
 2.
2.
The Authors.
This open access article is licenced under Creative Commons Attribution 4.0 International (CC BY 4.0). https://creativecommons.org/licenses/by-nc/4.0/

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Ward WH, Farma JM, editors. Cutaneous Melanoma: Etiology and Therapy [Internet]. Brisbane (AU): Codon Publications; 2017 Dec 21. doi: 10.15586/codon.cutaneousmelanoma.2017.ch9
Matthew P. Rausch and Karen Taraszka Hastings.
Author Information and AffiliationsImmune checkpoint blockade has revolutionized the treatment of patients with advanced melanoma and many other cancers. Blockade of inhibitory receptors, CTLA-4 and PD-1, enhances T-cell-mediated antitumor immune responses, leading to improved survival and durable responses in patients. Based on their mechanism of action, immune checkpoint inhibitors can also induce immune-related adverse events that require careful monitoring and prompt treatment. Despite these successes, only a fraction of patients benefit from immune checkpoint blockade. Basic science approaches and clinical experience are defining predictive biomarkers to identify patients most likely to respond to therapy as well as mechanisms of resistance that limit responses in certain tumors or shorten the duration of response. New approaches and combination therapies are under development to broaden the clinical impact of immune checkpoint blockade by overcoming resistance to therapy and limiting adverse events.
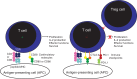
The development of immune checkpoint inhibitors has transformed the treatment of melanoma (1). Immune checkpoint inhibitors were the first class of therapy shown to improve the overall survival for patients with advanced melanoma. In fact, long-term, durable tumor regression has become a reality for some patients. However, only a subset of melanoma patients respond to immune checkpoint inhibitors, highlighting the need to identify biomarkers that are predictive of response and to develop strategies that overcome resistance. T-cell activation is a complex process that begins with the binding of a specific T-cell receptor (TCR) to its cognate peptide-MHC complex presented on the surface of an antigen-presenting cell (APC). Full T-cell activation requires co-stimulatory signals. CD28 is the major co-stimulatory receptor on T-cells, and by interacting with B7 family ligands CD80 and CD86 on APCs, CD28 promotes enhanced proliferation, IL-2 production, and T-cell survival (2) (Figure 1). In addition, T-cell activation involves the carefully balanced integration of a number of co-inhibitory signals delivered by immune checkpoint receptors. Immune checkpoints are a critical control mechanism to turn off T-cell responses and prevent destructive inflammation. The most extensively studied immune checkpoint receptors are cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and programmed cell death protein-1 (PD-1).
Initially cloned in the 1980s, CTLA-4 is a member of the immunoglobulin (Ig) gene superfamily with homology to CD28 (3). CTLA-4 is expressed on the surface of activated T-cells and regulatory T (Treg) cells. CTLA-4 inhibits T-cell activation during the priming phase of immunity (4–6) (Figure 1). Like CD28, CTLA-4 binds to the B7 ligands CD80 and CD86 on APCs, but unlike its homologue, CTLA-4 binds these ligands with a much higher affinity and does not deliver a positive signal (4, 6–8). Thus, CTLA-4 competitively inhibits the CD28:B7 interaction, leading to attenuation of co-stimulatory signaling. In addition, CTLA-4-expressing cells have been shown to capture and degrade CD80 and CD86 from the APC surface (9). The mechanism of action of CTLA-4-mediated T-cell suppression involves the inhibition of IL-2 production and blockade of cell cycle progression in T-cells following initial activation (5).
The expression of CTLA-4 is tightly regulated and dependent on T-cell activation. Unlike CD28, which is constitutively expressed by all T-cells, CTLA-4 expression is absent from naïve T-cells (10). CTLA-4 is only expressed after T-cell activation with transcript levels becoming detectable 1 h after TCR stimulation (10) and cell surface expression at the immunological synapse showing up 24–48 h post stimulation (6). Furthermore, the strength of T-cell stimulation is directly proportional to the level of CTLA-4 expression (11). In this way, CTLA-4 functions as a T-cell intrinsic inhibitory feedback mechanism that plays a vital role in shutting down T-cell-mediated immune responses. The critical importance of CTLA-4 in the control of T-cell-mediated immunity has been demonstrated in knockout animals, where CTLA-4-deficient mice develop a fatal lymphoproliferative disorder characterized by rapid T-cell proliferation and extensive tissue damage, resulting in death at 4 weeks of age (12, 13).
