accommodate a 100 mg unit dose. Patients were required to swallow the tablets intact.
Patients who were stable on concomitant medications for the treatment of ADHD, anxiety, depression, or other neuropsychiatric illness were to continue the medications unchanged unless medically warranted. The Investigator was able to prescribe additional medications during the trial, as long as the prescribed medication was not prohibited by the protocol. Patients with mental health and/or behavioural issues were allowed to continue psychotherapy or psychosocial counselling as part of their management plan. Patients were to remain stable on all such therapies unless medically warranted.
Patients were not to deviate from their typical dietary Phe intake consumed prior to entering the trial. The only exception was for those patients who demonstrated a significant decrease in blood Phe concentration of < 120 µmol/L. If the blood Phe level decreased to < 120 µmol/L, the Investigator was notified and the patient managed according to the medical judgment of the Investigator.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬).
In the SPARK study, patients initially received SAP 10 mg/kg/day plus a Phe-restricted diet or a Pherestricted diet alone. If after approximately four weeks, a patient’s Phe tolerance had not increased by more than 20% compared with baseline, the SAP dose could be increased in a single step to 20 mg/kg/day. To administer the appropriate dose of SAP, patients received the corresponding number of SAP tablets (i.e., based on the dose calculated according to the patient’s weight and rounded to the closest number of tablets), which were dissolved in the protocol-defined volume of water (20 to 120 mL depending upon body weight) and given to the child during breakfast. The solution was to be ingested within 15 to 20 minutes after dissolution.
For those patients randomized to a Phe-restricted diet alone during the 26-week study period who continued in the open-label extension, their starting SAP dose was 10 mg/kg/day with a maximum dose increase to 20 mg/kg/day permitted during the extension period. Patients who were randomized to the Phe-restricted diet arm received only the Phe-restricted diet and no matched placebo to SAP.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
4.2.4. Outcomes
In PKU-016, there were two primary end points:
- Symptoms of ADHD as measured by the change in the ADHD Rating Scale (i.e., ADHD-RS for patients eight to 17 years of age) or the ASRS (i.e., ASRS for patients ≥ 18 years of age) total score and the two separate components of the total score: the Inattention and Hyperactivity-Impulsivity subscale scores from baseline to week 13 in Phe responders with symptoms of ADHD.
- Global function as measured by the proportion of patients with CGI-I scale rating of 1 (very much improved) or 2 (much improved) at week 13 in Phe responders, with or without ADHD symptoms.
a) ADHD Rating Scale and ADHD Self-Report Scale
The ADHD-RS is an 18-item scale based on Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV) criteria that was developed and standardized for caregivers and educators of children to rate ADHD symptoms. The adult ASRS also includes 18 questions about the frequency of symptoms of adult ADHD. Details of both instruments are provided in Appendix 5. In PKU-016, the ADHD-RS was completed by the same parent or legal guardian for patients aged eight to 17 years, at every visit throughout the study to maintain consistency, whereas the ASRS was self-administered by patients aged 18 years and older. For both instruments, higher scores indicate worse severity of ADHD.
In PKU-016, the ADHD-RS and ASRS were combined to create a total score (ADHD-RS/ASRS) to reflect PKU-associated ADHD symptomatology in both children and adults. As the total possible scores for the ASRS and ADHD-RS are 72 and 54, respectively, due to the different rating scales, a correction factor or multiplier of 0.75 was applied to the ASRS score. Combining the scales was based on a clinical rationale that both scales include 18 questions, and each question assesses a specific DSM-IV—defined clinical symptom or behaviour. According to the CSR, the combination of both parent and self-rated scores allows analysis of all patients together, regardless of age, and increased the size of the analysis population.10
b) Clinical Global Impression
The Clinical Global Impression—Improvement (CGI-I) is a seven-point scale that requires the clinician to assess how much the patient’s illness has improved or worsened relative to a baseline state at the beginning of the intervention. It is rated as follows: 1, very much improved; 2, much improved; 3, minimally improved; 4, no change; 5, minimally worse; 6, much worse; or 7, very much worse. In PKU-016, the CGI rater was a qualified clinician who had access to all rating scales administered at the same visit (e.g., ADHD-RS or ASRS, HAM-A, HAM-D, BRIEF, etc.). Following a review of these rating instruments, patient interviews, and discussions with other health care practitioners who had seen the patient, the CGI rater assessed the CGI score. Raters were required to be trained and certified prior to assessing patients in the study and the same rater was to conduct the CGI at all visits for a subject.
