

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Hypertriglyceridemia (HTG) can result from a variety of causes. Mild to moderate HTG tracks along with the metabolic syndrome, obesity and diabetes. HTG can be the result of multiple small gene variants or secondary to several diseases and drugs. Severe HTG with plasma triglyceride (TG) levels >1000-1500 mg/dL typically results from: (1) rare variants in the lipoprotein lipase (LPL) complex, where it is termed the familial chylomicronemia syndrome (FCS), and (2) the co-existence of genetic and secondary forms of HTG, termed the multifactorial chylomicronemia syndrome (MFCS), which is a much more common cause of severe HTG. Mild to moderate HTG is associated with an increased risk of premature cardiovascular disease (CVD), while severe HTG can lead to pancreatitis as well as an increased risk of premature CVD. Appropriate management of the patient with HTG requires knowledge of the likely cause of the HTG, to prevent its complications. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
PHYSIOLOGY
A detailed overview of lipoprotein physiology is provided in the Endotext chapter on Lipoprotein Metabolism (1). Here we will briefly review some aspects the metabolism of the triglyceride (TG)-rich lipoproteins, very low-density lipoproteins (VLDL) and chylomicrons (CM) of particular relevance to this chapter.
Secretion of TG-rich Lipoproteins Into Plasma
TGs are transported through plasma as VLDL), which transport TGs primarily made in the liver, and as CM, which transport dietary (exogenous) fat. VLDL secretion by the liver is regulated in several ways. Each VLDL particle has one ApoB100 molecule, making ApoB100 availability a key determinant of the number of VLDL particles, and hence, TG secretion by the liver. In addition to one molecule of ApoB-100, each VLDL particle contains multiple copies of other apolipoproteins, together with varied amounts of TGs, cholesteryl esters, and phospholipids. The extent of TG synthesis is in part determined by the flux of free fatty acids (FFA) to the liver. The addition of TG to the developing VLDL particle in the endoplasmic reticulum is mediated by the enzyme microsomal triglyceride transfer protein (MTTP). The pool of ApoB100 in the liver is not typically regulated by its level of synthesis, which is relatively constant, but by its level of degradation, which can occur in several proteolytic pathways (2). Insulin also plays a role in the regulation of VLDL secretion - it decreases hepatic VLDL production by limiting fatty acid influx into the liver and decreases the stability of, and promotes the posttranslational degradation of ApoB100 (3). Recent studies have shown that ApoC-III, an Apolipoprotein thought to primarily play a role in inhibiting TG removal (see below), also is involved in the assembly and secretion of VLDL (4). VLDL particles (containing ApoB100) also increase in plasma in the postprandial state as well as CM that contain ApoB48 (5).
Consumption of dietary fat results in the formation of CM by enterocytes. Fatty acids and monoacylglycerols that result from digestion of dietary TGs by acid and pancreatic lipases are transported into enterocytes by mechanisms that are not completely understood. In the enterocyte, monoacylglycerol and fatty acids are resynthesized into TGs by the action of the enzymes acyl-coenzyme A: monoacylglycerol acyltransferase and acyl-coenzyme A: diacylglycerol acyltransferase 1 and 2 (DGAT 1 and 2). The resulting TGs are packaged with ApoB48 to form CM, a process also mediated by MTTP (6). CM then pass into the thoracic duct from where they enter plasma and acquire additional apolipoproteins. Of particular relevance to their clearance from plasma is the acquisition of ApoC-II and ApoC-III.
Catabolism of the TG-rich Lipoproteins
TGs in both VLDL and CM are hydrolyzed by the lipoprotein lipase (LPL) complex. LPL is synthesized by several tissues, including adipose tissue, skeletal muscle, and cardiac myocytes. After secretion by adipocytes, the enzyme is transported by glycosylphosphatidylinositol-anchored high-density lipoprotein–binding protein 1 (GPIHBP1) to the luminal side of the capillary endothelium, where it becomes tethered to glycosaminoglycans (GAGs). This pool of LPL is referred to as “functional LPL”, since it is available to hydrolyze TGs in both VLDL and chylomicrons. LPL can be liberated from these GAG binding sites by heparin injection. Several other proteins, reviewed in (7), regulate LPL activity. These include ApoC-II, which activates LPL, and ApoC-III, which inhibits LPL in addition to its effect on VLDL secretion alluded to earlier. Both are produced by the liver and are present on TG-rich lipoproteins. ApoC-III also inhibits the turnover of TG-rich lipoproteins through a hepatic clearance mechanism involving the LDL receptor/LDL receptor-related protein 1 (LDLR/LRP1) axis (8). ApoE also is present on the TG-rich lipoproteins and plays an important role in the uptake and clearance of the remnants of the TG-rich lipoproteins that result from hydrolysis of TGs in these lipoproteins. Other activators of LPL include ApoA-IV (9), ApoA-V (10-12) and lipase maturation factor 1 (LMF1) (13, 14). In addition, several members of the angiopoietin-like (ANGPTL) protein family play a role in regulating LPL activity. ANGPTL3 is produced by the liver and is an endocrine regulator by inhibiting LPL in peripheral tissues (7, 15, 16). ANGPTL4 is produced in several tissues (7), where it inhibits LPL in a paracrine fashion (7, 17). Both ANPGTL3 and ANGPTL4 delay the clearance of the TG-rich lipoproteins (7).
The core TGs in VLDL and chylomicrons are hydrolyzed by ApoC-II activated LPL; FFA thus formed are taken up by adipocytes and re-incorporated into TGs for storage, or in skeletal and cardiac muscle, utilized for energy. Hydrolysis of chylomicron- and VLDL-TG results in TG-poor, cholesteryl ester and ApoE-enriched particles called chylomicron and VLDL remnants, respectively, which under physiological conditions are removed by the liver by binding to LDL receptors, LDL receptor related protein, and cell surface proteoglycans (12, 18). Hepatic TG lipase and ApoA-V also are involved in the remnant clearance process (10-12, 19, 20).
The clearance of TGs from plasma is saturable when plasma TGs exceed ~500-700 mg/dL (21). When removal mechanisms are saturated, additional chylomicrons and VLDL entering plasma cannot readily be removed and hence accumulate in the plasma. As a result, plasma TGs can increase dramatically, resulting in very high levels and the accumulation of chylomicrons in plasma obtained after an overnight fast.
