

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

The κ opioid receptors (KOR) provide desirable substrates for targeted therapy and unraveling pathways responsible for mediating addictive behavior. Kappa opioid agonists offer a means to modulate the effects of stimulant drugs. However, there currently are no available approved agents or compounds for these purposes. Thus, the specific aim of this project was to identify subtype specific small molecule agonists of the human KOR. Such agonists might be useful in elucidating different signaling modalities for kappa receptors, but more importantly, they might also be partial agonists that will facilitate the discovery of new molecular scaffolds for kappa antagonists. In this report, we describe the discovery and optimization of two novel agonists for the KOR, the small molecular probes ML139 (CID-5236771) and ML138 (CID-44601470), each of which are greater than 100-fold selective over the mu-(µ) and delta (δ) opioid subtype receptors. Furthermore, these probes and their analogs represent novel chemical classes compared to current literature agonists, with potentially unique pharmacology that may serve as interesting tools to advance addiction research.
Probe project: Selective KOP Receptor agonists
Assigned Assay Grant #: 1X01MH084153-01
Screening Center Name & PI: Burnham Center for Chemical Genomics (BCCG) & John C. Reed (renamed Conrad Prebys Center for Chemical Genomics at Sanford|Burnham Medical Research Institute in 2010 but still referred to for NIH Roadmap Purposes as BCCG above)
Chemistry Center Name & PI: Kansas Specialized Chemistry Center (KSCC) & Jeffrey Aubé
Assay Submitter & Institution: Lawrence Barak, Duke University Medical Center. Collaborating PI: (Laura M. Bohn, The Scripps Research Institute, FL)
PubChem Summary Bioassay Identifier (AID): AID-1786
Probe(s) Structure & Characteristics
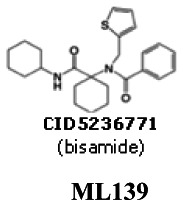
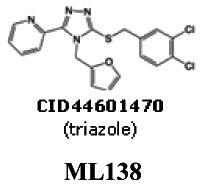
This joint KU & Burnham Center Probe Report describes two selective agonists for the kappa (κ) opioid receptor [KOP receptor or KOR] representing two novel scaffolds or chemical series: (1) bisamides exemplified by CID5236771 and (2) triazoles exemplified by CID44601470.
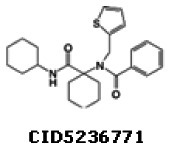
| CID | Target Name | IC50/EC50 (nM) [SID, AID] | Anti- target Name(s) | IC50/EC50 (μM) [SID, AID] | Selectivity | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID5236771 (bisamides) ML139 | KOR κ-opioid receptor | 120 nM IC50 SID-87218782 AID-2284 | MOR μ-opioid receptor DOR δ-opioid receptor | >32 μM EC50
SID-87218782
AID-2352 >32 μM EC50 SID-87218782 AID-2370 | > 270X (Dx/HCS) >530X (HCS/HCS) > 270X (Dx/HCS) >530X (HCS/HCS) | KOR Transflour <60 nM EC50 [SID-87218782, AID-2359] MOR Transflour >32,000 nM EC50 [SID-87218782, AID-2352] DOR Transflour >32,000 nM EC50 [SID-87218782, AID-2370] |
| 44601470 (triazoles) ML138 | KOR κ-opioid receptor | 870 nM IC50 SID-87334039 AID-2284 | MOR μ-opioid receptor DOR δ-opioid receptor | >32 μM EC50
SID-87334039
AID-2352 >32 μM EC50 SID-87334039 AID-2370 | > 37X (Dx/HCS) >91X (HCS/HCS) > 37X (Dx/HCS) >91X (HCS/HCS) | KOR Transfluor 350 nM EC50 [SID-87334039, AID-2359] MOR Transfluor >32000 nM EC50 [SID-87334039, AID-2352] DOR Transfluor >32000 nM EC50 [SID-87334039, AID-2370] |
Recommendations for the scientific use of this probe
The probe candidates described in this report, by selectively activating the human kappa opioid receptor subtype, would provide a scientific tool useful in helping to elucidate individual brain pathways that underlie addictive behavior, thus enabling improved understanding of the molecular basis of dependency and potentially providing a basis for therapeutic development.
1. Scientific Rationale for Project
Specific Aims
The identification of small molecules, each able to activate or block a distinct receptor underlying an addiction will provide a means to untangle the many pathways resulting in addictive behavior and create detailed pharmacological maps for designing novel targeted treatments. This project proposes screening a G protein-coupled receptor relevant to drug abuse and to the study and treatment of addiction, in a fashion that affords the unique opportunity to discriminate between G protein and arrestin-based signaling modalities. This project will hopefully contribute to understanding and treating addiction by providing chemical probes for dissecting the individual brain pathways that underlie addictive behavior thus enabling improved understanding of the molecular basis of addiction and potentially providing targeted therapeutics for this affliction.
The specific aims of this project are to identify subtype specific small molecule agonists of the human kappa opioid (KOP) receptor. Such agonists may be useful in elucidating different signaling modalities for kappa receptors, but more importantly may be partial agonists that will facilitate the discovery of new molecular scaffolds for kappa antagonists. Subtype selectivity is defined as selective for KOR but not active against μ (MOR) or δ (DOR) subtypes
Background and Significance
For normal activities that produce rewards, there is a rapid habituation of the circuits involved and the behaviors will wane. However, for addictive drugs habituation does not occur and dopamine release persists despite repetitive trials. Upon withdrawal of the drug, a decrease of dopamine levels in the nucleus accumbens results, and this has been observed for opioids, cannabinoids, alcohol, amphetamines, and nicotine (1). This loss of dopamine accounts for the withdrawal syndromes observed with these drugs. The prototype opioid drug is morphine. It produces many effects typical of most opioids including analgesia, euphoria, nausea, and respiratory depression. Repeated use of opioids produces physical dependence and tolerance. These manifestations of opioid use are due to the three recognized types of opioid receptors that are members of the GPCR family, the mu (μ), delta (δ), and kappa (κ) subtype receptors. While stimulation of the mu and delta receptors increases dopamine release in the nucleus accumbens, κ receptor activation by its endogenous ligand dynorphin-A reduces extracellular dopamine. It has been suggested that stimulation of κ receptors by endogenous opioids like dynorphins should produce an aversive state that can antagonize the effects of other addictive compounds like alcohol, cocaine and nicotine. Moreover, exogenous κ agonists have also been observed to attenuate drug-taking behavior (2–6). Interestingly, kappa opioid antagonists have also been considered for use in treatment of cocaine abuse and appetite suppression (6).
