

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
The pituitary gland, or hypophysis, and adjacent structures of the sellar region can be affected by a wide range of pathologies leading to endocrine and neurological disorders. These include neoplasms arising from the adenohypophysis, such as pituitary adenomas associated with distinctive endocrine disorders such as acromegaly or Cushing’s disease; cysts or neoplasms derived from remnants of Rathke’s pouch (Rathke’s cleft cyst, craniopharyngioma); tumours of the neurohypophysis and pituitary stalk (pituicytoma, granular cell tumour) and neoplasms of the parasellar bone (chordoma). Further, conditions like lymphocytic or granulomatous hypophysitis may mimic pituitary neoplasms. Here, we provide an overview of the molecular pathogenesis and neuropathological features of these common lesions. For complete coverage of this and related areas of Endocrinology, please visit our free web –book, www.endotext.org.
PITUITARY ADENOMAS
Definition
Pituitary adenomas are benign clonal neoplasms of the neuroendocrine epithelial cells of the adenohypophysis.
General Features
Pituitary adenomas share characteristics with other adenomas of endocrine glands: granular cytoplasm, round nuclei with finely dispersed chromatin and multiple distinct nucleoli; they generally also express both markers of neurosecretory granules (synaptophysin, chromogranin) as well as epithelial differentiation (cytokeratins). However, they may present with a wide range of morphological features depending on hormonal or genetic subtype, or as a result of treatment effect. Although called benign, pituitary adenomas can be locally invasive and destructive, or clinically malignant due to the metabolic consequences of excess hormone secretion.
Pituitary adenomas are common intracranial neoplasms and may be clinically silent, detected incidentally on MRI scans of the brain (~ 22%), or found at autopsy (~ 14%)(1). It has been estimated that they represent approximately 25% of all clinically manifest intracranial neoplasms.
WHO Classification
Pituitary adenomas can be classified in various ways, according to size, clinically functional or silent manifestation, hormone or cytokeratin expression profile, defining somatic mutations, and histologic features. The 2004 edition of the WHO classification of endocrine tumours uses markers of cytodifferentiation as the principal classifier. In addition to the category of ‘typical pituitary adenoma’ and ‘pituitary carcinoma’, it also introduced the concept of ‘atypical pituitary adenoma’. However, the latter is controversial (2), as the criteria are to some degree subjective and the clinical significance of ‘atypia’ as currently defined remains to be determined in longitudinal studies (3). The current classification is summarised in Table 3b.1
Table 3b1 Classification of pituitary adenomas (Adapted from reference (4))
| Adenoma type | Transcription Factors | Hormones | Cytokeratin |
| GH-producing adenomas | |||
| Densely granulated somatotroph adenoma | Pit-1 | GH, a-SU | diffuse |
| Sparsely granulated somatotroph adenoma | Pit-1 | GH | dot-like |
| Mammosomatotroph adenoma | Pit-1, ER | GH, PRL, a-SU | diffuse |
| Mixed somatotroph and lactotroph andenoma | Pit-1, ER | GH, PRL, a-SU | diffuse |
| PRL-producing adenomas | |||
| Sparsely granulated lactotroph adenoma | Pit-1, ER | PRL (Golgi) | diffuse |
| Densely granulated lactotroph adenoma | Pit-1, ER | PRL (diffuse) | diffuse |
| Acidophil stem-cell adenoma | Pit-1, ER | PRL (diffuse), GH | rare dot-like |
| TSH-producing adenoma | |||
| Thyrotroph adenoma | Pit-1, GATA-2 | b-TSH, a-SU | diffuse |
| ACTH-producing adenomas | |||
| Densely granulated corticotroph adenoma | Tpit | ACTH | diffuse |
| Sparsely granulated corticotroph adenoma | Tpit | ACTH | diffuse |
| Crooke’s cell adenoma | Tpit | ACTH | ring-like |
| Gonadotropin-producing adenoma | |||
| Gonadotroph adenoma | SF-1, GATA-2, ER | b-FSH, b-LH, a-SU | diffuse |
| Plurihormonal adenomas | |||
| Silent type III adenoma | Pit-1 (?), ER | multiple | diffuse |
| Unusual plurihormonal adenoma (NOS) | multiple | multiple | n/a |
| Hormone negative adenoma | |||
| Null cell adenoma | none | none | diffuse |

Figure 3b-1
Principles of pituitary adenoma classification. Clinical and neuropathological classification schemes vary in their emphasis. This chapter will use a pathological / cell-lineage based approach. Compared with other intracranial neoplasms, molecular genetic, epigenetic or proteomic classification schemes that influence therapeutic decisions are only beginning to emerge.
Gh-Producing Adenoma
Definition
Benign lesions arising from Pit-1 lineage cells of the anterior pituitary that express, store and secrete growth hormone. The classical cause of acromegaly or gigantism.
Pathology
Somatotroph adenomas (SA) occur in the anterior pituitary, arising from growth hormone-producing cells, often in the lateral wings of the gland. They account for 10-15% of pituitary adenomas. T1-weighted MRI imaging shows a sellar structure that is hypointense relative to normal gland. Invasion of the sphenoid or cavernous sinus or suprasellar extension to give the characteristic snowman shape may be seen (5). Lesions are non- or slowly enhancing. Macroscopically, somatotroph adenomas are soft tan-to-grey lesions. Microscopically, somatotroph adenomas occur as two major variants: densely and sparsely granulated.
Densely Granulated Somatotroph Adenoma
Densely granulated somatotroph adenomas (DGSAs) are the most common finding and are composed of large, round, eosinophilic cells with spherical nuclei and prominent nucleoli that closely resemble somatotroph cells. They are diffusely and strongly immunopositive for growth hormone and may also variably express prolactin and less frequently, thyroid-stimulating hormone. Nuclei are strongly immunopositive for Pit1. Ultrastructurally, they contain a well-developed endoplasmic reticulum, a prominent Golgi complex and numerous, large (300-600nm) secretory granules containing growth hormone that are distributed throughout the cytosol. Growth hormone is expressed throughout the lesion. Immunostaining with CAM5.2 antibody against cytokeratin (predominantly cytokeratin 8) reveals a diffuse cytosolic pattern.
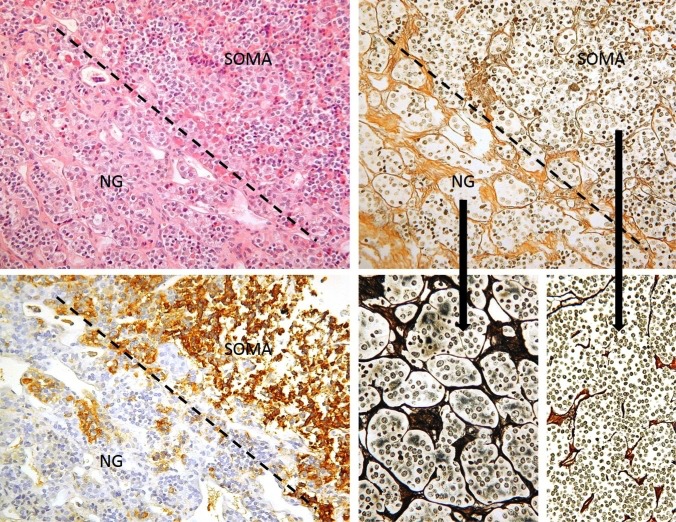
Figure 3b-2
Normal anterior gland (NG) and somatotroph adenoma (SOMA) interface. This figure illustrates histological principles of distinction of adenoma from normal gland, which may be difficult on routine HE stains (top left), but is greatly aided by a reticulin stain (top and bottom right). The normal adenohypophysis consists of very well demarcated cell nests separated by dense septa. Bottom left: Serial section to the top row images stained for GH. Dashed line: Border between normal gland and adenoma.
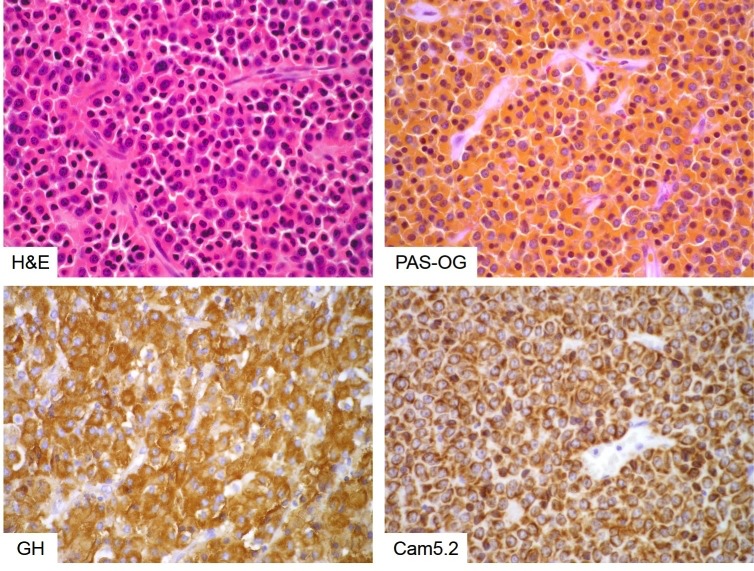
Figure 3b-3
Densely granulated somatotroph adenoma. Densely granulated adenomas consist of monomorphic cells that are eosinophilic on HE stain (top left) and intensely orangeophilic on PAS-OG histochemistry (top right). They show strong, diffuse GH expression (bottom left) and an evenly distributed, cell-membrane-anchored keratin cytoskeleton (bottom right).
Sparsely Granulated Somatotroph Adenoma
Sparsely granulated somatotroph adenomas (SGSAs) are less common and are composed of sheets of poorly cohesive, chromophobic cells often containing eccentric, pleiomorphic nuclei. SGSAs are weakly and focally immunopositive for growth hormone and nuclei are immunopositive for Pit1. They may also variably express prolactin and less frequently, thyroid-stimulating hormone. SGSAs contain dense juxtanuclear deposits of low-molecular weight cytokeratin, termed fibrous bodies that appear as pale spherical inclusions on H&E staining and are strongly immunopositive with CAM5.2 antibodies against cytokeratin (predominantly cytokeratin 8). Ultrastructurally, SGSAs contain few, small (100-250nm) growth-hormone containing granules that align along the plasma membrane. The distribution of cytokeratin and growth hormone-containing granules co-segregate with tumour variant type, so the presence of fibrous bodies is a diagnostic feature of SGSA.
There is a growing body of evidence that DGSAs and SGSAs behave differently with SGSAs being larger, more common in younger, female patients, more proliferative (higher MIB1 indices) and with a greater capacity to invade surrounding structures (6-11). Some studies have found that SGSAs are more poorly responsive to somatostatin treatment than DGSAs (7) although the extent of the impact of tumour subtype on behaviour is unclear.

Figure 3b-4
Sparsely granulated somatotroph adenoma. Sparsely granulated somatotroph adenoma cells are pleomorphic and chromophobe on HE stain (top left) and PAS-OG histochemistry (top right). Fibrous bodies can be seen as pale discs in the cytoplasm on routine stains (arrows). As the name implies, sparsely granulated cells show weak, patchy GH expression (bottom left) and their keratin cytoskeleton is disrupted and condensed into a paranuclear globular structure – the fibrous body (bottom right).
Mixed Pattern Somatotroph Adenoma
SAs that contain cells of both the densely granulated and sparsely granulated type are not uncommon and if more than 30% of cells differ from the predominant cell type, a diagnosis of mixed pattern is required. Very occasionally, SAs that are not immunopositive for low molecular weight cytokeratin are seen although their clinical significance is not known.

