Study design
The DEVELOP-UK study was designed as a multicentre, unblinded, non-randomised, non-inferiority observational study with an adaptive design, to evaluate the clinical and economic effectiveness of EVLP in assessing and reconditioning donor lungs for transplantation compared with standard lung transplantation. The study also includes an embedded qualitative substudy.
Primary outcome measure
Survival during the first 12 months following lung transplantation was chosen as the primary outcome measure in the study. It is a robust, well-recognised, clinically relevant outcome that is used in the Royal College of Surgeons national audit of UK cardiothoracic transplant activity and in the ISHLT lung transplant registry. A dichotomous outcome such as survival ‘yes/no’ at 30 or 90 days would be less informative, and would omit valuable information about potentially differing survival patterns between the two study groups.
Secondary outcome measures
The secondary outcome measures in this study were all deemed to be important, clinically relevant, patient-centred outcomes that are influenced by the effectiveness of lung transplantation, contribute to the health-care costs and impact on health-related quality of life (HRQoL).
Primary graft dysfunction is a clinical entity that reflects the development of early acute lung injury after lung transplantation. PGD was first defined by a working group (which included a number of the study investigators) of the ISHLT in 2005.39 Its severity is graded between 0 and 3 and it is measured at 0–6, 24, 48 and 72 hours after lung transplantation. The grade is determined by the degree of gas exchange impairment, and by the presence of infiltrate on the post-operative chest radiograph. The PGD grade has been validated in both retrospective and prospective studies, and presence of PGD grade 3 at 72 hours is associated with a reduced early survival. A full PGD score was requested to be determined for all patients in the study.
The durations of invasive ventilation and ITU stay after lung transplantation were collected for all study participants, and provide a valuable source of a range of complications in early post-operative course. In addition, the duration of hospital stay before first discharge home gives a good indication of how effectively the patient is rehabilitating after their lung transplant. These measurements also provided useful information on health resource utilisation for economic evaluation.
The presence of specific post-operative complications was also collected as a secondary outcome measure. These complications included anastomotic complications scored using the recognised and validated Couraud Classification (see Appendix 1),40 which scores airway complications including dehiscence or stricture requiring dilatation or stent placement. Episodes of infection requiring treatment with or without associated hospital admission during the first year, and episodes of acute rejection of ISHLT grade A2 or higher, B1 or higher, or clinically diagnosed acute rejection requiring treatment during the first year, were also collected.
Details of lung function measurements by forced expiratory volume in 1 second (FEV1) and vital capacity at 1, 3, 6 and 12 months post transplant were collected to demonstrate changes in lung allograft function in the first year. Data on chest radiograph appearance at the same time points as lung function were collected to look for any persistent abnormalities such as effusions, cavitation or chronic scarring from the time of transplantation.
Patient survival rate at 90 days post transplantation was collected as an internationally recognised outcome measure in lung transplantation that can be benchmarked against outcomes reported in both the UK and international (ISHLT) registries.
An assessment of HRQoL using the Short Form questionnaire-36 items (SF-36) was collected at three time points in the study (while participants were waiting for transplant and again at 90 days and 1 year post lung transplantation) allowing comparison of HRQoL measured while on the waiting list with that measured post transplant. The HRQoL scores have allowed health state utility scores to be determined using the Short Form questionnaire-6 Dimensions (SF-6D) as part of the economic evaluation.
Health economic assessment
In addition, the full economic impact of using EVLP-reconditioned lungs was assessed, allowing policy-makers to consider these costs in comparison with benefits of increased donor utilisation and reduced waiting list mortality. We aimed to determine whether or not EVLP is a cost-effective intervention for the NHS to support as standard care within UK lung transplant centres in the future.
Patients’ attitudes and experiences
To gain an understanding of the potential impact of EVLP provision to service users, we explored attitudes towards EVLP in patients awaiting lung transplantation, and the experiences of patients receiving EVLP-reconditioned lungs, in a qualitative interview substudy.