The CGI-S and CGI-I are two separate, but related instruments that assess improvement over time following the intervention (CGI-I) and the severity of disease (CGI-S). The CGI-S is also a seven-point scale that requires the clinician to rate the severity of the patient’s mental illness at the time of assessment, relative to the clinician’s past experience with patients who have the same diagnosis. Considering total clinical experience, a patient is assessed on severity of mental illness at the time of rating as follows: 1, normal, not at all ill; 2, borderline ill; 3, mildly ill; 4, moderately ill; 5, markedly ill; 6, severely ill; or 7, among the most extremely ill.
The secondary end points in PKU-016 were:
- Change in HAM-D total score, HAM-A total score, BRIEF Global Executive Composite (GEC) T score, BRIEF Behavior Regulation Index (BRI) T score, BRIEF Metacognition Index (MI) T score, and CGI-S scores from baseline to week 13
- Proportion of patients with CGI-I scale of 1 or 2 at week 26
- Change in ADHD-RS/ASRS total score, ADHD-RS/ASRS Inattention subscale score, ADHD-RS/ASRS Hyperactivity-Impulsivity subscale score, HAM-D total score, HAM-A total score, BRIEF-GEC T score, BRIEF-BRI T score, BRIEF-MI T score, and CGI-S scores from week 13 to week 26
- Change in ADHD-RS/ASRS total score, ADHD-RS/ASRS Inattention subscale score, ADHD-RS/ASRS Hyperactivity-Impulsivity subscale score, HAM-D total score, HAM-A total score, BRIEF-GEC T score, BRIEF-BRI T score, BRIEF-MI T score, and CGI-S score from baseline to week 26.
The HAM-A and HAM-D scores in PKU-016 were primarily analyzed for the Phe responder study population, regardless of age. The rationale was that by combining the age populations, this allowed analysis in a meaningful population size as PKU is a rare disease.
c) Hamilton Anxiety and Depression Rating Scales
The HAM-A is a clinician-administered scale designed to assess anxiety symptoms not specific to any disorder. It has 14 items, each measuring specific anxiety symptom clusters (e.g., tension, insomnia, respiratory). Each item is given a five-point score, as follows: 0, absent; 1, mild; 2, moderate; 3, severe; or 4, incapacitating. The HAM-D is a 17-item depression rating scale and nine of the items are scored on a five-point scale, as follows: 0, absence of the depressive symptom being measured; 1, doubt concerning the presence of the symptom; 2, mild symptoms; 3, moderate symptoms; or 4, severe symptoms. The remaining eight items are scored on a three-point scale as follows: 0, absence; 1, doubt on the presence of the symptom; or 2, clear presence of symptoms. The clinicians administering the HAM-A and HAM-D were required to be trained and certified prior to assessing patients and the same clinician was to conduct the HAM-A and HAM-D at all visits. Detailed interview guides were provided to the qualified clinicians to assist with administering the HAM-D. Question 3 of the HAM-D is an assessment of suicidal impulses. Every site was required to have a defined process by which a study patient at risk for suicide would be referred for expert consultation in the event it was deemed appropriate.
d) Behaviour Rating Inventory of Executive Function Scale
The BRIEF—Adult Version (BRIEF-A) is a self-reported questionnaire for patients aged 18 years and older and the Parent Form of the BRIEF (BRIEF-Parent) is a parent- or legal guardian—reported questionnaire for patients younger than 18 years. If the patient was 17 at the start of the study, but turned 18 during the course of the study, the Parent Form of the BRIEF continued to be parent- or guardian-reported for the remainder of the study. A higher score on the BRIEF or index subscales indicates greater frequency of the behaviour.
Study site personnel were to remain available for questions during administration of the BRIEF. A quiet, private, and non-distracting environment was to be provided during the tests.
e) Phe Blood Levels
In PKU-016, Phe blood levels were measured throughout the study for safety monitoring and to determine the efficacy analysis population based on the level of Phe reduction after SAP treatment. Laboratory assessments of Phe levels by blood spot testing were to be performed at screening, at baseline (unless the screening and baseline visits were combined), and at weeks 4, 8, and 13 of the randomized treatment period and at weeks 17, 21, and 26 of the open-label treatment period. Weekly home Phe level testing by blood spot test was to be performed at weeks 1, 2, and 3 during the randomized treatment period and at weeks 14, 15, and 16 during the open-label period for the purpose of determining the primary efficacy population (Phe responders).