Figure 3b-5
Mixed densely and sparsely granulated somatotroph adenoma. A proportion of somatotroph adenomas demonstrate a clearly segregated mixed densely-sparsely phenotype. The respective cells remain true to their ‘pure’ counterparts: The sparsely granulated cells can be identified as chromophobe islands amongst orangeophilic densely granulated cells (left) and their contrasting cytokeratin pattern is absolutely clear following incubation with Cam5.2 antibody (right).
Somatotroph Adenoma With Neuronal Differentiation
A rare but pathologically intriguing subtype of SA, always associated with acromegaly and usually presenting as macroadenoma with or without hypothalamic involvement, shows sparsely granulated GH-producing cells admixed with large atypical ganglion cells. These resemble tumour cells seen in gangliocytomas and represent truly metaplastic tumour cells, as they express a mixture of lineage markers that otherwise are virtually never co-expressed (synaptophysin, neurofilament, cytokeratin and GH). This is of no known clinical relevance and the mechanisms of transdifferentiation remain unexplored.
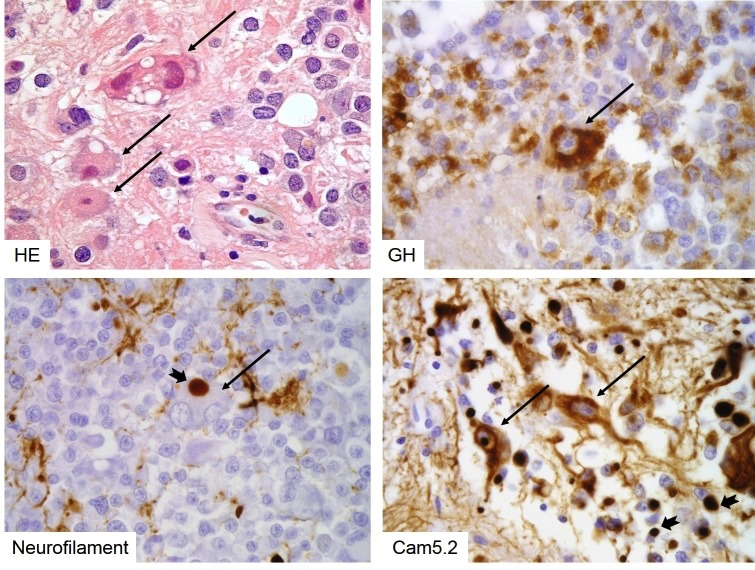
Figure 3b-6
Sparsely granulated somatotroph adenoma with neuronal metaplasia. Atypical large, neoplastic ganglion cells can be seen in rare sparsely granulated somatotroph adenomas (long arrows). These cells show true metaplasia, expressing GH (top right), neurofilament (bottom left) and cytokeratins (bottom right). Note that both types of intermediate filaments aggregate in fibrous bodies (notched short arrows).
Somatostatin Analogue Effect On Somatotroph Adenomas
Densely granulated somatotroph adenomas tend to respond better to somatostatin analogue treatment than sparsely granulated tumours. This results in a distinct perivascular hyaline / fibrous reaction. The reaction of somatotroph adenomas to somatostatin analogues is morphologically distinct to that of prolactinomas to dopamine agonists (see figure 3b-9 later in this chapter).
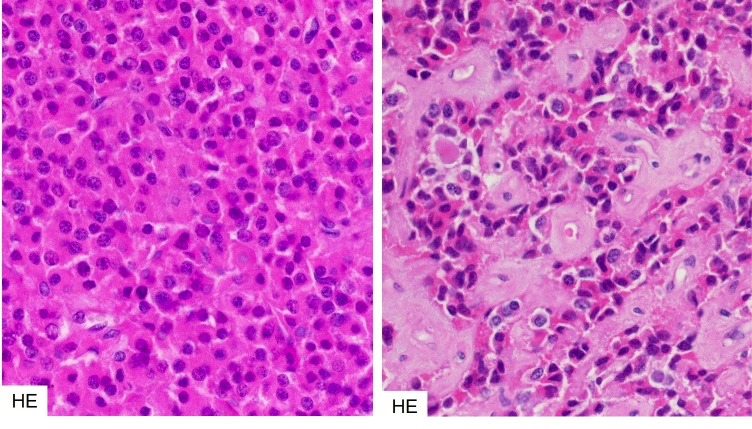
Figure 3b-7Somatostatin analogue effect in densely granulated somatotroph adenomas.
Densely granulated somatotroph adenomas that have responded to somatostatin analogues tend to show perivascular hyaline/fibrous degeneration. Left – untreated, right – treated.
Molecular Genetics
G protein α-subunit
One of the earliest mutations to be associated with sporadic somatotroph adenomas was at the GNAS complex locus. This locus contains four alternative promoters and 5’ exons and has a complex, imprinted expression pattern. Different isoforms of the G protein alpha subunit arise as a result of alternative splicing. The GNAS gene encodes the G protein alpha subunit Gsα, which couples seven-transmembrane receptors to adenylyl cyclase (12). Mutation at either Arg201 or Gln227, destroys GTPase activity (13). Gsα mutation leads to constitutive activation of adenylyl cyclase (termed the gsp oncogene) resulting in increased cAMP synthesis. Mutations in GNAS have been identified in 15-58% of somatotroph adenomas (6,8,11,13-19) (6,8,11,13-19)This mutation may promote tumorigenesis since cAMP can function as a mitogenic signal.
The functional implications of gsp mutation are not determined. Elevated cAMP may be countered by increases in the activity of phosphodiesterase enzymes (PDEs) especially PDE4, which is 7-fold more active in adenomas with a mutation in Gsα. Inhibition of this enzyme resulted in an increase in cellular cAMP (20). However, the effect of Gsα mutation on downstream target genes induced by CREB is uncertain and although increases in the expression of these genes were observed in some cases, they were not consistent (21). No association was observed between mutation in gsp and the granulation pattern of somatotroph adenomas (6,8,22)
Ghrelin And Receptor
There is growing evidence to suggest a role for ghrelin in somatotroph adenomas. Ghrelin (GHS) is a growth hormone secretagogue that acts on the pituitary and has been associated with increased cell migration and proliferation in certain cancers (23,24). Two forms of the ghrelin receptor (GHSR) GHSR1α and the non-functional splice-variant GHSR1β which contains all of exon 1 and some of the following intron are differentially expressed in normal somatotrophs compared to somatotroph adenomas, although there is disagreement concerning which isoform mRNA is more abundant in adenomas compared to normal pituitary (25,26). In different studies, GHSR1α mRNA has been shown to be both reduced (27) and increased (28) in somatotroph adenomas that have a GNAS mutation compared to wild type adenomas. It is unclear whether GHS/GHSR1α. expression is related to adenoma subtype.
Somatostatin receptor
Differing expression of the somatostatin receptor between adenoma subtypes has been observed and this pattern can be influenced by somatostatin analogue (SSA) treatment. A positive correlation has been observed between SSTR2 expression and reduction in GH after SSA treatment (19,29,30). Greater expression of SSTR2 has also been associated with densely granulated adenomas (31), while SSTR5 was associated with sparsely granulated tumours (15,22,30). One study suggests that the proportion of cells expressing SSTR2 is a more reliable indicator of response to SSA than overall expression level (7,32)
Aryl hydrocarbon interacting protein
Mutations in AIP associated with FIPA are covered elsewhere (Section 11a1, Stiles and Korbonits).
Mutations in AIP (aryl hydrocarbon interacting protein) are most frequently associated with somatotroph adenomas. They are generally truncations or nonsense mutations leading to loss of function, which has resulted in the classification of AIP as a tumour suppressor gene, although the mechanism by which it functions is not yet known. Consistent with its purported tumour suppressor role, multiple different mutations are seen in AIP, with some “hotspots” (33-41). Among patients with acromegaly, germline mutation in AIP is rare, but is relatively more common in the young; the reported incidence of AIP mutations in sporadic somatotroph adenomas varies from 4.2% (patients < 40 years) (42) to 5.5-13% (patients <30 years) (43,44). Mutation in AIP in somatotroph adenomas is associated with larger tumours and more invasive behaviour and more recurrences (45). Furthermore patients with AIP mutations are relatively resistant to treatment with somatostatin analogues although the mechanism of this resistance remains to be clarified (35,39). Treatment with SSAs leads to and is associated with upregulation of AIP expression, (34,46). The mechanism for this upregulation is not fully understood, but some authors have proposed that it is ZAC1- (zinc finger regulator of apoptosis and cell cycle arrest) mediated. ZAC1 induces G1 cell cycle arrest and apoptosis (47-49). Low levels of AIP expression have been linked to tumour invasiveness (46) suggesting that patients with AIP mutation require more stringent follow-up.
Gpr101 Mutations And X-Lag
A study of early childhood onset gigantism with growth hormone hypersecretion found heritable microduplications on chromosome Xq26.3. The condition was termed X-LAG or x-linked acrogigantism (50). Analysis of the expression of the genes encoded in this region in a small number of patients showed that GPR101 mRNA was upregulated by up to 1000-fold. In a screen of 248 patients with sporadic acromegaly, there were no microduplications at Xq26.3, but in 11 (4.4%) patients, a mutation in GPR101 (c.924G-C; pE308D) was found that was not present in control samples. In 3 cases, the mutation was also observed in blood and presumed to be germline; in one case, it was a somatic mutation. In a screens of 263 patients with gigantism or acromegaly and 579 patients with acromegaly, the incidence of GPR101 mutation was shown to be 1.1% and 0.69% respectively (51,52)
GPR101 encodes an orphan G-protein-coupled receptor that is predicted to bind the stimulatory G protein and regulate activation of adenylyl cyclase. Overexpression of this mutated form of GPR101 in rat GH3 somatotroph cells resulted in increased proliferation and growth hormone secretion, along with increased cAMP signalling. In rare cases of sporadic acromegaly, mutation in GPR101 may upregulate cAMP signalling and promote growth hormone secretion and tumorigenesis.
Micro Rna In Somatotroph Adenomas
Recently, miRNA profiling of pituitary adenomas has shown that miR-23a, miR-23b, and miR-24-2 expression were increased in these somatotroph adenomas along with prolactinomas (53). The function of these miRNAs is unknown. Microarray analysis of somatotroph adenomas and normal pituitary gland showed significant downregulation of miR-34b, miR-326, miR-432, miR-548c-3p, miR-570 and miR-603 in adenomas. Among the targets of these miRNAs are high-mobility group A1 (HMGA1), HMGA2 and E2F1, genes whose activation plays a role in pituitary tumorigenesis. Overexpression of these miRNAs resulted in reduced growth of pituitary adenoma cell lines (54).
Epigenetic Regulators Of Somatotroph Adenoma Progression
A number of studies propose an epigenetic mechanism of pituitary somatotroph tumorigenesis. The expression of the adherens junction component E-cadherin has been shown to be significantly lower in sparsely than densely granulated adenomas and lower levels of E-cadherin correlate with larger tumour size, invasiveness, GH and IGF-1 levels and poor acute response to SSAs (55). A regulator of alternative splicing that promotes the epithelial phenotype (ESRP1) was found to be expressed at much lower levels in tumours that did not express E-cadherin (56). The role of ESRP1 in somatotroph adenomas is yet to be clarified, but tumours expressing low levels of ESRP1 also expressed low levels of proteins involved in regulation of the SNARE complex, vesicle trafficking and calcium signalling (56).
Somatotroph Adenomas Are Not Associated With Recurrent Genetic Alterations
Whole genome and exome sequencing of somatotroph adenomas has not identified recurrent genetic alterations other than those in Gsα. Pathway analysis has suggested that mutation events were associated with the cAMP pathway and calcium signalling pathway (57,58).
PRL-PRODUCING ADENOMA
Definition
A Pit-1-lineage derived adenoma expressing mostly prolactin and containing characteristic ultrastructural secretory granules demonstrating ‘misplaced exocytosis’.
Pathology
Lactotroph adenomas are the most common hormone-secreting pituitary adenomas. Two types are distinguished according to their granularity – sparsely and densely granulated. A third, very rare subtype, is the so-called acidophil stem cell adenoma. Prolactinomas in women are often detected at younger age and smaller size than in men. This has been attributed to the clinical syndrome associated with these tumours in women, but some observations suggest that lactotroph macroadenomas in men may be biologically different and behave more aggressively. The typical functional lactotroph adenoma consists of sheets of either acidophilic or chromophobe cells, which are smaller than in other adenomas (even in patients not exposed to dopaminergic agonists). In drug-responders morphological effects may be striking, resulting in reduced granularity, shrunken cytoplasm and condensed, hyperchromatic nuclei. Most tumours are of the sparsely granulated subtype characterised by chromophobe cytoplasm and restriction of prolactin immunohistochemistry to the Golgi apparatus, resulting in a polarised or cap-like prolactin pattern. Densely granulated tumours show a diffuse pattern and are acidophil. The sparsely granulated tumour may be associated with spherical calcifications (psammoma bodies) or amyloid deposition.
The acidophil stem-cell adenoma is rare and its nosological status remains to be further defined. It shows eosinophilia on H&E due to accumulation of mitochondria (oncocytic change) and distinct clear cytoplasmic vacuoles may be seen on light microscopy. Occasional perinuclear dot-like fibrous bodies may be seen with cytokeratin stains. The acidophil stem-cell adenoma is considered to be more prone to recurrence than other adenomas.
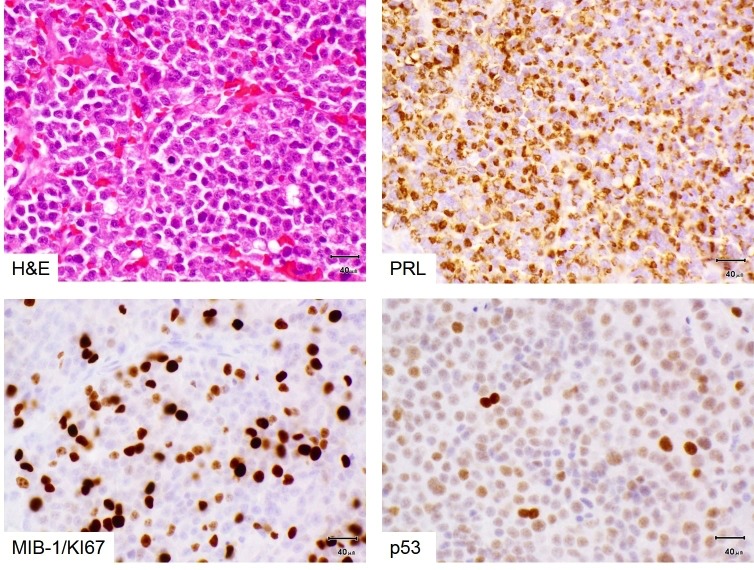
Figure 3b-8
Prolactinoma with atypia and resistance to dopamine agonist treatment. Prolactinomas consist of sheets of relatively small, monomorphic cells (top left) with a cap-like (‘Golgi-pattern’) staining for prolactin (top right). Most pituitary adenomas of any lineage, including lactotroph adenomas, do not show any mitotic figure in routine stains. Some lactotroph adenomas in men can be resistant to dopamine agonists and histologically atypical, as in this example: Mitotic figures are common, the Ki67/MIB-1 proliferation index is high (10-20% [usually <3%], bottom left) and nuclear p53 is overexpressed (bottom right). This indicates a high likelihood of tumour recurrence if incompletely excised (as proved to be the case in this instance).
Figure 3b-9
Histology of dopamine agonist response in a prolactinoma. Bromocriptine treated prolactinomas respond with characteristic dense fibrosis and condensation of the cytoplasm and nucleus of the neoplastic cells. Left – untreated, right treated.
Molecular Genetics
To date, no mutational events have been unequivocally associated with prolactinomas. However, management of prolactinomas is predominantly medical, using dopamine agonists, with a high proportion of patients achieving disease control or remission, so surgery is not often indicated. Consequently, surgical specimens are few and represent patients who do not tolerate or respond to medical treatment. These tumours are likely to have acquired multiple alterations that allow escape from apoptosis or unrestrained replicative potential which could complicate analysis of mechanisms involved in dopamine agonist-responsive PRLomas.
Oestrogen Receptor Aib1 And Aromatase
A significant correlation between oestrogen receptor ERα mRNA and PRL level, tumour volume and TGFβ1 mRNA was observed in prolactinomas (59), suggesting a role for both ERα and TGFβ1 in prolactinoma tumorigenesis, but the mechanism by which this may occur is unclear.
AIB1 (Amplified in breast cancer) is a member of the p160/SRC family of nuclear co-activators and is a co-activator of the oestrogen receptor. It integrates extracellular signals from growth factors and – through MAPK activation - relays them to the oestrogen receptor, enhancing its transcriptional activity (60). Overexpression of AIB1 was observed in prolactinomas and was associated with expression of ERα and aromatase. In addition, subcellular distribution of AIB1 was linked to cell cycle phase and viability. Nuclear AIB1 expression correlated with nuclear PCNA (a marker of cell proliferation) and cytosolic expression correlated with caspase-3 activation (a marker of apoptosis) (61).
Increased expression of aromatase cytochrome P450 (an enzyme that converts androgens to oestrogens) was observed in prolactinoma relative to normal pituitary but its expression did not correlate with resistance to dopamine agonists or remission (62).
Dopamine Receptor And Downstream Signalling
Dopamine agonists are the first choice of treatment for PRLomas and act by increasing the inhibition of prolactin release mediated by dopamine signalling. The major dopamine receptors expressed in pituitary are D1R (adenylyl cyclase-stimulating) and D2R (adenylyl cyclase- inhibiting). Expression of D2R is more prevalent. The dopamine receptor D2 is expressed as long (D2L) and short (D2S) isoforms, with D2L being the predominant isoform. In patients who were poor responders and those with secondary DA resistance, levels of D2L were significantly reduced (63).
The molecular mechanism of resistance to dopamine agonists is not fully understood. There have been no reported mutations in dopamine receptors in prolactinoma, however, studies have been few (64). Downregulation of the dopamine receptor (D2R) and alterations in the downstream signalling pathway are thought to be involved (65) and binding of PREB (prolactin regulatory element-binding protein) has been shown to be essential for dopamine-mediated inhibition of PRL gene expression – mutation of the PREB consensus sequence in the PRL promoter of GH3 cells prevented cabergoline-induced suppression of PRL expression (66). Further support for the role of dopamine receptor in DA resistance came from studies in mice. In xenografts of prolactin-secreting GH3 cells, those overexpressing the short form of the dopamine receptor (D2S) showed increased sensitivity to bromocriptine in the form of reduced tumour growth (67).
Filamin –A (FLNA) is a cytoskeletal protein that is widely expressed and associates with D2R. FLNA is important for D2R signalling and targeting. In PRLomas with differing responses to DAs, the effect of FLNA on D2R expression and signalling was investigated. Silencing of FLNA in DA-sensitive PRLoma primary cultures resulted in reduced D2R expression and signalling, which could be restored by FLNA overexpression, however, in cells that do not express D2R, overexpression of FLNA did not induce D2R expression, suggesting a more complex mechanism of regulation of D2R expression and signalling (68).
Nerve Growth Factor
A series of studies of PRLomas that were totally resistant to DA therapy and lacked D2 receptors expressed NGFR (nerve growth factor receptor) and cells from these tumours could be induced to differentiate and express D2R upon NGF treatment, furthermore, this expression persisted after cessation of NGF treatment, a feature that was accompanied by reduced tumour growth (69). In female patients with microprolactinoma, hyperprolactinaemia correlated with increased serum NGF, suggesting that release of both molecules is regulated by a D2R-mediated mechanism (70). Further insight into the mechanism of NGF-mediated suppression of DA-resistant PRLoma growth came from the observation that in DA-resistant cells, p53 adopted a different conformation that prevented its nuclear translocation. Treatment with NGF restored p52 conformation and DNA-binding ability, an effect mediated by trkA through activation of PI-3-K (71).
Egfr Receptor Family
Signalling through ErbB and other EGFR family tyrosine kinase receptors occurs upstream of PRL synthesis. The subtype and distribution of expression of these receptors was correlated with therapeutic reduction of prolactin levels in DA resistant prolactinomas in patients receiving lapatinib treatment. Increased expression of ErbB3 was associated with optic chiasm compression, suprasellar extension and carotid artery encasement. Higher ErbB3 expression was also associated with increased response to DA therapy (72)
High mobility group A2 (HMGA2) is an abundant, non-histone DNA-binding protein that mediates the assembly of nucleoprotein complexes involved in the determination of chromatin architecture, transcriptional regulation and RNA processing. HMGA2 is involved in many aspects of cell function, including proliferation and tumour progression, but the exact role of HMGA2 is still not understood (reviewed in (50,73)). The HMGA2 gene was found to be amplified and overexpressed in PRLomas, which often have trisomy of chromosome 12 (containing the HMGA2 gene) (74,75). HMGA2 is thought to promote the activity of transcription factor E2F1, which is required for entry of cells into S-phase. In non-proliferating cells, this activity is repressed by interaction of E2F1 with retinoblastoma protein (pRB) (67). Expression of HMGA2 and HMGA1B have been shown to correlate with expression of PIT1, a transcription factor that regulates expression of PRL (along with GH, GHRHR and Pit1 itself), HMGA2 and HMGA1B bind the Pit1 promoter and enhance Pit1 expression, implicating HMGA2 (and HMGA1B) in pituitary tumorigenesis (76).
E-cadherin, α, β and γ catenins and p120
An immunohistochemical comparison of the expression of E-cadherin, α, β and γ catenins and p120 in normal pituitary, indolent and invasive prolactinomas showed that expression of these proteins was membranous and strong in normal pituitary, decreased in prolactinoma and markedly decreased or absent in invasive prolactinoma, with the exception of γ-catenin, which was expressed more highly in invasive prolactinoma (77). The expression of E-cadherin was inversely proportional to invasiveness, proliferation index (Ki67) and tumour size in prolactinoma. E-cadherin is a suppressor of invasion and participates in the formation of adherens junctions and a decrease in its expression is often seen associated with tumour invasiveness (reviewed in (78,79)).
Micro Rna In Prolactinomas
Little is known about the involvement of miRNAs in prolactinoma pathogenesis. A study examining the expression profiles of miRNAs in prolactinomas that had been treated with bromocriptine or were treatment naïve showed upregulation of miR-206, miR-516b and miR-550 and downregulation of miR-671-5p was shown to be associated with bromocriptine treatment (80). A study examining miRNA expression profiles in bromocriptine-resistant and bromocriptine-sensitive prolactinomas showed that resistance was associated with increased expression of Hsa-mir-93, hsa-mir-17, hsa-mir-22*, hsa-mir-126*, hsa-mir-142-3p, hsa-mir-144*, hsa-mir-486-5p, hsa-mir-451, and hsa-mir-92a and decreased expression of hsa-mir-30a, hsa-mir-382, and hsa-mir-136 (81). The functional significance of this change in expression pattern is not understood, but silencing of mir-93 was shown to suppress p21 expression.
THS-PRODUCING ADENOMA
Definition
A Pit-1-lineage derived neoplasm that mostly expresses TSH and contains typical TSH-type granules on electron microscopy.
Pathology
Thyrotroph adenomas are rare (~1% of all pituitary adenomas). They arise usually in the 5th decade and present as functional macroadenomas resulting in diffuse goitre and hyperthyroidism. Longstanding primary hypothyroidism may lead to thyrotroph adenomas via thyrotroph hyperplasia. Histologically they comprise sheets of angulated or elongated, chromophobe cells, often accompanied by fibrosis. Staining for beta-TSH is usually patchy; tumour cells also express GATA-2 and Pit-1.