Predicting ex vivo lung perfusion success or failure
The DEVELOP-UK study provided a unique opportunity to better understand the donor- and procedure-related clinical determinants of successful or failed EVLP donor lung reconditioning. Objective clinical and physiological indices in the donor lungs before and during EVLP can therefore be correlated with the decision of whether or not to accept the donor lungs for transplant and with clinical outcomes in recipients of EVLP donor lungs.
Sample collection and storage
To add significant value to the DEVELOP-UK study, standardised protocols for BAL, perfusate and lung tissue sampling during EVLP and subsequent storage have been developed. The collection and storage of samples during EVLP was part of the DEVELOP-UK study, and allowed complementary mechanistic studies of EVLP to be performed from the data set. Details of the laboratory-based mechanistic work are, however, not included in this report, as this element of the study was funded from sources other than the National Institute for Health Research (NIHR) Health Technology Assessment (HTA) programme.
Justification for non-randomised design
This is a non-randomised study, as randomisation between EVLP and standard lung transplantation was not considered a viable option. The matching of potential donor lungs to potential recipients is dictated by a number of independent factors, including donor and recipient size, blood group and, if applicable, human leucocyte antigen (HLA) tissue matching to avoid any pre-formed HLA antibodies in the recipient. It was, therefore, not logistically possible to randomise recipients to receive either standard or EVLP donor lungs as part of the study. Furthermore, any attempt to randomly pre-allocate patients on the waiting list to an EVLP or standard group could give rise to a situation where a recipient may not be able to access a well-matched donor organ because it did not fall into his or her pre-allocated group, which would not be ethically acceptable. Randomisation would be possible only if all donor organs were being randomly allocated to EVLP or control, but this is a different research question and was not an objective of this study.
Lung donations from donors with brain death and DCD donors were considered in both arms of this study. The number of DCD donors is increasing year on year in the UK.41 Evidence has emerged that, when lungs from these donors are transplanted, outcomes in recipients are comparable to those achieved with lungs from donation after brain death (DBD) donors.42 However, only a fraction of the UK DCD donors, currently about 5%, have their lungs used for standard transplantation.41 Frequently, there are insufficient data available to be able to objectively assess the function of the lungs from DCD donors, or there is a prolonged warm ischaemic phase after withdrawal of life support that renders the lungs unusable for standard transplantation. EVLP does, however, provide the potential to assess and potentially recondition lungs from DCD donors that cannot be used for standard transplantation.
It was anticipated that a direct result of the DEVELOP-UK study would be an increase in the proportion of DCD donor lungs being used, as DCD donor lungs are often deemed unusable because functional information about the organs is unavailable, which is an indication for use of EVLP assessment. It was considered likely that as the number of DCD donors increases, more lungs from this cohort of donors would be transplanted in the EVLP arm of the study than in the standard arm. This reflects the potential for EVLP to significantly increase the use of lungs from DCD donors. To ensure that the possible higher proportion of DCD donor lungs in the EVLP arm of the study did not bias the results, we planned to use the donor type (DCD or DBD) as a covariate in the multiple regression analysis of the primary and secondary outcome measures to determine their influence.
Justification for adaptive study design
The study statistics and trial methodology teams, in consultation with the clinical investigators, made the decision to use an adaptive design for the DEVELOP-UK study, to allow for the possibility of stopping the trial early should non-inferiority in our primary outcome be determined at an interim analysis, and to allow for re-evaluation of the sample size requirements on the basis of a potentially improved standard of care. It was felt that a total of three analyses, two interim and one final, would achieve a suitable balance between allowing for early stopping and ensuring that sufficient data were collected on secondary outcome variables to make these meaningful. The plan was for the interim analyses to be carried out once a prespecified number of patients had been recruited to each arm (see Power calculation and definition of non-inferiority). The O’Brien–Fleming critical values for the analyses during our study were chosen so that the overall study would have sufficient power to detect our target differences at a significance level of 0.05 once allowance had been made for the interim analyses.