NORMAL RANGE FOR PLASMA TRIGLYCERIDES AND DEFIJITION OF HYPERTRIGLYCERIDEMIA
Plasma TG levels reflect the TG content of multiple lipoprotein particles, primarily chylomicrons and VLDL. Fasting TG levels less than 150 mg/dL has been generally accepted as “normal” (22, 23). A non-fasting TG of 175mg/dL represents ~75th percentile of the normal range, and levels of ~400mg/dL represent the 97th percentile (24). Plasma TGs are heavily skewed to the right in the general population with a tail towards highest levels and vary depending upon the population mix (25). The full range of TG extends from 30mg/dL to 10,000mg/dL (22). TG levels are different between sexes, being higher in males than in females, and increase with age and development of other coexisting conditions such as central adiposity, metabolic syndrome, and diabetes (24). TG levels also vary between geographic areas, among people of different ethnic backgrounds, with higher levels observed in certain populations such as Mexicans and South Asians. Lower TG levels have been observed in people of African descent and African-Americans but this may be changing due to adoption of urban lifestyles (26). Because of this skewed distribution, logarithmic transformation is required to establish statistical normal ranges of TG levels. There is no current widely accepted definition of elevated non-fasting TG levels, but some groups have utilized 175mg/dL as a cut point (27, 28). Due to high variability of TG levels, precise definitions for non-fasting levels are difficult to establish. It is worthwhile noting that post-prandial TG levels rarely exceed 400mg/dL even after a high fat challenge.
Normal Range Based on Risk of Complications of Hypertriglyceridemia
The major complications of hypertriglyceridemia (HTG) are (1) acute pancreatitis and (2) increased risk of atherosclerotic cardiovascular disease (ASCVD). These two complications occur at different levels of TGs, the risk of pancreatitis occurring at much higher TG levels than the risk of premature ASCVD and are discussed in detail later in this chapter.
Normal Range According to Guidelines
Despite concerns regarding establishment of an upper limit of normal for TGs, most guidelines define values for HTG, often without a strong biological rationale. Definitions for the diagnosis of HTG provided in several guidelines are shown in Table 1.
Cut points for HTG were first defined by the National Cholesterol Education Program Adult Treatment Panel (NCEP-ATP). The terms mild, moderate and severe have been used based on degree of TG elevation (table 1). In general, mild to moderate HTG reflects TG levels under 500mg/dL. Severe hypertriglyceridemia (sHTG) has been arbitrarily defined by different national guidelines as either TG levels ≥500 mg/dL by the American Heart Association (AHA)/American College of Cardiology (ACC), Multispecialty Cholesterol and Canadian Cardiovascular Society Guidelines (29, 30) or TG levels ≥880 mg/dL according to the European Society of Cardiology guidelines (31). The Endocrine Society has used severe HTG for 1000 to 1999 mg/dL and very severe HTG for values >2000 mg/dL (23).
Table 1.
Definition of Hypertriglyceridemia According to Various Clinical Guidelines
| Guideline | Classification | Triglyceride Levels |
|---|---|---|
| NCEP/ ATP III (32) American Heart Association (33) National Lipid Association (34) | Normal Borderline-high TGs High TGs Very high TGs | <150 mg/dL (< 1.7 mmol/L) 150-199 mg/dL (1.7-2.3 mmol/L) 200-499 mg/dL (2.3-5.6 mmol/L) ≥500 mg/dL (≥5.6 mmol/L) |
| The Endocrine Society (35) | Normal Mild HTG Moderate HTG Severe HTG Very severe HTG | <150 mg/dL (< 1.7 mmol/L) 150-199 mg/dL (1.7-2.3 mmol/L) 200-999 mg/dL (2.3-11.2 mmol/L) 1000-1999 mg/dL (11.2-22.4 mmol/L) ≥2000 mg/dL (≥22.4 mmol/L) |
| European Society of Cardiology/European Atherosclerosis Society (36) | Normal Mild-moderate HTG Severe HTG | <1.7 mmol/L (<150mg/dL) >1.7-< 10mmol/L (150-880 mg/dL) > 10 mmol/L (> 880mg/dL) |
| Hegele (22) | Normal Mild to moderate Severe | <2.0 mmol/L (<175 mg/dL) 2.0-10 mmol/L (175- 885 mg/dL) >10 mmol/dL (>885 mg/dL) |
In summary, establishing a precise definition of what constitutes abnormal TG values is fraught with difficulty. An acceptable level for the prevention of pancreatitis is likely to be quite different from that at which CVD risk might be increased. The impact of HTG on CVD risk needs to be evaluated in the context of the family history of premature CVD, associated abnormalities of lipids and lipoproteins, and other CVD risk factors, particularly those associated with the metabolic syndrome (see below).
CAUSES AND CLASSIFICATION OF HYPERTRIGLYCERIDEMIA
In general, HTG has been classified as primary, when a genetic or familial basis is suspected, or secondary, where other conditions that raise TG levels can be identified. However, this classification is likely overly simplistic. It has become clear in the past decade that the spectrum of plasma TG levels, ranging from mild elevation to very severe HTG, is modulated by a multitude of genes working in concert with non-genetic secondary and environmental contributors. Thus, in the vast majority of individuals, mutations in multiple genes with interaction from non-genetic factors result in altered TG-rich lipoprotein synthesis and catabolism and subsequent HTG.
Historical Perspective
Phenotypic heterogeneity among patients with HTG has been historically defined by qualitative and quantitative differences in plasma lipoproteins. In the pre-genomic era, the Fredrickson classification of hyperlipoproteinemia was based on electrophoretic patterns of lipoprotein fractions (37). The phenotypes are distinguished based on the specific class or classes of accumulated TG-rich lipoprotein particles, including chylomicrons, VLDL and VLDL-remnants. This classification included 6 phenotypes, five of which included HTG in their definition (except for Frederickson type 2 A hyperlipoproteinemia, which equates with genetic primary hypercholesterolemia). It has now become apparent that except for type 1 hyperlipoproteinemia (FCS), the HTG phenotypes, particularly Frederickson type 4 and type 5 hyperlipoproteinemia, are due to the accumulation of polygenic traits predisposing to HTG. However, this classification system is dated, has neither improved clinical or scientific insight, and therefore does not find wide use at this time (22).
In 1973, Goldstein and colleagues characterized a variable pattern of lipid abnormalities in families of survivors of myocardial infarction that they termed familial combined hyperlipidemia (FCHL) (38). At the same time, this phenotype of mixed or combined hyperlipidemia was observed in another cohort, where it was called multiple-type familial hyperlipoproteinemia (39). Affected family members can present with hypercholesterolemia alone, HTG alone, or with elevations in both TGs and LDL. This pattern was estimated to have a population prevalence of 1-2% (40), making it the most common inherited form of dyslipidemia.
In the aforementioned study, a pattern of isolated HTG, historically called familial HTG (FHTG) also was described (38). This condition was characterized by increased TG synthesis, with secretion of normal numbers of large TG-enriched VLDL particles (41), elevated VLDL levels, but normal levels of LDL and HDL cholesterol (42). FHTG did not appear to be associated with an increased risk of premature CVD in an early study (43), but baseline TG levels predicted subsequent CVD mortality after 20 years of follow up among relatives in families classified as having FHTG (44, 45).