Figure 3b-10
Histology of thyrotroph adenoma. These rare tumours contain interlacing, relatively plump spindle cells (left) with strong, patchy TSH expression (right).
Molecular Genetics
The pathogenetic mechanisms of thyroid-hormone-producing adenomas (TSHomas) are not well understood. This may be in part due to the rarity of the lesion (thyrotroph adenomas are estimated to represent 1-3% of pituitary adenomas (82)). No mutations have so far been associated with TSHomas. Experiments that sequenced TSHomas show no mutations in G-protein subunits or the TRH receptor (83) . Pit 1 is overexpressed in these tumours, but not mutated. Expression of somatostatin receptors SSTR2A and SSTR5 was found in TSHomas (31). A high ratio of expression of SSTR5 to SSTR2 might indicate a better response to long-term treatment with somatostatin analogues in TSHomas (84,85) but this is not a consistent finding (86).
ACTH-PRODUCING ADENOMA
Definition
Corticotroph pituitary adenomas are Tpit-lineage derived tumours producing ACTH stored in ultrastructurally typical ACTH granules. They are the defining neoplasms of Cushing’s disease.
Pathology
Corticotroph adenomas associated with manifest Cushing’s disease are composed of deeply basophilic (PAS-positive) cells with granular cytoplasm and round nuclei; these comprise the densely granulated subtype of ACTH adenoma. Most tumours arise in women in the 4th or 5th decade (F:M = 8:1); prepubertal tumours are rare and equally distributed between the sexes, with a slight male predominance. Sparsely granulated tumours are weakly basophilic or chromophobe, and individuals may lack an overt Cushing’s phenotype (‘silent corticotroph adenomas’). The cytokeratin pattern in typical Cushing’s adenomas is diffuse. However, rare neoplasms may display ‘Crooke’s hyaline change’, classically interpreted as a morphological manifestation of intact feedback inhibition by excess systemic cortisol on non-neoplastic corticotrophs. This change is therefore seen in intact acini adjacent to a typical corticotroph adenoma and results in a ring-like accumulation of cytokeratins. If present in many adenoma cells, these tumours are called ‘Crooke’s cell adenomas’, possibly representing a subgroup with an adverse outcome and silent presentation. Immunohistochemistry of all corticotroph adenomas shows strong nuclear T-pit positivity. Densely granulated tumours show strong diffuse cytoplasmic ACTH expression, whilst chromophobe tumours show only patchy positivity. Following detection of somatic Usp8 mutations in a subgroup of ACTH-producing adenomas (see below), it has been suggested that nuclear translocation of Usp8 may represent an immunohistochemically detectable surrogate marker of these mutations (87). USP8-mutated corticotroph adenomas are more commonly microadenomas compared to USP8-wild-type Cushing’s adenomas. The ‘minimal pathological unit’ of Cushing’s disease is corticotroph hyperplasia. This is defined as a distention of normal adenohypophyseal acini by a homogeneous population of corticotrophs that does not lead to complete breakdown of the acinar reticulin border. The described morphological entities associated with Cushing’s disease are illustrated below in figures 3b-11 to 3b-14.
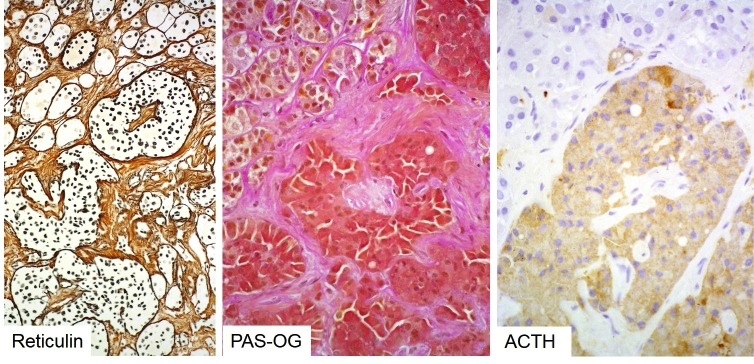
Figure 3b-11
Histology of corticotroph hyperplasia causing Cushing’s disease. Note distended but still intact reticulin network of hyperplastic pituitary acini (left) populated by a homogeneous population of deeply basophilic cells (center) expressing ACTH (right).

Figure 3b-12
Histology of a corticotroph microadenoma. These (often USP8-mutated) Cushing’s adenomas represent microscopic nodules well-demarcated from normal gland (NG). Reticulin stain is often essential for their detection (top left) and discrimination from corticotroph hyperplasia: a corticotroph adenoma results in completed destruction of the reticulin network as seen here (right side of the dashed line in top left image). Typical Cushing’s adenomas are deeply basophilic (top right) and show strong diffuse ACTH positivity (bottom left) and an intact keratin cytoskeleton (bottom right).

Figure 3b-13
Crooke’s hyaline change in non-neoplastic corticotroph cells in response to hypercortisolaemia. The physiological response of normal corticotrophs to exposure of excess cortisol (of any source, neoplastic or iatrogenic) is downregulation of ACTH synthesis and development of hyaline, cytokeratin-rich perinuclear rings: Crooke’s hyaline degeneration (named after the English endocrinologist Arthur Carleton Crooke).

Figure 3b-14
Histology of Crooke’s cell adenoma. In these corticotroph adenomas Crooke’s cell change is seen in the tumour cells, rather than non-neoplastic corticotrophs. This is clearly evident in PAS-OG histochemistry, where the hyaline ring displaces the deeply basophilic granules (left). It is also reflected in the dense ring-like cytokeratin pattern (right).
Molecular Genetics
ACTH-producing adenomas causing Cushing’s disease are associated with both an excess of corticotroph releasing hormone (CRH) and a loss of negative feedback inhibition by glucocorticoids. However, no mutations in either the CRH receptor or the glucocorticoid receptor have been reported.
USP8
Exome sequencing of corticotroph adenomas from patients with Cushing’s disease revealed recurrent heterozygous somatic mutations in the deubiquitinase USP8 in one third of cases (87,88), although one study has estimated the prevalence to be 62% (89). Mutations were clustered within the 14-3-3 binding motif of USP8: a highly evolutionarily conserved region that is rarely mutated in other human cancers. Mutations in USP8 were found to be more common in adult than paediatric cases and more common in females than males (ratio 5:2). Patients with a USP8 mutation were less likely to develop postoperative adrenal insufficiency (88). Tumours with a mutation in USP8 were also found to be smaller and to produce more ACTH than their wild-type counterparts (89)
USP8 is a ubiquitin-specific protease that regulates the fate of numerous cellular proteins. Conjugated ubiquitin molecules target a protein for degradation and these modifications are removed by deubiquitinases (DUBs). USP8 specifically targets the pathway whereby activated EGFR is targeted for lysosomal degradation and leads to increased cellular concentrations of EGFR and sustained levels of EGFR signalling. However, one study of 60 corticotroph adenomas did not find any association between USP8 mutation and EGFR expression; there was, however significantly higher expression of POMC, SSTR5 and MGMT (90). Binding of 14-3-3 proteins to Usp8 has a suppressive effect and so mutations in the 14-3-3 binding motif of USP8 that perturb this interaction lead to a gain of function of USP8 and increased EGFR signalling. Additionally, mutations in the 14-3-3 binding motif that abrogate 14-3-3 binding result in exposure of a cleavage site and an increase in proteolytic cleavage of USP8 between Lys714 and Arg715. This results in a shorter 40kDa C-terminal fragment of USP8 with increased deubiquitinase activity. Mutant USP8 also results in increased activation of the POMC promoter in the AtT-20 corticotroph adenoma mouse cell line.
Cyclins And Cyclin-Dependent Kinases
A study investigating the role of cell-cycle regulators and related transcription factors in ACTH-secreting and silent corticotroph adenomas found that CDKN2A expression was four times greater in ACTH-expressing than silent corticotroph adenomas, while cyclins D1, E1 and B1 were suppressed. It is suggested that the upregulation of a cell-cycle inhibitor combined with the downregulation of cyclins may restrict growth of ACTH-producing adenomas compared to their silent counterparts (91).
11β-Hydroxysteroid Dehydrogenase
Cortisol and inactive cortisone are interconverted by 11β-hydroxysteroid dehydrogenase. This enzyme exists as two isoforms: 11β-HSD1 and 11β-HSD2. Studies investigating the expression ratio of these two isoforms in ACTH-expressing adenomas found that Isoform 1 is downregulated in ACTHomas and 2 is upregulated compared to normal pituitary (92). The role of increased expression of 11β-HSD2 in ACTHoma tumorigenesis is unclear and findings are inconsistent (93).
Somatostatin Receptors
A comparison of the expression of somatostatin receptor subtypes SSTR2 and SSTR5 in silent corticotroph adenomas and adenomas responsible for Cushing’s disease showed that mRNA encoding SSTR1 and 2 was expressed in greater quantities in silent corticotrophs (SSTR2 5-fold increase), whereas in Cushing’s disease, SSTR5 was expressed more highly (14-fold increase) (94). Although the implications of this difference in expression are not fully understood, it may be that treatments that selectively target SSTR5 could be useful for ACTHoma treatment.
In a series of ACTH-secreting pituitary adenomas, levels of miR-26a were assessed by RT-qPCR. This micro-RNA was upregulated in all ACTHomas compared to normal pituitary. The putative target of this miRNA, PRKCD, was downregulated in tumours with elevated miR-26a (95). PRKCD encodes protein kinase C delta, a serine/threonine kinase involved in a diverse range of signalling pathways including regulation of growth, apoptosis and differentiation.
GONADOTROPHIN-PRODUCING ADENOMA
Definition
Pituitary adenomas derived from SF-1 expressing adenohypophyseal cells producing mainly FSH or LH and typical secretory granules.
Pathology
Classic gonadotroph adenomas are chromophobe adenomas with a growth pattern that may include papillae and perivascular pseudorosettes. Although all tumour cells express nuclear SF-1, FSH and LH are restricted to clusters of cells often demonstrating striking polarisation towards vascular lumina in well-differentiated examples. Gonadotroph adenomas are usually endocrinologically silent and therefore present as macroadenomas with compression of the optic chiasm or invasion of the cavernous sinus. They are often called non-functioning adenomas (NFAs) but it should be noted that adenomas of other lineages may also be clinically ‘non-functional’ (e.g. silent corticotroph or somatotroph adenomas). Rare functionally active tumours in females of reproductive age may be associated with ovarian hyperstimulation syndrome.

