

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some Drugs and Herbal Products. Lyon (FR): International Agency for Research on Cancer; 2016. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 108.)

4.1. Absorption, distribution, metabolism, and excretion
4.1.1. Humans
(a) Absorption, distribution, metabolism, and excretion
The metabolic scheme for sulfasalazine in humans is shown in Fig. 4.1 (Das & Dubin, 1976; NTP, 1997a).
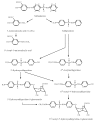
Fig. 4.1
Metabolic pathways of sulfasalazine in humans
Sulfasalazine is not absorbed to any significant extent from the stomach (Das & Dubin, 1976). Slow absorption of small amounts (~10–30%) via the small intestine has been reported before enterohepatic recycling, and with the majority of unchanged drug reaching the colon (Das & Dubin, 1976; Azad Khan et al., 1982).
The sulfasalazine molecule comprises 5-ASA and sulfapyridine moieties, linked by an azo bond, which is cleaved by bacterial azoreductases in the colon, releasing 5-ASA and sulfapyridine (Azad Khan et al., 1982). This cleavage is the rate-limiting step for clearance of sulfasalazine (Das & Dubin, 1976). Most of the 5-ASA is excreted; approximately 50% directly in the faeces, and at least 25% via the kidneys (after absorption and acetylation in the liver) (Das & Dubin, 1976; Azad Khan et al., 1982). In contrast, sulfapyridine is almost completely absorbed. In the liver, sulfapyridine undergoes hydroxylation and/or N-acetylation to 5′-hydroxysulfapyridine, N4-acetylsulfapyridine, and N4-acetyl-5′-hydroxysulfapyridine subsequently forming glucuronic acid conjugates, before excretion mainly in the urine (Das & Dubin, 1976; Azad Khan et al., 1982).
In studies of serum from 10 healthy male volunteers given single oral doses of 4 g of sulfasalazine, parent drug was detectable at 1.5 hours after dosing, and at maximum concentrations at 3–5 hours in nine subjects, and after 7 hours in one subject (Schröder & Campbell, 1972). Metabolites (sulfapyridine, and acetylated and glucuronidated derivatives) were detected in the serum at 3–5 hours after dosing (Schröder & Campbell, 1972). The pharmacokinetics of rectally administered sulfasalazine have been studied in three healthy male Japanese volunteers (Tokui et al., 2002). Sulfasalazine (6.5 mmol), given as a single suppository, reached maximum plasma concentration (2.5 ± 0.4 µM) in 5 hours (Tmax), and an area under the curve (AUC) of 27.4 ± 4.8 µM.h. Parent drug was almost completely hydrolysed in the colon, and the urinary recovery was only approximately 0.2%. Maximum plasma concentration (Cmax) of the metabolite N-acetyl-5-aminosalicylic acid was 0.5 ± 0.2 µM, reached in 12 hours, while that of sulfapyridine was 1.2 ± 0.4 µM, reached in 5 hours. 5-ASA was not detected in the serum. Administration of an enema containing 6.5 mmol of 5-ASA, resulted in Cmax 5.8 ± 2.0 µM in 1 hour and an AUC of 29.4 ± 11.1 µM.h. In the urine, approximately 0.3% was recovered unchanged. The Cmax for the acetylated metabolite, N-acetyl-5-aminosalicylic acid, was 13.3 ± 3.6 µM in 7 hours. More than 10% of 5-ASA was excreted in the urine as acetyl-5-aminosalicylic acid, suggesting that absorption of 5-ASA is favoured when administered rectally (Tokui et al., 2002).
A study to compare the absorption and metabolism of oral preparations of sulfasalazine, mesalazine (5-ASA) and olsalazine (a dimer of 5-ASA) used regularly by patients (n = 12, 13, and 8, respectively) for treatment of ulcerative colitis, showed considerably greater absorption of 5-ASA and less acetylation, in patients receiving mesalazine than in those receiving olsalazine or sulfasalazine (Stretch et al., 1996).
(b) Variation in absorption, distribution, metabolism, and excretion
(i) IBD and rheumatoid arthritis
The characteristics of absorption, metabolism, and excretion of the parent drug in four patients with IBD (ulcerative colitis or Crohn disease) were similar to those in four healthy subjects, each given a single oral dose of sulfasalazine (3 or 4 g). However, absorption and urinary excretion of the metabolite, sulfapyridine, was decreased in patients with IBD. The metabolism of sulfasalazine was markedly reduced in patients taking antibiotics and after removal of the large bowel (Azad Khan et al., 1982).
Pharmacokinetic studies of sulfasalazine and its principal metabolites in 13 patients with rheumatoid arthritis and 8 patients with IBD given sulfasalazine as a single oral dose of 2 g (Astbury et al., 1990), showed that patients with rheumatoid arthritis had a significantly higher (and more sustained) plasma concentration of sulfapyridine than did patients with IBD (medians of 14.0 µg/mL and 7.4 µg/mL, respectively). Two factors may have contributed to high peak plasma concentrations of sulfapyridine in patients with rheumatoid arthritis: firstly, the metabolism of sulfapyridine may be impaired, and secondly, a larger quantity of sulfasalazine may reach the lower bowel leading to higher concentrations of the subsequent cleavage compounds. The pharmacokinetics of sulfasalazine were variable among patients; maximum plasma concentrations of sulfapyridine ranged from 8 to 22 µg/mL in patients with rheumatoid arthritis, and from 5 to 18 µg/mL in patients with IBD. The elimination half-life of sulfasalazine ranged from 3 to 8 hours in patients with rheumatoid arthritis, and from 4 to 9 hours in patients with IBD (Astbury et al., 1990).