Power calculation and definition of non-inferiority
In the standard arm, the initial best available estimate for survival to 30 days was 94.2%, for survival to 90 days was 91.2% and for survival to 1 year was 78.7%. These data were determined from the Royal College of Surgeons’ UK national audit of lung transplant outcomes. Our aim was to demonstrate that using reconditioned EVLP lungs does not increase the hazard rate of death during the first year by a factor of > 2. A doubling of the hazard rate would imply that survival rates on EVLP would be 88.7% for 30 days, 83.2% for 90 days and 61.9% for 1 year. It was considered that such a difference is not clinically significant and still represents an advantage over waiting longer for a transplant.
It was anticipated that over the predicted 3 years of the study, about 100 EVLP lungs would be transplanted and ≥ 300 normal lung transplants would take place. If both treatment arms matched the standard 78.7% rate of survival over 12 months, then approximately 85 deaths would occur within 1 year of transplantation. Using a fixed sample design, this would be sufficient to ensure 80% power of claiming a significant finding of non-inferiority (at a one-sided 5% level) if both treatment groups actually have the same survival pattern.31 The study was therefore powered to detect a difference of 2, meaning that non-inferiority is assumed to have been achieved if the hazard rate of 12-month survival is not doubled by the use of EVLP.
To obtain sample sizes for an adaptive design, we took the standard sample size and multiplied it by the appropriate inflation factor (which depends on the choices of critical values, number of analyses, significance level and power). For our choices, the inflation factor was 1.0128, resulting in a sample size of 304 in the standard arm and 102 in the EVLP arm. We increased the sample size to 306 in the standard arm while keeping it at 102 in the EVLP arm so that the sample size in both arms would be divisible by 3, to allow for equally spaced interim analyses. This resulted in a required minimum total sample size of 408 with interim analyses after 12-month survival data were available from 102 and 204 patients in the standard arm (34 and 68 in the EVLP arm).
Risks and anticipated benefits for study participants, NHS and society
There is a huge discrepancy between the supply of usable donor lungs and the number of patients with end-stage lung disease who could potentially benefit from lung transplantation surgery in terms of extended longevity and improved quality of life. As a result, many patients die on the waiting list before suitable donor lungs become available. EVLP allows otherwise unusable donor lungs to be meticulously assessed and potentially reconditioned for successful transplantation. The study would also help to understand better how to optimise the use of lungs procured from DCD donors. This technology, therefore, has the potential to expand the donor pool and increase UK lung transplant activity, thereby shortening time spent on the waiting list and reducing waiting list deaths.
The primary risk for the individual participant awaiting lung transplantation is that if they are enrolled in the EVLP arm they may receive a lung or lungs that do not function well, but that risk also exists for standard donor lungs accepted by the current assessment methods. Compared with standard criteria organs, it was not anticipated that EVLP should expose recipients to any different risk profile in terms of microbiological exposure, intensity of induction and maintenance immunosuppression or early post-transplant complications. This was based on reported worldwide experience with EVLP at the time (2010–11) the study was designed and launched. Patients awaiting lung transplantation have severe, often complex, morbidity and place a heavy resource burden on both health and social services. Data from the ISHLT registry clearly demonstrate that nearly 80% of successful lung transplant recipients have no or little functional limitation and around 40% return to either full- or part-time employment, the rest being close to or over retirement age.43 Fewer than 20% of transplant recipients require inpatient treatment related to their lung disease post hospital discharge following the transplant procedure. Thus, by increasing the numbers of successful transplants, EVLP may help to reduce the UK health- and social-care costs of patients awaiting lung transplantation. Furthermore, by assessing the economic impact of using EVLP-reconditioned lungs, the study results should allow policy-makers to balance these costs against the benefits of increased donor utilisation and reduced waiting time mortality. The study aimed to help determine if EVLP is a cost-effective use of tax-payers’ money and an intervention applicable to NHS lung transplant services.
Study population
The DEVELOP-UK study was a UK national multicentre study involving all five officially designated NHS lung transplant centres: Freeman Hospital, Newcastle upon Tyne; Harefield Hospital, London; Papworth Hospital, Cambridge; Wythenshawe Hospital, Manchester; and Queen Elizabeth Hospital, Birmingham. These five centres provide all adult lung transplant activity to potential recipients with end-stage chronic lung disease in England, Scotland, Wales and Northern Ireland.