The κ opioid receptors provide desirable substrates for targeted therapy and unraveling pathways responsible for mediating addictive behavior. Kappa opioid agonists offer a means to modulate the effects of stimulant drugs. However, there are currently no approved agents or compounds for these purposes (2).
In this probe report, we describe the discovery and optimization of novel agonists for the kappa-opioid (κ-opioid) receptor [KOP receptor or KOR] that is greater than 100-fold selective over the mu- (μ) [MOP receptor or MOR] and delta- (δ) [DOP receptor or DOR] opioid receptors (as measurable within the limits of solubility). We sent the probe for broad GPCR paneling and these data are discussed below (see Table 9). Furthermore, these probes and their analogs represent a novel chemical classes compared to current literature agonists with potentially novel pharmacology that may serve as interesting tools to advance addiction research.
Table 9
Comparative summary of two current probes CID5236771 & CID44601470 to literature examples.
Follow up studies subsequent to the probe report to investigate the properties of the new probes will be required and will employ confirmatory assays to validate that the compounds are interacting directly with the KOR and to characterize compound signaling. To demonstrate subtype specific binding in competitive assays a cohort of radiolabeled compounds are available commercially. [3H]-Diprenorphine has been used to demonstrate high affinity and selectivity (Kd = 0.1 nM) toward KOR and MOR. [3H]-U-69,593, [Phenyl-3,4-–H] can be used to demonstrate high affinity and selectivity (Kd = 1 nM) toward KOR. Likewise, [3H]-DAMGO, [Tyrosyl-3,5–3H(N)] is also available for MOR displacement binding and Naltrindole [5′,7′-3H] or [3H]-Enkephalin are available for DOR binding studies. Additionally, these receptors couple through a multiplicity of G proteins and signaling pathways that can be studied by available assays including determination of the changes in cAMP, calcium, and certain phospho-kinase levels in cells.
2. Project Description
a. Original goal for probe characteristics
The original goal from the CPDP was to find agonists different from current literature probes, with potencies of less than 1 µM for the kappa(κ)-opioid receptor (KOR), but with 100X selectivity against the mu(μ)-opioid receptor (MOR) and 100X selectivity against the delta(δ)-opioid receptor (DOR).
b. Information for each Assay Implemented and Screening Run
i. PubChem Bioassay Name(s), AID(s), Assay-Type (Primary, DR, Counterscreen, Secondary)
Table 1Listing of all Assays and AIDs for this project
| PubChemBioAssay Name | AID | Probe Type | Assay Type | Assay Format | Assay Detection & well format |
|---|---|---|---|---|---|
| Summary of small molecule agonists of the kappa opioid receptor via a luminescent beta-arrestin assay [Summary] | 1786 | Agonist | Summary | N/A | N/A |
| uHTS identification of small molecule agonists of the kappa opioid receptor via a luminescent beta-arrestin assay [Confirmatory] | 1777 | Agonist | Primary | Cell-based | Luminescence -DiscoveRx β-arrestin & 1536 |
| SAR analysis of small molecule agonists of the kappa opioid receptor via a luminescent beta-arrestin assay [Confirmatory] | 2284 | Agonist | SAR | Cell-based | Luminescence -DiscoveRx β-arrestin & 1536 |
| HTS Dose response counterscreen for assays utilizing the enzyme, b- galactosidase [Confirmatory] | 1966 | Agonist | Counterscreen | Biochemical | Luminescence &1536 |
| HTS Image-Based Screen for Selective Agonists of the KOR Receptor [Confirmatory] | 2133 | Agonist | Alternate | Cell-based | HCS – Transfluor & 384 |
| HTS Image-Based Screen for Agonists of the MOR Receptor [Confirmatory] | 2344 | Agonist | Selectivity | Cell-based | HCS – Transfluor & 384 |
| HTS Image-Based Screen for Agonists of the DOR Receptor [Confirmatory] | 2343 | Agonist | Selectivity | Cell-based | HCS – Transfluor & 384 |
| SAR analysis of Agonists of the Kappa Opioid Receptor (KOR) using an Image-Based Assay [Confirmatory] | 2359 | Agonist | Alternate | Cell-based | HCS – Transfluor & 384 |
| SAR Analysis of Agonists of the MOR Receptor using an Image-Based Assay [Confirmatory] | 2352 | Agonist | Selectivity | Cell-based | HCS – Transfluor & 384 |
| SAR Analysis of Agonists of the DOR Receptor using an Image-Based Assay [Confirmatory] | 2370 | Agonist | Selectivity | Cell-based | HCS – Transfluor & 384 |
ii. Assay Rationale & Description
Unlike imaging or other second messenger assays, the DiscoveRx β-Arrestin assay allows a direct measure of GPCR activation by detection of β-Arrestin binding to the KOR1 receptor. In this system, β-Arrestin is fused to an N-terminal deletion mutant of β-gal (termed the enzyme acceptor of EA) and the GPCR of interest is fused to a smaller (42 amino acids), weakly complementing fragment termed ProLink™. In cells that stably express these fusion proteins, ligand stimulation results in the interaction of β-Arrestin and the Prolink-tagged GPCR, forcing the complementation of the two β-gal fragments and resulting in the formation of a functional enzyme that converts substrate to detectable signal.
The primary screening protocol is described below.
Assay materials
- OPRK1 beta-Arrestin (DiscoveRx)
- Assay Medium: Opti-MEM Medium supplemented with 1% hiFBS, 1X Pen/Strep/Glu, 125 ug/mL Hygromycin (1/2 recommended), 250 ug/mL Geneticin (1/2 recommended)
- Growth Medium: MEM supplemented with 10% hiFBS, 1X Pen/Strep/Glu, 125 ug/mL Hygromycin (1/2 recommended), 250 ug/mL Geneticin (1/2 recommended)
Table 2Reagents used for the uHTS experiments
| Reagent | Vendor |
|---|---|
| OPRK1 beta-Arrestin Cell Line | DiscoveRx |
| Assay Medium: Opti-MEM Medium supplemented with 1% hiFBS, 1X Pen/Strep/Glu, 125 ug/mL Hygromycin, 250 ug/mL Geneticin | Invitrogen |
| Growth Medium: MEM supplemented with 10% hiFBS, 1X Pen/Strep/Glu, 125 ug/mL Hygromycin, 250 ug/mL Geneticin | Invitrogen |
uHTS protocol
The following uHTS protocol was implemented at single point concentration confirmation:
Day 1
- Harvest cells using Enzyme-Free Dissociation Buffer (Invitrogen Cat#13151-14). Add 500 cells/well in 5 uL of media to each well of a white, 1536 well plate.
- Spin cells at 500 rpm for 1 min, then wrap plates in Saran Wrap.
- Incubate overnight at 37C with 5% CO2.