Figure 3
b-15.1+15.2: MRI and macroscopic pathology of gonadotroph pituitary macroadenomas. Gonadotroph adenomas are usually clinically silent and thus tend to present as space occupying lesions compressing the optic chiasm, pituitary stalk or hypothalamus. 3b-15.1: Sagittal MRI (left) and post-mortem view of the same tumour. Note the suprasellar extension and compression of the hypothalamus. 3b-15.2: Two further gonadotroph macroadenomas seen in situ in the skull base (left) and the base of the brain (right) compressing the optic chiasm. Note in the left image the anatomical relationship to the sphenoid wings, right optic nerve and basal vessels of the brain (one of which is stuck to the rostral surface of the macroadenoma). The adenoma (asterisk) in the right image has grown through the diaphragma sellae and therefore is attached to dura mater – which is used to pull the adenoma away from the chiasmatic cistern to reveal the chiasm (arrow) and optic nerves.

Figure 3b-16
Histology of a gonadotroph (non-functioning) adenoma. In their typical form these adenomas have a distinct architecture comprising perivascular rosettes of neuroendocrine cells with a distinct polarity of their processes towards the vascular lumen (left) and always patchy and focal, rather than diffuse expression of FSH and LH (right). Note a rare mitosis in the HE image (left, arrow).
Molecular Genetics
Micorarray studies comparing functional gonadotroph tumours to normal post mortem pituitary found that downstream p53 target genes RPRM, p21/CDKN1A and PMAIP1 were consistently downregulated (96). These genes are mediators of cell cycle arrest and apoptosis. Members of the GADD45 family were differentially expressed, with GADD45β downregulated in gonadotroph adenomas compared to normal gland. Overexpression of GADD45β in gonadotroph cells inhibited proliferation and activated apoptosis in the absence of growth factor, however, the authors found no evidence of hypermethylation of GADD45β (96) and so the mechanism of downregulation remains unknown.
A whole-exome sequencing study of histologically typical, clinically non-functioning gonadotroph adenomas revealed 24 somatic variants in independent genes, none of which were recurrent. There were no mutations that had been previously associated with pituitary tumorigenesis and the authors conclude that mechanisms other than somatic mutation may be involved in sporadic NFPA tumorigenesis (74).
NULL CELL ADENOMA
Definition
Null cell adenomas are neoplasms derived from adenohypophyseal endocrine cells that cannot be assigned to any specific subtype based on transcription factor, hormone or ultrastructural features.
Pathology
These tumours are chromophobe and show usually a diffuse growth pattern. Increasing sensitivity and specificity of immunohistochemical techniques for detection of pituitary transcription factors and hormones make this a shrinking diagnostic subgroup. Distinction of this subtype from rare endocrine tumours not derived from adenohypophyseal cells (paraganglioma, metastatic endocrine carcinoma) is important but can be difficult.
Molecular Genetics
As tumours previously designated ‘null-cell’ or ‘non-functioning’ may actually represent SF-1 lineage tumours, interpretation of molecular studies is difficult. A study examining the expression of E-cadherin (CDH1), slug (SNAI2) and oestrogen receptor ERα and ERβ in invasive compared to non-invasive non-functioning pituitary adenomas (NFPAs) showed that E-cadherin is downregulated in more invasive tumours, while its repressor, slug, is upregulated. Expression of slug was positively correlated with ERα expression, while E-cadherin was positively correlated with ERβ expression. The relevance of these findings for patient prognosis and treatment has yet to be determined (75). MicroRNA profiling of NFPAs compared to normal pituitary showed that miRNAs predicted to target components of the TGFβ signalling pathway and result in their downregulation are overexpressed. This pathway is known to have a role in tumorigenesis, but the nature of its role in the pathogenesis of NFPAs is not well understood (97). Another pathway commonly disrupted in tumorigenesis, the Notch signalling pathway was investigated in NFPAs. Upregulation of Notch3 (a regulator of cell proliferation and apoptosis) and its ligand, Jagged1, was observed in NFPAs compared to normal gland. Owing to the complexity of Notch pathway regulation, the consequence of this upregulation is not yet clear (98,99). The pathogenic mechanisms of NFPAs are largely unclear and targeted treatments are not available.
PITUITARY CARCINOMA
Definition
Pituitary carcinoma is defined as a neoplasm of adenohypophyseal endocrine cells with cerebrospinal or systemic dissemination.
Pathology
Pituitary carcinoma is very rare, comprising approximately ~0.2% of operated pituitary neoplasms. Most pituitary carcinomas develop from recurrent endocrinologically functioning, invasive macroadenomas with a highly variable lag period. The majority represents corticotroph or lactotroph neoplasms. Bilateral adrenalectomy in the setting of Cushing’s syndrome with an undetected pituitary microadenoma may predispose to pituitary carcinoma (Nelson’s syndrome). It has also been suggested that silent corticotroph adenomas or Crooke’s cell adenomas may pose a risk, but data are scant. Histologically, pituitary carcinomas may show remarkably little pleomorphism; however, an increased MIB-1 index and p53 overexpression are usually present. Despite the introduction of the ‘atypical pituitary adenoma’ category, no reliable diagnostic markers are available that allow prediction of carcinomatous behaviour before dissemination has occurred. The prognosis is poor once systemic metastases are present. Treatment with temozolomide should be considered.
Molecular Genetics
Molecular studies of pituitary carcinomas are scant, presumably due to the rarity of the lesion. One study has observed a mutation in H-ras in a PRL-producing carcinoma. Unlike other pituitary tumours, pituitary carcinomas show aggressive tendencies and metastasise (100). A microarray study comparing expression levels in pituitary adenomas relative to an ACTH pituitary carcnioma identified the LGALS3 (galactin 3) gene as being upregulated in pituitary carcinomas (101)
PITUITARY BLASTOMA
Definition
Pituitary blastoma is a rare pediatric neoplasm of the anterior gland composed of primitive follicular structures of endocrine cells admixed with folliculo-stellate cells. It is pathognomonic of germline DICER1 syndrome or pleuropulmonary blastoma-familial tumor and dysplasia syndrome [online Mendelian inheritance in man (OMIM) #601200] (102)
Pathology
Tumours are variably cellular, likely reflecting different degrees of maturation, and consist of cells arranged in rosettes and glandular structures reminiscent of Rathke’s epithelium, undifferentiated cells (blastema) and larger granular (secretory cells) (103). Ultrastructurally FS-like cells may also be seen. There is usually ACTH-positivity in a few cells and GH may also be seen.
Mitoses are present but the MIB-1 index may be very variable. The designation as ‘blastoma’ reflects the original view that these tumours are highly malignant with a natural history similar to other embryonal neoplasms; however, more recent evidence suggests that the prognosis is not uniformly poor (102).

Figure 3b-17
Histology of a pituitary blastoma. These are primitive tumours resembling ‘small-blue round-cell’ neoplasms.
Reproduced from Acta Neuropathologica ”Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations” volume 128, 2014 pages 111-122 de Kock L, et al. (102) with permission of Springer. Fig. 3a case 13, T1-weighted post-contrast midline sagittal MR image showing pituitary region mass (red arrow). b case 4, hematoxylin and eosin (H&E) staining ×250: three enlarged follicles lined by stem cells. c Immunohistochemical staining I case 10, growth hormone (GH) immunostaining ×400: enlarged GH/alpha subunit cells immunopositive for GH. II case 10, ACTH immunostaining ×400: small vessel surrounded by stem cells. Some cells display ACTH immunoreactivity
Molecular Genetics
The precise mechanisms driving tumorigenesis remain to be defined. The morphological evidence of stem-cell-like features of pituitary blastoma cells and known roles of micro RNAs in regulation of stem cell differentiation make it plausible that profound abnormalities in micro RNA profiles following mutations of Dicer, a key regulator of micro RNA maturation, are causative. Mutations occur in highly conserved regions of DICER1, particularly the RNase IIIb domain, resulting in predicted loss of function, following Knudson’s dual hit model.
CRANIOPHARYNGIOMA
Definition
Usually benign, but invasive epithelial lesions of the supra-sellar region or third ventricle that exist as two variants: adamantinomatous and papillary. The variants have distinct clinicopatholoigcal and genetic features which may represent different pathogenic mechanisms.
Pathology
Craniopharyngiomas occur with an incidence of 0.13 per 100 000 person years. There are two variants: adamantinomatous (aCP) and papillary (pCP) that occur in the ratio ~9:1 with no sex differences. aCP usually occurs in childhood (mean age 5-14 years) while the pCP is almost exclusively seen in adults (mean age 65-74 years). Craniopharyngiomas are complex, epithelial neoplasms that arise in the sellar region along the vestigial craniopharyngeal tract. Although some overlapping features have been observed, the two variants represent clinicopathologically distinct lesions.
Adamantinomatous Craniopharyngioma
aCPs are located predominantly in the suprasellar region although infrequently they have an intrasellar component. Rare locations include the sphenoid sinus and cerebello-pontine angle. They are multi-lobulated and often multi-cystic masses. On T1 weighted MRI imaging they are hypo- or iso-intense with areas of hyperintensity, corresponding to the cystic components. Enhancement is strong and heterogeneous.
Macroscopically, aCP are firm, lobular lesions with an irregular, but sharp interface strongly adherent to and invading surrounding structures. Cyst contents are variable and may contain necrotic or inflammatory debris or a dark, cholesterol-rich fluid resembling motor oil. Calcification is often present.
Microscopically, the architecture of aCP shows a well circumscribed, multicystic lesion with finger-like protrusions into surrounding brain parenchyma (Figure 3.b.18). The lesion is composed of a peripheral palisading epithelium surrounding a loose core of stellate reticulum. Nodules of anuclear “ghost cells” containing wet keratin are commonly found and are pathognomonic for this tumour type. Near the tumour invading edge, epithelial whorls of cells that often show translocation of beta-catenin from membrane to cytosol/nucleus are common. Degenerative changes include intra-cystic squamous debris, chronic inflammation and the appearance of cholesterol clefts and extensive calcification. These changes can elicit a granulomatous inflammatory response and brain invasion may cause Rosenthal fibre gliosis.

Figure 3b-18
Histology of adamantinomatous craniopharyngioma. This highly distinctive neoplasm consists of loosely arranged ‘stellate’ reticular stroma, palisading peripheral epithelium and nodules of ‘ghost’ cells (degenerated keratinocytes, arrow in the left image). Finger-like protrusions of neoplastic epithelium commonly invade the hypothalamic brain parenchyma resulting in a dense, hypereosinophilic Rosenthal fibre gliosis (arrow and bottom half of right image).
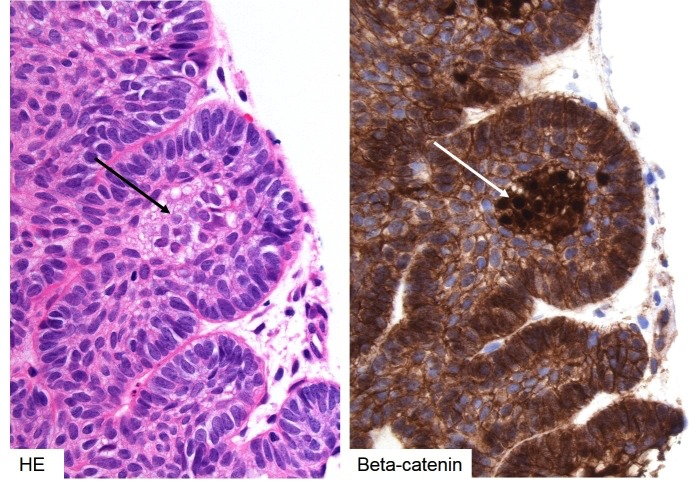
Figure 3b-19
Histology of adamantinomatous craniopharyngioma – beta-catenin. Nodules or whorls of tinctorially distinct epithelium is often seen in the invading edge of the adamantinomatous subtype (left, black arrow). The cells in these nests demonstrate nuclear translocation of beta-catenin (right, white arrow), indicating activated WNT signalling.
Papillary Craniopharyngioma
pCPs are located in the suprasellar region or within the third ventricle. They are usually more solid than aCPs but may have a cystic component. On T1-weighted MRI imaging, they appear hypointense with enhancement of the cyst wall.
Macroscopically, pCP are discrete, well circumscribed and often solid lesions with little adherence to surrounding structures. When cystic, the cyst contents are clear without cholesterol crystals. Clacification is not seen.
Microscopically, pCPs are composed of squamous, well differentiated, non-keratinizing epithelium. There is no stellate reticulum; these lesions have a fibrovascular stroma without a palisading layer. The lesions form pseudopapillae as a result of epithelial dehiscence and do not contain wet keratin. In these lesions, beta-catenin retains its membranous location. There may be scant foci of goblet or ciliated cells that resemble Rathke’s cleft cyst

Figure 3b-20
Histology of papillary craniopharyngioma. Papillary craniopharyngiomas lack ’stellate reticular’ stroma and keratin ‘ghost’ cell nests and nodules or whorls in the advancing edge. They have a more solid growth pattern prone to artifactual ‘cracking’ (arrow, left) giving it a (pseudo-) papillary appearance.

Figure 3b-21
Histology of papillary craniopharyngioma – beta-catenin and BRAFV600E. Papillary craniopharyngiomas lack any nuclear translocation of beta-catenin; it remains restricted to the adherens junction (left). Instead, the tumours are always positive for mutated BRAFV600E protein (right).
Molecular Genetics
Two alternate theories have been proposed to explain the pathogenesis of craniopharyngiomas. The embryogenetic theory states that CPs arise from neoplastic transformation of ectopic embryonic remnants of Rathke’s pouch. The metaplastic theory states that differentiated squamous epithelium that forms part of the anterior pituitary or pituitary stalk undergoes metaplastic transformation. In addition, a dual theory has also been proposed that the adamantinomatous type is formed via the embryogenetic mechanism while the papillary type follows the metaplastic route.
Mutations have been found in craniopharyngiomas that may co-segregate with subtype. Mutations in exon 3 of CTNNB1, the gene encoding β-catenin have been observed in around 70% of aCP cases, although estimates of frequency vary (104-106). Β-catenin is a mediator of the Wnt signaling pathway and exon 3 of CTNNB1 contains critical Ser and Thr residues S33, S37, T41 and S45 (107) that are phosphorylated during formation of the β-catenin degradation complex. This complex regulates Wnt signalling by targeting β-catenin for ubiquitination and degradation in the absence of receptor-bound Wnt ligand. Mutations in these critical Ser and Thr residues prevent formation of the β-catenin destruction complex, leading to a constitutively active Wnt signal and accumulation of β-catenin in the cytosol and nucleus. Nuclear and cytosolic β-catenin was observed in 90-100% of adamantinomatous craniopharyngiomas, but never in papillary craniopharyngiomas or other tumours of the sellar region (pituitary adenomas, arachnoid cysts, Rathke’s cleft cysts and xanthogranulomas), (104,108-110). A study that selectively expressed mutant CTNNB1 in developing mouse pituitary showed disrupted Pit1 lineage differentiation, hypopituitarism and large, cystic tumours resembling adamantinomatous craniopharyngiomas (111,112), suggesting that CTNNB1 mutation may be sufficient for aCP formation.
Despite the presence of a β-catenin mutation in all cells, nuclear and cytosolic accumulation of β-catenin is found only in small clusters of cells near the infiltrating edge of the tumour (104,108,109,113-115). It has been shown that these cells possess stem-cell-like properties and may perform a paracrine function by secretion of members of the SHH, BMP and FGF family that promote division of the surrounding tumour cells (113,116) These cells also show reduced expression of fascin and increased phosphorylation and activation of EGFR, suggesting increased capacity for migration (117-119).
Mutations in the protein kinase BRAF (V600E) have been shown to be associated with pCPs in 81-95% of cases (106,110). BRAF is a component of the MAP kinase signalling cascade and mutations in this pathway are associated with numerous neoplasms including melanoma, for which treatment with BRAF inhibitors is common. There have been two reports of targeted treatment of pCP. Dabrafenib (150mg, orally twice daily) and trametinib (2mg, orally twice daily), resulted in 85% reduction in tumour volume after 35 days (120). Vemurafenib (960mg twice daily) for three months resulted in significant reduction in tumour volume, but the tumour recurred within 6 weeks upon cessation of treatment (121).
The two subtypes of craniopharyngiomas are clinicopathologically distinct, but do have some overlapping features. This observation has led to the hypothesis that craniopharyngiomas fall on a histopathological continuum with other cystic epithelial sellar lesions (109,122-124). It has been suggested that papillary craniopharyngiomas represent an intermediate entity between Rathke’s cleft cysts and adamantinomatous craniopharyngiomas, as they have been found to contain ciliated epithelial cells and goblet cells characteristic of Rathke’s cleft cysts (122,125-127). Craniopharyngiomas, particularly the papillary form, can arise after treatment for Rathke’s cleft cysts although the possibility of coexisting lesions cannot be excluded (128,129).
RATHKE’S CLEFT CYST
Definition
A benign, non-neoplastic epithelial cyst arising from accumulation of mucinous material in remnants of Rathke’s pouch.
Pathology
Classical Rathke’s cleft cysts consist of a monolayer of cuboidal cells on with microvilli and scattered columnar and goblet cells. Cyst contents consist usually of amorphous eosinophilic material. Squamous metaplasia of the lining epithelium is common and may result in the differential diagnosis of craniopharyngioma. Xanthogranulomas with chronic inflammation and cholesterol crystals may also occur. Rathke’s cleft cysts have no neoplastic potential but may recur following incomplete excision.

