7.1. Introduction
Ca2+ signals control a vast array of cellular processes and are mediated by the concerted effort of a spectrum of Ca2+ channels, transporters, and pumps present in the plasma membrane (PM) and endoplasmic reticulum (ER) membrane [1,2]. In nonexcitable cell types, store-operated channels (SOCs) are the major means through which extracellular Ca2+ enters cells to generate Ca2+ signals. The two major components of SOCs, STIM1 and Orai1, were identified a decade ago [3–8], and extensive studies have focused on the mechanisms of how STIM1 becomes activated in response to store depletion, how Orai1 subunits assemble to form the channel, and how the STIM1 molecule interacts with Orai1 to achieve channel gating [1,9–13] (discussed in Chapters 2 and 3). The physical interaction between STIM1 and Orai1 has been one of the most important parameters in order to understand the stoichiometry and gating mechanism of the STIM1/Orai1 complex. The physical interaction between STIM and Orai1 has been studied extensively using Förster (fluorescence) resonance energy transfer (FRET) imaging technology [13–16]. FRET allows the visualization and quantification of macromolecular interactions in living cells by measuring light energy transfer between closely associated fluorescently tagged proteins [17]. Since STIM1 and Orai1 are in different membranes and interact at discrete ER-PM junctions, FRET is a highly effective means for assessing this interaction. FRET occurs in a short range (5–10 nm) across which energy is transferred from an excited donor to acceptor fluorophore [18]. The efficiency of energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor fluorophores; hence, FRET measurements give extremely sensitive information on the distance separating the pair. The mechanism and uses of FRET are well described in other reviews [19–23].
Despite intense study, the molecular nature of the coupling interaction between the activated STIM1 protein and the Orai1 channel remains elusive. Our approach to studying this interaction is to use a fragment of the STIM1 protein that itself is able to mediate full activation of the Orai1 channels. This 100-amino acid fragment is known as SOAR (STIM-Orai-activating region) [24,25] (see Chapter 2). In a previous report, Shen et al. [26] purified and crystallized this fragment from STIM1 and revealed that it can exist as a dimer. Indeed, the SOAR fragment appears to be an important “core” structure within the STIM1 protein contributing to dimerization of the whole STIM1 protein [12]. We have been able to express the SOAR protein as a concatenated dimer construct and therefore genetically manipulate the exact dimeric composition of SOAR expressed in cells [13]. We revealed that a single point mutation (F394H) in the Orai1 binding site of the SOAR fragment from STIM1 can completely prevent STIM1 binding to and activation of Orai1 channels [16]. Importantly, since the SOAR dimer contains two of these sites, we can modify either one or both of these sites to study the requirements for interaction with the Orai1 channel. Using a set of concatemer-dimers of SOAR containing one or two F394H mutations, we are able to study how the SOAR dimer interacts with Orai1 and whether each SOAR unit within a dimer is equivalent. The aim of this chapter is to provide a detailed protocol to assess the STIM/Orai interaction by FRET. We describe how to ensure the FRET assay is reliable and consistent and how to analyze and how to interpret the FRET data.
7.2. Strategy for Quantitative FRET Measurement
We first introduce the term “E-FRET,” which is a fundamental “instrument-independent” measurement of FRET, representing the “FRET efficiency” or what percentage of donor emission is quenched by the acceptor [27]. Any microscope that can simultaneously collect 3-channel images is usable for such FRET experiments. Some imaging software programs have already been embedded with E-FRET formulation capability. It is also possible to use either MATLAB® or Excel to manually calculate the E-FRET values. The current chapter will focus on E-FRET analysis methodology since it is the most widely used in our field and, importantly, the values of E-FRET are directly comparable between different labs and imaging systems [27]. In this procedure, 3-channel image collection is undertaken for FRET measurements using a traditional epifluorescence microscope. It is important to correct for cross-channel “bleed-through” of fluorescence. The following formula is used to calculate the corrected FRET (FRETC):
- IDD, IAA, and IDA represent the intensity of the background-subtracted CFP, YFP, and FRET images, respectively (these are defined further later)
- FRETC represents the corrected energy transfer
- a represents the measured coefficient of the acceptor (YFP) bleed-through the FRET filter cube
- d is the measured coefficient of the donor (CFP) bleed-through the FRET filter cube

Figure 7.1
FRET measurements and the relationship with donor and acceptor expression levels. (a and b) Assessing the interaction between protein X and protein Y by FRET. X is labeled with CFP (donor) and Y is labeled with YFP (acceptor). The ratio of the expression (more...)
