

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Centers for Disease Control and Prevention (US); National Center for Chronic Disease Prevention and Health Promotion (US); Office on Smoking and Health (US). How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention (US); 2010.

How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General.
Show detailsIntroduction
This chapter reviews the epidemiology of smoking-induced cardiovascular disease (CVD) and the mechanisms by which tobacco smoke is thought to cause CVD. The discussion includes use of biomarkers to diagnose smoking-induced CVD and treatment implications of the pathophysiology of the disease. The link between secondhand smoke and CVD has been reviewed in the 2006 report of the Surgeon General, The Health Consequences of Involuntary Exposure to Tobacco Smoke (U.S. Department of Health and Human Services [USDHHS] 2006), so discussion of secondhand smoke in this report is limited.
Tobacco Use and Cardiovascular Disease
Cigarette smoking is a major cause of CVD, and past reports of the Surgeon General extensively reviewed the relevant evidence (U.S. Department of Health, Education, and Welfare [USDHEW] 1971 1979; USDHHS 1983, 2001, 2004). Cigarette smoking has been responsible for approximately 140,000 premature deaths annually from CVD (USDHHS 2004). More than 1 in 10 deaths worldwide from CVD in 2000 were attributed to smoking (Ezzati et al. 2005). In the United States, smoking accounted for 33 percent of all deaths from CVD and 20 percent of deaths from ischemic heart disease in persons older than 35 years of age (Centers for Disease Control and Prevention 2008). Cigarette smoking also influences other cardiovascular risk factors, such as glucose intolerance and low serum levels of high-density lipoprotein cholesterol (HDLc). However, studies have reported that smoking increases the risk of CVD beyond the effects of smoking on other risk factors. In other words, the risk attributable to smoking persisted even when adjustments were made for differences between persons who smoke and nonsmokers in levels of these other risk factors (Friedman et al. 1979; USDHHS 1983, 2001, 2004; Shaper et al. 1985; Criqui et al. 1987; Ragland and Brand 1988; Shaten et al. 1991; Neaton and Wentworth 1992; Freund et al. 1993; Cremer et al. 1997; Gartside et al. 1998; Wannamethee et al. 1998; Jacobs et al. 1999a). For example, in one study, the effect of cigarette smoking on the risk of coronary heart disease (CHD) was evident even among persons with low serum levels of cholesterol (Blanco-Cedres et al. 2002).
Beyond its status as an independent risk factor, smoking appears to have a multiplicative interaction with the other major risk factors for CHD—high serum levels of lipids, untreated hypertension, and diabetes mellitus (USDHHS 1983). For instance, if the presence of smoking alone doubles the level of risk, the simultaneous presence of another major risk factor is estimated to quadruple the risk (2 × 2). The presence of two other risk factors with smoking results in approximately eight times the risk (2 × 2 × 2) of persons with no risk factors. Cigarette smoking also is a cause of peripheral arterial disease (PAD), aortic aneurysm, CHD, and cerebrovascular disease, but the relative risk (RR) of disease varies with the vascular bed (USDHEW 1971, 1979; USDHHS 1983, 2001, 2004). The highest RRs are observed for diseases of peripheral arteries in the lower extremities, and the lowest are for stroke; RRs are intermediate for CHD and aortic aneurysm.
The general mechanisms by which smoking results in cardiovascular events include development of atherosclerotic changes with narrowing of the vascular lumen and induction of a hypercoagulable state, which create risk of acute thrombosis (USDHHS 1983, 2004). The rapid decline in risk of a recurrent myocardial infarction (MI) after smoking cessation (USDHHS 1990) supports the role of smoking in thrombosis. In addition, abundant evidence demonstrates that smoking contributes to development of atherosclerotic plaque (Strong and Richards 1976; Auerbach and Garfinkel 1980; Solberg and Strong 1983; USDHHS 1983, 2004).
Estimation of Risk
The risk of CHD from cigarette smoking can be described in terms of RR and excess risk (Thun et al. 1997). The RR is the ratio of CHD rates for populations of smokers to rates for lifetime nonsmokers. Excess risk is the difference between the rates of disease for smokers and nonsmokers.
These two estimates of risk can lead to conflicting impressions of the changes in smoking-related CHD risks with advancing age. The RRs and excess death rates for CHD are shown by age group in data from Cancer Prevention Study II (CPS-II) (Thun et al. 1997), sponsored by the American Cancer Society (see Figure 6.1 for data on men). The RRs were highest at younger ages (35 to 54 years) and declined steeply with advancing age. This finding leaves the false impression that the disease burden of CHD from smoking declined with age or was low among older smokers. However, excess CHD death rates for smokers by age group presented different evidence. The high RR for CHD at younger ages can be explained in part because mortality rates are low for death from CHD at those ages and because coronary events in young people occur primarily among smokers. Even though RR declined with increasing age, because the absolute rate of deaths from CHD increased markedly, the magnitude of the CHD burden produced by smoking increased with advancing age.
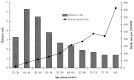
Figure 6.1
Relative risk and excess death rate for coronary heart disease among men, by age group. Source: Burns 2003. Adapted from Thun et al. 1997 with permission from Elsevier, © 2003. Note: Data are from the American Cancer Society’s Cancer Prevention (more...)
The age at onset of substantial excess risk differs by disease. For smokers, age-specific excess death rates attributable to CHD, lung cancer, cerebrovascular disease, and chronic obstructive pulmonary disease (COPD) are illustrated by data from CPS-II (Figure 6.2 shows data for men) (Thun et al. 1997). For persons younger than age 45 years, CHD was the dominant cause of increased mortality attributable to cigarette smoking. Excess rates for death from lung cancer increased steeply after age 50 years, and excess death rates from COPD were largely confined to the seventh and eighth decades of life. Late in life, excess deaths from COPD matched and those from lung cancer exceeded the excess death rates attributable to CHD. The RR for death from a cerebrovascular disease among smokers was substantially elevated among younger smokers (RR = 4 to 5; data not shown). However, the absolute rate of stroke at these younger ages was low, and this finding resulted in a low excess mortality rate. At older ages, the death rate from stroke in the general population increased and the RR among smokers declined, thus moderating the excess death rate attributable to smoking.
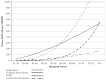
Figure 6.2
Age-specific excess death rates among male smokers for coronary heart disease, lung cancer, chronic obstructive pulmonary disease (COPD), and cerebrovascular disease. Source: Burns 2003. Adapted from Thun et al. 1997 with permission from Elsevier, © (more...)
Coronary Heart Disease
Cigarettes Smoked per Day
Studies showed increased risk of having CHD at all levels of cigarette smoking, and increased risks were evident even for persons who smoked fewer than five cigarettes per day (Rosengren et al. 1992; Prescott et al. 2002; Bjartveit and Tverdal 2005). Prospective mortality studies conducted in the 1960s and 1970s showed a clear increase in CHD mortality with an increase in the number of cigarettes smoked per day, regardless of the actual number (Doll and Peto 1976; USDHHS 1983). Other studies suggested that risk increased up to at least 40 cigarettes per day (Miettinen et al. 1976; Willett et al. 1987). However, more recent data appeared to show an increase in CHD risk with more cigarettes smoked per day only up to about 25 cigarettes; the risk increased relatively little even with further increases in cigarette consumption (Neaton and Wentworth 1992; Rosengren et al. 1992; Thun et al. 1997).
Law and Wald (2003), who conducted a meta-analysis of five large studies of smoking and CHD, demonstrated a nonlinear dose-response relationship between the number of cigarettes smoked per day and the RR of disease (Figure 6.3). The researchers suggested that the effect of cigarette smoking on risk of CHD may have a low threshold and that the dose-response characteristics of the risk relationship are less steep at higher doses. This hypothesis was used to explain the seeming anomaly of a high RR of CHD associated with relatively low exposure to secondhand smoke. By using serum levels of cotinine (a metabolite of nicotine) as biomarkers of exposure, Whincup and colleagues (2004) explored the dose response relationship between exposure to cigarette smoke and CHD in persons involuntarily exposed to cigarette smoke. More than 2,000 men who said they did not smoke had blood levels of cotinine measured in 1978–1980 and then had follow-up for 20 years. Nicotine exposure was examined by quartiles of blood cotinine as follows: less than or equal to 0.7 nanograms per milliliter (ng/mL), 0 to 1.4, 1.5 to 2.7, and 2.8 to 14.0. Hazard ratios for CHD, which included deaths and non- fatal MIs, were significantly increased at all upper quartiles (hazard ratios, 1.43 to 1.57) compared with the lowest exposure quartile, after adjustment for established CHD risk factors. Hazard ratios were also higher at the first and second five-year follow-ups (3.73 to 10.58 and 1.95 to 2.48, respectively) than those at later follow-ups. The substantial cardiovascular risk attributable to involuntary exposure to cigarette smoke (USDHHS 2006) and the practice in most CVD studies of not excluding from the control group persons who had secondhand smoke exposure have resulted in underestimation, in many research reports, of the effects of active smoking compared with no exposure to cigarette smoke.

Figure 6.3
Dose-response relationship between number of cigarettes smoked per day and relative risk of ischemic heart disease. Source: Law and Wald 2003. Reprinted with permission from Elsevier, © 2003. Note: The dose-response relationship between exposure (more...)
The data on secondhand smoke and CHD risk indicate that the dose-response relationship between exposure to smoke and cardiovascular effects is nonlinear. Another consideration is that the number of cigarettes smoked per day may not provide a linear measure of exposure to tobacco smoke. When carboxyhemoglobin or serum cotinine levels were used as measures of the smoke taken in, persons who smoked more cigarettes per day had higher levels of these biologic substances (Benowitz 1996). Even so, the carboxyhemoglobin and cotinine levels were substantially lower than those predicted by linear extrapolation from data on persons who reported smoking 1 to 20 cigarettes per day (Law et al. 1997). Among smokers of 40 or more cigarettes per day, the levels of these biomarkers were 35 percent lower than those predicted by linear extrapolation from data on persons who reported smoking fewer than 20 cigarettes per day.
At the same reported number of cigarettes per day, cotinine levels varied substantially (Benowitz 1996). Smokers titrate cigarette smoke to achieve a consistent intake of nicotine by altering the number of cigarettes smoked per day or by changing the puffing pattern—that is, by taking deeper, faster, more, or longer puffs (National Cancer Institute [NCI]2001). When the sesmoking behaviors fail to restore the level of nicotine intake, as they may with cigarettes that have very low machine-measured yields, smokers may increase the number of cigarettes per day to maintain the same level of nicotine intake.
These observations suggest that the number of cigarettes smoked per day may have become a less precise measure of exposure to tobacco smoke with the introduction of cigarettes with low machine-measured yields of tar and nicotine. This diminished precision of cigarettes smoked per day as a measure of exposure may account for some of the discordance between studies that define increased risk by an increase of more than one pack per day in the number of cigarettes smoked. By using serum cotinine as an indicator of nicotine intake and exposure to tobacco smoke in the population demonstrates a linear increase for 10 to 15 cigarettes per day. However, as use exceeds 10 to 15 cigarettes per day, a progressively smaller increment in serum cotinine for each increment in the number of cigarettes smoked per day is observed (Caraballo et al. 1998; O’Connor et al. 2006). This flattening of the relationship between exposure and cigarettes smoked per day was similar to flattening of the relationship between the RR of CHD and the number of cigarettes smoked per day. Thus, researchers should be cautious about defining the absence of a continuing increase in risk among smokers of more than 20 cigarettes per day as evidence that increases in actual exposure are not accompanied by increases in risk.
Duration of Smoking
Researchers have not always demonstrated a significant relationship between duration of cigarette smoking and CHD risk when adjustment was made for other risk factors and the number of cigarettes smoked per day (Kuller et al. 1991; Tverdal 1999). Variation in the number of cigarettes smoked per day and in the products smoked during the lifetime of a smoker is often substantial, but this variable is not well captured in epidemiologic studies.
Age is colinear with duration of smoking, because the two variables grow in tandem after a person starts to smoke and the RRs for smoking and CHD decline with advancing age. Furthermore, most smokers begin to smoke during adolescence, which promotes the colinearity. These realities make it difficult to estimate the independent contributions of age and duration of smoking to risk of CHD in multivariate models. However, the two studies of the American Cancer Society are a good source of data, because each study consists of more than 1 million men and women (Burns et al. 1997; Thun et al. 1997). Analyses of these data stratified by age and the number of cigarettes smoked per day showed steady increases in CHD mortality rates with increasing duration of smoking for persons younger than age 70 years. Using data from CPS-I, investigators calculated the risk of developing CHD by age and duration of smoking (see Table 6.1 for data on White men) (Burns et al. 1997). For almost all age groups younger than age 70 years, RRs increased with increasing duration of smoking. Data from CPS-II on men (Thun et al. 1997) also demonstrated a pattern of increasing RR with age-specific mortality due to CHD and increasing duration of smoking for each level of cigarettes smoked per day (Table 6.2). Even though data in these analyses were not adjusted for potential differences in other cardiovascular risk factors, the findings presented a convincing picture of increasing risk of CHD with longer duration of smoking.
Table 6.1
Rate ratios for coronary heart disease among White men, by age and duration of cigarette smoking.
Table 6.2
Death rates and rate ratios for death from coronary heart disease among men, by age and duration of smoking by number of cigarettes smoked per day.
Smoking Cessation
The risks of MI and death from CHD are lower among former smokers than among continuing smokers in many studies, including those with data adjusted for levels of other risk factors (Gordon et al. 1974; Åberg et al. 1983; USDHHS 1990; Kuller et al. 1991; Frost et al. 1996). The risk fell rapidly, decreasing about one-half in one year (Lightwood and Glantz 1997). Risks appear to remain slightly elevated for more than a decade after persons stopped smoking, but in some studies this increased risk was not statistically significant (Dagenais et al. 1990; Omenn et al. 1990; Kawachi et al. 1993a, 1994; Jacobs et al. 1999a; Qiao et al. 2000). Among smokers who had MI or angiographically documented CHD, persons who stopped smoking had a substantially lower rate of reinfarction than did those who continued to smoke. Reduction in risk was evident within the first year after MI. Risk continued to be lower among former smokers than among continuing smokers for prolonged periods after the first MI (Daly et al. 1983; Omenn et al. 1990). Studies also demonstrated rapid reduction in risk after persons stopped smoking among populations at high risk for CHD (Ockene et al. 1990) and among women (Kawachi et al. 1993a, 1994).
Patients with angiographically documented CHD who stopped smoking at the diagnosis of CHD (Vlietstra et al. 1986) or before diagnosis (Hermanson et al. 1988) had lower death rates from MI or CHD than did continuing smokers. In addition, the benefit of stopping smoking did not decline with advancing age.
In the 16-year follow-up of the Multiple Risk Factor Intervention Trial Research Group (1990, 1996), mortality from CHD was 11.4 percent lower in the “special intervention” group than in the “usual care” group. This result may illustrate the benefit of stopping smoking, because one of the interventions targeted smoking cessation. A trial of advice to civil servants in London, England, to stop smoking demonstrated an 18-percent reduction in mortality from CHD in the intervention group versus the control group after 10 years of follow-up (Rose et al. 1982). Suskin and colleagues (2001) reported that in addition to benefits for CHD, stopping smoking also reduces morbidity and mortality in patients with left ventricular dysfunction. In this study, the benefits of stopping smoking on mortality and recurrent congestive heart failure requiring hospitalization were similar to the benefits from treatments with angiotensin-converting-enzyme (ACE) inhibiting drugs, β-blockers, or spironolactone, which are mainstays for the treatment of heart failure.
Women
Women have lower absolute rates of CHD than do men. However, cigarette smoking has been associated with higher RR of MI (Njølstad et al. 1996) and higher CHD mortality (Kawachi et al. 1994; Thun et al. 1997) among women than among men. The absolute increase in risk of CHD from smoking is similar for men and women (USDHHS 1983, 2001).
A prospective evaluation of fatal and nonfatal CVD events among women in the Nurses’ Health Study (Willett et al. 1987) found that smoking was an independent cause of CVD. Age-adjusted risks of disease increased progressively with more cigarettes smoked per day up to 45 or more per day. Even when the combined risks of fatal CHD and nonfatal MI were adjusted for levels of other risk factors, risks increased with increasing numbers of cigarettes per day.
Researchers have demonstrated a rapid decline in excess risk of CHD in women after they stopped smoking cigarettes. Even so, 10 to 14 years of nonsmoking are required before risks approach those of lifetime nonsmokers (Kawachi et al. 1993a, 1994).
Race and Ethnicity
In 2004, heart disease mortality was higher among African Americans than among Whites (National Heart, Lung, and Blood Institute [NHLBI] 2007). From 1999 through 2004, the prevalence of acute MI was higher for African Americans than for Whites aged 35 through 54 years; however, for ages 55 years and older, the prevalence of acute MI was higher among Whites (NHLBI 2007).
The INTERHEART study is a case-control investigation of acute MI in 52 countries in Africa, Asia, Australia, the Middle East Crescent, and North and South America (Teo et al. 2006). The odds ratio (OR) for acute MI in smokers was 2.95 for this large multiethnic population compared with lifetime nonsmokers. In addition, the risk of MI was higher among persons who smoked bidis than among nonsmokers in countries where use of this form of tobacco is common.
Researchers also identified cigarette smoking as a significant risk factor for CHD among Hispanic populations (Mendelson et al. 1998) and Asian populations (Kiyohara et al. 1990; Miyake et al. 2000; Lam et al. 2002).
Sudden Death
Most sudden death is due to CVD. In many epidemiologic studies, RRs for sudden cardiac death were higher than RRs for CHD or MI among persons who smoked. The RRs for sudden death among current smokers, compared with lifetime nonsmokers, often exceeded 3.0 (USDHEW 1971, 1979; Dawber 1980; Kannel and Thomas 1982; USDHHS 1983; Wannamethee et al. 1995; Sexton et al. 1997). In multivariate analyses of the combined data from the Framingham Heart Study and the Albany Study, which examined sudden cardiac death in men aged 45 through 64 years, cigarette smoking was the risk factor with the highest statistical significance (Kannel et al. 1975). In a study of data from the 1986 National Mortality Followback Survey among persons with no history of CHD, cigarette smoking was the only modifiable risk factor associated with sudden coronary death and it was one factor associated with increased risk of sudden coronary death among persons with known CHD (Escobedo and Zack 1996; Escobedo and Caspersen 1997). Cigarette smoking was also associated with risk of sudden cardiac death in the 18-year follow-up of the Honolulu Heart Program (Kagan et al. 1989) and the 28-year follow-up of the Framingham Heart Study (Cupples et al. 1992).
Peters and colleagues (1995) found an association between smoking cessation and reduction in death from cardiac arrhythmia for patients with left ventricular dysfunction after MI. Finally, the risk of recurrent cardiac arrest among smokers surviving out-of-hospital cardiac arrest was lower among persons who then stopped smoking than among those who continued to smoke (Hallstrom et al. 1986).
Stroke
After adjustment of data for other risk factors, cigarette smokers have higher risk of stroke and higher mortality from cerebrovascular disease than do lifetime nonsmokers, and a dose-response relationship is evident (USDHHS 1983, 2001, 2004; Neaton et al. 1984; Colditz et al. 1988; Wolf et al. 1988; Kannel and Higgins 1990; Kuller et al. 1991; Freund et al. 1993; Hames et al. 1993; Håheim et al. 1996; Tanne et al. 1998; Jacobs et al. 1999a; Sharrett et al. 1999; Djoussé et al. 2002). In addition, in the 20-year follow-up of a prospective study of mortality that controlled for other cardiovascular risk factors, cigarette smoking increased the risk of death from stroke and mortality rates grew the number of cigarettes smoked increased (Hart et al. 1999).
In a meta-analysis of data from 32 studies, the overall RR for stroke associated with cigarette smoking was 1.5 (95 percent confidence interval [CI], 1.4–1.6) (Shinton and Beevers 1989). The RRs varied with the stroke subtypes: 1.9 for cerebral infarction, 0.7 for cerebral hemorrhage, and 2.9 for subarachnoid hemorrhage. The researchers reported a dose-response relationship between the number of cigarettes smoked per day and the RR. The data suggested a sustained higher risk of stroke among former smokers younger than age 75 years than the risk for nonsmokers in the same age group. For all ages combined, RR for former smokers was 1.2.
During the 26-year follow-up of the cohort in the Framingham Heart Study, cigarette smoking was a significant risk factor for stroke (Wolf et al. 1988). The risk declined, however, among smokers who had stopped smoking for two years and was similar to that of lifetime nonsmokers after five years of abstinence from smoking. In the 12-year follow-up of the Nurses’ Health Study (Kawachi et al. 1993b), RR for stroke among current smokers was 2.58 compared with nonsmokers, but it was 1.34 among former smokers compared with nonsmokers. Once those who stopped smoking had abstained for two to four years, their risk for stroke could not be distinguished from that of lifetime nonsmokers. In addition, the pattern of decline in total risk for stroke after stopping smoking remained the same after adjustments for other risk factors.
Aortic Aneurysm
Mortality studies consistently demonstrated higher risk of death from abdominal aortic aneurysm among cigarette smokers than among nonsmokers (Hammond and Horn 1958; Weir and Dunn 1970; USDHHS 1983, 2004; Strachan 1991; Nilsson et al. 2001). In addition, the risk rose with an increasing number of cigarettes smoked per day (Kahn 1966; Hammond and Garfinkel 1969; Burns et al. 1997; Blanchard et al. 2000; Vardulaki et al. 2000).
Studies have demonstrated an association of cigarette smoking with prevalence of aortic aneurysm or aortic dilation, as determined by ultrasonography in cohorts of men and women, even after adjustment for a large number of known risk factors (Alcorn et al. 1996; Lee et al. 1997; Wilmink et al. 1999; Jamrozik et al. 2000; Lederle et al. 2001). The U.S. Preventive Services Task Force (2005) recommended a one-time screening by ultrasonography for abdominal aortic aneurysm among men aged 65 to 75 years who had ever smoked. Cigarette smoking has been associated with increased growth of abdominal aortic aneurysms (Brady et al. 2004). This finding suggests that more frequent monitoring of smokers for this condition is necessary. With increasing duration of abstinence from smoking, the risk of developing an abdominal aneurysm appears to slowly decline (Wilmink et al. 1999).
Peripheral Arterial Disease
Cigarette smoking and diabetes are well established as the major risk factors for PAD, and a strong dose-response relationship for smoking was observed even after adjustment for other CVD risk factors (Weiss 1972; Kannel and Shurtleff 1973; USDHHS 1983; Wilt et al. 1996; Price et al. 1999; Meijer et al. 2000; Ness et al. 2000). Data from the Framingham Heart Study demonstrated increased risk of PAD among both young and older male and female cigarette smokers after adjustment for other cardiovascular risk factors. In addition, this risk increased with the increase in the number of cigarettes smoked per day, and this result was statistically significant (Freund et al. 1993). The Framingham Offspring Study reported a similar finding (Murabito et al. 2002). Finally, researchers have observed a significantly higher rate of late arterial occlusion in patients who continued to smoke after peripheral vascular surgery than in those who stopped smoking (Wray et al. 1971; Ameli et al. 1989; Wiseman et al. 1989). Among smokers with claudication, progression to critical limb ischemia is reduced in those who stopped smoking (Jonason and Bergström 1987).
Pipes, Cigars, and Low-Tar Cigarettes
“Low-tar” or “light” cigarettes were designed to produce low machine-measured yields of tar and nicotine (NCI 2001). Design characteristics of low-tar cigarettes include increased ventilation and more rapid cigarette burn rate. By changing the way they smoke or the number of cigarettes smoked, persons who smoke these products can obtain as much nicotine as from “regular” or “full- flavored” cigarettes, thereby satisfying their addiction (USDHEW 1979; NCI 2001). Comprehensive reviews of this issue concluded that use of low-tar cigarettes has not resulted in meaningful reduction in the risk of CVD (NCI 2001; Stratton et al. 2001; Scientific Advisory Committee on Tobacco Product Regulation 2002; USDHHS 2004).
Compared with persons who smoke cigarettes, smokers who exclusively smoke pipes or cigars have lower risk for many smoking-related diseases (NCI 1998). Smoke from pipes and cigars contains the same toxic substances as cigarette smoke, but those who use a pipe or cigar usually smoke at lower intensity; observation indicates that they tend not to inhale the smoke, thus reducing their exposure to its toxic substances (USDHEW 1979; NCI 1998; Shanks et al. 1998). Most current cigar users are young males who often smoke less than one cigar daily (NCI 1998); no data on risk for this population are available. For older adults who regularly use cigars, particularly those who smoke more than one cigar per day or inhale the smoke, risk of CHD is modestly higher than that for nonsmokers (NCI 1998; Iribarren et al. 1999; Jacobs et al. 1999b; Baker et al. 2000). Studies have reported similar increases in risks for CHD and cerebrovascular disease for persons who smoke a pipe exclusively (Henley et al. 2004).
Summary
Cigarette smoking and involuntary exposure to cigarette smoke are major causes of CHD, stroke, aortic aneurysm, and PAD. The risk is seen both as an increased risk of acute thrombosis of narrowed vessels and as an increased degree of atherosclerosis in the blood vessels involved. The cardiovascular risks attributable to cigarette smoking increase with the number of cigarettes smoked and with the duration of smoking. However, risk is substantially increased even by exposure to low levels of cigarette smoke as with exposure to secondhand smoke or smoking a few cigarettes per day. Risks are not reduced by smoking cigarettes with lower machine-measured yields of tar and nicotine. Smokers of only pipes or cigars seem to have lower risks of CVD than do cigarette smokers. However, cigarette smokers who switch to pipes or cigars often inhale the tobacco smoke and may not experience the lower CVD risk of persons who primarily smoke a pipe or cigar. Stopping cigarette smoking and eliminating exposure to secondhand smoke rapidly and substantially reduce risks of various CVDs.
Secondhand Tobacco Smoke and Cardiovascular Disease
The 2006 Surgeon General’s report on involuntary exposure to tobacco smoke (USDHHS 2006) and Barnoya and Glantz (2005) extensively reviewed risks of CVD among nonsmokers exposed to secondhand tobacco smoke. They found a causal relationship among both men and women between exposure to secondhand smoke and increased risks of CHD morbidity and mortality. Pooled RRs from meta-analyses indicated a 25- to 30-percent increase in risk of CHD from exposure to secondhand smoke. The study by Whincup and associates (2004), which was based on blood levels of cotinine in men, suggested a 50- to 60-percent increase in risk of CHD from exposure to secondhand smoke. The risk of acute MI appeared to decline rapidly after cessation of exposure to secondhand smoke, as evidenced by a decline in hospital admissions for MI after smoke-free laws were put in place (Dinno and Glantz 2007; Lightwood and Glantz 2009; Meyers et al. 2009). As for stroke, the evidence was insufficient to infer a causal relationship between increased risk of CHD morbidity and mortality and exposure to secondhand smoke. Studies of the effects of secondhand smoke on subclinical vascular disease, particularly thickening of the walls of the carotid arteries, also suggest a causal relationship between exposure to secondhand smoke and atherosclerosis. As mentioned previously, the substantial CVD risk associated with involuntary exposure to cigarette smoke indicates that the risks estimated in most studies of active smoking are biased downward because the control groups generally included large numbers of persons with exposure to secondhand smoke.
Pathophysiology
This section on pathophysiology focuses primarily on mechanisms by which cigarette smoking may increase risk of CVD.
Cigarette Smoke Constituents and Cardiovascular Disease
Three constituents of cigarette smoke have received the greatest attention as potential contributors to CVD: nicotine, carbon monoxide (CO), and oxidant gases. Some research also investigated the contributions of polycyclic aromatic hydrocarbons (PAHs), particulate matter, and other constituents of tobacco smoke to the pathophysiology of CVD including atherogenesis (Brook et al. 2004; Vermylen et al. 2005; Bhatnagar 2006).
Nicotine, which is absorbed rapidly from cigarette smoke, was found in arterial blood levels of 40 to 100 ng/mL after each cigarette was smoked (Henningfield et al. 1993). The typical dose of nicotine systematically absorbed from each cigarette is 1 to 2 milligrams (mg). Although plasma nicotine levels peaked sharply after each cigarette, trough values also rose during the first six to eight hours of regular smoking during the day (Benowitz et al. 1982a). This accumulation pattern was consistent with an elimination half-life for nicotine of two hours (Benowitz et al. 1982a). In persons who smoke regularly, venous plasma levels of nicotine reached a plateau in early afternoon and remained at that level until bedtime (Figure 6.4). Significant levels of nicotine were in the smoker’s venous blood even on waking in the morning. Thus, these findings indicate that the regular smoker is exposed to significant levels of nicotine 24 hours per day.
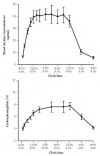
Figure 6.4
Plasma nicotine and carboxyhemoglobin concentrations throughout a day of cigarette smoking. Source: Benowitz 2003. Adapted from Benowitz et al. 1982b with permission from Elsevier, © 2003. Note: Mean (± standard error of measurement) blood (more...)
Nicotine is a sympathomimetic drug that releases catecholamines both locally from neurons and systemically from the adrenal gland. In studies of the pharmacodynamics of nicotine, the intensity of its maximal effect was greater with more rapid delivery (Porchet et al. 1987). Pharmacodynamic studies also indicated that although tolerance to the effects of nicotine developed rapidly, tolerance was incomplete (Porchet et al. 1987). In one study, a constant intravenous infusion of nicotine increased the heart rate even though nicotine levels in the blood were relatively low. As the infusion continued, the heart rate reached a plateau despite a progressive rise in blood levels of nicotine (Benowitz et al. 1982a). The same phenomenon was observed in comparisons of acceleration of heart rate with level of blood nicotine during regular cigarette smoking throughout the day (Benowitz et al. 1984).
In another study, heart rate measured by ambulatory monitoring was higher throughout the day when persons were smoking than when they were not smoking (Benowitz et al. 1984). The extent of elevation was independent of the blood level of nicotine absorbed from the cigarettes. The researchers concluded that the elevated heart rate reflected persistent stimulation of the sympathetic nervous system, a possible contributing factor to CVD. Nicotine may also contribute to endothelial dysfunction, lipid abnormalities, and insulin resistance (Benowitz 2003).
CO is a major constituent of cigarette smoke. In regular smokers, carboxyhemoglobin levels average about 5 percent, compared with 10 percent or higher in heavy smokers (Benowitz et al. 1982b). These values compare with levels of 0.5 to 2 percent in nonsmokers, depending on exposure to automobile exhaust. Like nicotine levels, elevated carboxyhemoglobin levels persist for 24 hours a day in smokers (Figure 6.4).
CO exposure can aggravate ischemia and worsen symptoms in persons with vascular disease, although it is not clear that CO contributes directly to atherosclerosis (Benowitz 2003). CO binds avidly to hemoglobin, reducing the amount of hemoglobin available to carry oxygen and impeding release of oxygen by hemoglobin. In some studies, inhalation of CO at levels comparable to those in cigarette smokers reduced exercise tolerance in patients with angina pectoris, intermittent claudication, or COPD (Calverley et al. 1981; Allred et al. 1989). Another study reported that CO exposure in persons with obstructive coronary disease resulted in a greater degree of exercise-induced ventricular dysfunction and an increase in the number and complexity of ventricular arrhythmias during exercise (Sheps et al. 1990). Inhaling CO reduced the threshold for ventricular fibrillation in animals (DeBias et al. 1976).
Long-term CO exposure in smokers resulted in greater red blood cell mass and reduced the oxygen- carrying capacity of red blood cells, resulting in relative hypoxemia (Benowitz 2003). In response to hypoxemia, red blood cell masses increased to maintain the amount of oxygen needed by organs in the body. The increase in red blood cell mass increased blood viscosity and may contribute to hypercoagulation in smokers.
Cigarette smoke delivers a high level of oxidizing chemicals to smokers, including oxides of nitrogen and many free radicals from both the gas and tar phases of cigarette smoke (Church and Pryor 1985). Exposure to oxidant chemicals in smoke was associated with depletion of endogenous levels of antioxidants, manifested as lower blood levels of vitamin C in smokers than in nonsmokers (Lykkesfeldt et al. 2000). Cigarette smoking also was reported to increase levels of lipid peroxidation products in the plasma and urine of smokers (Morrow et al. 1995). Study results also indicated that oxidant stress contributes to several potential mechanisms of CVD, including inflammation, endothelial dysfunction, lipid abnormalities such as oxidation of low-density lipoprotein (LDL), and platelet activation (Burke and FitzGerald 2003).
Acrolein, a reactive aldehyde produced by endogenous lipid peroxidation, is present at high levels in cigarette smoke. Acrolein binds covalently to form protein adducts, and acrolein-induced modification of proteins has been implicated in atherogenesis. Acrolein modifies apolipoprotein A-I (APO A-I), the major protein in HDL (Shao et al. 2005). HDL protects against atherosclerosis. Acrolein-protein adducts co-localize with APO A-I in macrophages in the intima of human atheromatous blood vessels (Szadkowski and Myers 2008).
Acrolein also oxidized thioredoxins 1 and 2 in endothelial cells. Thioredoxins are prominent antioxidant proteins that regulate the oxidation-reduction balance critical for normal cell function. These results suggest that oxidation of thioredoxins can result in dysfunction and death of endothelial cells, contributing to atherosclerosis. In addition, acrolein induces production of the enzyme cyclooxygenase-2 (COX-2) in human endothelial cells in vitro (Park et al. 2007). This finding is relevant because COX-2 is expressed in atherosclerotic lesions and may participate in atherogenesis. Acrolein may contribute to thrombogenicity in smokers by inhibiting antithrombin activity (Gugliucci 2007). Finally, acrolein induces hypercontraction in isolated human arteries and could contribute to smoking-induced coronary vasospasm (Conklin et al. 2006).
Cigarette smoke contains a number of metals, including aluminum, cadmium, copper, lead, mercury, nickel, and zinc. Metals in cigarette smoke catalyze the oxidation of cellular proteins (Bernhard et al. 2005). This reaction may lead to structural damage, endothelial dysfunction, and detachment of endothelial cells from the walls of blood vessels. Mixtures of metals and oxidants may be particularly damaging to endothelial cells. Cadmium levels are higher in serum of smokers, and cadmium accumulates in the aortic walls of smokers (Abu-Hayyeh et al. 2001). Epidemiologic evidence indicates an association between serum levels of cadmium and lead and CVD, including hypertension and MI (Abu-Hayyeh et al. 2001).
PAHs found in the tar fraction of cigarette smoke reportedly accelerated atherosclerosis in experimental animals. Weekly injections of benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene, at doses below those that produce tumors, increased development of atherosclerotic plaque in the aortas of cockerels (Penn and Snyder 1988). Similarly, inhaled butadiene, a component of the vapor phase of cigarette smoke, increased the amount of atherosclerotic plaque in the same animal model (Penn and Snyder 1996). The researchers speculated that one mechanism of atherogenesis is a mutation, followed by hyperproliferation of smooth muscle or other cells that may contribute to growth of atherosclerotic plaque.
Studies of the cardiovascular effects of smokeless tobacco may be informative for understanding the pathophysiology of smoking-induced CVD. Oral and nasal smokeless tobacco products have been used for centuries around the world (International Agency for Research on Cancer [IARC] 2007). Traditional smokeless tobacco products vary widely among countries; however, similar to Sweden, forms of oral snuff are the most common types of products used in the United States (Substance Abuse and Mental Health Services Administration 2009). These products contain a large array of chemicals, including nicotine, nitrosamines, nitrosamine acids, PAHs, aldehydes, and metals (IARC 2007). A recent systematic review reported that studies from both the United States and Sweden showed an increased risk of death from MI and stroke related to the frequency and duration of use of smokeless tobacco products (Boffetta and Straif 2009). This review relied heavily (85–89 percent of the weight) on results of a large U.S. cohort study conducted in two waves between 1959–1971 and 1982–1988 and may not represent risk associated with products currently marketed in the United States and Europe. As in cigarettes, nicotine is the principal alkaloid in smokeless tobacco products, and the concentrations of nicotine (mg/gram [g] tobacco) are similar between cigarettes and the types of oral snuff sold in the United States (Djordjevic and Doran 2009). An analysis comparing the effects of using oral snuff with those of smoking cigarettes provided insights into the role of nicotine versus the effects of other toxins from tobacco smoke on CVD and cardiovascular risk factors (Benowitz et al. 1988, 1989). In addition clinical trials of nicotine patches in patients with known CVD have not shown that transdermal nicotine increased cardiovascular risk (Working Group for the Study of Transdermal Nicotine in Patients with Coronary Artery Disease 1994; Joseph et al. 1996). In the study of 3,094 middle-aged smokers with chronic obstructive lung disease, the U.S. Lung Health Study found no evidence of increased cardiovascular risk in subjects who quit smoking by using nicotine gum versus those who quit without use of nicotine gum (Murray et al. 1996). These studies and related evidence suggest that chemicals other than nicotine may contribute to the elevated risk of death from MI and stroke. In the INTERHEART study, the OR of acute MI was 2.23 among those who used only smokeless tobacco compared with those who used no tobacco. The OR was comparable to that of current cigarette smokers (OR = 2.95) compared with those who used no tobacco (Teo et al. 2006). In addition, the risk of acute MI among smokers who also used smokeless tobacco was the highest risk related to tobacco use (OR = 4.09), suggesting that some of the toxicants involved in the elevated cardiovascular risk could be contained in both tobacco smoke and smokeless products. Smokeless tobacco products have been found to have significant amounts of numerous other toxicants and carcinogens, particularly tobacco-specific nitrosamines as well as volatile aldehydes and PAHs (Stepanov et al. 2008). Additional research on these and other toxicants in smokeless tobacco, such as heavy metals like cadmium, is needed to understand the observed cardiovascular risks among users of smokeless tobacco products.
Mechanisms
Cigarette smoking produces acute myocardial ischemia by adversely affecting the balance of demand for myocardial oxygen and nutrients with myocardial blood supply (Figure 6.5). The increase in demand for oxygen in the myocardium is a consequence of nicotine stimulation of the sympathetic nervous system and the heart. Cigarette smoking acutely increases levels of plasma norepinephrine and epinephrine and enhanced 24-hour urinary excretion of these catecholamines (review by Benowitz and Gourlay 1997). Regular smoking increases the heart rate both in the short term (up to 20 beats per minute) and throughout the day (average increase, 7 beats per minute), as measured during ambulatory monitoring. Nicotine also increases heart rate, blood pressure, and myocardial contractility. These hemodynamic changes result in increases in myocardial work that in turn require increased myocardial blood flow.
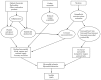
Figure 6.5
Overview of mechanisms by which cigarette smoking causes an acute cardiovascular event. Source: Benowitz 2003. Reprinted with permission from Elsevier, © 2003.
In healthy persons, cigarette smoking increases coronary blood flow in response to increases in myocardial work. In smokers, the response in coronary blood flow to increased myocardial demand was impaired (i.e., reduced coronary vasodilatory reserve) (Czernin and Waldherr 2003). Cigarette smoking played a direct role by constricting coronary arteries through nicotine-mediated action on α-adrenergic receptors and by induction of endothelial dysfunction by nicotine and oxidizing chemicals (Nicod et al. 1984; Puranik and Celermajer 2003). In addition, oxidant chemicals contribute to platelet activation and thrombogenesis (Burke and FitzGerald 2003).
Exposure to CO may also contribute to the adverse hemodynamic effects of cigarette smoking. By producing functional anemia, CO increases the need for coronary blood flow, especially during physical exertion. An in-adequate vasodilatory flow reserve produced by cigarette smoking, in the face of need for increased coronary blood flow mediated by carbon dioxide, could contribute to myocardial ischemia with exercise in smokers.
In addition to the mechanisms described in Figure 6.5, cigarette smoking has effects on inflammation, insulin sensitivity, and lipid abnormalities that most likely contribute to smoking-induced CVD.
Hemodynamic Effects
Blood Pressure and Heart Rate
In 1907, Erich Hesse published “The Influence of Smoking on the Circulation,” which documents his observations on the effects of smoking on heart rate and blood pressure (Hesse 1907). In most study participants, both heart rate and blood pressure increased immediately after smoking. Hesse observed a greater response in blood pressure after smoking in persons who smoked than in non-smokers. Speculating that the increases in blood pressure and heart rate reflected stimulation of the heart or nervous system, he instituted a rule prohibiting patients with a “heart weakness” from smoking, to avoid unnecessary strain and stress for the heart muscle. Many investigators confirmed Hesse’s observations on the hemodynamic effects of cigarette smoking (Deanfield et al. 1986; Czernin et al. 1995; Barutcu et al. 2004).
The positive chronotropic, inotropic, and blood pressure effects of smoking are explained by nicotine-induced activation of the sympathetic nervous system (review by Benowitz 2003). Nicotine promotes the release of epinephrine and norepinephrine from the adrenal medulla and terminal nerve endings, resulting in increased heart rate and greater contractility through stimulation of myocardial β1 receptors. Peripheral vascular resistance increases through α-receptor mediated vasoconstriction that in turn increases blood pressure. Coronary β2 and α2 receptors are also stimulated: stimulation of β2 receptors promoted vasodilation, and stimulation of α2 receptors promoted vasoconstriction (Cryer et al. 1976; Benowitz 2003).