Day 2
- Using a Highres Biosolutions pintool pin 25 nL to wells Columns 1–2 should be DMSO only (− Control), Columns 3–4 should contain Dynorphin A (+ Control). Working concentration = 200 µM, FAC = 1 µM and Columns 5–48 contain test compounds (10µM final in well concentration).
- Immediately following pintool addition, spin plates at 500 rpm for 1 min, return assay plates to an incubator at 37C
- Incubate for 1hr and 30 minutes.
- During test incubation, prepare Detection Reagent Solution from DiscoveRx (1 part Galacton Star: 5 parts Emerald II and 19 parts Cell Assay Buffer)
- Add 2.5ul of detection reagent solution to each well.
- Incubate at room temperature for 60 min in the dark
- Read plates in a Perkin Elmer Envision using a luminescence protocol
The average Z′ for the screen was 0.56, the signal to background (S/B) was 5.02, signal to noise (S/N) was 33.79 and signal window was 5.03.
Rationale for confirmatory, counter and selectivity assays
The initial frontline counterscreen that was performed shortly following dose response confirmations on both the agonist and antagonist KOR1 primary screens was the β-galactosidase dose response assay. Each confirmed hit (EC50 < 10 µM) was run in a β-gal dose response assay. Because the primary screen is based upon the formation of a functional β-gal enzyme upon β-arrestin migration to the GPCR, we wanted to rule out compound interaction, either stimulatory or inhibitory, with the β-gal enzyme in the absence of GPCR interaction.
The High-Content Imaging-based confirmatory (KOR) and selectivity assays (MOR, DOR) which are based upon the translocation of β-arrestin linked to GFP to other receptor subtypes were developed and performed to confirm agonist activity in the KOR agonist primary assay, as well as to ascertain the selectivity of compounds for the KOR receptor vs. the MOR and DOR receptor sub-types.
Improved potency for KOR and increased selectivity against MOR and DOR will be primary drivers for compound selection and optimization.
Confirmation assays
The initial confirmatory assays were performed in full dose-response for compounds from solvated DPI stock solutions to confirm activity seen first in test agents from screening the library in the initial primary screen. Active compounds were then tested in an alternative format for GPCR activation, via the imaging-based KOR High-Content Transfluor Agonist Assay. In the Transfluor assay, GPCR activation is measured indirectly by via the detection of β-Arrestin-GFP redistribution from the cytosolic compartment to the plasma membrane to coated pits and finally endosomal vesicles. The image-based KOR assays allowed for independent confirmation of KOR activation utilizing an alternative technology.
The following are confirmation assays for this project:
Assay 1: uHTS identification of small molecule agonists of the kappa opioid receptor via a luminescent beta-arrestin assay (AID 1777)
Assay 2: SAR analysis of small molecule agonists of the kappa opioid receptor via a luminescent beta-arrestin assay (AID-2284)
Assay 3: HTS Image-Based Screen for Selective Agonists of the KOR Receptor (AID-2133)
Assay 4: SAR analysis of Agonists of the Kappa Opioid Receptor (KOR) using an Image-Based Assay (AID-2359)
Counterscreen assays
The β-Galactosidase Counterscreen Assay was utilized to ascertain possible enzyme activation, which might present the opportunity for false positives from the initial primary assay. The enhancement of activity of the β-galactosidase fragment complementation in the primary KOR1 β-Arrestin Assay in the presence of test agent could lead to increased signal formation and therefore a false positive result. This counterscreen assay would allow for the detection of these artifactual compounds.
Assay 1: HTS Dose response counterscreen for assays utilizing the enzyme, β-galactosidase (AID 1966)
Selectivity assays
The imaging-based MOR and DOR High-Content Transfluor Agonist Assays provide for the determination of KOR receptor selectivity. The probe criteria specifies the necessity of at least 100-fold selectivity against MOR and 10-fold against DOR, or within the reasonable limitations imposed for testing compounds at high concentrations, i.e. 100 µM selectivity for a 1 µM compound.
Assay 1: HTS Image-Based Screen for Agonists of the MOR Receptor (AID 2344)
Assay 2: HTS Image-Based Screen for Agonists of the DOR Receptor (AID-2343)
Assay 3: SAR Analysis of Agonists of the MOR Receptor using an Image-Based Assay (AID-2352)
Assay 4: SAR Analysis of Agonists of the DOR Receptor using an Image-Based Assay (AID-2370)
iii. Summary of Results
The following flowchart summarizes the compound triage and decision tree for advancement of compounds:


A library of approximately 290,000 compounds was tested in the KOR1 DiscoveRx β-Arrestin primary screen. Upon data analysis, 292 hits with activity >50% at a single concentration point of 10 µM were identified. Liquid samples were then ordered through DPI and 276 compounds were received.
The compound solutions resupplied by the MLSMR were first confirmed in 10 µM single-point duplicate in the KOR1 DiscoveRx β-Arrestin primary assay. Of these, 37 compounds were confirmed to have at least 50% activity at a 10 µM assay concentration. The confirmed compounds were further tested in dose response in the KOR1 DiscoveRx β-Arrestin primary assay to obtain EC50 values and were also tested in a β-galactosidase Counterscreen assay to assess the possibility that these compounds might somehow activate the enzyme. All compounds were confirmed as active in the KOR1 β-Arrestin dose response assay and none of the compounds were found to have activated β-galactosidase.
The active, confirmed compounds were then tested in the KOR1 High-Content Transfluor Agonist assay for further confirmation, then in the MOR and DOR High-Content Transfluor Agonist assays to determine subtype selectivity.
Chemistry and cheminformatics resources were then employed in the selection of both novel and chemically tractable molecules to pursue for a KOR1 selective probe. Structures of interest and analogs thereof were either purchased as commercial dry powders. In total, 30 structures were ordered though commercial vendors. From these experiments, a diverse SAR emerged and several compounds were discovered that met our established probe criteria.
Additional medicinal chemistry/SAR testing of re-constituted powders encompassed dose response testing of compounds in the four assays: KOR1 DiscoveRx β-Arrestin assay, the KOR1 High-Content Transfluor Agonist assay, and the MOR and DOR High-Content Transfluor Agonist assays.
c. Probe Optimization
i. Description of SAR & chemistry strategy (including structure and data) that led to the probe
The screening of the MLPCN compound collection at the Sanford-Burnham Center for Chemical Genomics identified several promising chemotypes (Figure 1) with potencies of the lead exemplar noted from solution reconfirmation. This probe report describes the optimization of probes from the first two chemotypes, bisamides (series 1) and triazoles (series 2). The third series did not yield sufficiently compelling SAR and was set aside.