7.3. FRET Measurements to Quantitate SOAR-Orai1 Interactions
7.3.1. Generation of Stable Cell Lines
We recently constructed a number of concatemer-dimers of SOAR as described in Figure 7.2a in order to study the STIM1-Orai1 coupling interaction. As described in our recent study [13], we hypothesize that the wild-type SOAR homodimer (YFP-S-S) has two Orai1 binding sites, while the F394H-mutated SOAR heterodimers (YFP-SH-S or YFP-S-SH) have only one Orai1 binding site. When both SOAR monomers are mutated to F394H, there is no interaction with Orai1. One important aspect of these studies was to assess FRET between each concatemer and Orai1 channels. As described in Figure 7.1c and d, it is important to ensure an excess of donor is expressed in cells (e.g., Orai1-CFP or a CFP-tagged STIM1-binding fragment of Orai1, as described later). In addition, it is necessary that cell lines are generated that stably express the donor proteins to ensure accuracy and reproducibility of FRET measurements. In contrast, YFP-tagged acceptor proteins (SOAR or SOAR concatemers) are transiently expressed at relatively low levels.

Figure 7.2
Schematic diagram of the four SOAR concatemers. YFP-S-S is the homodimer concatemer of wild-type SOAR concatemer-dimer, YFP-SH-S is the YFP-tagged heterodimer concatemer of SOAR containing one SOAR-F394H unit and one wild-type SOAR unit, YFP-S-SH is the (more...)
The process of making stable cells usually takes considerable time (1–3 months or longer). We have successfully expedited this process by modifying the protocol described earlier [28]. Our protocol is fast and inexpensive and can be completed in 1 month or less. There are several key parameters for the successful generation of stable cell lines: (1) ensuring cells are optimally healthy with regular medium changes prior to the initial transfection and (2) prior to cloning of cells, ensuring to freeze stocks of multiclonal cells and any clones with good expression levels as soon as possible. Large numbers of cells per vial are not necessary for frozen stocks, but backups are often crucial.
7.3.1.1. Electroporation for Initial Transfection
Although there are many methods to introduce DNA into mammalian cells, electroporation and application of cationic lipids are the two most widely used methods for cell lines. They both have very high transfection efficiency in human embryonic kidney (HEK 293) cells. Electroporation is undertaken at 180 V, 25 ms in 4 mm cuvettes (Molecular BioProducts) using the Bio-Rad Gene Pulser Xcell system in 500 μL OPTI-MEM medium. Since the number of cells used to make stable cell lines varies based on the transfection efficiency, it is important to gauge the transfection efficiency for each plasmid prior to initiating stable cell line generation. The quantity of cells needed to initiate stable cell line generation is important. For G418 (100 μg/mL) selection of constructs, we use about 10% of cells from a single 10 cm dish (about 80% confluence) or approximately 150,000 cells. G418 usually takes 1 or 2 weeks to kill the nonexpressing cells. For puromycin (2 μg/mL) selection of cells, about 30% of cells from a single dish (∼450,000 cells) should be used since the transfection efficiency can be lower and puromycin kills untransfected cells in 1 or 2 days.
7.3.1.2. Selection of Stably Expressing Cells
Two days after transfection, the cell culture medium is changed to include selection reagents to kill off untransfected cells. For G418 selection, cells are detached by pipetting up and down after a medium change, which helps kill nonexpressing cells. Thereafter, cell condition is assessed every 2 days with medium changes to remove dead cells if necessary. After keeping cells in selection medium for 7 days, there should be many clones visible in the cell dish. After clones begin to appear, cells are trypsinized and reattached to the same dish to avoid any loss of cells. Minimal trypsinization is important—treatment for too long kills the cells very rapidly. The following day, cell status should be checked. If cells are 5%–10% confluent and there are few floating dead cells, then single-cell clones can be isolated.