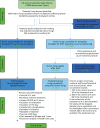
The target population for the study was adult patients aged ≥ 18 years with advanced lung disease, who had already been accepted (at study inception) onto an active lung transplant waiting list in one of the five UK centres, plus any new adult patients who were added to the active waiting list during the study recruitment period of April 2012 to June 2014. The full network coverage means all patients awaiting lung transplantation in the UK, at any one time approximately 250, had the opportunity to take part in the study, and our previous pilot experience suggested that > 90% would consent to take part. The study was designed to have no effect on how potential lung transplant recipients were assessed or selected, or the timing of when they were added to the active transplant waiting list. The flow chart in shows the planned recruitment targets and summary of data collection for the DEVELOP-UK study.
Diagram to explain planned recruitment targets, study end points and planned analysis. QoL, quality of life.
Study inclusion criteria
Male or female adult patients (aged ≥ 18 years) who were either already on or added to the active waiting list for their first lung transplant while the DEVELOP-UK study was in its recruitment phase were eligible to participate; patients provided informed consent for participation in the DEVELOP-UK study at the time of study commencement or time of listing for transplant and reconfirmed informed consent for the DEVELOP-UK study on the day of lung transplant.
Study exclusion criteria
Patients aged < 18 years and adult patients listed for lung retransplantation, heart–lung transplantation, multiorgan transplantation including lung or live donor lobar transplantation were excluded. Patients not in possession of the patient information sheets for the DEVELOP-UK study prior to the day of lung transplantation or those not reconfirming consent for the DEVELOP-UK study on the day of lung transplant were excluded. Patients in the ITU requiring invasive ventilation, extracorporeal membrane oxygenation (ECMO) or interventional lung assist (iLA) support when a donor lung became available were excluded. Patients enrolled in other trials within the preceding 12 months of signing an expression of interest (EOI) or giving full consent had to be discussed with the principal investigator (PI) and chief investigators before being excluded on this basis.
Inclusion and exclusion criteria for interview substudy
All patients who were eligible for the DEVELOP-UK study at The Newcastle Hospitals NHS Foundation Trust and the Royal Brompton and Harefield NHS Foundation Trust were eligible for the interview study. All patients who consented to the DEVELOP-UK study, as a whole, at the above centres were eligible to take part in the interview substudy regardless of whether or not they received a transplant. All patients who consented to the DEVELOP-UK study from Manchester, Papworth and Birmingham sites were excluded from the qualitative study.
Concomitant medications
All standard prescribed medications taken by patients on the waiting list for lung transplantation were permitted in the study. Some medications are stopped at the time of transplant or in the perioperative period. These changes are in line with standard clinical processes and were felt to be equally likely to occur in lung transplant recipients in both arms of the study.
Peri- and post-transplant immunosuppression, including any induction therapy and maintenance immunosuppression, may vary slightly between centres, but continued as per usual practice during the study. In any of the centres, patients in both the EVLP and standard arms of the study got the same standard routine immunosuppressive approach normally used in that centre. The immunosuppressive regimes could, however, be changed, intensified or reduced in line with standard transplant clinical management of the individual patient and his or her circumstances. It was possible that patients awaiting lung transplantation might already be enrolled in a clinical trial of investigational medicinal product (CTIMP) for their underlying disease. Such medications were stopped at the time of transplant and participation in the CTIMP was censored as an event and, therefore, the participation of these patients in the DEVELOP-UK study was not affected.
Patients enrolled in the DEVELOP-UK study who underwent lung transplant in either the standard or EVLP arm should not have been enrolled in any other interventional study in their first 12 months post transplant that might have an effect on 12-month survival. If there was any question of this, then the local PI discussed this with the DEVELOP-UK study chief investigator, who then liaised with the chief investigator of the other study and reported back to the trial steering committee. Observational non-interventional studies were allowable but, again, the local PI had to check with the chief investigator to make sure that there was no interference between the studies. Participants were free to be entered in interventional studies started after their first 12 months post lung transplantation.
