KCNT1-Related Epilepsy
Tracy Gertler, MD, PhD, David Bearden, MD, MSCE, Arin Bhattacharjee, PhD, and Gemma Carvill, PhD.
Author Information and AffiliationsInitial Posting: September 20, 2018.
Estimated reading time: 23 minutes
Summary
Clinical characteristics.
KCNT1-related epilepsy is most often associated with two phenotypes: epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE).
EIMFS is characterized by seizures, typically focal and asynchronous, beginning in the first six months of life with associated developmental plateau or regression. Autonomic manifestations (e.g., perioral cyanosis, flushing, apnea) are common. Seizures are intractable to multiple anticonvulsants and progress to become nearly continuous by age six to nine months.
ADNFLE is characterized by clusters of nocturnal motor seizures that vary from simple arousals to hyperkinetic events with tonic or dystonic features. Individuals with KCNT1-related ADNFLE are more likely to develop seizures at a younger age, have cognitive comorbidity, and display psychiatric and behavioral problems than individuals with ADNFLE resulting from other causes.
Less common seizure phenotypes in individuals with KCNT1-related epilepsy include West syndrome, Ohtahara syndrome, early myoclonic encephalopathy, leukodystrophy and/or leukoencephalopathy, focal epilepsy, and multifocal epilepsy. Additional neurologic features include hypotonia, microcephaly developing by age 12 months, strabismus, profound developmental delay, and additional movement disorders. Other systemic manifestations including pulmonary hemorrhage caused by prominent systemic-to-pulmonary collateral arteries or cardiac arrhythmia have been reported.
Diagnosis/testing.
The diagnosis of KCNT1-related epilepsy is established in a proband with intractable epilepsy and a heterozygous pathogenic variant in KCNT1 identified by molecular genetic testing.
Management.
Treatment of manifestations: KCNT1-related epilepsy is often refractory to conventional anticonvulsants; stiripentol, benzodiazepines, levetiracetam, and the ketogenic diet have all been well tolerated with limited success; quinidine has been used as an off-label anticonvulsant with success in some individuals; in rare cases of pulmonary hemorrhage as a result of systemic pulmonary collaterals, embolization has been recommended; developmental support is appropriate.
Surveillance: EEG at intervals determined by seizure frequency and progression, for evaluation of new involuntary movements or unexplained, paroxysmal changes in vital signs, or following adjustments to an anticonvulsant regimen; monitoring of development.
Agents/circumstances to avoid: For individuals with ADNFLE, activities in which a sudden loss of consciousness could lead to injury or death should be avoided (e.g., bathing, swimming, driving, or working/playing at heights).
Pregnancy management: For women with ADNFLE, a discussion of the risks and benefits of using a given anti-seizure medication during pregnancy should ideally take place before conception. Transitioning to a lower-risk medication prior to pregnancy may be possible.
Genetic counseling.
KCNT1-related epilepsy is inherited in an autosomal dominant manner. The majority of affected individuals represent simplex cases (i.e., a single occurrence in a family) resulting from a de novo KCNT1 pathogenic variant. The proportion of cases caused by a de novo pathogenic variant varies by phenotype. All individuals diagnosed with KCNT1-related epilepsy of infancy with migrating focal seizures (EIMFS) have the disorder as the result of a de novo pathogenic variant or an inherited variant from an unaffected parent with somatic and/or germline mosaicism. Some individuals diagnosed with KCNT1-related autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) have an affected parent. Each child of an individual with KCNT1-related epilepsy has a 50% chance of inheriting the pathogenic variant, and intrafamilial clinical variability and reduced penetrance have been reported. Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible if the pathogenic variant in the family is known.
Diagnosis
No formal diagnostic criteria for KCNT1-related epilepsy have been published to date.
KCNT1-related epilepsy is most often associated with two phenotypes: epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). Less often, KCNT1 pathogenic variants are associated with epilepsy with variable presentation.