7.3.1.3. Selection of Single Cells for Cloning Using the Patch-Clamp Rig
There are several strategies for single-cell screening. Fluorescence-activated cell sorting is highly efficient but expensive and may produce false-positive cell lines, for example, cells that have fluorescence but the localization of fluorescent-tagged protein is wrong. Limiting dilution for obtaining single-cell clones is cheap but is an inefficient and also time-consuming process. We have developed an easy and inexpensive strategy to obtain single-cell clones using the patch-clamp rig, as described in the following protocol:
- After 7–10 days of culture in medium containing selection reagent (G418 or puromycin), cells are treated with trypsin and resuspended in fresh selection medium at a low density of 5000 cells/mL. The cells are plated on 12 mm glass coverslips (Warner) and allowed to attach for about 40 min.
- Preparation of glass electrodes for single-cell collection. Thin wall glass capillaries (World Precision Instruments, Cat#: TW150F-4) are pulled using a P-97 puller (Sutter). The diameter of the glass pipette tip is about 60–80 μm. Polishing is not needed for this purpose. About 30 glass electrodes are usually sufficient for one cell line.
- The glass coverslip containing the cells of interest is placed in the patch-clamp chamber on the microscope taking care not to displace the cells. The MPC-200 micromanipulator system (Sutter) is used to move the headstage holding the glass pipette. It is important to set up the working position and home position for the headstage so that the glass electrode can be withdrawn quickly to avoid collecting any other cells in addition to the cell selected for removal.
- The key to the effectiveness of our method is to select single cells that not only have positive fluorescence (e.g., CFP) but also express the tagged protein at the correct location. For example, expression of Orai1-CFP can be determined by observing whether the fluorescence is confined to the PM. Make sure you have the correct filter cube in place (e.g., for CFP). It is important that when you select a single cell, there are no nonfluorescent cells nearby—thus, it is necessary to look at a bright-field view prior to harvesting the cell of interest.
- Center the cell of interest in the field. Carefully move the electrode close to the targeted cell without any positive or negative pressure. With one hand ready on the “home” position button, the other hand is holding the syringe to give a little negative pressure to suck the cell of interest into the glass electrode. Once the cell enters the electrode, push the home button immediately.
- Unmount the glass pipette by hand and using a 200 μL tip on an Eppendorf pipetter, apply 100 μL of culture medium to the electrode to flush out the single cell into one well of a 24-well plate already containing 1 ml of culture medium. Mount a new glass electrode on the holder and put it into ready state by pressing working position button.
- Repeat steps 4–6 to select at least 24 cells.
7.3.1.4. Checking Fluorescence and Functional Tests of Individual Clones
Five days after cells are seeded into 24-well plates, wells are examined under the microscope, and positive wells labeled. Once clones occupy almost one entire field of view with a 20x objective, they are ready for expanding into 6-well plates. Cells should not become confluent (usually they reach confluence in 4 days). Contaminated wells should be emptied by aspiration. After confluence is reached, aspirate the old medium out, add 1.5 mL fresh medium, detach the cells by pipetting up and down to separate cells (9–12 times is sufficient; avoid bubble formation), and transfer all the cells into a 10 cm dish containing 10 mL medium. Let the cells grow until they reach confluence, trypsinize and resuspend cells, and utilize some for functional tests (e.g., Fura-2 Ca2+ imaging or electrophysiology). If three or four clones test positive for desired Ca2+ signaling and/or patch-clamp results, these clones can be grown further and the rest discarded. For selected clones, cells are grown in 10 cm dishes and frozen in approximately 10 vials.