Cryer and colleagues (1976) elucidated the mechanisms behind observed hemodynamic changes during smoking. They observed more than a 150-percent increase in plasma epinephrine levels (from 44 to 113 picograms [pg]/mL) at 10 minutes after participants started to smoke a cigarette. Norepinephrine values increased to a smaller degree (from 227 to 315 pg/mL) at 12.5 minutes after the start of smoking. These increases were associated with significant increases in heart rate and blood pressure. Pretreatment with α-receptor blockers and β-receptor blockers had little effect on the increase in plasma levels of catecholamines, but increases in blood pressure and heart rate were eliminated. This study confirmed that smoking-induced increases in blood pressure and heart rate are attributable to adrenergic mechanisms.
The hemodynamic effects of cigarette smoking are mediated primarily by nicotine, although oxidizing chemicals in tobacco smoke also affect vascular function. Intravenous nicotine, nicotine nasal spray, and nicotine chewing gum all increased the heart rate up to 10 to 15 beats per minute and raised systolic blood pressure by up to 5 to 10 millimeters of mercury (mm Hg), responses similar to the effects of cigarette smoking (Gourlay and Benowitz 1997). Nicotine increased cardiac output by increasing both heart rate and myocardial contractility. Different vascular beds express different types and ratios of adrenergic receptors. Therefore, not all vascular responses to nicotine or tobacco smoke are the same. For example, nicotine constricts some vascular beds, such as the skin, and cutaneous vasoconstriction explains the reduced temperature of the fingertip observed with administration of nicotine (Benowitz et al. 1982a). Conversely, nicotine appears to dilate other vascular beds, such as skeletal muscle (Diana et al. 1990). Vasodilation of skeletal muscle may partly result from the increase in cardiac output, although the release of epinephrine from nerve terminals may also contribute. The net result of increases in heart rate, blood pressure, and myocardial contractility is an increase in myocardial work, followed by increased myocardial blood flow.
Coronary Blood Flow
An important hemodynamic consequence of cigarette smoking is its effect on blood flow in the coronary arteries. Cigarette smoking acutely increased coronary blood flow by up to 40 percent, apparently a response to the increase in myocardial work (review by Czernin and Waldherr 2003).
In anesthetized dogs, coronary blood flow showed a biphasic response to nicotine. Initially, researchers hypothesized that increases in coronary blood flow—in the large coronary vessels as well as the smaller vessels—resulted from an increase in myocardial metabolic demand.
Cigarette smoking impairs the response of coronary blood flow to an increase in myocardial demand for oxygen; that is, it reduces the coronary vasodilatory flow reserve. Thus, the increase in coronary blood flow based on the level of myocardial work is less than would be expected in the absence of exposure to tobacco smoke. Considerable evidence indicates that cigarette smoking causes dysfunction of the coronary arterial endothelium (see “Endothelial Injury or Dysfunction” later in this chapter).
Cigarette smoking may also be associated with coronary vasoconstriction. Although cigarette smoking increases coronary blood flow in a person who does not have CHD, it may decrease coronary blood flow in the presence of coronary disease. Regan and colleagues (1960) measured coronary sinus blood flow in seven male volunteers with documented CHD before and after they smoked two cigarettes during a period of about 25 minutes; cardiac work increased by about 30 percent during smoking. Even so, in response to smoking, coronary blood flow fell in three patients, did not change in three patients, and increased in one patient. Similar paradoxical responses were observed after long-term smokers were exposed to cold by testing with cold pressors (Campisi et al. 1998). In the testing, the hand is immersed in ice water for one to two minutes. This painful procedure evokes a mixed adrenergic response involving coronary receptors α1 and α2 (vasoconstriction), β2 (vasodilation), and myocardial β1 (indirect coronary vasodilation). Endothelial α2 receptors that promote indirect coronary vasodilation may also be involved. In healthy volunteers, cold-induced increases of about 30 percent in the product of heart rate and blood pressure were associated with appropriate and similar increases in blood flow. In contrast, smokers showed no measurable increases in blood flow in response to the cold. Campisi and colleagues (1998) ascribed this observation to coronary endothelial dysfunction.
Kaufmann and colleagues (2000) provided additional evidence for the association between smoking and endothelial dysfunction. Using quantitative positron emission tomography, they found that coronary microvascular function was abnormal in smokers but could be restored by infusion of vitamin C. Earlier, Zeiher and colleagues (1995) used quantitative coronary angiography to measure the effects of long-term smoking on the diameter of the coronary artery at study entry, during flow-mediated coronary vasodilation, and after intracoronary administration of nitroglycerin. The study population consisted of patients with or without mild disease of the left anterior descending coronary artery and no other cardiac problems. Flow-mediated vasodilation was markedly blunted in long-term smokers. In a study of smokers with CHD, cigarette smoking increased coronary vascular resistance, an effect that can be blocked by α-adrenergic blockers. This finding indicates that the mechanism of increased coronary artery resistance is at least partly due to stimulation of the sympathetic nervous system by nicotine.
Intracoronary measurements by Doppler ultrasonography demonstrated that cigarette smoking constricts epicardial arteries and increases total coronary vascular resistance. This result indicates that impairment of coronary blood flow by cigarette smoking results from constriction of both epicardial and resistance blood vessels. In one study, after pretreatment with calcium-channel-blocking agents or nitroglycerin, cigarette smoking increased coronary blood flow in patients with CHD who had manifested no increase after cigarette smoking alone (Winniford et al. 1987). This finding that coronary vasodilator drugs, which block chemically mediated vasoconstriction, permit the usual increase in coronary blood flow in response to increased myocardial work supports the hypothesis that cigarette smoking directly produces coronary vasoconstriction. In another study, chewing 4 mg of nicotine gum by healthy nonsmokers blunted the increase in coronary blood flow that occurs with increased heart rate produced by cardiac pacing (Kaijser and Berglund 1985). This result confirmed that even low doses of nicotine can directly constrict coronary arteries in humans.
Another study found that nicotine worsened myocardial dysfunction in “regionally stunned” ischemic myocardium of anesthetized dogs (Przyklenk 1994). In a placebo-controlled experiment, transient ischemia was induced in dogs by clamping the left anterior descending coronary artery for 15 minutes. Segmental shortening of the myocardium recovered to only 29 percent of the preischemic baseline values in dogs pretreated with nicotine, compared with 54 percent in saline-treated control dogs. The doses of nicotine administered to the animals did not alter heart rate, blood pressure, or blood flow or cause myocyte necrosis.
In patients with vasospastic angina, cigarette smoking is associated with increased occurrence of the condition and a poorer response to medication compared with the response in nonsmokers (Caralis et al. 1992). The researchers observed that cigarette smoking during coronary angiography (cardiac catheterization) produced an acute coronary vasospasm.
Schelbert and colleagues (1979) extensively studied the relationship of coronary vasomotion, endothelial function, and myocardial blood flow to potential reversibility of coronary vasomotor abnormalities. They adopted a noninvasive approach by measuring blood flow with positron emission tomography using 13N ammonia: (1) at rest during testing with cold pressors to probe endothelium-dependent coronary vasomotion and (2) during dipyridamole-induced hyperemia to assess endothelium-independent coronary vasomotion. Using this protocol, Czernin and associates (1995) investigated the effects of short- and long-term smoking on myocardial blood flow and flow reserve in smokers. The investigators sought to determine whether abnormalities in coronary vasomotion in response to cold could be reversed in response to l-arginine infusion (Campisi et al. 1998, 1999). The findings indicated that short-term and long-term smokers had normal coronary vasodilatory capacity. Short-term smoking, however, reduced flow reserve in both short- and long-term smokers. Also, exposure of long-term smokers to cold resulted in abnormal blood flow. The smoking-associated abnormalities in vasomotion were restored by intravenous l-arginine, and this result further implicates the endothelium as the target of toxic substances contained in cigarette smoke. Campisi and colleagues (1999) quantified myocardial blood flow during exposure to cold while l-arginine, the substrate of ENOS, was infused intravenously for 45 minutes at a dose of 30 g as 10 percent arginine hydrochloride. This infusion produced significant improvement in responses of myocardial blood flow to cold. In addition to active smoking, exposure to secondhand smoke for 30 minutes abruptly reduces coronary blood flow velocity in nonsmokers, as assessed by echocardiography (Otsuka et al. 2001).
Summary
Cigarette smoking impairs the vascular endothelial function and activates the sympathetic nervous system. These effects can result in inappropriate reduction in or failure to increase coronary blood flow in response to increases in myocardial demand. Together with long-term atherosclerotic damage from smoking, these effects contribute to ischemic cardiac events. Coronary endothelial dysfunction clearly increases the risk for cardiovascular events. The smoking-induced alterations in vasomotor function appear to be substantially reversible, which underscores the importance of smoking cessation programs and policies to promote a smoke-free environment.
Smoking and the Endothelium
Endothelial Injury or Dysfunction
Endothelial injury and dysfunction are thought to contribute to the initiation of atherogenesis and to have a major role in acute cardiovascular events. Cigarette smoking produces endothelial injury and dysfunction in both peripheral and coronary arteries. Other cardiovascular risk factors such as hypercholesterolemia, diabetes, and hypertension also produce endothelial dysfunction.
The healthy endothelium is a diaphanous film of tissue that invests the luminal surface of all blood and lymphatic vessels. In larger-conduit vessels, the endothelium forms a monolayer between the circulating blood and the vessel wall. The tissue capillaries, which are the smallest conduits for blood and lymphatic flow, are composed exclusively of endothelial cells. Because of the ubiquity of endothelial cells, the surface area of the endothelium in a human weighing 70 kilograms (kg) is 1,000 to 4,000 square meters—equivalent to two to four tennis courts—with a weight of approximately 1 kg (Wolinsky 1980). The endothelium produces a variety of paracrine factors that regulate vascular homeostasis, including proteins, lipids, and small molecules that (1) can relax or activate the underlying vascular smooth muscle, (2) regulate the interaction of the vessel wall with circulating blood elements, and (3) modulate vessel structure (Aird 2005). In healthy persons, the endothelium primarily exerts a vasodilator influence that reduces vascular resistance and maintains blood flow. The endothelium maintains the blood’s fluidity by elaborating anticoagulant substances and generally resists adherence of platelets and infiltration of immune cells.
Regeneration of Endothelium
With aging, the normal functioning of the endothelium requires replacement of apoptotic or injured cells. Normally, the turnover of endothelial cells is low, on the order of 0.1 percent of the cells undergoing mitosis at any time (Wright 1972). The rate of endothelial turnover increases, however, in areas of disturbed flow (bends, branches, or bifurcations of blood vessels). The length of chromosome telomeres documents that endothelial aging occurs more rapidly in these areas (Chang and Harley 1995). Furthermore, the accelerated aging in these areas may lead to focal senescence, which is demonstrated by impaired endothelium-dependent vasodilation (McLenachan et al. 1990).
Persons who smoke may have impaired ability to regenerate the endothelium. The endothelial monolayer is regenerated in part from circulating endothelial progenitor cells derived from bone marrow, and the supply of these cells may be a key determinant of endothelial health. The number of circulating endothelial progenitor cells, which is estimated by ex vivo colony counts or by analysis using fluorescence-activated cell sorting, is directly associated with the ability of the endothelium to induce vasodilation (Hibbert et al. 2003; Hill et al. 2003). Smokers have reduced numbers of circulating endothelial progenitor cells and impaired endothelium-dependent vasodilation (Vasa et al. 2001, Hill et al. 2003). In addition, smoking cessation was associated with a rebound in the number of circulating endothelial progenitor cells and improvement in endothelium-dependent vasodilation (Moreno et al. 1998; Kondo et al. 2004).
Endothelial Dysfunctions
A variety of endothelial dysfunctions may contribute to disorders of vessel tone and structure that precede clinical vascular disease. Cardiovascular risk factors such as hypercholesterolemia, hypertension, diabetes, and use of tobacco cause endothelial aberrations long before clinical vascular disease becomes evident. Endothelial dysfunction is the first step in vascular disease, because it leads to vascular inflammation, cell proliferation, and thrombosis, which contribute to progression of vascular disease.
Endothelial generation of adhesion molecules increases in smokers, as evidenced by higher plasma levels of soluble adhesion molecules (Blann et al. 1997, 1998). These molecules include soluble forms of the vascular cell adhesion molecule (sVCAM) and the intercellular adhesion molecule (sICAM). The soluble adhesion molecules, which are shed from the endothelium, reflect the increased endothelial production of these adhesion molecules in the context of vascular inflammation. Endothelial adhesion molecules are required for adherence to blood leukocytes and their infiltration into the vessel wall (Gimbrone 1995). The increased elaboration of adhesion molecules is an endothelial dysfunction that promotes leukocyte infiltration, vascular inflammation, and progression of atherosclerosis. Studies have associated elevated levels of either sVCAM or sICAM with increased risk of cardiovascular events (Blankenberg et al. 2001).
Smoking also impairs the ability of the endothelium to resist thrombosis. Compared with nonsmokers, smokers have higher levels of von Willebrand factor protein (MacCallum 2005) and tissue factor (Matetzky et al. 2000; Sambola et al. 2003), which may be generated by the endothelium. Tissue factor activates the coagulation cascade, and von Willebrand factor protein mediates adherence of platelets to the vessel wall (MacCallum 2005). Furthermore, study findings indicate that smoking impairs capacity to lyse the thrombus that is formed. Plasma levels of tissue plasminogen activator (tPA), a thrombolytic protein produced by the endothelium, are reduced in smokers (Newby et al. 2001). In contrast, smoking increases levels of plasminogen activator inhibitor-1 (PAI-1) (Simpson et al. 1997). By interfering with the function of tPA, PAI-1 reduces thrombolytic capacity (MacCallum 2005). Imbalance in thrombolytic capacity attributable to higher PAI-1 values or reduction in tPA levels is associated with occurrence of adverse cardiovascular events.
The healthy endothelium elaborates vasodilator substances such as nitric oxide (NO), prostacyclin, atrial natriuretic peptide, endothelium-derived hyperpolarizing factor, and adrenomedullin (Chen and Burnett 1998; Busse and Fleming 2003; Brain and Grant 2004). In doing so, the healthy endothelium increases the diameter of the blood vessels and reduces resistance to blood flow. When the endothelium becomes diseased, synthesis and bioactivity of the vasodilators are reduced, and the balance tips in favor of endothelium-derived vasoconstrictors such as endothelin and thromboxane (Vanhoutte et al. 2005). This derangement in endothelial function has clinical consequences. Because vasodilator function is impaired, coronary vascular resistance increases, and ischemia can result. Furthermore, endothelial vasodilator dysfunction in the coronary arteries of humans is associated with reversible myocardial perfusion defects, which are associated with other vascular abnormalities (Hasdai et al. 1997b). These abnormalities include expression of adhesion molecules, adherence and infiltration of leukocytes, and proliferation of smooth muscle cells.
Most of the endothelium-derived vasodilators also oppose key processes involved in atherogenesis (cell adhesion, proliferation, and inflammation) (Cooke and Dzau 1997a,b). Thus, by reducing the generation or bioactivity of endothelial vasodilators, exposure to tobacco can accelerate atherosclerosis. This mechanistic explanation for tobacco-related CVD is supported by the finding that dysfunction of endothelial vasodilators is an independent predictor of vascular events (Schächinger et al. 2000; Suwaidi et al. 2000; Gokce et al. 2003). The role of these mechanisms involving NO is vascular protection, which is impaired by exposure to tobacco.
Nitric Oxide and Vascular Homeostasis
NO induces vasodilation by stimulating soluble guanylate cyclase to produce cyclic guanosine monophosphate (Ignarro et al. 1984). NO, which has a short half-life, avidly interacts with sulfhydryl-containing proteins, heme proteins, and oxygen-derived free radicals. By virtue of its ability to nitrosylate proteins, NO may change their activity or behavior (Hess et al. 2005). The significant increase in vascular resistance induced in animals and humans exposed to pharmacologic antagonists of ENOS reflects the physiological importance of this endothelium-derived vasodilator (Rees et al. 1989; Vallance et al. 1989).
Endothelium-derived NO also inhibits adherence of platelets and leukocytes to the vessel wall (Kubes et al. 1991; Tsao et al. 1994). This effect is mediated partly by activation of cyclic guanosine monophosphate and phosphorylation of intracellular signaling proteins such as vasodilator-stimulated phosphoprotein (Smolenski et al. 1998). In addition, NO suppresses expression of adhesion molecules and chemokines that regulate endothelial interactions with circulating blood elements (Tsao et al. 1996, 1997). These observations suggest that NO is an endogenous antiatherogenic molecule. Impairment of ENOS contributes to the pathologic alterations in vascular reactivity and structure observed in atherosclerosis (Cooke and Dzau 1997a,b). The pharmacologic inhibition or genetic deficiency of ENOS inhibits endothelium- dependent vasodilation, impairs blood flow in tissues, and raises blood pressure (Huang et al. 1995; Kielstein et al. 2004). Furthermore, NO deficiency promotes adherence to and intimal accumulation of mononuclear cells and accelerates formation of lesions in animal models of atherosclerosis (Kuhlencordt et al. 2001). In contrast, enhancing production of NO in the vessel wall slows or even reverses atherogenesis and restenosis (Cooke et al. 1992; von der Leyen et al. 1995; Candipan et al. 1996). NO is a survival factor for endothelial cells, and it induces apoptosis of macrophages and proliferation of vascular smooth muscle cells (Wang et al. 1999).
Certain polymorphisms of the ENOS gene predict development of CHD (Ichihara et al. 1998; Tsukada et al. 1998; Yoshimura et al. 1998). The ENOS gene GLU298ASP polymorphism is more prevalent in patients with variant angina, essential hypertension, and acute MI (Hibi et al. 1998; Miyamoto et al. 1998; Shimasaki et al. 1998; Yoshimura et al. 1998). Intriguingly, this polymorphism is associated with greater sensitivity to the effects of smoking on endothelial vasodilator function. Young men who are carriers of the ENOS *ASP298 allele have increased susceptibility to smoking-associated reduction in endothelial function (Leeson et al. 2002). Similarly, a quadruple repeat of a sequence of 27 base pairs in *intron 4 of the ENOS gene (allele *a) is associated with increased risk of CHD and acute MI (Ichihara et al. 1998). Smokers who are homozygous for the ENOS allele *a are at risk for more severe CHD than are those who are not homozygous (Wang et al. 1996).
Endothelium-Dependent Vasodilation
The effect of exposure to tobacco on endothelium-dependent vasodilation in humans was assessed by observing its effect on flow-mediated vasodilation. As blood flow through a vessel is increased, the vessel relaxes. In animal models, this flow-mediated vasodilation was abolished by removing the endothelium (Pohl et al. 1986). When a pharmacologic antagonist of ENOS was used, flow- mediated vasodilation in the rabbit iliac artery depended on the endothelial release of NO (Cooke et al. 1991). Celermajer and colleagues (1992) used duplex ultrasonography to record flow-mediated vasodilation of the brachial artery in response to hyperemic vasodilation of the forearm. The investigators induced vasodilation by using a blood pressure cuff inflated to suprasystolic pressures to transiently occlude blood flow in the forearm. Joannides and colleagues (1995) extended this finding by showing that flow-mediated vasodilation of the brachial artery could be abolished by pharmacologic antagonism of ENOS. Subsequently, numerous studies used this approach to document impairment of flow-mediated, endothelium-dependent vasodilation in smokers and in persons exposed to secondhand smoke (Celermajer et al. 1993, 1996; Barua et al. 2001). Researchers also observed that tobacco use impaired endothelium-dependent vasodilation in the coronary microcirculation. Intracoronary infusion of acetylcholine induced vasodilation that was partly attributable to release of NO, and this response was blunted in persons who smoked (Kugiyama et al. 1996).
Impairment of Endothelium-Dependent Vasodilation
Many factors contribute to the ability of tobacco to impair endothelial function. Tobacco use adversely affects the ENOS pathway. Exposure of cultured endothelial cells derived from human coronary arteries or umbilical veins to sera from smokers reduced expression and activity of ENOS (Barua et al. 2001, 2003). The researchers attributed this effect partly to the oxygen-derived free radicals in tobacco smoke. In addition, the half-life of NO was markedly shortened by oxidative stress (Rubanyi and Vanhoutte 1986).
Superoxide anion reacts avidly with NO to form a peroxynitrite (ONOO−) anion, which itself is a highly reactive free radical (Beckman and Koppenol 1996). Other sources of free radicals that may inactivate NO and impair endothelial vasodilator function include activated leukocytes, xanthine oxidase, the mitochondrial electron-transport chain, and uncoupling of ENOS itself (Heitzer et al. 2000; Barua et al. 2003; Kayyali et al. 2003; Sydow and Münzel 2003; Guthikonda et al. 2003). Investigators postulated that this uncoupling occurs when amounts of the NOS cofactor tetrahydrobiopterin or the NO precursor l-arginine are insufficient. These conditions involve transfer of electrons from ENOS to oxygen and thus formation of a superoxide anion.
Oxidative stress also impairs the NOS pathway by increasing accumulation of asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of ENOS produced by all cells during degradation of methylated nuclear proteins (Vallance et al. 1992; Tran et al. 2003). Most of the ADMA produced is degraded by the enzyme dimethylarginine dimethylaminohydrolase. Oxidative stress impairs the activity of this enzyme and leads to accumulation of ADMA and suppression of ENOS (Cooke 2004).
Blood levels of antioxidant vitamins are lower than normal in smokers, reflecting endogenous consumption of these vitamins in response to ongoing oxidant stress (Lykkesfeldt et al. 2000). Administration of vitamin C reverses the impairment of endothelium-mediated vasodilation in smokers, a finding consistent with an oxidant mechanism of endothelial dysfunction (Heitzer et al. 1996).
Nicotine itself may injure endothelial cells. Studies showed that in levels similar to those found in the blood of cigarette smokers, nicotine altered structural and functional characteristics of cultured vascular smooth muscle and endothelial cells (Csonka et al. 1985; Thyberg 1986). In one study, oral nicotine administered to rats to achieve blood levels comparable to those in human smokers produced myointimal thickening of the aorta after an experimental injury (denudation of the endothelium with a balloon catheter) (Krupski et al. 1987). The excessive myointimal thickening in nicotine-treated animals is consistent with persistent injury to endothelial cells. Studies reported increased numbers of circulating endothelial cells in the venous blood, reflecting endothelial injury, and decreased platelet aggregate ratios, reflecting platelet aggregation in persons who had never smoked but who, for experimental purposes, smoked cigarettes containing tobacco (Davis et al. 1985). These results were not observed in nonsmokers who smoked cigarettes that did not contain tobacco. These findings further support a role for nicotine in injury to endothelial cells.
Nicotine may influence endothelial function in other ways. In studies of cultured endothelial cells, nicotine enhanced release of the basic fibroblast growth factor and inhibited production of transforming growth factor β1 (Villablanca 1998; Cucina et al. 1999). Nicotine also increased DNA synthesis, mitogenic activity, and endothelial proliferation. Nicotine has been shown to induce endothelial dysfunction (Chalon et al. 2000; Sarabi and Lind 2000; Neunteufl et al. 2002).
Pathologic Angiogenesis
Another endothelial function influenced by exposure to tobacco is development of new blood vessels (angiogenesis). Angiogenesis requires activation of endothelial cells by an angiogenic cytokine, followed by endothelial cell proliferation and migration and the formation of tubes. Exposure to secondhand smoke promotes tumor angiogenesis and growth (Zhu et al. 2003). Human lung cancer cells implanted in a subcutaneous or orthotopic location grew more rapidly in mice when they were exposed to secondhand tobacco smoke. Furthermore, these mice had higher plasma levels of vascular endothelial growth factor, and capillary density in their tumor nodules was greater than that in control mice. Mecamylamine, an antagonist of the nicotinic acetylcholine receptor (nAChR), abolished the effects of exposure to secondhand smoke. These observations indicate that secondhand smoke increases tumor angiogenesis and growth, at least partly through a nicotine-mediated mechanism.
Researchers observed the effect of nicotine in promoting pathologic angiogenesis in numerous murine models of tobacco-related diseases, including lung cancer, atherosclerosis, and retinopathy (Heeschen et al. 2001, 2002; Natori et al. 2003; Shin et al. 2004; Suñer et al. 2004). Tumor angiogenesis was required for tumor growth, and correspondingly, promotion of tumor angiogenesis accelerated tumor growth (Folkman 2003). Conversely, antiangiogenic agents inhibit progression of cancer and are now approved as treatment for some advanced human malignant diseases (Jain 2005). The effect of nicotine on promotion of tumor angiogenesis may be attributable to a direct effect on endothelial cells. In clinically relevant levels, nicotine promoted endotheliaprocesses that may be involved in tumor angiogenesis. At these doses, nicotine promoted survival, proliferation, and migration of endothelial cells (Heeschen et al. 2001, 2002). Nicotine also induced elaboration and release of angiogenic factors, including NO, prostacyclin, vascular endothelial growth factor, and fibroblast growth factor (Carty et al. 1996; Heeschen et al. 2001a; Lane et al. 2005). These effects of nicotine were mediated by nAChRs on the endothelium (Heeschen et al. 2002). Conversely, pharmacologic inhibition or genetic deficiency of endothelial nAChRs reduced angiogenic response to nicotine. A variety of human lung cancer cells synthesized acetylcholine or expressed nAChRs. Nicotine increased growth of these cells in vitro, but this effect was inhibited by antagonists of the nAChRs (Schuller 2002).
The importance of pathologic angiogenesis in growth of tumors is well known. Less widely recognized, however, is the role of neovascularization in progression of atherosclerotic plaque. Large atherosclerotic plaques in human coronary arteries are well vascularized by microvessels originating from the vasa vasorum (Barger et al. 1984). In mice with hypercholesterolemia, growth of atheroma can be inhibited by antiangiogenic agents, and in the same murine models, nicotine promoted neovascularization of plaque and its progression in the aorta (Heeschen et al. 2001; Moulton et al. 2003). The effect of nicotine in increasing neovascularization and progression of plaque may partially explain increased risk of atherosclerotic disease in persons who smoke.
Similarly, in a model of age-related macular degeneration in mice, nicotine stimulated retinal neovascularization (Suñer et al. 2004). This effect was antagonized by hexamethonium, another antagonist of nAChRs. In a clinical study, the most virulent form of age-related maculopathy is associated with retinal neovascularization that contributes to visual deterioration, and tobacco smokers are at greater risk of age-related macular degeneration than are nonsmokers (Christen et al. 1996; Seddon et al. 1996). Thus, a variety of tobacco-related diseases are characterized by pathologic neovascularization, an effect that may be promoted by nicotine.
Notwithstanding nicotine’s effect as a promoter of neovascularization, long-term exposure to tobacco may impair therapeutic angiogenesis. In a murine model of hindlimb ischemia, short-term exposure to nicotine paradoxically increased capillary density and improved regional blood flow in the ischemic hindlimb (Heeschen et al. 2001, 2003). However, long-term exposure to nicotine for 16 weeks (about one-third of the life span of a mouse) before induction of ischemia obliterated angiogenic response to nicotine (Konishi et al. 2010).
The relevance of animal models for research on nicotine and angiogenesis to human smokers is not clear. It is important to differentiate studies that show effects of pure nicotine from those in which the exposure is to tobacco smoke. In mice, effects of nicotine on angiogenesis depended on release of NO, but the net effect of smoking in humans seems to be impaired release of NO. Also, most studies on angiogenesis involve short-term administration of nicotine, although tolerance to the effects of nicotine may develop with long-term use.
Inhibition of ACE normalized impaired bradykinin-mediated, endothelium-dependent venodilation in smokers (Chalon et al. 1999). Furthermore, coronary vasomotor responses to acetylcholine in patients with CHD improved in response to the ACE inhibitor quinapril to a much greater extent in smokers than in nonsmokers (Schlaifer et al. 1999). ACE inhibitors have an antioxidant activity, which could contribute to the clinical benefit in smokers.
Summary
The endothelium, a delicate monolayer of cells that invests all blood vessels, is a major regulator of vascular reactivity and structure. Healthy endothelium maintains vascular homeostasis by promoting fluidity of the blood, vasodilation, and relaxation of the underlying vascular smooth muscle. Endothelial dysfunction regularly accompanies and promotes vascular disease. Endothelial vasodilator dysfunction is an independent risk factor for major adverse cardiovascular events and mortality. Active smoking and involuntary exposure to cigarette smoke injure endothelial cells and impair endothelial vasodilation. Thus, each type of exposure to tobacco or tobacco smoke contributes to the development of CVD.
Thrombogenic Effects
This section on thrombogenic effects reviews the state of knowledge of mechanisms by which smoking or secondhand smoke exposure may predispose a person to thrombosis, a pathologic reaction that commonly results in smoking-related MI or stroke. Smoking-mediated thrombosis appears to be a major factor in the pathogenesis of acute cardiovascular events.
Epidemiologic evidence indicates that cigarette smoking increases risk of acute MI and sudden death more than it increases risk of angina pectoris. Researchers hypothesize that risk of acute MI and sudden death is mediated by thrombosis, whereas angina is mediated primarily by hemodynamic factors. Successful revascularization in patients with MI after treatment with thrombolysis is more likely in smokers than in non-smokers (Bowers et al. 1996). At the time of MI, smokers are younger and have fewer cardiac risk factors and less severe underlying coronary disease than do nonsmokers (Metz and Waters 2003). Enhanced thrombosis superimposed on less severely stenotic arteries best explains these observations. In men who died suddenly, a history of cigarette smoking was significantly more likely when pathologic examination showed acute thrombosis (75 percent of cases) than when the finding was plaque with no thrombosis (41 percent) (Burke et al. 1997). Conversely, stable plaque with no thrombosis was the more common finding in nonsmokers.
Thrombosis occurs when fibrinogen is converted to fibrin, a process involving interaction of platelets, blood-borne proteins, endothelial cells, and subendothelial vascular tissue. An endogenous antithrombotic mechanism involves these same components. Imbalance in these pathways results in predisposition to thrombosis. Virchow’s triad, first described in 1856 and modified by Aschoff in 1924, provides a framework for risk factors for thrombosis that is still valid. These authorities described the cardinal risk factors as “alterations in blood coagulation,” “alterations in blood flow,” and “alterations in the blood vessel wall.”
Alterations in Blood
Blood contains platelets, red blood cells, and leukocytes suspended in plasma. Plasma in turn contains a variety of coagulation proteins and lipids that also contribute to the clotting process. Smokers tend to develop MI at a lower burden of atheroma than do nonsmokers. This finding suggests a greater role for formed elements of blood or for cardiac electrical instability in cardiovascular events in smokers.
Platelets
Platelets, although only a minor component of the solid phase of blood, are critical to the coagulation process and are important mediators of the impact of smoking on cardiovascular outcomes (Figure 6.6).
Figure 6.6
Potential sites of actions and mechanisms of effects of smoking on platelets. Note: Smoking decreases NO-mediated inhibition of platelet activation and increases platelet activation through oxidative stress and other mechanisms. NO = nitric oxide.
Turnover and Activation
Studies have reported that sudden cardiac death is 2.5 times higher in smokers than in nonsmokers (Kannel et al. 1984; Goldenberg et al. 2003). Research findings broadly implicated activation of platelets and subsequent focal ischemia in sudden cardiac death among smokers. The turnover of platelets is accelerated in cigarette smokers (Fuster et al. 1981). Researchers related the number of newly formed reticulated platelets, rather than absolute platelet count, to incidence of thrombotic events (Rinder et al. 1998).
Urinary excretion of thromboxane metabolites (TxMs), such as 2,3-dinor thromboxane B2 (TxB2) and 11-dehydro TxB2, which are markers of platelet activation in vivo, increases in smokers in a dose-dependent manner (Murray et al. 1985). Studies demonstrated increases in levels of urinary TxM among smokers who were monozygotic twins but divergent for smoking behaviors (Lassila et al. 1988). These increases were also observed in young Swedish army recruits who smoked but had no apparent vascular disease (Wennmalm et al. 1991). Studies also showed that mainstream and sidestream smoke directly promoted platelet activation and enhanced activation induced by shear stress (Rubenstein et al. 2004).
Elevated levels of TxM observed in persons who smoke decreased substantially within days of smoking cessation, although they did not reach the levels found in nonsmokers (Rangemark et al. 1993; Saareks et al. 2001). In an ex vivo study, platelets from smokers showed greater aggregation than those from nonsmokers (Takajo et al. 2001). A decline in platelet aggregation during 14 days of abstinence from smoking was reversed rapidly after smoking was resumed (Morita et al. 2005). Ex vivo aggregability, however, is a crude index of in vivo platelet activation and aggregation. Some studies reported diminished ex vivo aggregability in smokers. This finding suggests that partial activation in vivo resulted in the harvest of a less responsive subset of platelets for study ex vivo. Increased levels of urinary TxMs in smokers may be attributable to activation of both platelets and macrophages. Low-dose aspirin, which inhibits COX activity in platelets, substantially depressed the increment of TxM in smokers, leading investigators to hypothesize that TxM was principally derived from the activity of platelet COX-1 rather than macrophage COX-2 (Nowak et al. 1987; McAdam et al. 2005).
Generation of Nitric Oxide
Platelets constitutively express ENOS. Although platelet-derived NO is a modest inhibitor of platelet activation in vitro, it may be critical to inhibit recruitment of platelets to a growing thrombus (Freedman et al. 1997). Aggregating platelets obtained from patients with acute coronary syndromes produced less NO than did those from patients with stable angina (Freedman et al. 1998). Such findings must be interpreted with caution, however, because results reflect in vivo platelet activity (see “Turnover and Activation” earlier in this chapter). In one study, levels of platelet-derived NO were lower in smokers than in nonsmokers (Takajo et al. 2001). This finding was associated with lower levels of intraplatelet-reduced glutathione—a marker of oxidative stress and increased platelet aggregation. In another study, both the platelet-derived release of NO and the glutathione levels recovered in a time-dependent manner after smoking cessation, but they rapidly decreased again when smoking was resumed (Morita et al. 2005). Abstinence from smoking was also associated with decreased agonist-induced platelet aggregation ex vivo. Furthermore, levels of intraplatelet nitrotyrosine and urinary 8-hydroxy-2′-deoxyguanosine, which are markers of oxidative stress, were also depressed after smoking cessation. One study showed that supplementation with vitamin C restored NO levels and platelet aggregation in current smokers to levels observed in non-smokers (Takajo et al. 2001). Another study demonstrated that the normal morning increase in platelet sensitivity to NO ex vivo was lost in smokers, leaving platelets potentially more susceptible to activation during early morning hours, when MIs are most common (Sawada et al. 2002). In yet another study, platelets from smokers were less sensitive to administration of nitroglycerin, a documented NO donor (Haramaki et al. 2001).
Oxidative Stress and Platelet Function
Cigarette smoke has been shown to be an abundant source of free radicals (Church and Pryor 1985). Levels of isoprostanes—quantitative indices of in vivo oxidative stress—are higher in smokers than in nonsmokers (Reilly et al. 1996; Chehne et al. 2001; Dietrich et al. 2002), and they decrease with smoking cessation (Reilly et al. 1996; Praticò et al. 1997). In addition to serving as biomarkers of oxidative stress, isoprostanes may serve as secondary messengers that exert biologic effects, at least in vitro.
Studies demonstrated elevated production of isoprostanes and decreased levels of reduced glutathione in the platelets of smokers (Takajo et al. 2001). Intraplatelet levels of nitrotyrosine, which is a marker of modification of proteins induced by oxidative stress, decreased with smoking abstinence but increased rapidly when smoking was resumed, together with return of increased sensitivity to agonist-induced platelet aggregation ex vivo (Morita et al. 2005). In another study, products from activated platelets induced oxidative stress in vascular smooth muscle cells, which is associated with increased expression of tissue factor, a highly thrombogenic protein (Görlach et al. 2000). Other investigators showed that administration of antioxidants to persons with diabetes, just as to smokers, decreased production of isoprostane and urinary TxM (Davì et al. 1999) and decreased platelet aggregation ex vivo (Salonen et al. 1991).
Isoprostanes and Platelet Function
In one study, platelets oxidized ex vivo demonstrated increased aggregation induced by shear stress, an effect that is only partly inhibited by administration of aspirin (Chung et al. 2002). In another study, oxidation of platelet membranes was associated with reduced expression of glycoprotein Ib, a receptor for the von Willebrand factor that is critical to platelet activation and aggregation under conditions of shear stress (Escolar and White 2000). The researchers postulated that decreased expression of glycoprotein Ib indicates a highly reactive status of platelets.
Some evidence indicates that isoprostanes may act as platelet and vascular agonists through ligation of the thromboxane A2 receptor (TP) (Audoly et al. 2000). To date, no molecular evidence exists for a distinct isoprostane receptor (Praticò et al. 1996). One study reported that infusion of isoprostanes elevated blood pressure and activated platelets—effects that are lost in mice lacking TP. Binding of isoprostane iPF2α-III to TP promoted change in platelet shape and facilitated response to other proaggregatory stimuli (Praticò et al. 1996). In another study, however, isoprostane alone did not induce platelet activation and partially blocked the proaggregatory effects of TP agonists and high-dose collagen (Cranshaw et al. 2001). Also, isoprostane iPF2α-III was reported to decrease the antiplatelet activity of NO (Minuz et al. 1998). Some researchers speculated that this isoprostane has a role in the resistance to low-dose aspirin observed in patients with CVD (Csiszar et al. 2002). It is not known, however, how these effects, which are demonstrable in vitro, relate to endogenous levels of isoprostanes attained locally in vivo under conditions of oxidative stress.
Summary
Thus, platelets from smokers demonstrate a dose-dependent increase in activity and adhesiveness that rapidly decreases with smoking abstinence. Findings suggest that the inhibitory NO pathway is impaired and responsiveness to other agonists is increased at least partially through the mediation of TP.
Red Blood Cells
Hematocrit in Adults
Smoking is associated with an increase in hematocrit or red blood cell mass attributable to increased levels of CO and carboxyhemoglobin. Hematocrit decreases with smoking cessation, but increased blood viscosity and deformability of red blood cells may persist (Haustein et al. 2004). Increases in hematocrit and blood viscosity are associated with increasing risk of CVD. It is unclear, however, whether these risk factors are independent of smoking and other conventional risk factors, particularly in women (Lowe et al. 1997; Irace et al. 2003; Woodward et al. 2003). When data are adjusted for smoking, hypertension, and high cholesterol, viscosity remains a significant risk factor for stroke and for PAD but not for ischemic heart disease. This finding suggests that the effect of high viscosity may be an independent risk factor for stroke or PAD (Lee et al. 1996; Lowe et al. 1997).
Hematocrit in Neonates
Researchers showed that the effects of smoking on hematocrit were transmitted to the fetus during pregnancy. Some studies reported a dose-dependent increase in hemoglobin levels in infants of mothers who smoked (al-Alawi and Jenkins 2000; Habek et al. 2002). Furthermore, infants born to mothers who smoked more than 20 cigarettes per day had higher rates of fetal hypoxia, polycythemia, and neurological complications than did infants of nonsmoking mothers.
Leukocytes
Polymorphonuclear Leukocytes
In one study, persons who smoked had higher numbers of circulating polymorphonuclear leukocytes than did nonsmokers (Sela et al. 2002). In other research, neutrophils from smokers had higher levels of myeloperoxidase (Bridges et al. 1985) and increased expression of integrins CD11b, CD15, and CD63, which are markers of leukocyte activation (Gustafsson et al. 2000). Furthermore, when stimulated, these leukocytes released superoxide at a faster rate than did leukocytes from nonsmokers (Sela et al. 2002), which further increased local oxidative stress. Studies documented a greater variety of circulating cellular adhesion molecules, including ICAM-1, VCAM, P-selectin, and E-selectin, in smokers than in nonsmokers (Mazzone et al. 2001; Bermudez et al. 2002). An increase in these cellular adhesion molecules (monocytes) may facilitate recruitment of inflammatory cells to sites of vascular injury (Figure 6.7).
Figure 6.7
Potential sites of effects of smoking on thrombosis through oxidative stress and other mechanisms. Note: 1. Increased numbers and activation of polymorphonuclear leukocytes; increased production of superoxide radicals; and increased expression of integrins (more...)
Monocytes
Monocytes from smokers demonstrated increased expression of the integrins CD11b and CD18, which augment adhesiveness of monocytes to endothelial cells, at least in vitro (Weber et al. 1996). This process is thought to be mediated by activation of protein kinase C (Kalra et al. 1994) and is attenuated by supplementation with vitamin C (Weber et al. 1996). In one study, isoprostane iPF2α-III inhibited adhesion of monocytes to cultured dermal cells or renal endothelial cells in rats but paradoxically increased adhesion of monocytes to human endothelial cells in the umbilical vein (Leitinger et al. 2001; Kumar et al. 2005). Adhesion of monocytes to endothelial cells may increase their access into the subendothelium, where they differentiate into macrophages and promote atherogenesis (Ross 1999). Differentiation of monocytes into macrophages depends on production of intracellular reactive oxygen species induced by nicotinamide adenine dinucleotide phosphate, but no evidence exists for a role of cigarette smoking in this differentiation (Barbieri et al. 2003).