(ii) Pregnancy
Sulfasalazine and its primary metabolites are able to cross the placenta (Azad Khan & Truelove, 1979; Järnerot et al., 1981). In five patients with ulcerative colitis treated with sulfasalazine (0.5 g, four times per day) throughout and after pregnancy, sulfasalazine was detected in the umbilical cord blood (mean concentration, 50% of that in maternal serum) and at very low concentrations in the amniotic fluid (Azad Khan & Truelove, 1979). Analyses of metabolites showed that total concentrations of sulfapyridine were equal in maternal and cord sera, but concentrations of free sulfapyridine were significantly lower (P < 0.02) in cord sera. Total concentrations of acetylated sulfapyridine were significantly higher (P < 0.025) in cord sera than maternal sera. Total concentrations of 5-ASA were very low in all fluids analysed (Azad Khan & Truelove, 1979). In the study by Järnerot et al. (1981) of 11 pregnant patients with IBD treated with sulfasalazine (1 g daily), concentrations of sulfasalazine were almost identical in cord and maternal serum. Seven of the women were analysed at a later date (4–24 months) after pregnancy; plasma concentrations of sulfasalazine remained the same, but concentrations of sulfapyridine had increased, probably reflecting different extents of protein binding of these compounds, and the different distribution volume in pregnancy (Järnerot et al., 1981).
Small quantities of sulfasalazine and sulfapyridine have also been detected in breast milk (Azad Khan & Truelove, 1979; Järnerot & Into-Malmberg, 1979). Mean concentrations of sulfasalazine and total sulfapyridine in milk, compared with concentrations in maternal serum, were approximately 30% and 50%, respectively, as reported by Azad Khan & Truelove (1979), and negligible and 40%, respectively as reported by Järnerot & Into-Malmberg (1979). The various metabolites of sulfapyridine were present in approximately the same proportions as in maternal serum (Azad Khan & Truelove, 1979). It was estimated that an infant would receive sulfapyridine at dose of 3–4 mg/kg bw, after a maternal dose of 2 g of sulfasalazine per day (Järnerot & Into-Malmberg, 1979). Sulfapyridine and its acetylated and glucuronidated metabolites have been shown to be excreted by babies, 1–2.5 months after maternal dosing (Järnerot & Into-Malmberg, 1979).
(c) Genetic polymorphisms
(i) N-Acetyltransferases
The sulfasalazine molecule may be considered as a slow-release carrier for sulfapyridine, but there is large inter-individual variation in the rate of metabolism of sulfapyridine, which can affect steady-state serum concentrations (Das & Dubin, 1976). The rate of N-acetylation of 5-ASA to N-acetyl-5-aminosalicylic acid is under genetic control (Das & Dubin, 1976). The cytosolic N-acetyltransferase (NAT) family comprises NAT1 and NAT2, which catalyse the transfer of acetyl groups from activated acetyl-coenzyme A to the nitrogen of primary amines, hydrazines, or hydrazides (Kuhn et al., 2010). Single nucleotide polymorphisms have been detected in NAT2. The wildtype has been designated NAT2*4, but NAT2*5, NAT2*6, and NAT2*7 alleles encode N-acetyltransferase enzymes with amino-acid changes that cause reduced activity (slow acetylating function). Patients with a “slow” acetylator phenotype generally show significantly higher, and more sustained plasma concentrations of sulfapyridine and its non-acetylated metabolites. The elimination half-life of sulfasalazine in patients with a slow-acetylator phenotype may be approximately 50–100% longer than in those with a fast-acetylator phenotype (Taggart et al., 1992).
A 1–3 year study of 185 patients with ulcerative colitis undergoing daily treatment with sulfasalazine (2 g per day) demonstrated that serum concentrations of sulfapyridine (both free and total) were higher in patients with a slow-acetylator phenotype (Azad Khan et al., 1983). Concentrations of sulfasalazine and of 5-ASA were not significantly different in fast and slow acetylators. These findings were confirmed by later studies (Taggart et al., 1992).
The frequency of polymorphism in NAT2 varies in different racial or ethnic populations (Ma et al., 2009). Studies have shown that about 60% of patients with IBD, studied in Edinburgh, Scotland, are slow acetylators (Das et al., 1973), and a similar proportion was found in healthy volunteers in a study in Liverpool, England (Schröder & Evans, 1972). In Germany, a study of NAT2 genotype and acetylation, using sulfasalazine as probe drug, showed that 24 out of 44 healthy volunteers (54.5%) were slow acetylators, in accordance with the “slight prevalence of slow acetylators in central European (Caucasian) populations” which has been reported in several studies (Kuhn et al., 2010).