FCHL and FHTG were initially believed to be monogenic disorders (38). However, more recent genetic characterization of individuals with familial forms of HTG indicates that these are not disorders associated with variation within a single gene, but rather polymorphisms in multiple genes associated with HTG, as detailed below. Therefore, classification of FCHL and FHTG is potentially misleading. Nevertheless, it is important to note that FCHL as originally described is associated with a very high prevalence of premature CVD (43, 44, 46).
Genetic Forms of Hypertriglyceridemia
It is now evident that clinically relevant abnormalities of plasma TG levels appear to require a polygenic foundation of common or rare genetic variants (22). Common small-effect gene variants confer a background predisposition that interact with rare large-effect heterozygous variants in genes that govern synthesis or catabolism of TG-rich lipoproteins, or nongenetic secondary factors, leading to the expression of a more severe TG phenotype (47). Recently, the most prevalent genetic feature underlying severe HTG was shown to be the polygenic accumulation of common (rather than rare) variants—more specifically, the accumulation of TG-raising alleles across multiple SNP loci (48).
Thus, mild to moderate hypertriglyceridemic states are complex, genetically heterogeneous disorders. Mild-to-moderate HTG is typically polygenic and results from the cumulative burden of common and rare variants in more than 30 genes, as quantified by genetic risk scores. All genetic forms can be exacerbated by non-genetic factors. Because they are a consequence of interaction between multiple susceptibility genes and lifestyle factors, individuals with moderate HTG should be considered as a single group without distinction, irrespective of concomitant lipoprotein disturbances (22). Because of the complexity of these disorders, routine genetic testing is not recommended.
Pathogenesis of Genetic Forms of Hypertriglyceridemia
Genetic forms of HTG without other lipoprotein disturbances (i.e., pure HTG) are characterized by increased TG synthesis, where normal numbers of large TG-enriched VLDL particles are secreted (41, 49-51). Reduced TG clearance also has been observed in some individuals (50-52). Affected people have elevated VLDL levels, but normal levels of LDL, and are generally asymptomatic unless clinical CVD or severe HTG develops.
A variety of metabolic defects that differ among families are associated with the combined hyperlipidemia phenotype. The characteristic lipoprotein abnormalities are increased ApoB levels and increased number of small dense LDL particles (42), a phenotype similar to that seen in the metabolic syndrome and type 2 diabetes (53). These primary defects occur due to 1) hepatic overproduction of VLDL particles (41) due to increased ApoB synthesis in the setting of disordered adipose metabolism (54, 55), insulin resistance (41, 56-58), and liver fat accumulation, and, 2) impaired clearance of ApoB containing particles (59, 60). Increased VLDL secretion results in an elevated plasma ApoB and HTG (56). Long residence time of VLDL particles favors the formation of small dense LDL (59). An abundance of small dense LDL particles traditionally is associated with the presence of HTG; however, these LDL characteristics remain even after correction of the HTG by treatment with fibrates (61, 62).
In addition to Apo B abnormalities, other lipoprotein disturbances include abnormal expression of ApoA-II, ApoC-III, and PCSK9. VLDL-TG levels in combined hyperlipidemia are modulated by ApoA-II and ApoC-III (63). Plasma PCSK9 levels are higher in these patients, and levels correlate with TG and Apo B levels (64).
Visceral adiposity appears to be an important determinant of insulin resistance, which occurs commonly in subjects with both isolated HTG (65) and combined hyperlipidemia (65-69). Other abnormalities that have been reported in clinical FCHL include impaired lipolysis due to decreased cyclic AMP dependent signaling (54, 69), abnormal adipocyte TG turnover (70), fatty liver (71), increased arterial stiffness (72), and increased carotid intimal-medial thickness (73).
In all of the phenotypes described above, severe HTG can occur when secondary causes of HTG such as untreated diabetes, marked weight gain, or use of TG-raising drugs are present concurrently, leading to the Multifactorial Chylomicronemia Syndrome (MFCS), described later (74).
Secondary Forms of Hypertriglyceridemia
These are described in greater detail in the chapters on Secondary Disorders of Lipid and Lipoprotein Metabolism (75-78). However, in the section where we describe MFCS we will briefly touch on some aspects of secondary forms of HTG, since they assume importance in the pathogenesis of the severe HTG seen in the MFCS, where they often co-exist in individuals with genetic forms of HTG. In our experience, the commonest secondary forms of HTG that interact with genetic forms of HTG are type 2 diabetes (usually as part of the metabolic syndrome), obesity, recent weight gain, excessive alcohol consumption, the use of drugs that can raise TGs, and chronic kidney disease (CKD)(74, 79, 80). (Table 3)
Severe Hypertriglyceridemia and the Chylomicronemia Syndrome
In the late 1960s Fredrickson, Levy and Lees (37) classified HTG into types dependent on the pattern of lipoproteins on paper electrophoresis and the presence or absence of chylomicrons in fasting plasma. They recognized that acute pancreatitis and eruptive xanthomata occurred in the presence of chylomicronemia that accumulate in what they termed Type I and Type V hyperlipoproteinemia. Chylomicrons are present in the post-prandial state, and usually are present in fasting plasma when TG levels exceed 800 mg/dL, but absent in fasting plasma below that value (81). The term chylomicronemia syndrome was first used to describe a constellation of clinical findings such as abdominal pain, acute pancreatitis, eruptive xanthoma and lipemia retinalis that occurred in association with very high TG levels (82). Two groups of conditions can lead to severe HTG and clinical manifestations of the chylomicronemia syndrome; (1) familial chylomicronemia syndrome (FCS) due to variants in the LPL complex, and (2) multifactorial chylomicronemia syndrome (MFCS), in which genetic predisposition and secondary forms of HTG co-exist.
FAMILIAL CHYLOMICRONEMIA SYNDROME (FCS)
FCS is a monogenic disorder due to variants in one or more genes of the LPL complex that affect chylomicron catabolism. FCS incidence is very rare, with an estimated prevalence ranging from 1 in 20,000 to 300,000 (83).
Genetics: Biallelic loss of function variants in five canonical genes lead to impaired hydrolysis of TG-rich lipoproteins, with subsequent increases in chylomicron particle numbers and markedly increased TG concentrations. The most common gene affected in FCS is LPL itself, in which patients are homozygous or compound heterozygous for two defective LPL alleles. Over 180 variants that result in LPL deficiency have been described with some clustered mutations due to founder effects (84-87). Loss of function variants account for over 90% of cases (83). Many are missense variants, some in catalytically important sites and some in regions that predispose to instability of the homodimeric structure of LPL required for enzyme activity (88). However, many common LPL gene variants have been described that have no clinical phenotype (89). Variants in the APOC2 gene, encoding ApoC-II, an activator of LPL, is another cause of FCS. Variants have been described in several families (90, 91). In FCS thus far there is no known gene variant that affects synthesis or production of TG-rich lipoproteins.