It was hypothesized that CTLA-4 could inhibit T-cell-mediated antitumor immune responses by attenuating tumor-specific T-cell activation before these T-cells have been able to eradicate tumors, and that blockade of CTLA-4 would enhance T-cell-mediated antitumor immunity by removing this inhibitory signal. In mice, antibody-mediated blockade of CTLA-4 induces complete tumor rejection and immunologic memory in several murine models of cancer (14). In addition, preclinical murine studies have shown that CTLA-4 blockade synergizes with radiation therapy (15), chemotherapy (16), molecularly targeted therapy (17), and tumor vaccination (18) to eradicate established tumors. Mechanistic studies in mice have shown that CTLA-4 blockade increases the ratio of effector T-cells to Foxp3+ Treg cells in tumors (19). Blockade of CTLA-4 on Treg cells is critical to CTLA-4 blocking antibody therapy. CTLA-4 plays a major role in Treg-cell-mediated immunosuppression. Genetic ablation of CTLA-4 on Treg cells results in fatal autoimmunity and is sufficient to induce tumor regression in some models (20). In addition, maximal antitumor activity of CTLA-4 blockade requires engagement of CTLA-4 on both effector and Treg-cell populations (21). Furthermore, anti-CTLA-4 monoclonal antibodies of particular isotypes, such as IgG1, induce depletion of intra-tumoral Foxp3+ Treg cells through antibody-dependent cell-mediated cytotoxicity by Fcγ receptor-expressing macrophages within the tumor microenvironment (22). This activity likely contributes to antitumor efficacy.
Based on the promising antitumor activity of CTLA-4 inhibition in preclinical cancer models, several CTLA-4-blocking antibodies have been developed. Ipilimumab is a fully human monoclonal antibody of the IgG1 isotype that binds CTLA-4, preventing it from interacting with its ligands (23). Based on encouraging results in early clinical studies of ipilimumab for metastatic melanoma, ipilimumab was advanced into Phase III trials. In the first Phase III study, previously treated patients with unresectable Stage III or Stage IV melanoma were treated with ipilimumab alone, ipilimumab with a glycoprotein 100 (gp100) melanoma-specific peptide vaccine, or gp100 alone (24). This study demonstrated improved overall survival in patients receiving ipilimumab (10.1 months for ipilimumab alone and 10.0 months for ipilimumab and gp100, compared with 6.4 months for gp100 alone) and led to the FDA approval of ipilimumab for patients with late stage, unresectable melanoma. The overall response rate, including complete and partial responses, was 10.9% for ipilimumab, 5.7% for ipilimumab and gp100, and 1.5% for gp100 alone. A subsequent study demonstrated a median overall survival benefit of ipilimumab plus dacarbazine compared to placebo and dacarbazine (11.2 months vs. 9.1 months) in previously untreated metastatic melanoma patients (25). Overall response rates were 15.2% for ipilimumab and dacarbazine versus 10.3% for placebo and dacarbazine. In addition, ipilimumab therapy has demonstrated promising results in a Phase II study of melanoma patients with brain metastases, who have historically been a difficult patient population to treat (26). Pooled analysis of overall survival data of Phase II and Phase III trials including previously treated and treatment naïve advanced melanoma patients revealed a median overall survival of 11.4 months with a plateau in the survival curve at 22% at 3 years, demonstrating the durability of responses to ipilimumab (27). Ipilimumab is also efficacious in the adjuvant therapy of Stage III melanoma patients with pathological involvement of regional lymph nodes. In a Phase III study of Stage III melanoma patients who have undergone complete surgical resection, ipilimumab improved both the 5-year recurrence-free survival (40.8% vs. 30.3% with placebo) and the 5-year overall survival (65.4% vs. 54.4% with placebo) (28), resulting in the FDA approval for ipilimumab for the adjuvant therapy of melanoma.
Based upon preclinical studies discussed above, the mechanism of action of ipilimumab is enhancing T-cell-mediated antitumor immunity through blocking an inhibitory receptor on effector T-cells and depleting Treg cells. Analysis of pre- and posttreatment TCR expression from melanoma patients reveals that ipilimumab treatment leads to the expansion of T-cell clones not detected before therapy and only rarely boosts the expansion of T-cell clones present before therapy (29). In this way, ipilimumab is thought to broaden the repertoire of responding melanoma-specific T-cells. In addition, IFN-γ is central to the antitumor activity in CTLA-4 blockade, and anti-CTLA-4 treatment increases IFN-γ production by T-cells in both mouse models and patients (30, 31).
A second CTLA-4-blocking antibody, tremelimumab, has been developed. Tremelimumab is a fully human anti-CTLA-4 monoclonal antibody of the IgG2 isotype. Despite promising early clinical data in melanoma, tremelimumab failed to hit its primary endpoint of improved overall survival in comparison to standard of care chemotherapy for patients with previously untreated, unresectable Stage III or Stage IV melanoma (32). As a result, clinical development for melanoma was halted, but evaluation of tremelimumab in other cancers is currently ongoing.