The NAT2 genotype has also been investigated in Asian populations. Studies in 21 Japanese subjects (8 healthy subjects and 13 patients with IBD) given a single oral dose of sulfasalazine at 40 mg/kg bw demonstrated generally good correlation between three NAT2 genotypes (rapid, intermediate, slow acetylators) and the plasma or urinary concentrations of sulfapyridine and N4-acetyl sulfapyridine (Tanigawara et al., 2002). Similar analyses in seven healthy Japanese subjects after 8 days of continued (“multiple dosing”) oral doses of 1 g of sulfasalazine once per day also demonstrated correlation with genotype (Kita et al., 2001). In 18 healthy Chinese men given 1 g of sulfasalazine as a single oral dose, the NAT2 gene was shown to be an important determinant of metabolite profiles; the frequency of slow acetylators in the Chinese population was lower than in Caucasians, but higher than in the Japanese population (Ma et al., 2009).
The effects of age and acetylator status on the pharmacokinetics of sulfasalazine were compared in patients with rheumatoid arthritis (8 young and 12 elderly, with equal numbers of slow and fast acetylators in both age groups), each receiving sulfasalazine at 2 g per day for 21 days (Taggart et al., 1992). In the elderly, the elimination half-life of sulfasalazine was increased [possibly partly due to slow cleavage of the azo bond (Tett, 1993)], and steady-state serum concentrations of N-acetyl-5-aminosalicylic acid were higher. The pharmacokinetics of sulfapyridine were unchanged with age, but were influenced by acetylation status, in particular, with increased steady-state serum concentrations in slow acetylators. Although age is a determinant of the steady-state concentration of salicylate moieties, the acetylator phenotype seems to play the greater role in determining serum concentrations of sulfapyridine (Taggart et al., 1992).
(ii) Role of transporter proteins
An efflux ATP-binding cassette (ABC) transporter, the breast cancer resistance BCRP protein (encoded by the ABCG2 gene), has a role in the pharmacokinetics of various drugs, including sulfasalazine. The BCRP protein is expressed at the luminal membrane of cells with key functions in drug transport, namely, placental trophoblast cells, hepatocyte bile canaliculi, kidney cells, and enterocytes.
The poor bioavailability of sulfasalazine has long been attributed to its low solubility and poor permeability (Das & Dubin, 1976). However, treatment of human T-cells with sulfasalazine was shown to cause cellular drug resistance that was mediated by induction of BCRP (ABCG2), suggesting that sulfasalazine may be a substrate for human BCRP (van der Heijden et al., 2004; Urquhart et al., 2008).
A subsequent study in vitro, using data on expression in human tissue, indicated an association between reduced cell surface expression of the 421C > A variant and reduced BCRP-mediated transport of sulfasalazine in patients carrying an A allele at position 421 of the BCRP [ABCG2] gene (Urquhart et al., 2008).
Variation in the ABCG2 gene may impair the transport of drugs that are substrates for the ABC transporter, leading to increased intestinal absorption, and/or decreased biliary excretion, and result in high plasma concentrations, as demonstrated in 37 healthy Japanese people selected according to ABCG2 and NAT2 genotype and given a single oral dose of 2 g of sulfasalazine (Yamasaki et al., 2008). They showed that in ABCG2-A/A subjects, mean plasma AUC0–48h and Cmax values for sulfasalazine were significantly higher, and the AUC for sulfapyridine was lower (except in those who also had the slow-acetylator NAT2 genotype) than in individuals without the ABCG2 variant. The increased AUC for sulfasalazine in ABCG2-A/A subjects may result from increased oral availability and/or decreased hepatic clearance, since BCRP is expressed on enterocytes and hepatocytes. The ratios of AUCacetylsulfapyridine/AUCsulfapyridine were significantly higher in subjects with the rapid-acetylator NAT2 phenotype than in those with intermediate or slow genotypes, demonstrating that inter-individual variability in the pharmacokinetics of sulfasalazine can be attributed to genetic polymorphism in drug transport and metabolism (Yamasaki et al., 2008).
4.1.2. Experimental systems
(a) Absorption, distribution, metabolism, and excretion
Experimental studies in Sprague-Dawley rats given diets containing sulfasalazine have shown that most of the sulfasalazine is reductively cleaved by intestinal bacteria to two compounds, 5-ASA (which is poorly absorbed) and sulfapyridine (which is well absorbed) (Peppercorn & Goldman, 1972). After metabolism by mammalian enzymes, 5-ASA and sulfapyridine are excreted mainly in the faeces, and in the urine, respectively (Peppercorn & Goldman, 1972).
In male and female B6C3F1 mice given sulfasalazine as an intravenous dose at 5 mg/kg bw, plasma concentrations of the parent drug rapidly declined with a mean elimination half-life of 0.5 hour in males and 1.2 hour in females (Zheng et al., 1993). The sex-specific differences in clearance rate were reflected in AUCsulfasalazine values (9.21 µM.h-1 and 21.39 µM.h-1, in males and females, respectively).