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
f) Phe Tolerance
In the SPARK study, the primary outcome was dietary Phe tolerance after 26 weeks (six months), which was defined as the daily amount of Phe (mg/kg/day) that could be ingested in the diet while maintaining average Phe blood levels within the specified therapeutic target range (defined as ≥ 120 to < 360 µmol/L). ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬. The assessment of Phe tolerance at the final visit (week 26) was used for the primary efficacy analysis, unless the patient was assessed to be not maintaining control at that visit. If this occurred, the last visit at which the patient was assessed to be maintaining overall control was used for the analysis.
Safety parameters included AEs, SAEs and deaths, vital signs, findings on physical examination and changes in clinical laboratory tests (e.g., chemistry, hematology, and urinalysis) in both included trials.
4.2.5. Statistical Analysis
In PKU-016, the planned enrolment was approximately 200 total patients with 100 patients in each treatment group in order to enrol approximately 50 patients who had a blood Phe level reduction of ≥ 20% after SAP treatment in each treatment group. At least 20 of these 50 patients in each treatment group were to have symptoms of ADHD with enrolment extended to include a minimum of 20 patients (out of the expected 50 patients with a ≥ 20% drop in Phe) in each treatment group with symptoms of ADHD. Based on a type I error rate of 0.05 (two-sided) and assuming a mean improvement in ADHD-RS/ASRS score of 13 in the SAP-treated patients, a mean improvement of 5 in the placebo-treated patients, and with a common SD of 9, a total of 20 patients in each treatment group who had a blood Phe level reduction after SAP treatment and also had symptoms of ADHD provided approximately 80% power to detect the estimated difference between SAP and placebo-treated patients.
This sample size calculation assumed that approximately 40% of the patients had a blood Phe level reduction after SAP treatment and have symptoms of ADHD, and there would be a minimum of 50 patients who had a blood Phe level reduction after SAP treatment enrolled in each treatment group. A sample size of 50 patients who had a blood Phe level reduction after SAP treatment in each group yielded approximately 80% power to detect a 30% difference in the proportion of patients with a CGI-I scale rating of 1 or 2 between the two treatment groups. The proportion of patients with a CGI-I scale rating of 1 or 2 was assumed to be 60% in the SAP-treated patients and 30% in the placebo-treated patients in this power calculation.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬. For the primary efficacy outcome (i.e., ADHD-RS or ASRS and separate Inattention and Hyperactivity-Impulsivity subscale scores), the changes from baseline to week 13 were analyzed by the analysis of covariance (ANCOVA) with treatment group, ADHD medication, age group at study entry (< 18 or ≥ 18 years), and ADHD symptoms as factors and baseline total score as a covariate. For the analysis of change in ADHDRS/ASRS total score, the total score was rescaled by multiplying 0.75 to reduce the heterogeneity between the variances of ADHD-RS total score and ASRS total score in order to make them comparable. The stratified Wilcoxon rank-sum test (van Elteren test) was performed as supportive analyses for the change from baseline in ADHD-RS/ASRS total scores across strata (age, ADHD symptom, and ADHD medication). The proportion of patients with a CGI-I scale rating of 1 or 2 in the two treatment groups was compared by the Cochran—Mantel—Haenszel (CMH) test adjusted for age group, ADHD symptom, and ADHD medication, as appropriate. The 95% CIs were provided for the proportion by treatment group. Statistical analysis of the secondary end points was similar to that used for the primary efficacy end point. For the within-treatment comparison, a pair-wise two-sided t test was used. There was no type I error rate adjustment for multiple tests for the secondary end points. For each treatment arm, additional exploratory analyses were performed on ADHD-RS/ASRS total score by subgroup to determine the possible interaction of subgroups with treatment using the ANCOVA model of the primary analysis. These subgroups were age (< 18 years versus ≥ 18 years), gender, whether on ADHD medication at baseline, and presence of ADHD symptoms at baseline.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
In the SPARK study, the calculation of sample size for the study period was based on the assumption that at the week 26 (month 6) visit, the dietary Phe tolerance for the SAP-treated group would be 75% greater than the dietary Phe tolerance for the group treated with dietary therapy alone. ▬ ▬ ▬ ▬ ▬▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ a sample size of 23 patients per group would yield 80% power for testing the null hypothesis of no treatment difference between the treatment groups.