Summary
In summary, smoking induced changes in the numbers and activity of polymorphonuclear leukocytes and monocytes. In addition, it promoted expression of chemoattractant and adhesion molecules and integrins, which would be expected to increase recruitment of activated leukocytes to areas of oxidative stress, including sites of platelet deposition after vascular injury.
Circulating Proteins
In addition to its effects on the cellular elements of blood, smoking alters the proteins involved in the coagulation pathway by changing procoagulant factors in the circulation and anticoagulation factors derived from the endothelium.
Fibrinogen
Study findings indicate that circulating levels of fibrinogen increase in smokers and decrease with smoking cessation (Thomas et al. 1995; Hunter et al. 2001; Tuut and Hense 2001). Also, research suggests that elevated fibrinogen values are an independent risk factor for CHD (Paramo et al. 2004) and deep-vein thrombosis (Vayá et al. 2002). For CVD, the predictive effect was reportedly similar and additive to traditional cardiovascular risk factors (Woodward et al. 1998). The effect of fibrinogen on CVD is partly attributable to smoking and seems to be mediated through alterations in rates of synthesis by the liver. Use of snuff is not associated with increased fibrinogen levels (Eliasson et al. 1995).
Nitration of tyrosine residues, a marker of NO-dependent damage, is increased in smokers (Petruzzelli et al. 1997). Presence of these residues depends strictly on availability of nitrogen dioxide radicals that are in turn derived from ONOO− (Kirsch et al. 2002). Tyrosine nitration modifies a variety of proteins, including fibrinogen (Petruzzelli et al. 1997; Pignatelli et al. 2001). Nitrogenated fibrinogen is more reactive and thrombogenic than is native fibrinogen (Gole et al. 2000), a fact that seems attributable to accelerated formation of clots without modification in plasmin-induced thrombolysis (Vadseth et al. 2004). The antioxidants glutathione and vitamin C protect against formation of nitrogenated fibrinogen by interfering with interaction of nitrate radicals with tyrosine (Kirsch and de Groot 2000; Kirsch et al. 2001). ONOO− is likely to derive from interaction of NO from cigarette smoke with superoxide radicals from pulmonary macrophages (Deliconstantinos et al. 1994). In studies of both animal models and human volunteers, even brief exposure to tobacco smoke induced prolonged production (>30 minutes) of ONOO−, apparently from pulmonary macrophages (Deliconstantinos et al. 1994).
Plasmin
Circulating plasminogen is activated to plasmin by fibrin, thrombin, and tPA. Plasminogen has fibrinolytic and collagenase activities. In vitro studies showed that levels of ONOO− increased in smokers and that ONOO− induced nitration of plasminogen in a concentration- dependent manner and reduced proteolytic activity of plasmin (Nowak et al. 2004). This effect is partially reduced by glutathione.
C-Reactive Protein
Chronic, low-level inflammation—reflected by elevated levels of C-reactive protein (CRP) and other biomarkers—is an important risk factor for atherosclerosis (Koenig et al. 1999). Investigators reported that levels of CRP, which likely contributes to both oxidative stress and mitogenic and fibrogenic characteristics of atherosclerotic plaque, are higher in smokers than in nonsmokers in a dose-dependent manner (Bakhru and Erlinger 2005). This increase persisted even after adjustment for diabetes, lipid profile, and CVD, as well as age, gender, and race. More important, five years after smoking cessation, CRP levels were decreased to levels similar to those in lifetime nonsmokers. This finding suggests vascular healing. The timeframe is consistent with that observed in the multinational monitoring of trends and determinants of CVD (MONICA) (Dobson et al. 1991) and in the Northwick Park Heart studies (Meade et al. 1987). In those studies, cardiovascular risk was reduced at two to five years after a person stopped smoking.
Oxidized Low-Density Lipoprotein Cholesterol
In vitro research on oxidative modification of LDL cholesterol (LDLc) by extracts from cigarette smoke showed a significant increase in atherogenicity (Chisolm and Steinberg 2000). In one study, LDL isolated after participants smoked six or seven cigarettes was more susceptible to ex vivo oxidation than was LDL isolated after 24 hours of abstinence from smoking (Harats et al. 1989). In another study, oxidizability of LDL ex vivo decreased with smoking cessation (Sasaki et al. 1997). Oxidized LDLc, but not native LDL, interacted with scavenger receptors on lipid-laden lung cells (foam cells) and was readily incorporated into atherosclerotic plaque. Findings in other studies suggest that oxidized LDL prompts migration and degranulation of neutrophils (Sedgwick et al. 2003) and increases expression of the Toll-like receptor, a trans-membrane protein in macrophages, thus promoting their activation (Xu et al. 2001).
Clinical studies produced conflicting data on the ability of cigarette smoke to oxidize LDLc and on the role of cigarette smoke in the process in vivo. Findings in several studies (Scheffler et al. 1992; Mahfouz et al. 1995; Kagota et al. 1996; GouaŸe et al. 1998; Yamaguchi et al. 2005), but not all studies (Princen et al. 1992; Siekmeier et al. 1996; Marangon et al. 1997; van den Berkmortel et al. 2000), suggest that smoking increases oxidation of LDLc. Studies of an animal model suggest a similar oxidative effect (Yamaguchi et al. 2002, 2004). In one study, modification of LDLc by cigarette smoke was diminished in a dose-dependent manner by administration of fluvastatin (Yamaguchi et al. 2002; Franzoni et al. 2003). Fluvastatin is a potent scavenger of the peroxyl radical. A similar clinical study showed no decrease in oxidized LDL with administration of atorvastatin, despite improvement in endothelial function (Beckman et al. 2004). It is unclear whether failure to demonstrate an association between improved endothelial function and oxidative stress in the study of atorvastatin reflects a true independence of these effects, a distinction from fluvastatin, or limitations attributable to the small sample size.
Thus, cigarette smoking may induce changes in both coagulation and fibrinolytic pathways to promote a prothrombotic state. In addition to its effects on inflammatory cells, smoking promotes production of inflammatory markers and acute-phase reactants.
Alterations in Blood Vessels
Nitric Oxide
Cigarette smoking has injurious effects on the vascular endothelium (see “Smoking and the Endothelium” earlier in this chapter). Abnormalities in the release of chemical mediators occur as a consequence of endothelial dysfunction and are likely to contribute to the prothrombotic condition of smokers. Examples include decreases in NO-mediated inhibition of interactions between platelets and the blood vessel wall, in platelet-induced NO, and in inhibition of platelet activation (see “Platelets” earlier in this chapter). Blood vessel tone is more sensitive to low NO levels than is platelet function (Loscalzo 2001). The importance of NO deficiency mediated by oxidative stress in thrombosis is suggested by familial childhood stroke resulting from deficiency in glutathione peroxidase. This condition decreases NO levels in association with both increased expression of P-selectin in platelets and platelet aggregation and activation (Kenet et al. 1999).
Prostacyclin Production
In vitro studies showed impaired production of prostacyclin by vascular cells exposed to cigarette smoke extracts. In vivo studies, however, showed increased biosynthesis of prostacyclin that is presumed to be reactive to accelerated interactions of platelets and neutrophils with vessel walls in smokers (Murray et al. 1990; Lassila and Laustiola 1992). Consistent with this concept, levels of markers of platelet and leukocyte activation were greater in smokers than in nonsmokers. Thus, the augmented biosynthesis of prostacyclin is likely to reflect a compensatory reaction by the vascular endothelium.
von Willebrand Factor
Studies reported higher circulating levels of von Willebrand factor in smokers than in nonsmokers (Blann and McCollum 1993; Smith et al. 1993), and findings indicated that these levels may precede clinically overt atherosclerosis (Prisco et al. 1999). The von Willebrand factor is essential for initial adhesion of activated platelets to the vessel wall and for expansion of thrombi. It is unclear to what degree high levels of von Willebrand factor might contribute to the increased thrombosis and atherogenesis observed in smokers.
Tissue Factor
Tissue factor is a prothrombotic protein made by numerous cells in the blood vessel wall, predominantly macrophages but also vascular smooth muscle and endothelial cells. Tissue factor is released when the endothelium is injured and can start the clotting cascade. Studies assessing the effects of smoking on tissue factor yielded mixed results (Barua et al. 2002; Sambola et al. 2003). The immunoreactivity of tissue factor was higher in specimens of plaque obtained during endarterectomy from smokers than in those from nonsmokers (Matetzky et al. 2000). In mice with APO E deficiency that were fed high-cholesterol diets, exposure to cigarette smoke resulted in higher levels of tissue factor and VCAM-1 and higher macrophage counts in atherosclerotic plaques than in those of unexposed mice (Lykkesfeldt et al. 2000). Aspirin treatment of cigarette smokers and of mice exposed to smoke was associated with lower levels of tissue factor in plaque. This finding indicates that aspirin may have a protective role for smokers.
Tissue Plasminogen Activator
Vascular endothelial cells and other tissues secrete tPA. This protein has a central role in fibrinolysis, which limits expansion of clots during thrombosis and eventually dissolves the clot during thrombolysis. Evidence from the Physicians’ Health Study indicates that high levels of tPA are an independent risk factor for stroke and suggests that activation of the endogenous fibrinolytic system occurs years before arterial vessels become occluded (Ridker et al. 1994). PAI-1, which is also secreted by vascular endothelial cells, opposes the actions of tPA.
Data on the effects of smoking on tPA and PAI-1 are conflicting (Blann et al. 2000; Enderle et al. 2000; Matetzky et al. 2000; Newby et al. 2001). Using an in vitro model, researchers showed that serum from smokers impaired tPA production by human umbilical vein endothelial cells but that PAI-1 production was unchanged (Barua et al. 2002). In an in vivo model, bradykinin (Newby et al. 1999; Pretorius et al. 2002) and substance P (Newby et al. 1999; Pretorius et al. 2002) stimulated the production of tPA, which had decreased in smokers, but there was no effect on the release of methacholine-induced tPA. Vitamin C failed to ameliorate this decrease (Pellegrini et al. 2004). Studies showed that levels of PAI-1 may decrease after smoking cessation (Simpson et al. 1997).
Thus, the evidence suggests that smoking decreases the production of tPA and perhaps also increases the amount of PAI-1 produced. These changes would be expected to impair fibrinolytic activity. Alternatively, a population-based study demonstrated increased levels of fibrinogen, but fibrinolytic activity in smokers did not differ from that in nonsmokers (Eliasson et al. 1995). Another study suggested that differences between smokers and nonsmokers in thrombolysis may be evident only in older adults (Ikarugi et al. 2003).
Summary
A variety of abnormalities can be observed in endothelial cell function among smokers compared with nonsmokers. These abnormalities affect the ability of the endothelium to modulate vascular tone, platelet function, thrombogenesis, and thrombolysis. Administration of antioxidants mitigates some but not all of these abnormalities. The relative importance of these abnormalities and their interactions in clinical settings remains to be elucidated.
Exposure to Secondhand Tobacco Smoke
In one study, levels of fibrinogen and coagulation factor VII were higher in nonsmoking adolescent offspring of smokers than in nonsmoking adolescent offspring of nonsmokers (Stavroulakis et al. 2000). This finding is in keeping with a prothrombotic state in smokers. In addition, levels of PAI-1 were lower in nonsmoking offspring of smokers. This result suggests decreased fibrinolysis, but there was no difference in tPA levels. Levels of thrombomodulin, which is produced by the vascular endothelium and has anticoagulant effects, were higher in nonsmoking offspring of smokers, but levels of von Willebrand factor were unchanged. Another study demonstrated that TxM levels increased in one exposure to secondhand smoke; after six hours of exposure, levels approached those observed in smokers (Schmid et al. 1996). In other research, exposure to secondhand smoke decreased sensitivity of platelets to the inhibitory effects of prostacyclin in vitro (Burghuber et al. 1986).
Thus, many of the effects from active smoking can be observed in persons involuntarily exposed to cigarette smoke. The magnitude of the effect of secondhand smoke is relatively large considering the low systemic exposure to tobacco smoke for nonsmokers compared with that for active smokers and supports the finding of high cardiovascular risk at low levels of exposure to smoke.
Nicotine and Thrombosis
Nicotine replacement therapy (NRT) does not seem to increase the acute risk of thrombosis, even in patients with established cardiac disease (Joseph et al. 1996). However, investigators have not fully studied cardiovascular effects of extended administration of nicotine, which might be used as an adjunct for persons trying to stop smoking (Stratton et al. 2001). The high urinary excretion of TxM in smokers declines rapidly after smoking cessation. In one study, this decline did not occur in smokers who were using NRT, suggesting that nicotine may be contributing to platelet activation (Saareks et al. 2001). However, other studies in which smokers switched to nicotine patches found a decline in eicosanoid excretion and that long-term use of smokeless tobacco, which results in nicotine exposure similar to that of cigarette smokers, does not increase urinary excretion of TxM (Wennmalm et al. 1991; Benowitz et al. 1993). These findings suggest that nicotine per se does not activate platelets. When nicotine or cotinine was added to the platelet-rich plasma of nonsmokers, platelet-dependent formation of thrombin increased (Hioki et al. 2001). The magnitude of the effect was similar to that observed in smokers, even though the basal nicotine levels in smokers were higher than those in nonsmokers. When cultured endothelial cells from the human brain were exposed to nicotine, tPA levels were unchanged (Zidovetzki et al. 1999). Rather, PAI-1 messenger RNA and protein expression increased, favoring a prothrombotic state. Alternatively, when transdermal nicotine was administered to nonsmokers, the release of tPA induced by substance P was greater than that in nonsmokers who had received placebo patches (Pellegrini et al. 2001). This finding suggests a more favorable effect in vivo.
A study of cardiovascular biomarkers indicates that smokeless tobacco produced neither the inflammatory reaction found in smokers nor endothelial dysfunction, activation of platelets, or evidence of oxidant stress (Axelsson et al. 2001). Leukocyte counts; levels of CRP, fibrinogen, and antioxidant vitamins; and lipid profiles were similar in users of smokeless tobacco and in persons who did not use tobacco.
Summary
Multiple factors produced in the blood and released from the vasculature determine the likelihood of a clinically significant thrombosis. Cigarette smoke and components of the smoke stimulate formation or activity of factors that favor the development of thrombosis. It remains to be seen whether biomarkers of individual cardiovascular risk among smokers will emerge and which genetic variants might particularly influence these risks in smokers. The implications of the hypercoagulable state are observed both in the epidemiology of active and involuntary smoking-related cardiovascular events and in the rapid rate of decline in the major component of excess risk for those events after smoking cessation. A hypercoagulable state can result in acute MI in persons who have less severe underlying coronary disease, so smokers who stop smoking have a better prognosis than do nonsmokers after MI. A more gradual decline of residual risk may reflect resolution of smoking-induced vascular injury, which in turn stimulates platelet activation.
Inflammation
Studies demonstrate that cigarette smoking results in a chronic inflammatory state, evidenced by increased counts of circulating leukocytes, CRP, and acute-phase reactants such as fibrinogen (Tracy et al. 1997; Jensen et al. 1998; Tuut and Hense 2001). Cigarette smoking also activates monocytes and enhances recruitment and adhesion of leukocytes to blood vessel walls, an integral step in vascular inflammation (Lehr et al. 1994). Research indicates that inflammation contributes to atherogenesis, because high leukocyte counts and high levels of CRP and fibrinogen are all powerful predictors of future cardiovascular events (Libby et al. 2002).
However, the mechanisms by which cigarette smoking promotes inflammation are not completely elucidated. As discussed previously, oxidant stress appears to be a critical factor; oxidized LDL is a proinflammatory stimulus (see “Cigarette Smoke Constituents and Cardiovascular Disease” earlier in this chapter). Studies also show that the products of lipid peroxidation are proinflammatory, acting in part on the receptor for platelet-activating factor (PAF). In hamsters, the antioxidant vitamin C prevented adhesion of leukocytes to the endothelium and leukocyte-platelet aggregation (Lehr et al. 1994). In the same animal model, adhesion of leukocytes and formation of leukocyte-platelet aggregates were mediated by PAF-like agonists (Lehr et al. 1997). This PAF-like factor was derived from oxidative modification of phospholipids and was distinct from biosynthetic PAF. Treatment with vitamin C inhibited generation of PAF-like lipids. In contrast, oral l-arginine but not vitamin C reversed the effect of sera from smokers by promoting monocyte-endothelial cell adhesion, which is associated with higher levels of ICAM (Newby et al. 2001). The study findings suggest that smoking-related impairment of NO release is an important determinant of increased adhesion of monocytes to endothelial cells.
Nicotine may contribute to inflammation by acting as a chemotactic agent for migration of neutrophils (Nicod et al. 1984). One study indicates that nicotine enhanced leukocyte-endothelium interactions, resulting in greater leukocyte rolling and adhesion in the cerebral microcirculation of mice (Nitenberg et al. 1993). Nicotine reportedly acts on human monocyte-derived dendritic cells to stimulate an inflammatory response (Nowak et al. 1987). Dendritic cells, which were detected in the walls of arteries and in atherosclerotic lesions, present antigens and are thus required for the start of adaptive immunity. Studies showed that nicotine is a potent inducer of expression of a variety of co-stimulatory molecules and that it increases secretion of the proinflammatory cytokine interleukin-12 in cultured dendritic cells (Aicher et al. 2003). Nicotine augmented the capacity of dendritic cells to stimulate proliferation of T cells and cytokines. Finally, intravenous injection of nicotine increased the movement of dendritic cells into atherosclerotic lesions in vivo in mice deficient in APO E. This line of research suggests that nicotine could contribute to adaptive immunity, which may have a role in atherogenesis. However, switching from smoking to transdermal nicotine resulted in a significant decline in the leukocyte count (Benowitz et al. 1993). In addition, use of smokeless tobacco did not produce higher leukocyte counts or higher CRP levels than are seen in persons who do not use tobacco. These observations suggest that nicotine is not the main determinant of the inflammatory response in smokers.
Smoking and Diabetes
Cigarette smoking is widely known to increase the risks of CVD. Even so, this knowledge does not appear to influence smoking behaviors among patients with diabetes, who bear a higher risk of cardiovascular morbidity and mortality than those who do not have diabetes (Haffner et al. 1998). Surveys found that smoking patterns were similar in patients with diabetes and comparable populations without that disorder (Ford et al. 1994; Gill et al. 1996).
Numerous experimental studies demonstrated that smoking had negative effects on the metabolism of glucose and lipids in persons with or without diabetes. Investigators reported that cigarette smoking in patients with diabetes was associated with deterioration of metabolic control (Madsbad et al. 1980; Bott et al. 1994) and increased risk of microvascular and macrovascular complications and death (Chase et al. 1991; Morrish et al. 1991). Furthermore, cigarette smoking increases risk of type 2 diabetes in the general population (Will et al. 2001). This risk may be mediated through direct metabolic effects alone or in combination with a metabolically unfavorable lifestyle.
Risk
In several prospective studies, cigarette smoking was associated with increased risk of type 2 diabetes in both men and women (Willi et al. 2007). Generally, these prospective studies were large and population based. Most of the information was collected by mailing participants a questionnaire, which in some cases, was supplemented with information from medical records. Most of these studies included follow-up of more than 10 years. Results were generally presented after adjustments for possible covariates.
In the Health Professionals Follow-Up Study, the RR of developing diabetes among men who smoked 25 or more cigarettes per day was 1.94 (95 percent CI, 1.25–3.03) when nonsmokers were the reference group (Rimm et al. 1995). In a smaller British study, the risk of diabetes was 50 percent higher among smokers, but this finding was not independent of other risk factors, such as obesity and low levels of physical activity (Perry et al. 1995). Another British study demonstrated that RR for diabetes in men who smoked was approximately 1.7, after adjustments for the effects of age, body mass index, physical activity, alcohol intake, social class, undiagnosed CHD, and treatment for hypertension (Wannamethee et al. 2001). Mean follow-up was almost 17 years. The increased risk of diabetes was lower 5 years after smoking cessation, and the risk normalized 20 years later. A Japanese study found similar results and also reported a positive correlation between use of tobacco products and risk of diabetes (Uchimoto et al. 1999).
Almost identical results were presented in the Physicians’ Health Study (Manson et al. 2000). During 12 years (255,830 person-years1) of follow-up, the risk of diabetes in men who smoked more than 20 cigarettes per day was 70 percent higher than that in nonsmokers, after adjustment for multiple variables. The results also showed a significant positive association between the risk of developing diabetes and higher consumption of cigarettes (Manson et al. 2000). In addition, the Insulin Resistance Atherosclerosis Study monitored a cohort of 906 study participants for five years with equal representation of African Americans, Hispanics, and Whites (Foy et al. 2005). For all persons studied, current smoking was associated with development of diabetes: the adjusted OR was 2.66 (95 percent CI, 1.49–4.77). Among participants who had normal glucose tolerance at baseline, the OR was 5.27 (95 percent CI, 2.11–13.16).
At least three other studies of men confirmed the main results of these prospective studies (Feskens and Kromhout 1989; Kawakami et al. 1997; Ko et al. 2001). There have been fewer studies with women, but two major prospective surveys yielded similar results. In the Nurses’ Health Study (114,247 women; 1,277,589 person-years of follow-up), RR for diabetes in heavy smokers was 1.42, after adjustments for other risk factors (Rimm et al. 1993).
In an analysis of data from the Nurses’ Health Study after 16 years of follow-up, Hu and colleagues (2001) showed that the strongest predictors of diabetes were being overweight or obese. In addition, poor diet, smoking, abstinence from alcohol, and low levels of physical activity were all independently associated with the risk of developing diabetes. Adjusted RR for developing diabetes was approximately 1.4 for smokers compared with nonsmokers.
Data from CPS-I, a prospective cohort study conducted between 1959 and 1972, were used to analyze the correlation between tobacco use and risk of diabetes in both men and women (Will et al. 2001). In comparison with the risk of developing diabetes for nonsmokers, the risks were higher for men who smoked more than one pack of cigarettes per day (RR = 1.19) or two packs per day (RR = 1.45). Risks were also higher for women who smoked more than one pack per day (RR = 1.21) or two packs per day (RR = 1.74). After smoking cessation, the risk returned to normal after 5 years for women and after 10 years for men (Will et al. 2001).
Only a few studies, all from the 1970s or 1980s, failed to demonstrate a positive association between smoking and diabetes, likely due to inadequate study design or lack of power in the study to test this hypothesis (Medalie et al. 1975; Keen et al. 1982; Wilson et al. 1986).
It is generally accepted that risk increases more for type 2 diabetes than for type 1, because type 1 diabetes is relatively rare among the age groups in the studies. Risk for type 2 diabetes is also consistent with the adverse metabolic effects of smoking (see “Insulin Resistance” later in this chapter). Type 1 diabetes is insulin deficiency caused by autoimmune destruction of pancreatic beta cells; in type 2 diabetes, insulin resistance is combined with impaired secretion of insulin (Reaven 1988; Kahn 2001).
Metabolic Control
Some studies examined the effects of cigarette smoking on the body’s requirement for insulin and metabolic control in patients with diabetes. In a cross-sectional study, Madsbad and colleagues (1980) investigated the relationship between insulin doses and related variables in patients treated with injections of insulin. Insulin doses and serum levels of triglycerides were significantly higher in the 114 persons who smoked than in the 163 who did not smoke, and these values increased in a dose-dependent manner in relation to the number of cigarettes smoked. Hemoglobin A1c (HbA1c)—a marker of long-term glucose elevation—was not measured in this study, but blood and urine levels of glucose did not differ between the two groups. This finding suggests that in patients who smoke, a larger insulin dose is needed to achieve metabolic control similar to that in patients who do not smoke.
In a cross-sectional study of 192 patients with type 1 diabetes, smoking was more common in those with higher HbA1c values (Lundman et al. 1990). Other differences between the smokers and nonsmokers were in attitudes toward diabetes, psychological well-being, and similar factors.
In a relatively large prospective study that examined the effects of intensified insulin treatment and of an educational program, smoking was the most consistent determinant of HbA1c levels in relatively young articipants treated with insulin (Bott et al. 1994). The investigators performed a three-year follow-up on 697 patients with diabetes who had no debilitating late complications. HbA1c levels were higher throughout the study in smokers but eventually improved to levels similar to those in nonsmokers, presumably because of the educational program.
Insulin Resistance
The metabolic effects of smoking have been generally studied in persons who did not have diabetes. Insulin sensitivity was usually determined by using the euglycemic hyperinsulinemic clamp technique (DeFronzo et al. 1979). This technique or a slightly modified version of it is considered to be the gold standard in metabolic studies.
In 1993, Attvall and colleagues showed that short-term smoking caused impaired insulin sensitivity in healthy young men. Separately, two cross-sectional studies of men compared uptake of insulin-mediated glucose (insulin sensitivity) in smokers and nonsmokers (Facchini et al. 1992; Eliasson et al. 1997a). Insulin sensitivity was significantly lower (by 10 to 40 percent) in smokers. The degree of insulin resistance was positively correlated with tobacco use, and in long-term users of nicotine gum, with serum cotinine values (Eliasson et al. 1994, 1996). Because cotinine is a metabolite of nicotine, serum and urine levels of cotinine reflect the amount of nicotine use. Insulin resistance in smokers normalized eight weeks after smoking cessation, despite a weight gain of 2.7 kg (Eliasson et al. 1997a).
Smokers in these two cross-sectional studies had signs of insulin-resistance syndrome, such as significantly high serum levels of free fatty acids (FFAs) and triglycerides and low levels of HDLc (Facchini et al. 1992; Eliasson et al. 1997b). In the study by Eliasson and colleagues (1997b), smokers had a high proportion of atherogenic small and dense LDL particles, high fibrinogen levels, and high PAI-1 activity compared with those of nonsmokers. PAI-1 activity among long-term users of nicotine gum was similar, but effects on lipids were not as pronounced as those in smokers (Eliasson et al. 1996).
One aspect of insulin-resistance syndrome that attracted attention was postprandial hypertriglyceridemia, a phenomenon associated with CVD and insulin resistance (Patsch et al. 1992; Jeppesen et al. 1995). This phenomenon is also observed in smokers (Axelsen et al. 1995; Eliasson et al. 1997b), but its cause is unknown. One possible explanation is the inability of smokers to adequately clear triglyceride-rich chylomicrons and their remnants from the body (Mero et al. 1997).
Other studies that did not use exact measurements of insulin sensitivity reported changes in glucose metabolism in smokers compared with nonsmokers. Compared with nonsmokers, smokers were hyperinsulinemic and relatively glucose intolerant (Eliasson et al. 1991; Zavaroni et al. 1994; Frati et al. 1996; Ronnemaa et al. 1996). A large cross-sectional study showed that after adjustments for confounding factors, smoking behaviors were clearly correlated with HbA1c values in persons who did not have diabetes (Sargeant et al. 2001), but researchers have debated the importance of HbA1c in persons who do not have diabetes. Even so, these findings add support to the hypothesis that use of tobacco exerts adverse effects on glucose homeostasis.
Results in a few studies did not support these findings. Godsland and Walton (1992) found no differences in insulin sensitivity between women smokers and nonsmokers, but this result may be attributable to lower levels of tobacco use. In addition, the results were from analysis of data obtained to test a different hypothesis. In a study of metabolic changes in patients with or without hypertension, no differences in insulin sensitivity between smokers and nonsmokers were detected (Nilsson et al. 1995). However, the study design likely did not enable discrimination between metabolic changes caused by hypertension and those caused by smoking.
In patients with type 1 diabetes, Helve and colleagues (1986) examined cross-sectional and short-term effects of smoking on insulin sensitivity. Despite elevated levels of circulating epinephrine, cortisol, growth hormone, and glucagon after smoking, no effect of smoking on insulin sensitivity was observed. The investigators concluded that fluctuations in blood glucose and metabolic control disguised the influence of smoking in these patients with diabetes. In a study of 28 smokers and 12 nonsmokers with type 2 diabetes, the researchers measured insulin sensitivity by using euglycemic clamps (Targher et al. 1997). Smokers had higher insulin resistance and glucose intolerance than did nonsmokers. The researchers concluded that smoking markedly and in a dose-dependent manner aggravated insulin resistance observed in patients with type 2 diabetes (Targher et al. 1997).
Axelsson and colleagues (2001) reported that nicotine administered intravenously to nonsmokers caused a marked reduction (about 30 percent) in insulin sensitivity in those with type 2 diabetes but not in healthy control participants. These results suggest that nicotine and possibly tobacco use or other environmental factors may have particularly adverse effects in persons susceptible to diabetes but not in those who are healthy (insulin sensitive).
Microvascular Complications
Microvascular complications in diabetes (retinopathy, nephropathy, and neuropathy) are linked to metabolic control in both type 1 and type 2 disease (New England Journal of Medicine 1993; Lancet 1998). The mechanisms for development of microvascular complications are not fully understood, although several pathogenetic pathways have been suggested (Brownlee et al. 1984; Tomlinson 1999; Cai and Boulton 2002). Hyperglycemia has a central role as a trigger for subsequent events, such as conversion of glucose to sorbitol by aldose reductase; nonenzymatic glycosylation of proteins and receptors in susceptible tissues; increased exposure to oxidative stress; and activation of protein kinase C and mitogen-activated protein kinases. Researchers have suggested that these pathogenetic pathways lead to the disturbances in morphology and function found in diabetic nephropathy, retinopathy, and neuropathy (Brownlee et al. 1984; Tomlinson 1999; Cai and Boulton 2002).
Nephropathy
Some studies showed that smoking increased risk of microvascular complications in diabetes. Several studies of patients with type 1 diabetes reported negative effects of tobacco use on albuminuria and renal function. Chase and colleagues (1991), for example, showed that the albumin excretion rate was 2.8 times higher in smokers than in nonsmokers, after statistical corrections for glycemic control, duration of diabetes, age, gender, and blood pressure. In addition, albuminuria progressed at a more rapid rate in smokers than in nonsmokers.
Smoking promoted progression of renal disease in persons with type 2 diabetes (Biesenbach et al. 1997; Chuahirun and Wesson 2002; Chuahirun et al. 2003). Biesenbach and colleagues (1997) studied only 36 patients, but follow-up lasted 13 years. At study entry, smokers and nonsmokers had similar clinical and laboratory characteristics, but progression of nephropathy and development of atherosclerotic disease progressed more rapidly in the smokers than in the nonsmokers. Multiple regression analysis showed that only tobacco use and blood pressure levels were independently associated with impairment in renal function. This finding underscored the roles of smoking and vascular disease in susceptibility to renal disease (Biesenbach et al. 1997). In two prospective studies by Chuahirun and colleagues (2002, 2003), the effects of cigarette smoking on acceleration of nephropathy in patients with type 2 diabetes were confirmed even in those who had optimal therapy for hypertension. Research presented further evidence of functional and structural changes in the glomeruli of patients with type 2 diabetes who smoke (Baggio et al. 2002). In a study of 96 patients who had biopsy of the kidney, electron and light microscopy demonstrated significant changes in glomeruli and basal membranes that corresponded to impaired glomerular filtration rates in the smokers.
Retinopathy
Generally, investigators have not considered smoking to be a substantial risk factor for diabetic retinopathy (Porta and Bandello 2002). Findings in fairly large studies with mixed populations showed no strong support for such an association, except in older adults with certain conditions (Walker et al. 1985; Moss et al. 1991). At least two studies of patients with type 1 diabetes, however, suggest that smoking does predispose these patients to retinopathy (Mulhauser et al. 1986, 1996). In addition, Chase and colleagues (1991) showed that retinopathy was more common in patients with type 1 diabetes who smoked than in those who did not smoke, but after adjustments for covariates, differences were not statistically significant. The study also reported accelerated progression of retinopathy in patients who smoked. Thus, smoking may be a risk factor for diabetic retinopathy, but only in certain subgroups.
Neuropathy
The role of tobacco in the development of diabetic neuropathy is relatively difficult to examine because of methodologic problems and the frequent prevalence of confounding factors (Westerman et al. 1992). Diabetic neuropathy usually develops during a long period, and it may affect different sensory, motor, and autonomic nerve fibers in varying degrees in individuals. This variation makes it difficult to standardize study methods. One case-control study reported that risk of neuropathy was three times higher in patients with type 1 diabetes who smoked than in those who did not smoke (Mitchell et al. 1990). Smoking was not related to neuropathy in patients with type 2 diabetes. In a study of young patients treated with insulin, independent risk factors for progression of distal sensory neuropathy, apart from poor glycemic control, were cigarette smoking, greater height, and female gender (Christen et al. 1999). Other studies in patients with type 1 diabetes confirmed the roles of glycemic control and smoking behaviors in development of clinical neuropathy (Maser et al. 1989; Reichard 1992).
Macrovascular Complications
The multiple effects of smoking on the vascular and hemostatic systems and on inflammation are reviewed elsewhere in this chapter (see “Hemodynamic Effects,” “Smoking and the Endothelium,” “Nicotine and Thrombosis,” and “Inflammation” earlier in this chapter). Diabetes patients are particularly susceptible to some effects of smoking, because their risk of cardiovascular morbidity and mortality is elevated (Jarrett et al. 1982; Manson et al. 1991; Morrish et al. 1991).
In a study cohort in London, England, in the prospective Multinational Study of Vascular Disease in Diabetes, sponsored by the World Health Organization, smokers with type 1 or type 2 diabetes had significantly increased risk of CHD, but not stroke, during the eight-year follow-up (Morrish et al. 1991). In the Diabetes Control and Complications Trial (New England Journal of Medicine 1993), designed to study the role of intensive insulin treatment and optimized glycemic control in type 1 diabetes, smoking was not a significant risk factor for macrovascular complications. Because the participants were relatively young, this trial was not optimally designed to study the role of tobacco use. Other studies with slightly older participants who had type 1 diabetes reported that smoking increased risk of CHD (Moy et al. 1990; Sinha et al. 1997).
Among patients with type 2 diabetes in the United Kingdom Prospective Diabetes Study, cigarette smoking was a significant and independent risk factor for CHD (Turner et al. 1998), stroke (Kothari et al. 2002), and PAD (Adler et al. 2002). Also, an analysis of data from the Nurses’ Health Study demonstrated that for women with type 2 diabetes, a dose-effect relationship existed between smoking behaviors and mortality (Al-Delaimy et al. 2001). Compared with nonsmokers, risk of mortality from all causes was 1.64 for women who smoked 15 to 34 cigarettes per day and 2.19 for women who smoked more than 34 cigarettes per day. Ten years after smoking cessation, risk of mortality had normalized. Researchers published similar data on smoking and CHD risk in the same cohort (Al-Delaimy et al. 2002).
A relatively large prospective study that analyzed the effects of smoking cessation on cardiovascular risk in persons with diabetes compared mortality risk for former smokers with that for lifetime nonsmokers (Chaturvedi et al. 1997). Compared with mortality risk for lifetime non-smokers, risk of death from all causes was approximately 50 percent higher for patients who had stopped smoking during the past one to nine years and 25 percent higher for those who had not smoked for more than nine years. Smoking cessation reduced mortality risk among persons with diabetes, but risks remained high several years after smoking cessation and were highly dependent on the duration of smoking.
Pathophysiological Mechanisms
Having diabetes, even for nonsmokers, is associated with long-term exposure to oxidative stress, impaired endothelial function, and dyslipidemia (Brownlee et al. 1984; Turner et al. 1998; Dogra et al. 2001; Cai and Boulton 2002; Komatsu et al. 2002). The causes of type 2 diabetes are still not fully understood, although the main metabolic aberrations are well characterized. Research showed that type 2 diabetes is caused by insulin resistance in combination with relative impairment of insulin secretion (Reaven 1988; Kahn 2001). Published studies have not demonstrated a significant impairment in insulin secretion among cigarette smokers (Epifano et al. 1992; Facchini et al. 1992; Persson et al. 2000), but several studies documented a negative effect of smoking on insulin sensitivity (Facchini et al. 1992; Eliasson et al. 1997a,b).
Cigarette smoking and intake of nicotine increase the circulating levels of insulin-antagonistic hormones (i.e., catecholamines, cortisol, and growth hormone) (Kershbaum and Bellet 1966; Cryer et al. 1976; Wilkins et al. 1982; Kirschbaum et al. 1992). Smoking also activates the sympathetic nervous system (Niedermaier et al. 1993; Lucini et al. 1996). Nicotine likely impairs insulin sensitivity directly or indirectly through these and possibly other mechanisms. An additional negative factor for insulin-mediated glucose uptake is high circulating levels of FFAs, secondary to increased lipolysis (Bergman and Ader 2000). Research has shown that smoking acutely elevates circulating FFA levels (Kershbaum and Bellet 1966).
Researchers have proposed, but not fully elucidated, the potential role of endothelial dysfunction or inflammation in development of insulin resistance and type 2 diabetes.
Summary
Many clinical and experimental studies have found significant associations between cigarette smoking and development of diabetes, impaired glycemic control, and diabetic complications (microvascular and macrovascular). A different lifestyle of smokers, in contrast to that maintained by nonsmokers, may also contribute to these effects. Most of the reviewed studies, however, either attempted to statistically adjust for confounding factors or were designed to examine short-term effects of tobacco and nicotine.
The development of type 2 diabetes is another harmful consequence of cigarette smoking, one that adds to the heightened risks of CVD. In diabetes care, smoking cessation is crucial to facilitating glycemic control and limiting development of complications.
Lipid Abnormalities
Cigarette smoking is associated with an atherogenic lipid profile likely to contribute to risk of CVD.
Epidemiologic Observations
Several observations are central to the relationship between cigarette smoking and lipids. Compared with nonsmokers, smokers have higher levels of triglycerides associated with very-low-density lipoprotein (VLDL), total triglycerides, and APO B, in addition to modest increases in LDLc and lower levels of plasma HDLc and APO A-I (Billimoria et al. 1975; Criqui et al. 1980; Wilson et al. 1983; Craig et al. 1989; Muscat et al. 1991; Freeman and Packard 1995; Villablanca et al. 2000). These findings are robust and are reported in numerous survey studies. Researchers observed a dose-response relationship between the number of cigarettes smoked per day and plasma lipid levels (Muscat et al. 1991). In contrast, plasma lipid and lipoprotein levels in former smokers typically are similar to those in nonsmokers.
The ratio of LDLc to HDLc, which is used as a measure of atherogenic risk, is higher in smokers than in nonsmokers. Cigarette smoking is thought to raise the LDLc to HDLc ratio by 15 to 20 percent. Increased levels of plasma triglycerides are associated with lower HDLc levels, but reduction in HDLc from cigarette smoking persists even after corrections for levels of total triglycerides. Early epidemiologic studies, such as the Lipid Research Clinics Program Prevalence Study and the Framingham Heart Study (Criqui et al. 1980; Muscat et al. 1991; Freeman and Packard 1995), emphasized lower HDLc values as the primary effect of cigarette smoking.
Researchers have estimated, however, that these effects of cigarette smoking on plasma lipids and lipoproteins account for only 10 percent of the observed 70 percent increase in risk of vascular disease associated with cigarette smoking (Craig et al. 1989). In the Edinburgh Artery Study, for example, adjusting for known CHD risk factors reduced the RR of CHD in heavy smokers from 3.94 to 2.72 and the RR in moderate smokers from 2.72 to 1.70 (Price et al. 1999). However, cigarette smoking still accounted for 75 percent of the risk of developing PAD, after adjustment for other known risk factors, such as hyperlipidemia and type 2 diabetes (Lu and Creager 2004). Other researchers reported similar findings (Cullen et al. 1998). Cigarette smoking thus appears to have atherogenic effects that are not explained by traditional CHD risk factors, including abnormal levels of blood lipids.
Analyses of mechanisms related to lipid and lipoprotein metabolism may be required for understanding the atherogenicity of cigarette smoking. Such mechanisms include lipid oxidation; changes in composition of lipoproteins; alterations in plasma- and lipoprotein-associated lipid transfer enzymes; changes in metabolism of fatty acids; effects on levels of postprandial lipids; and changes in cholesterol fluxes, particularly reverse cholesterol transport (RCT). The following discussion reviews the effects of cigarette smoking on these potential underlying mechanisms.
Lipoprotein Composition and Apolipoprotein Levels
Cigarette smoking clearly reduces APO A-I and the ratio of A-I to A-II (Mero et al. 1998). APO A-I is a major component of HDL particles. The reduction in A-I levels observed in smokers is similar to, although perhaps somewhat lower than, the reduction in HDLc levels seen in this population. For example, Mero and colleagues (1998) documented that levels of plasma APO A-I were 4 to 6 percent lower and HDLc values were 6 to 9 percent lower in moderate-to-heavy smokers. The effects of cigarette smoking on APO B and other APOs are also well documented (Billimoria et al. 1975; Craig et al. 1989; Muscat et al. 1991; Villablanca et al. 2000).