In male and female B6C3F1 mice given sulfasalazine by gavage at various doses (67.5, 675, 1350, or 2700 mg/kg bw), the bioavailability of the parent drug was approximately 17% (range, 16–18%) at the lowest dose (67.5 mg/kg bw), and was lower (range, 3–9%) at the higher doses. Both sulfapyridine and N4-acetylsulfapyridine were identified in the plasma. Sulfapyridine was eliminated more slowly than the parent compound, and thus accumulated in all mice given multiple doses of sulfasalazine. The differential between plasma concentrations of sulfapyridine and sulfasalazine varied, however, between males and females: the AUCs for sulfapyridine, at all four doses of sulfasalazine, were higher than those of parent drug by 21–32 times in males and by 5–25 times in females, while maximum plasma concentrations were higher than those of the parent drug by 6–8 times in male mice, and up to 4 times in females. Plasma concentrations of N4-acetylsulfapyridine were very low compared with those of sulfapyridine. [This indicated slow acetylation of sulfapyridine by B6C3F1 mice, comparable to that found in humans with the slow-acetylator phenotype.] The pharmacokinetic pattern in B6C3F1 mice given multiple oral doses of sulfasalazine (i.e. daily doses for three consecutive days) was similar to that in mice given a single oral dose, but accumulation of sulfapyridine was evident, producing greater AUCsulfapyridine values in the multiple-dose study than in the single-dose study (Zheng et al., 1993).
In a study by the NTP (1997a), male F344/N rats were given sulfasalazine as an intravenous dose at 5 mg/kg bw, and pharmacokinetic parameters were compared with those in the study in male B6C3F1 mice by Zheng et al. (1993). The rats retained sulfasalazine longer than mice; the AUC for sulfasalazine in rats was double that in mice, while values for systemic clearance and apparent volume of distribution in mice were double those in rats. The rate of elimination of sulfasalazine was similar in rats and mice; plasma elimination rate constants were 1.47 per hour and 1.28 per hour, respectively, and elimination half-lives, 0.53 hour and 0.54 hour, respectively. In F344/N rats given a low oral dose of 67.5 mg/kg bw, sulfasalazine and its metabolites were undetectable; however, after a higher dose (675 mg/kg bw), plasma concentrations of parent compound were detectable within 12 hours (NTP, 1997a).
(b) Role of transporter proteins
BCRP is a member of the ATP-dependent efflux transporters, which includes P-glycoprotein or multi-drug resistance protein 1 (MDR1/ABCB1) and multidrug resistance-associated protein 2 (MRP2/ABCC2). These transporter proteins have significant roles in the processes of drug absorption, distribution, and clearance, and are expressed at the apical membrane of cells in the liver, kidney, brain, placenta, colon, and intestine. From the latter location, on the villus tip of the apical brush-border membrane of intestinal enterocytes, they actively cause efflux of drugs from gut epithelial cells back into the intestinal lumen.
In experiments in Bcrp−/− [Abcg2−/−] knockout mice given sulfasalazine, the AUC for sulfasalazine was greater than in wildtype mice by 13-fold after an intravenous dose (5 mg/kg bw) and 111-fold after an oral dose (20 mg/kg bw) (Zaher et al., 2006). This treatment in the mdr1a [Abcb1a] knockout mouse did not significantly change the AUC for sulfasalazine. Furthermore, studies in wildtype mice treated with an inhibitor of Bcrp (gefitinib) before an oral dose of sulfasalazine resulted in a 13-fold increase in the AUC of plasma sulfasalazine compared with nontreated controls. This work thus demonstrated that Bcrp has a key role in controlling [i.e. maintaining a low (Dahan & Amidon, 2010)] oral bioavailability of sulfasalazine (Zaher et al., 2006).
In Caco-2 cells, sulfasalazine normally exhibits a basolateral-to-apical permeability that is 19 times higher than apical-to-basolateral permeability, indicative of net mucosal secretion (Dahan & Amidon, 2009). In this study of three ATP-dependent efflux transporters (in Caco-2 cells and rat jejunum), specific inhibitors of BCRP and of MRP2 were shown to disrupt the normal direction of sulfasalazine permeability. The presence of both MRP2 and BCRP inhibitors produced an efflux ratio of 1, indicating no efflux of sulfasalazine. Inhibitors of P-glycoprotein had no effect on the movement of sulfasalazine. The results thus suggested that efflux transport of sulfasalazine is mediated by BCRP and MRP2 (Dahan & Amidon, 2009). A more recent study of sulfasalazine absorption has shown that curcumin is a potent inhibitor of human BCRP. Curcumin not only increased the plasma AUC0–8h eightfold in wildtype mice (but not in Bcrp–/– mice), but also increased the plasma AUC0–24h by twofold at microdoses of sulfasalazine and by 3.2-fold at therapeutic doses in humans (Kusuhara et al., 2012).