7.3.2. Calibration of FRET Imaging System
We use a Leica DMI6000B automatic turret-equipped microscope and a 40x oil objective (N.A.1.35; Leica) with the following filter cubes: CFP (438Ex/483Em), YFP (500Ex/542Em), and FRET (438Ex/542Em). We collect images using a Hamamatsu ORCA-Flash4.0 camera controlled by SlideBook 6.0 software (Intelligent Imaging Innovations; Denver, CO) as described in detail elsewhere [13]. We use a xenon lamp and Lambda 10-3 shutter wheel (Sutter), although the shutter wheel is not used for FRET measurements. Before any FRET imaging measurements are taken, the instrument must be calibrated to determine the following parameters:
- The ratio of fluorescence intensity for equimolar donor and acceptor used
- Bleed-through coefficients for donor and acceptor
- The G constant for E-FRET analysis
7.3.2.1. Assessment of Equimolar Fluorescence Intensity of Donor and Acceptor
The fluorescence intensities of YFP and CFP tags are not the same even when the expression level of YFP and CFP are equal. These intensities also vary with different platforms and settings. It is necessary to calculate the ratio of the fluorescence intensity of the same molarity of YFP to CFP. This information is particularly important in assessing whether there is excess donor or acceptor in the region of interest for FRET analysis. To accomplish this goal, we have designed a specific STIM1-derived construct as shown in Figure 7.2b. We added a YFP tag to the N-terminus of the STIM1 molecule that is located in the ER lumen and a CFP tag to the STIM1 C-terminus located in the cytosol. When this construct is expressed, it will ensure that in the same cell there is an identical level of expression of YFP and CFP. With resting STIM1, although the N- and C-termini are separated across the ER membrane, their distance is unknown, and there may be some FRET between them. However, after store depletion, the protein unfolds and stretches out particularly in the ER-PM junctions, so that the distance between N- and C-termini is about 20 nm. The maximal distance for FRET between fluorophores to obtain measurable FRET is approximately 10 nm [27]. Since we measure fluorescence exclusively at the cell periphery, and since all the STIM1 molecules in this region are adjacent to one another in ER-PM junctions, all the signal comes from YFP and CFP molecules that are undergoing minimal FRET. Thus, to avoid FRET signals, we express YFP-STIM1-CFP in cells and treat them with ionomycin to empty stores and assure that STIM1 molecules move into ER-PM junctions. Ionomycin can be used up to 2.5 μM to selectively release Ca2+ from stores but not directly move Ca2+ into the cell across the PM. Alternatively, the ER Ca2+ pump inhibitor, thapsigargin, can be applied to cells to empty stores that cause slightly slower Ca2+ release. The CFP tag is brought close to PM in junctions, but the intermembrane distance of the junctions is 12–17 nm [29,30], and the CFP-YFP distance will be larger than the 10 nm maximum for FRET signals (Figure 7.2b).
At the beginning of a FRET imaging experiment, all microscope parameters are fixed, including exposure times for YFP, CFP, and FRET channels. We randomly select different fields and collect the images with the YFP or CFP channels. Under our normal settings, using the YFP-STIM1-CFP probe with equimolar YFP and CFP, the ratio of fluorescence intensity is about 1.33 ± 0.04. In our FRET experiments, if the ratio is smaller than 1.33, this indicates there is excess of CFP donor-tagged protein. All the parameters of light source output, microscope settings, and camera settings in the software should be kept exactly the same for the calibration and data collection as follows.
7.3.2.2. Calibration of Bleed-Through Coefficients
As shown in formula (7.1), the accuracy of the corrected energy transfer, FRETC, depends on determination of the bleed-through parameters. The bleed-through parameters need to be recalibrated if the xenon light bulb is changed or anything in the light pathway is replaced. Although only two bleed-through parameters (d and a) were considered in formula (7.1), in fact there are four bleed-through coefficients:

Figure 7.3
Three-channel demonstration of the partial receptor YFP-photobleaching method to determine the value of the G parameter using the YFP-CFP construct. Left, IDD is the CFP fluorescence intensity from images collected through the CFP filter cube; middle, (more...)
For these calibrations, it is important to use the protein of interest tagged with CFP or YFP, but not the CFP or YFP empty vector constructs since these would be expressed at much higher levels. The following is a detailed protocol for obtaining the bleed-through coefficients in our experiments on Orai1-CFP and the YFP-SOAR dimers:
- HEK cells are transfected with the Orai1-CFP construct alone or the YFP-S-S construct alone for 24 h.
- Using the settings that were optimized in Section 7.3.2.1, two sets of images (CFP and FRET) are collected from different fields at room temperature using HEK cells transiently expressing Orai1-CFP. Another two sets of images (YFP and FRET) are collected from different fields using HEK cells expressing YFP-S-S.