Limiting the potential for bias
As a non-randomised, non-blinded study, it was important that the potential for bias in the selection of recipients to receive donor lungs from the EVLP or standard arms was considered and carefully monitored. There was, however, no a priori reason to expect a systematic difference to exist in characteristics between the recipients in the two arms of the study. This is because the donor–recipient match was established before the clinical decision on the usability of the donor lungs was made, meaning that recipient selection should not be influenced by whether EVLP-conditioned or standard lung donation occurs. In particular, there was no evidence to suggest that sicker recipients, whose transplant might be seen as more urgent, would be more likely to receive EVLP-reconditioned lungs than standard donor lungs.
Only when donor lungs were available that had more than one potentially matching recipient was urgency taken into account by the transplant centre. This scenario would be likely to happen as frequently in the standard transplant arm as in the EVLP arm. The two arms of the study were monitored carefully to ensure that no systematic differences occurred in the recipient characteristics. Additionally, it was planned that recognised covariates that are known from the international registry to influence outcomes after lung transplantation would be adjusted for in the statistical analysis. Our pilot experience of transplants performed using EVLP-reconditioned lungs across the UK centres indicated that patients with a range of disease indications, ages, disease severity and both single and bilateral transplants have been included, reflecting the variability that exists on the lung transplant waiting list.
Interventions common to experimental (ex vivo lung perfusion) and control (standard) groups
Donor pathway
Any potential offer of donor lungs was communicated to the transplant centres by standard procedures via the specialist nurses for organ donation (SNODs). Each of the five centres was then responsible for making an initial assessment of the suitability of the donor lungs for transplant, and for determining if they had an appropriately matched potential recipient on their waiting list. If a centre did not have a suitably matched recipient, then the donor lungs were offered to another centre in a controlled rotational manner as part of the standard donor organ placement protocol by NHS Blood and Transplant (NHSBT). The donor lung indices were compared against the donor lung selection criteria for the study and, if suitable for potential transplantation, then the NHSBT zonal organ retrieval team were dispatched to the donor hospital to further assess the donor lungs. After careful assessment, a decision was made using the donor lung acceptance criteria whether the lungs could be used immediately for standard transplantation, should undergo EVLP assessment and reconditioning or were contraindicated completely for transplantation. If appropriate for transplant, the donor lungs were then transported back to the transplant centres in accordance with standard practice.
Donor lung procurement for all lungs in the DEVELOP-UK study
A standard lung procurement procedure was followed for donor lungs used for EVLP in the study. In brief, the organs were antegradely flushed with supplemented PERFADEX® (XVIVO Perfusion AB, Gothenburg, Sweden) [3.3 ml of 3.6% trometamol (THAM), 0.6 ml of calcium chloride (CaCl2) ± 2.5 ml of prostacyclin/l], initially at room temperature and then the remainder at 4 °C. A minimum volume of 60 ml/kg was given. After the antegrade dose, 200 ml was given down each pulmonary vein as a final retrograde flush. An adequate portion of main pulmonary artery, left atrial cuff and, particularly, at least 4 cm of trachea was taken by the retrieval surgeon.
Donor and next of kin consent
Consent for potential donor lungs to be used for lung transplantation was obtained from the donor’s next of kin at the donor hospital by the SNODs, who were employed by NHSBT. This process is standardised nationally and was performed completely independently of the DEVELOP-UK study.
If standard consent for lung donation was granted, the SNODs also asked the next of kin for generic research consent, which is a standard part of the donor consent process. This allowed the study team to collect and store samples from the donor lung before and after EVLP, as described in Appendix 2, for parallel mechanistic studies even if the donor lungs were not deemed transplantable after EVLP. If the donor’s next of kin did not provide generic research consent, then only clinical data measured during the EVLP process were collected and used in the study, and no lung tissue samples were taken for mechanistic work. This did not compromise the delivery of the primary and secondary end points of the study.