Figure 3b-22
Histology of Rathke’s cleft cyst. Symptomatic Rathke’s cleft cysts are lined by a ciliated epithelium (top left) that is cytokeratin-positive (top right) and often contains PAS-positive vacuoles (bottom left). It often undergoes attenuation or squamous metaplasia (bottom right).
DIFFERENTIAL DIAGNOSIS OF CYSTIC LESIONS
A series of observations that note similarities between cystic sellar lesions has led to the hypothesis that there exists a histopathological continuum that includes epithelial, epidermoid and dermoid cysts, Rathke’s cleft cysts and both papillary and adamantinomatous craniopharyngiomas (109,122,125,130,131). Although experimental evidence is lacking there are reports of transitional lesions that lend support to this idea. Due to the rarity of these lesions and the paucity of material available for study, so far no genetic event has been unequivocally associated with the development of non-neoplastic cystic lesions arising in the sellar region. Immunohistochemistry and sequencing for BRAFV600E mutations in Rathke’s cleft cysts was negative (132).
TUMOURS OF THE NEUROHYPOPYSIS: GRANULAR CELL TUMOUR, PITUICYTOMA, SPINDLE CELL ONCOCYTOMA: TTF-1 FAMILY OF PITUITARY NEOPLASMS
Definition
Rare endocrinologically silent neoplasms of the posterior pituitary or infundibulum that share the expression of thyroid-transcription factor 1 (TTF-1).
Pathology
The neurohypophysis is derived from the floor of the diencephalon. The development of its specialised glial cells, termed pituicytes, is controlled in part by the expression of TTF-1, which is maintained throughout adulthood. Pituicytes are thought to provide structural and functional support for the axonal processes and neurosecretory terminals of oxytocin and vasopressin producing cells whose cell bodies are located in the hypothalamus. Electron microscopic studies have suggested that there are five different types of pituicytes, which (simplified) can be described as: light, dark, granular, ependymal and oncocytic (133). Neoplastic transformation of these cells is thought to give rise to three distinct neoplasms, termed granular cell tumour, pituicytoma and spindle cell oncocytoma. However, the precise relationship of these lesions remains to be defined, as some studies suggest that the spindle cell oncocytoma is arising from folliculo-stellate cells of the adenohypophysis.
Granular Cell Tumour
These lesions may be found incidentally at autopsy as microscopic nodules along the pituitary stalk. Clinically relevant lesions present as slow-growing, solid space-occupying tumours that mimic pituitary macroadenomas on preoperative imaging. Microscopically they are characterised by sheets of relatively large cells with eosinophilic, granular cytoplasm. Nuclei are round, sometimes eccentric and contain inconspicuous nucleoli. There are generally no mitoses. The cytoplasmic granules remain periodic-acid-Schiff (PAS) positive after diastase treatment. Ultrastructurally, the granules correspond to membrane-bound lysosomal organelles. This is reflected in patchy immunostaining with PGM-1 antibody against CD68, a membrane epitope belonging to the lysosomal/endosomal-associated membrane glycoprotein (LAMP) family. Tumour cells are also usually S100-positive but negative for cytokeratins, synaptophysin and pituitary hormones. In our experience granular cell tumours of the sellar region consistently show strong nuclear TTF-1 expression. The proliferation fraction is low (<5%) but tumours with mitoses and multiple recurrences have been described. Surgery is the preferred treatment modality.
Pituicytoma
This variant of posterior pituitary or infundibular TTF-1-positive neoplasm consists of fascicles of elongated yet plump bipolar cells that are pale eosinophilic and usually lack granularity (134). There may be moderate nuclear hyperchromasia but mitoses are generally absent. Electron microscopy demonstrates intermediate filaments and no secretory granules. Tumour cells express vimentin and S100 and show variable GFAP positivity. Proliferation is low. Again, surgery is the main treatment
Spindle Cell Oncocytoma
Spindle cell oncocytoma may share light-microscopic appearances with pituicytoma, particularly if accumulation of mitochondria, a defining feature, is not fully developed. Tumour cells are elongated, spindle-shaped, sometimes arranged in fascicles or epithelioid. Nuclei may be moderately pleomorphic and hyperchromatic but mitoses are again rare, although a few reports documented atypical variants with an increased recurrence rate. Scattered lymphocytic infiltration may be seen. Apart from abundant mitochondria, ultrastructural features that help to distinguish these tumours morphologically are well-formed desmosomes (135). Tumours show strong nuclear TTF-1 positivity and cytoplasmic annexin-1 expression. The latter is shared with folliculo-stellate cells of the adenohypophysis (but may also be seen in pituicytes). EMA may be expressed and is usually absent from pituicytomas and granular cell tumours. GFAP, cytokeratins and neuroendocrine markers are generally negative. The proliferation fraction is usually low but may reach 25% in recurrent tumours.

Figure 3b-23
Histology of TTF-1 positive neoplasms of the neurohypophysis. Granular cell tumour (left) and pituicytoma (right) are morphologically distinct but share nuclear TTF-1 expression, confirming their origin from specialised glial cells of the posterior gland or infundibulum. Both entities are S100 positive but only the pituicytoma expresses GFAP.
Molecular Genetics
No pathognomonic molecular genetic features have been identified for these neoplasms. Comparative genomic hybridization on one case demonstrated losses on chr 1p, 14q and 22q and gains on 5p (136). Presumed glial origin prompted examination of the IDH1 R132H and BRAF V600E mutations and BRAF-KIAA fusion gene in a recent study of 14 cases comprising all three pathologies (137). Systematic genomic and epigenomic analysis may clarify the aetiologic relationship of these tumours.
PRIMARY NEOPLASMS OF THE SELLAR REGION THAT MAY MIMIC PITUITARY ADENOMAS
Germinoma
Intracranial germ cell neoplasms have a predilection for midline structures and commonly involve the infundibular region. The most common form of this rare tumour is germinoma.
Definition
An extragonadal germ cell tumour arising in or above the pituitary fossa with histological features resembling gonadal seminoma.
Pathology
Tumours may present as large compressive lesions or subtle thickening of the (posterior) pituitary stalk. Diabetes insipidus is a classic presentation but delayed puberty due to hypopituitarism is also seen. Pituitary germinomas commonly manifest in children or young adults, mostly males. Historically, the incidence is far higher in East Asia than Western countries. CSF/blood tumour markers (alpha-fetoprotein and human chorionic gonadotrophin) that can be diagnostic in germ cell tumours with yolk-sac or choriocarcinoma components may not be helpful in pure germinoma, resulting in biopsy. Histological diagnosis may be difficult because some germinomas elicit a profound inflammatory or even granulomatous reaction that can obscure the neoplastic cells. Typical examples show a biphasic architecture of large tumour cells with vesicular nuclei, prominent nucleoli and mitoses accompanied by reactive lymphocytes.
Tumour cells express placental alkaline phosphatase (PLAP), CD117 (KIT) and the transcription factor POU5F1 (Oct3/4).
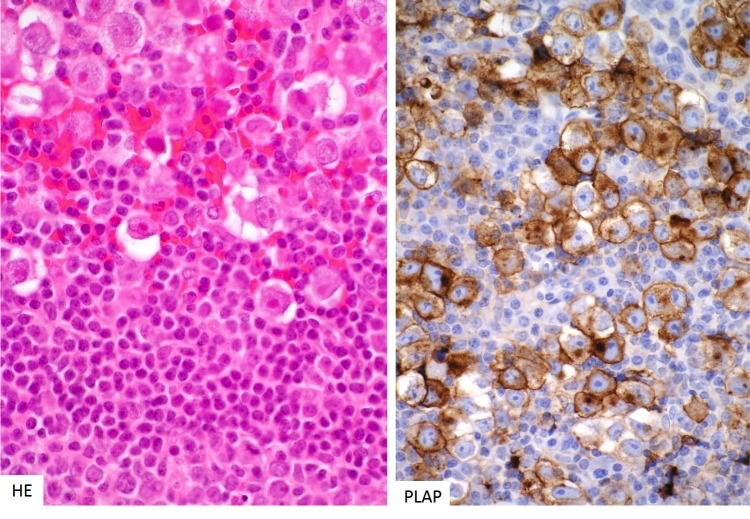
Figure 3b-24
Histology of germinoma of the pituitary gland. Pituitary germinomas share their morphological phenotype with gonadal seminomas. They consist of large anaplastic cells accompanied by a dense lymphocytic infiltrate (left). The tumour cells strongly express c-kit, PLAP (right) and Oct3/4 transcription factors.
Molecular Genetics
Intracranial germinomas are thought to arise from displaced primordial germ cells. Until recently, little was known about the molecular pathogenesis of these rare tumours. Comprehensive genomic and transcriptomic analyses revealed that pure germinomas are associated with mutually exclusive mutations in KIT and RAS in the majority of cases (138). These mutations result in the constitutive activation of the KIT-driven MAPK pathway, consistent with the observation of strong KIT expression by these tumours. Another study, employing next-generation sequencing, found additional somatic mutations in CBL, a negative regulator of KIT, as well as copy number gains at the AKT1 locus resulting in mTOR pathway activation (139) This study also found germ line variants in JMJD1C among Japanese patients, a possible explanation for the skewed incidence rates described above (139). Treatment for localised pure germinoma consists of radiotherapy; chemotherapy is an effective strategy to reduce the radiation dose (140).
CHORDOMA OF THE CLIVUS
Definition
A slow-growing but highly destructive neoplasm arising from remnants of the rostral notochord.
Pathology
Intracranial chordomas are almost exclusively located at the dorsum sellae. They may result in compression of the pituitary and destruction of the pituitary fossa. Tumours are soft, gelatinous lesions with a striking cytopathology. Typical tumours contain large, vacuolated (‘physaliphorous’) cells surrounded by a mucinous matrix. They are arranged in anastomosing cords or sheets. Occasional mitoses are found. Tumours express S100 and low-molecular weight keratins and epithelial membrane antigen (EMA). Brachyury, or transcription factor T, serves as a highly sensitive and specific marker for the diagnosis of chordoma, facilitating distinction from chondrosarcomas, (chordoid) meningiomas or metastases. It is physiologically expressed in undifferentiated notochord of the axial skeleton. The neoplasm slowly invades along neurovascular bundles and may be surgically incurable. Proton beam therapy is commonly applied in order to preserve neurological function (141). Dedifferentiation upon recurrence may rarely occur, resulting in a poor prognosis.

Figure 3b-25
Histology of sellar chordoma. These tumours arise from remnants of the notochord and are highly characteristic in appearance, comprising pleomorphic cells with very large vacuoles and intracellular PAS-positive mucin, floating in an alcian blue-positive matrix.
Molecular Genetics
The discovery of gene duplications involving the transcription factor T gene (brachyury) in familial chordoma strongly supported the idea of notochordal origin of these tumours (142). However, no recurrent somatic mutations in T or its promotor have been described that could explain the consistently high expression of brachyury by tumour cells. More recent analysis has revealed that a common single nucleotide variant in T (SNP rs2305089) is strongly associated with chordomas in apparently non-mendelian cases (143). The risk allele variant leads to increased expression of T, providing a plausible pathogenetic link and target for future molecular therapies.
SECONDARY NEOPLASMS OF THE SELLAR REGION THAT MAY MIMIC PITUITARY ADENOMAS
Definition
These lesions are here defined as neoplasms that arise at extracranial sites and colonise the pituitary, usually via haematogenous spread; i.e. pituitary metastases.
Pathology
The rich vascularity of the pituitary gland facilitates haematogenous seeding of micrometastases. Deposits from breast, lung and gastrointestinal carcinomas are most common. Autopsy series have suggested a relatively high incidence of between 3-27% in the setting of disseminated malignancy. However, many may represent asymptomatic micrometastases. In clinically manifest examples neuroimaging features can be very similar to pituitary adenomas. Even biopsy appearances can be deceptive, particularly in the setting of TTF-1-positive neuroendocrine carcinomas of the lung. However, the degree of cytological atypia and mitoses usually point to the right diagnosis. Judicial use of immunohistochemistry helps to narrow down the possible site of origin if the pituitary metastasis is the first manifestation of an occult malignancy.
NON-NEOPLASTIC LESIONSOF THE PITUITARY THAT MAY MIMIC ADENOMAS: LYMPHOCYTIC HYPOPHYSITIS
Definition
A rare lymphocytic inflammatory disorder of the pituitary gland of autoimmune aetiology.
Pathology
Classic lymphocytic hypophysitis consists of a dense, sometimes follicular, lymphoplasmacytoid infiltrate of the gland that in some cases may result in fibrosis and permanent hypopituitarism. All parts of the gland may be affected to variable degrees, resulting in distinction of adenohypophysitis, infundibuloneurohypophysitis or panhypophysitis. In the acute phase symmetrical swelling of the gland may lead to headaches or diabetes insipidus, a presenting symptom often associated with radiologically detectable swelling of the pituitary stalk. Hypogonadotropic hypogonadism represents a common deficit of anterior pituitary function. The inflammatory infiltrate consists predominantly of T-cells with a CD4/CD8 ratio of 2:1 or more. Lymphoid follicles may occasionally be observed.