Further studies of the absorption characteristics of sulfasalazine in the isolated mouse intestine, have indicated that both influx and efflux transporters are involved in the intestinal absorption of sulfasalazine (Tomaru et al., 2013). OATP2B1 is a multispecific organic anion influx transporter. Like BCRP, it is localized at the brush-border membrane of intestinal epithelial cells, and mediates uptake of many endogenous and xenobiotic substrates from the lumen. Like BCRP, sulfasalazine is a substrate for OATP2B1 (Kusuhara et al., 2012; Tomaru et al., 2013). The study by Kusuhara et al. (2012) was inspired by the finding that pharmacokinetic data (plasma AUC for parent drug) from human subjects given sulfasalazine, at either a microdose (100 µg suspension) or a therapeutic dose (2 g as tablets), demonstrated nonlinearity between doses in the AUC of plasma sulfasalazine (Kusuhara et al., 2012). Investigations of sulfasalazine transport were performed in three systems in vitro, namely: (i) ATP-dependent uptake of sulfasalazine by membrane vesicles expressing human BCRP; (ii) oral bioavailability of sulfasalazine in vivo, in wildtype and Bcrp–/– mice; and (iii) uptake of sulfasalazine in HEK293 cells transfected with the influx transporter OATP2B1 (SLCO2B1). The results indicated that sulfasalazine is a substrate for OATP2B1, and that saturation of the influx transporter OATP2B1 at the therapeutic dose is a possible mechanism underlying nonlinearity in the dose–exposure relationship for sulfasalazine (Kusuhara et al., 2012).
The nonsteroidal anti-inflammatory drug indomethacin (an inhibitor of the MRP family that includes MRP2) has been shown to change, in a concentration-dependent manner, the direction of membrane permeability to sulfasalazine in Caco-2 cells; high concentrations of indomethacin substantially reduced efflux of sulfasalazine. Efflux was not however abolished, due to the contribution of BCRP to the control of absorption. Additionally, an indomethacin-induced increase in sulfasalazine permeability through the gut wall was also shown in the rat jejunal perfusion model. The concomitant intake of indomethacin and sulfasalazine may lead to increased absorption of sulfasalazine in the small intestine, reducing its concentration in the colon, and potentially altering its therapeutic effect (Dahan & Amidon, 2010).
4.2. Genetic and related effects
Several studies, particularly those conducted in vivo, have demonstrated genotoxicity associated with sulfasalazine and some of its metabolites. The mutagenicity of sulfasalazine and its two major metabolites, sulfapyridine and 5-ASA, was reviewed by Iatropoulos et al. (1997).
4.2.1. Humans
See Table 4.1
Table 4.1
Genetic and related effects of sulfasalazine.
Mitelman et al. (1982) reported that there was no clear evidence of chromosomal damage in lymphocytes of patients receiving sulfasalazine for 1 month or 4 months at 3 g per day, although an effect could not be ruled out.
Increased frequencies of micronucleus formation and sister chromatid exchange in patients with IBD receiving sulfasalazine have been reported, but confounding factors were apparent in the study (Erskine et al., 1984).
4.2.2. Experimental systems
See Table 4.1
(a) Mutagenicity
Sulfasalazine was not mutagenic in assays for gene mutation in bacteria, including a variety of strains of Salmonella typhimurium, Escherichia coli, and Klebsiella pneumoniae, in a variety of protocols, with or without metabolic activation (Voogd et al., 1980; Zeiger et al., 1988; Iatropoulos et al., 1997). In addition, treatment with sulfasalazine did not result in an increase in mutations conferring 6-thioguanine resistance in mouse lymphoma L5178Y cells, with or without metabolic activation (Iatropoulos et al., 1997).
(b) Chromosomal damage
Mackay et al. (1989) reported positive results in a test for induction of sister chromatid exchange in cultured human lymphocytes treated with sulfasalazine at a concentration of 20–160 μg/mL in the absence of metabolic activation. In contrast, Bishop et al. (1990) observed no increase in the frequency of sister chromatid exchange in Chinese hamster ovary cells treated with sulfasalazine at up to 1000 μg/mL, with or without metabolic activation. An increase in the formation of micronuclei in cultured human lymphocytes after treatment with sulfasalazine (effective concentration range, 40–160 μg/mL) in the absence of metabolic activation was reported by Mackay et al. (1989). No significant increases in the frequency of chromosomal aberration were observed in cultured Chinese hamster ovary cells (Bishop et al., 1990), or cultured human lymphocytes (Iatropoulos et al., 1997), treated with sulfasalazine (concentration, up to 1000 or 100 μg/mL, respectively). Thus the results of tests for chromosomal damage in vitro after treatment with sulfasalazine were generally negative, although sporadic positive results were reported.
In vivo, consistent with results reported in assays in vitro, no increases in the frequency of chromosomal aberration were observed in male mice or male and female rats treated with sulfasalazine by gavage at doses of up to 4000 mg/kg bw per day (Bishop et al., 1990; Iatropoulos et al., 1997; NTP, 1997a).