▬ To compensate for possible dropouts, a total of 50 patients (25 per treatment group) were to be randomized to treatment. The rationale for the above estimates was based on the results of trial PKU-006 Part 2, which evaluated the effects of SAP therapy on Phe tolerance in children aged four to 12 years who were under adequate control of Phe blood levels.
The dietary Phe tolerance during the study period was to be analyzed using a repeated-measures ANCOVA on the observed records applying direct likelihood method. ▬ ▬ ▬▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
The adjusted means and their 95% CIs were to be derived for each time point within each treatment group. Additionally, the adjusted treatment difference (SAP plus Phe-restricted diet and Phe-restricted diet alone) at week 26, its two-sided 95% CI, and the associated P value were derived.
Changes from baseline in Phe blood levels, blood pressure, physical growth parameters, and age-related neuromotor developmental milestones and standardized neurodevelopmental results were analyzed using repeated-measures ANCOVA. ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
a) Analysis Populations
In PKU-016, the enrolled population or intention-to treat (ITT) population included all patients randomized to treatment. The safety population included all patients who were randomized to treatment, received any study drug (either SAP or placebo), and had any safety data collected after first dose.
The primary efficacy population comprised patients who had a blood Phe level reduction of ≥ 20% from baseline within four weeks of SAP treatment (Phe responders) and had symptoms of ADHD at screening. For patients randomized to SAP treatment during the first 13 weeks, Phe levels obtained weekly during the first four weeks of treatment were used to determine the primary efficacy population based on the level of Phe level reduction after SAP treatment. For patients randomized to placebo who completed the randomized treatment period, Phe levels obtained weekly during the first four weeks of SAP treatment in the open-label treatment period (weeks 14 to 17) were used to determine the primary efficacy population based on the level of Phe level reduction from baseline after SAP treatment.
A 20% blood Phe level decrease was defined as the difference between 1) the mean of the baseline and screening values, and 2) the mean of three out of the four lowest values taken during the first four weeks after starting SAP. The lowest three values were chosen to best represent clinically stable conditions. For example, a markedly elevated Phe level observed during an acute febrile illness would thus be disregarded. Similarly, if the patient’s Phe level decreased abruptly to below the locally acceptable safety threshold and supplemental dietary Phe was administered, a value obtained immediately following the administration of supplemental Phe would similarly be discarded.
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
In the SPARK study, the analysis populations were defined as follows:
ITT population: consisted of all patients who were randomized at the start of the study period and analyzed according to the group allocated.
Per-protocol (PP) population: consisted of those patients from the ITT population without a major protocol deviation.
Safety population: consisted of all patients who had some safety assessment data available (at least one visit in vital signs, AE, or laboratory results) in the study period and:
- who received at least one dose of SAP in the study period, or
- who were randomized to the Phe-restricted diet alone.
▬ ▬ ▬ ▬ ▬ ▬
▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬ ▬
4.3. Patient Disposition
Patient disposition data are provided in Table 5 in both the PKU-016 and SPARK trials; the proportion of patients who discontinued in either treatment arm did not exceed approximately 10%. The majority of patients withdrew in PKU-016 due to loss to follow-up during the open-label treatment phase or withdrawal by the patient in the SPARK study. In the PKU-specific analysis populations for Study PKU-016, more patients in the SAP + diet arm (62%) compared with the placebo + diet arm (53%) were considered to be Phe responders (i.e., based on a 20% reduction in Phe levels from baseline following a SAP challenge). It should be noted that the Phe-responder population for the placebo + diet arm includes patients initially randomized to placebo during the randomized treatment phase who were then crossed over to SAP during the open-label treatment phase. Of the patients who were Phe responders, only about 18% to 19% also had ADHD symptoms at baseline.
Table 5
Patient Disposition.
4.4. Exposure to Study Treatments
In PKU-016, exposure to study drug in the ITT population (safety population) was similar between patients in each treatment group and each period, as detailed in Table 6. In addition, the mean daily dose was also similar between treatment arms and study periods. The mean ± SD daily dose on a mg/kg basis adjusted by planned exposure days was 18.8 ± 3.7 mg/kg/day for patients in the placebo group and 19.2 ± 2.5 mg/kg/day for patients in the SAP treatment group.
Table 6
Exposure to Study Drug (Intention-to-Treat Population).