FCS can also occur from biallelic loss of function variants in other components of the LPL complex, namely APOA5, LMF1, and GPIHBP1 genes (Table 2), each of which plays an important role in determining LPL function (92). The lipoprotein phenotype in these mimics that seen in classical LPL deficiency. Loss of function variants in GPIHBP1, which directs transendothelial LPL transport and helps anchor chylomicrons to the endothelial surface near LPL, thereby providing a platform for lipolysis, has been described in several families (83). Autoantibodies to GPIHBP1 also can lead to chylomicronemia (93). A small number of individuals with homozygous variants in Apo A-V, which stabilizes the lipoprotein–enzyme complex thereby enhancing lipolysis (10), have been described (94). Variants in LMF1, an endoplasmic reticulum chaperone protein required for post-translational activation of LPL, have also been identified in a few individuals (95).
Clinical presentation: FCS usually manifests in childhood or early adolescence with nausea, vomiting, failure to thrive and recurrent abdominal pain in infancy and childhood. Occasionally it can present in adulthood (87) but this is often due to delayed diagnosis with median age at diagnosis being due to unfamiliarity in most healthcare providers (96). Adults may report “brain fog” or transient confusion.
Classical clinical findings include eruptive xanthomas (often seen on buttocks, back, extensor surfaces of upper limbs), lipemia retinalis, and hepatosplenomegaly. Less common symptoms of FCS can include intestinal bleeding, anemia, and neurological features such as irritability and seizures. Patients present with TG levels ≥1000 mg/dL and often much higher, due to abnormal accumulation of chylomicrons, which can be detected by the appearance of lipemic/milky plasma. Despite prolonged overnight fasting, plasma TG levels are >1000mg/dL due to the presence of chylomicrons in the circulation as a result of impaired clearance. The most serious concern, however, is the development of acute pancreatitis, which can lead to systemic inflammatory response syndrome, multi-organ failure, and death.
Table 2.
Genetic Disorders Resulting in Familial Chylomicronemia Syndromes (FCS)
| Disorder | Inheritance | Incidence | Lipid Phenotype | Underlying Defect | Clinical Features |
|---|---|---|---|---|---|
| LPL deficiency | Autosomal Recessive | 1 in 1,000,000 | Marked HTG/ chylomicronemia in infancy or childhood | Very low or absent LPL activity; circulating inhibitor of LPL | Hepato-splenomegaly; severe chylomicronemia |
| Apo C-II deficiency | Autosomal Recessive | Rare | Marked HTG/ chylomicronemia in infancy or childhood | Absent Apo C-II | Hepato-splenomegaly; severe chylomicronemia |
| Apo A-V mutation | Autosomal Recessive | Rare | Marked HTG/ chylomicronemia in adulthood | Defective or absent Apo A-V | Chylomicronemia |
| GPIHBP1 mutation | Autosomal Recessive | Rare | Marked HTG/ chylomicronemia in adulthood | Defective or absent GPIHBP1 | Chylomicronemia |
| LFM1 mutation | Autosomal Recessive | Rare | Marked HTG/ chylomicronemia in adulthood | Defective or absent LFM1 | Chylomicronemia |
Adapted from Ref (83).
MULTIFACTORIAL CHYLOMICRONEMIA SYNDROME (MFCS)
In contrast to FCS, MFCS is more common and complex. The prevalence of MFCS is much higher than FCS and estimated to be ~1:600-1000 (84).
Genetics: MFCS has a genetic basis, but unlike FCS (where recessive or biallelic variants in the affected genes are causative), the genetic alteration does not always result in the phenotypic expression of the trait but only increases the possibility of the risk of developing the condition. Other factors, including non-genomic effects (epigenetics, methylation), gene-gene, or gene-environment interactions can also contribute. MFCS develops due to two main types of genetic factors that increase the odds that a patient will develop very high TG levels. First, heterozygous rare large-effect variants in one of the five canonical TG metabolism genes (LPL, Apo C-II, Apo A5, LMF-1 and GPIHBP-1) can contribute to TG elevations. These variants have variable penetrance, i.e.- clinical presentation can vary from normal to severe hypertriglyceridemia.
Secondly, the presence of a high burden of common small-effect TG-raising SNPs; cumulatively, these common SNP alleles increase susceptibility for developing hypertriglyceridemia. These SNPs may have an indirect impact of the metabolism of TG-rich Lipoproteins. There is incomplete understanding of how an excess burden of SNPs contributes to TG levels, but their prevalence in patients with severe hypertriglyceridemia has been consistently demonstrated.
Several polygenic risk scores (PRS) for TG levels have been published (97). A recent study found that 32.0% of patients had a high polygenic score of TG-raising alleles across 16 loci compared to only 9.5% of normolipidemic controls (25). When the PRS is high, there is a significantly increased risk of developing HTG but this is not diagnostic or definitive.
Secondary Causes Contributing to Severe Hypertriglyceridemia in MFCS: The most common secondary cause in the past was undiagnosed or untreated diabetes (74), although earlier detection of diabetes may be making the association of marked hyperglycemia of untreated diabetes with very severe HTG less common. In addition, individuals with the metabolic syndrome and obesity have mild to moderate HTG which can become severe HTG; weight regain following successful weight loss can lead to marked HTG (23, 84). These patients almost always have relatives with genetic forms of HTG, whose TG levels are considerably lower than the index patient with severe HTG, in whom secondary forms of HTG also are present (74). MFCS can result from the addition of specific drugs in patients with a genetic predisposition (23). These drugs include beta-adrenergic blocking agents (selective and non-selective) and/or diuretics (thiazides and loop-diuretics such as furosemide) used for hypertension, retinoid therapy for acne, oral estrogen therapy for menopause or birth control, selective estrogen receptor modulators (particularly raloxifene) for osteoporosis or breast cancer, protease inhibitors for HIV/AIDS, atypical anti-psychotic drugs, alcohol, and possibly sertraline (84). Rarer causes of very severe HTG include autoimmune disease (sometimes with LPL- or GPIHBP1- specific antibodies), asparaginase therapy for acute lymphoblastic leukemia (98), (99) and bexarotene, a RXR agonist used in the treatment of cutaneous T cell lymphoma (100).
Table 3.