The results of assays for micronucleus formation in male and female mice treated with sulfasalazine were uniformly positive when multiple treatments (at least two) were employed (Bishop et al., 1990; Witt et al., 1992a, NTP, 1997a). Further investigation of the nature of the induced micronuclei revealed that the majority were kinetochore-positive, suggesting that the micronuclei contained whole chromosomes rather than fragments, and were primarily due to aneuploidy events rather than chromosome breakage (Witt et al., 1992a). This observation was consistent with the negative results in assays for chromosomal aberration with sulfasalazine in vitro and in vivo (Mitelman et al., 1980; Bishop et al., 1990; Iatropoulos et al., 1997; NTP, 1997a). The negative results of one test in male mice given a single dose of sulfasalazine at 4000 mg/kg bw underscored the need for multiple treatments to induce an observable increase in micronucleus formation (NTP, 1997a).
In addition to the necessity for multiple treatments, sulfasalazine may also demonstrate selective activity in mice; the results of a study on micronucleus formation in bone marrow of male rats given three doses of sulfasalazine (highest dose, 3000 mg/kg bw) were judged to be equivocal (NTP, 1997a); in this assay, an initial trial gave a positive response at the highest dose of 2700 mg/kg bw, but a second trial, with a highest dose of 3000 mg/kg bw, gave negative results. This apparent preferential activity in mice was consistent with the observation that mice have a greater systemic exposure than rats to sulfapyridine, the active moiety, after administration of similar doses (Zheng et al., 1993).
In one study, no evidence for genotoxicity was obtained for sulfasalazine when tested for the induction of micronuclei in mouse bone marrow, with or without pretreatment with folate. Likewise, no evidence for formation of DNA adducts was detected by 32P-postlabelling in rat and mouse liver and urinary bladder (Iatropoulos et al., 1997). [The nuclease P1 enrichment procedure was used in the 32P-postlabelling method. Assuming that N-hydroxylation of sulfapyridine occurred in vivo, adducts derived from this metabolite would probably be lost.]
4.2.3. Genotoxicity of sulfasalazine metabolites
See Table 4.2
Table 4.2
Genetic and related effects of metabolites of sulfasalazine.
Sulfasalazine has two major metabolites, sulfapyridine (a carrier molecule that allows transport of sulfasalazine to the intestine, where it is activated) and 5-ASA, the therapeutically active moiety.
(a) 5-ASA
5-ASA has not shown activity in any assay for genotoxicity in vitro or in vivo. It does not induce mutations in any of a variety of Salmonella typhimurium strains, with or without metabolic activation or in Klebsiella pneumoniae in the absence of metabolic activation (Voogd et al., 1980). No induction of sister chromatid exchange, micronucleus formation, or chromosomal aberration has been reported in human lymphocytes or Chinese hamster ovary cells in vitro (Mackay et al., 1989; Witt et al., 1992b). In vivo, no increase in the frequency of micronucleated polychromatic erythrocytes was observed in the bone marrow of male mice treated with 5-ASA (dose range, 125–250 mg/kg bw per day for 3 days) by intraperitoneal injection (Witt et al., 1992b).
(b) Sulfapyridine
Sulfapyridine has been reported to induce sister chromatid exchange in Chinese hamster ovary cells and cultured human lymphocytes in the absence of metabolic activation (Mackay et al., 1989; Witt et al., 1992b); no increase in the frequency of sister chromatid exchange was noted in the presence of metabolic activation in Chinese hamster ovary cells (Witt et al., 1992b). Sulfapyridine did not induce chromosomal aberration in Chinese hamster ovary cells, with or without metabolic activation (Witt et al., 1992b). Mackay et al. (1989) reported that sulfapyridine did not induce micronucleus formation in cultured human lymphocytes in the absence of metabolic activation, at concentrations that reached 400 µg/mL. Sulfapyridine induced a strong, dose-related increase in the frequency of micronucleated polychromatic erythrocytes when administered either as multiple intraperitoneal injections (Witt et al., 1992b) or by gavage (Witt et al., 1992a). As with sulfasalazine, the majority of micronucleated erythrocytes induced by sulfapyridine in mice were shown to contain kinetochores (Witt et al., 1992a), implying that sulfapyridine-induced micronucleus formation resulted from failure of mitotic chromosomal segregation, rather than chromosome breakage.
(c) Metabolites of 5-ASA and sulfapyridine
Mackay et al. (1989) also tested four acetylated and/or hydroxylated metabolites of sulfapyridine and 5-ASA for their ability to induce sister chromatid exchange and micronucleus formation in cultured human lymphocytes. N4-acetylsulfapyridine was capable of inducing both sister chromatid exchange and micronucleus formation, while N4-acetyl-5′-hydroxysulfapyridine only induced sister chromatid exchange. 5′-Hydroxysulfapyridine and N4-acetyl-5-aminosalicylic acid did not induce either sister chromatid exchange or micronucleus formation at the concentrations tested.
4.3. Other mechanistic data relevant to carcinogenicity
4.3.1. Adverse effects
In humans, sulfasalazine is associated with a wide range of adverse side-effects that include agranulocytosis (Kaufman et al., 1996), hepatotoxicity (de Abajo et al., 2004; Jobanputra et al., 2008), nephrotoxicity (Gisbert et al., 2007), neurotoxicity (Liedorp et al., 2008), and pulmonary toxicity (Parry et al., 2002). Sulfasalazine is also associated with reversible infertility in men and in male experimental animals (O’Moráin et al., 1984). Reactions to sulfasalazine may result from an idiosyncratic delayed-type hypersensitivity reaction that may affect internal organs in variable ways (Jobanputra et al., 2008).