Researchers have associated abnormalities in different subfractions of HDL with different risks of CHD. Cigarette smoking reduced different HDL subfractions in different studies (Billimoria et al. 1975; Craig et al. 1989; Muscat et al. 1991). Even so, the true atherogenicity of different HDL subfractions remains controversial. The role of altered HDL subfractions in arterial disease associated with cigarette smoking requires further study.
Plasma- and Lipoprotein-Associated Lipid Transfer Enzymes
Several enzymes in plasma—either free or associated with lipoproteins—are involved in transport and use of lipids. Lipoprotein lipase (LPL) activity is involved in clearance of total triglycerides from triglyceride-rich lipoproteins (TGRLs), particularly the chylomicra formed in persons after a meal containing fat. Cigarette smoking reportedly reduced plasma LPL activity after a mixed meal (Freeman et al. 1998). Reduced LPL activity may contribute to the reduced clearance of TGRLs reported for total triglycerides, APO B, and retinyl-ester components of TGRLs (Mero et al. 1998). In another study, the cholesterol ester transfer protein (CETP) received considerable attention as a therapeutic target for raising HDLc levels (Brousseau et al. 2004; Ruggeri 2005). CETP mediates transfer of cholesterol esters between HDL and other lipoproteins (VLDL and LDL). However, controversy exists as to whether CETP activity is beneficial or deleterious to the process of RCT. Moreover, the proatherogenic or antiatherogenic consequences of changing HDLc by the CETP mechanism remain uncertain (Ruggeri 2005). Studies have reported both increases and decreases in plasma CETP activity in smokers (Dullaart et al. 1994; Zaratin et al. 2004).
Some studies measured other enzymes in smokers. For example, cigarette smoking did not appear to markedly alter lecithin cholesterol acyltransferase activity (McCall et al. 1994).
Oxidized Lipoproteins
Many investigators hypothesized that oxidized LDL is highly atherogenic (Dullaart et al. 1994; Zaratin et al. 2004), and studies reported increased oxidative damage to LDL in smokers (Ambrose and Barua 2004). However, attribution of a precise atherogenic contribution from oxidative damage to LDL remains speculative. The inadequacy of the metrics of pro-oxidative and antioxidative status in vivo needs to be resolved before the role of oxidative damage to LDL can be adequately evaluated.
Postprandial Lipid Changes
Traditionally, plasma lipid and lipoprotein measurements used to evaluate CHD risks have, for largely technical reasons, been performed in the fasting (postabsorptive) state. Plasma levels of metabolites are more easily characterized in steady-state conditions than in a nonsteady state. From a pathophysiological perspective, however, events in the postprandial state may be critical in atherogenesis (Zilversmit 1979). Some studies explored effects of cigarette smoking on postprandial lipid metabolism (Mero et al. 1998). For example, total triglycerides increased to higher levels after a mixed meal in smokers than in nonsmokers. These researchers observed increases in plasma APO B level and reductions in levels of APO A-I, lipoprotein A-I, HDLc, and LDL-APO B after a meal. Other investigators postulated that the mechanisms underlying altered postprandial lipid changes in smokers include lower LPL activity (Freeman et al. 1998), but higher endogenous production of VLDL-total triglycerides by the liver has not been excluded. The postprandial effects of cigarette smoking in particular and their role in atherogenesis in general are not completely understood.
Metabolism of Free Fatty Acids
Changes in FFAs (nonesterified fatty acids) are attributed to an increase in adipocyte lipolysis, and they represent the most well-characterized mechanistic action of cigarette smoking in the context of alterations in lipids and lipoproteins. Many studies reported higher plasma levels of FFAs in smokers than in nonsmokers (Kershbaum et al. 1963; Bizzi et al. 1972; Walsh et al. 1977; Hellerstein et al. 1994; Neese et al. 1994).
Using stable (nonradioactive) isotopes to measure FFA kinetics, researchers demonstrated that cigarette smoking immediately and markedly increased influx of FFAs into the bloodstream and thereby raised plasma levels of FFA (Hellerstein et al. 1994; Neese et al. 1994). Plasma FFAs are primarily derived from adipose tissue by lipolytic breakdown of stored triglycerides. Catecholamines stimulate hormone-sensitive lipase activity in adipose tissue and oppose various antilipolytic actions of insulin. Increases in FFA levels and flux induced by smoking were temporally correlated with increases in plasma epinephrine levels (Watts 1960; Bizzi et al. 1972; Arcavi et al. 1994; Hellerstein et al. 1994; Neese et al. 1994). These increases are prevented by β-adrenergic blockers. Nicotine increases the adrenal medullary release of epinephrine in persons with nontolerance of nicotine (Arcavi et al. 1994). Therefore, the model implicated as the cause of increases in plasma FFA levels induced by cigarette smoking seems clear: cigarette smoking→nicotine→increased plasma epinephrine→increased lipolysis in adipose tissue→increased release of FFAs into plasma→increased plasma levels of FFA.
The fate of FFAs released into the bloodstream in response to cigarette smoking is also relevant. Cigarette smoking increases expenditure of energy through the activity of nicotine and catecholamines (Ilebekk et al. 1975; Perkins et al. 1989; Hellerstein et al. 1994; Neese et al. 1994). However, most of the FFAs released in response to cigarette smoking are not oxidized but taken up and reesterified to triglycerides in tissues, particularly the liver. This conclusion was partly based on kinetic studies that compared the rates at which plasma FFA appeared with whole-body rates of fat oxidation (Hellerstein et al. 1994; Neese et al. 1994). These studies demonstrated that the rate of FFA influx into plasma in response to cigarette smoking greatly exceeded changes in whole-body fat oxidation.
Accordingly, the catabolic effects of cigarette smoking on total adipose triglycerides do not directly promote oxidation of body fat (weight loss); instead, the primary result is overproduction of VLDL-total triglycerides (Hellerstein et al. 1994; Neese et al. 1994). This “futile cycle,” a substrate cycle in which adipose triglycerides are converted to hepatic VLDL triglycerides, is modestly wasteful of energy. It accounts for about 5 percent of the thermogenic effects of long-term cigarette smoking—for example, fewer than 10 kilocalories (kcal)/day if cigarette smoking increases total energy expenditure by 200 kcal/day. This cycle, however, may be the central driving force behind the atherogenic dyslipidemia associated with cigarette smoking. Overproduction of VLDL-total triglycerides typically results in elevated plasma levels of VLDL-total triglycerides and APO B, as well as increased numbers of LDL particles (Sniderman et al. 2001). Furthermore, high VLDL-total triglycerides contribute to lowering of HDLc through CETP-mediated transfer of cholesterol-ester from HDL to VLDL particles (Brousseau et al. 2004; Ruggeri et al. 2005). Influx of FFAs into the liver for reesterification and secretion of total triglycerides is most likely a major reason for the low HDLc levels observed in smokers, but perhaps it does not represent the entire effect of smoking on HDLc (Criqui et al. 1980; Muscat et al. 1991; Freeman and Packard 1995).
If nicotine-stimulated release of catecholamine is responsible for the hypertriglyceridemia and low HDLc levels observed in smokers, NRT as an adjunct to smoking cessation should logically prevent improvements in plasma lipids and lipoproteins after smoking cessation. Moffatt and colleagues (2000) reported that nicotine-patch therapy prevented the normalization of HDLc levels observed with smoking cessation in the absence of the nicotine patch. The patch also prevented weight gain after smoking cessation (Allen et al. 2005), a finding consistent with the hypothesis that shared catecholamines are the basis for two important effects of cigarette smoking: weight reduction and dyslipidemia. Other studies, however, did not confirm that use of the nicotine patch as an agent for smoking cessation prevents improvements in HDL levels (Allen et al. 1994). In addition, lipid profiles are similar in persons who use smokeless tobacco and in those who do not use any form of tobacco. To the extent that dyslipidemia contributes to vascular disease associated with cigarette smoking, it is important to determine the full range of effects of NRT on lipid and lipoprotein metabolism.
Reverse Cholesterol Transport
RCT refers to the pathway by which cholesterol is mobilized from tissues, carried through the blood, and excreted from the body. HDL and its associated membrane receptors (e.g., SR-B1, ABC-A1, and ABC-G1), plasma enzymes (e.g., CETP and phospholipid transfer protein), APOs (e.g., APO A-I), and hepatobiliary enzymes (e.g., cholesterol 7α-hydroxylase) constitute a system that mediates the complex process of RCT through pathways that are increasingly well characterized in molecular terms (Neese et al. 1994; Tall 1998). RCT is generally accepted as the leading explanation for the cardioprotective activity of HDL, although other actions of HDL (e.g., antioxidative and anti-inflammatory) may also be involved.
Flux through the RCT pathway and, thus, antiatherogenic activity cannot be predicted simply from plasma levels of HDL or APO A-I (Tall 1998). Thus, changes in levels of HDLc, VLDL-total triglycerides, and other lipoproteins may not fully capture the effects of cigarette smoking on pro-atherogenic or antiatherogenic fluxes such as RCT. Until more recently, however, there were no viable techniques for measuring RCT fluxes. Therefore, this question could be addressed only indirectly—for example, through changes in lipid transfer proteins in plasma that may influence the efficiency of RCT. Studies reported decreases in activity of CETP and phospholipid transfer protein in smokers after they had smoked a cigarette (Zaratin et al. 2004).
Reduced capacity for remodeling HDL particles in the vascular compartment could alter these fluxes in a manner not reflected by HDLc levels. This possibility will remain speculative, however, until RCT fluxes are measured in humans. The ability to directly measure effects of cigarette smoking on the cardioprotective process of RCT could provide a major tool for advancing understanding of the role of lipids in causing vascular disease associated with cigarette smoking.
Effects of Smoking Cessation
Research on smoking cessation largely confirmed the associations observed in smokers (Gordon et al. 1975; Rabkin 1984; Stamford et al. 1986; Critchley and Capewell 2004). Many studies documented the return of normal levels of plasma lipids and lipoproteins after cessation of cigarette smoking.
Therapeutic Implications of Pathogenic Mechanisms
If stimulation of lipolysis underlies the atherogenic dyslipidemia associated with cigarette smoking, inhibition of lipolysis might be an effective therapeutic strategy to improve blood lipid profiles in smokers or persons receiving NRT. This strategy is an attractive approach in one sense because inhibition of lipolysis does not block the thermogenic actions of nicotine. The cycle of lipolysis and reesterification accounts for less than 5 percent of the increase in energy expenditure observed in cigarette smokers (Hellerstein et al. 1994; Neese et al. 1994). Another consideration is that if FFAs released by lipolysis are involved in the insulin resistance reportedly associated with cigarette smoking (Facchini et al. 1992), lipolysis inhibitors may have an additional therapeutic use.
Niacin, a hypolipidemic agent, is thought to act at least partly by inhibiting total triglyceride lipolysis in adipose tissue (Meyers et al. 2004). The use of niacin in smokers who are at high risk for CHD has not been fully investigated. The side effects of niacin, including cutaneous flushing and worsening of insulin resistance in some persons, perhaps from effects on pancreatic islet function, may discourage its clinical use. The impact of niacin and its analogs on lipolysis is complex. They induce a rebound overshoot of lipolysis after initial inhibition, but niacin does reduce production of VLDL-total triglycerides (Wang et al. 2001).
Other strategies for use of lipolysis inhibitors to prevent CHD related to cigarette smoking may require development of specific antilipolytic agents that are well tolerated. One possibility is the thiazolidinedione class of insulin-sensitizing drugs. In one study, such drugs reduced lipolysis in adipose tissue, perhaps by activating glyceroneogenesis and thereby promoting intra-adipocytic reesterification of FFAs (Chen et al. 2005). No studies are known to have tested the efficacy of thiazolidinediones in smokers to determine effects on lipid abnormalities or sensitivity to insulin. However, recent research indicates that rosiglitazone (a thiazolidinedione) increases CHD risk, although pioglitazone, another drug in the same class, does not increase this risk (Lincoff et al. 2007; Nissen and Wolski 2007). Thus, it is unclear whether the possible benefits of this class of drugs in smokers will be pursued.
Summary
Effects of cigarette smoking on standard measures of blood lipids and lipoproteins are well characterized. The most important effects are to lower levels of HDLc and increase levels of total triglycerides. The metabolic mechanisms underlying these changes are known to some extent, particularly the catecholamine-mediated increase in adipocyte lipolysis, changes in plasma levels of FFAs, and reesterification of FFAs by the liver. However, the predicted effect from changes in standard lipid risk factors for vascular disease associated with cigarette smoking appears to be modest. Future research will reveal whether this estimation of a modest effect is a true estimate of the pathogenic importance of smoking-induced changes in blood lipid levels or an inability to measure the full effects of cigarette smoking on atherogenesis.
Cardiovascular Biomarkers
Biomarkers of smoking-related CVD risk are useful for stratifying individual risk and, perhaps, for assessing product risk. Biomarkers for CVD risk can be divided into three categories: (1) constituents of cigarette smoke that contribute to CVD, (2) physiological changes involving potential mechanisms of CVD, and (3) chemical biomarkers of cardiovascular dysfunction and disease (Table 6.3). Studies showed that cigarette smoking altered many of the CVD biomarkers, as evidenced by comparisons of smokers with nonsmokers and former smokers. However, fewer studies prospectively examined reversal of such changes after smoking cessation. More important, to date, there are no data on how changes in smoking-related biomarkers predict risk of disease.
Table 6.3
Biomarkers of risk for cardiovascular disease from exposure to cigarette smoke.
Three constituents of cigarette smoke received the greatest attention as potential contributors to CVD: CO measured as exhaled CO or as blood carboxyhemoglobin, nicotine, and oxidant chemicals (Benowitz 2003). These constituents are used as general biomarkers of exposure to tobacco or tobacco smoke. Apparently, no direct measures of levels of oxidizing chemicals in the body have been developed, but numerous measures of the biologic consequences of exposure to oxidizing chemicals exist. Exposure to particulate matter in cigarette smoke is likely to contribute to CVD in smokers (Brook et al. 2004; Vermylen et al. 2005; Bhatnagar 2006), but no direct biomarkers of particulate exposure are available. Particulate matter appears to affect oxidative stress, coagulability, and inflammation, for which biomarkers are available. A lesser body of research suggests that PAHs and other constituents of tobacco smoke may also contribute to atherogenesis (Penn and Snyder 1988, 1996). Urine levels of PAH metabolites can also be measured in smokers; 1-hydroxy-pyrene is most widely used for this purpose.
Cigarette smoke exposes the smoker to high levels of potentially oxidizing chemicals (Burke and FitzGerald 2003). In one study, cigarette smoking increased levels of lipid peroxidation products, such as F2-isoprostanes, in the plasma and urine (Nowak et al. 1987). Other markers of oxidative stress in smokers included higher plasma levels of oxidized LDL and oxidized fibrinogen, higher urine levels of substances reactive with thiobarbituric acid, and reduced plasma levels of antioxidant vitamins such as E, C, and beta-carotene.
The hemodynamic effects of cigarette smoking can be observed while a person smokes a cigarette. These effects include elevation in heart rate, blood pressure, and cardiac output. Coronary blood flow, as assessed by coronary perfusion studies, may increase or decrease with smoking, depending on underlying atherosclerosis and endothelial function (Czernin and Waldherr 2003).
Researchers have proposed numerous biomarkers for measuring endothelial dysfunction, and many of these biomarkers are affected by cigarette smoking. The functional assessment most widely used is flow-mediated arterial vasodilation (Puranik and Celermajer 2003), a test that measures the diameter of the brachial artery in response to changes in forearm blood flow. The brachial artery is imaged by using Doppler ultrasonography before and after release of a blood pressure cuff that is inflated over the forearm to occlude arterial blood flow. With release of the cuff, the increase in blood flow triggers an increase in the diameter of the brachial artery that is mediated by release of NO and prostacyclin by endothelial cells. Many researchers demonstrated impairment of flow-mediated dilation in populations of active smokers and persons exposed involuntarily to cigarette smoke, but estimates of impairment in persons with no exposure to smoke overlapped considerably with those for the other two groups. Other potential markers of endothelial dysfunction that can be measured in the blood include ADMA, von Willebrand factor, tPA, E-selectin, and P-selectin. Prostacyclin metabolites can be measured in the urine (Cooke 2000). Selectins are adhesion molecules released by both endothelial cells and platelets (Ley 2003).
Markers of the hypercoagulable state include increased urine levels of thromboxane A2 metabolites. Thromboxane A2 is released when platelets aggregate in vivo, and its metabolites in urine are a useful noninvasive measure of the point of activation (Nowak et al. 1987). Other relevant biomarkers of a hypercoagulable state include fibrinogen, red blood cell mass, blood viscosity, tPA, PAI-1, homocysteine, and P-selectin (Benowitz 2003).
Biomarkers used to assess an inflammatory state include total leukocyte and neutrophil counts and levels of CRP, fibrinogen, and interleukin-6 (Pearson et al. 2003). In addition, the counts of several cell-surface adhesion molecules increased in inflammatory states. These molecules included ICAM, sVCAM-1, and monocyte chemoattractant protein-1.
Another study found several markers to be useful for assessing insulin resistance (Eliasson 2003). For example, in persons with insulin resistance, levels of plasma glucose were likely to be elevated in fasting status and two hours after eating. HbA1c levels, which reflect plasma glucose levels throughout the day, were elevated in persons in a hyperglycemic state. The ratio of insulin to glucose after glucose loading was useful as an index of insulin sensitivity. The most definitive investigations were glucose-clamping studies, in which insulin levels were measured when the glucose level was constant or vice versa.
Numerous standard markers of lipids may be altered in cigarette smokers. These markers include HDLc, LDLc, the ratio of total cholesterol to HDL, and serum triglyceride levels.
Nuclear coronary perfusion studies with or without physical exercise are among several functional studies for diagnosing cardiovascular dysfunction or disease. They indicate that cigarette smoking reduces cardiac perfusion in patients with coronary disease (Czernin and Waldherr 2003). Reserve in endothelial function can be assessed by studying flow-mediated dilation (see “Endothelium-Dependent Vasodilation” earlier in this chapter). Findings in another study indicate that vascular disease can be assessed by measuring intima-media thickness of the carotid and femoral arteries by ultrasonography, which provides a direct measure of early atherosclerotic changes in blood vessels (de Groot et al. 2004).
Numerous cardiovascular biomarkers that might be used to assess the effects of cigarette smoking and involuntary smoking and that are expected to increase the risk of CVD are discussed here. Many biomarkers, however, do not reflect causal pathways related to development of CVD. Instead, they reflect the pathophysiological effects of the constituents of cigarette smoke. In addition, many biomarkers are influenced by processes and risk factors that are independent of cigarette smoking. Many of the same abnormalities produced by smoking are also produced by diabetes, hypercholesterolemia, and hypertension. Thus, it is unclear which biomarkers are most specific to cigarette smoking. It is also unclear which bio-markers best predict the risk of CVD attributable to cigarette smoke. Also, a given biomarker profile can indicate any of several marked differences in a person’s susceptibility to CVD.
The potential exists to develop improved biomarkers for CVD by using advances in high-throughput genomics and by examining the relationships of gene polymorphisms or alterations in protein expression or activity to smoking-induced disease (Zhang et al. 2001). Emerging genomic and proteomic technologies may cast light on the signaling pathways activated by smoking and the constituents of tobacco smoke that culminate in cardiovascular dysfunction. Such approaches may also contribute to an understanding of individual differences in susceptibility to the cardiovascular complications of smoking.
Numerous studies of clinical genetics examined differences in susceptibility to smoking-induced CVD as a function of different genetic variants (Wang et al. 2003). Such studies, combined with genomic and proteomic approaches, may provide mechanistic information on pathogenesis and, in combination with other biomarkers, may result in better predictions of cardiovascular risk in smokers.
Smoking Cessation and Cardiovascular Disease
Smoking cessation reduces the risk of cardiovascular morbidity and mortality for smokers with or without CVD (USDHHS 1990). Smoking directly accelerates atherogenesis, causes acute cardiovascular events, and contributes to and acts synergistically with other risk factors, such as hyperlipidemia and diabetes (see “Smoking and Diabetes” earlier in this chapter). Although cigarette smoking does not cause hypertension, smoking is associated with higher blood pressure in persons with hypertension and enhances the likelihood of complications, including progression of renal disease in patients with hypertension (Green et al. 1986; Mann et al. 1991; McNagny et al. 1997; Regalado et al. 2000). One study demonstrated that cigarette smoking was a substantial contributor to morbidity and mortality in patients with left ventricular dysfunction (Suskin et al. 2001). In such patients, the benefit of reducing the likelihood of death by smoking cessation is equal to or greater than the benefit of therapy with inhibitors of ACE, β-blockers, or spironolactone. Smoking cessation is particularly important in patients with diabetes. For these patients, smoking markedly increases cardiovascular risks, including the risk that diabetic nephropathy will progress. Smoking also increases insulin resistance and increases the difficulty of controlling diabetes. For these and other reasons, smoking cessation in patients with CVD is an essential therapeutic intervention.
The 1990 Surgeon General’s report on smoking cessation (USDHHS 1990) outlines the evidence that stopping smoking helps to prevent CVD, and subsequent research has reinforced this concept (Hasdai et al. 1997a; van Domburg et al. 2000; Wilson et al. 2000). Estimates in case-control and cohort studies indicate that most risk reduction for mortality occurred in the first one to three years after smoking cessation, and approximately one-half of the risk of smokers for a nonfatal MI was eliminated in the first year after cessation. It takes about three to five years of abstinence from smoking for most of the excess CVD risk to be gone (USDHHS 1990; Lightwood and Glantz 1997). Smoking cessation after MI reduces the risk of cardiovascular morbidity and mortality by 36 to 50 percent (USDHHS 1990; Kumanan et al. 2000; Wilson et al. 2000; Rea et al. 2002; Critchley and Capewell 2003).
Smoking cessation is highly cost-effective (Krumholz et al. 1993; Lightwood 2003) and is recommended in professional guidelines for prevention of recurrent cardiovascular events in persons with known CVD (Smith et al. 2001). Evidence supports the central roles of smoking cessation and eliminating exposure to secondhand smoke in preventing development and progression of CVD (USDHHS 1990, 2006; Benowitz and Gourlay 1997; Hasdai et al. 1997a; van Domburg et al. 2000; Wilson et al. 2000; Goldenberg et al. 2003).
Methods
Tobacco use and dependence are determined by complex physiological and psychological factors. Use of nicotine causes tolerance, physical dependence, and a withdrawal syndrome when smoking is stopped (USDHHS 1988). Use of tobacco is a learned behavior that becomes part of the daily routine of a smoker and is often used to cope with stress, anxiety, anger, and depression (USDHHS 1988; Rigotti 2002).
Interventions to achieve smoking cessation target both the physiological and psychological factors that contribute to tobacco use. Evidence from randomized controlled clinical trials of cessation methods has been summarized in meta-analyses conducted independently by two groups—the U.S. Public Health Service (PHS) and the Cochrane Database of Systematic Reviews (the Cochrane Library). These reviews document the efficacy of both psychosocial counseling and pharmacologic agents for cessation (Fiore et al. 2000, 2008). Combination of the two methods is the most effective strategy. Interventions in psychosocial counseling range from brief counseling by the physician to intensive, cognitive-behavioral counseling interventions during several weeks. There is a dose-response relationship between behavioral treatment and smoking cessation; that is, the efficacy of counseling interventions increases with increased intensity and duration of the program (Fiore et al. 2000, 2008; USDHHS 2000). The U.S. Food and Drug Administration (FDA) has approved pharmacotherapy for tobacco dependence. The pharmacotherapy includes five types of NRT (gum, transdermal patch, nasal spray, vapor inhaler, and lozenge), sustained-release bupropion, and varenicline. PHS designated these medications as first-line therapies for smoking cessation in Treating Tobacco Use and Dependence: Clinical Practice Guidelines (Fiore et al. 2000). Two other drugs—nortriptyline and clonidine—were efficacious in randomized controlled trials and were shown to be effective in the Cochrane review and in meta-analyses conducted for development of the PHS guidelines (Fiore et al. 2000, 2008; Gourlay et al. 2004; Hughes et al. 2004b). These drugs have not been approved by FDA for use in smoking cessation, and in the PHS report, they are designated as second-line interventions. There is no evidence to support use of alternative therapies such as acupuncture and hypnosis for smoking cessation (Abbot et al. 1998; Fiore et al. 2000, 2008; White et al. 2002).
Interventions
Multiple randomized controlled clinical trials demonstrated the benefits of counseling patients with CVD on smoking cessation (Table 6.4) (Thomson and Rigotti 2003). In contrast, relatively few clinical trials tested the safety or efficacy of pharmacotherapy for treating smokers with CVD (Table 6.5). Researchers raised concerns about the safety of NRT and sustained-release bupropion in patients with CVD, because both agents can have sympathomimetic activity and can theoretically increase myocardial work, and NRT might also reduce the myocardial oxygen supply through coronary vasoconstriction by aggravating endothelial dysfunction (Benowitz and Gourlay 1997).
Table 6.4
Randomized controlled trials of counseling for smokers hospitalized with cardiovascular disease.
Table 6.5
Randomized controlled trials of pharmacologic interventions for smoking cessation in patients with cardiovascular disease.
Counseling
Several randomized controlled clinical trials demonstrated the efficacy of counseling for patients hospitalized with CVD (Table 6.4). The evidence for efficacy is strongest for patients who had acute MI (Pozen et al. 1977; Taylor et al. 1990; Ockene et al. 1992; DeBusk et al. 1994; Dornelas et al. 2000). In one study, an intensive, nurse-managed intervention to achieve smoking cessation for 173 smokers hospitalized with an acute MI doubled the cessation rates at one year (61 versus 32 percent, p <0.001) (Taylor et al. 1990). At the bedside, the nurse delivered a 30-minute cognitive-behavioral counseling session focused on self-efficacy and prevention of relapse. Additional counseling was delivered by telephone at one, two, and three weeks after discharge and then every month for four months. A second study of the same patients expanded the nurse-delivered counseling model to target multiple cardiac risk factors (DeBusk et al. 1994). This study also reported rates of smoking abstinence higher than those in the first study (Taylor et al. 1990). A third trial assigned 100 consecutive smokers admitted with MI to either minimal care or bedside counseling with seven follow-up telephone calls (Dornelas et al. 2000). Smoking cessation rates at one year were higher in the intervention group than in the minimal care group (55 versus 34 percent, p <0.05). A more recent study of 240 smokers admitted to the hospital for MI, unstable angina, or cardiac bypass surgery demonstrated that counseling by cardiac nurses untrained in counseling and then follow-up counseling during the next five months reduced smoking rates at one year (50 versus 37 percent, absolute risk reduction, 13 percentage points [95 percent CI, 0 to 26 percentage points]) (Quist-Paulsen and Gallefoss 2003).
Other studies that lacked the same intensity of follow-up after hospital discharge produced less impressive results. One study examined a multicomponent behavioral smoking intervention delivered to 267 patients having coronary angiography (Ockene et al. 1992). Compared with patients who did not receive the intervention, those with angiography had higher rates of validated smoking abstinence at 6 months (45 versus 34 percent) and 12 months (35 versus 28 percent), but these differences were not statistically significant. Two randomized trials (Rigotti et al. 1994; Hajek et al. 2002) and one partially randomized trial (Bolman et al. 2002) examined the effects of inpatient counseling with minimal follow-up in cardiac patients. These studies showed no improvement in abstinence rates with the counseling intervention versus usual care.
The most successful counseling interventions for cardiac inpatients include high-intensity baseline counseling with sustained contacts after discharge for prevention of relapse. However, even with the most successful counseling interventions, at least 40 percent of smokers who have cardiac disease resume smoking within one year. Guidelines for smoking cessation recommend addition of pharmacotherapy to counseling (Fiore et al. 2000, 2008). Pharmacotherapy has the potential to improve smoking cessation rates in smokers with CVD (see “Nicotine Replacement Therapy” and “Bupropion” later in this chapter).
Nicotine Replacement Therapy
NRT helps smokers stop smoking and also reduces nicotine withdrawal symptoms, which begin a few hours after the last cigarette is smoked and can last up to four weeks (Hughes et al. 1992). The typical withdrawal syndrome is characterized by agitation, anxiety, depressed mood, difficulty concentrating, increased appetite, insomnia, irritability, restlessness, and an intense craving to smoke. Most smokers who stop smoking relapse to smoking within the first week, when withdrawal symptoms are strongest.
In multiple clinical trials, all the NRT products approximately doubled rates of smoking abstinence compared with rates for participants receiving a placebo (Fiore et al. 2000; Silagy et al. 2004). Meta-analyses conducted for PHS demonstrated ORs of 1.9 (95 percent CI, 1.7–2.2) for the nicotine patch and 1.5 (95 percent CI, 1.3–1.8) for nicotine gum. Meta-analyses from the Cochrane Library found similar results; ORs were 1.74 (95 percent CI, 1.57–1.93) for the patch and 1.66 (95 percent CI, 1.52–1.81) for the gum (Silagy et al. 2004). For smokers with greater dependence on nicotine, the 4-mg dose of nicotine gum was more effective than the 2-mg dose. Maximum effectiveness depended on correct chewing techniques. Similar ORs were reported after meta-analyses of data on the nicotine inhaler and nicotine nasal spray (Fiore et al. 2000). Only one study compared the efficacy of four forms of NRT (patch, gum, inhaler, and nasal spray); they demonstrated similar efficacy rates after 12 weeks of follow-up (Hajek et al. 1999).
Nicotine directly affects the cardiovascular system by multiple mechanisms (see “Cigarette Smoke Constituents and Cardiovascular Disease” earlier in this chapter). The various effects lead to increased heart rate, blood pressure, and myocardial contractility, and reduced coronary blood flow. Nicotine may also contribute to insulin resistance and development of a more atherogenic lipid profile. The nicotine dose in NRT products is usually lower than the dose from smoking, but there have been concerns about the safety of NRT in patients with CVD. Case reports in the medical literature described atrial fibrillation, MI, and stroke in patients receiving NRT (Joseph and Fu 2003). It is difficult to assess the cardiovascular risk from NRT on the basis of these reports, because of the inability to control for individual risk factors for these events, especially that these persons were smokers (Benowitz and Gourlay 1997; Joseph and Fu 2003).
To date, three randomized controlled trials of transdermal use of nicotine have been conducted in patients with stable CVD. The first study enrolled 156 patients with CHD and randomly assigned them to receive either 14-mg nicotine patches or a placebo for five weeks (Working Group for the Study of Transdermal Nicotine in Patients with Coronary Artery Disease 1994). The dose was increased to 21 mg if smoking persisted. Smoking abstinence was achieved at five weeks by 36 percent in the patch group and 22 percent in the placebo group (p <0.05). Patients recorded all episodes of angina, palpitations, and other cardiac symptoms in daily diaries and had a 12-lead electrocardiogram at three time points. The nicotine patch did not affect the frequency of angina, arrhythmias, or depression of the ST segment (isoelectric period) on electrocardiograms during the five weeks of treatment, even in patients who smoked intermittently.
A second randomized trial of transdermal nicotine included 584 outpatients with CVD from 10 Veterans Affairs hospitals (Joseph et al. 1996). The participants were randomly assigned to receive 21-mg patches or a placebo for 10 weeks. Primary cardiovascular endpoints during 14 weeks of follow-up included MI, cardiac arrest, death, and hospital admission for angina, dysrhythmia, or congestive heart failure. The two groups did not differ in the proportion of patients who reached at least one cardiovascular endpoint (5.4 versus 7.9 percent, p = 0.23). Concomitant use of the nicotine patch and smoking was not associated with an increase in adverse events. Although use of the patch was safe in this population, no improvement was observed in rates of short-term or long-term smoking abstinence in comparisons with the placebo group (Joseph et al. 1996; Joseph and Antonuccio 1999).
A third trial tested use of the nicotine patch in 106 smokers with CHD (Tzivoni et al. 1998). Patients were randomly assigned to receive nicotine patches or placebo patches for two weeks. All patients had ambulatory electrocardiogram monitoring and exercise testing at study entry, after the first application of the patch, and at two weeks. No difference was observed at any of the three time points, between the patch and placebo groups, in resting heart rate, blood pressure, the number or duration of ischemic episodes, frequency of arrhythmias, exercise duration, or time to 1-mm depression of the ST segment on an electrocardiogram. In a randomized study of 234 patients with both cardiovascular and respiratory diseases, no increase in adverse events was observed in patients assigned to the nicotine patch or the placebo (Campbell et al. 1996).
In a case-control study, Kimmel and colleagues (2001) found no increased risk of a first MI with use of the nicotine patch in persons who stopped smoking or those who continued to smoke. Using a computerized database for general practice in the United Kingdom, Hubbard and colleagues (2005) studied the relative incidence of MI and stroke in four two-week periods before and after the first prescription for NRT. They found a progressive increase in risk in the 56 days after the first NRT prescription but no evidence of increased cardiovascular events or mortality in the 56 days after the NRT prescription.
Meine and colleagues (2005) addressed the safety of transdermal nicotine in the setting of acute CHD. These investigators analyzed data in the Duke University Cardiovascular Databank for 9,991 smokers who had cardiac catheterization after hospital admission for unstable angina or non-ST-segment MI. This retrospective observational study compared outcomes of patients who did or did not receive transdermal nicotine during hospitalization. The study identified 194 patients who had been treated with transdermal nicotine during the hospital stay. The investigators used a “propensity score analysis” to compare patients receiving transdermal nicotine with matched patients from the database who did not receive the medication. Because patients were not randomly assigned to receive transdermal nicotine, selection bias could have confounded these results. In an attempt to reduce this bias, patients who did or did not receive transdermal nicotine were matched on demographic characteristics, diagnosis, cardiac risk factors, and mean cardiac ejection fraction. Rates of cardiac outcomes in the two groups were compared. No differences in 7-day, 30-day, or one-year mortality rates were observed. Patients receiving transdermal nicotine were not more likely to have coronary artery bypass grafting or percutaneous transluminal coronary angioplasty during hospitalization. It was not possible to control for the dose of medication received, the amount of smoking before hospital admission, or relapse to smoking after hospitalization. These results provide some evidence that NRT was safe in the setting of acute CVD, but randomized controlled trials are needed to establish the safety of NRT in patients who have unstable cardiac disease.
Other studies examined markers of exposure to tobacco smoke related to cardiovascular risk as surrogate endpoints for cardiovascular events in clinical trials. There was no evidence that NRT raised blood pressure in any of the efficacy trials, but these trials typically excluded patients with poor control of hypertension. In one small trial of 30 smokers with or without hypertension, NRT increased mean arterial pressure in smokers with normal blood pressure but not in smokers who had hypertension (Tanus-Santos et al. 2001).
To date, two studies have tested the effect of NRT on coronary circulation in smokers with CHD. One study examined the size of defects in myocardial perfusion in 36 patients with baseline CHD who were treated with nicotine patches (Mahmarian et al. 1997). Participants continued to smoke but at reduced levels. The researchers concluded that use of the patch, even with concomitant smoking and higher plasma levels of nicotine, resulted in reduction of exercise-induced ischemia in a comparison with baseline values. This finding suggested that components of tobacco smoke other than nicotine are responsible for impaired coronary blood flow. The second study investigated the effect of nicotine gum on coronary perfusion in former cigarette smokers having angiography (Nitenberg and Antony 1999). The findings demonstrated that the gum did not reduce the surface area of normal or diseased segments of the coronary artery. Other studies of the effects of smoking cessation on lipids and thrombosis reported improvements in these markers, even among smokers using NRT (Allen et al. 1994; Lúdvíksdóttir et al. 1999; Eliasson et al. 2001; Haustein et al. 2002).
In summary, despite anecdotal reports of cardiovascular events attributable to use of NRT, data from multiple clinical trials of smokers with or without CVD show no evidence for increased cardiovascular risk when NRT is used to treat tobacco dependence. However, the safety of NRT has not been tested in a more acute setting, such as during hospitalization for a cardiovascular event. Observational data suggest that use of the nicotine patch in patients with unstable cardiac disease is probably safe (Meine et al. 2005), but randomized trials are needed to confirm these findings. Current PHS guidelines recommend that NRT be used with caution in smokers with unstable angina, MI in the past two weeks, or serious arrhythmia (Fiore et al. 2000).
Bupropion
Bupropion is an aminoketone approved by FDA in 1989 for treatment of depression and in 1997 for smoking cessation. The drug is included in national guidelines as first-line therapy for smoking cessation (Fiore et al. 2000, 2008). Its mechanism of action is not fully understood, but researchers think it acts by inhibiting neuronal uptake of norepinephrine and dopamine. Bupropion may also block activity of nAChRs. The mechanism of action for smoking cessation appears to be unrelated to the antidepressant effects of bupropion. A preparation of the drug for sustained release provides a better safety profile and more convenient dosing than does the immediate-release form.
Evidence from several randomized controlled trials shows that bupropion doubled the smoking cessation rates obtained with a placebo. Meta-analyses of data on bupropion for smoking cessation conducted by PHS and the Cochrane Library yielded ORs of 2.1 (95 percent CI, 1.5–3.0) and 2.06 (95 percent CI, 1.77–2.40), respectively (Fiore et al. 2000; Hughes et al. 2004b). One trial compared use of bupropion, a nicotine patch, bupropion plus a patch, and a placebo among 893 participants (Jorenby et al. 1999). Smoking abstinence rates at one year were 15.6 percent in the placebo group, 16.4 percent in the nicotine-patch group, 30.3 percent in the bupropion group (p <0.0001 versus the placebo group), and 35.5 percent in the bupropion-plus-patch group (p <0.0001 versus the placebo group; p = 0.06 versus the bupropion-alone group).
The major risk of bupropion is that it lowers a person’s seizure threshold. The risk of seizure from the sustained-release formulation is 0.1 percent, which is no different from that for other antidepressants (Hughes et al. 1999; Rigotti 2002). No seizures were reported in any of the clinical trials that tested sustained-release bupropion for smoking cessation.
As with NRT, early case reports of serious cardiovascular events with sustained-release bupropion raised questions about the safety of this agent in patients with CVD. These reports, which were mostly in Canada and England, included cardiac deaths, chest pain, MI, and myocarditis (Joseph and Fu 2003). Assessment of the contribution of bupropion to these events is difficult because evaluation of other cardiac risk factors in these patients was not possible.
To date, none of the efficacy trials of bupropion for smoking cessation has reported a significant increase in cardiovascular events. Two randomized controlled trials enrolled only smokers with CVD. The first trial enrolled 629 outpatients with stable CVD—that is, MI or an interventional cardiac procedure more than three months earlier and stable angina pectoris, PAD, or congestive heart failure (Tonstad et al. 2003). Patients were randomly assigned to receive bupropion or a placebo for seven weeks. This study found no differences in the number of deaths in the two groups—two in the bupropion group and two in the placebo group. Overall, 38 participants (6 percent) reported a single adverse cardiovascular event—24 in the bupropion group and 14 in the placebo group. The most common cardiovascular events were angina pectoris, hypertension, and palpitations; 13 events occurred in the bupropion group versus 8 events in the placebo group. No statistical tests were performed on the rates of adverse events. Patients who took bupropion were more likely to have stopped smoking at one year than were patients who took the placebo (27 versus 12 percent, p <0.001).
A second trial enrolled 248 smokers hospitalized with acute CVD, including acute MI, unstable angina, or other cardiovascular conditions (Rigotti et al. 2006). Patients were randomly assigned to receive sustained-release bupropion or a placebo for 12 weeks, and all patients received intensive counseling during hospitalization and follow-up. At the one-year follow-up, the difference between death rates in the bupropion group (no deaths) and in the placebo group (two deaths) was not statistically significant. During the 12 weeks of drug treatment, the difference between the number of cardiovascular events in the bupropion group (20 events) and the placebo group (17 events) was also not significant. Cardiovascular events included death, nonfatal MI, unstable angina, congestive heart failure, stroke, and coronary revascularization procedure. At the one-year follow-up, the number of cardiovascular events in the bupropion group (32 events, 26 percent) exceeded the number in the placebo group (22 events, 18 percent), but this difference was not significant. In addition, at one year, the difference between the smoking abstinence rates for the bupropion group (25 percent) versus the placebo group (21 percent) was not significant. However, the results after 12 weeks of drug treatment suggested that bupropion had short-term efficacy (37 versus 27 percent, p = 0.08).
In summary, sustained-release bupropion is effective and safe for treating smokers with stable CVD. The drug appears to be less efficacious in smokers hospitalized with acute CVD than in other groups of patients. Bupropion is the only medication for treating tobacco dependence that has been tested in patients with acute CVD, and it appears to be safe for those with either stable or acute disease.