Secondary Causes That Can Contribute to Severe HTG
| Conditions |
| Hypothyroidism Suboptimally managed or new onset diabetes Obesity Sudden weight gain, weight regain after weight loss Chronic kidney disease Nephrotic syndrome Pregnancy Acute hepatitis Sepsis Inflammatory disorders Cushing syndrome Autoimmune chylomicronemia Systemic lupus erythematosis Anti-LPL antibodies GPIHBP-1 antibodies |
| Rare Genetic Causes |
| Glycogen storage disorders Lipodystrophies Congenital- generalized or partial Acquired- HIV, autoimmune |
| Drugs |
| Alcohol ingestion Beta blockers Thiazide diuretics Oral estrogens Selective estrogen reuptake modulators - tamoxifen, raloxifene, clomiphene Androgens Glucocorticoids Atypical anti-psychotics Sertraline Bile acid resins Sirolimus, tacrolimus Cyclosporine RXR agonists -bexarotene, isotretinoin, acetretin HIV Protease inhibitors L- asparaginase Alpha-interferon Propofol Lipid emulsions |
Following correction of treatable secondary forms of HTG in the MFCS, TG levels usually decrease to the moderately elevated levels seen in their affected relatives (101, 102).
OTHER CONDITIONS RESULTING IN HYPERTRIGLYCERIDEMIA
Familial Dysbetalipoproteinemia (FDB or Remnant Removal Disease)
Familial dysbetalipoproteinemia, also referred to as remnant removal disease or type III hyperlipoproteinemia, is a rare autosomal recessive disorder that can present with elevated TG levels. This disorder is characterized by the accumulation of remnant lipoproteins.
PATHOGENESIS AND GENETICS
Remnant removal disease requires homozygosity for the ApoE2 genotype or a rare heterozygosity for a variant in the ApoE gene, which results in pathologic accumulation of remnant lipoproteins in the circulation due to impaired hepatic uptake of ApoE-containing lipoproteins (103). ApoE is a glycoprotein synthesized in the liver, brain and tissue macrophages and present on chylomicrons, VLDL and HDL. Apo E through interaction with the LDLR and heparan sulphate proteoglycans promotes the hepatic clearance of remnants of chylomicrons and VLDL (104); it also facilitates cholesterol efflux from macrophages to HDL (105). In humans, there are 3 common isoforms of ApoE , ApoE2, ApoE3, and ApoE4 (106). Each differs in isoelectric point by one charge unit, ApoE4 being the most basic isoform and ApoE2 the most acidic. ApoE3 (Cys112→Arg158) is the commonest isoform. ApoE2 (Arg158→Cys) and ApoE4 (Cys112→Arg) differ from ApoE3 by single amino acid substitutions at positions 158 and 112, respectively (107). In the majority of cases (90%), remnant removal disease is associated with the E2/E2 genotype and results from impaired binding to the Apo E receptor. It is an autosomal recessive disorder with the prevalence of ApoE2 homozygosity in Caucasian populations estimated to be about 1% (108). Rarer ApoE variants such as ApoE3-Leiden (109) and ApoE2 (Lys1463Gln) that also can cause remnant accumulation are dominantly inherited (110) and account for 10% of cases (111, 112). Rare APOE variants in the population, other than the APOE2 and APOE4 alleles, play an important role in the development of isolated hypercholesterolemia (113) and mixed hyperlipidemia, with and without familial dysbetalipoproteinemia (114). Thus it is becoming apparent that two different DBL phenotypes may exist- a genetic Apo E dysfunction and a multifactorial form (115). Modern prevalence of FDB is estimated at 1-2% (116).
In the absence of additional genetic, hormonal, or environmental factors, remnants do not accumulate to a degree sufficient to cause hyperlipidemia in ApoE2 homozygotes; in fact, lipid levels are commonly low. Remnant accumulation results when the E2/2 genotype is accompanied by a second genetic or acquired defect that causes overproduction of VLDL such as obesity or diabetes (117) (111, 118) , a decrease in remnant clearance, or a reduction in LDL receptor activity (e.g., hypothyroidism (119)). Thus, full phenotypic expression requires the presence of other environmental or genetic factors (120). In these circumstances, the reduced uptake of remnant lipoproteins by the liver results in reduced conversion of VLDL and intermediate density lipoproteins to LDL, with subsequent accumulation of remnant lipoproteins (121, 122), hence the term remnant removal disease.
DIAGNOSIS
Patients with remnant removal disease have roughly equivalent elevations in plasma cholesterol and TGs. The disease rarely manifests before adulthood, and in some individuals never manifests clinically. It is more common in men than in women, where expression seldom occurs before menopause, since estrogen has a protective effect in women who are ApoE2 homozygotes (108). Palmar xanthomas (Figure 1), orange lipid deposits in the palmar or plantar creases, are pathognomonic of remnant removal disease but are not always present (123). Tuberoeruptive xanthomas can be found at pressure sites on the elbows, knees and buttocks. The presence of remnant removal disease should be suspected when total cholesterol and TG levels range from 300 to 1000 mg/dL and are roughly equal in magnitude. Special diagnostic tests such as beta-quantification or lipoprotein electrophoresis are often required and are time consuming and not widely available. VLDL particles are cholesterol- enriched, which can be determined by isolation of VLDL by ultracentrifugation and by the demonstration of beta migrating VLDL on lipoprotein electrophoresis. A VLDL-cholesterol/plasma TG ratio of <0.30 is usually observed (124). A low ApoB/total cholesterol ratio of <0.33 also can be helpful in making the diagnosis (125). Simplified criteria for the diagnosis of DBL using a 3-step process has been proposed (126). The diagnosis of remnant removal disease should be confirmed by demonstrating the presence of the E2/E2 genotype. If the genotype result is not E2/E2, an autosomal dominant variant of APOE should be suspected. There is a high prevalence of premature coronary artery disease (127-129) and peripheral arterial disease (130-132). Occasionally severe HTG and an increased risk of pancreatitis can develop in the presence of a concomitant secondary form of HTG or TG-raising drugs.
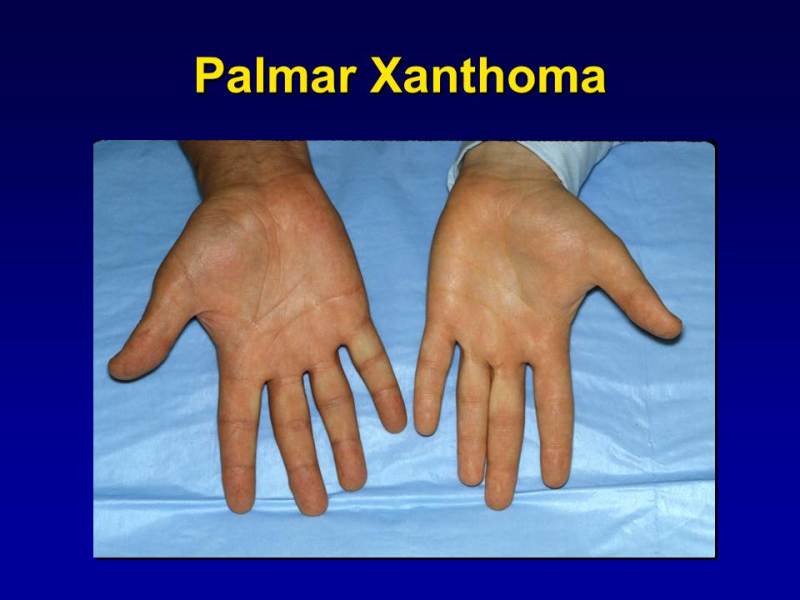
Figure 1.