- Formulas (7.2) and (7.3) are used to calculate the bleed-through coefficients using Excel. We use SlideBook software to automatically calculate the bleed-through coefficients after the images have been collected.
7.3.3. Determination of G Constant Number
As described earlier, G is a constant parameter for a specified imaging system and a pair of FRET fluorophores. It is described in more detail elsewhere [27]. G is pivotal for the calculation of FRET efficiency (Eapp):
We made a specific construct designed for the determination of the G constant. This is the YFP-CFP construct that we described in a recent paper [13]. CFP was flanked with HindIII and BamHI restriction sites and subcloned into the pEYFP vector. The expressed YFP-CFP proteins will result in maximum FRET between CFP and YFP fluorophores as shown in Figure 7.3.
The following is our protocol for determining the G constant:
- HEK cells are transfected with the pEYFP-CFP construct by electroporation.
- Using the settings that were optimized in Section 7.3.2.1, three sets of images (CFP, YFP, and FRET) are collected at room temperature for HEK cells transiently expressing the YFP-CFP protein. This step is called pre-photobleaching, and FRETC and IDD are obtained from this step, as shown in Figure 7.3a.
- The photobleaching step can vary in effectiveness and how confined the area of bleaching is, depending on the microscope/software platforms. The process described here should apply to most platforms. To bleach the YFP fluorophore in this field, the output of the light source is set to 100% and exposure time for YFP is set to 5 min (this time can increase if 5 min is insufficient).
- After photobleaching, it is important to restore the settings back to step 2.
- A further three sets of images (CFP, YFP, and FRET) are collected in the same field at room temperature for HEK cells transiently expressing the YFP-CFP protein. This step is called post-photobleaching, and are obtained from this step, as shown in Figure 7.3b. At this step, the YFP image should be dim compared to that obtained in step 2. If the difference in intensity is hard to tell, repeat step 3 and increase the exposure time to 15 min.
- The value of the G constant that was determined using formula (7.8) for the data in Figure 7.4 was 1.9 ± 0.1 (n = 32 cells) using formula (7.8).
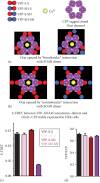
Figure 7.4
Evidence for a “unimolecular” interaction between the SOAR dimer and the Orai1 subunit within hexamer. Two possible models for the SOAR-Orai1 interaction: (a) The bimolecular binding of a homomeric SOAR dimer (red) to two adjacent Orai1 (more...)
7.3.4. Data Collection
A useful example of our measurement of E-FRET is given in Figure 7.4 in which we investigate whether the two SOAR units within the SOAR dimer are equivalent. Thus, an important question was whether the active SOAR dimer unit (the active site within the dimeric STIM1 molecule) functions in a bimolecular manner in which each of the two SOAR units in the dimer together associate with each of the two adjacent Orai1 subunits within the hexameric Orai1 channel, as shown in Figure 7.4a. Alternatively, each SOAR unit in the SOAR dimer may interact independently with a single Orai1 subunit in a unimolecular manner, as shown in Figure 7.1b.
To investigate this further, we designed a specific Orai1-derived construct that expresses only the strong STIM1-binding C-terminus of Orai1 (aa 267–301). This construct, named PM-CFP-Orai1CT, includes a PM-directed transmembrane helix, which assures that it is expressed only in the PM [13] (Figure 7.4a). We generated a stable HEK cell line expressing the PM-CFP-Orai1CT construct comparable to the Orai1-CFP-expressing line described earlier. HEK-PM-CFP-Orai1CT stable cell lines were transfected with YFP-S-S, YFP-SH-S, YFP-S-SH, YFP-SH-SH, or YFP vector, 24 h prior to experiments. Transfected cells are seeded on 25 mm round glass coverslips.
- To ensure consistent data collection, images for each coverslip are obtained under identical conditions. During experiments, unused coverslips are kept in 6-well plates in the incubator until utilized and assembled in the chamber.
- Fields are selected with more YFP-positive cells, using the settings that were optimized in Section 7.3.2.1. Sets of images (CFP, YFP, and FRET) are collected at room temperature. Further sets of images (CFP, YFP, and FRET) are collected again from different fields. The number of views will depend on cell density. If transfected cells are sparse, further views are needed until the total cell count is 30–40 cells or more.