Other Pharmacotherapy
Varenicline, a partial agonist of the α4β2 nAChR, has been marketed for the treatment of tobacco dependence but its use in smokers with CVD has not yet been studied (Coe et al. 2005). The drug produces approximately 50 percent of the receptor stimulation provided by nicotine, but it blocks the effects of any nicotine taken in from cigarette smoking. Clinical trials have found it superior to bupropion in promoting smoking cessation, and prolonged administration has been shown to reduce relapse in smokers who had been abstinent 12 weeks after initial therapy (Gonzalez et al. 2006; Jorenby et al. 2006; Tonstad et al. 2006). Two other medications have been demonstrated to be effective for smoking cessation: nortriptyline, a tricyclic antidepressant, and clonidine, a central α-agonist antihypertension agent. However, neither drug has been approved by FDA for smoking cessation. Both agents have potential cardiovascular side effects, and the safety profile of these drugs should be considered carefully before use in smokers with CVD.
Meta-analyses of data on use of clonidine for treating smokers resulted in ORs for smoking abstinence of 2.1 (95 percent CI, 1.4–3.2) (Fiore et al. 2000) and 1.89 (95 percent CI, 1.30–2.74) (Gourlay et al. 2004). To date, no safety data for patients with CVD are available. However, clonidine is known to cause orthostatic hypotension, rebound hypertension from abrupt cessation of the drug, and rarely, atrioventricular nodal blockade. Nortriptyline is also effective in promoting smoking cessation; meta-analyses yielded ORs of 3.2 (95 percent CI, 1.8–5.7) (Fiore et al. 2000) and 2.79 (95 percent CI, 1.70–4.59) (Hughes et al. 2004b). Nortriptyline was designated a second-line drug for smoking cessation in PHS clinical guidelines because of a smaller evidence base of support and greater side effects than those of other medications for smoking cessation. In general, tricyclic antidepressants are avoided in patients with CVD because of concerns about increased risks for arrhythmias and depression of myocardial contractility (Joseph and Fu 2003).
Although the focus of the preceding section (“Methods”) was on clinical interventions to reduce smoking, it is important to recognize that policy-based interventions, such as smoke-free environments and community and statewide tobacco control programs, are also important elements in a strategy to improve cardiovascular health. For example, smoke-free workplaces are a highly cost-effective approach to promoting smoking cessation with an impact on cardiovascular health (Ong and Glantz 2004). Decreases in admissions to hospitals have been observed after smoke-free laws have gone into effect (Dinno and Glantz 2007). The California Tobacco Control Program substantially accelerated the decline in the heart disease death rate in the state (Fichtenberg and Glantz 2000).
It should be noted that although long-term smoking quit rates after various interventions in cardiovascular patients may appear to be low (most less than 30 percent), smoking cessation therapy has an important impact on CVD, and the cost per life saved is lower than that of many other therapeutic interventions for CHD that are considered to be the standard (such as treatment of hypertension and hyperlipidemia) (Lightwood 2003).
Summary
Smoking cessation is a key element in both primary and secondary prevention of CVD. Guidelines from PHS recommend counseling, NRT, sustained-release bupropion, and varenicline as first-line treatments to achieve smoking cessation. Studies show that NRT and bupropion are effective in patients with CVD, although not all trials demonstrated efficacy. Several studies have demonstrated the safety of NRT in patients with stable CVD, but randomized trials are needed to establish the safety of this treatment for patients hospitalized with acute disease. Bupropion appears to be safe in patients with stable or unstable CVD, but it is less effective in patients with acute disease. Varenicline is a partial agonist of the α4β2 nAChR that is effective in treating tobacco dependence, but it has not yet been studied in smokers with CVD. The development of more effective pharmacotherapies to aid smoking cessation that are safe in persons with CVD is a high research priority.
Methods to Reduce Exposure
Evidence-based interventions for treating smokers include behavioral and pharmacological treatments, which significantly increase rates for long-term abstinence from smoking. Even so, absolute rates of abstinence are modest; they range from 8 to 25 percent, depending on the study population and the treatment. In addition, only a small proportion of smokers are interested in treatment at any given time. Interest in smoking cessation and success in achieving long-term abstinence are greater among patients with CVD than in the general population of smokers (Thomson and Rigotti 2003). Nevertheless, abstinence rates remain disappointingly low, particularly in light of the important health benefits for this population when they do stop smoking (USDHHS 1990; Burns 2003).
Suboptimal treatment outcomes prompted interest in testing interventions that might decrease the risk of smoking among those who continue to use tobacco. These strategies are often termed “harm reduction” interventions, although data are limited as to whether harm is really lessened with reduced exposure to tobacco. To date, the effect of methods for reducing exposure on risk factors for CVD and on development of CVD has been evaluated in a limited number of clinical trials, prospective cohort studies, and epidemiologic studies.
Endpoints with respect to CVD that have been measured in studies of reduced exposure include measures of exposure to tobacco constituents (e.g., nicotine and CO); biomarkers of inflammation (e.g., CRP, leukocyte counts, and fibrinogen); thrombosis (e.g., fibrinogen and PAI-1); lipid abnormalities (e.g., levels of total cholesterol, HDLc, LDLc, triglycerides, APOs A-I and B, and HDLc to LDLc ratio); oxidative stress that reflects and may contribute to cardiovascular risk (e.g., F2-isoprostanes); and clinical outcomes (blood pressure, heart rate, angina, exercise tolerance, MI, other adverse events, and death) (Table 6.3).
Reduced Smoking
Important methodologic considerations in evaluating studies of reduced smoking include the extent and duration of smoking reduction, use of nicotine replacement products, doses, and timing of endpoint measurements (Table 6.6). In addition, some studies report outcome data on the basis of intention to treat all participants, regardless of whether treatment was successful. Others report only on subgroups who achieve specific goals for smoking reduction.
Table 6.6
Smoking reduction and cardiovascular disease endpoints: biomarkers and clinical outcomes.
Eliasson and colleagues (2001) tested the effect of nicotine nasal spray on achieving smoking reduction and abstinence among 58 healthy adult smokers in an open-label cohort study. The primary goal for the first eight weeks of the study was to reduce daily smoking by 50 percent; participants were asked to stop smoking after eight weeks. Cardiovascular risk factors were evaluated at baseline, at eight weeks, and after eight weeks of abstinence. The 33 study participants provided data at all three time points. After participants completed eight weeks of smoking reduction, mean cigarette use decreased by 50.2 percent, expired CO dropped 17 percent, and plasma thiocyanate decreased by 20.1 percent. Significant improvements included fibrinogen levels (from 2.9 g/liter [L] to 2.65 g/L, p = 0.011); hemoglobin values (from 13.8 to 13.3 g/L, p <0.001); leukocyte counts (from 7.0 to 6.2 x 109/L, p = 0.005); and HDLc to LDLc ratio (from 0.33 to 0.37, p <0.005). Eight weeks of abstinence from smoking was associated with further improvements in hemoglobin levels, leukocyte counts, and HDL and LDL levels, and significant reduction in PAI-1 activity. These researchers did not observe improvements in HDLc and LDLc levels with reduction in smoking.
Hurt and colleagues (2000) conducted a small, open-label cohort study to test the effect of a nicotine inhaler on biomarkers of exposure to cigarette smoke among 23 heavy smokers. Levels of blood thiocyanate, several urine carcinogens, and expired CO were measured at study entry and at 4, 8, 12, and 24 weeks. Despite an average reduction among participants from 40 or more to 10 cigarettes per day, expired CO decreased only from 30.4 to 26.0 parts per million (ppm), reflecting compensatory smoking or misreporting of reduction in smoking. This change was not statistically significant.
In the U.S. Lung Health Study, 5,887 male and female smokers were randomly assigned to one of three groups: intervention for smoking cessation, including nicotine gum, plus bronchodilator therapy; intervention plus a placebo; or usual care (Hughes et al. 2004a). Among 3,923 participants in the intervention at the one-year follow-up, 1,722 continued to smoke daily. Reduction in the number of cigarettes smoked was not an objective of the intervention, but 16 percent of those who continued to smoke daily smoked 1 to 24 percent fewer cigarettes than at baseline; another 27 percent reduced smoking by 25 to 49 percent; 19 percent, by 50 to 74 percent; and 11 percent, by 75 to 99 percent. The mean reduction in cigarettes smoked was 29 percent (from 32 to 22 cigarettes per day), and the mean reduction in CO levels was 24 percent (from 34 to 26 ppm). Thus, more than 80 percent of those who did not stop smoking achieved some level of reduction. However, the reduction in CO was not as large as the reduction in cigarettes per day. This finding again suggests compensatory smoking.
Hatsukami and colleagues (2005) examined the effect of smoking reduction on cardiovascular risk factors among 151 cigarette smokers interested in stopping smoking but not in reducing their smoking. Nicotine patches and gum were used to assist with reduction of smoking. The cardiovascular risk factors (CO and cholesterol levels, leukocyte counts, blood pressure, and heart rate) were measured for 12 weeks after study entry. Biomarkers did not change among persons who continued to smoke ad libitum, but the 61 persons who reduced smoking achieved significant improvements. Smokers who reduced the self-reported number of cigarettes smoked per day by 40 percent or more had significantly reduced leukocyte counts (from 7.39 to 6.98 x 109/L, p <0.001); higher HDLc levels (from 50.3 to 52.8 mg per deciliter [dL], p <0.0167); improved HDL to LDL ratios (from 0.47 to 0.49, p <0.0167); lower APO B levels (from 103.7 to 103.0 mg/dL, p <0.0167); lower systolic blood pressure (from 123.0 to 120.3 mm Hg, p <0.0167); and lower heart rate (from 75.7 to 70.2 beats per minute, p <0.001). Although some of these changes were statistically significant, they are modest, and the clinical importance is undetermined. Levels of triglycerides, total cholesterol, APO A-I, and diastolic blood pressure did not change, and LDLc levels increased from 122.1 to 124.4 mg/dL (p <0.0167).
To date, only one randomized controlled trial of an intervention for smoking reduction among persons with known CVD has been conducted (Joseph et al. 2005). Treatment included behavioral counseling and NRT with patches and gum. The goal of this study of 152 participants was at least a 50-percent reduction in cigarettes smoked per day; usual care was the standard for comparison. At study entry, participants smoked an average of 27.4 cigarettes per day. At six months, the intervention group had reduced cigarette use by 39 percent, versus a decline of 25 percent in the usual care group, but this difference was not statistically significant. Biomarkers for carcinogenesis and CVD were measured, as were clinical outcomes. No significant differences between the treatment groups were observed for levels of CO, fibrinogen, F2-isoprostanes, or CRP, or for leukocyte counts. The groups did not differ in clinical outcomes, including body weight, distance completed in a six-minute walking test, the proportion of participants completing that test, the prevalence and frequency of angina, the need for urgent cardiac care, and other serious adverse events. Because no significant differences between treatment groups were observed and because both groups achieved significant reductions in cigarette use from baseline, results for the entire cohort at six months were compared with the baseline data. There were no significant differences in biomarkers of cardiovascular risk, including leukocyte counts and levels of F2-isoprostanes or CRP, but CO had decreased by 6.0 ppm (p = 0.007).
Godtfredsen and colleagues (2003) conducted a prospective cohort study in Denmark to examine changes in the incidence of MI after spontaneous reductions in cigarette use. This study included 10,956 men and 8,467 women who provided detailed information on smoking behavior during two examinations. Mortality registers and hospital registers were searched for an incident of hospital admission or a death attributable to MI. A sample consisting of pooled data from three population studies yielded 643 participants who were heavy smokers at study entry and were evaluated for the effects of reduced smoking.
These persons reported unassisted reductions in tobacco use by at least 50 percent and were compared with 1,379 persons who reported abstinence from smoking. Outcomes were adjusted for baseline cardiovascular risk factors. Smoking cessation was associated with a hazard ratio for MI of 0.71 (95 percent CI, 0.59–0.85), but smoking reduction was not associated with a statistically significant reduction in risk of MI (hazard ratio, 1.15 [95 percent CI, 0.94–1.40]). A subgroup analysis demonstrated significant reductions in levels of expired CO among persons who reduced cigarette smoking. The investigators concluded that the results were consistent with a short-term thrombogenic effect of tobacco exposure rather than with a cumulative effect of exposure. They speculated that an approximate 50-percent reduction (from 20 g to 10 g of tobacco smoked per day) was not sufficient to improve cardiovascular risk. These epidemiologic data make an important contribution because they are population based and come from a large cohort of smokers who reduced their smoking during a period longer than the usual timeframe for clinical trials.
In summary, these studies show that a significant reduction in cigarette use, even to levels as low as 10 cigarettes per day, results in reduction of exposure to CO from tobacco smoke that is smaller than expected. This finding most likely reflects compensatory smoking. Some but not all studies showed reduced exposure to nicotine, as well as improvements in values for hemoglobin, leukocyte counts, and fibrinogen and cholesterol levels. However, the improvements in values are relatively minor compared with those observed with abstinence from smoking. Many of these improvements occurred in study participants who were using NRT (see “Nicotine Replacement Therapy” below). None of the studies showed improvements in clinical outcomes of heart disease, consistent with evidence presented earlier that low levels of smoke exposure trigger many of the adverse cardiovascular effects of smoke (see “Exposure to Secondhand Tobacco Smoke” earlier in this chapter).
Nicotine Replacement Therapy
In addition to providing data on smoking reduction, the Lung Health Study of 3,094 persons offered a unique opportunity to examine the natural history and safety of prolonged use of nicotine gum among thousands of people during a five-year follow-up (Murray et al. 1996). Persistent smoking, but not use of nicotine gum, predicted fatal and nonfatal cardiovascular events and elevation of diastolic blood pressure.
In general, trials of smoking cessation and smoking reduction showed improvements in lipid profiles, even with NRT. Eliasson and colleagues (2001) observed that use of nicotine nasal spray for smoking reduction or cessation yielded significant improvements in HDLc to LDLc ratios at 9 weeks, with further improvements if smokers abstained from smoking for 17 weeks (see “Reduced Smoking” earlier in this chapter). In addition, significant improvements in fibrinogen levels and leukocyte counts were observed. A clinical trial of smoking cessation also reported significant decreases in hemoglobin values, leukocyte counts, and total and LDLc concentrations (Lúdvíksdóttir et al. 1999). In addition, HDLc to LDLc ratios improved among participants who abstained from smoking after three months of treatment with a nicotine nasal spray. Allen and colleagues (1994) also observed significantly increased HDLc levels in participants treated with the nicotine patch.
Investigators have noted improvements in markers of thrombogenesis among persons who abstained from smoking and were using medicinal nicotine. In one study, plasma fibrinogen levels were reduced among 164 men using a combination of a nicotine patch and gum for 12 weeks to stop smoking (Haustein et al. 2002). In another study, use of transdermal nicotine appeared to activate platelet aggregation less than smoking did (Benowitz et al. 1993).
Mahmarian and colleagues (1997) used single photon emission computed tomography to measure the combined effects of smoking and use of nicotine patches on myocardial perfusion. In 36 patients with known CHD who were treated with nicotine patches, the amount of heart muscle deprived of normal blood flow (size of perfusion defects) decreased despite increased serum nicotine levels. The baseline size of defects and changes in CO levels, but not in nicotine levels, predicted the size of perfusion defects. The researchers concluded that the reduction in the size of defects resulted from reduction in smoking that was facilitated by NRT and from decreased exposure to CO. They also concluded that nicotine patches were safe to use in smokers with heart disease.
Nitenberg and Antony (1999) used angiography to examine short-term effects of use of nicotine gum on perfusion of the coronary arteries in former cigarette smokers at study entry, after a test with immersion of the hand in cold water (cold pressor test) before and after administration of the gum. The gum did not augment the result of the cold pressor test, which constricted normal and diseased segments of the coronary artery, reducing their cross-sectional area, and it did not reduce the surface area of the arterial segments at rest or under conditions of sympathetic stimulation.
These results suggest that reduction of exposure to tobacco smoke through use of NRT or with abstinence from smoking is associated with improvements in biomarkers of cardiovascular risk.
Summary
Epidemiologic studies demonstrate a strong dose-response relationship between the number of cigarettes smoked per day and cardiovascular risk. The relationship is not linear, however, and even low levels of exposure to tobacco, such as a few cigarettes per day, occasional smoking, or exposure to secondhand tobacco smoke are sufficient to substantially increase risk of cardiac events. Some interventions have accomplished significant reductions in the number of cigarettes smoked per day, but the reductions in levels of biomarkers of exposure and biomarkers of cardiovascular risk factors are not proportional, probably because of compensatory smoking by study participants and the nonlinear dose-response relationship. The limited data on clinical outcome do not confirm reduction in cardiovascular events due to reduced smoking. Other methods for reducing exposure, including NRT with abstinence from smoking, are associated with more improvement in risk factors for CVD than is smoking reduction with or without pharmacologic support. Accordingly, these methods hold more potential for reducing risk.
Implications
These findings suggest that to lower cardiac risk, interventions would have to reduce exposure to tobacco smoke to extremely low levels or eliminate the exposure. Studies of smoking reduction to date suggest that goals would be difficult to accomplish. Reducing exposure by reducing smoking, therefore, appears to have limited promise for improving cardiac risk unless this method contributes to eventual smoking cessation (Hughes 2000). Because smoking cessation is associated with marked improvements in the risk of MI, sudden death, and stroke, it should be stressed as the goal for interventions dealing with dependence on tobacco. The safety and efficacy of long-term NRT use to reduce cardiovascular risk by maintaining smoking cessation have not been established.
Evidence Summary
Exposure to tobacco smoke is associated with accelerated atherosclerosis and an increased risk of acute MI, stroke, PAD, aortic aneurysm, and sudden death. Smoking appears to have both causal relationships and multiplicative interactions with other major risk factors for CHD, including hyperlipidemia, hypertension, and diabetes mellitus.
The cardiovascular risk attributable to cigarette smoking increases sharply at low levels of cigarette consumption and with exposure to secondhand smoke. The risk then tends to plateau at higher levels of smoking. This finding indicates a low threshold for effect and a nonlinear dose-response relationship. Some of the nonlinearity of the relationship between the number of cigarettes smoked per day and CVD risk may be due to impreciseness of this measure of actual exposure to smoke. However, the data on risk associated with exposure to secondhand smoke indicate a true nonlinear relationship between exposure and CVD risk. Cardiovascular risk is not reduced by smoking cigarettes of lower machine-delivered yields of nicotine or tar.
The constituents of tobacco smoke believed to be responsible for cardiovascular disease include oxidizing chemicals, nicotine, CO, and particulate matter. Oxidizing chemicals, including oxides of nitrogen and many free radicals, increase lipid peroxidation and contribute to several potential mechanisms of CVD, including inflammation, endothelial dysfunction, oxidation of LDL, and platelet activation.
Nicotine is a sympathomimetic drug that increases heart rate and cardiac contractility, transiently increasing blood pressure and constricting coronary arteries. Nicotine may also contribute to endothelial dysfunction, insulin resistance, and lipid abnormalities. However, international epidemiologic evidence and data from clinical trials of nicotine patches suggest that chemicals other than nicotine contribute to an elevated risk of death from MI and stroke. CO reduces the delivery of oxygen to the heart and other tissues and can aggravate angina pectoris or PAD and can lower the threshold for arrhythmias in the presence of CHD. Exposure to particulates is associated with oxidant stress and cardiovascular autonomic disturbances that potentially contribute to acute cardiovascular events.
Cigarette smoking causes acute cardiovascular events such as MI and sudden death by adversely affecting the balance of myocardial demand for oxygen and nutrients and coronary blood flow. Smoking results in increased myocardial work, reduced coronary blood flow, and enhanced thrombogenesis. Enhancement of thrombogenesis appears to be particularly important in that smokers with acute MI have less severe underlying coronary artery disease than do nonsmokers with MI, but smokers have a greater burden of thrombus.
Several potential mechanisms appear to contribute to the effects of smoking in accelerating atherosclerosis. These mechanisms include inflammation, endothelial dysfunction, impaired insulin sensitivity, and lipid abnormalities. Cigarette smoking is a risk factor for diabetes and aggravates insulin resistance in persons with diabetes. The mechanism appears to involve both the effects of oxidizing chemicals in the smoke and the sympathomimetic effects of nicotine.
Conclusions
- There is a nonlinear dose response between exposure to tobacco smoke and cardiovascular risk, with a sharp increase at low levels of exposure (including exposures from secondhand smoke or infrequent cigarette smoking) and a shallower dose-response relationship as the number of cigarettes smoked per day increases.
- Cigarette smoking leads to endothelial injury and dysfunction in both coronary and peripheral arteries. There is consistent evidence that oxidizing chemicals and nicotine are responsible for endothelial dysfunction.
- Tobacco smoke exposure leads to an increased risk of thrombosis, a major factor in the pathogenesis of smoking-induced cardiovascular events.
- Cigarette smoking produces a chronic inflammatory state that contributes to the atherogenic disease processes and elevates levels of biomarkers of inflammation, known powerful predictors of cardiovascular events.
- Cigarette smoking produces an atherogenic lipid profile, primarily due to an increase in triglycerides and a decrease in high-density lipoprotein cholesterol.
- Smoking cessation reduces the risk of cardiovascular morbidity and mortality for smokers with or without coronary heart disease.
- The use of nicotine or other medications to facilitate smoking cessation in people with known cardiovascular disease produces far less risk than the risk of continued smoking.
- The evidence to date does not establish that a reduction of cigarette consumption (that is, smoking fewer cigarettes per day) reduces the risks of cardiovascular disease.
- Cigarette smoking produces insulin resistance and chronic inflammation, which can accelerate macrovascular and microvascular complications, including nephropathy.
References
- Abbot NC, Stead LF, White AR, Barnes J. Hypnotherapy for smoking cessation. Cochrane Database of Systematic Reviews. 1998;(2) Art. No.: CD001008. [PubMed: 10796583] [CrossRef]
- Åberg A, Bergstrand R, Johansson S, Ulvenstam G, Vedin A, Wedel H, Wilhelmsson C, Wilhelmsen L. Cessation of smoking after myocardial infarction: effects on mortality after 10 years. British Heart Journal. 1983;49(5):416–22. [PMC free article: PMC481326] [PubMed: 6838729]
- Abu-Hayyeh S, Sian M, Jones KG, Manuel A, Powell JT. Cadmium accumulation in aortas of smokers. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(5):863–7. [PubMed: 11348888]
- Adler AI, Stevens RJ, Neil A, Stratton IM, Boulton AJM, Holman RR. UKPDS 59: hyperglycemia and other potentially modifiable risk factors for peripheral vascular disease in type 2 diabetes. Diabetes Care. 2002;25(5):894–9. [PubMed: 11978687]
- Aicher A, Heeschen C, Mohaupt M, Cooke JP, Zeiher AM, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003;107(4):604–11. [PubMed: 12566374]
- Aird WC. The endothelium as an organ. Endothelial Cells in Health and Disease. Aird WC, editor. Boca Raton (FL): Taylor and Francis Group; 2005. pp. 1–31.
- al-Alawi E, Jenkins D. Does maternal smoking increase the risk of neonatal polycythaemia? Irish Medical Journal. 2000;93(6):175–6. [PubMed: 11105440]
- Alcorn HG, Wolfson SK Jr, Sutton-Tyrrell K, Kuller LH, O’Leary D. Risk factors for abdominal aortic aneurysms in older adults enrolled in the Cardiovascular Health Study. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(8):963–70. [PubMed: 8696960]
- Al-Delaimy WK, Manson JE, Solomon CG, Kawachi I, Stampfer MJ, Willett WC, Hu FB. Smoking and risk of coronary heart disease among women with type 2 diabetes mellitus. Archives of Internal Medicine. 2002;162(3):273–9. [PubMed: 11822919]
- Al-Delaimy WK, Willett WC, Manson JE, Speizer FE, Hu FB. Smoking and mortality among women with type 2 diabetes: the Nurses’ Health Study cohort. Diabetes Care. 2001;24(12):2043–8. [PubMed: 11723080]
- Allen SS, Hatsukami D, Brintnell DM, Bade T. Effect of nicotine replacement therapy on post-cessation weight gain and nutrient intake: a randomized controlled trial of postmenopausal female smokers. Addictive Behaviors. 2005;30(7):1273–80. [PubMed: 16022925]
- Allen SS, Hatsukami D, Gorsline J. Transdermal Nicotine Study Group. Cholesterol changes in smoking cessation using the transdermal nicotine system. Preventive Medicine. 1994;23(2):190–6. [PubMed: 8047525]
- Allred EN, Bleecker ER, Chaitman BR, Dahms TE, Gottlieb SO, Hackney JD, Pagano M, Selvester RH, Walden SM, Warren J. Short-term effects of carbon monoxide exposure on the exercise performance of subjects with coronary artery. New England Journal of Medicine. 1989;321(21):1426–32. [PubMed: 2682242]
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. Journal of the American College of Cardiology. 2004;43(10):1731–7. [PubMed: 15145091]
- Ameli FM, Stein M, Provan JL, Prosser R. The effect of postoperative smoking on femoropopliteal bypass grafts. Annals of Vascular Surgery. 1989;3(1):20–5. [PubMed: 2713228]
- Arcavi L, Jacob P III, Hellerstein M, Benowitz NL. Divergent tolerance to metabolic and cardiovascular effects of nicotine in smokers with low and high levels of cigarette consumption. Clinical Pharmacology and Therapeutics. 1994;56(1):55–64. [PubMed: 8033495]
- Attvall S, Fowelin J, Lager I, Von Schenck H, Smith U. Smoking induces insulin resistance—a potential link with the insulin resistance syndrome. Journal of Internal Medicine. 1993;233(4):327–32. [PubMed: 8463765]
- Audoly LP, Rocca B, Fabre J-E, Koller BH, Thomas D, Loeb AL, Coffman TM, FitzGerald GA. Cardiovascular responses to the isoprostane iPF2α-III and iPE2α-III are mediated via the thromboxane A2 receptor in vivo. Circulation. 2000;101(24):2833–40. [PubMed: 10859290]
- Auerbach O, Garfinkel L. Atherosclerosis and aneurysm of aorta in relation to smoking habits and age. Chest. 1980;78(6):805–9. [PubMed: 7449458]
- Axelsen M, Eliasson B, Joheim E, Lenner RA, Taskinen MR, Smith U. Lipid intolerance in smokers. Journal of Internal Medicine. 1995;237(5):449–55. [PubMed: 7738484]
- Axelsson T, Jansson P-A, Smith U, Eliasson B. Nicotine infusion acutely impairs insulin sensitivity in type 2 diabetic patients but not in healthy subjects. Journal of Internal Medicine. 2001;249(6):539–44. [PubMed: 11422660]
- Baggio B, Budakovic A, Vestra MD, Saller A, Bruseghin M, Fioretto P. Effects of cigarette smoking on glomerular structure and function in type 2 diabetic patients. Journal of the American Society of Nephrology. 2002;13(11):2730–6. [PubMed: 12397043]
- Baker F, Ainsworth SR, Dye JT, Crammer C, Thun MJ, Hoffmann D, Repace JL, Henningfield JE, Slade J, Pinney J, et al. Health risks associated with cigar smoking. JAMA: the Journal of the American Medical Association. 2000;284(6):735–40. [PubMed: 10927783]
- Bakhru A, Erlinger TP. Smoking cessation and cardiovascular disease risk factors: results from the Third National Health and Nutrition Examination Survey. PLoS Medicine. 2005;2(6):e160. [PMC free article: PMC1160573] [PubMed: 15974805] [CrossRef]
- Barbieri SS, Eligini S, Brambilla M, Tremoli E, Colli S. Reactive oxygen species mediate cyclooxygenase-2 induction during monocyte to macrophage differentiation: critical role of NADPH oxidase. Cardiovascular Research. 2003;60(1):187–97. [PubMed: 14522422]
- Barger AC, Beeuwkes R 3rd, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries: a possible role in the pathophysiology of atherosclerosis. New England Journal of Medicine. 1984;310(3):175–7. [PubMed: 6197652]
- Barnoya J, Glantz S. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111(20):2684–98. [PubMed: 15911719]
- Barua RS, Ambrose JA, Eales-Reynolds LJ, DeVoe MC, Zervas JG, Saha DC. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation. 2001;104(16):1905–10. [PubMed: 11602492]
- Barua RS, Ambrose JA, Saha DC, Eales-Reynolds LJ. Smoking is associated with altered endothelial-derived fibrinolytic and antithrombotic factors: an in vitro demonstration. Circulation. 2002;106(8):905–8. [PubMed: 12186791]
- Barua RS, Ambrose JA, Srivastava S, DeVoe MC, Eales-Reynolds LJ. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107(18):2342–7. [PubMed: 12707237]
- Barutcu I, Esen AM, Degirmenci B, Acar M, Kaya D, Turkmen M, Melek M, Onrat E, Esen OB, Kirma C. Acute cigarette smoking-induced hemodynamic alterations in the common carotid artery—a transcranial Doppler study. Circulation Journal. 2004;68(12):1127–31. [PubMed: 15564695]
- Bazzano LA, He J, Muntner P, Vupputuri S, Whelton PK. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Annals of Internal Medicine. 2003;138(11):891–7. [PubMed: 12779299]
- Beckman JA, Liao JK, Hurley S, Garrett LA, Chui D, Mitra D, Creager MA. Atorvastatin restores endothelial function in normocholesterolemic smokers independent of changes in low-density lipoprotein. Circulation Research. 2004;95(2):217–23. [PMC free article: PMC2633456] [PubMed: 15178637]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. American Journal of Physiology–Cell Physiology. 1996;271(5Pt1):C1424–C1437. [PubMed: 8944624]
- Benowitz NL. Biomarkers of cigarette smoking. The FTC Cigarette Test Method for Determining Tar, Nicotine, and Carbon Monoxide Yields of US Cigarettes Report of the NCI Expert Committee, Smoking and Tobacco Control Monograph No 7. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1996. pp. 93–111. NIH Publication No. 96-4028.
- Benowitz NL. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Progress in Cardiovascular Diseases. 2003;46(1):91–111. [PubMed: 12920702]
- Benowitz NL, Fitzgerald GA, Wilson M, Zhang Q. Nicotine effects on eicosanoid formation and hemostatic function: comparison of transdermal nicotine and cigarette smoking. Journal of the American College of Cardiology. 1993;22(4):1159–67. [PubMed: 7691912]
- Benowitz NL, Gourlay SG. Cardiovascular toxicity of nicotine: implications for nicotine replacement therapy. Journal of the American College of Cardiology. 1997;29(7):1422–31. [PubMed: 9180099]
- Benowitz NL, Hall SM, Herning RI, Jacob P III, Jones RT, Osman AL. Smokers of low-yield cigarettes do not consume less nicotine. New England Journal of Medicine. 1983;309(3):139–42. [PubMed: 6866013]
- Benowitz NL, Hansson A, Jacob P III. Cardiovascular effects of nasal and transdermal nicotine and cigarette smoking. Hypertension. 2002;39(6):1107–12. [PubMed: 12052850]
- Benowitz NL, Jacob P III. Daily intake of nicotine during cigarette smoking. Clinical Pharmacology and Therapeutics. 1984;35(4):499–504. [PubMed: 6705448]
- Benowitz NL, Jacob P III, Jones RT, Rosenberg J. Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. Journal of Pharmacology and Experimental Therapeutics. 1982a;221(2):368–72. [PubMed: 7077531]
- Benowitz NL, Jacob P III, Yu L. Daily use of smokeless tobacco: systemic effects. Annals of Internal Medicine. 1989;111(2):112–6. [PubMed: 2742246]
- Benowitz NL, Kuyt F, Jacob P III. Circadian blood nicotine concentrations during cigarette smoking. Clinical Pharmacology and Therapeutics. 1982b;32(6):758–64. [PubMed: 7140139]
- Benowitz NL, Kuyt F, Jacob P III. Influence of nicotine on cardiovascular and hormonal effects of cigarette smoking. Clinical Pharmacology and Therapeutics. 1984;36(1):74–81. [PubMed: 6734053]
- Benowitz NL, Porchet H, Sheiner L, Jacob P III. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clinical Pharmacology and Therapeutics. 1988;44(1):23–8. [PubMed: 3391001]
- Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends in Endocrinology and Metabolism. 2000;11(9):351–6. [PubMed: 11042464]
- Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. American Journal of Cardiology. 2002;89(9):1117–9. [PubMed: 11988205]
- Bernhard D, Csordas A, Henderson B, Rossmann A, Kind M, Wick G. Cigarette smoke metal-catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. FASEB Journal. 2005;19(9):1096–107. [PubMed: 15985533]
- Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circulation Research. 2006;99(7):692–705. [PubMed: 17008598]
- Biesenbach G, Grafinger P, Janko O, Zazgornik J. Influence of cigarette-smoking on the progression of clinical diabetic nephropathy in type 2 diabetic patients. Clinical Nephrology. 1997;48(3):146–50. [PubMed: 9342485]
- Billimoria JD, Pozner H, Metselaar B, Best FW, James DC. Effect of cigarette smoking on lipids, lipoproteins, blood coagulation, fibrinolysis and cellular components of human blood. Atherosclerosis. 1975;21(1):61–76. [PubMed: 1131301]
- Bizzi A, Tacconi MT, Medea A, Garattini S. Some aspects of the effect of nicotine on plasma FFA and tissue triglycerides. Pharmacology. 1972;7(4):216–24. [PubMed: 5077308]
- Bjartveit K, Tverdal A. Health consequences of smoking 1–4 cigarettes per day. Tobacco Control. 2005;14(5):315–20. [PMC free article: PMC1748107] [PubMed: 16183982]
- Blanchard JF, Armenian HK, Friesen PP. Risk factors for abdominal aortic aneurysm: results of a case-control study. American Journal of Epidemiology. 2000;151(6):575–83. [PubMed: 10733039]
- Blanco-Cedres L, Daviglus ML, Garside DB, Liu K, Pirzada A, Stamler J, Greenland P. Relation of cigarette smoking to 25-year mortality in middle-aged men with low baseline serum cholesterol: the Chicago Heart Association Detection Project in Industry. American Journal of Epidemiology. 2002;155(4):354–60. [PubMed: 11836200]
- Blankenberg S, Rupprecht HJ, Bickel C, Peetz D, Hafner G, Tiret L, Meyer J. Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation. 2001;104(12):1336–42. [PubMed: 11560847]
- Blann AD, Amiral J, McCollum CN, Lip GYH. Differences in free and total tissue factor pathway inhibitor, and tissue factor in peripheral artery disease compared to healthy controls. Atherosclerosis. 2000;152(1):29–34. [PubMed: 10996336]
- Blann AD, McCollum CN. Adverse influence of cigarette smoking on the endothelium. Thrombosis and Haemostasis. 1993;70(4):707–11. [PubMed: 8116001]
- Blann AD, Seigneur M, Steiner M, Miller JP, McCollum CN. Circulating ICAM-1 and VCAM-1 in peripheral artery disease and hypercholesterolaemia: relationship to the location of atherosclerotic disease, smoking, and in the prediction of adverse events. Thrombosis and Haemostasis. 1998;79(6):1080–5. [PubMed: 9657427]
- Blann AD, Steele C, McCollum CN. The influence of smoking on soluble adhesion molecules and endothelial cell markers. Thrombosis Research. 1997;85(5):433–8. [PubMed: 9076900]
- Boffetta P, Straif K. Use of smokeless tobacco and risk of myocardial infarction and stroke: systematic review with meta-analysis. BMJ (British Medical Journal). 2009;339:b3060. [PMC free article: PMC2728803] [PubMed: 19690343] [CrossRef]
- Bolman C, de Vries H, van Breukelen G. A minimal-contact intervention for cardiac inpatients: long-term effects on smoking cessation. Preventive Medicine. 2002;35(2):181–92. [PubMed: 12200104]
- Bott U, Jorgens V, Grusser M, Bender R, Muhlhauser I, Berger M. Predictors of glycaemic control in type 1 diabetic patients after participation in an intensified treatment and teaching programme. Diabetic Medicine. 1994;11(4):362–71. [PubMed: 8088108]
- Bowers TR, Terrien EF, O’Neill WW, Sachs D, Grines CL. Effect of reperfusion modality on outcome in non-smokers and smokers with acute myocardial infarction (a Primary Angioplasty in Myocardial Infarction [PAMI] substudy). PAMI Investigators. American Journal of Cardiology. 1996;78(5):511–5. [PubMed: 8806333]
- Brady AR, Thompson SG, Fowkes FGR, Greenhalgh RM, Powell JT. UK Small Aneurysm Trial Participants. Abdominal aortic aneurysm expansion: risk factors and time intervals for surveillance. Circulation. 2004;110(1):16–21. [PubMed: 15210603]
- Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiological Reviews. 2004;84(3):903–34. [PubMed: 15269340]
- Bridges RB, Fu MC, Rehm SR. Increased neutrophil myeloperoxidase activity associated with cigarette smoking. European Journal of Respiratory Disease. 1985;67(2):84–93. [PubMed: 2996923]
- Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC Jr, et al. Air pollution and cardiovascular disease: a statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 2004;109(21):2655–71. [PubMed: 15173049]
- Brousseau ME, Schaefer EJ, Wolfe ML, Bloedon LT, Digenio AG, Clark RW, Mancuso JP, Rader DJ. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. New England Journal of Medicine. 2004;350(15):1505–15. [PubMed: 15071125]
- Brownlee M, Vlassara H, Cerami A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Annals of Internal Medicine. 1984;101(4):527–37. [PubMed: 6383165]
- Burghuber OC, Punzengruber C, Sinzinger H, Haber P, Silberbauer K. Platelet sensitivity to prostacyclin in smokers and non-smokers. Chest. 1986;90(1):34–8. [PubMed: 3522121]
- Burke A, FitzGerald GA. Oxidative stress and smoking-induced vascular tissue injury. Progress in Cardiovascular Diseases. 2003;46(1):79–90. [PubMed: 12920701]
- Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. New England Journal of Medicine. 1997;336(18):1276–82. [PubMed: 9113930]
- Burns DM. Epidemiology of smoking-induced cardiovascular disease. Progress in Cardiovascular Diseases. 2003;46(1):11–29. [PubMed: 12920698]
- Burns DM, Shanks T, Choi W, Thun M, Heath C, Garfinkel L, The American, Cancer Society, Cancer Prevention, Study I. 12-year followup of 1 million men and women, Changes in Cigarette-Related Disease Risks and Their Implication for Prevention and Control. Smoking and Tobacco Control Monograph No 8. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1997. pp. 113–304. NIH Publication No. 97-4213.