Cheminformatics and Medicinal Chemistry Analysis
Probe optimization/SAR
Bisamide series
Compound 1 CID5236771 (MLS-0254632) (entry 1, Table 3) was identified through a high-throughput screening campaign on 284,076 compounds as an active and selective scaffold. After confirmation of the initial results, the hit-to-probe process was initiated by both analog-by-catalog approach and collaborative medicinal chemistry effort with KSCC. Probe candidate CID5236771 (MLS-0254632, entry 1) and analogs (entries 2–9) were obtained from commercial sources. Several additional analogs were prepared using a protocol similar to the route shown in section 4h (entries 10 – 14 in Table 3). The structure-activity data are presented Table 3 below:
Table 3
SAR Analysis for Selective κ–opioid receptor agonist for the Bisamide Scaffold.
SAR Analyses for CID5236771 (bisamide series)
The above data set illustrate several emerging SAR trends for this chemotype. Foremost, the requirement for substitution on the methylene bridge between amides is demonstrated from the inactive glycine-derived analogs CID1458208 (MLS-0435478), CID44483222 (MLS-0435480), CID44483224 (MLS-0435479), CID44483221 (MLS-0435482) and CID44483223 (MLS-0435481) (entries 10–14). While the necessity for alkyl substitution is clear, the ideal composition of the R2/R3 groups has not been completely optimized for this chemotype The exact nature of the side group on the secondary amide on the left hand side of the general structure appears to have a less dramatic effect on potency than the R2/R3 substitution. While a range of hydrophobic groups afforded highly potent analogs, the most potent analog was obtained when R1 was cyclohexyl (CID5236771 (MLS-0254632), entry 1). The single case where a direct comparison is possible suggests the thiophene-containing side chain to be advantageous over the furan-containing side chain (CID5236771 and CID3950926, entries 1 and 3). Finally, a clear advantage was observed for the 2-pyridyl amides over the 2-pyrazinyl amides, as exemplified by CID5236771 compared to CID4014737 (entries 1 and 2 ) compared to CID3242935 (entries 9 and 8).
Triazole series
The triazole (Series 2) chemotype originally contained four compounds with potency around 2 μM and one example at 6.7 μM. This scaffold was pursued in a short and intense analog campaign to increase potency by at least two fold. The synthesis and purchase of a total of 43 analog compounds provided not only a compound exceeding the necessary increase in potency, but also less potent compounds possessing super agonist efficacies (Emax >100%): cmps 1 – 3, 5 – 11 in Table 4 below, which also details the SAR.
Table 4
SAR Analysis for Selective κ–opioid receptor agonist for the Triazole Scaffold.
While a limited selection of analogs for the series 2 chemotype are commercially available, limited substitution on the phenyl ring and no available thiophene-containing analogs led us to adopt an entirely synthetic approach. Beginning with the appropriate isothiocyanate and 2-picolynyl hydrazide, we synthesized the 1,2,4-triazole-3-thione scaffolds in two steps and excellent yields (77–82% overall yields) without chromatographic separations (section 4 h). The coupling with a wide range of benzyl halides proceeded smoothly in acetone facilitated by K2CO3 to furnish the 43 analogs described in Table 4.
In conjunction with canonical SAR substitution strategies (7,8), our approach toward the optimization of the Series 2 compounds benefited from the observation in the Series 1 work that in some cases a switch from the furan to thiophene moiety afforded at least a two-fold increase in potency. For example see: CID5236771 (MLS-0254632)/CID3964693 (MLS-0080731) (earlier section). For this chemotype, the thiophene and furan moieties afforded roughly equipotent analogs within the experimental standard deviation. A more productive approach was varying the substitution on the phenyl ring, producing the two most potent analogs CID44601470 (MLS-0435589) and CID44601475 (MLS-0435599) (entries 1 and 11), both bearing a 3,4-dichlorophenyl moiety. Other phenyl ring substitution also bore interesting results not in potency, but in the Emax values. Six compounds (entries 2, 3, 5, 6, 7 and 9 in Table 4) synthesized displayed 150% or greater activation at 20 μM. This unexpected (and to our knowledge unprecedented) super agonistic activity provides a strong rationale for further explorations into this chemotype.
3. Probe(s)
a. Chemical name of probe compound (s) (Chemical IUPAC)
Probe 1 (bisamide - series 1): N-[1-(cyclohexylcarbamoyl)cyclohexyl]-N-(thiophen-2-ylmethyl)pyridine-2-carboxamide [ML139]
Probe 2 (triazole – series 2): 2-(5-(3,4-dichlorobenzylthio)-4-(furan-2-ylmethyl)-4H-1,2,4-triazol-3-yl)pyridine [ML138]
b. Probe chemical structure(s) including stereochemistry

c. Structural Verification Information of probe SID
The probe SIDs are 87218782 (corresponding to CID5236771) and 87334039 (corresponding to CID44601470)


Mass Spec
SID-87218782: HRMS (ESI) m/z calcd for C24H32N3O2S ([M+H]+), 426.2215, found 426.2207.
SID-87334039: HRMS (ESI) m/z calcd for C19H15Cl2N4OS ([M+H]+), 417.0338, found 417.0339.
NMR Purity
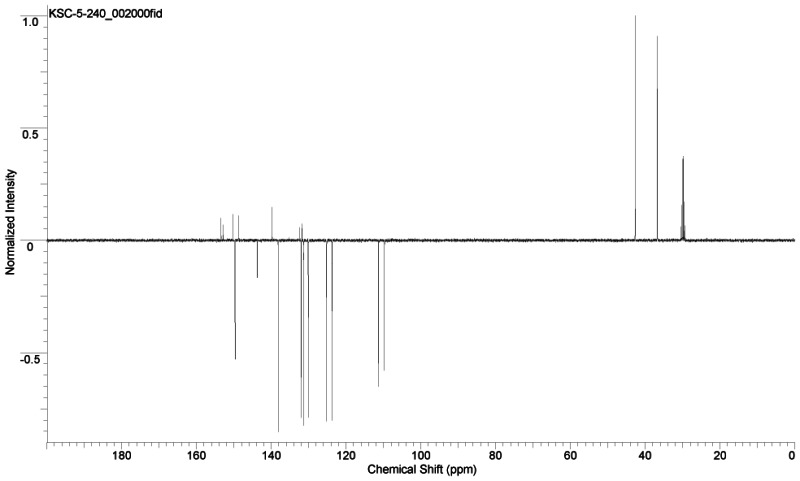
(1) SID-87218782: >95% (1H-NMR): 1H NMR (400 MHz, CDCl3) δ 1.06–1.22 (m, 3 H), 1.28–1.38 (m, 2 H), 1.42–1.83 (complex, 11 H), 2.14–2.26 (m, 4 H), 3.62–3.71 (m, 1 H), 4.98 (s, 2 H), 6.78 (d, J = 2.8 Hz, 1 H), 6.83 (t, J = 4.4 Hz, 1 H), 6.95 (br s, 1 H), 7.14 (d, J = 5.2 Hz, 1 H), 7.33 (dd, J = 0.8, 4.8 Hz, 1 H), 7.59 (d, J = 8.0 Hz, 1 H), 7.75 (dt, J = 1.6, 7.6 Hz, 1 H), 8.57 (d, J = 4.8 Hz, 1 H). 13C (100 MHz, CDCl3, APT pulse sequence) δ d (CH, CH3) 48.0, 124.3, 124.7, 125.2, 126.5, 126.6, 137.1, 148.0; u (C, CH2) 22.4, 24.7, 25.6, 25.7, 32.8, 33.1, 44.9, 66.9, 141.8, 155.1, 171.7, 172.7.