Case reports of serious hepatotoxicity associated with sulfasalazine are frequent and occur predominantly within the first month of starting therapy; the pattern of liver injury can be hepatocellular or cholestatic, and may lead to liver failure. Serious hepatotoxicity, which could be a part of the DRESS (drug rash, eosinophilia and systemic symptoms) syndrome is described in approximately 0.1% of users, but the estimated incidence is higher (0.4%) in patients with inflammatory arthritis (de Abajo et al., 2004; Jobanputra et al., 2008).
Renal toxicity associated with sulfasalazine treatment may be irreversible. Although the sulfapyridine moiety is thought to be responsible for most of the adverse effects of sulfasalazine, several case reports in patients with IBD indicate that renal toxicity in humans may occur from treatment with both sulfasalazine and 5-ASA (Gisbert et al., 2007). Clinically, 5-ASA-associated nephrotoxicity is typically expressed as interstitial nephritis, glomerulonephritis, nephritic syndrome, and acute renal failure (Barbour & Williams, 1990; Birketvedt et al., 2000; Augusto et al., 2009). The incidence of clinically restrictive renal impairment has been estimated at < 1 per 500 patients (World et al., 1996). The mechanism is unclear, although both a delayed cell-mediated response, and a dose-dependent effect have been considered (Corrigan & Stevens, 2000). 5-ASA-related nephrotoxicity appeared to be dose-related in female rats given a single intravenous injection of the sodium salt of 5-ASA at doses of up to 5.7 mmol/kg bw (Calder et al., 1972); however, dose-dependency may require the administration of doses much higher than those given to humans. Of note, 5-ASA combines the structural features of a salicylate and a phenacetin, both of which have well documented nephrotoxic potential (Corrigan & Stevens, 2000).
It has been hypothesized that oxidative stress may be a factor in sulfasalazine-induced renal and hepatic injury. Treatment-related alterations in the levels of biomarkers of oxidative stress were detected in kidney and liver tissues of male Sprague-Dawley rats given sulfasalazine as daily oral doses at 0, 300, or 600 mg/kg bw for 14 days. At the highest dose, there were significant decreases in the activities of renal and hepatic superoxide dismutase, and significant increases in catalase activity, thiobarbituric acid-reactive substances, and in the oxidized/reduced glutathione ratio (Linares et al., 2009).
Sulfasalazine can cause haemolytic anaemia (Das et al., 1973; Mechanick, 1985) and methaemoglobinaemia (Miller et al., 1971; Kater, 1974; Azad Khan et al., 1983). In a group of 50 patients receiving sulfasalazine at 2.5 g per day as maintenance therapy for ulcerative colitis, approximately 40% had elevated levels of methaemoglobin (Pounder et al., 1975). Although sulfonamide-induced haemolysis can be severe in patients with glucose-6-phosphate dehydrogenase deficiency, this study showed that sulfasalazine-induced erythrocyte damage also occurred in patients with normal levels of this enzyme (Pounder et al., 1975).
The role of metabolites in sulfasalazine-mediated toxicity was investigated in vitro, using human erythrocytes and mononuclear leukocytes as target cells in the presence of human liver microsomes; methaemoglobin formation and cytotoxicity were selected as toxicity end-points. In addition to sulfasalazine, the study included the metabolites 5-ASA, sulfapyridine, and 5′-hydroxysulfapyridine (Pirmohamed et al., 1991). Bioactivation by human liver microsomes that are dependent on reduced nicotinamide adenine dinucleotide phosphate (NADPH) to a species that caused methaemoglobinaemia and cytotoxicity was only observed with sulfapyridine. Chromatographic analysis demonstrated that sulfapyridine was converted to a short-lived intermediate (t1/2, 8.1 minutes at pH 7.4) with elution characteristics identical to those of synthetic sulfapyridine hydroxylamine. This hydroxylamine (10–500 µM) caused a concentration-dependent increase in both methaemoglobinaemia (2.9–24.4%) and cytotoxicity in the absence of a microsomal system; neither sulfasalazine nor any of the other test metabolites had such effects. When the microsomal incubations were conducted in the presence of micromolar concentrations of reducing agents (e.g. ascorbic acid, glutathione, or N-acetylcysteine), sulfapyridine-induced cytotoxity was decreased in mononuclear leukocytes, but there was no effect upon the levels of methaemoglobinaemia. This suggested that sulfapyridine hydroxylamine could readily penetrate erythrocytes, where it may undergo redox cycling to nitrososulfapyridine, the species ultimately responsible for the production of methaemoglobin. These observations further suggested that N-hydroxylation of sulfapyridine may account for some of the adverse effects associated with sulfasalazine (Pirmohamed et al., 1991). [Of note, N-hydroxylation is thought to play a key role in the bioactivation of aromatic amine carcinogens, such as 4-aminobiphenyl (IARC, 2012).]
4.3.2. Effects upon folate pathways
Sulfasalazine has been shown to inhibit the activity of dihydrofolate reductase, methylenetetrahydrofolate reductase, and serine transhydroxymethylase, and also the cellular uptake of folate (Selhub et al., 1978; Jansen et al., 2004; Urquhart et al., 2010).