- Busse R, Fleming I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends in Pharmacological Sciences. 2003;24(1):24–9. [PubMed: 12498727]
- Cai J, Boulton M. The pathogenesis of diabetic retinopathy: old concepts and new questions. Eye. 2002;16(3):242–60. [PubMed: 12032713]
- Calverley PM, Leggett RJ, Flenley DC. Carbon monoxide and exercise tolerance in chronic bronchitis and emphysema. BMJ (British Medical Journal). 1981;283(6296):878–80. [PMC free article: PMC1507111] [PubMed: 6793157]
- Campbell IA, Prescott RJ, Tjeder-Burton SM. Transdermal nicotine plus support in patients attending hospital with smoking-related diseases: a placebo-controlled study. Respiratory Medicine. 1996;90(1):47–51. [PubMed: 8857326]
- Campisi R, Czernin J, Schöder H, Sayre JW, Marengo FD, Phelps ME, Schelbert HR. Effects of long-term smoking on myocardial blood flow, coronary vasomotion, and vasodilator capacity. Circulation. 1998;98(2):119–25. [PubMed: 9679717]
- Campisi R, Czernin J, Schöder H, Sayre JW, Schelbert HR. L-arginine normalizes coronary vasomotion in long-term smokers. Circulation. 1999;99(4):491–7. [PubMed: 9927394]
- Candipan RC, Wang B-Y, Buitrago R, Tsao PS, Cooke JP. Regression or progression: dependency on vascular nitric oxide. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(1):44–50. [PubMed: 8548425]
- Caraballo RS, Giovino GA, Pechacek TF, Mowery PD, Richter PA, Strauss WJ, Sharp DJ, Eriksen MP, Pirkle JL, Maurer KR. Racial and ethnic differences in serum cotinine levels of cigarette smokers: Third National Health and Nutrition Examination Survey, 1988–1991. JAMA: the Journal of the American Medical Association. 1998;280(2):135–9. [PubMed: 9669785]
- Caralis DG, Deligonul U, Kern MJ, Cohen JD. Smoking is a risk factor for coronary spasm in young women. Circulation. 1992;85(3):905–9. [PubMed: 1537126]
- Carty CS, Soloway PD, Kayastha S, Bauer J, Marsan B, Ricotta JJ, Dryjski M. Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cells. Journal of Vascular Surgery. 1996;24(6):927–34. [PubMed: 8976346]
- Celermajer DS, Adams MR, Clarkson P, Robinson J, McCredie R, Donald A, Deanfield JE. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. New England Journal of Medicine. 1996;334(3):150–4. [PubMed: 8531969]
- Celermajer DS, Sorensen KE, Georgakopoulos D, Bull C, Thomas O, Robinson J, Deanfield JE. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88(5 Pt 1):2149–55. [PubMed: 8222109]
- Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–5. [PubMed: 1359209]
- Centers for Disease Control and Prevention. Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000–2004. Morbidity and Mortality Weekly Report. 2008;57(45):1226–8. [PubMed: 19008791]
- Chalon S, Moreno H Jr, Benowitz NL, Hoffman BB, Blaschke TF. Nicotine impairs endothelium-dependent dilatation in human veins in vivo. Clinical Pharmacology and Therapeutics. 2000;67(4):391–7. [PubMed: 10801248]
- Chalon S, Moreno H Jr, Hoffman BB, Blaschke TF. Angiotensin-converting enzyme inhibition improves venous endothelial dysfunction in chronic smokers. Clinical Pharmacology and Therapeutics. 1999;65(3):295–303. [PubMed: 10096262]
- Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(24):11190–4. [PMC free article: PMC40597] [PubMed: 7479963]
- Chase HP, Garg SK, Marshall G, Berg CL, Harris S, Jackson WE, Hamman RE. Cigarette smoking increases the risk of albuminuria among subjects with type I diabetes. JAMA: the Journal of the American Medical Association. 1991;265(5):614–7. [PubMed: 1987411]
- Chaturvedi N, Stevens L, Fuller JH. Which features of smoking determine mortality risk in former cigarette smokers with diabetes: the World Health Organization Multinational Study Group. Diabetes Care. 1997;20(8):1266–72. [PubMed: 9250452]
- Chehne F, Oguogho A, Lupattelli G, Budinsky AC, Palumbo B, Sinzinger H. Increase of isoprostane 8-epi-PGF2α after restarting smoking. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2001;64(6):307–10. [PubMed: 11427039]
- Chen HH, Burnett JC Jr. C-type natriuretic peptide: the endothelial component of the natriuretic peptide system. Journal of Cardiovascular Pharmacology. 1998;32(Suppl 3):S22–S28. [PubMed: 9883743]
- Chen JL, Peacock E, Samady W, Turner SM, Neese RA, Hellerstein MK, Murphy EJ. Physiologic and pharmacologic factors influencing glyceroneogenic contribution to triacylglyceride glycerol measured by mass isotopomer distribution analysis. Journal of Biological Chemistry. 2005;280(27):25396–402. [PubMed: 15888453]
- Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radical Biology & Medicine. 2000;28(12):1815–26. [PubMed: 10946223]
- Christen WG, Glynn RJ, Manson JE, Ajani UA, Buring JE. A prospective study of cigarette smoking and risk of age-related macular degeneration in men. JAMA: the Journal of the American Medical Association. 1996;276(14):1147–51. [PubMed: 8827967]
- Christen WG, Manson JE, Bubes V, Glynn RJ. Risk factors for progression of distal symmetric polyneuropathy in type 1 diabetes mellitus: Sorbinil Retinopathy Trial Research Group. American Journal of Epidemiology. 1999;150(11):1142–51. [PubMed: 10588075]
- Chuahirun T, Khanna A, Kimball K, Wesson DE. Cigarette smoking and increased urine albumin excretion are interrelated predictors of nephropathy progression in type 2 diabetes. American Journal of Kidney Diseases. 2003;41(1):13–21. [PubMed: 12500217]
- Chuahirun T, Wesson DE. Cigarette smoking predicts faster progression of type 2 established diabetic nephropathy despite ACE inhibition. American Journal of Kidney Diseases. 2002;39(2):376–82. [PubMed: 11840380]
- Chung TW, Tyan YC, Hsieh JH, Wang SS, Chu SH. Shear stress-induced aggregation of oxidized platelets. Thrombosis Research. 2002;105(4):325–9. [PubMed: 12031827]
- Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environmental Health Perspectives. 1985;64:111–26. [PMC free article: PMC1568603] [PubMed: 3007083]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, et al. Varenicline: an α4β2 nicotinic receptor partial agonist for smoking cessation. Journal of Medicinal Chemistry. 2005;48(10):3474–7. [PubMed: 15887955]
- Colditz GA, Bonita R, Stampfer MJ, Willett WC, Rosner B, Speizer FE, Hennekens CH. Cigarette smoking and risk of stroke in middle-aged women. New England Journal of Medicine. 1988;318(15):937–41. [PubMed: 3352685]
- Conklin DJ, Bhatnagar A, Cowley HR, Johnson GH, Wiechmann RJ, Sayre LM, Trent MB, Boor PJ. Acrolein generation stimulates hypercontraction in isolated human blood vessels. Toxicology and Applied Pharmacology. 2006;217(3):277–88. [PMC free article: PMC3487162] [PubMed: 17095030]
- Cooke JP. Does ADMA cause endothelial dysfunction? Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(9):2032–7. [PubMed: 10978245]
- Cooke JP. Asymmetrical dimethylarginine: the Uber marker? Circulation. 2004;109(15):1813–8. [PubMed: 15096461]
- Cooke JP, Dzau VJ. Derangements of the nitric oxide synthase pathway, l-arginine, and cardiovascular diseases. Circulation. 1997a;96(2):379–82. [PubMed: 9244195]
- Cooke JP, Dzau VJ. Nitric oxide synthase: role in the genesis of vascular disease. Annual Review of Medicine. 1997b;48:489–509. [PubMed: 9046979]
- Cooke JP, Rossitch E Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. Journal of Clinical Investigation. 1991;88(5):1663–71. [PMC free article: PMC295698] [PubMed: 1719029]
- Cooke JP, Singer AH, Tsao P, Zera P, Rowan RA, Billingham ME. Antiatherogenic effects of l-arginine in the hyper-cholesterolemic rabbit. Journal of Clinical Investigation. 1992;90(3):1168–72. [PMC free article: PMC329981] [PubMed: 1522225]
- Craig WY, Palomaki GE, Haddow JE. Cigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published data. BMJ (British Medical Journal). 1989;298(6676):784–8. [PMC free article: PMC1836079] [PubMed: 2496857]
- Cranshaw JH, Evans TW, Mitchell JA. Characterization of the effects of isoprostanes on platelet aggregation in human whole blood. British Journal of Pharmacology. 2001;132(8):1699–706. [PMC free article: PMC1572736] [PubMed: 11309241]
- Cremer P, Nagel D, Mann H, Labrot B, Müller-Berninger R, Elster H, Seidel D. Ten-year follow-up results from the Goettingen Risk, Incidence and Prevalence Study (GRIPS). I: risk factors for myocardial infarction in a cohort of 5790 men. Atherosclerosis. 1997;129(2):221–30. [PubMed: 9105565]
- Criqui MH, Cowan LD, Tyroler HA, Bangdiwala S, Heiss G, Wallace RB, Cohn R. Lipoproteins as mediators for the effects of alcohol consumption and cigarette smoking on cardiovascular mortality: results from the Lipid Research Clinics Follow-up Study. American Journal of Epidemiology. 1987;126(4):629–37. [PubMed: 3631053]
- Criqui MH, Wallace RB, Heiss G, Mishkel M, Schonfeld G, Jones GT. Cigarette smoking and plasma high-density lipoprotein cholesterol: the Lipid Research Clinics Program Prevalence Study. Circulation. 1980;62(4 Pt 2):IV70–76. [PubMed: 7418146]
- Critchley J, Capewell S. Smoking cessation for the secondary prevention of coronary heart disease. Cochrane Database of Systematic Reviews. 2004;4 Art. No.: CD003041. [PubMed: 14583958] [CrossRef]
- Critchley JA, Capewell S. Mortality risk reduction associated with smoking cessation in patients with coronary heart disease: a systematic review. JAMA: the Journal of the American Medical Association. 2003;290(1):86–97. [PubMed: 12837716]
- Cryer PE, Haymond MW, Santiago JV, Shah SD. Nor-epinephrine and epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. New England Journal of Medicine. 1976;295(11):573–7. [PubMed: 950972]
- Csiszar A, Stef G, Pacher P, Ungvari Z. Oxidative stress-induced isoprostane formation may contribute to aspirin resistance in platelets. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2002;66(5–6):557–8. [PubMed: 12144879]
- Csonka E, Somogyi A, Augustin J, Haberbosch W, Schettler G, Jellinek H. The effect of nicotine on cultured cells of vascular origin. Virchows Archiv A, Pathological Anatomy and Histopathology. 1985;407(4):441–7. [PubMed: 3931344]
- Cucina A, Corvino V, Sapienza P, Borrelli V, Lucarelli M, Scarpa S, Strom R, Santoro-D’Angelo L, Cavallaro A. Nicotine regulates basic fibroblastic growth factor and transforming growth factor β1 production in endothelial cells. Biochemical and Biophysical Research Communications. 1999;257(2):306–12. [PubMed: 10198208]
- Cullen P, Schulte H, Assmann G. Smoking, lipoproteins and coronary heart disease risk: data from the Munster Heart Study (PROCAM). European Heart Journal. 1998;19(11):1632–41. [PubMed: 9857915]
- Cupples LA, Gagnon DR, Kannel WB. Long- and short-term risk of sudden coronary death. Circulation. 1992;85(1 Suppl):I11–I18. [PubMed: 1370216]
- Czernin J, Sun K, Brunken R, Böttcher M, Phelps M, Schelbert H. Effect of acute and long-term smoking on myocardial blood flow and flow reserve. Circulation. 1995;91(12):2891–7. [PubMed: 7796497]
- Czernin J, Waldherr C. Cigarette smoking and coronary blood flow. Progress in Cardiovascular Diseases. 2003;45(5):395–404. [PubMed: 12704596]
- Dagenais GR, Robitaille NM, Lupien PJ, Christen A, Gingras S, Moorjani S, Meyer F, Rochon J. First coronary heart disease event rates in relation to major risk factors: Quebec Cardiovascular Study. Canadian Journal of Cardiology. 1990;6(7):274–80. [PubMed: 2224616]
- Daly LE, Mulcahy R, Graham IM, Hickey N. Long term effect on mortality of stopping smoking after unstable angina and myocardial infarction. BMJ (British Medical Journal). 1983;287(6388):324–6. [PMC free article: PMC1548591] [PubMed: 6409291]
- Davì G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonni E, Bucciarelli T, Constantini F, et al. In vivo formation of 8-isoprostaglandin F2α and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99(2):224–9. [PubMed: 9892587]
- Davis JW, Shelton L, Eigenberg DA, Hignite CE, Watanabe IS. Effects of tobacco and non-tobacco cigarette smoking on endothelium and platelets. Clinical Pharmacology and Therapeutics. 1985;37(5):529–33. [PubMed: 3987176]
- Dawber TR. The Framingham Study: The Epidemiology of Atherosclerotic Disease. Cambridge (MA): Harvard University Press; 1980.
- de Groot E, Hovingh GK, Wiegman A, Duriez P, Smit AJ, Fruchart JC, Kastelein JJ. Measurement of arterial wall thickness as a surrogate marker for atherosclerosis. Circulation. 2004;109(23 Suppl 1):III33–38. [PubMed: 15198964]
- Deanfield JE, Shea MJ, Wilson RA, Horlock P, de Landsheere CM, Selwyn AP. Direct effects of smoking on the heart: silent ischemic disturbances of coronary flow. American Journal of Cardiology. 1986;57(13):1005–9. [PubMed: 3486583]
- DeBias DA, Banerjee CM, Birkhead NC, Greene CH, Scott SD, Harrer WV. Effects of carbon monoxide inhalation on ventricular fibrillation. Archives of Environmental Health. 1976;31(1):42–6. [PubMed: 812430]
- DeBusk RF, Miller NH, Superko R, Dennis CA, Thomas RJ, Lew HT, Berger WE III, Heller RS, Rompf J, Gee D, et al. A case-management system for coronary risk factor modification after acute myocardial infarction. Annals of Internal Medicine. 1994;120(9):721–9. [PubMed: 8147544]
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. American Journal of Physiology – Endocrinology and Metabolism. 1979;237(3):E214–E223. [PubMed: 382871]
- Deliconstantinos G, Villiotou V, Stavrides JC. Scavenging effects of hemoglobin and related heme containing compounds on nitric oxide, reactive oxidants and carcinogenic volatile nitrosocompounds of cigarette smoke: a new method for protection against the dangerous cigarette constituents. Anticancer Research. 1994;14(6B):2717–26. [PubMed: 7872707]
- Diana JN, Qian SF, Heesch CM, Barron KW, Chien CY. Nicotine-induced skeletal muscle vasodilation is mediated by release of epinephrine from nerve terminals. American Journal of Physiology. 1990;259( 6 Pt 2):H1718–H1729. [PubMed: 1979721]
- Dietrich M, Block G, Hudes M, Morrow JD, Norkus EP, Traber MG, Cross CE, Packer L. Antioxidant supplementation decreases lipid peroxidation biomarker F2-isoprostanes in plasma of smokers. Cancer Epidemiology, Biomarkers & Prevention. 2002;11(1):7–13. [PubMed: 11815395]
- Dinno A, Glantz S. Clean indoor air laws immediately reduce heart attacks. Preventive Medicine. 2007;45(1):9–11. [PMC free article: PMC2693058] [PubMed: 17499350]
- Djordjevic MV, Doran KA. Nicotine content and delivery across tobacco products. Nicotine Psychopharmacology Handbook of Experimental Pharmacology. Henningfield JE, London ED, Pogun S, editors. Vol. 192. Berlin: Springer; 2009. pp. 61–92. [PubMed: 19184646]
- Djoussé L, Myers RH, Province MA, Hunt SC, Eckfeldt JH, Evans G, Peacock JM, Ellison RC. Influence of apolipoprotein E, smoking, and alcohol intake on carotid atherosclerosis: National Heart, Lung, and Blood Institute Family Heart Study. Stroke. 2002;33(5):1357–61. [PubMed: 11988615]
- Dobson AJ, Alexander HM, Heller RF, Lloyd DM. How soon after quitting smoking does risk of heart attack decline? Journal of Clinical Epidemiology. 1991;44(11):1247–53. [PubMed: 1941018]
- Dogra G, Rich L, Stanton K, Watts GF. Endothelium-dependent and independent vasodilation studies at normoglycaemia in type I diabetes mellitus with and without microalbuminuria. Diabetologia. 2001;44(5):593–601. [PubMed: 11380077]
- Doll R, Peto R. Mortality in relation to smoking: 20 years’ observations on male British doctors. BMJ (British Medical Journal). 1976;2(6051):1525–36. [PMC free article: PMC1690096] [PubMed: 1009386]
- Dornelas EA, Sampson RA, Gray JF, Waters D, Thompson PD. A randomized controlled trial of smoking cessation counseling after myocardial infarction. Preventive Medicine. 2000;30(4):261–8. [PubMed: 10731452]
- Dullaart RP, Hoogenberg K, Dikkeschei BD, van Tol A. Higher plasma lipid transfer protein activities and unfavorable lipoprotein changes in cigarette-smoking men. Arteriosclerosis and Thrombosis. 1994;14(10):1581–5. [PubMed: 7918308]
- Eliasson B. Cigarette smoking and diabetes. Progress in Cardiovascular Diseases. 2003;45(5):405–13. [PubMed: 12704597]
- Eliasson B, Attvall S, Taskinen MR, Smith U. The insulin resistance syndrome in smokers is related to smoking habits. Arteriosclerosis and Thrombosis. 1994;14(12):1946–50. [PubMed: 7981184]
- Eliasson B, Attvall S, Taskinen MR, Smith U. Smoking cessation improves insulin sensitivity in healthy middle-aged men. European Journal of Clinical Investigation. 1997a;27(5):450–6. [PubMed: 9179554]
- Eliasson B, Hjalmarson A, Kruse E, Landfeldt B, Westin Å. Effect of smoking reduction and cessation on cardiovascular risk factors. Nicotine & Tobacco Research. 2001;3(3):249–55. [PubMed: 11506768]
- Eliasson B, Mero N, Taskinen MR, Smith U. The insulin resistance syndrome and postprandial lipid intolerance in smokers. Atherosclerosis. 1997b;129(1):79–88. [PubMed: 9069521]
- Eliasson B, Taskinen MR, Smith U. Long-term use of nicotine gum is associated with hyperinsulinemia and insulin resistance. Circulation. 1996;94(5):878–81. [PubMed: 8790020]
- Eliasson M, Asplund K, Evrin PE, Lundblad D. Relationship of cigarette smoking and snuff dipping to plasma fibrinogen, fibrinolytic variables and serum insulin: the Northern Sweden MONICA Study. Atherosclerosis. 1995;113(1):41–53. [PubMed: 7755654]
- Eliasson M, Lundblad D, Hagg E. Cardiovascular risk factors in young snuff-users and cigarette smokers. Journal of Internal Medicine. 1991;230(1):17–22. [PubMed: 2066707]
- Enderle MD, Pfohl M, Kellermann N, Haering HU, Hoffmeister HM. Endothelial function, variables of fibrinolysis and coagulation in smokers and healthy controls. Haemostasis. 2000;30(3):149–58. [PubMed: 11014965]
- Epifano L, Di Vincenzo A, Fanelli C, Porcellati F, Perriello G, De Feo P, Motolese M, Brunetti P, Bolli GB. Effect of cigarette smoking and of a transdermal nicotine delivery system on glucoregulation in type 2 diabetes mellitus. European Journal of Clinical Pharmacology. 1992;43(3):257–63. [PubMed: 1425888]
- Escobedo LG, Caspersen CJ. Risk factors for sudden coronary death in the United States. Epidemiology. 1997;8(2):175–80. [PubMed: 9229210]
- Escobedo LG, Zack MM. Comparison of sudden and non-sudden coronary deaths in the United States. Circulation. 1996;93(11):2033–6. [PubMed: 8640979]
- Escolar G, White JG. Changes in glycoprotein expression after platelet activation: differences between in vitro and in vivo studies. Thrombosis and Haemostasis. 2000;83(3):371–86. [PubMed: 10744140]
- Ezzati M, Henley SJ, Thun MJ, Lopez AD. Role of smoking in global and regional cardiovascular mortality. Circulation. 2005;112(4):489–97. [PubMed: 16027251]
- Facchini FS, Hollenbeck CB, Jeppesen J, Chen YD, Reaven GM. Insulin resistance and cigarette smoking. Lancet. 1992;339(8802):1128–30. [PubMed: 1349365]
- Feskens EJ, Kromhout D. Cardiovascular risk factors and the 25-year incidence of diabetes mellitus in middle-aged men: the Zutphen Study. American Journal of Epidemiology. 1989;130(6):1101–8. [PubMed: 2589303]
- Fichtenberg CM, Glantz SA. Association of the California Tobacco Control Program with declines in cigarette consumption and mortality from heart disease. New England Journal of Medicine. 2000;343(24):1772–7. [PubMed: 11114317]
- Fiore MC, Bailey WC, Cohen SJ, Dorfman SF, Goldstein MG, Gritz ER, Heyman RB, Jaén CR, Kottke TE, Lando HA, et al. Treating Tobacco Use and Dependence, Clinical Practice Guideline. Rockville (MD): U.S. Department of Health and Human Services, Public Health Service; 2000.
- Fiore MC, Jaén CR, Baker TB, Bailey WC, Benowitz NL, Curry SJ, Dorfma SF, Froelicher ES, Goldstein MG, Heaton CG, et al. Treating Tobacco Use and Dependence: 2008 Update, Clinical Practice Guideline. Rockville (MD): U.S. Department of Health and Human Services, Public Health Service; 2008.
- Folkman J. Fundamental concepts of the angiogenic process. Current Molecular Medicine. 2003;3(7):643–51. [PubMed: 14601638]
- Ford ES, Malarcher AM, Herman WH, Aubert RE. Diabetes mellitus and cigarette smoking: findings from the 1989 National Health Interview Survey. Diabetes Care. 1994;17(7):688–92. [PubMed: 7924778]
- Foy GC, Bell RA, Farmer DF, Goff DC Jr, Wagenknecht LE. Smoking and incidence of diabetes among U.S. adults: findings from the Insulin Resistance Atherosclerosis Study. Diabetes Care. 2005;28(10):2501–7. [PubMed: 16186287]
- Franzoni F, Quiñones-Galvan A, Regoli F, Ferrannini E, Galetta F. A comparative study of the in vitro antioxidant activity of statins. International Journal of Cardiology. 2003;90(2–3):317–21. [PubMed: 12957768]
- Frati AC, Iniestra F, Ariza CR. Acute effect of cigarette smoking on glucose tolerance and other cardiovascular risk factors. Diabetes Care. 1996;19(2):112–8. [PubMed: 8718429]
- Freedman JE, Loscalzo J, Barnard MR, Alpert C, Keaney JF Jr, Michelson AD. Nitric oxide released from activated platelets inhibits platelet recruitment. Journal of Clinical Investigation. 1997;100(2):350–6. [PMC free article: PMC508197] [PubMed: 9218511]
- Freedman JE, Ting B, Hankin B, Loscalzo J, Keaney JF Jr, Vita JA. Impaired platelet production of nitric oxide predicts presence of acute coronary syndromes. Circulation. 1998;98(15):1481–6. [PubMed: 9769300]
- Freeman DJ, Caslake MJ, Griffin BA, Hinnie J, Tan CE, Watson TD, Packard CJ, Shepherd J. The effect of smoking on post-heparin lipoprotein and hepatic lipase, cholesteryl ester transfer protein and lecithin: cholesterol acyl transferase activities in human plasma. European Journal of Clinical Investigation. 1998;28(7):584–91. [PubMed: 9726040]
- Freeman DJ, Packard CJ. Smoking and plasma lipoprotein metabolism. Clinical Science (London). 1995;89(4):333–42. [PubMed: 7493432]
- Freund KM, Belanger AJ, D’Agostino RB, Kannel WB. The health risks of smoking. The Framingham Study: 34 years of follow-up. Annals of Epidemiology. 1993;3(4):417–24. [PubMed: 8275219]
- Friedman GD, Dales LG, Ury HK. Mortality in middle-aged smokers and nonsmokers. New England Journal of Medicine. 1979;300(5):213–7. [PubMed: 759867]
- Frost PH, Davis BR, Burlando AJ, Curb JD, Guthrie GP Jr, Isaacsohn JL, Wassertheil-Smoller S, Wilson AC, Stamler J. Coronary heart disease risk factors in men and women aged 60 years and older: findings from the Systolic Hypertension in the Elderly Program. Circulation. 1996;94(1):26–34. [PubMed: 8964114]
- Fuster V, Chesebro JH, Frye RL, Elveback LR. Platelet survival and the development of coronary artery disease in the young adult: effects of cigarette smoking, strong family history and medical therapy. Circulation. 1981;63(3):546–51. [PubMed: 7460239]
- Gartside PS, Wang P, Glueck CJ. Prospective assessment of coronary heart disease risk factors: the NHANES I Epidemiologic Follow-up Study (NHEFS) 16-year follow-up. Journal of the American College of Nutrition. 1998;17(3):263–9. [PubMed: 9627913]
- Gill GV, Rolfe M, MacFarlane IA, Huddle KR. Smoking habits of black South African patients with diabetes mellitus. Diabetic Medicine. 1996;13(11):996–9. [PubMed: 8946160]
- Gimbrone MA Jr. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. American Journal of Cardiology. 1995;75(6):67B–70B. [PubMed: 7532351]
- Godsland IF, Walton C. Insulin resistance and cigarette smoking [letter] Lancet. 1992;340(8819):607. [PubMed: 1355174]
- Godtfredsen NS, Osler M, Vestbo J, Andersen I, Prescott E. Smoking reduction, smoking cessation, and incidence of fatal and non-fatal myocardial infarction in Denmark 1976–1998: a pooled cohort study. Journal of Epidemiology and Community Health. 2003;57(6):412–6. [PMC free article: PMC1732492] [PubMed: 12775785]
- Gokce N, Keaney JF Jr, Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, Vita JA. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. Journal of the American College of Cardiology. 2003;41(10):1769–75. [PubMed: 12767663]
- Goldenberg I, Jonas M, Tenenbaum A, Boyko V, Matetzky S, Shotan A, Behar S, Reicher-Reiss H. Current smoking, smoking cessation, and the risk of sudden cardiac death in patients with coronary artery disease. Archives of Internal Medicine. 2003;163(19):2301–5. [PubMed: 14581249]
- Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust RF III, Finkel B, Lanken PN, Ischiropoulos H. Plasma proteins modified by tyrosine nitration in acute respiratory distress syndrome. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2000;278(5):L961–L967. [PubMed: 10781426]
- Gonzales D, Rennard SI, Nides M, Oncken C, Azoulay S, Billing CB, Watsky EJ, Gong J, Williams KE, Reeves KR, et al. Varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA: the Journal of the American Medical Association. 2006;296(1):47–55. [PubMed: 16820546]
- Gordon T, Kannel WB, Dawber TR, McGee D. Changes associated with quitting cigarette smoking: the Framingham Study. American Heart Journal. 1975;90(3):322–8. [PubMed: 1163424]
- Gordon T, Kannel WB, McGee D, Dawer TR. Death and coronary attacks in men after giving up cigarette smoking: a report from the Framingham Study. Lancet. 1974;2(7893):1345–8. [PubMed: 4143310]
- Görlach A, Brandes RP, Bassus S, Kronemann N, Kirchmaier CM, Busse R, Schini-Kerth VB. Oxidative stress and expression of p22phox are involved in the up-regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB Journal. 2000;14(11):1518–28. [PubMed: 10928986]
- GouaŸe V, Dousset N, Dousset J-C, Valdiguié P. Effect of nicotine and cotinine on the susceptibility to in vitro oxidation of LDL in healthy non smokers and smokers. Clinica Chimica Acta. 1998;277(1):25–37. [PubMed: 9776043]
- Gourlay SG, Benowitz NL. Arteriovenous differences in plasma concentration of nicotine and catecholamines and related cardiovascular effects after smoking, nicotine nasal spray, and intravenous nicotine. Clinical Pharmacology and Therapeutics. 1997;62(4):453–63. [PubMed: 9357397]
- Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database of Systematic Reviews. 2004;3 Art. No.: CD000058. [PMC free article: PMC7038651] [PubMed: 15266422] [CrossRef]
- Green MS, Jucha E, Luz Y. Blood pressure in smokers and nonsmokers: epidemiologic findings. American Heart Journal. 1986;111(5):932–40. [PubMed: 3706114]
- Gugliucci A. Antithrombin activity is inhibited by acrolein and homocysteine thiolactone: protection by cysteine. Life Sciences. 2007;82(7–8):413–8. [PubMed: 18206177]
- Gustafsson A, Åsman B, Bergström K. Cigarette smoking as an aggravating factor in inflammatory tissue-destructive diseases: increase in tumor necrosis factor-alpha priming of peripheral neutrophils measured as generation of oxygen radicals. International Journal of Clinical & Laboratory Research. 2000;30(4):187–90. [PubMed: 11289709]
- Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107(3):416–21. [PubMed: 12551865]
- Habek D, Habek JC, Ivanišević M, Djelmiš J. Fetal tobacco syndrome and perinatal outcome. Fetal Diagnosis and Therapy. 2002;17(6):367–71. [PubMed: 12393968]
- Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New England Journal of Medicine. 1998;339(4):229–34. [PubMed: 9673301]
- Håheim LL, Holme I, Hjermann I, Leren P. Smoking habits and risk of fatal stroke: 18 years follow up of the Oslo Study. Journal of Epidemiology and Community Health. 1996;50(6):621–4. [PMC free article: PMC1060377] [PubMed: 9039379]
- Hajek P, Taylor TZ, Mills P. Brief intervention during hospital admission to help patients to give up smoking after myocardial infarction and bypass surgery: randomized controlled trial. BMJ (British Medical Journal). 2002;324(7329):87–9. [PMC free article: PMC64504] [PubMed: 11786452]
- Hajek P, West R, Foulds J, Nilsson F, Burrows S, Meadow A. Randomized comparative trial of nicotine polacrilex, a transdermal patch, nasal spray, and an inhaler. Archives of Internal Medicine. 1999;159(17):2033–8. [PubMed: 10510989]
- Hallstrom AP, Cobb LA, Ray R. Smoking as a risk factor for recurrence of sudden cardiac arrest. New England Journal of Medicine. 1986;314(5):271–5. [PubMed: 3941718]
- Hames CG, Rose K, Knowles M, Davis CE, Tyroler HA. Black-white comparisons of 20-year coronary heart disease mortality in the Evans County Heart Study. Cardiology. 1993;82(2–3):122–36. [PubMed: 8324775]
- Hammond EC, Garfinkel L. Coronary heart disease, stroke, and aortic aneurysm: factors in the etiology. Archives of Environmental Health. 1969;19(2):167–82. [PubMed: 5381329]
- Hammond EC, Horn D. Smoking and death rates: report on forty-four months of follow-up of 187,783 men. II: death rates by cause. JAMA: the Journal of the American Medical Association. 1958;166(11):1294–308. [PubMed: 12308037]
- Haramaki N, Ikeda H, Takajo Y, Katoh A, Kanaya S, Shintani S, Haramaki R, Murohara T, Imaizumi T. Long-term smoking causes nitroglycerin resistance in platelets by depletion of intraplatelet glutathione. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(11):1852–6. [PubMed: 11701477]
- Harats D, Ben-Naim M, Dabach Y, Hollander G, Stein O, Stein Y. Cigarette smoking renders LDL susceptible to peroxidative modification and enhanced metabolism by macrophages. Atherosclerosis. 1989;79(2–3):245–52. [PubMed: 2597232]
- Hart CL, Hole DJ, Smith GD. Risk factors and 20-year stroke mortality in men and women in the Renfrew/Paisley Study in Scotland. Stroke. 1999;30(10):1999–2007. [PubMed: 10512898]
- Hasdai D, Garratt KN, Grill DE, Lerman A, Holmes DR Jr. Effect of smoking status on the long-term outcome after successful percutaneous coronary revascularization. New England Journal of Medicine. 1997a;336(11):755–61. [PubMed: 9052653]
- Hasdai D, Gibbons RJ, Holmes DR Jr, Higano ST, Lerman A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation. 1997b;96(10):3390–5. [PubMed: 9396432]
- Hatsukami DK, Kotlyar M, Allen S, Jensen J, Li S, Le C, Murphy S. Effects of cigarette reduction on cardiovascular risk factors and subjective methods. Chest. 2005;128(4):2528–37. [PubMed: 16236919]
- Hatsukami DK, Slade J, Benowitz NL, Giovino GA, Gritz ER, Leischow S, Warner KE. Reducing tobacco harm: research challenges and issues. Nicotine & Tobacco Research. 2002;4(Suppl 2):S89–S101. [PubMed: 12573171]
- Haustein K-O, Krause J, Haustein H, Rasmussen T, Cort N. Effects of cigarette smoking or nicotine replacement on cardiovascular risk factors and parameters of haemorheology. Journal of Internal Medicine. 2002;252(2):130–9. [PubMed: 12190888]
- Haustein K-O, Krause J, Haustein H, Rasmussen T, Cort N. Changes in hemorheological and biochemical parameters following short-term and long-term smoking cessation induced by nicotine replacement therapy (NRT). International Journal of Clinical Pharmacology, Therapy, and Toxicology. 2004;42(2):83–92. [PubMed: 15180168]
- Hecht SS, Murphy SE, Carmella SG, Zimmerman CL, Losey L, Kramarczuk I, Roe MR, Puumala SS, Li YS, Le C, et al. Effects of reduced cigarette smoking on uptake of a tobacco-specific lung carcinogen. Journal of the National Cancer Institute. 2004;96(2):107–15. [PubMed: 14734700]
- Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nature Medicine. 2001;7(7):833–9. [PubMed: 11433349]
- Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. Journal of Clinical Investigation. 2002;110(4):527–36. [PMC free article: PMC150415] [PubMed: 12189247]
- Heeschen C, Weis M, Cooke JP. Nicotine promotes arteriogenesis. Journal of the American College of Cardiology. 2003;41(3):489–96. [PubMed: 12575981]
- Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Münzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circulation Research. 2000;86(2):E36–E41. [PubMed: 10666424]
- Heitzer T, Just H, Munzel T. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation. 1996;94(1):6–9. [PubMed: 8964118]
- Hellerstein MK, Benowitz NL, Neese RA, Schwartz JM, Hoh R, Jacob P III, Hsieh J, Faix D. Effects of cigarette smoking and its cessation on lipid metabolism and energy expenditure in heavy smokers. Journal of Clinical Investigation. 1994;93(1):265–72. [PMC free article: PMC293761] [PubMed: 8282797]
- Helve E, Yki-Järvinen H, Koivisto VA. Smoking and insulin sensitivity in type I diabetic patients. Metabolism. 1986;35(9):874–7. [PubMed: 3528745]
- Henley SJ, Thun MJ, Chao A, Calle EE. Association between exclusive pipe smoking and mortality from cancer and other diseases. Journal of the National Cancer Institute. 2004;96(11):853–61. [PubMed: 15173269]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug and Alcohol Dependence. 1993;33(1):23–9. [PubMed: 8370337]
- Hermanson B, Omenn GS, Kronmal RA, Gersh BJ. Beneficial six-year outcome of smoking cessation in older men and women with coronary artery disease: results from the CASS registry. New England Journal of Medicine. 1988;319(21):1365–9. [PubMed: 3185646]
- Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cell Biology. 2005;6(2):150–66. [PubMed: 15688001]
- Hesse E. Der einfluss des rauchens auf den kreislauf (The influence of smoking on the circulation) [German] Archiv fur Klinische Medizin. 1907;89:565–75.
- Hibbert B, Olsen S, O’Brien E. Involvement of progenitor cells in vascular repair. Trends in Cardiovascular Medicine. 2003;13(8):322–6. [PubMed: 14596947]
- Hibi K, Ishigami T, Tamura K, Mizushima S, Nyui N, Fujita T, Ochiai H, Kosuge M, Watanabe Y, Yoshii Y, et al. Endothelial nitric oxide synthase gene polymorphism and acute myocardial infarction. Hypertension. 1998;32(3):521–6. [PubMed: 9740620]
- Hill JM, Zalos G, Halcox JPJ, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. New England Journal of Medicine. 2003;348(7):593–600. [PubMed: 12584367]
- Hioki H, Aoki N, Kawano K, Homori M, Hasumura Y, Yasumura T, Maki A, Yoshino H, Yanagisawa A, Ishikawa K. Acute effects of cigarette smoking on platelet-dependent thrombin generation. European Heart Journal. 2001;22(1):56–61. [PubMed: 11133210]
- Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. New England Journal of Medicine. 2001;345(11):790–7. [PubMed: 11556298]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377(6546):239–42. [PubMed: 7545787]
- Hubbard R, Lewis S, Smith C, Godfrey C, Smeeth L, Farrington P, Britton J. Use of nicotine replacement therapy and the risk of acute myocardial infarction, stroke, and death. Tobacco Control. 2005;14(6):416–21. [PMC free article: PMC1748112] [PubMed: 16319366]
- Hughes JR. Reduced smoking: an introduction and review of the evidence. Addiction. 2000;95(Suppl 1):S3–S7. [PubMed: 10723815]
- Hughes JR, Goldstein MG, Hurt RD, Shiffman S. Recent advances in the pharmacotherapy of smoking. JAMA: the Journal of the American Medical Association. 1999;281(1):72–6. [PubMed: 9892454]
- Hughes JR, Gulliver SB, Fenwick JW, Valliere WA, Cruser K, Pepper S, Shea P, Solomon L, Flynn BS. Smoking cessation among self-quitters. Health Psychology. 1992;11(5):331–4. [PubMed: 1425551]
- Hughes JR, Lindgren PG, Connett JE, Nides MA. Smoking reduction in the Lung Health Study. Nicotine & Tobacco Research. 2004a;6(2):275–80. [PubMed: 15203800]
- Hughes JR, Stead LF, Lancaster T. Antidepressants for smoking cessation. Cochrane Database of Systematic Reviews. 2004b;(4) Art. No.: CD000031. [PubMed: 15494986] [CrossRef]
- Hunter KA, Garlick PJ, Broom I, Anderson SE, McNurlan MA. Effects of smoking and abstention from smoking on fibrinogen synthesis in humans. Clinical Science (London). 2001;100(4):459–65. [PubMed: 11256988]
- Hurt RD, Croghan GA, Wolter TD, Croghan IT, Offord KP, Williams GM, Djordjevic MV, Richie JP Jr, Jeffrey AM. Does smoking reduction result in reduction of biomarkers associated with harm: a pilot study using a nicotine inhaler. Nicotine & Tobacco Research. 2000;2(4):327–36. [PubMed: 11197312]
- Ichihara S, Yamada Y, Fujimura T, Nakashima N, Yokota M. Association of a polymorphism of the endothelial constitutive nitric oxide synthase gene with myocardial infarction in the Japanese population. American Journal of Cardiology. 1998;81(1):83–6. [PubMed: 9462613]
- Ignarro LJ, Burke TM, Wood KS, Wolin MS, Kadowitz PJ. Association between cyclic GMP accumulation and acetylcholine-elicited relaxation of bovine intrapulmonary artery. Journal of Pharmacology and Experimental Therapeutics. 1984;228(3):682–90. [PubMed: 6323677]
- Ikarugi H, Yamashita T, Aoki R, Ishii H, Kanki K, Yamamoto J. Impaired spontaneous thrombolytic activity in elderly and in habitual smokers, as measured by a new global thrombosis test. Blood Coagulation & Fibrinolysis. 2003;14(8):781–4. [PubMed: 14614361]
- Ilebekk A, Miller NE, Mjos OD. Effects of nicotine and inhalation of cigarette smoke on total body oxygen consumption in dogs. Scandinavian Journal of Clinical and Laboratory Investigation. 1975;35(1):67–72. [PubMed: 1129594]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Smokeless Tobacco and Some Tobacco-specific N-Nitrosamines. Vol. 89. Lyon (France): International Agency for Research on Cancer; 2007. [PMC free article: PMC4781254] [PubMed: 18335640]
- Irace C, Ciamei M, Crivaro A, Fiaschi E, Madia A, Cortese C, Gnasso A. Hematocrit is associated with carotid atherosclerosis in men but not in women. Coronary Artery Disease. 2003;14(4):279–84. [PubMed: 12826926]
- Iribarren C, Tekawa IS, Sidney S, Friedman GD. Effect of cigar smoking on the risk of cardiovascular disease, chronic obstructive pulmonary disease, and cancer in men. New England Journal of Medicine. 1999;340(23):1773–80. [PubMed: 10362820]
- Jacobs DR Jr, Adachi H, Mulder I, Kromhout D, Menotti A, Nissinen A, Blackburn H. Cigarette smoking and mortality risk: twenty-five-year follow-up of the Seven Countries Study. Archives of Internal Medicine. 1999a;159(7):733–40. [PubMed: 10218754]
- Jacobs EJ, Thun MJ, Apicella LF. Cigar smoking and death from coronary heart disease in a prospective study of US men. Archives of Internal Medicine. 1999b;159(20):2413–8. [PubMed: 10665889]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. [PubMed: 15637262]
- Jamrozik K, Norman PE, Spencer CA, Parsons RW, Tuohy R, Lawrence-Brown MM, Dickinson JA. Screening for abdominal aortic aneurysm: lessons from a population-based study. Medical Journal of Australia. 2000;173(7):345–50. [PubMed: 11062788]
- Jarrett RJ, McCartney P, Keen H. The Bedford survey: ten year mortality rates in newly diagnosed diabetics, borderline diabetics and normoglycaemic controls and risk indices for coronary heart disease in borderline diabetics. Diabetologia. 1982;22(2):79–84. [PubMed: 7060853]
- Jensen EJ, Pedersen B, Frederiksen R, Dahl R. Prospective study on the effect of smoking and nicotine substitution on leucocyte blood counts and relation between blood leucocytes and lung function. Thorax. 1998;53(9):784–9. [PMC free article: PMC1745328] [PubMed: 10319062]
- Jeppesen J, Hollenbeck CB, Zhou M-Y, Coulston AM, Jones C, Chen Y-DI, Reaven GM. Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipemia. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995;15(3):320–4. [PubMed: 7749841]
- Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Luscher TF. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91(5):1314–9. [PubMed: 7867167]
- Jonason T, Bergström R. Cessation of smoking in patients with intermittent claudication: effects on the risk of peripheral vascular complications, myocardial infarction and mortality. Acta Medica Scandinavica. 1987;221(3):253–60. [PubMed: 3591463]
- Jorenby DE, Hays JT, Rigotti NA, Azoulay S, Watsky EJ, Williams KE, Billing CB, Gong J, Reeves KR. Varenicline Phase 3 Study Group. Efficacy of varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA: the Journal of the American Medical Association. 2006;296(1):56–63. [PubMed: 16820547]
- Jorenby DE, Leischow SJ, Nides MA, Rennard SI, Johnston JA, Hughes AR, Smith SS, Muramoto ML, Daughton DM, Doan K, et al. A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. New England Journal of Medicine. 1999;340(9):685–91. [PubMed: 10053177]
- Joseph A, Hecht S, Murphy S, Gross M, Lando H, Bliss R, Le C, Hatsukami D. A randomized controlled trial of smoking reduction in heart disease patients. Abstract presented at the Society for Research on Nicotine and Tobacco’s 11th Annual Meeting; March 20–23, 2005; Prague. [accessed: October 30, 2006]. < http://www
.srnt.org/pubs/abstract.html>. - Joseph AM, Antonnucio DO. Lack of efficacy of transdermal nicotine in smoking cessation. New England Journal of Medicine. 1999;341(15):1157–8. [PubMed: 10515763]
- Joseph AM, Fu SS. Safety issues in pharmacotherapy for smoking in patients with cardiovascular disease. Progress in Cardiovascular Diseases. 2003;45(6):429–41. [PubMed: 12800126]
- Joseph AM, Norman SM, Ferry LH, Prochazka AV, Westman EC, Steele BG, Sherman SE, Cleveland M, Anton-nucio DO, Hartman N, et al. The safety of transdermal nicotine as an aid to smoking cessation in patients with cardiac disease. New England Journal of Medicine. 1996;335(24):1792–8. [PubMed: 8943160]
- Kagan A, Yano K, Reed DM, MacLean CJ. Predictors of sudden cardiac death among Hawaiian-Japanese men. American Journal of Epidemiology. 1989;130(2):268–77. [PubMed: 2750726]
- Kagota S, Yamaguchi Y, Shinozuka K, Kwon YM, Kunitomo M. Cigarette smoke-modified low density lipoprotein impairs endothelium-dependent relaxation in isolated rabbit arteries. General Pharmacology. 1996;27(3):447–81. [PubMed: 8723530]
- Kahn HA. The Dorn study of smoking and mortality among US veterans: report on 8 and one-half years of observation. Epidemiological Approaches to the Study of Cancer and Other Chronic Diseases. National Cancer Institute Monograph No 19. Haenszel W, editor. Bethesda (MD): U.S. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health, National Cancer Institute; 1966. pp. 1–125. [PubMed: 5905668]
- Kahn SE. Beta cell failure: causes and consequences. International Journal of Clinical Practice Supplement. 2001;(123):13–8. [PubMed: 11594291]
- Kaijser L, Berglund B. Effect of nicotine on coronary blood-flow in man. Clinical Physiology. 1985;5(6):541–52. [PubMed: 4092414]
- Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD. Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. Journal of Cellular Physiology. 1994;160(1):154–62. [PubMed: 7517402]
- Kannel WB, Doyle JT, McNamara PM, Quickenton P, Gordon T. Precursors of sudden coronary death: factors related to the incidence of sudden death. Circulation. 1975;51(4):606–13. [PubMed: 123182]
- Kannel WB, Higgins M. Smoking and hypertension as predictors of cardiovascular risk in population studies. Journal of Hypertension Supplement. 1990;8( Suppl 5):S3–S8. [PubMed: 2286855]
- Kannel WB, McGee DL, Castelli WP. Latest perspectives on cigarette smoking and cardiovascular disease: the Framingham Study. Journal of Cardiac Rehabilitation. 1984;4(7):267.