(2) SID-87334039: >90% pure. 1H NMR (acetone-d6) δ 4.54 (s, 2 H), 5.91 (s, 2 H), 6.24 (d, J = 2.8 Hz, 1 H), 6.29 (dd, J = 2.0, 3.2 Hz, 1 H), 7.40–7.51 (complex, 4 H), 7.70 (d, J = 2.0 Hz, 1 H), 7.96 (dt, J = 1.6, 7.6 Hz, 1 H), 8.24 (d, J = 8.0 Hz, 1 H), 8.72 (d, J = 4.4 Hz, 1 H); 13C NMR (acetone-d6, APT pulse sequence) δ d (CH, CH3) 109.7, 111.3, 123.7, 125.2, 130.1, 131.4, 132.0, 138.2, 143.8, 149.7; u (C, CH2) 36.7, 42.7, 131.7, 132.5, 139.7, 148.8, 150.3, 152.8, 153.4; (neat) 1701, 1589, 1463, 1446 cm−1

d. PubChem CID(s) (corresponding to the SID)
PubChem CIDs are CID5236771 (corresponding to SID-87218782) and CID44601470 (corresponding to SID-87334039)
e. Availability from a vendor
- The bisamide probe, CID5236771 is commercially available in milligram quantities from Chem Div (catalog # C094-1951), Aurora Screening Library (catalog # kcd-303814), Ambinter Screening Collection (catalog # STK142410), New Chemistry Horizons Laboratories Screening Library (catalog # NCHSC1-21611), Princeton Gold Collection I (catalog # OSSK_529711) and AKOS Screening Library (catalog # AKG-C094-1951). However, KSCC is depositing 50 mg of newly synthesized material with DPI.
- The triazole probe, CID44601470 is not commercially available. However, KU SCC has deposited 50 mg of synthesized material with the MLSMR.
f. MLS#'s of probe molecule and five related samples that were submitted to the SMR collection
(see Table 5)
Table 5
Probe and Analog Submissions to the MLMR (BioFocus DPI).
g. Mode of action for biological activity of probe
Both probes were submitted to Bryan Roth’s Psychoactive Drug Screening Program (PDSP) GPCR Panels and the results are summarized in Table 6 below. The first bisamide probe CID5236771 [ML139] was indeed very clean with no activity < 10,000 nM except for the KOP, MOP and DOP receptors, with values of 0.79, 671.1, and 1,315 nM, respectively. These yield selectivity values of 850-fold (MOR/KOR) and 1,660-fold (DOR/KOR) in the PDSP assays which compare favorably with those obtained by our DiscoveRx (>270-fold for MOR & DOR) and HCS (>530-fold for MOR & DOR) assays (see “Probe(s) Structure & Characteristics:” table on p.1).
Table 6
Receptor Paneling of KOP agonist Probes 1 [ML139] CID5236771 & 2 [ML138] CID44601470 through the PDSP (Bryan Roth, PI).
The second triazole probe CID44601470 [ML138] was not as “clean” and showed some weak activity (1200 – 9600 nM Kis) against 5HT2b, 5HT5a, α2A, α2B, b3, D1, and D5 receptors, in addition to stronger activities against the KOR, MOR and DOR receptors, with values of 0.23, 773.3, and 484.1 nM, respectively. In light of this potent binding at the KOR, even the most potent binding to other receptors in this panel translates to an impressive selectivity of greater than 5,000 fold. These yield selectivity values of 2,100-fold (MOR/KOR) and 3,360-fold (DOR/KOR) in the PDSP assays which compare much more favorably with those obtained by our DiscoveRx (>37-fold for MOR & DOR) and HCS (>91-fold for MOR & DOR) assays.
Is should be noted the that the subnanomolar binding Kis of CID5236771 (0.79 nM) and CID44601470 (0.23 nM) for the KOR receptor are the markedly more potent than those determined by either functional assay (see table on p. 1) for CID5236771 (120 nM DiscoveRx; <60 nM HCS) and CID44601470 (870 nM DiscoveRx; 350 nM HCS) and the rank order is inverted. Although corrrelation of the functional EC50 data to equilibrium ligand binding values (Ki) is not straightforward, these potencies and selectivites compare very favorably with even the most potent previously reported compounds in the literature and augue favorably for the utility of the presently reported probes or their analogs in the future. For example, the comparative Ki data for salvinorin A (also obtained in the PDSP under analogous conditions), a KOR agonist of extremely high contemporary interest, are 7.4 nM for KOR, 1370 nM for the MOR, and >10,000 for the DOR.
G protein coupling assays using the 35S-GTPγS binding assay were undertaken to demonstrate single point efficacy for the probes and select derivatives. Full concentration response curves were pursued to determine agonist potencies, which are shown in Figure 2. Interestingly, the efficacy and potency of the current compounds in the G protein coupling assay was highly correlative with the observations made in the beta-arrestin translocation assay further reinforcing this assay as a sensitive and selective means to identify novel agonists.

Figure 2
35S-GTPγS binding assay – Secondary functional assays for KOR agonist probes and selected analogs.
Since agonists can differentially activate GPCRs to engage diverse signaling cascades, a secondary, downstream Map kinase assay (Erk1/2 activation) was used to evaluate agonist activity in Figure 3. Single point efficacy curves reveal a tendency for CID44601470 to be more efficacious in this assay, which was confirmed by full concentration response curve analysis.

Figure 3
Downstream MAP Kinase Activation - Secondary functional assays for KOR agonist probes and selected analogs.