- After imaging, glass coverslips are discarded and the chamber is washed, and the subsequent coverslip readied for imaging. It is important to try to ensure that each coverslip is treated as consistently as possible.
7.3.5. Data Analysis
To analyze FRET images and export the data, we use SlideBook 6.0 (Intelligent Imaging Innovations). The FRET module comes with this software. For other platforms, the detailed process will be different in exporting the raw data. If the software being used does not have the module for E-FRET calculation, then raw data for IDD, IAA, and IDA are exported manually and calculations for E-FRET are made in Excel using formulas (7.1) and (7.6) with the given bleed-through coefficients (a and d) and G parameter. After obtaining all the raw data for all groups, it is important to exclude those cells that have abnormal expression of YFP-tagged concatemer. As mentioned in Sections 7.2 and 7.3.2.1, only cells that have similar expression levels of YFP-tagged concatemer and also have YFP/CFP ratios that are smaller than 1.33 should be analyzed. To do this in an unbiased manner:
- Export the data into an Excel file.
- For each cell, calculate the YFP/CFP ratio using the values of IAA/IDD.
- Each cell will have two important parameters: E-FRET and YFP/CFP. Sort the Excel sheet based on the value of YFP/CFP and then exclude all the cells with the value larger than 1.33. To ensure there is an excess of donor, we only select cells that have YFP/CFP around 1 or smaller as shown in Figure 7.4d and g. Thus, the E-FRET values are analyzed from cells with a narrow range of YFP/CFP ratios.
The strong binding site for STIM1 lies in the helical cytosolic Orai1 C-terminus (amino acids 267–301). Therefore, we made a new construct (PM-CFP-Orai1CT) in which we expressed this strong binding site attached via CFP to a PM-targeted transmembrane helix (Figure 7.4e). We used this probe to determine whether the SOAR dimer molecule could simultaneously bind to two Orai1 binding sites for STIM1. We examined this by measuring E-FRET. As shown in Figure 7.4f and g, the E-FRET between PM-CFP-Orai1CT and YFP-S-S is almost double that between PM-CFP-Orai1CT and either YFP-SH-S or YFP-S-SH. In contrast, the E-FRET level with YFP-SH-SH was very close to background nonspecific FRET observed with cells expressing YFP (Figure 7.4f and g). We interpret these data in light of the models shown in Figures 7.1d and 7.4h. Thus, each YFP-S-S dimer is able to bind two PM-CFP-Orai1CT molecules, whereas YFP-SH-S or YFP-S-SH is only able to bind one PM-CFP-Orai1CT. This is almost twice as much CFP donor that can be quenched by the YFP acceptor. These data suggest that the two SOAR units within the SOAR dimer are equivalent and can each independently bind to one Orai1 subunit within the hexameric channel. This provides important new information on the molecular binding stoichiometry of the STIM1-Orai1 coupling interface, as we recently described [13].
7.4. Conclusions
FRET is one of the most powerful techniques for studying protein interactions. We detail here that meaningful quantitative analysis of protein-protein interactions can be achieved; however, careful awareness of the expression levels and ratio of donor and acceptor are crucial to such determination. The STIM and Orai coupling interaction is particularly suited to analysis by FRET. Thus, each protein resides in a separate, distinct membrane (ER or PM), and the proteins only interact within a “FRETable” distance (<10 nm) when they enter ER-PM junctions. Thus, there is little, if any, STIM1-Orai1 FRET in resting cells with full Ca2+ stores in which STIM has not been activated and has not been trapped within ER-PM junctions. For full-length STIM1, although FRET only occurs in the ER-PM region, the excess of Orai1 or STIM1 outside of the ER-PM region will affect the FRET analysis as described in Figure 7.1.
The use of SOAR instead of the full-length STIM1 molecule provides several strong advantages to study the coupling interaction with the Orai1 channel. SOAR is cytoplasmic and therefore is not restricted to the ER; hence, it can bind to any Orai1 in the PM where FRET will occur. By using a series of SOAR concatemers with either one or two F394H mutations expressed in the stable HEK-Orai1-CFP cell line, we were able to show that only one site in the SOAR dimer was required for activation of the Orai1 channel. Our FRET analysis was important in revealing that the interaction of the SOAR dimer with Orai1 required only one of the two Orai1 binding sites within the dimer. This revealed our “unimolecular coupling” model [13] that argues against the bimolecular coupling model that had been predicted earlier (see Chapter 2).