- Kannel WB, Shurtleff D. The Framingham Study: cigarettes and the development of intermittent claudication. Geriatrics. 1973;28(2):61–8. [PubMed: 4683662]
- Kannel WB, Thomas HE Jr. Sudden coronary death: the Framingham Study. Annals of the New York Academy of Sciences. 1982;382:3–21. [PubMed: 7044245]
- Kaufmann PA, Gnecchi-Ruscone T, di Terlizzi M, Schäfers KP, Lüscher TF, Camici PG. Coronary heart disease in smokers: vitamin C restores coronary microcirculatory function. Circulation. 2000;102(11):1233–8. [PubMed: 10982536]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Hunter DJ, Hennekens CH, Speizer FE. Smoking cessation in relation to total mortality rates in women: a prospective cohort study. Annals of Internal Medicine. 1993a;119(10):992–1000. [PubMed: 8214996]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. Smoking cessation and decreased risk of stroke in women. JAMA: the Journal of the American Medical Association. 1993b;269(2):232–6. [PubMed: 8417241]
- Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. Smoking cessation and time course of decreased risks of coronary heart disease in middle-aged women. Archives of Internal Medicine. 1994;154(2):169–75. [PubMed: 8285812]
- Kawakami N, Takatsuka N, Shimizu H, Ishibashi H. Effects of smoking on the incidence of non-insulin-dependent diabetes mellitus: replication and extension in a Japanese cohort of male employees. Amercan Journal of Epidemiology. 1997;145(2):103–9. [PubMed: 9006306]
- Kayyali US, Budhiraja R, Pennella CM, Cooray S, Lanzillo JJ, Chalkley R, Hassoun PM. Upregulation of xanthine oxidase by tobacco smoke condensate in pulmonary endothelial cells. Toxicology and Applied Pharmacology. 2003;188(1):59–68. [PubMed: 12668123]
- Keen H, Jarrett RJ, McCartney P. The ten-year follow-up of the Bedford survey (1962–1972): glucose tolerance and diabetes. Diabetologia. 1982;22(2):73–8. [PubMed: 7060852]
- Kenet G, Freedman J, Shenkman B, Regina E, Brok-Simoni F, Holzman F, Vavva F, Brand N, Michelson A, Trolliet M, et al. Plasma glutathione peroxidase deficiency and platelet insensitivity to nitric oxide in children with familial stroke. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(8):2017–23. [PubMed: 10446087]
- Kershbaum A, Bellet S. Smoking as a factor in atherosclerosis: a review of epidemiological, pathological, and experimental studies. Geriatrics. 1966;21(12):155–70. [PubMed: 5333109]
- Kershbaum A, Khorsandian R, Caplan RF, Bellet S, Feinberg LJ. The role of catecholamines in the free fatty acid response to cigarette smoking. Circulation. 1963;28(1):52–7. [PubMed: 14032101]
- Kielstein JT, Impraim B, Simmel S, Bode-Böger SM, Tsikas D, Frölich JC, Hoeper MM, Haller H, Fliser D. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation. 2004;109(2):172–7. [PubMed: 14662708]
- Kimmel SE, Berlin JA, Miles C, Jaskowiak J, Carson JL, Strom BL. Risk of acute first myocardial infarction and use of nicotine patches in a general population. Journal of the American College of Cardiology. 2001;37(5):1297–302. [PubMed: 11300438]
- Kirsch M, de Groot H. Ascorbate is a potent antioxidant against peroxynitrite-induced oxidation reactions: evidence that ascorbate acts by re-reducing substrate radicals produced by peroxynitrite. Journal of Biological Chemistry. 2000;275(22):16702–8. [PubMed: 10748119]
- Kirsch M, Korth HG, Sustmann R, de Groot H. The patho-biochemistry of nitrogen dioxide. Biological Chemistry. 2002;383(3–4):389–99. [PubMed: 12033430]
- Kirsch M, Lehnig M, Korth H-G, Sustmann R, de Groot H. Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry. 2001;7(15):3313–20. [PubMed: 11531117]
- Kirschbaum C, Wust S, Strasburger CJ. ‘Normal’ cigarette smoking increases free cortisol in habitual smokers. Life Sciences. 1992;50(6):435–42. [PubMed: 1734161]
- Kiyohara Y, Ueda K, Fujishima M. Smoking and cardiovascular disease in the general population in Japan. Journal of Hypertension. 1990;8(Suppl 5):S9–S15. [PubMed: 2286856]
- Ko GTC, Chan JCN, Tsang LWW, Critchley JAJH, Cockram CS. Smoking and diabetes in Chinese men. Postgraduate Medical Journal. 2001;77(906):240–3. [PMC free article: PMC1742000] [PubMed: 11264486]
- Koenig W, Sund M, Fröhlich M, Fischer H-G, Löwel H, Döring A, Hutchinson WL, Pepys MB. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99(2):237–42. [PubMed: 9892589]
- Komatsu M, Kawagishi T, Emoto M, Shoji T, Yamada A, Sato K, Hosoi M, Nishizawa Y. ecNOS gene polymorphism is associated with endothelium-dependent vasodilation in type 2 diabetes. Journal of Physiology - Heart and Circulatory Physiology. 2002;283(2):H557–H561. [PubMed: 12124201]
- Kondo T, Hayashi M, Takeshita K, Numaguchi Y, Kobayashi K, Iino S, Inden Y, Murohara T. Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(8):1442–7. [PubMed: 15191940]
- Konishi H, Wu J, Cooke JP. Chronic exposure to nicotine impairs cholinergic angiogenesis. Vascular Medicine. 2010;15(1):47–54. [PMC free article: PMC3142172] [PubMed: 19778953]
- Kothari V, Stevens RJ, Adler AI, Stratton IM, Manley SE, Neil HA, Holman RR. UKPDS 60: risk of stroke in type 2 diabetes estimated by the UK Prospective Diabetes Study risk engine. Stroke. 2002;33(7):1776–81. [PubMed: 12105351]
- Krumholz HM, Cohen BJ, Tsevat J, Pasternak RC, Weinstein MC. Cost-effectiveness of a smoking cessation program after myocardial infarction. Journal of the American College of Cardiology. 1993;22(6):1697–702. [PubMed: 8227841]
- Krupski WC, Olive GC, Weber CA, Rapp JH. Comparative effects of hypertension and nicotine on injury-induced myointimal thickening. Surgery. 1987;102(2):409–15. [PubMed: 3616923]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(11):4651–5. [PMC free article: PMC51723] [PubMed: 1675786]
- Kugiyama K, Yasue H, Ohgushi M, Motoyama T, Kawano H, Inobe Y, Hirashima O, Sugiyama S. Deficiency in nitric oxide bioactivity in epicardial coronary arteries of cigarette smokers. Journal of the American College of Cardiology. 1996;28(5):1161–7. [PubMed: 8890810]
- Kuhlencordt PJ, Cyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104(4):448–54. [PubMed: 11468208]
- Kuller LH, Ockene JK, Meilahn E, Wentworth DN, Svendsen KH, Neaton JD. Multiple Risk Factor Intervention Trial Research Group. Cigarette smoking and mortality. Preventive Medicine. 1991;20(5):638–54. [PubMed: 1758843]
- Kumanan W, Gibson N, Willan A, Cook D. Effect of smoking cessation on mortality after myocardial infarction: meta-analyses of cohort studies. Archives of Internal Medicine. 2000;160(7):939–44. [PubMed: 10761958]
- Kumar A, Kingdon E, Norman J. The isoprostane 8-iso-PGF2α suppresses monocyte adhesion to human microvascular endothelial cells via two independent mechanisms. FASEB Journal. 2005;19(3):443–5. [PubMed: 15640282]
- Lam TH, He Y, Shi QL, Huang JY, Zhang F, Wan ZH, Sun CS, Li LS. Smoking, quitting, and mortality in a Chinese cohort of retired men. Annals of Epidemiology. 2002;12(5):316–20. [PubMed: 12062918]
- Lancet. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837–53. [PubMed: 9742976]
- Lane D, Gray EA, Mathur RS, Mathur SP. Up-regulation of vascular endothelial growth factor-C by nicotine in cervical cancer cell lines. American Journal of Reproductive Immunology. 2005;53(3):153–8. [PubMed: 15727570]
- Lassila R, Laustiola KE. Cigarette smoking and platelet-vessel wall interactions. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 1992;46(2):81–6. [PubMed: 1502254]
- Lassila R, Seyberth HW, Haapanen A, Schweer H, Koskenvuo M, Laustiola KE. Vasoactive and atherogenic effects of cigarette smoking: a study of monozygotic twins discordant for smoking. BMJ (British Medical Journal). 1988;297(6654):955–7. [PMC free article: PMC1834683] [PubMed: 3142565]
- Law MR, Morris JK, Watt HC, Wald NJ. The dose-response relationship between cigarette consumption, biochemical markers and risk of lung cancer. British Journal of Cancer. 1997;75(11):1690–3. [PMC free article: PMC2223525] [PubMed: 9184188]
- Law MR, Wald NJ. Environmental tobacco smoke and ischemic heart disease. Progress in Cardiovascular Diseases. 2003;46(1):31–8. [PubMed: 12920699]
- Lederle FA, Johnson GR, Wilson SE. Abdominal aortic aneurysm in women. Journal of Vascular Surgery. 2001;34(1):122–6. [PubMed: 11436084]
- Lee AJ, Fowkes FG, Carson MN, Leng GC, Allan PL. Smoking, atherosclerosis and risk of abdominal aortic aneurysm. European Heart Journal. 1997;18(4):671–6. [PubMed: 9129900]
- Lee AJ, Fowkes FG, Rattray A, Rumley A, Lowe GD. Haemostatic and rheological factors in intermittent claudication: the influence of smoking and extent of arterial disease. British Journal of Haematology. 1996;92(1):226–30. [PubMed: 8562400]
- Leeson CPM, Hingorani AD, Mullen MJ, Jeerooburkhan N, Kattenhorn M, Cole TJ, Muller DPR, Lucas A, Humphries SE, Deanfield JE. Glu298Asp endothelial nitric oxide synthase gene polymorphism interacts with environmental and dietary factors to influence endothelial function. Circulation Research. 2002;90(11):1153–8. [PubMed: 12065317]
- Lehr H-A, Frei B, Arfors KE. Vitamin C prevents cigarette smoke-induced leukocyte aggregation and adhesion to endothelium in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(16):7688–92. [PMC free article: PMC44467] [PubMed: 7519784]
- Lehr H-A, Weyrich AS, Saetzler RK, Jurek A, Arfors KE, Zimmerman GA, Prescott SM, McIntyre TM. Vitamin C blocks inflammatory platelet-activating factor mimetics created by cigarette smoking. Journal of Clinical Investigation. 1997;99(10):2358–64. [PMC free article: PMC508074] [PubMed: 9153277]
- Leitinger N, Huber J, Rizza C, Mechtcheriakova D, Bochkov V, Koshelnick Y, Berliner JA, Binder BR. The isoprostane 8-iso-PGF2α stimulates endothelial cells to bind monocytes: differences from thromboxane-mediated endothelial activation. FASEB Journal. 2001;15(7):1254–6. [PubMed: 11344105]
- Ley K. The role of selectins in inflammation and disease. Trends in Molecular Medicine. 2003;9(6):263–8. [PubMed: 12829015]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43. [PubMed: 11877368]
- Lightwood J. Economics of smoking and cardiovascular disease. Progress in Cardiovascular Diseases. 2003;46(1):39–78. [PubMed: 12920700]
- Lightwood JM, Glantz SA. Short-term economic and health benefits of smoking cessation: myocardial infarction and stroke. Circulation. 1997;96(4):1089–96. [PubMed: 9286934]
- Lightwood JM, Glantz SA. Declines in acute myocardial infarction after smoke-free laws and individual risk attributable to secondhand smoke. Circulation. 2009;120(14):1373–9. [PMC free article: PMC2967202] [PubMed: 19770392]
- Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA: the Journal of the American Medical Association. 2007;298(10):1180–8. [PubMed: 17848652]
- Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circulation Research. 2001;88(8):756–62. [PubMed: 11325866]
- Lowe GD, Lee AJ, Rumley A, Price JF, Fowkes FG. Blood viscosity and risk of cardiovascular events: the Edinburgh Artery Study. British Journal of Haematology. 1997;96(1):168–73. [PubMed: 9012704]
- Lu JT, Creager MA. The relationship of cigarette smoking to peripheral arterial disease. Reviews in Cardiovascular Medicine. 2004;5(4):189–93. [PubMed: 15580157]
- Lucini D, Bertocchi F, Malliani A, Pagani M. A controlled study of the autonomic changes produced by habitual cigarette smoking in healthy subjects. Cardiovascular Research. 1996;31(4):633–9. [PubMed: 8689656]
- Lúdvíksdóttir D, Blöndal T, Franxon M, Gudmundsson TV, Säwe U. Effects of nicotine nasal spray on atherogenic and thrombogenic factors during smoking cessation. Journal of Internal Medicine. 1999;246(1):61–6. [PubMed: 10447226]
- Lundman BM, Asplund K, Norberg A. Smoking and metabolic control in patients with insulin-dependent diabetes mellitus. Journal of Internal Medicine. 1990;227(2):101–6. [PubMed: 2299302]
- Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. American Journal of Clinical Nutrition. 2000;71(2):530–6. [PubMed: 10648268]
- MacCallum PK. Markers of hemostasis and systemic inflammation in heart disease and atherosclerosis in smokers. Proceedings of the American Thoracic Society. 2005;2(1):34–43. [PubMed: 16113467]
- Madsbad S, McNair P, Christensen MS, Christiansen C, Faber OK, Binder C, Transbol I. Influence of smoking on insulin requirement and metabolic status in diabetes mellitus. Diabetes Care. 1980;3(1):41–3. [PubMed: 6996967]
- Mahfouz MM, Hulea SA, Kummerow FA. Cigarette smoke increases cholesterol oxidation and lipid peroxidation of human low-density lipoprotein and decreases its binding to the hepatic receptor in vitro. Journal of Environmental Pathology, Toxicology and Oncology. 1995;14(3–4):181–92. [PubMed: 9003696]
- Mahmarian JJ, Moyé LA, Nasser GA, Nagueh SF, Bloom MF, Benowitz NL, Verani MS, Byrd WG, Pratt CM. Nicotine patch therapy in smoking cessation reduces the extent of exercise-induced myocardial ischemia. Journal of American College of Cardiology. 1997;30(1):125–30. [PubMed: 9207632]
- Mann SJ, James GD, Wang RS, Pickering TG. Elevation of ambulatory systolic blood pressure in hypertensive smokers. JAMA: the Journal of the American Medical Association. 1991;265(17):2226–8. [PubMed: 2013955]
- Manson JE, Ajani UA, Liu S, Nathan DM, Hennekens CH. A prospective study of cigarette smoking and the incidence of diabetes mellitus among US male physicians. American Journal of Medicine. 2000;109(7):538–42. [PubMed: 11063954]
- Manson JE, Colditz GA, Stampfer MJ, Willett WC, Krolewski AS, Rosner B, Arky RA, Speizer FE, Hennekens CH. A prospective study of maturity-onset diabetes mellitus and risk of coronary heart disease and stroke in women. Archives of Internal Medicine. 1991;151(6):1141–7. [PubMed: 2043016]
- Marangon K, Herbeth B, Artur Y, Esterbauer H, Siest G. Low and very low density lipoprotein composition and resistance to copper-induced oxidation are not notably modified in smokers. Clinica Chimica Acta. 1997;265(1):1–12. [PubMed: 9352124]
- Maser RE, Steenkiste AR, Dorman JS, Nielsen VK, Bass EB, Manjoo Q, Drash AL, Becker DJ, Kuller LH, Greene DA. Epidemiological correlates of diabetic neuropathy: report from Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes. 1989;38(11):1456–61. [PubMed: 2620781]
- Matetzky S, Tani S, Kangavari S, Dimayuga P, Yano J, Xu H, Chyu K-Y, Fishbein MC, Shah PK, Cercek B. Smoking increases tissue factor expression in atherosclerotic plaques: implications for plaque thrombogenicity. Circulation. 2000;102(6):602–4. [PubMed: 10931797]
- Mazzone A, Cusa C, Mazzucchelli I, Vezzoli M, Ottini E, Ghio S, Tossini G, Pacifici R, Zuccaro P. Cigarette smoking and hypertension influence nitric oxide release and plasma levels of adhesion molecules. Clinical Chemistry and Laboratory Medicine. 2001;39(9):822–6. [PubMed: 11601680]
- McAdam BF, Byrne D, Morrow JD, Oates JA. Contribution of cyclooxygenase-2 to elevated biosynthesis of thromboxane A2 and prostacyclin in cigarette smokers. Circulation. 2005;112(7):1024–9. [PubMed: 16087791]
- McCall MR, van den Berg JJ, Kuypers FA, Tribble DL, Krauss RM, Knoff LJ, Forte TM. Modification of LCAT activity and HDL structure: new links between cigarette smoke and coronary heart disease risk. Arteriosclerosis and Thrombosis. 1994;14(2):248–53. [PubMed: 8305416]
- McLenachan JM, Vita J, Fish DR, Treasure CB, Cox DA, Ganz P, Selwyn AP. Early evidence of endothelial vasodilator dysfunction at coronary branch points. Circulation. 1990;82(4):1169–73. [PubMed: 2401058]
- McNagny SE, Ahluwalia JS, Clark WS, Resnicow KA. Cigarette smoking and severe uncontrolled hypertension in innercity African Americans. American Journal of Medicine. 1997;103(2):121–7. [PubMed: 9274895]
- Meade TW, Imeson J, Stirling Y. Effects of changes in smoking and other characteristics on clotting factors and the risk of ischaemic heart disease. Lancet. 1987;2(8566):986–8. [PubMed: 2889958]
- Medalie JH, Papier CM, Goldbourt U, Herman JB. Major factors in the development of diabetes mellitus in 10,000 men. Archives of Internal Medicine. 1975;135(6):811–7. [PubMed: 1130926]
- Meijer WT, Grobbee DE, Hunink MGM, Hofman A, Hoes AW. Determinants of peripheral arterial disease in the elderly: the Rotterdam Study. Archives of Internal Medicine. 2000;160(19):2934–8. [PubMed: 11041900]
- Meine TJ, Patel MR, Washam JB, Pappas PA, Jollis JG. Safety and effectiveness of transdermal nicotine patch in smokers admitted with acute coronary syndromes. American Journal of Cardiology. 2005;95(8):976–8. [PubMed: 15820167]
- Mendelson G, Aronow WS, Ahn C. Prevalence of coronary artery disease, atherothrombotic brain infarction, and peripheral arterial disease: associated risk factors in older Hispanics in an academic hospital-based geriatrics practice. Journal of the American Geriatric Society. 1998;46(4):481–3. [PubMed: 9560072]
- Mero N, Syvänne M, Eliasson B, Smith U, Taskinen M-R. Postprandial elevation of ApoB-48-containing triglyceride-rich particles and retinyl esters in normolipemic males who smoke. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(10):2096–102. [PubMed: 9351377]
- Mero N, Van Tol A, Scheek LM, Van Gent T, Labeur C, Rosseneu M, Taskinen M-R. Decreased postprandial high density lipoprotein cholesterol and apolipoproteins A-I and E in normolipidemic smoking men: relations with lipid transfer proteins and LCAT activities. Journal of Lipid Research. 1998;39(7):1493–502. [PubMed: 9684753]
- Metz L, Waters DD. Implications of cigarette smoking for the management of patients with acute coronary syndromes. Progress in Cardiovascular Diseases. 2003;46(1):1–9. [PubMed: 12920697]
- Meyers CD, Kamanna VS, Kashyap ML. Niacin therapy in atherosclerosis. Current Opinion in Lipidology. 2004;15(6):659–65. [PubMed: 15529025]
- Meyers DG, Neuberger JS, He J. Cardiovascular effects of bans on smoking in public places: a systematic review and meta-analysis. Journal of the American College of Cardiology. 2009;54(14):1249–55. [PubMed: 19778665]
- Miettinen OS, Neff RK, Jick H. Cigarette-smoking and nonfatal myocardial infarction: rate ratio in relation to age, sex and predisposing conditions. American Journal of Epidemiology. 1976;103(1):30–6. [PubMed: 1247010]
- Minuz P, Andrioli G, Degan M, Gaino S, Ortolani R, Tommasoli R, Zuliani V, Lechi A, Lechi C. The F2-isoprostane 8-epiprostaglandin F2α increases platelet adhesion and reduces the antiadhesive and antiaggregatory effects of NO. Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18(8):1248–56. [PubMed: 9714131]
- Mitchell BD, Hawthorne VM, Vinik AI. Cigarette smoking and neuropathy in diabetic patients. Diabetes Care. 1990;13(4):434–7. [PubMed: 2318103]
- Miyake Y. Risk factors for non-fatal acute myocardial infarction in middle-aged and older Japanese: Fukuoka Heart Study Group. Japanese Circulation Journal. 2000;64(2):103–9. [PubMed: 10716523]
- Miyamoto Y, Saito Y, Kajiyama N, Yoshimura M, Shimasaki Y, Nakayama M, Kamitani S, Harada M, Ishikawa M, Kuwahara K, et al. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension. 1998;32(1):3–8. [PubMed: 9674630]
- Moffatt RJ, Biggerstaff KD, Stamford BA. Effects of the transdermal nicotine patch on normalization of HDL-C and its subfractions. Preventive Medicine. 2000;31(2 Pt 1):148–52. [PubMed: 10938215]
- Moreno H Jr, Chalon S, Urae A, Tangphao O, Abiose AK, Hoffman BB, Blaschke TF. Endothelial dysfunction in human hand veins is rapidly reversible after smoking cessation. American Journal of Physiology. 1998;275(3 Pt 2):H1040–H1045. [PubMed: 9724311]
- Morita H, Ikeda H, Haramaki N, Eguchi H, Imaizumi T. Only two-week smoking cessation improves platelet aggregability and intraplatelet redox imbalance of long-term smokers. Journal of the American College of Cardiology. 2005;45(4):589–94. [PubMed: 15708708]
- Morrish NJ, Stevens LK, Fuller JH, Jarrett RJ, Keen H. Risk factors for macrovascular disease in diabetes mellitus: the London follow-up to the WHO Multinational Study of Vascular Disease in Diabetics. Diabetologia. 1991;34(8):590–4. [PubMed: 1936663]
- Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ II. Increase in circulating products of lipid peroxidation (F2-iso-prostanes) in smokers: smoking as a cause of oxidative damage. New England Journal of Medicine. 1995;332(18):1198–203. [PubMed: 7700313]
- Moss SE, Klein R, Klein BE. Association of cigarette smoking with diabetic retinopathy. Diabetes Care. 1991;14(2):119–26. [PubMed: 2060413]
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo K-M, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4736–41. [PMC free article: PMC153625] [PubMed: 12682294]
- Moy CS, LaPorte RE, Dorman JS, Songer TJ, Orchard TJ, Kuller LH, Becker DJ, Drash AL. Insulin-dependent diabetes mellitus mortality: the risk of cigarette smoking. Circulation. 1990;82(1):37–43. [PubMed: 2364522]
- Muhlhauser I, Bender R, Bott U, Jorgens V, Grusser M, Wagener W, Overmann H, Berger M. Cigarette smoking and progression of retinopathy and nephropathy in type 1 diabetes. Diabetic Medicine. 1996;13(6):536–43. [PubMed: 8799657]
- Muhlhauser I, Sawicki P, Berger M. Cigarette-smoking as a risk factor for macroproteinuria and proliferative retinopathy in type 1 (insulin-dependent) diabetes. Diabetologia. 1986;29(8):500–2. [PubMed: 3758532]
- Multiple Risk Factor Intervention Trial Research Group. Mortality rates after 10.5 years for participants in the Multiple Risk Factor Intervention Trial: findings related to a priori hypotheses of the trial. JAMA: the Journal of the American Medical Association. 1990;263(13):1795–801. [PubMed: 2179590]
- Multiple Risk Factor Intervention Trial Research Group. Mortality after 16 years for participants randomized to the Multiple Risk Factor Intervention Trial. Circulation. 1996;94(5):946–51. [PubMed: 8790030]
- Murabito JM, Evans JC, Nieto K, Larson MG, Levy D, Wilson PWF. Prevalence and clinical correlates of peripheral arterial disease in the Framingham Offspring Study. American Heart Journal. 2002;143(6):961–5. [PubMed: 12075249]
- Murray JJ, Nowak J, Oates JA, FitzGerald GA. Biosynthesis of thromboxane A2 and prostacyclin during chronic smoking and withdrawal in man [abstract] Clinical Research. 1985;33:521A.
- Murray JJ, Nowak J, Oates JA, FitzGerald GA. Platelet-vessel wall interactions in individuals who smoke cigarettes. Advances in Experimental Medicine and Biology. 1990;273:189–98. [PubMed: 2288276]
- Murray RP, Bailey WC, Daniels K, Bjornson WM, Kurnow K, Connett JE, Nides MA, Kiley JP. Safety of nicotine polacrilex gum used by 3,094 participants in the Lung Health Study Research Group. Chest. 1996;109(2):438–45. [PubMed: 8620719]
- Muscat JE, Harris RE, Haley NJ, Wynder EL. Cigarette smoking and plasma cholesterol. American Heart Journal. 1991;121(1 Pt 1):141–7. [PubMed: 1985356]
- National Cancer Institute. Cigar Smoking in the United States: Health Effects and Trends, Smoking and Tobacco Control Monograph No 9. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1998. NIH Publication No 98-4302.
- National Cancer Institute. Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine, Smoking and Tobacco Control Monograph No13. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 2001. NIH Publication No 02-5047.
- National Heart.Lung, and Blood Institute. Morbidity & Mortality: 2007 Chart Book on Cardiovascular, Lung, and Blood Diseases. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Heart, Lung, and Blood Institute; Jun, 2007.
- Natori T, Sata M, Washida M, Hirata Y, Nagai R, Makuuchi M. Nicotine enhances neovascularization and promotes tumor growth. Molecules and Cells. 2003;16(2):143–6. [PubMed: 14651253]
- Neaton JD, Kuller LH, Wentworth D, Borhani NO. Total and cardiovascular mortality in relation to cigarette smoking, serum cholesterol concentration, and diastolic blood pressure among black and white males followed up for five years. American Heart Journal. 1984;108(3 Pt 2):759–69. [PubMed: 6475745]
- Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease: overall findings and differences by age for 316,099 white men. Multiple Risk Factor Intervention Trial Research Group. Archives of Internal Medicine. 1992;152(1):56–64. [PubMed: 1728930]
- Neese RA, Benowitz NL, Hoh R, Faix D, LaBua A, Pun K, Hellerstein MK. Metabolic interactions between surplus dietary energy intake and cigarette smoking or its cessation. American Journal of Physiology – Endocrinology and Metabolism. 1994;267(6 Pt 1):E1023–E1034. [PubMed: 7810617]
- Ness J, Aronow WS, Ahn C. Risk factors for symptomatic peripheral arterial disease in older persons in an academic hospital-based geriatrics practice. Journal of the American Geriatric Society. 2000;48(3):312–4. [PubMed: 10733059]
- Neunteufl T, Heher S, Kostner K, Mitulovic G, Lehr S, Khoschsorur G, Schmid RW, Maurer G, Stefenelli T. Contribution of nicotine to acute endothelial dysfunction in long-term smokers. Journal of the American College of Cardiology. 2002;39(2):251–6. [PubMed: 11788216]
- New England Journal of Medicine. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. Diabetes Control and Complications Trial Research Group. New England Journal of Medicine. 1993;329(14):977–86. [PubMed: 8366922]
- Newby DE, McLeod AL, Uren NG, Flint L, Ludlam CA, Webb DJ, Fox KA, Boon NA. Impaired coronary tissue plasminogen activator release is associated with coronary atherosclerosis and cigarette smoking: direct link between endothelial dysfunction and atherothrombosis. Circulation. 2001;103(15):1936–41. [PubMed: 11306520]
- Newby DE, Wright RA, Labinjoh C, Ludlam CA, Fox KAA, Boon NA, Webb DJ. Endothelial dysfunction, impaired endogenous fibrinolysis, and cigarette smoking: a mechanism for arterial thrombosis and myocardial infarction. Circulation. 1999;99(11):1411–5. [PubMed: 10086962]
- Nicod P, Rehr R, Winniford MD, Campbell WB, Firth BG, Hillis LD. Acute systemic and coronary hemodynamic and serologic responses to cigarette smoking in long-term smokers with atherosclerotic coronary artery disease. Journal of the American College of Cardiology. 1984;4(5):964–71. [PubMed: 6548482]
- Niedermaier ON, Smith ML, Beightol LA, Zukowska-Grojec Z, Goldstein DS, Eckberg DL. Influence of cigarette smoking on human autonomic function. Circulation. 1993;88(2):562–71. [PubMed: 8339419]
- Nilsson PM, Lind L, Pollare T, Berne C, Lithell HO. Increased level of hemoglobin A1c, but not impaired insulin sensitivity, found in hypertensive and normotensive smokers. Metabolism. 1995;44(5):557–61. [PubMed: 7752901]
- Nilsson S, Carstensen JM, Pershagen G. Mortality among male and female smokers in Sweden: a 33 year follow up. Journal of Epidemiology and Community Health. 2001;55(11):825–30. [PMC free article: PMC1763319] [PubMed: 11604439]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007;356(24):2457–71. [PubMed: 17517853]
- Nitenberg A, Antony I. Effects of nicotine gum on coronary vasomotor responses during sympathetic stimulation in patients with coronary artery stenosis. Journal of Cardiovascular Pharmacology. 1999;34(5):694–9. [PubMed: 10547086]
- Nitenberg A, Antony I, Foult J-M. Acetylcholine-induced coronary vasoconstriction in young, heavy smokers with normal coronary arteriographic findings. American Journal of Medicine. 1993;95(1):71–7. [PubMed: 8328499]
- Njølstad I, Arnesen E, Lund-Larsen PG. Smoking, serum lipids, blood pressure, and sex differences in myocardial infarction: a 12-year follow-up of the Finnmark Study. Circulation. 1996;93(3):450–6. [PubMed: 8565161]
- Nowak J, Murray JJ, Oates JA, FitzGerald GA. Biochemical evidence of a chronic abnormality in platelet and vascular function in healthy individuals who smoke cigarettes. Circulation. 1987;76(1):6–14. [PubMed: 3297389]
- Nowak P, Kolodziejczyk J, Wachowicz B. Peroxynitrite and fibrinolytic system: the effect of peroxynitrite on plasmin activity. Molecular and Cellular Biochemistry. 2004;267(1–2):141–6. [PubMed: 15663195]
- Ockene J, Kristeller JL, Goldberg R, Ockene I, Merriam P, Barrett S, Pekow P, Hosmer D, Gianelly R. Smoking cessation and severity of disease: the coronary artery smoking intervention study. Health Psychology. 1992;11(2):119–26. [PubMed: 1582380]
- Ockene JK, Kuller LH, Svendsen KH, Meilahn E. The relationship of smoking cessation to coronary heart disease and lung cancer in the Multiple Risk Factor Intervention Trial (MRFIT). American Journal of Public Health. 1990;80(8):954–8. [PMC free article: PMC1404774] [PubMed: 2368857]
- O’Connor RJ, Giovino GA, Kozlowski LT, Shiffman S, Hyland A, Bernert JT, Caraballo RS, Cummings KM. Changes in nicotine intake and cigarette use over time in two nationally representative cross-sectional samples of smokers. American Journal of Epidemiology. 2006;164(8):750–9. [PubMed: 16887891]
- Omenn GS, Anderson KW, Kronmal RA, Vlietstra RE. The temporal pattern of reduction of mortality risk after smoking cessation. American Journal of Preventive Medicine. 1990;6(5):251–7. [PubMed: 2268453]
- Ong MK, Glantz SA. Cardiovascular health and economic effects of smoke-free workplaces. American Journal of Medicine. 2004;117(1):32–8. [PubMed: 15210386]
- Otsuka R, Watanabe H, Hirata K, Tokai K, Muro T, Yoshiyama M, Takeuchi K, Yoshikawa J. Acute effects of passive smoking on the coronary circulation in healthy young adults. JAMA: the Journal of the American Medical Association. 2001;286(4):436–41. [PubMed: 11466122]
- Panagiotakos DB, Pitsavos C, Chrysohoou C, Skoumas J, Masoura C, Toutouzas P, Stefanadis C. Effect of exposure to secondhand smoke on markers of inflammation: the ATTICA study. American Journal of Medicine. 2004;116(3):145–50. [PubMed: 14749157]
- Paramo JA, Beloqui O, Roncal C, Benito A, Orbe J. Validation of plasma fibrinogen as a marker of carotid atherosclerosis in subjects free of clinical cardiovascular disease. Haematologica. 2004;89(10):1226–31. [PubMed: 15477208]
- Park YS, Kim J, Misonou Y, Takamiya R, Takahashi M, Freeman MR, Taniguchi N. Acrolein induces cyclo-oxygenase-2 and prostaglandin production in human umbilical vein endothelial cells: roles of p38 MAP kinase. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(6):1319–25. [PubMed: 17363696]
- Patsch JR, Miesenbock G, Hopferwieser T, Muhlberger V, Knapp E, Dunn JK, Gotto AM Jr, Patsch W. Relation of triglyceride metabolism and coronary artery disease: studies in the postprandial state. Arteriosclerosis and Thrombosis. 1992;12(11):1336–45. [PubMed: 1420093]
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499–511. [PubMed: 12551878]
- Pellegrini MP, Newby DE, Johnston NR, Maxwell S, Webb DJ. Vitamin C has no effect on endothelium-dependent vasomotion and acute endogenous fibrinolysis in healthy smokers. Journal of Cardiovascular Pharmacology. 2004;44(1):117–24. [PubMed: 15175566]
- Pellegrini MP, Newby DE, Maxwell S, Webb DJ. Short-term effects of transdermal nicotine on acute tissue plasminogen activator release in vivo in man. Cardiovascular Research. 2001;52(2):321–7. [PubMed: 11684081]
- Penn A, Snyder C. Arteriosclerotic plaque development is ‘promoted’ by polynuclear aromatic hydrocarbons. Carcinogenesis. 1988;9(12):2185–9. [PubMed: 3142695]
- Penn A, Snyder CA. 1,3 Butadiene, a vapor phase component of environmental tobacco smoke, accelerates arteriosclerotic plaque development. Circulation. 1996;93(3):552–7. [PubMed: 8565175]
- Perkins KA, Epstein LH, Marks BL, Stiller RL, Jacob RG. The effect of nicotine on energy expenditure during light physical activity. New England Journal of Medicine. 1989;320(14):898–903. [PubMed: 2927460]
- Perry IJ, Wannamethee SG, Walker MK, Thomson AG, Whincup PH, Shaper AG. Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men. BMJ (British Medical Journal). 1995;310(6979):560–4. [PMC free article: PMC2548938] [PubMed: 7888929]
- Persson P-G, Carlsson S, Svanström L, Östenson C-G, Efendic S, Grill V. Cigarette smoking, oral moist snuff use and glucose intolerance. Journal of Internal Medicine. 2000;248(2):103–10. [PubMed: 10947888]
- Peters RW, Brooks MM, Todd L, Liebson PR, Wilhelmsen L. Smoking cessation and arrhythmic death: the CAST experience. Journal of the American College of Cardiology. 1995;26(5):1287–92. [PubMed: 7594045]
- Petruzzelli S, Puntoni R, Mimotti P, Pulerá N, Baliva F, Fornai E, Giuntini C. Plasma 3-nitrotyrosine in cigarette smokers. American Journal of Respiratory and Critical Care Medicine. 1997;156(6):1902–7. [PubMed: 9412573]
- Pignatelli B, Li C-Q, Boffetta P, Chen Q, Ahrens W, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Fortes C, Constantinescu V, et al. Nitrated and oxidized plasma proteins in smokers and lung cancer patients. Cancer Research. 2001;61(2):778–84. [PubMed: 11212282]
- Pilz H, Oguogho A, Chehne F, Lupattelli G, Palumbo B, Sinzinger H. Quitting cigarette smoking results in a fast improvement of in vivo oxidation injury (determined via plasma, serum and urinary isoprostane). Thrombosis Research. 2000;99(3):209–21. [PubMed: 10944241]
- Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension. 1986;8(1):37–44. [PubMed: 3080370]
- Porchet HC, Benowitz NL, Sheiner LB, Copeland JR. Apparent tolerance to the acute effect of nicotine results in part from distribution kinetics. Journal of Clinical Investigation. 1987;80(5):1466–71. [PMC free article: PMC442405] [PubMed: 3680508]
- Porta M, Bandello F. Diabetic retinopathy: a clinical update. Diabetologia. 2002;45(12):1617–34. [PubMed: 12488951]
- Pozen MW, Stechmiller JA, Harris W, Smith S, Fried DD, Voigt GC. A nurse rehabilitator’s impact on patients with myocardial infarction. Medical Care. 1977;15(10):830–7. [PubMed: 909325]
- Praticò D, Reilly M, Lawson JA, FitzGerald GA. Novel indices of oxidant stress in cardiovascular disease: specific analysis of F2-isoprostanes. Agents and Actions Supplements. 1997;48:25–41. [PubMed: 9177098]
- Praticò D, Smyth EM, Viola F, FitzGerald GA. Local amplification of platelet function by 8-epi prostaglan-din F2α is not mediated by thromboxane receptor isoforms. Journal of Biological Chemistry. 1996;271(25):14916–24. [PubMed: 8663015]
- Prescott E, Scharling H, Osler M, Schnohr P. Importance of light smoking and inhalation habits on risk of myocardial infarction and all cause mortality: a 22 year follow up of 12,149 men and women in the Copenhagen City Heart Study. Journal of Epidemiology and Community Health. 2002;56(9):702–6. [PMC free article: PMC1732233] [PubMed: 12177089]
- Pretorius M, Rosenbaum DA, Lefebvre J, Vaughan DE, Brown NJ. Smoking impairs bradykinin-stimulated t-PA release. Hypertension. 2002;39(3):767–71. [PubMed: 11897760]
- Price JF, Mowbray PI, Lee AJ, Rumley A, Lowe GDO, Fowkes FGR. Relationship between smoking and cardiovascular risk factors in the development of peripheral arterial disease and coronary artery disease: Edinburgh Artery Study. European Heart Journal. 1999;20(5):344–53. [PubMed: 10206381]
- Princen HM, Van Poppel G, Vogelezang C, Buytenhek R, Kok FJ. Supplementation with vitamin E but not β-carotene in vivo protects low density lipoproteins from lipid peroxidation in vitro: effect of cigarette smoking. Arteriosclerosis and Thrombosis. 1992;12(5):554–62. [PubMed: 1576117]
- Prisco D, Fedi S, Brunelli T, Chiarugi L, Lombardi A, Gianni R, Santoro E, Cappelletti C, Pepe G, Gensini GF, et al. The influence of smoking on von Willebrand factor is already manifest in healthy adolescent females: the Floren-teen (Florence Teenager) Study. International Journal of Clinical & Laboratory Research. 1999;29(4):150–4. [PubMed: 10784376]
- Przyklenk K. Nicotine exacerbates postischemic contractile dysfunction of ‘stunned’ myocardium in the canine model: possible role of free radicals. Circulation. 1994;89(3):1272–81. [PubMed: 8124816]
- Puranik R, Celermajer DS. Smoking and endothelial function. Progress in Cardiovascular Diseases. 2003;45(6):443–58. [PubMed: 12800127]
- Qiao Q, Tervahauta M, Nissinen A, Tuomilehto J. Mortality from all causes and from coronary heart disease related to smoking and changes in smoking during a 35-year follow-up of middle-aged Finnish men. European Heart Journal. 2000;21(19):1621–6. [PubMed: 10988015]
- Quist-Paulsen P, Gallefoss F. Randomised controlled trial of smoking cessation intervention after admission for coronary heart disease. BMJ (British Medical Journal). 2003;327(7426):1254–7. [PMC free article: PMC286243] [PubMed: 14644967]
- Rabkin SW. Effect of cigarette smoking cessation on risk factors for coronary atherosclerosis: a control clinical trial. Atherosclerosis. 1984;53(2):173–84. [PubMed: 6517973]
- Ragland DR, Brand RJ. Coronary heart disease mortality in the Western Collaborative Group Study: follow-up experience of 22 years. American Journal of Epidemiology. 1988;127(3):462–75. [PubMed: 3341353]
- Rangemark C, Ciabattoni G, Wennmalm A. Excretion of thromboxane metabolites in healthy women after cessation of smoking. Arteriosclerosis and Thrombosis. 1993;13(6):777–82. [PubMed: 8499397]
- Rea TD, Heckbert SR, Kaplan RC, Smith NL, Lemaitre RN, Psaty BM. Smoking status and risk for recurrent coronary events after myocardial infarction. Annals of Internal Medicine. 2002;137(6):494–500. [PubMed: 12230350]
- Reaven GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607. [PubMed: 3056758]
- Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(9):3375–8. [PMC free article: PMC287135] [PubMed: 2497467]
- Regalado M, Yang S, Wesson DE. Cigarette smoking is associated with augmented progression of renal insufficiency in severe essential hypertension. American Journal of Kidney Diseases. 2000;35(4):687–94. [PubMed: 10739791]
- Regan TJ, Hellems HK, Bing RJ. Effect of cigarette smoking on coronary circulation and cardiac work in patients with arteriosclerotic coronary disease. Annals of the New York Academy of Sciences. 1960;190:186–9. [PubMed: 13740347]
- Reichard P. Risk factors for progression of microvascular complications in the Stockholm Diabetes Intervention Study (SDIS). Diabetes Research and Clinical Practice. 1992;16(2):151–6. [PubMed: 1600854]
- Reilly M, Delanty N, Lawson JA, FitzGerald GA. Modulation of oxidant stress in vivo in chronic cigarette smokers. Circulation. 1996;94(1):19–25. [PubMed: 8964113]
- Ridker PM, Hennekens CH, Stampfer MJ, Manson JE, Vaughan DE. Prospective study of endogenous tissue plasminogen activator and risk of stroke. Lancet. 1994;343(8903):940–3. [PubMed: 7909008]
- Rigotti NA. Treatment of tobacco use and dependence. New England Journal of Medicine. 2002;346(7):506–12. [PubMed: 11844853]
- Rigotti NA, McKool KM, Shiffman S. Predictors of smoking cessation after coronary artery bypass graft surgery: results of a randomized trial with 5-year follow-up. Annals of Internal Medicine. 1994;120(4):287–93. [PubMed: 8291821]
- Rigotti NA, Thorndike AN, Regan S, McKool K, Pasternak RC, Chang Y, Swartz S, Torres-Finnerty N, Emmons KM, Singer DE. Bupropion for smokers hospitalized with acute cardiovascular disease. American Journal of Medicine. 2006;119(12):1080–7. [PubMed: 17145253]
- Rimm EB, Chan J, Stampfer MJ. Prospective study of cigarette smoking, alcohol use, and the risk of diabetes in men. BMJ (British Medical Journal). 1995;310(6979):555–9. [PMC free article: PMC2548937] [PubMed: 7888928]
- Rimm EB, Manson JE, Stampfer MJ, Colditz GA, Willett WC, Rosner B, Hennekens CH, Speizer FE. Cigarette smoking and the risk of diabetes in women. American Journal of Public Health. 1993;83(2):211–4. [PMC free article: PMC1694562] [PubMed: 8427325]
- Rinder HM, Schuster JE, Rinder CS, Wang C, Schweidler HJ, Smith BR. Correlation of thrombosis with increased platelet turnover in thrombocytosis. Blood. 1998;91(4):1288–94. [PubMed: 9454759]
- Ronnemaa T, Ronnemaa EM, Puukka P, Pyorala K, Laakso M. Smoking is independently associated with high plasma insulin levels in nondiabetic men. Diabetes Care. 1996;19(11):1229–32. [PubMed: 8908385]
- Rose G, Hamilton PJS, Colwell L, Shipley MJ. A randomised controlled trial of anti-smoking advice: 10-year results. Journal of Epidemiology and Community Health. 1982;36(2):102–8. [PMC free article: PMC1052903] [PubMed: 7119652]
- Rosengren A, Wilhelmsen L, Wedel H. Coronary heart disease, cancer and mortality in male middle-aged light smokers. Journal of Internal Medicine. 1992;231(4):357–62. [PubMed: 1588259]
- Ross R. Atherosclerosis—an inflammatory disease. New England Journal of Medicine. 1999;340(2):115–26. [PubMed: 9887164]
- Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. American Journal of Physiology – Heart and Circulatory Physiology. 1986;250(5 Pt 2):H822–H827. [PubMed: 3010744]
- Rubenstein D, Jesty J, Bluestein D. Differences between mainstream and sidestream cigarette smoke extracts and nicotine in the activation of platelets under static and flow conditions. Circulation. 2004;109(1):78–83. [PubMed: 14691035]
- Ruggeri RB. Cholesteryl ester transfer protein: pharmacological inhibition for the modulation of plasma cholesterol levels and promising target for the prevention of atherosclerosis. Current Topics in Medicinal Chemistry. 2005;5(3):257–64. [PubMed: 15857309]
- Saareks V, Ylitalo P, Alanko J, Mucha I, Riutta A. Effects of smoking cessation and nicotine substitution on systemic eicosanoid production in man. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2001;363(5):556–61. [PubMed: 11383717]
- Salonen JT, Salonen R, Seppanen K, Rinta-Kiikka S, Kuukka M, Korpela H, Alfthan G, Kantola M, Schalch W. Effects of antioxidant supplementation on platelet function: a randomized pair-matched, placebo-controlled, double-blind trial in men with low antioxidant status. American Journal of Clinical Nutrition. 1991;53(5):1222–9. [PubMed: 1826987]
- Sambola A, Osende J, Hathcock J, Degen M, Nemerson Y, Fuster V, Crandall J, Badimon JJ. Role of risk factors in the modulation of tissue factor activity and blood thrombogenicity. Circulation. 2003;107(7):973–7. [PubMed: 12600909]
- Sarabi M, Lind L. Short-term effects of smoking and nicotine chewing gum on endothelium-dependent vasodilation in young healthy habitual smokers. Journal of Cardiovascular Pharmacology. 2000;35(3):451–6. [PubMed: 10710132]
- Sargeant LA, Khaw K-T, Bingham S, Day NE, Luben RN, Oakes S, Welch A, Wareham NJ. Cigarette smoking and glycaemia: the EPIC-Norfolk Study. International Journal of Epidemiology. 2001;30(3):547–54. [PubMed: 11416081]
- Sasaki A, Kondo K, Sakamoto Y, Kurata H, Itakua H, Ikeda Y. Smoking cessation increases the resistance of low-density lipoprotein to oxidation. Atherosclerosis. 1997;130(1–2):109–11. [PubMed: 9126654]
- Sawada M, Kishi Y, Numano F, Isobe M. Smokers lack morning increase in platelet sensitivity to nitric oxide. Journal of Cardiovascular Pharmacology. 2002;40(4):571–6. [PubMed: 12352319]
- Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899–906. [PubMed: 10779454]
- Scheffler E, Wiest E, Woehrle J, Otto I, Schulz I, Huber L, Ziegler R, Dresel HA. Smoking influences the atherogenic potential of low-density lipoprotein. Clinical Investigation. 1992;70(3–4):263–8. [PubMed: 1521040]
- Schelbert HR, Phelps ME, Hoffman EJ, Huang SC, Selin CE, Kuhl DE. Regional myocardial perfusion assessed with N-13 labeled ammonia and positron emission computerized axial tomography. American Journal of Cardiology. 1979;43(2):209–18. [PubMed: 760475]
- Schlaifer JD, Mancini GBJ, O’Neill BJ, Pitt B, Haber HE, Pepine CJ. Influence of smoking status on angiotensin-converting enzyme inhibition–related improvement in coronary endothelial function. Cardiovascular Drugs and Therapy. 1999;13(3):201–9. [PubMed: 10439882]
- Schmid P, Karanikas G, Kritz H, Pirich C, Stamatopoulos Y, Peskar BA, Sinzinger H. Passive smoking and platelet thromboxane. Thrombosis Research. 1996;81(4):451–60. [PubMed: 8907294]
- Schuller HM. Mechanisms of smoking-related lung and pancreatic adenocarcinomas development. Nature Reviews Cancer. 2002;2(6):455–63. [PubMed: 12189387]
- Scientific Advisory Committee on Tobacco Product Regulation. Recommendation on Health Claims Derived from ISO/FTC Method to Measure Cigarette Yield. Geneva: World Health Organization; 2002.