Lung recipient pathway pre and post transplantation
Patients referred to any of the five participating sites for consideration of lung transplantation over the course of the study recruitment phase underwent a standard clinical assessment. Those deemed eligible for, and who consented to, lung transplantation were added to the active lung transplant waiting list. Those on the transplant list at the time of study inception would already have been through the same assessment process.
At the time of listing for transplant, patients were offered the opportunity to take part in the DEVELOP-UK study. In addition, at the time of study inception, any patient who was already on the active lung transplant list was also offered the opportunity to take part in the DEVELOP-UK study. The consent process was performed in accordance with National Research Ethics Service guidance, as described in Lung recipient consent. As the period of waiting for lung transplantation can vary widely and commonly exceeds 12 months, it was necessary to reconfirm consent for the study at the time when a potential donor lung(s) became available and the study participant was called in for possible transplantation. However, if the original consent form had been signed on the day of transplant, reconfirmation of consent was not required.
Patients were told on the day of transplant whether they were to receive a donor lung that had undergone EVLP assessment and reconditioning or a standard donor lung. Patients received either standard donor lungs direct from a donor (standard transplant, control arm) or donor lungs after EVLP assessment and reconditioning (intervention arm) in accordance with donor organ–recipient matching. Transplanted lungs, whether ‘standard’ or EVLP reconditioned, always remain vulnerable to the possibility of rejection and one of the main risk factors is low immunosuppression levels. For this reason, patients were thoroughly counselled prior to being accepted onto the transplant list about the need for absolute concordance with their treatment and to attend all arranged post-transplant follow-up visits. As a result, during the multidisciplinary pre-transplant assessment, a considerable amount of time was spent explaining this aspect of care to the patients. If, despite these attempts, there remained evidence of likely non-compliance with treatment, these individuals were not usually offered the option of transplantation.
Lung recipient consent
Informed and voluntary consent was obtained via an iterative process, first at the initial discussion of the clinical and research aspects of the study, and then again, provided this occurred not less than 24 hours later, on the day of possible transplant. If, however, the consent form was signed on the day of transplant, reconsent was not required. Consent for the DEVELOP-UK study participation was sought separately from the standard consent for lung transplant surgery. No additional screening procedures, over and above those necessary to determine eligibility and suitability for lung transplant, were required to determine eligibility for the trial element of the DEVELOP-UK study. Therefore, all adult patients being considered for lung transplant who satisfied the inclusion criteria were approached to take part in the DEVELOP-UK study. Patients waiting for transplantation are desperately sick, very vulnerable and grasping at any lifeline. Securing genuinely informed consent was therefore an important consideration. The initial consent process took place well ahead of the time of transplant and the stressful environment that this generates. Consent was taken either at inception of the study for those already on the transplant waiting list or at the time of listing for transplant for those added to the active transplant list during the course of the study. A copy of the consent documentation is included in Appendices 3 and 4.
Consent was taken by the site PI or a member of the study team with appropriate designated responsibility on behalf of the local PI. In the consent process, care was taken not to unjustifiably inflate hope of a shorter waiting time for transplantation as a result of EVLP being available. A clear definition of what constitutes an unusable donor lung in the study was explained; definitions of acceptability of lungs for standard transplantation and for transplantation after EVLP were agreed and standardised across all centres. Patients were offered firm reassurance that if donor lungs did not improve sufficiently after EVLP reconditioning to satisfy acceptability criteria, they would not be used. Any potential recipient who decided not to participate in the DEVELOP-UK study continued to have equal access to donor lungs for standard transplant. Those choosing not to give consent were not obliged to give a reason, but if they provided a reason this was recorded in an anonymised way to inform the Trial Steering Committee.
Additional informed consent, using a separate participant information sheet and consent form, was sought from the subset of patients approached to take part in the qualitative interviews. Lack of consent to take part in this element of the study did not preclude participation in the trial.
For both the trial and the qualitative substudy, if a potential participant had the capacity to consent for him/herself, but was unable to provide written consent because of visual or motor impairments, or literacy problems, oral informed consent was taken in the presence of an independent witness, who initialled, signed and dated the consent form on the participant’s behalf.