The probe appears to bind reversibly to the opiate agonist binding site of the receptor on the basis of the beta-arrestin primary and secondary assays. As described in the introduction to the report, the site specificity will be tested through competitive assays with known KOR ligands. It is clear from these report findings that the probes are able to activate beta-arrestin. It will be of interest in the follow up studies whether they will demonstrate a similar efficacy for G protein signaling, since it is possible that biased agonism towards one or the other pathway produces a distinctive physiological response profile.
h. Detailed synthetic pathway for making probe
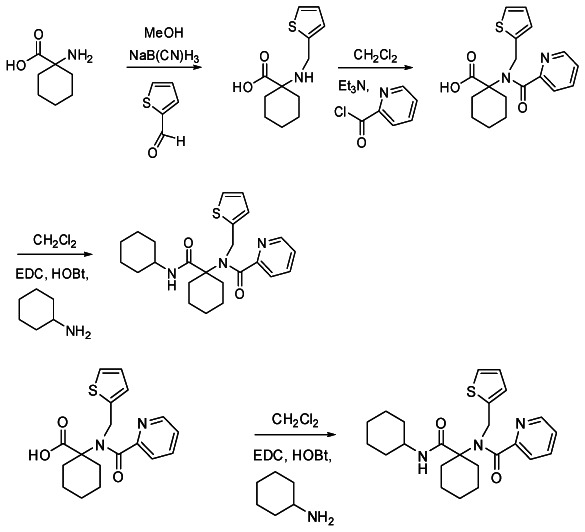
Bisamide Probe 1
N-(1-(cyclohexylcarbamoyl)cyclohexyl)-N-(thiophen-2-ylmethyl) picolinamide (SID-87218782). The carboxylici acid precursor [1-(N-(thiophen-2-ylmethyl) picolinamido)cyclohexanecarboxylic acid] (64 mg, 0.186 mmol) was combined in a Biotage microwave process vial (2–5 mL size) with HOBt (30 mg, 0.223 mmol), DCC (46 mg, 0.223 mmol) and cyclohexylamine (55 mg, 0.558 mmol) in MeCN (2 mL). The reaction was heat at 100 °C for 6 min using microwave irradiation. After cooling, the reaction mixture was adsorbed on Celite and chromatographed on silica to afford the bisamide product SID-87218782 (27 mg, 0.063 mmol, 34% yield).
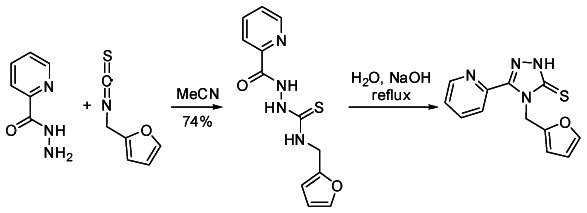
Triazole Probe 2
2-(5-(3,4-dichlorobenzylthio)-4-(furan-2-ylmethyl)-4H-1,2,4-triazol-3-yl)pyridine (SID-87334039). Following the general procedure listed below and silica gel chromatography, furan thione (93 mg, 0.36 mmol) and 3,4-dichlorobenzyl chloride (0.66 mL, 0.43 mmol) afforded the product as an off-white solid (116 mg, 0.278 mmol, 77% yield). Rf = 0.24 (1:1 Hexanes: EtOAc); mp = 99.0–99.5 °C; 1H NMR (acetone-d6) δ 4.54 (s, 2 H), 5.91 (s, 2 H), 6.24 (d, J = 2.8 Hz, 1 H), 6.29 (dd, J = 2.0, 3.2 Hz, 1 H), 7.40–7.51 (complex, 4 H), 7.70 (d, J = 2.0 Hz, 1 H), 7.96 (dt, J = 1.6, 7.6 Hz, 1 H), 8.24 (d, J = 8.0 Hz, 1 H), 8.72 (d, J = 4.4 Hz, 1 H); 13C NMR (acetone-d6) δ d 109.7, 111.3, 123.7, 125.2, 130.1, 131.4, 132.0, 138.2, 143.8, 149.7; u 36.7, 42.7, 131.7, 132.5, 139.7, 148.8, 150.3, 152.8, 153.4; (neat) 1701, 1589, 1463, 1446 cm−1; HRMS (ESI) m/z calcd for C19H15Cl2N4OS ([M+H]+), 417.0344., found 417.0353.
N-(Furan-2-ylmethyl)-2-picolinoylhydrazinecarbothioamide
2-Picolynyl hydrazide (410 mg, 2.99 mmol) and furfuryl isothiocyanate (416 mg, 2.99 mmol) in MeCN (15 mL) were stirred for 16 h at rt. The reaction mixture was filtered, the precipitate washed with additional MeCN (3 × 10 mL) and dried under vacuum to afford the thioamide as an off-white solid (610 mg, 2.21 mmol, 74% yield), which was used without further purification. IR (neat) 3135, 1673, 1533, 1500, 1465 cm−1; HRMS (ESI) m/z calcd for C12H13N4O2S ([M+H]+), 277.0759, found 277.0761.
4-(Furan-2-ylmethyl)-3-(pyridin-2-yl)-1H-1,2,4-triazole-5(4H)-thione
To a slurry of the above thioamide (530 mg, 1.92 mmol) in water (25 mL) was added NaOH (4.00 g, 100 mmol). The reaction was heated at reflux for 2 h, the starting thioamide dissolved promptly upon warming. The reaction was cooled to rt, diluted with aqueous HCl (1 N, 20 mL) and acidified to pH = 6 with concentrated HCl. The solid precipitate was filtered, washed with water (2 × 15 mL) and dried under vacuum to afford the thione as a white solid (478 mg, 1.85 mmol, 96% yield), which was used without further purification. IR (neat) 3021, 2894, 2766, 1585, 1550, 1502, 1463 cm−1; HRMS (ESI) m/z calcd for C12H11N4OS ([M+H]+), 259.0654, found 259.0642.
Synthesis of Series 2 thiophene scaffold:

N-(Thiophen-2-ylmethyl)-2-picolinoylhydrazinecarbothioamide
2-Picolynyl hydrazide (883 mg, 6.44 mmol) and thiophene isothiocyanate (1,000 mg, 6.44 mmol) in MeCN (20 mL) were stirred for 16 h at rt. The reaction mixture was filtered, the precipitate washed with additional MeCN (3 × 10 mL) and dried under vacuum to afford the thioamide as an off-white solid (1,642 mg, 5.62 mmol, 87% yield), which was used without further purification. IR (neat) 3141, 1672, 1527, 1499, 1466 cm−1; HRMS (ESI) m/z calcd for C12H13N4OS2 ([M+H]+), 293.0531, found 293.0516.