Suggestive Findings
KCNT1-related epilepsy of infancy with migrating focal seizures (EIMFS) should be suspected in individuals with the following history and findings:
Normal prenatal course and birth without history, clinical features, or imaging suggestive of traumatic, anoxic, vascular, or infectious injury
Sporadic, asynchronous focal seizures arising independently from either hemisphere with patterns of intracortical "migration" occurring by age six months, with subsequent escalation of seizure frequency
Developmental plateau or regression following the onset of seizures
Intractability to anticonvulsant medication
KCNT1-related autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) should be suspected in individuals with the following history and findings:
Frequent brief, nocturnal seizures
Mild-to-moderate intellectual disability
Psychiatric disease (e.g., depression, anxiety, suicidality, attention-deficit/hyperactivity disorder)
Family history of ADNFLE or EIMFS
KCNT1-related epilepsy has been less frequently identified in individuals with the following phenotypes:
West syndrome
Ohtahara syndrome (early-infantile epileptic encephalopathy)
Early myoclonic encephalopathy
Leukodystrophy/leukoencephalopathy
Focal epilepsy
Multifocal epilepsy
Establishing the Diagnosis
The diagnosis of KCNT1-related epilepsy is established in a proband with intractable epilepsy and a heterozygous pathogenic (or likely pathogenic) variant in KCNT1 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous KCNT1 variant of uncertain significance does not establish or rule out the diagnosis.
Because the phenotype of KCNT1-related epilepsy is indistinguishable from many other inherited disorders with epilepsy, recommended molecular genetic testing approaches include use of a multigene panel or comprehensive genomic testing.
Note: (1) Single-gene testing (sequence analysis of KCNT1) is rarely useful and typically NOT recommended. (2) KCNT1-related epilepsy is postulated to occur through a gain-of-function mechanism. Large intragenic deletions and duplication have not been reported; testing for intragenic deletions or duplication is not indicated.
A seizure
multigene panel that includes KCNT1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is another good option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in KCNT1-Related Epilepsy
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
KCNT1
| Sequence analysis 3 | 100% 4 |
| Gene-targeted deletion/duplication analysis 5 | None reported 4, 6 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
Clinical Characteristics
Clinical Description
KCNT1-related epilepsy encompasses a range of epilepsy syndromes. The most common phenotypes reported in individuals with KCNT1-related epilepsy are epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE).
Epilepsy Phenotype
EIMFS is an early-infantile epileptic encephalopathy characterized by seizures beginning in the first six months of life with associated developmental plateau or regression. The seizures are primarily focal motor, variably with secondary generalization, but also include tonic, clonic, tonic-clonic, myoclonic, and epileptic spasms [McTague et al 2013]. Autonomic manifestations (e.g., perioral cyanosis, flushing, apnea) are common. Seizures progress to become nearly continuous by age six to nine months. Seizures are intractable to multiple anticonvulsants. Rarely, status epilepticus at onset has been described [Zamponi et al 2008]. The characteristic feature on EEG is focal ictal discharges that migrate across contiguous cortical regions and arise independently at multiple foci. An increase in amplitude and frontal predominance over time with post-ictal and interictal suppression has been noted [McTague et al 2018].
Additional neurologic features reported in individuals with KCNT1-related EIMFS include hypotonia (axial>appendicular), decreased head growth with microcephaly developing by age 12 months, strabismus, and profound developmental delay with rare ability to ambulate or verbalize. Additional reported movement disorders include choreoathetosis, dyskinesias, and focal and generalized dystonia.
Prognosis for individuals with KCNT1-related EIMFS is currently unknown.
ADNFLE is characterized by clusters of nocturnal motor seizures that vary from simple arousals to hyperkinetic events with tonic or dystonic features (see Autosomal Dominant Nocturnal Frontal Lobe Epilepsy). Individuals with KCNT1-related ADNFLE are more likely to develop seizures before adolescence, have cognitive comorbidity, and display psychiatric and behavioral problems than are individuals with ADNFLE resulting from other causes.
Less common epilepsy phenotypes in individuals with a KCNT1 pathogenic variant include:
West syndrome
Ohtahara syndrome (early-infantile epileptic encephalopathy)
Early myoclonic encephalopathy
Leukodystrophy/leukoencephalopathy
Focal or multifocal epilepsy
Brain MRI and/or CT examination is often normal prior to seizure onset, though recent studies have noted variable delayed myelination, hippocampal volume loss, and cerebellar atrophy [McTague et al 2018]. Temporal lobe pathology as a cause versus consequence has been noted in two individuals with KCNT1-related temporal lobe epilepsy [Hansen et al 2017].
Other. Prenatal history, birth, and neonatal history prior to seizure onset are normal, with no notable dysmorphic features.