Palmar Xanthomas: Orange-yellow discoloration confined to the palmar creases.
Familial Partial Lipodystrophy (FPLD) Syndromes
A distinct entity that results in moderate and severe hypertriglyceridemia include partial lipodystrophy syndromes. Inherited lipodystrophies are a heterogeneous group of disorders considered to be rare, that manifest as complete or partial loss of white adipose tissue with accompanying severe metabolic dysregulation(133) and are reviewed elsewhere in the Endotext chapter on Lipodystrophies (134). Loss of fat can be either localized to small discrete areas, in some cases partial with loss from extremities, or generalized with fat loss from nearly the entire body. Inherited lipodystrophies, while rare, can be autosomal dominant or recessive. Some forms manifest at birth, while others become evident later in life.
Partial or generalized lipodystrophic disorders frequently are associated with significant metabolic derangements associated with severe insulin resistance, including HTG. The extent of fat loss sometimes determines the severity of metabolic complications (135). HTG is a common accompaniment of many lipodystrophies, often in conjunction with low HDL-C levels. The pathophysiology of hypertriglyceridemia in these subjects is possibly related to the reduced ability to deposit free fatty acids in adipose tissue due to its maldevelopment, and accelerated lipolysis with increased hepatic VLDL synthesis and delayed clearance (135).
Genetics: Several genes have been implicated in the manifestation of various forms including LMNA, PPARG, LIPE, CIDEC (136). In the Dunnigan variety, the most commonly identified genetic variant of FPLD, the commonest variants are in the LMNA gene and less frequently PPARG (133). No specific genetic defect has been identified in Köbberling’s FPLD, although recent evidence suggests a heavy polygenic burden in these individuals (137, 138).
Diagnosis: Congenital generalized lipodystrophy (CGL) is a rare autosomal recessive disorder in which near total absence of subcutaneous adipose tissue is evident from birth. HTG and hepatic steatosis are evident at a young age and are often difficult to control. Severe HTG, often associated with eruptive xanthoma and recurrent pancreatitis, can occur in patients with CGL. The prevalence of HTG in case series of CGL patients is over 70% (135, 139). Plasma TGs are normal or slightly increased during early childhood, with severe HTG manifesting at puberty along with onset of diabetes mellitus.
Familial partial lipodystrophies (FPLD) are complex metabolic disorders that are often not recognized clinically (140). Partial lipodystrophies are characterized by partial loss of adipose tissue and significant metabolic derangements. The Dunnigan variety of FPLD (FPLD type 2) is a rare autosomal dominant disorder in which fat loss mostly involves the extremities and the trunk. Onset of fat loss in the buttocks and extremities occurs at puberty or late adolescence, with gain of fat to the face and neck. Acanthosis nigricans, calf muscle hypertrophy, and phlebomegaly (prominent veins) due to lack of subcutaneous fat, can be observed. Significant metabolic dysfunction including diabetes, which is often very insulin resistant, resistant hypertension, and HTG often severe and difficult to treat, can occur. Myopathy, cardiomyopathy, and/or conduction system abnormalities can occur (141). ASCVD risk also is increased (142, 143).
Some lipodystrophies, where fat loss appears to be proportionate to loss of total and lean body mass, do not result in dyslipidemia. Elevated TG levels have been reported in patients with atypical progeroid syndrome due to LMNA mutations (144, 145). Of the acquired lipodystrophies, the HIV-associated form usually is characterized by more moderate HTG. HIV-associated lipodystrophy occurs in patients receiving protease inhibitor containing highly active anti-retroviral therapy regimens (146). Fat loss occurs in the face, buttocks, and extremities.
CONSEQUENCES OF HYPERTRIGLYCERIDEMIA
Atherosclerotic Cardiovascular Disease
EPIDEMIOLOGY
HTG has long been known to be a risk factor for ASCVD (33, 147-150), which has been confirmed in meta-analyses (45). However, HTG also is frequently associated with low levels of HDL-cholesterol and an accumulation of remnants of the TG-rich lipoproteins, both known risk factors for ASCVD. When adjusted for both HDL-C and non-HDL-C, which contains both remnants of the TG-rich lipoproteins and LDL, the association of TGs with ASCVD risk remained significant, although somewhat attenuated (151). Postprandial TGs are elevated throughout the day in subjects with HTG, and postprandial TG-rich lipoproteins and their remnants also have been hypothesized to be important in the pathogenesis of atherosclerosis (150). It is therefore of interest that non-fasting TGs have been associated with ASCVD risk (150, 152, 153), despite non-fasting TGs being quite variable. However, unlike the situation with elevated LDL-C levels, the magnitude of the TG elevation does not appear to correlate with the extent of ASCVD risk. In particular, very severe HTG per se does not always appear to confer increased ASCVD risk, possibly because the chylomicrons that accumulate are too large to enter the arterial intima (154, 155).
TRIGLYCERIDES IN THE PATHOGENESIS OF ASCVD
Although chylomicrons may be too large to enter the arterial intima, ApoE-and cholesterol-enriched remnants of the TG-rich lipoproteins can enter with ease (153) where they can bind to vascular proteoglycans, similar to LDL (156, 157). Modification of these retained lipoproteins by either oxidative damage or enzyme digestion of some of the lipid components can liberate toxic by-products, which have been hypothesized to play a role in atherogenesis by facilitating local injury, generation of adhesion molecule, and cytokine expression and inflammation (157). Remnants of the TG-rich lipoproteins also can be taken up by macrophages leading to the formation of foam cells, an important component of atherosclerotic plaques. HTG also is associated with a preponderance of small, dense LDL, particles, reduced levels of HDL-C, and in the metabolic syndrome, with abnormalities of HDL composition (see earlier). Small, dense LDL can traverse the endothelial barrier more easily than large, buoyant LDL particles (158), are retained more avidly than large, buoyant LDL (159), and also are more readily oxidized (160, 161), all of which may facilitate atherogenesis. HDL particles in some hypertriglyceridemic states, e.g., in association with the metabolic syndrome, might be dysfunctional with respect to their cholesterol efflux, anti-inflammatory, and anti-oxidant properties. Moreover, a hypercoagulable state has been reported in association with both HTG and the metabolic syndrome (162). Thus, HTG might accelerate atherosclerosis by several mechanisms, all of which could increase CVD risk.