4.5. Critical Appraisal
4.5.1. Internal Validity
The methods used for randomization and allocation concealment in PKU-016 were appropriate (i.e., IVRS and used of matched placebo tablets); however, in the SPARK study, the lack of a matched placebo to SAP may have introduced performance bias resulting from potential confounding factors associated with the administration of SAP.
A large placebo effect was observed in both the PKU-016 and the SPARK study for various outcomes. It is possible that more frequent clinic visits may have increased patient and provider attention to dietary treatment, which, in turn, contributed to improved Phe blood levels in both treatment groups. It is not uncommon for a large placebo effect to be observed in the evaluation of mental health outcomes (e.g., ADHD symptoms and executive functioning were assessed using parent-reported and self-reported measures, which could differ from assessments made by a health care professional). In addition, selfreporting of ADHD symptoms over time could have been affected by the patient’s improved ability to focus and monitor themselves over the course of treatment.9 It may have been advisable to also have an independent rater provide collateral information in this regard for self-reported outcomes.9 In addition, patients with mental health and/or behavioural issues were allowed to continue psychotherapy or psychosocial counselling as part of their management plan; therefore, such interventions could have contributed to the observed results.
Baseline demographic and disease characteristics were generally balanced between treatment arms in each trial, with the possible exception of more males in the SAP treatment arms of both trials and a greater proportion of older patients (aged ≥ 18 years) compared with < 18 years of age in PKU-016. It was not possible to compare patient populations between the two trials due to study design differences such as the age of enrolled patients (i.e., mean age of enrolled patients in PKU-016 was approximately 23 years compared with 21 months in the SPARK study), which precluded making comparisons.
The use of a population after the conclusion of the actual primary end point (i.e., week 13) to select the control groups may have introduced bias, because the responses of the treatment and control group are being determined at two different time points. This is based on the possibility that defining one group of responders during the double-blind phase and the other group during the open-label phase could potentially result in there being differences between the two subsets of patients, such as differences related to patient behaviour (e.g., adherence to diet).
In general, the statistical methods and analyses appeared to be appropriate; however, there was no control for multiplicity in the analyses of the two primary end points in PKU-016 or for the analyses of the secondary end points in both trials. Therefore, the risk of type 1 error is very high.
The small patient numbers in the primary efficacy populations (e.g., 19 patients in each treatment arm for the primary efficacy analysis population of Phe responders with ADHD symptoms at baseline in PKU-016) are expected to have had only limited statistical power, although based on the manufacturer’s sample size calculations detailed in section 4.2.5, PKU-016 should have had approximately 80% power to detect treatment differences.
In PKU-016, following the randomized treatment period, patients who were initially randomized to placebo + diet crossed over to open-label SAP + diet after week 13 in the open-label treatment period. Thus, comparisons of results at week 26 are between patients who were originally randomized to SAP + diet in the randomized treatment period and continued on this regimen in the open-label treatment period with patients who crossed over to open-label SAP + diet, thus complicating the interpretation of the results after week 13. Furthermore, because the analysis population is not the population that was randomized, the benefits of randomization are lost, thereby subjecting the results to the limitations commonly observed with non-randomized studies. For instance, any differences between the treatment and control groups may be driven by potential differences in patient characteristics as the benefit of randomization has been removed. Furthermore, interpreting patient-reported outcomes for the open-label phase is particularly problematic, as these outcomes are more susceptible to bias in open-label studies.
It does not appear that any of the outcomes used to assess ADHD symptoms or neurocognitive or psychological effects of treatment in the trials (with the exception of the ADHD-RS/ASRS Inattention subscale score) have been validated in patients with PKU or the minimal clinically important differences (MCIDs) for these outcomes established, which complicates interpretation of the results. Furthermore, for the primary end point analysis in the PKU-016 trial, the ADHD-RS (parent-reported in patients younger than 18 years) and ASRS (self-reported in patients aged 18 years and older) were combined to derive an overall score and the ASRS was corrected by multiplying by 0.75. The appropriateness of doing so is questionable, especially because it is a new approach that has not been used previously in PKU or in other disease areas.9
4.5.2. External Validity
The PKU-016 trial included clinical sites in Canada, whereas the SPARK study was conducted primarily in Europe. Nonetheless, based on the inclusion criteria and given the rare nature of PAH deficiency PKU, it is expected that the patient populations in the included trials would be similar to the target treatment population in Canada.