4.3.3. Urolithiasis
An increased incidence of transitional cell papilloma of the urinary bladder in male rats treated with sulfasalazine has been correlated (P < 0.01) with increased incidences of concretions (calculi) in the urinary bladder (NTP, 1997a; see also Section 3). In a subsequent study, there was decreased incidence of urinary hyperplasia in male rats subjected to caloric restriction and treated with sulfasalazine, and little evidence of urinary bladder concretion, compared with rats fed ad libitum (NTP, 1997b; see also Section 3). [These data suggested that chronic inflammation associated with urolithiasis may be a factor in sulfasalazine-induced carcinogenesis of the bladder in male rats.]
4.4. Susceptible populations
The adverse effects of sulfasalazine have been linked to sulfapyridine (Das et al., 1973). This metabolite, which is well absorbed from the colon, is inactivated by NAT2-mediated N-acetylation. NAT2 polymorphisms have been associated with different susceptibilities to the adverse effects of sulfasalazine. People with the slow-acetylator genotype have higher serum concentrations of free sulfapyridine and lower concentrations of acetylated sulfapyridine than fast acetylators (see also Section 4.1.1), and appear more likely to experience toxic symptoms when treated with equivalent doses of sulfasalazine (Das & Dubin, 1976; Azad Khan et al., 1983; Ricart et al., 2002; Tanaka et al., 2002; Kumagai et al., 2004; Chen et al., 2007; Soejima et al., 2008).
4.5. Mechanistic considerations
Sulfasalazine was reported to act as a co-carcinogen at a dose of 60 mg/kg bw per day in the 1,2-dimethylhydrazine model of colon carcinogenesis in rats. In the same study, 5-ASA, the active pharmacophore unit of sulfasalazine, acted as a co-carcinogen at a dose of 30 mg/kg bw per day, but not at 60 mg/kg bw per day, which suggested that 5-ASA exerts a protective effect on the colon mucosa, provided a sufficient amount of the compound reaches the colon (Davis et al., 1992). On the basis of early proposals that localized tissue folate deficiency may account for carcinogenesis (Lashner et al., 1989), it was hypothesized that sulfasalazine may be co-carcinogenic due to its anti-folate characteristics. Sulfasalazine inhibited dihydrofolate reductase, methylenetetrahydrofolate reductase, and serine transhydroxymethylase, and also the cellular uptake of folate (Selhub et al., 1978; Jansen et al., 2004; Urquhart et al., 2010). Reduced levels of S-adenosylmethionine or 5,10-methylenetetrahydrofolate, required for thymidine synthesis, might account for the effect; however, colonic cells may not be completely dependent on blood stream nutrients (Meenan, 1993). In the rat, colonic bacterial folate is incorporated in the hepatic folate pool (Rong et al., 1991), and this could counteract sulfasalazine-induced folate depletion (Meenan, 1993). In patients with ulcerative colitis, folate concentrations measured in colonic epithelial cells obtained from endoscopic colon biopsies were not decreased in sulfasalazine-treated patients compared with controls; this contrasted with serum concentrations of folate, which were reduced in patients receiving sulfasalazine (Meenan et al., 1996). These data suggested that the potentially protective effects of folate supplementation against colorectal carcinogenesis in patients with ulcerative colitis were not due to correction of localized folate deficiency.
Two-year studies in male and female F344/N rats given sulfasalazine by gavage indicated some evidence for carcinogenic activity on the basis of increased incidences of transitional cell papilloma of the urinary bladder, and clear evidence for carcinogenic activity in male and female B6C3F1 mice on the basis of increased incidences of hepatocellular adenoma and hepatocellular carcinoma (NTP, 1997a; see also Section 3). The data on mutagenicity of sulfasalazine and its metabolite, sulfapyridine, suggested that the parent drug and the metabolite are predominantly aneugens (Bishop et al., 1990; Witt et al., 1992a, b). Increased frequencies of micronucleus formation and sister chromatid exchange in patients with IBD receiving sulfasalazine have been reported, but confounding factors were apparent in the study (Erskine et al., 1984).
Folate deficiency was considered as a possible explanation for the induction of micronucleus formation by sulfasalazine in vivo. However, patients reported to have an elevated frequency of sister chromatid exchange and micronucleus formation had serum folate concentrations that were at the low end of the normal range, and the observation of reticulocytosis in a 90-day study in mice suggested an erythropoietic effect not characteristic of folate deficiency (Bishop et al., 1990).
The increased incidence of transitional cell papilloma of the urinary bladder in male rats treated orally with sulfasalazine was correlated with increased incidence of concretions (calculi) in the urinary bladder (NTP, 1997a). Chronic inflammation associated with urolithiasis may be a factor in sulfasalazine-induced carcinogenesis of the bladder in male rats.
- Mechanistic and Other Relevant Data - Some Drugs and Herbal ProductsMechanistic and Other Relevant Data - Some Drugs and Herbal Products
Your browsing activity is empty.
Activity recording is turned off.
See more...