4-(Thiophene-2-ylmethyl)-3-(pyridin-2-yl)-1H-1,2,4-triazole-5(4H)-thione
To a slurry of the above thioamide (602 mg, 2.06 mmol) in water (25 mL) was added NaOH (4.00 g, 100 mmol). The reaction was heated at reflux for 2 h, the starting thioamide dissolved promptly upon warming. The reaction was cooled to rt, diluted with aqueous HCl (1 N, 20 mL) and acidified to pH = 6 with concentrated HCl. The solid precipitate was filtered, washed with water (2 × 15 mL) and dried under vacuum to afford the thione as a white solid (530 mg, 1.93 mmol, 94% yield), which was used without further purification. IR (neat) 3019, 2896, 1584, 1549, 1501, 1462 cm−1; HRMS (ESI) m/z calcd for C12H11N4S2 ([M+H]+), 275.0475, found 275.0412.
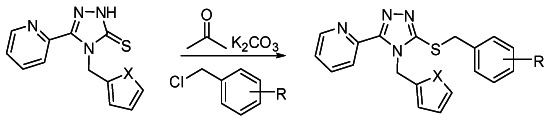
General procedure for the synthesis of series 2 analogs from thiones and benzyl halides
The thione scaffold (0.1 to 0.3 mmol), K2CO3 (2 equiv) and the benzyl halide (1.2 equiv) were combined in acetone (15 mL/mmol substrate) and stirred in a sealed vial. After 15 h, the solvent was removed and the residue washed with CH2Cl2 (2 × 3 mL) and filtered. The combined filtrates were evaporated down and either chromatographed on silica or subjected to mass-directed, reverse phase preparative HPLC purification.
i. Summary of probe properties (solubility, absorbance/fluorescence, reactivity, toxicity, etc
Both probes are novel chemical scaffolds and have demonstrated potent (< 1µM EC50) agonist activity in both the primary enzyme complementation assay and the image base orthogonal assay for β-arrestin mediated signaling and activation of the kappa opioid receptors, with selectivity over for activation of both the mu- and delta opioid receptors. The bisamide probe, CID5236771, has exceeded the criteria for probe as defined in the CPDP Chem Update document filed on October 28, 2009 and revised and refilled on November 21, 2009, and finally refilled after corrections requested by NIH on January 12, 2010 with the NIH PT: KOP receptors of less than 1 µM, and at least 100-fold selective over MOP and DOP receptors or as obtained by the achievable test concentrations, (e.g. solubility limited), regardless of whether this selectivity is calculated between the enzyme complementation/HCS or the HCS/HCS assays.
The triazole probe, CID44601470, formally misses the 100-fold selectivity of against MOR and DOR, though it approaches the desired selectivity when the HCS/HCS ratios are considered. However, all these compounds may not be able to achieve a higher than 30–40 µM test concentrations without some precipitation, and in most cases the dose-response curve is completely flat so the >32 µM is an under estimate of the true IC50, which is certainly >>32 µM, so well within a factor of 2–3 for an effective >100 –fold selectivity against MOR. The assay provider concurs that even at this pro forma lower ~40-fold estimate of selectivity this probe compound should be useful. Furthermore, we note the intriguing result that it is in the triazole scaffold series (Table 4) that the apparent super-agonism is experimentally seen, in contrast to the bisamide series (Table 3). While we do not currently have any mechanistic understanding of this difference, the clear pharmacologic difference between these two classes of compounds supports nomination of this a bonafide 2nd agonist probe for the kappa-opioid receptor. KU, SBCCG and the assay provider all concur on this point.
In Vitro Pharmacology Profiles of Probes CID5236771 [ML139] and CID44601470 [ML138] (See Table 7 below). Both probes had poor solubility at all pH’s tested, with the 2nd triazole probe (CID44601470) the poorest solubility.
Table 7
Summary of in vitro ADME/T Properties of Kappa Opioid Agonist probe(s).
The PAMPA (Parallel Artificial Membrane Permeability Assay) assay is used as an in vitro model of passive, transcellular permeability. An artificial membrane immobilized on a filter is placed between a donor and acceptor compartment. At the start of the test, drug is introduced in the donor compartment. Following the permeation period, the concentration of drug in the donor and acceptor compartments are measured using UV spectroscopy. In this assay, both probes CID5236771 [ML139] and CID44601470 [ML138] have excellent permeability. CID5236771 [ML139] had moderate brain barrier permeabilty while CID44601470 [ML138] had almost 3-fold selectivity in the blood brain PAMPA assay.
Plasma Protein Binding is a measure of a drug's efficiency to bind to the proteins within blood plasma. The less bound a drug is, the more efficiently it can traverse cell membranes or diffuse. Highly plasma protein bound drugs are confined to the vascular space, thereby having a relatively low volume of distribution. In contrast, drugs that remain largely unbound in plasma are generally available for distribution to other organs and tissues. CID5236771 [ML139] and CID44601470 [ML138] are both highly bound (90–99%) to both human and mouse plasma.
Plasma Stability is a measure of the stability of small molecules and peptides in plasma and is an important parameter, which strongly can influence the in vivo efficacy of a test compound. Drug candidates are exposed in plasma to enzymatic processes (proteinases, esterases), and they can undergo intramolecular rearrangement or bind irreversibly (covalently) to proteins. Both CID5236771 [ML139] and CID44601470 [ML138] shows excellent stability (>99%) in both human and mouse plasma.
The microsomal stability assay is commonly used to rank compounds according to their metabolic stability. This assay addresses the pharmacologic question of how long the parent compound will remain circulating in plasma within the body. Both CID5236771 [ML139] and CID44601470 [ML138] are rapidly metabolized in either Human or mouse microsomes. CID5236771 [ML139] shows high levels of toxicity toward human hepatocyctes in our assay, whereas CID 44601470 does not.
j. Probe properties
Table 8Calculated physical properties of Kappa opioid Agonist probe(s)
| Calculated Property | Probe Identity | |
|---|---|---|
| CID5236771 (bisamide) | CID44601470 (Triazole) | |
| MLS-0254632 | MLS-0435589 | |
| Molecular Weight [g/mol] | 425.58684 | 417.31166 |
| Molecular Formula | C24H31N3O2S | C19H14Cl2N4OS |
| XLogP3-AA | 4.6 | 4.3 |
| H-Bond Donor | 1 | 0 |
| H-Bond Acceptor | 3 | 4 |
| Rotatable Bond Count | 6 | 6 |
| Tautomer Count | 2 | 0 |
| Exact Mass | 425.2137 | 416.026537 |
| MonoIsotopic Mass | 425.2137 | 416.026537 |
| Topological Polar Surface Area | 62.3 | 56.7 |
| Heavy Atom Count | 30 | 27 |
| Formal Charge | 0 | 0 |
| Complexity | 587 | 477 |
| Isotope Atom Count | 0 | 0 |
| Defined Atom StereoCenter Count | 0 | 0 |
| Undefined Atom StereoCenter Count | 0 | 0 |
| Defined Bond StereoCenter Count | 0 | 0 |
| Undefined Bond StereoCenter Count | 0 | 0 |
| Covalently-Bonded Unit Count | 1 | 1 |
4. Comparative data showing probe specificity for target in biologically relevant assays
As described in the CPDP these further biological studies are now on-going in the assay provider’s and collaborating laboratories, as post-probe nomination research and we hope to publish jointly in the future.