Figure 3b-26
MRI and histology of lymphocytic hypophysitis. The MRI appearances may be mistaken for a non-functioning adenoma; however, there are distinctive features, including homogeneous enhancement with extension posteriorly and rostrally via the infundibulum (‘infundibulohypophysitis’). There is a dense, destructive lymphocytic infiltrate, leaving islands of residual anterior gland (eosinophilic cells, top right) that in this instance are lactotrophs (bottom left; serial section to top right). The lymphocytes are predominantly T-cells (CD3-positive, bottom left).
Pathogenesis
It is likely that different immunopathogenic mechanisms result in lymphocytic hypophysitis as the common endpoint. Historically, the disease was thought to be largely restricted to young females, temporally related to the late stages of pregnancy or early post-partum period. Shared placental and pituitary antigens have been implicated in these cases. An association with other autoimmune diseases has been reported in up to 50% of cases. Numerous studies have tried to pinpoint specific autoantibodies against pituitary or hypothalamic antigens (summarised in (144)). However, these assays are at present not as robust as those for other autoimmune diseases. Recent associations of lymphocytic hypophysitis with IgG4 disease and immune modulatory cancer therapies have led to novel insights. Hypophysitis in IgG4-related, multifocal systemic autoimmune disease is characterised by a relative increase of the plasma-cell population in the inflammatory infiltrate (145). These plasma cells are polyclonal and a significant proportion can be stained by monoclonal antibodies against IgG4. Patients may have raised serum IgG4 levels and coexistent lesions in other organs. The hypophysitis is exquisitely sensitive to steroids and surgery should be avoided (145). The administration of Ipilimumab, a blocker of T cell inhibitory molecule CTLA-4 that is successfully used in immunomodulatory therapy of advanced malignancies such as melanoma, induces lymphocytic hypophysitis in up to 4% of patients. Direct study of human pituitary tissue and experimental CTLA-4 blockade in mice suggested that off-target blockade of CTLA-4 expressed by pituitary endocrine cells, mostly lactotrophs and thyrotrophs, triggers inflammation (146). Specifically, binding of Ipilimumab to endocrine cells resulted in deposition of complement components, triggering a type II hypersensitivity reaction. In patients this was followed by production of anti-pituitary serum antibodies (146).
GRANULOMATOUS HYPOPHYSITIS
Definition
Inflammation of the pituitary gland characterised by the presence of well-formed granulomas with giant cells.
Pathology
The gland may be friable and swollen. Microscopically lymphoplasmacytic inflammation is associated with epithelioid histiocytes and multinucleated giant cells. Necrosis may or may not be present.
Pathogenesis
Granulomatous hypophysitis represents not a single entity and is even less common than lymphocytic hypophysitis. It may be idiopathic (primary), or a (secondary) manifestation of a systemic granulomatous disorder such as sarcoidosis or Wegener’s granulomatosis. Infectious aetiologies include tuberculosis, syphilis and fungal disease. The relationship of the idiopathic form to lymphocytic hypophysitis remains unclear. A recent review of 82 published cases noted a female sex bias but significantly later age at presentation than for lymphocytic hypophysitis (147). The authors speculate that idiopathic granulomatous hypophysitis may represent a chronic or late-stage manifestation of (initially subclinical) lymphocytic hypophysitis.
REFERENCES CHAPTER 3B
- Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE. The prevalence of pituitary adenomas: a systematic review. Cancer 2004; 101:613-619
- Laws ER, Jr., Lopes MB. The new WHO classification of pituitary tumors: highlights and areas of controversy. Acta neuropathologica 2006; 111:80-81
- Zada G, Woodmansee WW, Ramkissoon S, Amadio J, Nose V, Laws ER, Jr. Atypical pituitary adenomas: incidence, clinical characteristics, and implications. Journal of neurosurgery 2011; 114:336-344
- Al-Shraim M, Asa SL. The 2004 World Health Organization classification of pituitary tumors: what is new? Acta Neuropathol 2006; 111:1-7
- Zada G, Lin N, Laws ER, Jr. Patterns of extrasellar extension in growth hormone-secreting and nonfunctional pituitary macroadenomas. Neurosurg Focus 2010; 29:E4
- Bakhtiar Y, Hirano H, Arita K, Yunoue S, Fujio S, Tominaga A, Sakoguchi T, Sugiyama K, Kurisu K, Yasufuku-Takano J, Takano K. Relationship between cytokeratin staining patterns and clinico-pathological features in somatotropinomae. Eur J Endocrinol 2010; 163:531-539
- Bhayana S, Booth GL, Asa SL, Kovacs K, Ezzat S. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab 2005; 90:6290-6295
- Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol 2013; 168:491-499
- Mazal PR, Czech T, Sedivy R, Aichholzer M, Wanschitz J, Klupp N, Budka H. Prognostic relevance of intracytoplasmic cytokeratin pattern, hormone expression profile, and cell proliferation in pituitary adenomas of akromegalic patients. Clin Neuropathol 2001; 20:163-171
- Obari A, Sano T, Ohyama K, Kudo E, Qian ZR, Yoneda A, Rayhan N, Mustafizur Rahman M, Yamada S. Clinicopathological features of growth hormone-producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr Pathol 2008; 19:82-91
- Spada A, Arosio M, Bochicchio D, Bazzoni N, Vallar L, Bassetti M, Faglia G. Clinical, biochemical, and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab 1990; 71:1421-1426
- Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987; 330:566-568
- Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989; 340:692-696
- Adams EF, Brockmeier S, Friedmann E, Roth M, Buchfelder M, Fahlbusch R. Clinical and biochemical characteristics of acromegalic patients harboring gsp-positive and gsp-negative pituitary tumors. Neurosurgery 1993; 33:198-203; discussion 203
- Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (Oxf) 2012; 76:96-102
- Lyons J, Landis CA, Harsh G, Vallar L, Grunewald K, Feichtinger H, Duh QY, Clark OH, Kawasaki E, Bourne HR, et al. Two G protein oncogenes in human endocrine tumors. Science 1990; 249:655-659
- Yang I, Park S, Ryu M, Woo J, Kim S, Kim J, Kim Y, Choi Y. Characteristics of gsp-positive growth hormone-secreting pituitary tumors in Korean acromegalic patients. Eur J Endocrinol 1996; 134:720-726
- Freda PU, Chung WK, Matsuoka N, Walsh JE, Kanibir MN, Kleinman G, Wang Y, Bruce JN, Post KD. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007; 10:275-282
- Taboada GF, Luque RM, Neto LV, Machado Ede O, Sbaffi BC, Domingues RC, Marcondes JB, Chimelli LM, Fontes R, Niemeyer P, de Carvalho DP, Kineman RD, Gadelha MR. Quantitative analysis of somatostatin receptor subtypes (1-5) gene expression levels in somatotropinomas and correlation to in vivo hormonal and tumor volume responses to treatment with octreotide LAR. Eur J Endocrinol 2008; 158:295-303
- Lania A, Persani L, Ballare E, Mantovani S, Losa M, Spada A. Constitutively active Gs alpha is associated with an increased phosphodiesterase activity in human growth hormone-secreting adenomas. J Clin Endocrinol Metab 1998; 83:1624-1628
- Peri A, Conforti B, Baglioni-Peri S, Luciani P, Cioppi F, Buci L, Corbetta S, Ballare E, Serio M, Spada A. Expression of cyclic adenosine 3',5'-monophosphate (cAMP)-responsive element binding protein and inducible-cAMP early repressor genes in growth hormone-secreting pituitary adenomas with or without mutations of the Gsalpha gene. J Clin Endocrinol Metab 2001; 86:2111-2117
- Mayr B, Buslei R, Theodoropoulou M, Stalla GK, Buchfelder M, Schofl C. Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. European journal of endocrinology / European Federation of Endocrine Societies 2013; 169:391-400
- Jeffery PL, Murray RE, Yeh AH, McNamara JF, Duncan RP, Francis GD, Herington AC, Chopin LK. Expression and function of the ghrelin axis, including a novel preproghrelin isoform, in human breast cancer tissues and cell lines. Endocr Relat Cancer 2005; 12:839-850
- Chen JH, Huang SM, Chen CC, Tsai CF, Yeh WL, Chou SC, Hsieh WT, Lu DY. Ghrelin induces cell migration through GHS-R, CaMKII, AMPK and NF-kappaB signaling pathway in glioma cells. J Cell Biochem 2011;
- Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab 2002; 87:2988
- Korbonits M, Bustin SA, Kojima M, Jordan S, Adams EF, Lowe DG, Kangawa K, Grossman AB. The expression of the growth hormone secretagogue receptor ligand ghrelin in normal and abnormal human pituitary and other neuroendocrine tumors. J Clin Endocrinol Metab 2001; 86:881-887
- Kim K, Sanno N, Arai K, Takano K, Yasufuku-Takano J, Teramoto A, Shibasaki T. Ghrelin mRNA and GH secretagogue receptor mRNA in human GH-producing pituitary adenomas is affected by mutations in the alpha subunit of G protein. Clin Endocrinol (Oxf) 2003; 59:630-636
- Xu T, Ye F, Wang B, Tian C, Wang S, Shu K, Guo D, Lei T. Elevation of growth hormone secretagogue receptor type 1a mRNA expression in human growth hormone-secreting pituitary adenoma harboring G protein alpha subunit mutation. Neuro Endocrinol Lett 2010; 31:147-154
- Plockinger U, Albrecht S, Mawrin C, Saeger W, Buchfelder M, Petersenn S, Schulz S. Selective loss of somatostatin receptor 2 in octreotide-resistant growth hormone-secreting adenomas. J Clin Endocrinol Metab 2008; 93:1203-1210
- Kato M, Inoshita N, Sugiyama T, Tani Y, Shichiri M, Sano T, Yamada S, Hirata Y. Differential expression of genes related to drug responsiveness between sparsely and densely granulated somatotroph adenomas. Endocr J 2011;
- Chinezu L, Vasiljevic A, Jouanneau E, Francois P, Borda A, Trouillas J, Raverot G. Expression of somatostatin receptors, SSTR2A and SSTR5, in 108 endocrine pituitary tumors using immunohistochemical detection with new specific monoclonal antibodies. Human pathology 2014; 45:71-77
- Fougner SL, Borota OC, Berg JP, Hald JK, Ramm-Pettersen J, Bollerslev J. The clinical response to somatostatin analogues in acromegaly correlates to the somatostatin receptor subtype 2a protein expression of the adenoma. Clin Endocrinol (Oxf) 2008; 68:458-465
- Cazabat L, Libe R, Perlemoine K, Rene-Corail F, Burnichon N, Gimenez-Roqueplo AP, Dupasquier-Fediaevsky L, Bertagna X, Clauser E, Chanson P, Bertherat J, Raffin-Sanson ML. Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: mutations are found in a subset of young patients with macroadenomas. Eur J Endocrinol 2007; 157:1-8
- Chahal HS, Stals K, Unterlander M, Balding DJ, Thomas MG, Kumar AV, Besser GM, Atkinson AB, Morrison PJ, Howlett TA, Levy MJ, Orme SM, Akker SA, Abel RL, Grossman AB, Burger J, Ellard S, Korbonits M. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med 2011; 364:43-50
- Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, Murat A, Emy P, Gimenez-Roqueplo AP, Tamburrano G, Raverot G, Barlier A, De Herder W, Penfornis A, Ciccarelli E, Estour B, Lecomte P, Gatta B, Chabre O, Sabate MI, Bertagna X, Garcia Basavilbaso N, Stalldecker G, Colao A, Ferolla P, Wemeau JL, Caron P, Sadoul JL, Oneto A, Archambeaud F, Calender A, Sinilnikova O, Montanana CF, Cavagnini F, Hana V, Solano A, Delettieres D, Luccio-Camelo DC, Basso A, Rohmer V, Brue T, Bours V, Teh BT, Beckers A. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 2007; 92:1891-1896
- Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, Paschke R, Saeger W, van der Luijt RB, Sane T, Robledo M, De Menis E, Weil RJ, Wasik A, Zielinski G, Lucewicz O, Lubinski J, Launonen V, Vahteristo P, Aaltonen LA. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci U S A 2007; 104:4101-4105
- Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, Chapple JP, Trivellin G, Gueorguiev M, Guegan K, Stals K, Khoo B, Kumar AV, Ellard S, Grossman AB, Korbonits M, International FC. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Human mutation 2010; 31:950-960
- Jennings JE, Georgitsi M, Holdaway I, Daly AF, Tichomirowa M, Beckers A, Aaltonen LA, Karhu A, Cameron FJ. Aggressive pituitary adenomas occurring in young patients in a large Polynesian kindred with a germline R271W mutation in the AIP gene. Eur J Endocrinol 2009; 161:799-804
- Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, Chahal HS, Igreja SC, Jordan S, Rowe J, Stolbrink M, Christian HC, Wray J, Bishop-Bailey D, Berney DM, Wass JA, Popovic V, Ribeiro-Oliveira A, Jr., Gadelha MR, Monson JP, Akker SA, Davis JR, Clayton RN, Yoshimoto K, Iwata T, Matsuno A, Eguchi K, Musat M, Flanagan D, Peters G, Bolger GB, Chapple JP, Frohman LA, Grossman AB, Korbonits M. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab 2008; 93:2390-2401
- Vargiolu M, Fusco D, Kurelac I, Dirnberger D, Baumeister R, Morra I, Melcarne A, Rimondini R, Romeo G, Bonora E. The tyrosine kinase receptor RET interacts in vivo with aryl hydrocarbon receptor-interacting protein to alter survivin availability. J Clin Endocrinol Metab 2009; 94:2571-2578
- Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ, Launonen V, Karhu A, Aaltonen LA. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006; 312:1228-1230
- Preda V, Korbonits M, Cudlip S, Karavitaki N, Grossman AB. Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. Eur J Endocrinol 2014; 171:659-666
- Schofl C, Honegger J, Droste M, Grussendorf M, Finke R, Plockinger U, Berg C, Willenberg HS, Lammert A, Klingmuller D, Jaursch-Hancke C, Tonjes A, Schneidewind S, Flitsch J, Bullmann C, Dimopoulou C, Stalla G, Mayr B, Hoeppner W, Schopohl J. Frequency of AIP gene mutations in young patients with acromegaly: a registry-based study. J Clin Endocrinol Metab 2014:jc20142094