Given the ability of ipilimumab to enhance T-cell responses, ipilimumab treatment is associated with mechanism-based, immune-related adverse events. An early Phase II dosing study demonstrated a dose-dependent increase in immune-related adverse events with increasing ipilimumab dose (18% Grade 3 [severe] or Grade 4 [life-threatening] immune-related adverse events at 10 mg/kg vs. 5% Grade 3 or Grade 4 immune-related adverse events at 3 mg/kg) (33). Subsequent Phase III trials evaluated ipilimumab doses of 3 mg/kg and 10 mg/kg. The FDA-approved dose for melanoma treatment is 3 mg/kg every 3 weeks for four doses. In clinical trials, additional doses were given for stable disease or objective response. In the initial Phase III trial of ipilimumab at 3 mg/kg in patients with advanced melanoma, all immune-related adverse events developed during the induction and reinduction periods (24). Immune-related adverse events were generally reversible when managed with vigilant monitoring and systemic corticosteroids, as documented in the Risk Evaluation and Mitigation Strategy associated with the FDA approval. In the initial Phase III study of ipilimumab treatment of advanced melanoma, 17.4–22.9% of patients receiving ipilimumab experienced Grade 3 or Grade 4 treatment-related adverse events, with 10.2–14.5% of patients experiencing Grade 3 or Grade 4 immune-related adverse events. In addition, there were 14 treatment-related deaths, 7 of which were associated with immune-related adverse events. The most common sites for immune-related adverse events were the gastrointestinal tract and skin; 5.5–7.6% of ipilimumab-treated patients experienced Grade 3 or Grade 4 gastrointestinal immune-related adverse events, including diarrhea and colitis, and 1.5–2.3% of ipilimumab-treated patients had Grade 3 or Grade 4 skin immune-related adverse events, including pruritus, dermatitis, and vitiligo. Less frequently, patients experienced immune-related adverse events involving the endocrine system (hypothyroidism, hypopituitarism, hypophysitis, and adrenal insufficiency) or liver (hepatitis). Deaths associated with immune-related adverse events were a result of septicemia, bowel perforation, liver or multi-organ failure, or Guillain–Barre syndrome. In the second Phase III trial of ipilimumab treatment of advanced melanoma, treatment-related Grade 3 or Grade 4 adverse events occurring in 56.3% (41.7% due to immune-related adverse events) of patients receiving ipilimumab (10 mg/kg) and dacarbazine were increased compared with 27.5% (6% grade due to immune-related adverse events) of patients receiving dacarbazine and placebo (25). The FDA-approved dosing for the adjuvant therapy of melanoma is 10 mg/kg every 3 weeks for four doses followed by 10 mg/kg every 12 weeks for up to 3 years or until documented disease recurrence or unacceptable toxicity. In the Phase III trial of ipilimumab (10 mg/kg) in the adjuvant setting, there was an increased rate of adverse events in patients receiving ipilimumab versus placebo. Adverse events of Grade 3 or Grade 4 occurred in 54.1% (41.6% due to immune-related adverse events) and Grade 5 (death) occurred in 1.1% of patients receiving ipilimumab compared with 26.2% Grade 3 or Grade 4 (2.7% due to immune-related adverse events) in patients receiving placebo (28). The incidence of immune-related adverse events in this study of the adjuvant setting was higher than that observed with the same dose in pooled analysis involving the treatment of patients with advanced melanoma, and 40% of patients discontinued adjuvant therapy. Of note, systemic immunosuppression for the management of immune-related adverse events does not impact on antitumor activity, suggesting that the immune-related mechanisms responsible for these autoimmune side effects are uncoupled from the antitumor immune response (34). Table 1 summarizes the clinical efficacy and adverse events with ipilimumab in the treatment of advanced melanoma.
Ipilimumab Clinical Data in Advanced Melanoma Treatment.
Programmed cell death protein 1 (PD-1) is another immune checkpoint in the Ig superfamily (35, 36). Like CTLA-4, PD-1 inhibits T-cell activity and is expressed by activated T-cells (Figure 1). However, instead of competitively inhibiting co-stimulation by interfering with CD28/B7 ligand interaction, PD-1 negatively regulates TCR-signaling events. While CTLA-4 inhibits T-cells during the priming phase of immune responses, PD-1 is thought to inhibit activated T-cells at a later stage in peripheral tissues. In this way, PD-1 plays a critical role in the maintenance of peripheral T-cell tolerance. Consistent with the role of PD-1 in the prevention of autoimmunity, PD-1-deficient mice spontaneously develop late-onset autoimmunity, including lupus-like arthritis, glomerulonephritis, and cardiomyopathy, which is less severe, less frequent, and occurs later in life than CTLA-4-deficient mice (35–38).