The small number of patients in the primary efficacy populations in the trials affects the generalizability of results to a broader patient population and precludes identification of a subpopulation of patients who may benefit from SAP treatment. In PKU-016, the objective of the trial was to assess the impact of SAP + diet on ADHD symptoms in patients with PKU who are Phe responders; however, only approximately 30% of all enrolled patients had ADHD symptoms at baseline and only 8% to 9% were on ADHD medication at study entry.
Phe responders were defined differently in the two trials (i.e., 20% versus 30% reduction from baseline in Phe levels following SAP challenge, in PKU-016 and the SPARK study, respectively). In addition, patients in the SPARK study were required to be within the target Phe range of 120 to 360 µmol/L at study entry as opposed to the patients entering PKU-016 who were not well controlled by diet alone. According to the clinical expert consulted on this review, the per cent reduction thresholds for Phe blood levels are arbitrary, so the clinical significance of the threshold difference is uncertain.
The duration of the trials (26 weeks) is insufficient to appropriately assess the efficacy and safety of a medication intended for chronic, potentially lifelong administration.
4.6. Efficacy
Only those efficacy outcomes identified in the review protocol are reported below (section 3.2, Table 2). See Appendix 4 for detailed efficacy data.
4.6.1. Change in Phe Levels
In Study PKU-016, in the population of Phe responders, the mean (SD) Phe blood levels at baseline were 680,2 (435.44) |imol/L in the SAP + diet arm and 789.5 (464.97) μmol/L in the placebo + diet arm (Table 17). At week 13, levels in the SAP + diet arm decreased by approximately 30% from baseline to ▬μmol/L and remained largely unchanged in the placebo + diet arm (i.e., ▬(μmol/L). After week 13, levels remained relatively stable in the SAP + diet arm, but decreased in the placebo + diet arm as these patients crossed over to receive open-label SAP + diet during the open-label treatment period. At week 26, mean (SD) Phe levels were similar between the two groups: ▬) |μmol/L in ▬ μmol/L in the placebo + diet arm. No statistical comparisons were the SAP + diet arm and | conducted between the treatment groups.
In contrast to Study PKU-016, patients enrolled in the SPARK study were required to be within the Phe blood level target range of 120 to 360 µmol/L at study entry. The mean (SD) baseline values were ▬ (▬) µmol/L in the SAP + diet arm and ▬ (▬) µmol/L in the diet alone arm (Table 18). Phe blood levels remained relatively constant in both treatment arms throughout the duration of the study. At week 26, the mean (SE) change from baseline was —10.1 (▬) µmol/L in the SAP + diet arm and 23.1 (▬) µmol/L in the diet alone arm. The LS or adjusted mean difference between the treatment arms was not statistically significant (i.e., —33.2 [95% CI, —94.8 to 28.4]; P = 0.290). In the SPARK study, nine patients (33.3%) in the SAP + diet arm maintained Phe blood levels in the target range (120 to 360 µmol/L) throughout the study compared with three patients (10.3%) in the diet alone arm (Table 19).
4.6.2. Neuropsychiatric and Neurocognitive Effects
The effects of SAP treatment on neuropsychiatric and neurocognitive effects using various different instruments were investigated in Study PKU-016, whereas in the SPARK study, only effects of treatment on neuromotor developmental milestones were reported. The primary objective of PKU-016 was to evaluate the effect of SAP treatment on ADHD symptoms in patients who were Phe responders with ADHD symptoms at baseline (i.e., n = 19 patients in each treatment arm). The proportion of patients in the Phe responder population with CGI-I ratings of 1 (very much improved) or 2 (much improved) was the second primary end point in PKU-016. Other instruments included as secondary end points were the CGI-S, HAM-A, HAM-D, and the GEC, MI, and BRI index T scores of the BRIEF rating scale. In the SPARK study, neuromotor status assessment was performed using the standardized Bayley-III Scales of Infant and Toddler Development for patients younger than 3.5 years of age and the Wechsler Preschool and Primary Scale of Intelligence for patients between 3.5 and 4 years of age.
The first primary end point in PKU-016 was the change from baseline to week 13 in the ADHD-RS/ASRS total score and a higher score on either rating scale indicates greater severity of ADHD symptoms (Table 8). In both treatment groups, the mean (SE) ADHD-RS/ASRS total score decreased from baseline to week 13 (i.e., —9.1 [2.2] in the SAP + diet arm and —4.9 [2.0] in the placebo + diet arm), suggesting improvement, although the MCID is unknown. In each arm, the change from baseline to week 13 was statistically significant (▬), although the difference between arms was not (i.e., —4.2 [95% CI, —8.9 to 0.6]; P = 0.085). At week 26, the difference between arms was also not statistically significantly different (▬).