- Tichomirowa MA, Barlier A, Daly AF, Jaffrain-Rea ML, Ronchi C, Yaneva M, Urban JD, Petrossians P, Elenkova A, Tabarin A, Desailloud R, Maiter D, Schurmeyer T, Cozzi R, Theodoropoulou M, Sievers C, Bernabeu I, Naves LA, Chabre O, Montanana CF, Hana V, Halaby G, Delemer B, Aizpun JI, Sonnet E, Longas AF, Hagelstein MT, Caron P, Stalla GK, Bours V, Zacharieva S, Spada A, Brue T, Beckers A. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur J Endocrinol 2011; 165:509-515
- Daly AF, Tichomirowa MA, Petrossians P, Heliovaara E, Jaffrain-Rea ML, Barlier A, Naves LA, Ebeling T, Karhu A, Raappana A, Cazabat L, De Menis E, Montanana CF, Raverot G, Weil RJ, Sane T, Maiter D, Neggers S, Yaneva M, Tabarin A, Verrua E, Eloranta E, Murat A, Vierimaa O, Salmela PI, Emy P, Toledo RA, Sabate MI, Villa C, Popelier M, Salvatori R, Jennings J, Longas AF, Labarta Aizpun JI, Georgitsi M, Paschke R, Ronchi C, Valimaki M, Saloranta C, De Herder W, Cozzi R, Guitelman M, Magri F, Lagonigro MS, Halaby G, Corman V, Hagelstein MT, Vanbellinghen JF, Barra GB, Gimenez-Roqueplo AP, Cameron FJ, Borson-Chazot F, Holdaway I, Toledo SP, Stalla GK, Spada A, Zacharieva S, Bertherat J, Brue T, Bours V, Chanson P, Aaltonen LA, Beckers A. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab 2010; 95:E373-383
- Jaffrain-Rea ML, Rotondi S, Turchi A, Occhi G, Barlier A, Peverelli E, Rostomyan L, Defilles C, Angelini M, Oliva MA, Ceccato F, Maiorani O, Daly AF, Esposito V, Buttarelli F, Figarella-Branger D, Giangaspero F, Spada A, Scaroni C, Alesse E, Beckers A. Somatostatin analogues increase AIP expression in somatotropinomas, irrespective of Gsp mutations. Endocr Relat Cancer 2013; 20:753-766
- Pagotto U, Arzberger T, Theodoropoulou M, Grubler Y, Pantaloni C, Saeger W, Losa M, Journot L, Stalla GK, Spengler D. The expression of the antiproliferative gene ZAC is lost or highly reduced in nonfunctioning pituitary adenomas. Cancer Res 2000; 60:6794-6799
- Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, Igreja SC, Chapple JP, Jordan S, Lupp A, Schulz S, Ansorge O, Karavitaki N, Carlsen E, Wass JA, Grossman AB, Korbonits M. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab 2012; 97:E1411-1420
- Theodoropoulou M, Zhang J, Laupheimer S, Paez-Pereda M, Erneux C, Florio T, Pagotto U, Stalla GK. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res 2006; 66:1576-1582
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, Schernthaner-Reiter MH, Szarek E, Leal LF, Caberg JH, Castermans E, Villa C, Dimopoulos A, Chittiboina P, Xekouki P, Shah N, Metzger D, Lysy PA, Ferrante E, Strebkova N, Mazerkina N, Zatelli MC, Lodish M, Horvath A, de Alexandre RB, Manning AD, Levy I, Keil MF, Sierra Mde L, Palmeira L, Coppieters W, Georges M, Naves LA, Jamar M, Bours V, Wu TJ, Choong CS, Bertherat J, Chanson P, Kamenicky P, Farrell WE, Barlier A, Quezado M, Bjelobaba I, Stojilkovic SS, Wess J, Costanzi S, Liu P, Lupski JR, Beckers A, Stratakis CA. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 2014; 371:2363-2374
- Iacovazzo D, Caswell R, Bunce B, Jose S, Yuan B, Hernandez-Ramirez LC, Kapur S, Caimari F, Evanson J, Ferrau F, Dang MN, Gabrovska P, Larkin SJ, Ansorge O, Rodd C, Vance ML, Ramirez-Renteria C, Mercado M, Goldstone AP, Buchfelder M, Burren CP, Gurlek A, Dutta P, Choong CS, Cheetham T, Trivellin G, Stratakis CA, Lopes MB, Grossman AB, Trouillas J, Lupski JR, Ellard S, Sampson JR, Roncaroli F, Korbonits M. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta neuropathologica communications 2016; 4:56
- Kamenicky P, Bouligand J, Chanson P. Gigantism, acromegaly, and GPR101 mutations. N Engl J Med 2015; 372:1264
- Bottoni A, Zatelli MC, Ferracin M, Tagliati F, Piccin D, Vignali C, Calin GA, Negrini M, Croce CM, Degli Uberti EC. Identification of differentially expressed microRNAs by microarray: a possible role for microRNA genes in pituitary adenomas. J Cell Physiol 2007; 210:370-377
- D'Angelo D, Palmieri D, Mussnich P, Roche M, Wierinckx A, Raverot G, Fedele M, Croce CM, Trouillas J, Fusco A. Altered microRNA expression profile in human pituitary GH adenomas: down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J Clin Endocrinol Metab 2012; 97:E1128-1138
- Fougner SL, Lekva T, Borota OC, Hald JK, Bollerslev J, Berg JP. The expression of E-cadherin in somatotroph pituitary adenomas is related to tumor size, invasiveness, and somatostatin analog response. J Clin Endocrinol Metab 2010; 95:2334-2342
- Lekva T, Berg JP, Lyle R, Heck A, Ringstad G, Olstad OK, Michelsen AE, Casar-Borota O, Bollerslev J, Ueland T. Epithelial splicing regulator protein 1 and alternative splicing in somatotroph adenomas. Endocrinology 2013; 154:3331-3343
- Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, Schwarzmayr T, Sbiera S, Allolio B, Honegger J, Appenzeller S, Lania AG, Reincke M, Calebiro D, Spada A, Buchfelder M, Flitsch J, Strom TM, Fassnacht M. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol 2016; 174:363-372
- Valimaki N, Demir H, Pitkanen E, Kaasinen E, Karppinen A, Kivipelto L, Schalin-Jantti C, Aaltonen LA, Karhu A. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J Clin Endocrinol Metab 2015; 100:3918-3927
- Lv H, Li C, Gui S, Zhang Y. Expression of estrogen receptor alpha and growth factors in human prolactinoma and its correlation with clinical features and gender. J Endocrinol Invest 2012; 35:174-180
- Font de Mora J, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Molecular and cellular biology 2000; 20:5041-5047
- Carretero J, Blanco EJ, Carretero M, Carretero-Hernandez M, Garcia-Barrado MJ, Iglesias-Osma MC, Burks DJ, Font de Mora J. The expression of AIB1 correlates with cellular proliferation in human prolactinomas. Ann Anat 2013; 195:253-259
- Akinci H, Kapucu A, Dar KA, Celik O, Tutunculer B, Sirin G, Oz B, Gazioglu N, Ince H, Aliustaoglu S, Kadioglu P. Aromatase cytochrome P450 enzyme expression in prolactinomas and its relationship to tumor behavior. Pituitary 2013; 16:386-392
- Leung AW, Kent Morest D, Li JY. Differential BMP signaling controls formation and differentiation of multipotent preplacodal ectoderm progenitors from human embryonic stem cells. Developmental biology 2013; 379:208-220
- Friedman E, Adams EF, Hoog A, Gejman PV, Carson E, Larsson C, De Marco L, Werner S, Fahlbusch R, Nordenskjold M. Normal structural dopamine type 2 receptor gene in prolactin-secreting and other pituitary tumors. The Journal of clinical endocrinology and metabolism 1994; 78:568-574
- Pellegrini I, Rasolonjanahary R, Gunz G, Bertrand P, Delivet S, Jedynak CP, Kordon C, Peillon F, Jaquet P, Enjalbert A. Resistance to bromocriptine in prolactinomas. The Journal of clinical endocrinology and metabolism 1989; 69:500-509
- Zhang W, Murao K, Imachi H, Iwama H, Chen K, Fei Z, Zhang X, Ishida T, Tamiya T. Suppression of prolactin expression by cabergoline requires prolactin regulatory element-binding protein (PREB) in GH3 cells. Horm Metab Res 2010; 42:557-561
- Li Q, Su Z, Liu J, Cai L, Lu J, Lin S, Xiong Z, Li W, Zheng W, Wu J, Zhuge Q, Wu Z. Dopamine receptor D2S gene transfer improves the sensitivity of GH3 rat pituitary adenoma cells to bromocriptine. Mol Cell Endocrinol 2014; 382:377-384
- Peverelli E, Mantovani G, Vitali E, Elli FM, Olgiati L, Ferrero S, Laws ER, Della Mina P, Villa A, Beck-Peccoz P, Spada A, Lania AG. Filamin-A is essential for dopamine d2 receptor expression and signaling in tumorous lactotrophs. The Journal of clinical endocrinology and metabolism 2012; 97:967-977
- Missale C, Boroni F, Losa M, Giovanelli M, Zanellato A, Dal Toso R, Balsari A, Spano P. Nerve growth factor suppresses the transforming phenotype of human prolactinomas. Proceedings of the National Academy of Sciences of the United States of America 1993; 90:7961-7965
- Sigala S, Martocchia A, Missale C, Falaschi P, Spano P. Increased serum concentration of nerve growth factor in patients with microprolactinoma. Neuropeptides 2004; 38:21-24
- Facchetti M, Uberti D, Memo M, Missale C. Nerve growth factor restores p53 function in pituitary tumor cell lines via trkA-mediated activation of phosphatidylinositol 3-kinase. Molecular endocrinology 2004; 18:162-172
- Cooper O, Mamelak A, Bannykh S, Carmichael J, Bonert V, Lim S, Cook-Wiens G, Ben-Shlomo A. Prolactinoma ErbB receptor expression and targeted therapy for aggressive tumors. Endocrine 2013;
- Hock R, Furusawa T, Ueda T, Bustin M. HMG chromosomal proteins in development and disease. Trends Cell Biol 2007; 17:72-79
- Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M, Gregory L, Wass JA, Buck D, Karavitaki N, Grossman AB, McVean G, Ansorge O, Thakker RV. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 2013; 98:E796-800
- Zhou W, Song Y, Xu H, Zhou K, Zhang W, Chen J, Qin M, Yi H, Gustafsson JA, Yang H, Fan X. In nonfunctional pituitary adenomas, estrogen receptors and slug contribute to development of invasiveness. J Clin Endocrinol Metab 2011; 96:E1237-1245
- Palmieri D, Valentino T, De Martino I, Esposito F, Cappabianca P, Wierinckx A, Vitiello M, Lombardi G, Colao A, Trouillas J, Pierantoni GM, Fusco A, Fedele M. PIT1 upregulation by HMGA proteins has a role in pituitary tumorigenesis. Endocrine-related cancer 2012; 19:123-135
- Qian ZR, Li CC, Yamasaki H, Mizusawa N, Yoshimoto K, Yamada S, Tashiro T, Horiguchi H, Wakatsuki S, Hirokawa M, Sano T. Role of E-cadherin, alpha-, beta-, and gamma-catenins, and p120 (cell adhesion molecules) in prolactinoma behavior. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2002; 15:1357-1365
- Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci 2013; 116:317-336
- Stemmler MP. Cadherins in development and cancer. Mol Biosyst 2008; 4:835-850
- Wang C, Su Z, Sanai N, Xue X, Lu L, Chen Y, Wu J, Zheng W, Zhuge Q, Wu ZB. microRNA expression profile and differentially-expressed genes in prolactinomas following bromocriptine treatment. Oncology reports 2012; 27:1312-1320
- Wu ZB, Li WQ, Lin SJ, Wang CD, Cai L, Lu JL, Chen YX, Su ZP, Shang HB, Yang WL, Zhao WG. MicroRNA expression profile of bromocriptine-resistant prolactinomas. Mol Cell Endocrinol 2014; 395:10-18
- Mindermann T, Wilson CB. Thyrotropin-producing pituitary adenomas. J Neurosurg 1993; 79:521-527
- Dong Q, Brucker-Davis F, Weintraub BD, Smallridge RC, Carr FE, Battey J, Spiegel AM, Shenker A. Screening of candidate oncogenes in human thyrotroph tumors: absence of activating mutations of the G alpha q, G alpha 11, G alpha s, or thyrotropin-releasing hormone receptor genes. The Journal of clinical endocrinology and metabolism 1996; 81:1134-1140
- Gatto F, Barbieri F, Castelletti L, Arvigo M, Pattarozzi A, Annunziata F, Saveanu A, Minuto F, Castellan L, Zona G, Florio T, Ferone D. In vivo and in vitro response to octreotide LAR in a TSH-secreting adenoma: characterization of somatostatin receptor expression and role of subtype 5. Pituitary 2011; 14:141-147
- Gatto F, Barbieri F, Gatti M, Wurth R, Schulz S, Ravetti JL, Zona G, Culler MD, Saveanu A, Giusti M, Minuto F, Hofland LJ, Ferone D, Florio T. Balance between somatostatin and D2 receptor expression drives TSH-secreting adenoma response to somatostatin analogues and dopastatins. Clin Endocrinol (Oxf) 2012; 76:407-414
- Yoshihara A, Isozaki O, Hizuka N, Nozoe Y, Harada C, Ono M, Kawamata T, Kubo O, Hori T, Takano K. Expression of type 5 somatostatin receptor in TSH-secreting pituitary adenomas: a possible marker for predicting long-term response to octreotide therapy. Endocr J 2007; 54:133-138
- Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, Tanaka K, Wieland T, Graf E, Saeger W, Ronchi CL, Allolio B, Buchfelder M, Strom TM, Fassnacht M, Komada M. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nature genetics 2015; 47:31-38
- Perez-Rivas LG, Theodoropoulou M, Ferrau F, Nusser C, Kawaguchi K, Stratakis CA, Rueda Faucz F, Wildemberg LE, Assie G, Beschorner R, Dimopoulou C, Buchfelder M, Popovic V, Berr CM, Toth M, Ardisasmita AI, Honegger J, Bertherat J, Gadelha MR, Beuschlein F, Stalla G, Komada M, Korbonits M, Reincke M. The gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing Cushing's disease. J Clin Endocrinol Metab 2015:jc20151453
- Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, Mao Y, Li YM, Hu RG, Zhang ZY, Ye HY, Shen M, Shou XF, Li ZQ, Peng H, Wang QZ, Zhou DZ, Qin XL, Ji J, Zheng J, Chen H, Wang Y, Geng DY, Tang WJ, Fu CW, Shi ZF, Zhang YC, Ye Z, He WQ, Zhang QL, Tang QS, Xie R, Shen JW, Wen ZJ, Zhou J, Wang T, Huang S, Qiu HJ, Qiao ND, Zhang Y, Pan L, Bao WM, Liu YC, Huang CX, Shi YY, Zhao Y. Recurrent gain-of-function USP8 mutations in Cushing's disease. Cell research 2015; 25:306-317
- Hayashi K, Inoshita N, Kawaguchi K, Ibrahim Ardisasmita A, Suzuki H, Fukuhara N, Okada M, Nishioka H, Takeuchi Y, Komada M, Takeshita A, Yamada S. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing's disease. Eur J Endocrinol 2016; 174:213-226
- Tani Y, Inoshita N, Sugiyama T, Kato M, Yamada S, Shichiri M, Hirata Y. Upregulation of CDKN2A and suppression of cyclin D1 gene expressions in ACTH-secreting pituitary adenomas. Eur J Endocrinol 2010; 163:523-529
- Korbonits M, Bujalska I, Shimojo M, Nobes J, Jordan S, Grossman AB, Stewart PM. Expression of 11 beta-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. J Clin Endocrinol Metab 2001; 86:2728-2733
- Ebisawa T, Tojo K, Tajima N, Kamio M, Oki Y, Ono K, Sasano H. Immunohistochemical analysis of 11-beta-hydroxysteroid dehydrogenase type 2 and glucocorticoid receptor in subclinical Cushing's disease due to pituitary macroadenoma. Endocr Pathol 2008; 19:252-260