PD-1 expression is absent on resting T-cells and is upregulated following activation (39, 40). Persistent T-cell stimulation, as present during chronic viral infection and cancer, induces high levels of PD-1 expression, which subsequently induces a state of T-cell exhaustion where T-cells gradually lose effector functions. PD-1 has two ligands, namely, PD-L1 (35, 41) and PD-L2 (42). PD-L1 is constitutively expressed on a variety of immune cells, including T-cells, B-cells, dendritic cells (DC), NK cells, monocytes, and macrophages (43), as well as a number of nonhematopoietic cells, including vascular endothelial cells (44) and many tumor cells (45). PD-L1 expression can also be upregulated by pro-inflammatory cytokines such as IFN-γ (44, 46). PD-L2 is expressed on APC and can be induced on tumor cells, including ~2% of melanoma cases (47).
The mechanism by which PD-1 inhibits T-cell activation is distinct from CTLA-4. The intracellular region of the PD-1 protein contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) that play critical roles in PD-1-mediated suppression (48). Binding of PD-1 to its ligands triggers the phosphorylation of its ITIM and ITSM domains which subsequently induces the recruitment of Src homology region 2 domain-containing phosphatase-1 (SHP-1) and phosphatase-2 (SHP-2) (48, 49). These phosphatases then dephosphorylate members of the TCR-signaling complex, resulting in the inhibition of T-cell activation. Signaling through PD-1 inhibits TCR-induced proliferation, cytokine secretion, and expression of the pro-survival gene, Bcl-xL (35, 39).
The PD-1/PD-L1 axis represents a critical immune escape mechanism for cancer. In murine models of melanoma, PD-L1 expression correlates with diminished antitumor CD8+ T-cell activity, and antitumor T-cell activity can be restored by genetic deletion of PD-1 on T-cells or by treatment with PD-L1-blocking antibodies (50). Tumor-specific T-cell populations from melanoma patients often express high levels of PD-1, and melanoma tumor-infiltrating CD8+ T-cells often display functional impairment consistent with exhaustion (51–55). Elevated PD-L1 expression has been observed on both tumor cells and immune cell infiltrates in many different cancers, including melanoma (46, 56–59). Expression of PD-L1 in melanoma is associated with immune cell infiltration of tumors. PD-L1 expression is often located in close proximity to CD8+ T-cell infiltrates, and IFN-γ produced by these lymphocytes can lead to the upregulation of PD-L1 expression (56, 58, 59). These findings suggest that PD-1/PD-L1 functions as an adaptive tumor immune escape mechanism and that infiltrating T-cells may induce their own suppression through the production of pro-inflammatory cytokines. In a mouse model of chronic infection, anti-PD-L1 antibody treatment reinvigorates exhausted T-cells, but only produces minimal memory, and T-cells reacquire the exhausted phenotype with persistent antigen, suggesting a limited duration of antitumor T-cell responses to blockade of the PD-1/PD-L1 axis (60).
In addition to the well-established role of PD-1 on T-cells, a recent study demonstrates that PD-1 has an intrinsic effect in melanoma cells (61). A portion of human melanoma cells express PD-1. In in vitro studies, mouse models and human xenografts, PD-1 expression on melanoma cells promotes tumor growth, and inhibition of PD-1 reduces melanoma growth independent of the adaptive immune system. Furthermore, anti-PD1 treatment in human melanoma patients is associated with diminished PD-1 receptor signaling in melanoma cells, and a high frequency of PD-1 receptor signaling in melanoma cells pretreatment is associated with improved progression-free survival (PFS).
Based on preclinical animal studies showing that blockade of the PD-1/PD-L1 signaling axis can restore the function of exhausted T-cells to mediate antitumor immunity, several PD-1-blocking antibodies have been developed, including nivolumab and pembrolizumab. PD-L1 blockade is also being explored. Although antibodies blocking PD-L1 have been FDA-approved in the treatment of urothelial carcinoma, nonsmall cell lung cancer and Merkel cell carcinoma, to date no anti-PD-L1 antibodies have received FDA approval in melanoma.
Nivolumab is a fully human monoclonal antibody of the IgG4 isotype that binds to PD-1, preventing it from interacting with its ligands. Early clinical studies of nivolumab showed promising antitumor activity against a variety of tumor types, including melanoma. Based on these results, Phase III trials were initiated to test nivolumab against standard of care chemotherapy, first in previously treated patients and then as a first-line treatment. In a Phase III trial enrolling Stage III or Stage IV melanoma patients who had failed prior ipilimumab or BRAF inhibitor therapy, nivolumab demonstrated activity in patients with and without BRAF mutations and had an objective response rate of 31.7% compared to 10.7% for patients on chemotherapy (62). In another Phase III trial, nivolumab was tested in treatment-naïve melanoma patients with wild-type BRAF (63). In this study, patients were treated with either nivolumab or dacarbazine, and the nivolumab group demonstrated improved efficacy in terms of 1-year overall survival (72.9% vs. 42.1%), median PFS (5.1 months vs. 2.2 months), and objective response rate (40.0% vs. 13.9%).