Pulmonary Hemorrhage
Three individuals with KCNT1-related EIMFS were reported to have prominent systemic-to-pulmonary collateral artery formation and subsequent pulmonary hemorrhage that developed between age four and 19 months [Kawasaki et al 2017]. Evaluation for pulmonary hemorrhage should be considered if an individual develops acute respiratory failure, heart failure, or hemoptysis.
Cardiac Arrhythmia
Brugada syndrome was reported in one individual with a de novo KCNT1 variant [Juang et al 2014]. An individual with confirmed familial KCNT1-related epilepsy and an unspecified cardiac arrhythmia was reported by Møller et al [2015].
Genotype-Phenotype Correlations
There is some evidence for a genotype-phenotype correlation. However, disparate phenotypes (e.g., ADNFLE, EIMFS) have been identified in family members with the same pathogenic variant.
EIMFS. The majority of pathogenic variants associated with EIMFS occur in either the S5 transmembrane domain or the regulator of potassium conductance domains within the C-terminus.
ADNFLE-related pathogenic variants are concentrated in the NAD+ binding domain or more distal C-terminus.
Specific correlations between genetic variant and seizure burden, developmental impairment, or medication responsiveness have not yet been elucidated.
Nomenclature
In the initial description of EIMFS, Coppola et al [1995] described his cohort of globally arrested infants with frequent focal, "migrating" seizures that were medically intractable as malignant migrating partial seizures of infancy (MMPSI); it has also been variably referred to as migrating partial epilepsy of infancy (MPEI). In 2010, the International League Against Epilepsy reclassified this epilepsy syndrome as EIMFS [Berg et al 2010].
Prevalence
The prevalence of KCNT1-related epilepsy is unknown. To date, 88 probands with KCNT1-related epilepsy have been reported in the literature.
Differential Diagnosis
Phenotypic and EEG features associated with KCNT1 pathogenic variants are not sufficient to diagnose KCNT1-related epilepsy. All genes known to be associated with early-infantile epileptic encephalopathy (>30 have been identified; see OMIM Phenotypic Series) should be included in the differential diagnosis of KCNT1-related epilepsy including other genes less commonly associated with epilepsy of infancy with migrating focal seizures (SCN1A, SCN2A, SLC12A5, SLC25A22, TBC1D4, PLCB1) and autosomal dominant nocturnal frontal lobe epilepsy (CHRNA4, CHRNB2, DEPDC5, CRH).
Note: At seizure onset, it is most important to distinguish KCNT1-related epilepsy from potentially treatable causes of early infantile-onset epileptic encephalopathy, such as neurometabolic disorders, CNS infection, structural brain lesions, and other syndromes (see Table 2).
Table 2.
Treatable Disorders Associated with Early Infantile-Onset Epileptic Encephalopathy
View in own window
| Conditions | Gene(s) | MOI | Clinical Findings | Treatment |
|---|
Neuro-
metabolic
disorders
|
Pyridoxine-dependent epilepsy
|
ALDH7A1
| AR |
| Seizures/encephalopathy responsive to pyridoxine |
| Pyridoxamine 5'-phosphate oxidase deficiency (OMIM 610090) |
PNPO
| AR |
Lactic acidemia Hypoglycemia
| Seizures/encephalopathy responsive to pyridoxal 5-prime phosphate |
|
Biotinidase deficiency
|
BTD
| AR |
Deficient biotinidase enzyme activity in serum or plasma Ketolactic acidosis, organic aciduria, hyperammonemia Skin rash, alopecia, recurrent viral or fungal infections
| Lifelong biotin supplementation |
|
Glucose transporter 1 deficiency syndrome
|
SLC2A1
| AD
AR |
| Ketogenic diet |
|
Creatine deficiency syndromes
|
GAMT
GATM
SCL6A8
| AR
XL |
Cerebral creatine deficiency on brain MR spectroscopy Suggestive ratio of guanidinoacetate, creatine, &/or creatinine in plasma & urine
| Creatine monohydrate supplementation |
| Holocarboxylase synthetase deficiency (OMIM 253270) |
HLCS
| AR |
| Responsive to biotin |
|
Serine biosynthesis disorders
|
PHGDH
PSAT1
PSPH
| AR |
| L-serine & glycine supplementation can reduce seizures, improve psychomotor symptoms, & prevent progression depending on subtype |
|
Other
| Infection of the CNS | NA | | MRI, blood culture &/or lumbar puncture suggestive of infection | Antibiotic, antiviral, or antifungal therapy |
| Structural brain lesions | NA | | (Multi)focal lesions on brain MRI | |
|
Tuberous sclerosis complex
|
TSC1
TCS2
| |
MRI brain lesions (subependymal nodules, subependymal giant cell astrocytomas, tubers, focal cortical dysplasias) Cardiac rhabdomyoma, skin lesions, retinal lesions, renal lesions
| Consideration of mTOR inhibitor for astrocytoma, additional seizure reduction |
| ARX-associated encephalopathy (OMIM 308350) |
ARX
| | Enlarged ventricles & T2-weighted signals in basal ganglia on brain MRI | |
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; NA = not applicable; XL = X-linked
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with KCNT1-related epilepsy, the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Prolonged video EEG monitoring to evaluate electroclinical and electrographic seizure burden in consultation with a pediatric epileptologist
Evaluation by a movement disorder specialist if dictated by clinical presentation
Consideration of echocardiogram to evaluate for pulmonary collaterals
Electrocardiogram (EKG) to evaluate for cardiac rhythm abnormalities
Cognitive and behavioral assessment
Physical, occupational, and speech therapy evaluation
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Seizures.