For the ADHD-RS/ASRS subscale score of Inattention, in both treatment arms, the mean (SE) subscale score decreased from baseline to week 13 (i.e., —5.9 [1.4] in the SAP + diet arm and —2.5 [1.3] in the placebo + diet arm) (Table 9). In each arm, the change from baseline to week 13 was statistically significant (▬) and the difference between arms was also statistically significant (i.e., —3.4 [95% CI, —6.6 to —0.2]; P = 0.036), although the MCID is unknown. At week 26, the difference between arms was no longer statistically significantly different (▬).
For the ADHD-RS/ASRS subscale score of Hyperactivity-Impulsivity, in both treatment arms, the mean (SE) subscale score decreased from baseline to week 13 (i.e., —3.3 [1.1] in the SAP + diet arm and —2.3 [1.0] in the placebo + diet arm) (Table 10). The change from baseline to week 13, however, was statistically significant only in the SAP + diet arm (▬), but not in the placebo + diet arm (▬). The difference between arms was also not statistically significant at either week 13 (i.e., —1.0 [95% CI, —3.4 to 1.4]; P = 0.396) or week 26 (▬).
The second primary end point in PKU-016 was the proportion of patients with a rating of 1 or 2 in the CGI-I at week 13 in the population of Phe responders (Table 11). The proportion of patients with this outcome was 26.3% in the placebo + diet group and 21.7% in the SAP + diet group at week 13 and the difference was not statistically significantly different (i.e., 0.87 [95% CI, 0.46 to 1.64]; P = 0.670). At week 26, however, the proportion of patients with this outcome in the placebo + diet arm (which included patients who crossed over from placebo to open-label SAP) was ▬%compared to ▬%in the SAP + diet arm. The treatment difference was statistically significant in favour of the placebo + diet arm (▬)For the secondary outcome of CGI-S (where lower scores indicate improvement), the mean [SE] reduction in scores from baseline to week 13 in both treatment arms was statistically significant (i.e., -0.6 [0.2] in the SAP + diet arm and -0.5 [▬] in the placebo + diet arm; (▬) J); however, the difference between arms was not statistically significantly different at week 13 or week 26 (Table 12).
In PKU-016, the mean change (SE) from baseline to week 13 in the HAM-A in Phe responders was —3.2 (▬) in the SAP + diet arm and —3.6 (▬) in the placebo + diet arm, both of which were statistically significant (▬) (Table 13). A decline in the HAM-A or HAM-D score represents an improvement in symptoms. The difference between treatment arms, however, was not statistically significant (i.e., 0.4 [95% CI, —1.5 to 2.3]; P = 0.669). Similarly, the treatment difference at week 26 was also not statistically significant (i.e., —0.5 [95% CI, —2.4 to 1.4); P = 0.590). A similar pattern was observed for the HAM-D results in Phe responders. The mean change (SE) from baseline to week 13 in the HAM-D was —2.1 (▬) in the SAP + diet arm and —2.5 (▬) in the placebo + diet arm, both of which were statistically significant (▬) (Table 14). The difference between treatment arms, however, was not statistically significant (i.e., 0.4 [95% CI, —1.1 to 1.9]; P = 0.588). Similarly, the treatment difference at week 26 was also not statistically significant (▬).
In Study PKU-016, in Phe responders younger than 18 years of age, the BRIEF-Parent was used, which was completed by parents (Table 15). In those aged 18 years and older, the BRIEF-A was used, which was self-administered (Table 16). For each BRIEF assessment, results are reported separately for the three index scales (GEC, MI, and BRI). For the BRIEF-Parent results (i.e., patients younger than 18 years), the differences between treatments at week 13 were statistically significantly different for the GEC (i.e., —4.1 [95% CI, —7.9 to —0.3]; P = 0.034) and MI (i.e., —4.4 [95% CI, —8.5 to —0.2]; P = 0.038, but not for the BRI index scale (i.e., —3.4 [95% CI, —6.8 to 0.0]; P = 0.053), although the MCID is unknown. For the BRIEF-A results (i.e., patients ≥ 18 years), there were no statistically significant differences between groups for any of the three index scales, GEC, MI, or BRI.