CTLA-4 and PD-1 induce T-cell suppression through nonoverlapping mechanisms and likely impact different populations of T-cells during different phases of the immune response (CTLA-4 during priming and PD-1 during the effector phase), providing a mechanistic rationale for the combination of CTLA-4 and PD-1 blockade. A subsequent Phase III trial in previously untreated melanoma patients compared nivolumab and ipilimumab combination therapy, nivolumab alone, and ipilimumab alone (64). The median PFS was 11.5 months, 6.9 months, and 2.9 months, respectively. The objective response rate was 57.6, 43.7, and 19.0%, respectively. The median PFS and the objective response rate were significantly improved in both the nivolumab and ipilimumab combination and the nivolumab alone groups compared with the ipilimumab group. Based on these studies, nivolumab is FDA-approved as a monotherapy in advanced melanoma patients with wild-type BRAF and received accelerated approval for monotherapy in patients with BRAFV600E mutation and in combination with ipilimumab.
A second anti-PD-1-blocking antibody was developed called pembrolizumab. Like nivolumab, pembrolizumab is a fully human monoclonal antibody of the IgG4 isotype that binds to human PD-1 preventing ligand interaction. A Phase II trial of advanced melanoma patients, who progressed on ipilimumab therapy or BRAF and/or MEK inhibitors, demonstrated improved PFS with pembrolizumab at both 2 mg/kg and 10 mg/kg doses every 3 weeks compared with investigators’ choice of chemotherapy (65). A randomized Phase III trial compared pembrolizumab every 2 weeks, pembrolizumab every 3 weeks, and ipilimumab in the first-line treatment of advanced melanoma (66). Pembrolizumab every 2 weeks and every 3 weeks demonstrated improved efficacy compared with ipilimumab, in terms of 1-year overall survival (74.1 and 68.4% vs. 58.2%), median PFS (5.5 months and 4.1 months vs. 2.8 months), and objective response rate (33.7 and 32.9% vs. 11.9%). Based on these studies, pembrolizumab at 2 mg/kg every 3 weeks is FDA-approved for the treatment of advanced melanoma.
The most common adverse events observed following PD-1 blockade are fatigue, rash, diarrhea, pruritus, and nausea (62–64, 66). A similar pattern of mechanism-based, immune-related adverse events are seen with PD-1 blockade as with CTLA-4 blockade with ipilimumab. The vast majority of Grade 3 and Grade 4 immune-related adverse events resolve quickly with delay in treatment and/or administration of systemic corticosteroids using established safety management guidelines (62, 63). Consistent with mouse studies in which the autoimmune pathology of PD-1-deficient mice is decreased in severity compared to CTLA-4-deficient mice, the toxicity associated with PD-1 blockade is diminished in comparison to CTLA-4 blockade. In a Phase III trial of head to head comparison of pembrolizumab and ipilimumab, both pembrolizumab groups had a significantly lower incidence of Grade 3–Grade 5 adverse events compared with ipilimumab (10.1–13.3% in the pembrolizumab groups vs. 19.9% in the ipilimumab group), despite an approximately 3-fold longer duration of pembrolizumab therapy (66). Although the combination of ipilimumab and nivolumab therapy resulted in improved clinical efficacy, the combination therapy group had a higher incidence of Grade 3 and Grade 4 adverse events compared with either nivolumab or ipilimumab alone (55.5% vs. 16.3% or 27.3%, respectively) (64). Table 2 summarizes the clinical efficacy and adverse events of Phase III trials of PD-1 blockade as a first-line treatment of advanced melanoma.
PD-1 Blockade Clinical Data in the First-Line Treatment of Advanced Melanoma.
Taken together, PD-1 blockade has become the first-line therapy for advanced melanoma patients, given its improved clinical efficacy and improved safety profile compared with ipilimumab. It remains to be determined whether PD-1 blockade results in the same long-term duration of response as ipilimumab.
The clinical development of CTLA-4 and PD-1-/PD-L1-blocking antibodies has had a profound impact on the treatment of melanoma and several other cancers. However, despite this success, only a minority of advanced melanoma patients respond to checkpoint blockade, with a 10–40% objective response rate with monotherapy and up to 58% with combined ipilimumab and nivolumab. As a result, considerable effort is being invested in the identification of predictive biomarkers to identify patients most likely to benefit from checkpoint blockade and those at high risk for treatment failure who would benefit from more aggressive combination therapy in order to limit unnecessary exposure to immune-related adverse events. Early clinical experience with immune checkpoint blockade has identified several biomarkers associated with treatment efficacy, including tumor mutational burden, the presence of tumor-infiltrating lymphocytes, PD-L1 expression, and intestinal microbiota.