KCNT1-related epilepsy is often refractory to conventional anticonvulsants.
Quinidine. Seizure control and developmental progression with off-label use of quinidine was reported in an individual with
KCNT1 pathogenic variant p.Arg428Gln [
Bearden et al 2014], prompting subsequent treatment trials in individuals with the same and other pathogenic variants with negative results [
Mullen et al 2018] and pro-arrhythmic cardiotoxicity. Potential explanations for this variable responsiveness include genetic/epigenetic modifiers of
KCNT1 as well as polymorphisms in P-glycoprotein transporters, which actively shuttle quinidine across the blood-brain barrier [
Liu et al 2015]. The limited efficacy may also be narrowed by epilepsy type, as a small, randomized, placebo-controlled, crossover clinical trial of
KCNT1-related ADNFLE showed no efficacy [
Mullen et al 2018]. It has also been suggested that quinidine administered after age four years may be less effective [
Abdelnour et al 2018]. In addition, given the increased risk for arrhythmia associated with quinidine treatment, some individuals are not able to achieve adequate serum levels because of the development of life-threating cardiac rhythm abnormalities, thus limiting its utility.
Caregivers. For information on non-medical interventions and coping strategies for parents or caregivers of children diagnosed with epilepsy, see
Epilepsy Foundation Toolbox.
Pulmonary collaterals and pulmonary hemorrhage. Embolization of systemic pulmonary collateral arteries has been used with limited success [Kawasaki et al 2017].
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
Physical therapy is recommended to maximize mobility.
Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended for affected individuals who have difficulty feeding as a result of poor oral motor control.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and is typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications (e.g., to treat attention-deficit/hyperactivity disorder) when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
EEG is recommended at intervals determined by seizure frequency and progression, for evaluation of new involuntary movements or unexplained, paroxysmal changes in vital signs, or following adjustments to an anticonvulsant regimen.
Developmental evaluation and initiation of therapies is recommended at time of diagnosis if not already begun.
Following initial EKG and echocardiogram, there is no indication to repeat cardiac monitoring or cardiopulmonary imaging unless clinically indicated or following initiation of quinidine therapy.
Agents/Circumstances to Avoid
No anticonvulsants have been noted to exacerbate KCNT1-related epilepsy.
For individuals with ADNFLE, activities in which a sudden loss of consciousness could lead to injury or death should be avoided (e.g., bathing, swimming, driving, or working/playing at heights).
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic at-risk relatives of an affected individual by molecular genetic testing for the KCNT1 pathogenic variant in the family. Family members who are found to have a heterozygous KCNT1 pathogenic variant are at risk for seizures and cardiac arrhythmias, and thus appropriate screening should be performed.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
In general, women with epilepsy or a seizure disorder from any cause are at greater risk for mortality during pregnancy than pregnant women without a seizure disorder; use of anti-seizure medication during pregnancy reduces this risk. However, exposure to anti-seizure medication (e.g., valproate, phenobarbital, topiramate) may increase the risk for adverse fetal outcome (depending on the drug used, the dose, and the stage of pregnancy at which medication is taken). Nevertheless, the risk of an adverse outcome to the fetus from anti-seizure medication exposure is often less than that associated with exposure to an untreated maternal seizure disorder. Therefore, use of anti-seizure medication to treat a maternal seizure disorder during pregnancy is typically recommended. Discussion of the risks and benefits of using a given anti-seizure medication during pregnancy should ideally take place prior to conception. Transitioning to a lower-risk medication prior to pregnancy may be possible [Sarma et al 2016].