In the SPARK study, the only measure of neurodysfunction that was reported was the proportion of patients who were classified as either normal or abnormal with regard to neuromotor developmental milestones in four areas of assessment: fine motor, gross motor, language, and personal-social (Table 23). In all four areas, the majority of children were classified as normal and there were no statistically significant differences found between the SAP + diet and diet alone arm for any of the areas of assessment.
4.6.3. Growth Parameters
In the SPARK study, treatment differences in the change from baseline to week 26 were investigated for four different growth parameters: height, weight, BMI, and head circumference, all measured as standard deviation scores (SDS), as detailed in Table 22. For all four growth parameters, there were no statistically significant differences observed between the SAP + diet and diet alone arm.
4.6.4. Proportion of Responders
The proportion of Phe responders was not an outcome in either of the included trials; however, in PKU-016, those patients who demonstrated a 20% reduction from baseline in Phe blood levels following treatment with SAP 20 mg/kg/day for up to one month comprised the Phe responder population. This was the primary efficacy population for the co-primary end point of CGI-I, whereas those patients in the Phe responder population who also had ADHD symptoms at baseline comprised the primary efficacy population for the other co-primary end point of ADHD-RS/ASRS. Of the total patients randomized in PKU-016 (n = 206), 118 patients (57.3%) were Phe responders. Of these, 61 patients (62.2%) in the SAP + diet arm and 57 patients (52.8%) in the placebo + diet arm were Phe responders. In the SPARK study, this outcome was not applicable as patients entering the trial were required to be BH4 responders.
4.6.5. Change in Phe Tolerance
The primary outcome in the SPARK study was dietary Phe tolerance, which was defined as the prescribed amount of dietary Phe (mg/kg/day) tolerated while maintaining mean Phe blood levels within the target range of 120 to 360 µmol/L. At baseline, the mean (SE) Phe tolerance was 35.5 (3.8) mg/kg/day in the SAP + diet arm compared with 42.8 (4.1) mg/kg/day in the diet alone arm (Table 20). Phe tolerance steadily increased in the SAP + diet arm as opposed to the diet alone arm over the course of the trial. At week 26, the mean (SE) Phe tolerance was 80.6 (4.2) mg/kg/day in the SAP + diet arm and 50.1 (4.3) mg/kg/day in the diet alone arm. The difference between treatment arms at week 26 was statistically significant (i.e., 30.5 [95% CI, 18.7 to 42.3]; P < 0.001). Mean change in dietary Phe tolerance from baseline to week 26 was also statistically significant within each treatment arm (i.e., 36.9 [95% CI, 26.1 to 47.7]; P < 0.001 in the SAP + diet arm and 13.1 [95% CI, 5.4 to 20.9]; P = 0.002 in the diet alone arm) (Table 21).
There were no data reported in the included trials for the following outcomes identified in the review protocol: quality of life, health care resource utilization, and nutritional status.
4.7. Harms
Only those harms identified in the review protocol are reported below (see section 2.2.1, Protocol). See Appendix 4 for detailed harms data.
4.7.1. Adverse Events
The majority of patients in both trials (≥ 75%), regardless of treatment arm, experienced treatment-emergent AEs, as detailed in Table 7. In PKU-016, the frequency of AEs was similar in the randomized treatment arm and the open-label treatment arm. Overall, the most frequent AEs were headache, nasopharyngitis, and vomiting in Study PKU-016 and pyrexia, cough, decreased amino acid level, and vomiting in the SPARK study.
Table 7
Harms (Safety Population).
4.7.2. Serious Adverse Events
There were few patients with SAEs in either trial, as shown in Table 7. No one type of SAE occurred more frequently than in one patient in either of the trials.
4.7.3. Withdrawals Due to Adverse Events
There was only one patient with WDAE in Study PKU-016 who withdrew due to increased heart rate.
4.7.4. Mortality
There were no deaths reported in either trial.
Publication Details
Copyright
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Except where otherwise noted, this work is distributed under the terms of a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence (CC BY-NC-ND), a copy of which is available at http://creativecommons.org/licenses/by-nc-nd/4.0/
Publisher
Canadian Agency for Drugs and Technologies in Health, Ottawa (ON)
NLM Citation
Clinical Review Report: Sapropterin dihydrochloride (Kuvan) [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2017 Sep. 4, Results.