The primary mechanism of action of checkpoint inhibitor therapy involves the activation of antitumor T-cells. Many of the tumor-specific T-cells recognize tumor expressed “neoantigens” that are a product of mutational events in tumor cells (67, 68). Since these mutations arise through a random process, it is thought that tumors characterized by a high overall mutational load are more likely to result in the formation of immunogenic neoantigens. Whole exome sequencing of melanoma patients treated with anti-CTLA-4 therapy revealed that antitumor responses were associated with high mutational load, and strong responders expressed a specific antigen signature (69). Similarly, neoantigen load was shown to correlate with clinical response in a second cohort of melanoma patients treated with ipilimumab (70). The clonality of neoantigen expression has also been correlated with clinical response to checkpoint inhibitors. Melanoma and nonsmall cell lung cancer patients with neoantigens expressed in all tumor cells (clonal neoantigens) experienced long-term clinical benefit to anti-CTLA-4 and anti-PD-1 treatment (71). However, patients with neoantigens expressed in only a subset of their tumor cells (subclonal neoantigens) responded poorly to checkpoint blockade (71).
Preexisting tumor-infiltrating lymphocytes have been associated with clinical responses to PD-1 blockade. In melanoma, response to pembrolizumab is associated with a higher number of CD8+, PD-1+, and PD-L1+ cells within the tumor and at the invasive margin at baseline; the proximity of PD-1+ and PD-L1+ cells at baseline; and an increased density and proliferation of CD8+ T-cells on treatment, suggesting the need for preexisting T-cells in the tumor inhibited by PD-1/PD-L1 interaction (59). Flow cytometric analysis of melanoma tissue biopsies from patients undergoing treatment with pembrolizumab also showed that patients who responded to therapy had increased frequencies of tumor-infiltrating CD8+ memory T-cells compared to nonresponders (72).
Although early clinical trials showed an association between PD-L1 expression and objective response in patients with metastatic melanoma treated with PD-1 or PD-L1 blockade, patients whose tumor cells lacked PD-L1 expression still benefited from PD-1/PD-L1 blockade. Phase III trials of PD-1 blockade have demonstrated similar results. Although the subgroup of patients with tumor cells that were PD-L1+ had numerically higher objective response rates, patient subgroups with tumor cells that were PD-L1+ and PD-L1− both demonstrated improved overall survival and objective response rates when treated with nivolumab compared with dacarbazine (63). An overall survival benefit of pembrolizumab compared with ipilimumab was not observed in the subgroup of patients with PD-L1− tumor cells; however, the sample size of PD-L1− patients was too small to draw definitive conclusions (66). The patient subgroup with PD-L1+ tumor cells had the same PFS with combination ipilimumab and nivolumab therapy as with nivolumab alone, whereas PFS in the subgroup with PD-L1− tumor cells was improved with combination therapy versus nivolumab alone, suggesting that patients with PD-L1− tumor may have greater benefit from combination therapy (64). Yet, 41.3% of patients with PD-L1− tumors had an objective response to nivolumab alone (64). Analysis and interpretation of PD-L1 expression in tumors is complicated by multiple factors. Different trials used different anti-PD-L1 antibodies and immunohistochemical assays and different cutoff points for defining PD-L1 positivity. While most studies have assessed PD-L1 expression on tumor cells, PD-L1 expression on T-cells and macrophages may influence response to PD-1 blockade (57, 73). PD-L1 expression measured at one time point and in one metastasis is not representative, as PD-L1 expression is dynamic and differs in different metastases from the same patient (57, 66). Lastly, other PD-1 ligands may be involved in response to PD-1 blockade. In summary, clinical experience to date indicates that lack of PD-L1 expression in tumor cells is not a reason to withhold anti-PD-1 therapy.
In murine models, the presence of certain species of intestinal bacteria is associated with spontaneous antitumor immunity, and the presence of these bacteria can improve responses to CTLA-4 and PD-1 blockade (74, 75). In addition, T-cells, specific for some of these bacteria, are found in melanoma patients responding to anti-CTLA-4 treatment (75). The mechanism by which intestinal microbiota modulate antitumor immune responses is thought to involve the activation of innate immune cells, including DC, making them better able to stimulate T-cells. Alternatively, specific antigens from these bacteria may mimic antigens expressed by the tumor, leading to the activation of tumor cross-reactive T-cells.