See MotherToBaby for further information on medication use during pregnancy.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
KCNT1-related epilepsy is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Most individuals diagnosed with KCNT1-related epilepsy have the disorder as the result of a de novo pathogenic variant.
Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a
de novo pathogenic variant in the proband or germline mosaicism in an unaffected parent. Somatic and germline mosaicism have been reported [
Ohba et al 2015].
The family history of some individuals diagnosed with KCNT1-related epilepsy may appear to be negative because of failure to recognize the disorder in family members or reduced penetrance. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the parents of the proband.
Note: If the parent is the individual in whom the pathogenic variant first occurred, the parent may have somatic mosaicism for the variant and may be mildly/minimally affected.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
If a parent of the proband is known to be affected and/or is heterozygous for the
KCNT1 pathogenic variant, the risk to the sibs of inheriting the variant is 50%. Note: Intrafamilial clinical variability and reduced penetrance in some
KCNT1 seizure phenotypes has been observed (see
Genotype-Phenotype Correlations and
Penetrance) [
Møller et al 2015].
If the
KCNT1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline or somatic mosaicism [
Møller et al 2015,
Ohba et al 2015].
If the parents have not been tested for the KCNT1 pathogenic variant but are clinically unaffected, sibs of a proband are still at increased risk for KCNT1-related epilepsy because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism. (Note: Reduced penetrance has not been reported in KCNT1-related EIMFS.)
Offspring of a proband. Each child of an individual with KCNT1-related epilepsy has a 50% chance of inheriting the KCNT1 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the KCNT1 pathogenic variant, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the KCNT1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Epilepsy Society
Canadian Epilepsy Alliance
Canada
Phone: 1-866-EPILEPSY (1-866-374-5377)
Epilepsy Foundation
Phone: 800-332-1000; 866-748-8008
National Institute of Neurological Disorders and Stroke (NINDS)
Phone: 800-352-9424
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
KCNT1-Related Epilepsy: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
608167 | POTASSIUM CHANNEL, SUBFAMILY T, MEMBER 1; KCNT1 |
|
614959 | DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY 14; DEE14 |
|
615005 | EPILEPSY, NOCTURNAL FRONTAL LOBE, 5; ENFL5 |
Gene structure.
KCNT1 is located at chromosome 9q34.3. Although a long and short isoform have been reported in humans, the long isoform (Slack-B) composed of 31 exons is thought to encode the predominantly expressed functional protein and has been more extensively studied. With 1,235 amino acids, it is the largest potassium channel identified thus far. As studies in animals have revealed five splice isoforms with different expression patterns and physiologic properties, additional expression studies are necessary in humans to better define gene expression.
Pathogenic variants. All pathogenic variants reported to date are missense variants associated with an epilepsy phenotype, with the exception of two missense variants identified in individuals with cardiac conduction abnormalities [Juang et al 2014, Møller et al 2015]. Whereas most variants in neonates are associated with EIMFS, other phenotypes within the spectrum of infantile-onset epilepsy (e.g., West syndrome or EIEE not consistent with EIMFS) have been reported [Allen et al 2016, Fukuoka et al 2017].
The majority of ADNFLE-associated variants have also been observed in individuals with an EIMFS phenotype, consistent with variable expressivity [Heron et al 2012, Ishii et al 2013, Steinlein 2014, Møller et al 2015].
Table 3.