GENETIC EVIDENCE OF HYPERTRIGLYCERIDEMIA AND ATHEROSCLEROSIS
Recent human genetic studies have provided important insight into the contribution of TGs to ASCVD. Several genetic approaches, including candidate gene sequencing, GWAS of common DNA sequence variants, and genetic analysis of TG phenotypes have unraveled new proteins and gene variants involved in plasma TG regulation (163). Some genetic variants that influence TG levels appear to be associated with increased CVD risk even after adjusting for their effects on other lipid traits (164). GWAS have identified common noncoding variants of the LPL gene locus associated with TG and CVD risk (165, 166). A common gain-of-function mutation in the LPL gene, S447X (10% allele frequency), is associated with reduced TG levels and reduced risk of CVD (167) and an LPL variant associated with reduced TG and ApoB levels was associated with reduced CVD similar to LDL-C lowering variants, suggesting that the clinical benefit of lowering triglyceride and LDL-C levels may be proportional to the absolute change in ApoB (168). Conversely, several loss-of-function LPL variants linked with elevated TG levels are associated with increased CVD risk (169). Variants in the TRIB1 locus have been associated with LDL, HDL-C and TG levels (166), hepatic steatosis (170) and coronary artery disease (171). Mutations that disrupt APOC3 gene function and reduce plasma ApoC-III concentration are associated with lower TG levels and decreased risk of clinical CVD (172, 173). In contrast, carriers of rare mutations in APOA5, encoding ApoA-V, an activator of LPL, are associated with elevated TGs and with increased risk of myocardial infarction (174, 175). Loss of function variants in ANGPTL4 that had lower TG levels also were associated with reduced CVD risk (176, 177). Thus, exciting new human genetics findings have causally implicated TG and TG-rich lipoproteins in the development of CVD risk. In particular, the LPL pathway and its reciprocal regulators ApoC-III and ApoA-V appear to have an important influence on atherosclerotic CVD risk. However, despite this mountain of evidence demonstrating a causal relationship of TG with atherosclerosis, the possible involvement of a correlated trait, usually low HDL-C levels, or other unmeasured traits, cannot be ruled out.
CLINICAL TRIAL EVIDENCE OF TRIGLYCERIDE LOWERING AND ASCVD
In the pre-statin era, use of gemfibrozil monotherapy demonstrated cardiovascular benefit in men with coronary heart disease. However, since the advent of statins, drugs that specifically lower plasma triglyceride levels have not clearly been shown to have a benefit with cardiovascular risk reduction in clinical trials when added to background statin therapy. Reasons for this are unclear. In genetic studies to get a comparable reduction in Apo B and coronary heart disease (CHD) risk in clinical trials, a TG reduction of ~70mg/dL is required compared to a decrease in LDL-C of only 14mg/dL. Additionally, lipid alterations due to genetics are lifelong and result in much bigger reductions in CHD than a 5-year drug study. Based on genetic studies, the magnitude of TG reduction required to demonstrate cardiovascular benefit is quite large.
CARDIOVASCULAR DISEASE IN THE CHYLOMICRONEMIA SYNDROME
As described earlier, chylomicrons have been considered to be too large to penetrate the vascular endothelium and play a role in atherogenesis (152), although remnants of the TG-rich lipoproteins may be atherogenic (152, 178-181). The incidence of CVD is low in individuals with FCS (182), although premature atherosclerosis has been documented in well characterized subjects with this disorder (183). However, CVD risk clearly is increased in many patients with MFCS, although the exact frequency remains unclear. The frequency of CVD outcomes does not appear to relate to the magnitude of the TG elevations (184). It is not surprising that CVD is increased in MFCS considering the association between TGs and CVD that has been documented in many studies (reviewed in (150, 185, 186)). Many subjects develop severe HTG due to the co-existence of polygenic mutations that result in mild to moderate HTG (22) with secondary causes of HTG. Residual HTG due to these genetic disorders persists even after severely elevated TG levels have been reduced by treatment of the secondary forms of HTG and treatment of the HTG per se. Moreover, many patients with the MFCS have other CVD risk factors such as diabetes, reduced levels of HDL-C, and hypertension, the latter resulting in use of diuretics and beta-blockers, which play a role in raising their TGs to levels at which chylomicrons accumulate due to saturation of clearance mechanisms. Therefore, strategies to prevent CVD need to be undertaken once the TGs have been lowered to a level where pancreatitis is unlikely to recur.
Pancreatitis
Severe hypertriglyceridemia is the third most common cause of acute pancreatitis after alcohol and gallstones. The chylomicronemia syndrome describes a constellation of findings that occur with severe elevations of plasma TG levels. There is some lack of consensus as to what constitutes severe HTG, values >1000-1500 mg/dL are generally classified as severe, although some groups consider values in the 500-1000 mg/dL range also severe hypertriglyceridemia (187).
Individuals with both FCS and MFCS often present with hypertriglyceridemia induced acute pancreatitis, which can be recurrent if triglyceride levels remain elevated persistently. Women with genetic HTG can develop severe HTG and pancreatitis during pregnancy particularly during the third trimester (188).
The pancreatitis that occurs with severe HTG can be recurrent. In a prospective study of patients admitted with acute pancreatitis, the distribution of plasma TGs was bimodal when measured at the peak of the pain (101, 102). TG levels <880 mg/dL were associated with gall bladder disease and chronic alcoholism, while those above 2000 mg/dL were associated with the simultaneous presence of familial and secondary forms of HTG. It has been suggested that individuals become prone to the development of TG-induced pancreatitis at TG values between 1500-2000 mg/dL (189). TG-induced pancreatitis has been reported with TG levels lower than 500 mg/dL(190, 191), although in our experience this usually occurs when patients with severe HTG stopped eating some time prior to the blood draw. The frequency of severe HTG leading to acute pancreatitis varies widely from about 6-20% of subjects, possibly related to the type of patient presenting to different type of medical centers (192, 193). Pancreatitis often is recurrent if HTG is not appreciated to be the cause and if TG levels are not adequately controlled (87). With long term multiple episodes of acute, recurrent pancreatitis, exocrine pancreatic insufficiency or insulin deficient secondary diabetes may occur. A meta-analysis of observational studies suggests that TG-induced pancreatitis has worse outcomes than pancreatitis from other causes, with an approximate doubling of renal and respiratory failure, a nearly 4-fold increase of shock and a near doubling of mortality (194). Pancreatitis due to very severe HTG also may occur during infusion of lipid emulsions for parenteral feeding (195) or with use of the anesthetic agent propofol, which is infused in a 10% fat emulsion (196).