- Scott DA, Stapleton JA, Wilson RF, Sutherland G, Palmer RM, Coward PY, Gustavsson G. Dramatic decline in circulating intercellular adhesion molecule-1 concentration on quitting tobacco smoking. Blood Cells, Molecules & Diseases. 2000;26(3):255–8. [PubMed: 10950946]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA: the Journal of the American Medical Association. 1996;276(14):1141–6. [PubMed: 8827966]
- Sedgwick JB, Hwang YS, Gerbyshak HA, Kita H, Busse WW. Oxidized low-density lipoprotein activates migration and degranulation of human granulocytes. American Journal of Respiratory Cell and Molecular Biology. 2003;29(6):702–9. [PubMed: 12777245]
- Sela S, Shurtz-Swirski R, Awad J, Shapiro G, Nasser L, Shasha SM, Kristal B. The involvement of peripheral polymorphonuclear leukocytes in the oxidative stress and inflammation among cigarette smokers. Israel Medical Association Journal. 2002;4(11):1015–9. [PubMed: 12489494]
- Sexton PT, Walsh J, Jamrozik K, Parsons R. Risk factors for sudden unexpected cardiac death in Tasmanian men. Australian and New Zealand Journal of Medicine. 1997;27(1):45–50. [PubMed: 9079253]
- Shanks TG, Burns DM. Disease consequences of cigar smoking, Cigars: Health Effects and Trends. Smoking and Tobacco Control Monograph No 9. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1998. pp. 105–58. NIH Publication No. 98-4302.
- Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. Journal of Biological Chemistry. 2005;280(43):36386–96. [PubMed: 16126721]
- Shaper AG, Pocock SJ, Walker M, Phillips AN, Whitehead TP, Macfarlane PW. Risk factors for ischaemic heart disease: the prospective phase of the British Regional Heart Study. Journal of Epidemiology and Community Health. 1985;39(3):197–209. [PMC free article: PMC1052435] [PubMed: 4045359]
- Sharrett AR, Sorlie PD, Chambless LE, Folsom AR, Hutchinson RG, Heiss G, Szklo M. Relative importance of various risk factors for asymptomatic carotid atherosclerosis versus coronary heart disease incidence: the Atherosclerosis Risk in Communities Study. American Journal of Epidemiology. 1999;149(9):843–52. [PubMed: 10221321]
- Shaten BJ, Kuller LH, Neaton JD. Association between baseline risk factors, cigarette smoking, and CHD mortality after 10.5 years. Preventive Medicine. 1991;20(5):655–9. [PubMed: 1758844]
- Sheps DS, Herbst MC, Hinderliter AL, Adams KF, Ekelund LG, O’Neil JJ, Goldstein GM, Bromberg PA, Dalton JL, Ballenger MN, et al. Production of arrhythmias by elevated carboxyhemoglobin in patients with coronary artery disease. Annals of Internal Medicine. 1990;113(5):343–51. [PubMed: 2382916]
- Shimasaki Y, Yasue H, Yoshimura M, Nakayama M, Kugiyama K, Ogawa H, Harada E, Masuda T, Koyama W, Saito Y, et al. Association of the missense Glu298Asp variant of the endothelial nitric oxide synthase gene with myocardial infarction. Journal of the American College of Cardiology. 1998;31(7):1506–10. [PubMed: 9626827]
- Shin VY, Wu WK, Ye YN, So WH, Koo MW, Liu ES, Luo JC, Cho CH. Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis. 2004;25(12):2487–95. [PubMed: 15319299]
- Shinton R, Beevers G. Meta-analysis of relation between cigarette smoking and stroke. BMJ (British Medical Journal). 1989;298(6676):789–94. [PMC free article: PMC1836102] [PubMed: 2496858]
- Siekmeier R, Wulfroth P, Wieland H, Gross W, Marz W. Low-density lipoprotein susceptibility to in vitro oxidation in healthy smokers and non-smokers. Clinical Chemistry. 1996;42(4):524–30. [PubMed: 8605668]
- Silagy C, Lancaster T, Stead L, Mant D, Fowler G. Nicotine replacement therapy for smoking cessation. Cochrane Database of Systematic Reviews. 2004;3 Art. No.: CD000146. [PubMed: 15266423] [CrossRef]
- Simpson AJ, Gray RS, Moore NR, Booth NA. The effects of chronic smoking on the fibrinolytic potential of plasma and platelets. British Journal of Haematology. 1997;97(1):208–13. [PubMed: 9136967]
- Sinha RN, Patrick AW, Richardson L, Wallymahmed M, MacFarlane IA. A six-year follow-up study of smoking habits and microvascular complications in young adults with type 1 diabetes. Postgraduate Medical Journal. 1997;73(859):293–4. [PMC free article: PMC2431314] [PubMed: 9196703]
- Smith FB, Lowe GD, Fowkes FG, Rumley A, Rumley AG, Donnan PT, Housley E. Smoking, haemostatic factors and lipid peroxides in a population case control study of peripheral arterial disease. Atherosclerosis. 1993;102(2):155–62. [PubMed: 8251001]
- Smith SC, Blair SN, Bonow RO, Brass LM, Cerqueira MD, Dracup K, Fuster V, Gotto A, Grundy SM, Miller NH, et al. AHA/ACC guidelines for preventing heart attack and death in patients with atherosclerotic cardiovascular disease: 2001 update. A statement for healthcare professionals from the American Heart Association and the American College of Cardiology. Circulation. 2001;104(13):1577–9. [PubMed: 11571256]
- Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM, Walter U. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1998;358(1):134–9. [PubMed: 9721015]
- Sniderman AD, Scantlebury T, Cianflone K. Hypertriglyceridemic hyperapoB: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus. Annals of Internal Medicine. 2001;135(6):447–59. [PubMed: 11560458]
- Solberg LA, Strong JP. Risk factors and atherosclerotic lesions: a review of autopsy studies. Arteriosclerosis. 1983;3(3):187–98. [PubMed: 6342587]
- SRNT Subcommittee on Biochemical Verification. Biochemical verification of tobacco use and cessation. Nicotine & Tobacco Research. 2002;4(2):149–59. [PubMed: 12028847]
- Stamford BA, Matter S, Fell RD, Papanek P. Effects of smoking cessation on weight gain, metabolic rate, caloric consumption, and blood lipids. American Journal of Clinical Nutrition. 1986;43(4):486–94. [PubMed: 3962901]
- Stavroulakis GA, Makris TK, Hatzizacharias AN, Tsoukala C, Kyriakidis MK. Passive smoking adversely affects the haemostasis/fibrinolytic parameters in healthy non-smoker offspring of healthy smokers. Thrombosis & Haemostasis. 2000;84(5):923–4. [PubMed: 11127882]
- Stepanov I, Jensen J, Hatsukami D, Hecht SS. New and traditional smokeless tobacco: comparison of toxicant and carcinogen levels. Nicotine & Tobacco Research. 2008;10(12):1773–82. [PMC free article: PMC2892835] [PubMed: 19023828]
- Strachan DP. Predictors of death from aortic aneurysm among middle-aged men: the Whitehall Study. British Journal of Surgery. 1991;78(4):401–4. [PubMed: 2032097]
- Stratton K, Shetty P, Wallace R, Bondurant S, editors. Clearing the Smoke: Assessing the Science Base for Tobacco Harm Reduction. Washington: National Academy Press; 2001. [PubMed: 25057541]
- Strong JP, Richards ML. Cigarette smoking and atherosclerosis in autopsied men. Atherosclerosis. 1976;23(3):451–76. [PubMed: 1267863]
- Stubbe I, Eskilsson J, Nilsson-Ehle P. High-density lipoprotein concentrations increase after stopping smoking. BMJ (British Medical Journal). 1982;284(6328):1511–3. [PMC free article: PMC1498412] [PubMed: 6805587]
- Substance Abuse and Mental Health Services Administration. Results from the 2008 National Survey on Drug Use & Health: National Findings NSDUH Series H-36 Rockville (MD): US Department of Health and Human Services. Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2009. HHS Publication No SMA 09-4434.
- Suñer IJ, Espinosa-Heidmann DG, Marin-Castano ME, Hernandez EP, Pereira-Simon S, Cousins SW. Nicotine increases size and severity of experimental choroidal neovascularization. Investigative Ophthalmology & Visual Science. 2004;45(1):311–7. [PubMed: 14691189]
- Suskin N, Sheth T, Negassa A, Yusuf S. Relationship of current and past smoking to mortality and morbidity in patients with left ventricular dysfunction. Journal of the American College of Cardiology. 2001;37(6):1677–82. [PubMed: 11345383]
- Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101(9):948–54. [PubMed: 10704159]
- Sydow K, Münzel T. ADMA and oxidative stress. Atherosclerosis Supplements. 2003;(4):41–51. [PubMed: 14664902]
- Szadkowski A, Myers CR. Acrolein oxidizes the cytosolic and mitochondrial thioredoxins in human endothelial cells. Toxicology. 2008;243:1–2. 164–76. [PMC free article: PMC2220080] [PubMed: 18023956]
- Takajo Y, Ikeda H, Haramaki N, Murohara T, Imaizumi T. Augmented oxidative stress of platelets in chronic smokers: mechanisms of impaired platelet-derived nitric oxide bioactivity and augmented platelet aggregability. Journal of American College of Cardiology. 2001;38(5):1320–7. [PubMed: 11691502]
- Tall AR. An overview of reverse cholesterol transport. European Heart Journal. 1998;19(Suppl A):A31–A35. [PubMed: 9519340]
- Tanne D, Yaari S, Goldbourt U. Risk profile and prediction of long-term ischemic stroke mortality: a 21-year follow-up in the Israeli Ischemic Heart Disease (IIHD) Project. Circulation. 1998;98(14):1365–71. [PubMed: 9760289]
- Tanus-Santos JE, Toledo JCY, Cittadino M, Sabha M, Rocha JC, Moreno H Jr. Cardiovascular effects of trans-dermal nicotine in mildly hypertensive smokers. American Journal of Hypertension. 2001;14(7 Pt 1):610–4. [PubMed: 11465642]
- Targher G, Alberiche M, Zenere MB, Bonadonna RC, Muggeo M, Bonora E. Cigarette smoking and insulin resistance in patients with noninsulin-dependent diabetes mellitus. Journal of Clinical Endocrinology and Metabolism. 1997;82(11):3619–24. [PubMed: 9360516]
- Taylor CB, Houston-Miller N, Killen JD, DeBusk RF. Smoking cessation after acute myocardial infarction: effects of a nurse-managed intervention. Annals of Internal Medicine. 1990;113(2):118–23. [PubMed: 2360750]
- Teo KK, Ounpuu S, Hawken S, Pandey MR, Valentin V, Hunt D, Diaz R, Rashed W, Freeman R, Jiang L, et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: a case-control study. Lancet. 2006;368(9536):647–58. [PubMed: 16920470]
- Thomas AE, Green FR, Lamlum H, Humphries SE. The association of combined alpha and beta fibrinogen genotype on plasma fibrinogen levels in smokers and non-smokers. Journal of Medical Genetics. 1995;32(8):585–9. [PMC free article: PMC1051629] [PubMed: 7473646]
- Thomson CC, Rigotti NA. Hospital and clinic-based smoking cessation interventions for smokers with cardiovascular disease. Progress in Cardiovascular Diseases. 2003;45(6):459–79. [PubMed: 12800128]
- Thun MJ, Myers DG, Day-Lally C, Namboodin MM, Calle EE, Flanders WD, Adams SL, Heath CW Jr. Age and the exposure-response relationships between cigarette smoking and premature death in Cancer Prevention Study II, Changes in Cigarette-Related Disease Risks and Their Implications for Prevention and Control. Smoking and Tobacco Control Monograph No 8. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1997. pp. 383–475. NIH Publication No. 97-4213.
- Thyberg J. Effects of nicotine on phenotypic modulation and initiation of DNA synthesis in cultured arterial smooth muscle cells. Virchows Archiv B, Cell Pathology Including Molecular Pathology. 1986;52(1):25–32. [PubMed: 2881392]
- Tomlinson DR. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42(11):1271–81. [PubMed: 10550410]
- Tonstad S, Farsang C, Klaene G, Lewis K, Manolis A, Perruchoud AP, Silagy C, van Spiegel PI, Astbury C, Hider A, et al. Bupropion SR for smoking cessation in smokers with cardiovascular disease: a multicenter randomized study. European Heart Journal. 2003;24(10):946–55. [PubMed: 12714026]
- Tonstad S, Tønnesen P, Hajek P, Williams KE, Billing CB, Reeves KR. Varenicline Phase 3 Study Group. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA: the Journal of the American Medical Association. 2006;296(1):64–71. [PubMed: 16820548]
- Tracy RP, Psaty BM, Macy E, Bovill EG, Cushman M, Cornell ES, Kuller LH. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(10):2167–76. [PubMed: 9351386]
- Tran CTL, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atherosclerosis Supplements. 2003;4(4):33–40. [PubMed: 14664901]
- Tsao PS, Buitrago R, Chan JR, Cooke JP. Fluid flow inhibits endothelial adhesiveness: nitric oxide and transcriptional regulation of VCAM-1. Circulation. 1996;94(7):1682–9. [PubMed: 8840861]
- Tsao PS, McEvoy LM, Drexler H, Butcher EC, Cooke JP. Enhanced endothelial adhesiveness in hypercholesterolemia is attenuated by l-arginine. Circulation. 1994;89(5):2176–82. [PubMed: 8181143]
- Tsao PS, Wang BY, Buitrago R, Shyy JY-J, Cooke JP. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96(3):934–40. [PubMed: 9264504]
- Tsukada T, Yokoyama K, Arai T, Takemoto F, Hara S, Yamada A, Kawaguchi Y, Hosoya T, Igari J. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochemical and Biophysical Research Communication. 1998;245(1):190–3. [PubMed: 9535806]
- Turner RC, Millns H, Neil HAW, Stratton IM, Manley SE, Matthews DR, Holman RR. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ (British Medical Journal). 1998;316(7134):823–8. [PMC free article: PMC28484] [PubMed: 9549452]
- Tuut M, Hense H-W. Smoking, other risk factors and fibrinogen levels: evidence of effect modification. Annals of Epidemiology. 2001;11(4):232–8. [PubMed: 11306341]
- Tverdal A. Calculation of risk for the development of acute myocardial infarction in the normal population based on long-term follow-up studies: smokers compared with non-smokers. Journal of Cardiovascular Risk. 1999;6(5):287–91. [PubMed: 10534129]
- Tzivoni D, Keren A, Meyler S, Khoury Z, Lerer T, Brunel P. Cardiovascular safety of transdermal nicotine patches in patients with coronary artery disease who try to quit smoking. Cardiovascular Drugs and Therapy. 1998;12(3):239–44. [PubMed: 9784902]
- Uchimoto S, Tsumura K, Hayashi T, Suematsu C, Endo G, Fujii S, Okada K. Impact of cigarette smoking on the incidence of type 2 diabetes mellitus in middle-aged Japanese men: the Osaka Health Survey. Diabetic Medicine. 1999;16(11):951–5. [PubMed: 10588526]
- US Department of Health and Human Services. The Health Consequences of Smoking: Cardiovascular Disease A Report of the Surgeon General. Rockville (MD): U.S. Department of Health and Human Services, Public Health Service, Office on Smoking and Health; 1983. DHHS Publication No. (PHS) 84-50204.
- US Department of Health and Human Services. The Health Consequences of Smoking: Nicotine Addiction A Report of the Surgeon General. Atlanta: US Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 1988. DHHS Publication No. (CDC) 88-8406.
- US Department of Health and Human Services. The Health Benefits of Smoking Cessation A Report of the Surgeon General. Atlanta: US Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 1990. DHHS Publication No. (CDC) 90-8416.
- US Department of Health and Human Services. Reducing Tobacco Use A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2000.
- US Department of Health and Human Services. Women and Smoking: A Report of the Surgeon General. Rockville (MD): U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General; 2001. pp. 272–307.
- US Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004.
- US Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006.
- US Department of Health, Education, and Welfare. The Health Consequences of Smoking: A Report of the Surgeon General: 1971. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, Health Services and Mental Health Administration; 1971. DHEW Publication No. (HSM) 71-7513.
- US Department of Health, Education, and Welfare. Smoking and Health A Report of the Surgeon General. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, Office of the Assistant Secretary for Health, Office on Smoking and Health; 1979. DHEW Publication No. (PHS) 79-50066.
- U.S. Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Annals of Internal Medicine. 2005;142(3):198–202. [PubMed: 15684208]
- Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, Torbet J, Vilaire G, Bennett JS, Murciano J-C, et al. Prothrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. Journal of Biological Chemistry. 2004;279(10):8820–6. [PubMed: 14681238]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2(8670):997–1000. [PubMed: 2572793]
- Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339(8793):572–5. [PubMed: 1347093]
- van den Berkmortel FWJP, Demacker PNM, Wollersheim H, Thien T, Stalenhoef AFH. Smoking or its cessation does not alter the susceptibility to in vitro LDL oxidation. European Journal of Clinical Investigation. 2000;30(11):972–9. [PubMed: 11114959]
- van Domburg RT, Meeter K, van Berkel DFM, Veldkamp RF, van Herwerden LA, Bogers AJJC. Smoking cessation reduces mortality after coronary artery bypass surgery: a 20-year follow-up study. Journal of the American College of Cardiology. 2000;36(3):878–83. [PubMed: 10987614]
- Vanhoutte PM, Feletou M, Taddei S. Endothelium-dependent contractions in hypertension. British Journal of Pharmacology. 2005;144(4):449–58. [PMC free article: PMC1576026] [PubMed: 15655530]
- Vardulaki KA, Walker NM, Day NE, Duffy SW, Ashton HA, Scott RA. Quantifying the risks of hypertension, age, sex and smoking in patients with abdominal aortic aneurysm. British Journal of Surgery. 2000;87(2):195–200. [PubMed: 10671927]
- Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circulation Research. 2001;89(1):E1–E7. [PubMed: 11440984]
- Vayá A, Mira Y, Martínez M, Villa P, Ferrando F, Estellés A, Corella D, Aznar J. Biological risk factors for deep vein thrombosis. Clinical Hemorheology and Microcirculation. 2002;26(1):41–53. [PubMed: 11904470]
- Vermylen J, Nemmar A, Nemery B, Hoylaerts MF. Ambient air pollution and acute myocardial infarction. Journal of Thrombosis and Haemostasis. 2005;3(9):1955–61. [PubMed: 16102102]
- Villablanca AC. Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. Journal of Applied Physiology. 1998;84(6):2089–98. [PubMed: 9609804]
- Villablanca AC, McDonald JM, Rutledge JC. Smoking and cardiovascular disease. Clinics in Chest Medicine. 2000;21(1):159–72. [PubMed: 10763097]
- Vlietstra RE, Kronmal RA, Oberman A, Frye RL, Killip T III. Effect of cigarette smoking on survival of patients with angiographically documented coronary artery disease: report from the CASS registry. JAMA: the Journal of the American Medical Association. 1986;255(8):1023–7. [PubMed: 3945013]
- von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, Dzau VJ. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(4):1137–41. [PMC free article: PMC42653] [PubMed: 7532305]
- Walker JM, Cove DH, Beevers DG, Dodson PM, Leatherdale BA, Fletcher RF, Wright AD. Cigarette smoking, blood pressure and the control of blood glucose in the development of diabetic retinopathy. Diabetes Research (Edinburgh, Scotland). 1985;2(4):183–6. [PubMed: 4085172]
- Wallenfeldt K, Hulthe J, Bokemark L, Wikstrand J, Fagerberg B. Carotid and femoral atherosclerosis, cardiovascular risk factors and C-reactive protein in relation to smokeless tobacco use or smoking in 58-year-old men. Journal of Internal Medicine. 2001;250(6):492–501. [PubMed: 11902817]
- Walsh CH, Wright AD, Allbutt E, Pollock A. The effect of cigarette smoking on blood sugar, serum insulin and non esterified fatty acids in diabetic and non diabetic subjects. Diabetologia. 1977;13(5):491–4. [PubMed: 908473]
- Wang B-Y, Ho H-K, Lin PS, Schwarzacher SP, Pollman MJ, Gibbons GH, Tsao PS, Cooke JP. Regression of atherosclerosis: role of nitric oxide and apoptosis. Circulation. 1999;99(9):1236–41. [PubMed: 10069793]
- Wang W, Basinger A, Neese RA, Shane B, Myong S-A, Christiansen M, Hellerstein MK. Effect of nicotinic acid administration on hepatic very low density lipoprotein-triglyceride production. American Journal of Physiology – Endocrinology and Metabolism. 2001;280(3):E540–E547. [PubMed: 11171611]
- Wang XL, Raveendran M, Wang J. Genetic influence on cigarette-induced cardiovascular disease. Progress in Cardiovascular Diseases. 2003;45(5):361–82. [PubMed: 12704594]
- Wang XL, Sim AS, Badenhop RF, McCredie RM, Wilcken DE. A smoking-dependent risk of coronary artery disease associated with a polymorphism of the endothelial nitric oxide synthase gene. Nature Medicine. 1996;2(1):41–5. [PubMed: 8564837]
- Wannamethee G, Shaper AG, Macfarlane PW, Walker M. Risk factors for sudden cardiac death in middle-aged British men. Circulation. 1995;91(6):1749–56. [PubMed: 7882483]
- Wannamethee SG, Shaper AG, Perry IJ. Smoking as a modifiable risk factor for type 2 diabetes in middle-aged men. Diabetes Care. 2001;24(9):1590–5. [PubMed: 11522704]
- Wannamethee SG, Shaper AG, Walker M, Ebrahim S. Lifestyle and 15-year survival free of heart attack, stroke, and diabetes in middle-aged British men. Archives of Internal Medicine. 1998;158(22):2433–40. [PubMed: 9855381]
- Watts DT. The effect of nicotine and smoking on the secretion of epinephrine. Annals of the New York Academy of Sciences. 1960;90:74–80. [PubMed: 13783473]
- Weber C, Erl W, Weber K, Weber PC. Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation. 1996;93(8):1488–92. [PubMed: 8608614]
- Weir JM, Dunn JE Jr. Smoking and mortality: a prospective study. Cancer. 1970;25(1):105–12. [PubMed: 5410301]
- Weiss NS. The value of roentgenographic abdominal aortic calcification in predicting site of occlusion in arteriosclerosis obliterans. Angiology. 1972;23(3):136–9. [PubMed: 5018613]
- Wennmalm Å, Benthin G, Granström EF, Persson L, Petersson AS, Winell S. Relation between tobacco use and urinary excretion of thromboxane A2 and prostacyclin metabolites in young men. Circulation. 1991;83(5):1698–704. [PubMed: 2022025]
- Westerman RA, Lindblad LE, Wajnblum D, Roberts RG, Delaney CA. Confounding factors in non-invasive tests of neurovascular function in diabetes mellitus. Clinical and Experimental Neurology. 1992;29:149–60. [PubMed: 1343858]
- Whincup PH, Gilg JA, Emberson JR, Jarvis MJ, Feyerabend C, Bryant A, Walker M, Cook DG. Passive smoking and risk of coronary heart disease and stroke: prospective study with cotinine measurement. BMJ (British Medical Journal). 2004;329(7459):200–5. [PMC free article: PMC487731] [PubMed: 15229131]
- White AR, Rampes H, Ernst E. Acupuncture for smoking cessation. Cochrane Database of Systematic Reviews. 2002;(2) Art. No.: CD000009. [PubMed: 12076375] [CrossRef]
- Wilkins JN, Carlson HE, Van Vunakis H, Hill MA, Gritz E, Jarvik ME. Nicotine from cigarette smoking increases circulating levels of cortisol, growth hormone, and prolactin in male chronic smokers. Psychopharmacology. 1982;78(4):305–8. [PubMed: 6818588]
- Will JC, Galuska DA, Ford ES, Mokdad A, Calle EE. Cigarette smoking and diabetes mellitus: evidence of a positive association from a large prospective cohort study. International Journal of Epidemiology. 2001;30(3):540–6. [PubMed: 11416080]
- Willett WC, Green A, Stampfer MJ, Speizer FE, Colditz GA, Rosner B, Monson RR, Stason W, Hennekens CH. Relative and absolute excess risks of coronary heart disease among women who smoke cigarettes. New England Journal of Medicine. 1987;317(21):1303–9. [PubMed: 3683458]
- Willi C, Bodenmann P, Ghali WA, Faris PD, Cornuz J. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA: the Journal of the American Medical Association. 2007;298(22):2654–64. [PubMed: 18073361]
- Wilmink TBM, Quick CRG, Day NE. The association between cigarette smoking and abdominal aortic aneurysms. Journal of Vascular Surgery. 1999;30(6):1099–105. [PubMed: 10587395]
- Wilson K, Gibson N, Willan A, Cook D. Effect of smoking cessation on mortality after myocardial infarction: meta-analysis of cohort studies. Archives of Internal Medicine. 2000;160(7):939–44. [PubMed: 10761958]
- Wilson PW, Anderson KM, Kannel WB. Epidemiology of diabetes mellitus in the elderly: the Framingham Study. American Journal of Medicine. 1986;80(5A):3–9. [PubMed: 3706388]
- Wilson PW, Garrison RJ, Abbott RD, Castelli WP. Factors associated with lipoprotein cholesterol levels: the Framingham Study. Arteriosclerosis. 1983;3(3):273–81. [PubMed: 6573878]
- Wilt TJ, Davis BR, Meyers DG, Rouleau J-L, Sacks FM. Prevalence and correlates of symptomatic peripheral atherosclerosis in individuals with coronary heart disease and cholesterol levels less than 240 mg/dL: baseline results from the Cholesterol and Recurrent Events (CARE) Study. Angiology. 1996;47(6):533–41. [PubMed: 8678327]
- Winniford MD, Jansen DE, Reynolds GA, Apprill P, Black WH, Hillis LD. Cigarette smoking-induced coronary vasoconstriction in atherosclerotic coronary artery disease and prevention by calcium antagonists and nitroglycerin. American Journal of Cardiology. 1987;59(4):203–7. [PubMed: 3101478]
- Wiseman S, Kenchington G, Dain R, Marshall CE, McCollum CN, Greenhalgh RM, Powell JT. Influence of smoking and plasma factors on patency of femoropopliteal vein grafts. BMJ (British Medical Journal). 1989;299(6700):643–6. [PMC free article: PMC1837588] [PubMed: 2508847]
- Wolf PA, D’Agostino RB, Kannel WB, Bonita R, Belanger AJ. Cigarette smoking as a risk factor for stroke: the Framingham Study. JAMA: the Journal of the American Medical Association. 1988;259(7):1025–9. [PubMed: 3339799]
- Wolinsky H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circulation Research. 1980;47(3):301–11. [PubMed: 6996862]
- Woodward M, Lowe GDO, Rumley A, Tunstall-Pedoe H. Fibrinogen as a risk factor for coronary heart disease and mortality in middle-aged men and women: the Scottish Heart Health Study. European Heart Journal. 1998;19(1):55–62. [PubMed: 9503176]
- Woodward M, Rumley A, Tunstall-Pedoe H, Lowe GD. Does sticky blood predict a sticky end: associations of blood viscosity, haematocrit and fibrinogen with mortality in the West of Scotland. British Journal of Haematology. 2003;122(4):645–50. [PubMed: 12899720]
- Working Group for the Study of Transdermal Nicotine in Patients with Coronary Artery Disease. Nicotine replacement therapy for patients with coronary artery disease. Archives of Internal Medicine. 1994;154(9):989–95. [PubMed: 8179456]
- Wray R, DePalma RG, Hubay CH. Late occlusion of aortofemoral bypass grafts: influence of cigarette smoking. Surgery. 1971;70(6):969–73. [PubMed: 5124675]
- Wright HP. Mitosis patterns in aortic endothelium. Atherosclerosis. 1972;15(1):93–100. [PubMed: 5013281]
- Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu X-P, Rajavashisth TB, Yano J, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104(25):3103–8. [PubMed: 11748108]
- Yamaguchi Y, Haginaka J, Morimoto S, Fujioka Y, Kunitomo M. Facilitated nitration and oxidation of LDL in cigarette smokers. European Journal of Clinical Investigation. 2005;35(3):186–93. [PubMed: 15733073]
- Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Fluvastatin reduces modification of low-density lipoprotein in hyperlipidemic rabbit loaded with oxidative stress. European Journal of Pharmacology. 2002;436(1–2):97–105. [PubMed: 11834252]
- Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Peroxynitrite-mediated oxidative modification of low-density lipoprotein by aqueous extracts of cigarette smoke and the preventive effect of fluvastatin. Atherosclerosis. 2004;172(2):259–65. [PubMed: 15019535]
- Yoshimura M, Yasue H, Nakayama M, Shimasaki Y, Sumida H, Sugiyama S, Kugiyama K, Ogawa H, Ogawa Y, Saito Y, et al. A missense Glu298Asp variant in the endothelial nitric oxide synthase gene is associated with coronary spasm in the Japanese. Human Genetics. 1998;103(1):65–9. [PubMed: 9737779]
- Zaratin ÁCM, Quintão ECR, Sposito AC, Nunes VS, Lottenberg AM, Morton RE, de Faria EC. Smoking prevents the intravascular remodeling of high-density lipoprotein particles: implications for reverse cholesterol transport. Metabolism. 2004;53(7):858–62. [PubMed: 15254877]
- Zavaroni I, Bonini L, Gasparini P, Dall’Aglio E, Passeri M, Reaven GM. Cigarette smokers are relatively glucose intolerant, hyperinsulinemic and dyslipidemic. American Journal of Cardiology. 1994;73(12):904–5. [PubMed: 8184821]
- Zeiher AM, Schächinger V, Minners J. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation. 1995;92(5):1094–100. [PubMed: 7648652]
- Zhang S, Day I, Ye S. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis. 2001;154(2):277–83. [PubMed: 11166759]
- Zhu BQ, Heeschen C, Sievers RE, Karliner JS, Parmley WW, Glantz SA, Cooke JP. Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell. 2003;4(3):191–6. [PubMed: 14522253]
- Zidovetzki R, Chen P, Fisher M, Hofman FM. Nicotine increases plasminogen activator inhibitor-1 production by human brain endothelial cells via protein kinase C–associated pathway. Stroke. 1999;30(3):651–5. [PubMed: 10066866]
- Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60(3):473–85. [PubMed: 222498]
Footnotes
- 1
Person-year = the sum of the number of years that each member of a population has been smoking.
- Introduction
- Tobacco Use and Cardiovascular Disease
- Secondhand Tobacco Smoke and Cardiovascular Disease
- Pathophysiology
- Hemodynamic Effects
- Smoking and the Endothelium
- Thrombogenic Effects
- Inflammation
- Smoking and Diabetes
- Lipid Abnormalities
- Cardiovascular Biomarkers
- Smoking Cessation and Cardiovascular Disease
- Methods to Reduce Exposure
- Evidence Summary
- Conclusions
- References
- Cardiovascular Diseases - How Tobacco Smoke Causes Disease: The Biology and Beha...Cardiovascular Diseases - How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...