KCNT1 Variants Discussed in This GeneReview
View in own window
| Associated Phenotype | DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| EIMFS | c.769C>G | p.His257Asp |
NM_020822.2
NP_065873.2
|
| c.785G>A | p.Arg262Gln |
| c.808C>G | p.Gln270Glu |
| c.811G>T | p.Val271Phe |
| c.820C>A | p.Leu274Ile |
| c.1038C>G | p.Phe364Leu |
| c.1225C>T | p.Pro409Ser |
| c.1420C>T | p.Arg474Cys |
| c.1429G>A | p.Ala477Thr |
| c.1504T>G | p.Phe502Val |
| c.1546A>G | p.Met516Val |
| c.1885G>A | p.Lys629Glu |
| c.1887G>C | p.Lys629Asn |
| c.2280C>G | p.Ile760Met |
| c.2687T>A | p.Met896Lys |
| c.2771C>T | p.Pro924Leu |
| c.2797C>G | p.Arg933Gly |
| c.2800G>A | p.Ala934Thr |
| c.2839A>G | p.Lys947Glu |
| c.2849G>A | p.Arg950Gln |
| Temporal lobe epilepsy | c.398G>A | p.Arg133His |
| c.3320G>A | p.Arg1107His |
| ADNFLE | c.862G>A | p.Gly288Ser |
| c.2386T>C | p.Tyr796His |
| c.2688G>A | p.Met896Ile |
| ADNFLE, EIMFS | c.2849G>A | p.Arg950Gln |
| ADNFLE, EIMFS, multifocal epilepsy | c.1018G>A | p.Val340Met |
| c.1193G>A | p.Arg398Gln |
| EIMFS, multifocal epilepsy | c.1283G>A | p.Arg428Gln |
| EIEE | c.1799G>A | p.Arg600Gln |
| EIMFS, West syndrome, EIEE | c.1421G>A | p.Arg474His |
| EIMFS, West syndrome | c.1955G>T | p.Gly652Val |
| West syndrome, leukodystrophy | c.2718G>T | p.Gln906His |
| EIMFS, ADNFLE, focal epilepsy, cardiac arrhythmia | c.2782C>T | p.Arg928Cys |
| EIEE, delayed myelination, leukodystrophy | c.2794T>A | p.Phe932Ile |
| Multifocal epilepsy | c.2882G>A | p.Arg961His |
| Brugada syndrome | c.3317G>A | p.Arg1106Gln |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
KCNT1 encodes one of the two known sodium-activated potassium channels, termed the slack ("sequence like A calcium-activated K"; previously known as Slo2.2 and KCa4.1) channel [Joiner et al1998]. Current nomenclature now refers to KCNT1 as KNa1.1 [Kaczmarek et al 2017].
While KNa1.1 possesses six transmembrane domains and a pore domain between the S5 and S6 transmembrane domains, similar to other voltage-gated potassium channels, its C-terminus is disproportionately large. The C-terminus comprises:
Two regulators of conductance of K+ (RCK) domains that form a ring on the cytoplasmic face of the channel, which, in the presence of sodium, undergoes a conformational shift to expose the electrostatic channel pore [
Hite et al 2015];
An NAD+ binding domain in the C-terminus, which, in the presence of increased NAD+ concentrations, reduces the sodium requirement of the channel [
Tamsett et al 2009].
The C-terminus regulates channel opening by interaction with fragile X mental retardation protein (FMRP) [Zhang et al 2012].
KCNT1 expression is robust throughout the CNS in brain stem nuclei, the cerebellum, and the olfactory bulb and less strongly in the hippocampus and frontal cortex [Bhattacharjee et al 2002, Rizzi et al 2016].
Abnormal gene product. Of the variants studied functionally, all confer a gain-of-function channel phenotype regardless of the type of associated epilepsy [reviewed in Møller et al 2015, Lim et al 2016, McTague et al 2018], with the exception of p.Phe932Ile, which confers a loss-of-function channel phenotype [Vanderver et al 2014].
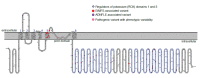
Although the majority of variants associated with KCNT1-related EIMFS and KCNT1-related ADNFLE cluster in different regions of the channel (see ), there is insufficient evidence to predict genotype-phenotype correlation. Clinical variability is expected.
Pathogenic variants identified in KCNT1-related epilepsy cluster in the S5 transmembrane and the Regulators of Potassium (RCK) domains of the channel protein. Figure generated from an image drawn using Protter open-source software [Omasits et al 2014] (more...)
Several mechanisms of channel dysfunction have been described. Some variants cause a shift in time spent in subconductance states, either secondary to loss of PKC-dependent regulation [Barcia et al 2012] or because of enhanced cooperativity of channels leading to increased open channel probability [Kim et al 2014]. Other pathogenic variants appear hypersensitive to intracellular sodium and thus more likely to be in an open state [Tang et al 2016]. Further research is needed to determine the impact of an enhanced potassium conductance on neuronal firing within regions of the developing brain susceptible to epileptogenesis.