- Tateno T, Kato M, Tani Y, Oyama K, Yamada S, Hirata Y. Differential expression of somatostatin and dopamine receptor subtype genes in adrenocorticotropin (ACTH)-secreting pituitary tumors and silent corticotroph adenomas. Endocrine journal 2009; 56:579-584
- Gentilin E, Tagliati F, Filieri C, Mole D, Minoia M, Rosaria Ambrosio M, Degli Uberti EC, Zatelli MC. miR-26a plays an important role in cell cycle regulation in ACTH-secreting pituitary adenomas by modulating protein kinase Cdelta. Endocrinology 2013; 154:1690-1700
- Michaelis KA, Knox AJ, Xu M, Kiseljak-Vassiliades K, Edwards MG, Geraci M, Kleinschmidt-DeMasters BK, Lillehei KO, Wierman ME. Identification of growth arrest and DNA-damage-inducible gene beta (GADD45beta) as a novel tumor suppressor in pituitary gonadotrope tumors. Endocrinology 2011; 152:3603-3613
- Butz H, Liko I, Czirjak S, Igaz P, Korbonits M, Racz K, Patocs A. MicroRNA profile indicates downregulation of the TGFbeta pathway in sporadic non-functioning pituitary adenomas. Pituitary 2011; 14:112-124
- Miao Z, Miao Y, Lin Y, Lu X. Overexpression of the Notch3 receptor in non-functioning pituitary tumours. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia 2012; 19:107-110
- Lu R, Gao H, Wang H, Cao L, Bai J, Zhang Y. Overexpression of the Notch3 receptor and its ligand Jagged1 in human clinically non-functioning pituitary adenomas. Oncology letters 2013; 5:845-851
- Karga HJ, Alexander JM, Hedley-Whyte ET, Klibanski A, Jameson JL. Ras mutations in human pituitary tumors. J Clin Endocrinol Metab 1992; 74:914-919
- Ruebel KH, Leontovich AA, Jin L, Stilling GA, Zhang H, Qian X, Nakamura N, Scheithauer BW, Kovacs K, Lloyd RV. Patterns of gene expression in pituitary carcinomas and adenomas analyzed by high-density oligonucleotide arrays, reverse transcriptase-quantitative PCR, and protein expression. Endocrine 2006; 29:435-444
- de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, Hamel N, Choi JH, Park SH, Deal CL, Kelsey MM, Dishop MK, Esbenshade A, Kuttesch JF, Jacques TS, Perry A, Leichter H, Maeder P, Brundler MA, Warner J, Neal J, Zacharin M, Korbonits M, Cole T, Traunecker H, McLean TW, Rotondo F, Lepage P, Albrecht S, Horvath E, Kovacs K, Priest JR, Foulkes WD. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 2014; 128:111-122
- Scheithauer BW, Horvath E, Abel TW, Robital Y, Park SH, Osamura RY, Deal C, Lloyd RV, Kovacs K. Pituitary blastoma: a unique embryonal tumor. Pituitary 2012; 15:365-373
- Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrubl F, Hahnen E, Kreutzer J, Fahlbusch R. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol 2005; 109:589-597
- Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y, Sakamoto M, Hirohashi S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol 2002; 161:1997-2001
- Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML, Lidov HG, Taha H, Martinez-Lage M, Santi M, Storm PB, Lee JY, Palmer JN, Adappa ND, Scott RM, Dunn IF, Laws ER, Jr., Stewart C, Ligon KL, Hoang MP, Van Hummelen P, Hahn WC, Louis DN, Resnick AC, Kieran MW, Getz G, Santagata S. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nature genetics 2014; 46:161-165
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002; 108:837-847
- Oikonomou E, Barreto DC, Soares B, De Marco L, Buchfelder M, Adams EF. Beta-catenin mutations in craniopharyngiomas and pituitary adenomas. J Neurooncol 2005; 73:205-209
- Hofmann BM, Kreutzer J, Saeger W, Buchfelder M, Blumcke I, Fahlbusch R, Buslei R. Nuclear beta-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and rathke cleft cysts: a clinico-pathologic approach. Am J Surg Pathol 2006; 30:1595-1603
- Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol 2014; 127:927-929
- Gaston-Massuet C, Andoniadou CL, Signore M, Sajedi E, Bird S, Turner JM, Martinez-Barbera JP. Genetic interaction between the homeobox transcription factors HESX1 and SIX3 is required for normal pituitary development. Developmental biology 2008; 324:322-333
- Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, Vernay B, Jacques TS, Taketo MM, Le Tissier P, Dattani MT, Martinez-Barbera JP. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A 2011; 108:11482-11487
- Andoniadou CL, Gaston-Massuet C, Reddy R, Schneider RP, Blasco MA, Le Tissier P, Jacques TS, Pevny LH, Dattani MT, Martinez-Barbera JP. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol 2012;
- Hassanein AM, Glanz SM, Kessler HP, Eskin TA, Liu C. beta-Catenin is expressed aberrantly in tumors expressing shadow cells. Pilomatricoma, craniopharyngioma, and calcifying odontogenic cyst. Am J Clin Pathol 2003; 120:732-736
- Holsken A, Kreutzer J, Hofmann BM, Hans V, Oppel F, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. Target gene activation of the Wnt signaling pathway in nuclear beta-catenin accumulating cells of adamantinomatous craniopharyngiomas. Brain Pathol 2009; 19:357-364
- Garcia-Lavandeira M, Saez C, Diaz-Rodriguez E, Perez-Romero S, Senra A, Dieguez C, Japon MA, Alvarez CV. Craniopharyngiomas express embryonic stem cell markers (SOX2, OCT4, KLF4, and SOX9) as pituitary stem cells but do not coexpress RET/GFRA3 receptors. J Clin Endocrinol Metab 2012; 97:E80-87
- Holsken A, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. Tumour cell migration in adamantinomatous craniopharyngiomas is promoted by activated Wnt-signalling. Acta Neuropathol 2010; 119:631-639
- Holsken A, Gebhardt M, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. EGFR signaling regulates tumor cell migration in craniopharyngiomas. Clin Cancer Res 2011; 17:4367-4377
- Yamashiro S, Yamakita Y, Ono S, Matsumura F. Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Molecular biology of the cell 1998; 9:993-1006
- Brastianos PK, Shankar GM, Gill CM, Taylor-Weiner A, Nayyar N, Panka DJ, Sullivan RJ, Frederick DT, Abedalthagafi M, Jones PS, Dunn IF, Nahed BV, Romero JM, Louis DN, Getz G, Cahill DP, Santagata S, Curry WT, Jr., Barker FG, 2nd. Dramatic Response of BRAF V600E Mutant Papillary Craniopharyngioma to Targeted Therapy. Journal of the National Cancer Institute 2016; 108
- Aylwin SJ, Bodi I, Beaney R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016; 19:544-546
- Crotty TB, Scheithauer BW, Young WF, Jr., Davis DH, Shaw EG, Miller GM, Burger PC. Papillary craniopharyngioma: a clinicopathological study of 48 cases. J Neurosurg 1995; 83:206-214
- Shin JL, Asa SL, Woodhouse LJ, Smyth HS, Ezzat S. Cystic lesions of the pituitary: clinicopathological features distinguishing craniopharyngioma, Rathke's cleft cyst, and arachnoid cyst. J Clin Endocrinol Metab 1999; 84:3972-3982
- Okada T, Fujitsu K, Ichikawa T, Mukaihara S, Miyahara K, Kaku S, Uryuu Y, Niino H, Yagishita S, Shiina T. Coexistence of adamantinomatous and squamous-papillary type craniopharyngioma: Case report and discussion of etiology and pathology. Neuropathology 2011;
- Harrison MJ, Morgello S, Post KD. Epithelial cystic lesions of the sellar and parasellar region: a continuum of ectodermal derivatives? J Neurosurg 1994; 80:1018-1025
- Oka H, Kawano N, Yagishita S, Kobayashi I, Saegusa H, Fujii K. Ciliated craniopharyngioma indicates histogenetic relationship to Rathke cleft epithelium. Clin Neuropathol 1997; 16:103-106
- Goodrich JT, Post KD, Duffy P. Ciliated craniopharyngioma. Surg Neurol 1985; 24:105-111
- Park YS, Ahn JY, Kim DS, Kim TS, Kim SH. Late development of craniopharyngioma following surgery for Rathke's cleft cyst. Clin Neuropathol 2009; 28:177-181
- Sato K, Oka H, Utsuki S, Kondo K, Kurata A, Fujii K. Ciliated craniopharyngioma may arise from Rathke cleft cyst. Clin Neuropathol 2006; 25:25-28
- Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. The EMBO journal 1995; 14:6107-6115
- Mao J, Wang J, Liu B, Pan W, Farr GH, 3rd, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 2001; 7:801-809
- Schweizer L, Capper D, Holsken A, Fahlbusch R, Flitsch J, Buchfelder M, Herold-Mende C, von Deimling A, Buslei R. BRAF V600E analysis for the differentiation of papillary craniopharyngiomas and Rathke's cleft cysts. Neuropathology and applied neurobiology 2015; 41:733-742
- Takei Y, Seyama S, Pearl GS, Tindall GT. Ultrastructural study of the human neurohypophysis. II. Cellular elements of neural parenchyma, the pituicytes. Cell and tissue research 1980; 205:273-287
- Brat DJ, Scheithauer BW, Staugaitis SM, Holtzman RN, Morgello S, Burger PC. Pituicytoma: a distinctive low-grade glioma of the neurohypophysis. Am J Surg Pathol 2000; 24:362-368
- Roncaroli F, Scheithauer BW, Cenacchi G, Horvath E, Kovacs K, Lloyd RV, Abell-Aleff P, Santi M, Yates AJ. 'Spindle cell oncocytoma' of the adenohypophysis: a tumor of folliculostellate cells? Am J Surg Pathol 2002; 26:1048-1055
- Phillips JJ, Misra A, Feuerstein BG, Kunwar S, Tihan T. Pituicytoma: characterization of a unique neoplasm by histology, immunohistochemistry, ultrastructure, and array-based comparative genomic hybridization. Arch Pathol Lab Med 2010; 134:1063-1069
- Mete O, Lopes MB, Asa SL. Spindle cell oncocytomas and granular cell tumors of the pituitary are variants of pituicytoma. Am J Surg Pathol 2013; 37:1694-1699
- Fukushima S, Otsuka A, Suzuki T, Yanagisawa T, Mishima K, Mukasa A, Saito N, Kumabe T, Kanamori M, Tominaga T, Narita Y, Shibui S, Kato M, Shibata T, Matsutani M, Nishikawa R, Ichimura K, Intracranial Germ Cell Tumor Genome Analysis C. Mutually exclusive mutations of KIT and RAS are associated with KIT mRNA expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol 2014; 127:911-925
- Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z, Doddapaneni H, Lewis L, Wang M, Suzuki T, Nishikawa R, Natsume A, Terasaka S, Dauser R, Whitehead W, Adekunle A, Sun J, Qiao Y, Marth G, Muzny DM, Gibbs RA, Leal SM, Wheeler DA, Lau CC. Novel somatic and germline mutations in intracranial germ cell tumours. Nature 2014; 511:241-245
- Murray MJ, Bartels U, Nishikawa R, Fangusaro J, Matsutani M, Nicholson JC. Consensus on the management of intracranial germ-cell tumours. The Lancet Oncology 2015; 16:e470-477
- Stacchiotti S, Sommer J, Chordoma Global Consensus G. Building a global consensus approach to chordoma: a position paper from the medical and patient community. The Lancet Oncology 2015; 16:e71-83
- Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li S, Goldstein AM, Parry DM, Kelley MJ. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nature genetics 2009; 41:1176-1178
- Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, Halai D, Berisha F, Cannon SR, Mead S, Kasperaviciute D, Palmen J, Talmud PJ, Kindblom LG, Amary MF, Tirabosco R, Flanagan AM. A common single-nucleotide variant in T is strongly associated with chordoma. Nature genetics 2012; 44:1185-1187
- Falorni A, Minarelli V, Bartoloni E, Alunno A, Gerli R. Diagnosis and classification of autoimmune hypophysitis. Autoimmunity reviews 2014; 13:412-416
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011; 96:1971-1980
- Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Science translational medicine 2014; 6:230ra245
- Hunn BH, Martin WG, Simpson S, Jr., McLean CA. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary 2014; 17:357-365
- ABSTRACT
- PITUITARY ADENOMAS
- PRL-PRODUCING ADENOMA
- THS-PRODUCING ADENOMA
- ACTH-PRODUCING ADENOMA
- GONADOTROPHIN-PRODUCING ADENOMA
- NULL CELL ADENOMA
- PITUITARY CARCINOMA
- PITUITARY BLASTOMA
- CRANIOPHARYNGIOMA
- RATHKE’S CLEFT CYST
- DIFFERENTIAL DIAGNOSIS OF CYSTIC LESIONS
- TUMOURS OF THE NEUROHYPOPYSIS: GRANULAR CELL TUMOUR, PITUICYTOMA, SPINDLE CELL ONCOCYTOMA: TTF-1 FAMILY OF PITUITARY NEOPLASMS
- PRIMARY NEOPLASMS OF THE SELLAR REGION THAT MAY MIMIC PITUITARY ADENOMAS
- CHORDOMA OF THE CLIVUS
- SECONDARY NEOPLASMS OF THE SELLAR REGION THAT MAY MIMIC PITUITARY ADENOMAS
- NON-NEOPLASTIC LESIONSOF THE PITUITARY THAT MAY MIMIC ADENOMAS: LYMPHOCYTIC HYPOPHYSITIS
- GRANULOMATOUS HYPOPHYSITIS
- Review Radiology of the Pituitary.[Endotext. 2000]Review Radiology of the Pituitary.Evanson J. Endotext. 2000
- Review Diabetes insipidus secondary to sellar/parasellar lesions.[J Neuroendocrinol. 2021]Review Diabetes insipidus secondary to sellar/parasellar lesions.Angelousi A, Mytareli C, Xekouki P, Kassi E, Barkas K, Grossman A, Kaltsas G. J Neuroendocrinol. 2021 Mar; 33(3):e12954.
- Rathke's cleft cysts: differentiation from other cystic lesions in the pituitary fossa by use of single-shot fast spin-echo diffusion-weighted MR imaging.[Acta Neurochir (Wien). 2007]Rathke's cleft cysts: differentiation from other cystic lesions in the pituitary fossa by use of single-shot fast spin-echo diffusion-weighted MR imaging.Kunii N, Abe T, Kawamo M, Tanioka D, Izumiyama H, Moritani T. Acta Neurochir (Wien). 2007 Aug; 149(8):759-69; discussion 769. Epub 2007 Jul 9.
- [Collision sellar lesions: pituitary adenoma and Rathke cleft cyst].[Medicina (B Aires). 2021][Collision sellar lesions: pituitary adenoma and Rathke cleft cyst].Padilla Lichtenberger F, Glerean M, Paissan A, Ajler P. Medicina (B Aires). 2021; 81(6):1069-1072.
- Review Sellar Pathologies Mimicking Pituitary Tumors.[Neurol India. 2020]Review Sellar Pathologies Mimicking Pituitary Tumors.Karmarkar VS, Deopujari CE. Neurol India. 2020 May-Jun; 68(Supplement):S154-S160.
- Pathology And Pathogenesis Of Pituitary Adenomas And Other Sellar Lesions - Endo...Pathology And Pathogenesis Of Pituitary Adenomas And Other Sellar Lesions - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...