As defined in the CPDP and the initial teleconference calls with the National Institute on Drug Abuse, this probe project was unusual as there are already many examples of very potent agonists and antagonists of the kappa-opioid receptors with low nanomolar EC50 and IC50s, that are very selective against the mu- and delta- opioid subtypes. As emphasized during those initial discussions, the overarching purpose was to find new chemical scaffolds that are chemically distinct from the rich literature of known agonists and antagonists as starting points for further synthesis and work by the assay provider and their collaborative chemists. As a comparative measure, the Table 9 summarizes our two agonist probes against the precedent state-of-art probes, and clearly these two scaffolds the bis- amides and the triazoles provide novel scaffolds as was the main goal of this project. Clearly potency and selectivity remain to be improved further, however, the SAR helps to define some areas to improve these. The bisamide probe is within the same range of potency as U69,593 a commonly used reference compound, but lacks its exquisite selectivity against MOR and DOR. It also has comparable or slightly better selectivity against MOR and DOR than TRK-820 has. The second triazole probe is a less selective and less potent than the first bisamide probe, however, as pointed out in a previous section, it demonstrates the novel pharmacology of super-agonism as compared to the bisamide probe or most of these literature agonists.
Additionally this probe compound was submitted for broad GPCR paneling at the PDSP (see Table 6 above) and were both shown to be fairly selective to the opioid receptors. Furthermore, the relatively good agreement among the GPCR panel, cell-based beta-arrestin mediated enzyme complementation assay and the more downstream image based beta-arrestin mediated G-protein redistribution with these compounds is notable.
In conclusion, we consider this probe project to be successful in defining novel chemical scaffold distinct form the current literature examples. Future work beyond the scope of the MLCPN is needed to verify whether the potentially novel pharmacological of these probes will provide added value to their utility.
5. Bibliography
- 1.
- Cami J, Farre M. Drug addiction. N Engl J Med. 2003;349:975. [PubMed: 12954747]
- 2.
- Prisinzano TE, Tidgewell K, Harding WW. Kappa opioids as potential treatments for stimulant dependence. Aaps J. 2005;7:E592. [PMC free article: PMC2751263] [PubMed: 16353938]
- 3.
- Xuei X, Dick D, Flury-Wetherill L, Tian HJ, Agrawal A, Bierut L, Goate A, Bucholz K, Schuckit M, Nurnberger J Jr, Tischfield J, Kuperman S, Porjesz B, Begleiter H, Foroud T, Edenberg HJ. Association of the kappa-opioid system with alcohol dependence. Mol Psychiatry. 2006;11:1016. [PubMed: 16924269]
- 4.
- Prisinzano TE. Life Sci. Vol. 78. 2005. Psychopharmacology of the hallucinogenic sage Salvia divinorum; p. 527. [PubMed: 16213533]
- 5.
- Hasebe K, Kawai K, Suzuki T, Kawamura K, Tanaka T, Narita M, Nagase H. Possible pharmacotherapy of the opioid kappa receptor agonist for drug dependence. Ann N Y Acad Sci. 2004;1025:404. [PubMed: 15542743]
- 6.
- Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704. [PMC free article: PMC2751273] [PubMed: 16353947]
- 7.
- Hajduk PJ, Sauer DR. Statistical analysis of the effects of common chemical substituents on ligand potency. J Med Chem. 2008;51:553. [PubMed: 18173228]
- 8.
- Topliss JG. A manual method for applying the Hansch approach to drug design. J. Med. Chem. 1977;20:463. [PubMed: 321782]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Antagonist for the Kappa Opioid Receptor.[Probe Reports from the NIH Mol...]Review Antagonist for the Kappa Opioid Receptor.Hedrick MP, Gosalia P, Li K, Frankowski KJ, Shi S, Prisinzano TE, Schoenen F, Aubé J, Su Y, Stonich D, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Selective KOP Receptor Antagonists: Probe 1.[Probe Reports from the NIH Mol...]Review Selective KOP Receptor Antagonists: Probe 1.Hedrick MP, Gosalia P, Frankowski K, Whipple DA, Shi S, Prisinzano TE, Schoenen F, Aubé J, Su Y, Vasile S, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Characterization of kappa opioid receptor mediated, dynorphin-stimulated [35S]GTPγS binding in mouse striatum for the evaluation of selective KOR ligands in an endogenous setting.[Neuropharmacology. 2015]Characterization of kappa opioid receptor mediated, dynorphin-stimulated [35S]GTPγS binding in mouse striatum for the evaluation of selective KOR ligands in an endogenous setting.Zhou L, Stahl EL, Lovell KM, Frankowski KJ, Prisinzano TE, Aubé J, Bohn LM. Neuropharmacology. 2015 Dec; 99:131-41. Epub 2015 Jul 6.
- Opioid receptor involvement in food deprivation-induced feeding: evaluation of selective antagonist and antisense oligodeoxynucleotide probe effects in mice and rats.[J Pharmacol Exp Ther. 2004]Opioid receptor involvement in food deprivation-induced feeding: evaluation of selective antagonist and antisense oligodeoxynucleotide probe effects in mice and rats.Hadjimarkou MM, Singh A, Kandov Y, Israel Y, Pan YX, Rossi GC, Pasternak GW, Bodnar RJ. J Pharmacol Exp Ther. 2004 Dec; 311(3):1188-202. Epub 2004 Aug 27.
- NPY-induced feeding: pharmacological characterization using selective opioid antagonists and antisense probes in rats.[Peptides. 2005]NPY-induced feeding: pharmacological characterization using selective opioid antagonists and antisense probes in rats.Israel Y, Kandov Y, Khaimova E, Kest A, Lewis SR, Pasternak GW, Pan YX, Rossi GC, Bodnar RJ. Peptides. 2005 Jul; 26(7):1167-75. Epub 2005 Feb 26.
- Selective KOP Receptor Agonists: Probe 1 & Probe 2 - Probe Reports from the NIH ...Selective KOP Receptor Agonists: Probe 1 & Probe 2 - Probe Reports from the NIH Molecular Libraries Program
- GSM1649616[Accession] (3)GEO DataSets
Your browsing activity is empty.
Activity recording is turned off.
See more...