Chapter Notes
Author Notes
The KCNT1 Registry includes individuals with epilepsy and known or suspected pathogenic variants in KCNT1. The registry is an ongoing natural history study of KCNT1-related epilepsy. Individuals interested in participating in the KCNT1 Registry should contact Dr David Bearden at david_bearden@urmc.rochester.edu.
References
Literature Cited
Abdelnour E, Gallentine W, McDonald M, Sachdev M, Jiang YH, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature.
Seizure. 2018;55:1–3. [
PubMed: 29291456]
Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, McCreary D, Magalhães TR, Ennis S, Lynch SA, King MD. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion.
Epilepsia. 2016;57:e12–7. [
PubMed: 26648591]
Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy.
Nat Genet. 2012;44:1255–9. [
PMC free article: PMC3687547] [
PubMed: 23086397]
Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine.
Ann Neurol. 2014;76:457–61. [
PubMed: 25042079]
Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, Moshé SL, Nordli D, Plouin P, Scheffer IE. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009.
Epilepsia. 2010;51:676–85. [
PubMed: 20196795]
Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system.
J Comp Neurol. 2002;454:241–54. [
PubMed: 12442315]
Caraballo RH, Fontana E, Darra F, Cassar L, Negrini F, Fiorini E, Arroyo H, Ferraro S, Fejerman N, Dalla Bernardina B. Migrating focal seizures in infancy: analysis of the electroclinical patterns in 17 patients.
J Child Neurol. 2008;23:497–506. [
PubMed: 18230844]
Cilio MR, Bianchi R, Balestri M, Onofri A, Giovannini S, Di Capua M, Vigevano F. Intravenous levetiracetam terminates refractory status epilepticus in two patients with migrating partial seizures in infancy.
Epilepsy Res. 2009;86:66–71. [
PubMed: 19520548]
Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest.
Epilepsia. 1995;36:1017–24. [
PubMed: 7555952]
Fukuoka M, Kuki I, Kawawaki H, Okazaki S, Kim K, Hattori Y, Tsuji H, Nukui M, Inoue T, Yoshida Y, Uda T, Kimura S, Mogami Y, Suzuki Y, Okamoto N, Saitsu H, Matsumoto N. Quinidine therapy for West syndrome with KCNTI mutation: a case report.
Brain Dev. 2017;39:80–3. [
PubMed: 27578169]
Hansen N, Widman G, Hattingen E, Elger CE, Kunz WS. Mesial temporal lobe epilepsy associated with KCNT1 mutation.
Seizure. 2017;45:181–3. [
PubMed: 28081520]
Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy.
Nat Genet. 2012;44:1188–90. [
PubMed: 23086396]
Hmaimess G, Kadhim H, Nassogne MC, Bonnier C, van Rijckevorsel K. Levetiracetam in a neonate with malignant migrating partial seizures.
Pediatr Neurol. 2006;34:55–9. [
PubMed: 16376281]
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, Shimizu T, Osawa M, Hirose S, Yamamoto T. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy.
Gene. 2013;531:467–71. [
PubMed: 24029078]
Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits.
Nat Neurosci. 1998;1:462–9. [
PubMed: 10196543]
Juang JM, Lu TP, Lai LC, Ho CC, Liu YB, Tsai CT, Lin LY, Yu CC, Chen WJ, Chiang FT, Yeh SF, Lai LP, Chuang EY, Lin JL. Disease-targeted sequencing of ion channel genes identifies de novo mutations in patients with non-familial Brugada syndrome.
Sci Rep. 2014;4:6733. [
PMC free article: PMC4206841] [
PubMed: 25339316]
Kaczmarek LK, Aldrich RW, Chandy KG, Grissmer S, Wei AD, Wulff H. International Union of Basic and Clinical Pharmacology. C. Nomenclature and properties of calcium-activated and sodium-activated potassium channels.
Pharmacol Rev. 2017;69:1–11. [
PMC free article: PMC11060434] [
PubMed: 28267675]
Kawasaki Y, Kuki I, Ehara E, Murakami Y, Okazaki S, Kawawaki H, Hara M, Watanabe Y, Kishimoto S, Suda K, Saitsu H, Matsumoto N. Three cases of KCNT1 mutations: malignant migrating partial seizures in infancy with massive systemic to pulmonary collateral arteries.
J Pediatr. 2017;191:270–4. [
PubMed: 28987752]
Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human slack potassium channel mutations increase positive cooperativity between individual channels.