Using HEK-PM-CFP-Orai1CT stable cell line that expresses the simple PM-CFP-Orai1CT construct (as opposed to the full-length CFP-Orai1 hexamer), our quantitative FRET approach allowed us to reveal that one YFP-SOAR dimer could simultaneously interact with two PM-CFP-Orai1CT molecules. However, this analysis required that the donor PM-CFP-Orai1CT be expressed in excess of the acceptor YFP-SOAR dimer. This result was important in drawing the conclusion that the active SOAR dimer within the STIM1 protein could undergo binding to two Orai1 channel subunits and suggests that STIM1 can actively cluster Orai1 channels by cross-linking between them [13]. This provides an important new understanding of the STIM1-Orai1 interface.
References
- 1.
- Soboloff, J., Rothberg, B. S., Madesh, M., and Gill, D. L. (2012) STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549-565. [PMC free article: PMC3458427] [PubMed: 22914293]
- 2.
- Kar, P., Nelson, C., and Parekh, A. B. (2012) CRAC channels drive digital activation and provide analog control and synergy to Ca2+-dependent gene regulation. Curr. Biol. 22, 242-247. [PubMed: 22245003]
- 3.
- Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., Jr., and Meyer, T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235-1241. [PMC free article: PMC3186072] [PubMed: 16005298]
- 4.
- Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., Safrina, O., et al. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435-445. [PMC free article: PMC2171946] [PubMed: 15866891]
- 5.
- Zhang, S. L., Yu, Y., Roos, J., Kozak, J. A., Deerinck, T. J., Ellisman, M. H., Stauderman, K. A., and Cahalan, M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902-905. [PMC free article: PMC1618826] [PubMed: 16208375]
- 6.
- Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., Hogan, P. G., Lewis, R. S., Daly, M., and Rao, A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179-185. [PubMed: 16582901]
- 7.
- Yeromin, A. V., Zhang, S. L., Jiang, W., Yu, Y., Safrina, O., and Cahalan, M. D. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226-229. [PMC free article: PMC2756048] [PubMed: 16921385]
- 8.
- Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230-233. [PubMed: 16921383]
- 9.
- McNally, B. A. and Prakriya, M. (2012) Permeation, selectivity and gating in store-operated CRAC channels. J. Physiol. 590, 4179-4191. [PMC free article: PMC3473277] [PubMed: 22586221]
- 10.
- Zhou, Y., Srinivasan, P., Razavi, S., Seymour, S., Meraner, P., Gudlur, A., Stathopulos, P. B., Ikura, M., Rao, A., and Hogan, P. G. (2013) Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 20, 973-981. [PMC free article: PMC3784406] [PubMed: 23851458]
- 11.
- Korzeniowski, M. K., Manjarres, I. M., Varnai, P., and Balla, T. (2010) Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 3, ra82. [PMC free article: PMC3408607] [PubMed: 21081754]
- 12.
- Covington, E. D., Wu, M. M., and Lewis, R. S. (2010) Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 21, 1897-1907. [PMC free article: PMC2877647] [PubMed: 20375143]
- 13.
- Zhou, Y., Wang, X., Wang, X., Loktionova, N. A., Cai, X., Nwokonko, R. M., Vrana, E., Wang, Y., Rothberg, B. S., and Gill, D. L. (2015) STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 6, 8395. [PMC free article: PMC4598629] [PubMed: 26399906]
- 14.
- Wang, Y., Deng, X., Zhou, Y., Hendron, E., Mancarella, S., Ritchie, M. F., Tang, X. D., et al. (2009) STIM protein coupling in the activation of Orai channels. Proc. Natl. Acad. Sci. USA 106, 7391-7396. [PMC free article: PMC2678612] [PubMed: 19376967]
- 15.
- Navarro-Borelly, L., Somasundaram, A., Yamashita, M., Ren, D., Miller, R. J., and Prakriya, M. (2008) STIM1-ORAI1 interactions and ORAI1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 586, 5383-5401. [PMC free article: PMC2655373] [PubMed: 18832420]
- 16.