We did not anticipate that any potential study participants would lack capacity to consent on initial recruitment to the study or at the point of reconfirming consent at the time a donor lung became available. It was, however, possible, although unlikely, that they could lose capacity over the follow-up period. For example, if as a result of transplant surgery, any participant were to lose capacity temporarily or permanently, such as by requiring prolonged ventilation in the ITU or by suffering a stroke, we planned to continue to collect outcome measures in relation to such patients, working with personal or nominated consultees and in line with the requirements of the Mental Capacity Act.44
We did not seek separate written consent from nominated consultees in the event of loss of capacity, as this scenario was included in the initial participant consent form and patients were specifically asked to give consent for continued collection of observational data as part of the study if they lost capacity after transplantation. As many of the data in the follow-up period were observational, their collection did not impact on the standard care that any participant who has lost capacity would expect to receive.
The original signed consent form and reconsent form were retained in the investigator site file, with a copy in the clinical notes and a copy provided to the participant. Participants were asked to consent explicitly to their general practitioner (GP) being informed of their participation in the trial element of the DEVELOP-UK study.
The right to refuse to participate without giving reasons was respected. Owing to the small subject population, the information sheet and consent form for the study were available only in English. Interpreters were available for all visits of patients who required them either for verbal translation to another language or for deaf subjects wishing to take part in the study, via local NHS arrangements.
Protocol compliance
The protocols determining the selection of donor lungs to undergo EVLP and indices that determine whether or not the lungs were suitable for transplant after EVLP were clearly described in the study protocol and are presented in an appendix to this report (see Appendix 2). To ensure compliance with the protocol, data were collected about the donor assessment and EVLP procedure. This allowed confirmation that the donor lung was appropriately allocated to undergo EVLP and that the decision on its suitability was correctly determined. If any instances were identified when the protocol was not followed, this was recorded as a protocol deviation and the site PI was asked to document why the protocol deviation occurred.
Ethics and regulatory issues
The conduct of this study was in accordance with the recommendations for physicians involved in research on human subjects adopted by the 18th World Medical Assembly, Helsinki, 1964, and later revisions.45 All members of the research team, the investigators and supporting staff at each of the participating sites received training in those aspects of good clinical practice appropriate to their role in the trial, in particular the processes for obtaining informed consent, including the requirements of the Mental Capacity Act,44 and were expected to operate to principles of good clinical practice.
A favourable ethical opinion from the National Research Ethics Service (reference number 11/NE/0342) and NHS research and development (R&D) approval was secured prior to commencement of the study. Local NHS approvals were secured before recruitment commenced at each site. The Newcastle Clinical Trials Unit, in its capacity as study co-ordination centre, obtained a written copy of local approval documentation before initiating each centre and accepting participants into the study.
Information sheets were provided to all eligible subjects, and written informed consent was obtained prior to any study procedures. Patients on the transplant waiting list who lived a significant distance from the transplant centre were given the opportunity to sign an EOI form that allowed them to subsequently consent when next attending the transplant centre (which might be on the day of transplant). Signing of the EOI form permitted completion of the first SF-36 questionnaire and collection of waiting list survival data. Copies of the patient information sheet and consent forms are included in Appendix 4.
We obtained informed and voluntary consent via an iterative process, providing adequate time (i.e. a period of not < 24 hours) for consideration and discussion of the clinical and research aspects of the study. For incident cases, initial consent was taken at the time a patient was listed for lung transplant. For those patients already on the transplant list at the time of study initiation, consent was sought when the study opened at their transplant centre. Reconsent on the day of transplant was sought only from patients initially consenting to the study prior to the day of lung transplant.
Assessments and data collection
All study-specific follow-up data were collected during the time of the clinical admission to hospital for the lung transplantation procedure and, subsequently, at study visits that were co-ordinated to coincide with routine post-lung transplantation clinic visits. The study research nurse ensured that routine clinic visits were mapped to the study visit requirements by liaison with study participants and the transplant outpatient facilities in each centre.