As antigen load is a key factor in T-cell exhaustion in preclinical models, more integrated strategies to predict the response to therapy are under investigation. These strategies incorporate immune status and tumor burden. A recent study found that the magnitude of reinvigorated, exhausted CD8+ T-cells in the peripheral blood on treatment with pembrolizumab in relationship to the pretreatment tumor burden correlated with clinical response, suggesting a clinically accessible on-treatment predictor of response (76). A more comprehensive strategy called the “cancer immunogram” incorporating the tumor mutational load, general immune status of the patient, immune cell infiltration of the tumor, absence of checkpoints, absence of soluble inhibitors, absence of inhibitory metabolism, and sensitivity to immune effectors is also under development (77).
Recent clinical experience has uncovered several resistance mechanisms to immune checkpoint blockade. These resistance mechanisms involve changes to the tumor microenvironment that limit T-cell activation, tumor infiltration, and effector-mediated destruction of tumor cells. A lack of tumor-associated antigens can impair tumor-specific T-cell activation and allows tumors to escape immune checkpoint blockade. Failure of tumor antigen presentation can occur as a result of outright antigen loss or from defects in components of antigen processing and presentation pathways. Failure of tumor antigen presentation is a major mechanism by which tumors escape from T-cell-mediated immune recognition (78, 79). Analysis of pretreatment and posttreatment tumor samples from patients with nonsmall cell lung cancer treated with checkpoint blockade revealed the loss of several neoantigens from treatment-refractory tumor cell clones (80). These neoantigens were capable of stimulating T-cell responses in vitro, and their loss coincided with the emergence of disease resistance. Mutations in β2-microglobulin, a protein required for the folding and transport of MHC Class I to the cell surface, have also been observed in melanoma patients at the time of anti-PD-1 treatment failure (81).
Mechanisms that inhibit T-cell trafficking to tumor tissue also cause resistance to immune checkpoint inhibitors. Mutations in BRAF and loss of PTEN expression both contribute to immune checkpoint blockade resistance in murine models and patients by inducing the production of a number of immunosuppressive proteins, including VEGF that limits T-cell trafficking to tumor sites and inhibits T-cell effector functions (82, 83). In addition, melanoma patients whose tumors had elevated signaling activity in the WNT/β-catenin pathway lacked infiltrating T-cells, and murine studies have shown that WNT signaling can promote anti-PD-L1/anti-CTLA-4 treatment failure in melanoma models (84).
Mutations in genes involved in the IFN-γ signaling pathway also contribute to both primary and acquired resistance to immune checkpoint blockade. IFN-γ signaling plays a critical role in T-cell-mediated antitumor immunity by enhancing MHC expression and subsequent tumor antigen presentation, inducing the recruitment of other immune cells, inhibiting tumor cell proliferation, and inducing tumor cell apoptosis (85). IFN-γ binds to the interferon gamma receptor 1 (IFNGR1) and interferon gamma receptor 2 (IFNGR2) and signals through the Janus-activated kinase 1 (JAK1) and Janus-activated kinase 2 (JAK2)/signal transducer and activator of transcription 1 (STAT1) signaling pathway (85). Ipilimumab-refractory melanoma tumors were insensitive to IFN-γ signaling due to mutations in IFNGR1, IFNGR2, JAK2, and interferon regulatory factor 1 (IRF1) which is responsible for the INF-γ-induced upregulation of PD-L1 (86). Mutations in JAK1 and JAK2 were also found in melanoma and colorectal cancer patients who failed to respond to anti-PD1 despite having tumors with high mutational load (87, 88). Similar mutations in JAK1 and JAK2 were also detected in relapsing tumors from melanoma patients who initially responded to anti-PD-1 therapy, indicating that loss of responsiveness to IFN-γ signaling may be a potential tumor escape mechanism contributing to relapse following immune checkpoint blockade (81).
Tumor-extrinsic mechanisms of resistance to immune checkpoint blockade have also been identified, including additional immune checkpoint receptors, immunosuppressive cytokines, and other factors present in the tumor microenvironment and immunosuppressive immune cell populations. In addition to CTLA-4 and PD-1, several other immune checkpoint receptors have been identified including lymphocyte activation gene 3 (LAG-3), T-cell immunoglobulin and mucin 3 (TIM-3), V-domain Ig-containing suppressor of T-cell activation (VISTA), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT) that are expressed by T-cells and negatively regulate immune responses (89). These checkpoints are often co-expressed by CTLA-4 and PD-1-expressing T-cells within tumors, and their expression can be upregulated following anti-CTLA-4 and anti-PD-1 treatment (89).