Cell Rep. 2014;9:1661–72. [
PMC free article: PMC4294418] [
PubMed: 25482562]
Lim CX, Ricos MG, Dibbens LM, Heron SE. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects.
J Med Genet. 2016;53:217–25. [
PubMed: 26740507]
Liu L, Collier AC, Link JM, Domino KB, Mankoff DA, Eary JF, Spiekerman CF, Hsiao P, Deo AK, Unadkat JD. Modulation of P-glycoprotein at the human blood-brain barrier by quinidine or rifampin treatment: a positron emission tomography imaging study.
Drug Metab Dispos. 2015;43:1795–804. [
PMC free article: PMC4613948] [
PubMed: 26354948]
McTague A, Appleton R, Avula S, Cross JH, King MD, Jacques TS, Bhate S, Cronin A, Curran A, Desurkar A, Farrell MA, Hughes E, Jefferson R, Lascelles K, Livingston J, Meyer E, McLellan A, Poduri A, Scheffer IE, Spinty S, Kurian MA, Kneen R. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum.
Brain. 2013;136:1578–91. [
PMC free article: PMC3634200] [
PubMed: 23599387]
McTague A, Nair U, Malhotra S, Meyer E, Trump N, Gazina EV, Papandreou A, Ngoh A, Ackermann S, Ambegaonkar G, Appleton R, Desurkar A, Eltze C, Kneen R, Kumar AV, Lascelles K, Montgomery T, Ramesh V, Samanta R, Scott RH, Tan J, Whitehouse W, Poduri A, Scheffer IE, Chong WKK, Cross JH, Topf M, Petrou S, Kurian MA. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy.
Neurology. 2018;90:e55–e66. [
PMC free article: PMC5754647] [
PubMed: 29196579]
Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, van Kempen MJ, Klinkenberg S, Andrews I, Kelley K, Ronen GM, Callen D, McMahon JM, Yendle SC, Carvill GL, Mefford HC, Nabbout R, Poduri A, Striano P, Baglietto MG, Zara F, Smith NJ, Pridmore C, Gardella E, Nikanorova M, Dahl HA, Gellert P, Scheffer IE, Gunning B, Kragh-Olsen B, Dibbens LM. Mutations in KCNT1 cause a spectrum of focal epilepsies.
Epilepsia. 2015;56:e114–20. [
PMC free article: PMC5915334] [
PubMed: 26122718]
Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, Nair U, Li M, Berkovic SF, Petrou S, Scheffer IE. Precision therapy for epilepsy due to KCNT1 mutations: a randomized trial of oral quinidine.
Neurology. 2018;90:e67–e72. [
PubMed: 29196578]
Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, Nabatame S, Azuma J, Fujii Y, Hara M, Tsurusawa R, Inoue T, Ogata R, Watanabe Y, Togashi N, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Saitsu H, Matsumoto N. De novo KCNT1 mutations in early-onset epileptic encephalopathy.
Epilepsia. 2015;56:e121–8. [
PubMed: 26140313]
Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data.
Bioinformatics. 2014;30:884–6. [
PubMed: 24162465]
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405–24. [
PMC free article: PMC4544753] [
PubMed: 25741868]
Steinlein OK. Genetic heterogeneity in familial nocturnal frontal lobe epilepsy.
Prog Brain Res. 2014;213:1–15. [
PubMed: 25194481]
Tang QY, Zhang FF, Xu J, Wang R, Chen J, Logothetis DE, Zhang Z. Epilepsy-related slack channel mutants lead to channel over-activity by two different mechanisms.
Cell Rep. 2016;14:129–39. [
PMC free article: PMC4706775] [
PubMed: 26725113]
Vanderver A, Simons C, Schmidt JL, Pearl PL, Bloom M, Lavenstein B, Miller D, Grimmond SM, Taft RJ. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy.
Pediatr Neurol. 2014;50:112–4. [
PMC free article: PMC4303471] [
PubMed: 24120652]
Zamponi N, Rychlicki F, Corpaci L, Cesaroni E, Trignani R. Vagus nerve stimulation (VNS) is effective in treating catastrophic 1 epilepsy in very young children.
Neurosurg Rev. 2008;31:291–7. [
PubMed: 18446391]
Zhang Y, Brown MR, Hyland C, Chen Y, Kronengold J, Fleming MR, Kohn AB, Moroz LL, Kaczmarek LK. Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels.
J Neurosci. 2012;32:15318–27. [
PMC free article: PMC3518385] [
PubMed: 23115170]