The scheduled outpatient study visits were at 1, 3, 6 and 12 months post transplant. A window of ± 10 days around each timetabled study visit was allowed. If a participant was unable to attend a study visit within the allowable window, for example because he or she was an inpatient at an external hospital that was not the study centre, then every effort was made to acquire the same study-specific information from the non-study hospital. The HRQoL questionnaire (SF-36) was self-completed by each study participant (or in conjunction with their nominated proxy).
Patients’ views and perceptions of EVLP were explored through qualitative interviews conducted by a trained researcher. When possible, these interviews were performed face to face at study visits after transplantation. Those interviewed prior to transplant were interviewed either in their own home or, more usually because of the large geographic spread of individuals, by telephone.
All clinical tests required to determine the success of EVLP assessment and reconditioning of donor lungs, including ABG analysis, glucose and lactate concentration measurement, and microbiological cultures, were performed in each study centre using local laboratories and equipment.
Standard blood profiles during follow-up were performed as part of the recipients’ routine clinic care in each participating centre’s certified NHS laboratories, and results were obtained from hospital data systems.
Data were collected by direct clinical observation, by clinical interpretation and from source patient records or NHS documentation by the study clinical research fellow and the study research nurse, and the required data fields were completed on the case report form (CRF) by the research nurse or a designated data manager in each centre under the supervision of the local PI. A paper CRF was initially used in the study, but in early 2014 an electronic CRF (MACRO; InferMed, Elsevier, London, UK) began to be used, in line with regulations at the sponsoring trust. The donor data required for the study (such as age, comorbidities and oxygenation, among others) were collected routinely by the SNODs, and were then captured electronically by linking to the core data data set collected by NHSBT centrally.
Serious adverse event reporting
Guidance on adverse event (AE) and serious adverse event (SAE) reporting, as well as determining the degree of relatedness and assessment of causality for SAEs that may be related to study participation, was provided in the study protocol.
As lung transplant recipients experience a significant number of AEs as part of their normal recovery from transplant surgery, the study protocol provided clear guidance on what constituted a SAE that required expedited reporting. This was to avoid a huge burden of reporting that had no relevance to this observational study (no CTIMP involved to monitor). Hospitalisations for elective treatment of a pre-existing condition, and hospitalisations as part of routine post-transplant surveillance did not need reporting as SAEs. Unrelated hospitalisations were elicited at the scheduled follow-up appointments and at all emergency appointments.
Serious adverse events requiring expedited reporting included death within 90 days of lung transplantation, severe PGD requiring ECMO/iLA support, bronchial anastomotic dehiscence or any unexpected SAE felt to be probably or definitely causally related to EVLP.
Some SAEs were excluded from expedited reporting to reduce the burden of reporting of events that are common in the transplant journey. These were death on the waiting list prior to transplant or later than 90 days after lung transplantation; PGD grades 1–3 not requiring ECMO/iLA support or severe sepsis associated with consolidation, necrosis or cavitation of lung tissue within 30 days of transplant; renal failure necessitating renal replacement therapy, gastrointestinal complications, central nervous system complications; and infections requiring an addition or change in antimicrobial therapy.
Medium- and longer-term outcomes that did not require reporting as urgent SAEs were bronchial strictures (whether or not they required bronchial stenting), acute rejection requiring augmented immunosuppression, development of post-transplant lymphoproliferative disease or obliterative bronchiolitis. Finally, deterioration of any pre-existing medical conditions both before and after transplantation did not require urgent reporting.
Public and patient involvement and engagement
The DEVELOP-UK investigators were committed to ensuring appropriate public and patient engagement throughout the study.
The CF Trust was approached to provide patient and service user expertise in the design of the study. Oli Lewington, who has previously undergone lung transplantation, agreed to join the study team in order to help prepare the application for funding, and to contribute to the study design and to the writing of the Plain English summary. The chief investigator presented the study proposal to the board of directors of the trust, and the concept of the study to the annual public meeting of the CF Trust. Following award of the funding, Mr Lewington assisted in producing the participant documentation and the final study report.
Lay members were appointed to the Trial Steering Committee to regularly review study progress and to provide valuable public input into key decision-making during the study.