Immunosuppressive factors in the tumor microenvironment produced by tumor cells and infiltrating immune cells may also cause immune checkpoint blockade resistance by inhibiting T-cell activity. TGF-β is an immunosuppressive cytokine produced by many different human tumor types that may limit the efficacy of checkpoint blockade by stimulating Treg cells and impairing T-cell function (90). In addition, indoleamine-pyrrole 2,3-dioxygenase (IDO), an enzyme responsible for the breakdown of tryptophan, is expressed in many tumors and may inhibit T-cell proliferation by depleting tryptophan (89). CD73, an ecto-enzyme responsible for mediating the catalysis of adenosine monophosphate to adenosine, is expressed by many tumors and is associated with anti-PD-1 resistance in murine models (91). Elevated levels of adenosine, as a result of CD73 expression, suppress T-cell activity by signaling through the adenosine receptor 2A (91).
Certain tumor-infiltrating immune cell populations also contribute to immune checkpoint blockade resistance. Myeloid-derived suppressor cells (MDSCs) are a CD11b+CD33+ myeloid cell population that plays an immunoregulatory role in a number of disease states, including cancer (92). MDSCs are immunosuppressive and contribute to angiogenesis, tumor invasion, and metastasis (93, 94). In addition, pretreatment MDSC frequencies are inversely correlated with clinical responses to ipilimumab and nivolumab in melanoma patients (95, 96).
These resistance mechanisms must be overcome in order to improve the clinical efficacy of immune checkpoint blockade for melanoma and other cancers. A number of strategies are currently being tested to target additional sources of immunosuppression in the tumor microenvironment for use in combination with immune checkpoint inhibitors. Tumor-specific peptide and cell-based vaccines are being tested in combination with CTLA-4 and PD-1-/PD-L1-blocking antibodies in order to boost antitumor T-cell responses (97). Molecularly targeted agents are also being combined with immune checkpoint inhibitors. BRAF inhibition, which is FDA-approved for the treatment of metastatic melanoma expressing the activating BRAFV600E mutation, has been shown to increase MHC expression, tumor antigen presentation, and T-cell infiltration (98–102). Similarly, MEK inhibitors have been shown to improve CD8+ T-cell activity in preclinical models in combination with PD-1 blockade (103). Given the clinical success of CTLA-4 and PD-1/PD-L1 inhibition, blocking antibodies have been developed to target additional immune checkpoints, including LAG-3, TIM-3, VISTA, and TIGIT, and these agents have entered clinical trials alone and in combination with anti-CTLA-4 and anti-PD-1/PD-L1 (89, 97). Furthermore, numerous treatments are being developed to target immunosuppressive cytokines and other factors present in the tumor microenvironment, including IDO inhibitors, CD73 blocking antibodies, and adenosine receptor 2A antagonists (97). Finally, strategies to deplete or reprogram MDSC are also under development for use in combination with immune checkpoint blockade. Signaling through the gamma isoform of phosphatidylinositide 3-kinase (PI3Kγ) is critical for the maintenance of myeloid cell immunosuppression in tumors, and genetic deletion or pharmacologic inhibition of PI3Kγ results in tumor-infiltrating myeloid cells with a more pro-inflammatory phenotype in murine models (104). In addition, the small molecule PI3Kγ inhibitor IPI-549 improved the ability of immune checkpoint blockade to induce tumor regression in preclinical murine models of melanoma, breast, and head and neck cancer (104, 105).
Immune checkpoint inhibitors have revolutionized the treatment of melanoma and many other cancers. Blocking antibodies to CTLA-4 and PD-1/PD-L1 have improved survival for many patients, and long-term durable responses have been observed in some patients. However, despite this promise, clinical benefit from immune checkpoint blockade is only seen in a minority of melanoma patients, and autoimmune toxicity, while manageable, requires careful monitoring. Clinical experience with anti-CTLA-4 and anti-PD-1/PD-L1 therapy has uncovered critical parameters that govern effective antitumor immune responses. This knowledge is leading to the identification of subsets of patients most likely to respond to therapy with immune checkpoint inhibitors. In addition, these insights have identified new immune targets that promise to expand the clinical reach of immunotherapy to more patients and cancer types.
Acknowledgments: This work was supported in part by National Institutes of Health grant R03-AR063259 and grants from the Valley Research Partnership at the University of Arizona College of Medicine—Phoenix and the Skin Cancer Institute at the University of Arizona Cancer Center.
Conflict of interest: Matthew P. Rausch is an employee of Surface Oncology Inc. and was an employee of Infinity Pharmaceuticals from 2015–2016. Karen Taraszka Hastings declares no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Copyright and permission statement: To the best of our knowledge, the materials included in this chapter do not violate copyright laws. All original sources have been appropriately acknowledged and/or referenced. Where relevant, appropriate permissions have been obtained from the original copyright holder(s).
This open access article is licenced under Creative Commons Attribution 4.0 International (CC BY 4.0). https://creativecommons.org/licenses/by-nc/4.0/
Your browsing activity is empty.
Activity recording is turned off.
See more...