- Wang, X., Wang, Y., Zhou, Y., Hendron, E., Mancarella, S., Andrake, M. D., Rothberg, B. S., Soboloff, J., and Gill, D. L. (2014) Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 5, 3183. [PMC free article: PMC3995141] [PubMed: 24492416]
- 17.
- Gandhi, C. S. and Isacoff, E. Y. (2005) Shedding light on membrane proteins. Trends Neurosci. 28, 472-479. [PubMed: 16043238]
- 18.
- Clegg, R. M. (1995) Fluorescence resonance energy transfer. Curr. Opin. Biotechnol. 6, 103-110. [PubMed: 7534502]
- 19.
- Jares-Erijman, E. A. and Jovin, T. M. (2006) Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 10, 409-416. [PubMed: 16949332]
- 20.
- Padilla-Parra, S. and Tramier, M. (2012) FRET microscopy in the living cell: Different approaches, strengths and weaknesses. Bioessays 34, 369-376. [PubMed: 22415767]
- 21.
- Arai, Y. and Nagai, T. (2013) Extensive use of FRET in biological imaging. Microscopy (Oxf) 62, 419-428. [PubMed: 23797967]
- 22.
- Heim, R. and Tsien, R. Y. (1996) Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 6, 178-182. [PubMed: 8673464]
- 23.
- Periasamy, A. (2001) Fluorescence resonance energy transfer microscopy: A mini review. J. Biomed. Opt. 6, 287-291. [PubMed: 11516318]
- 24.
- Yuan, J. P., Zeng, W., Dorwart, M. R., Choi, Y. J., Worley, P. F., and Muallem, S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337-343. [PMC free article: PMC2663385] [PubMed: 19182790]
- 25.
- Park, C. Y., Hoover, P. J., Mullins, F. M., Bachhawat, P., Covington, E. D., Raunser, S., Walz, T., Garcia, K. C., Dolmetsch, R. E., and Lewis, R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876-890. [PMC free article: PMC2670439] [PubMed: 19249086]
- 26.
- Yang, X., Jin, H., Cai, X., Li, S., and Shen, Y. (2012) Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 109, 5657-5662. [PMC free article: PMC3326449] [PubMed: 22451904]
- 27.
- Zal, T. and Gascoigne, N. R. (2004) Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 86, 3923-3939. [PMC free article: PMC1304294] [PubMed: 15189889]
- 28.
- Miller, M., Wu, M., Xu, J., Weaver, D., Li, M., and Zhu, M. X. (2011) High-throughput screening of TRPC channel ligands using cell-based assays. In TRP Channels (Zhu, M. X. ed.), Boca Raton, FL: CRC Press/Taylor & Francis.
- 29.
- Lur, G., Haynes, L. P., Prior, I. A., Gerasimenko, O. V., Feske, S., Petersen, O. H., Burgoyne, R. D., and Tepikin, A. V. (2009) Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr. Biol. 19, 1648-1653. [PMC free article: PMC2887489] [PubMed: 19765991]
- 30.
- Wu, M. M., Buchanan, J., Luik, R. M., and Lewis, R. S. (2006) Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 174, 803-813. [PMC free article: PMC2064335] [PubMed: 16966422]
Yandong Zhou
Department of Cellular and Molecular Physiology
Pennsylvania State University College of Medicine
Hershey, Pennsylvania
Youjun Wang
Beijing Key Laboratory of Gene Resources and Molecular Development
Beijing Normal University
Haidian, Beijing, People’s Republic of China
Donald L. Gill
Department of Cellular and Molecular Physiology
Pennsylvania State University College of Medicine
Hershey, Pennsylvania
Publication Details
Author Information and Affiliations
Authors
Yandong Zhou, Youjun Wang, and Donald L. Gill.Copyright
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
Publisher
CRC Press/Taylor & Francis, Boca Raton (FL)
NLM Citation
Zhou Y, Wang Y, Gill DL. Assessing the Molecular Nature of the STIM1/Orai1 Coupling Interface Using FRET Approaches. In: Kozak JA, Putney JW Jr., editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 7. doi: 10.1201/9781315152592-7