MECHANISM OF SEVERE HYPERTRIGLYCERIDEMIC PANCREATITIS
The mechanism by which very severe HTG leads to pancreatitis remains speculative. Suggested mechanisms include the local liberation of FFA from TGs and lysophosphatidylcholine from phosphatidycholine when pancreatic lipase encounters very high levels of TG-rich lipoproteins in the pancreatic capillaries (197). High local concentrations of FFA overwhelm the binding capacity of albumin with resultant aggregation into micellar structures with detergent properties. Both FFA and lysophosphatidylcholine have been shown to cause chemical pancreatitis when infused into pancreatic arteries in animal models (198-200). This leads to local liberation of more lipases from the damaged pancreatic acini, resulting in a vicious cycle (198, 201). It also has been hypothesized that increased plasma viscosity due to the presence of increased numbers of chylomicrons in the pancreatic microcirculation contributes to the development of pancreatitis (202). There also is recent evidence of gene associations in TG-induced pancreatitis; in a Chinese cohort with HTG, a CFTR variant and TNF alpha promoter polymorphism were found to be independent risk factors for developing pancreatitis (203), while another study found an increased frequency of ApoE4 (204).
DIAGNOSIS OF SEVERE HYPERTRIGLYCERIDEMIC INDUCED PANCREATITIS
The diagnosis of HTG-associated pancreatitis can be made by the presence of severely elevated TG levels in a patient with acute pancreatitis. Falsely low serum amylase levels can be encountered due to assay interference by the TG-rich lipoproteins (205). Pseudohyponatremia due to the presence of large numbers of TG-rich lipoproteins in plasma can be seen with very high TG levels. Interference with liver transaminase assays may also occur, giving spuriously high values making it difficult to exclude alcoholic liver disease (205).
With chronic chylomicronemia, patients may develop eruptive xanthomata (Figure 2). These xanthomas represent an inflammatory response to the deposition of chylomicron-associated lipids in tissues and are yellow-red papules that usually appear on the buttocks, back and extensor surfaces of the upper limbs. Histologically, these lesions contain lipid laden foamy macrophages (206).

Figure 2.
Eruptive Xanthomas. The commonest site is on the buttocks. The lesions are papular with an erythematous base. They often are itchy.
Lipemia retinalis, where the retinal vessels take on a whitish hue with pallor of the optic fundus and retina can be observed with very high TG levels (Figure 3). There is no associated visual impairment.
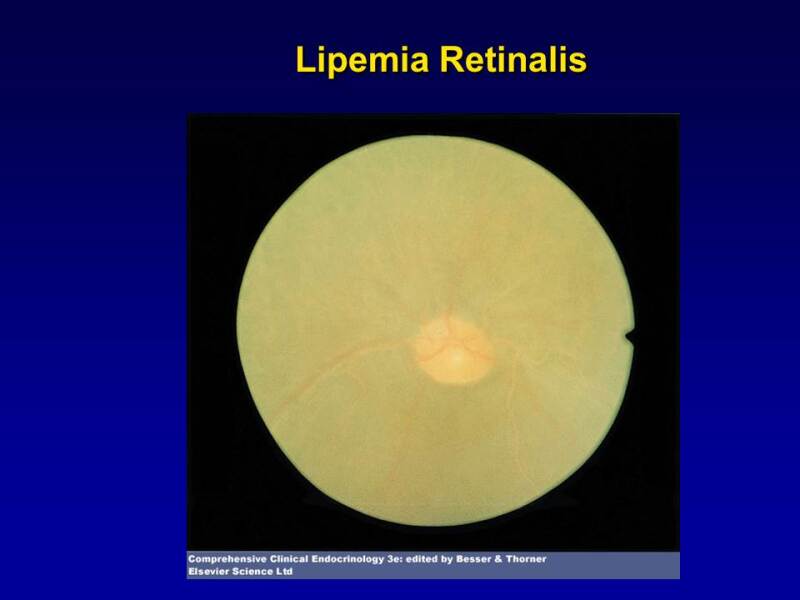
Figure 3.
Lipemia retinalis. Note the pale color of the retinal vessels.
Acute recent memory loss and mental fogginess (82) can also occasionally be seen, but has not been extensively studied. Symptoms such as fatigue, blurred vision, dysesthesias, and transient ischemic attacks have been suggested to be related to hyperviscosity resulting from high TG levels (207, 208). Hepatosplenomegaly is frequently present in FCS due to macrophage infiltration in response to the chylomicron accumulation. Fatty liver is a common finding on imaging in both FCS and MFCS.
MANAGEMENT OF SEVERE HYPERTRIGLYCERIDEMIA
Management of HTG by lifestyle and pharmacological means is discussed in detail in the Endotext chapters on The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels and Triglyceride Lowering Drugs (209, 210). However, in this section we will make a few points specifically relevant to this chapter.
Before initiating lifelong therapy for hypertriglyceridemia, evaluation for and treatment of reversible secondary disorders that can elevate plasma triglyceride levels is crucial. This includes appropriate management of diabetes and hypothyroidism and substituting drugs that can elevate triglyceride levels with lipid-neutral agents. Management of hypertension should include calcium channel blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and alpha-adrenergic blockers rather than beta-adrenergic blocking agents and diuretics.
Cardiovascular Disease Prevention
ASCVD risk in HTG is modulated by the presence of several other factors, including other lipoprotein abnormalities, other CVD risk factors, and family history of CVD, with some families with HTG appearing to have a greater risk of CVD than others (44). The role of TG lowering by pharmacological means remains controversial, but there is consensus that the presence of HTG imparts residual risk after LDL has been adequately lowered with statins.
Statins: The best clinical trial data currently available for the prevention of ASCVD in patients with HTG demonstrate that statins are likely to confer the most benefit, even though their primary mode of action is not to reduce plasma TGs, nor are they very effective in so doing (211). In patients with elevated TG levels statins will result in a significant decrease in TG levels. Based on the results of the IMPROVE-IT trial (212), the addition of ezetimibe may be of additional benefit.
Fibrates: Fibrates such as gemfibrozil and fenofibrate, are PPAR-α agonists, and very effective in lowering plasma TG levels (by up to 50%). Several studies have failed to demonstrate a benefit of fibrates on ASCVD events, either alone or in combination with statins. However, participants in these studies were not confined to individuals with HTG. Nonetheless, post-hoc analysis showed that subgroups of subjects who had mild HTG >200mg/dL and LDL-C <34mg/dL had a significant reduction of ASCVD events (213-216). In addition, the Action to Control Cardiovascular Risk in Diabetes (ACCORD)-LIPID trial, which was confined to subjects with diabetes, showed a similar outcome in the subgroups with HTG, although the trial was negative for all subjects (214). Recently a novel selective peroxisome proliferator-activated receptor α modulator, pemafibrate, that possesses unique PPARα activity and selectivity (217), was evaluated in individuals with HTG and diabetes in the Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts With diabetes (PROMINENT) trial. Patients with type 2 diabetes, triglyceride level 200 to 499 mg/dL and HDL-C of </=40 mg/dL were assigned to pemafibrate or placebo. Unfortunately, pemafibrate failed to demonstrate cardiovascular benefit in patients with type 2 diabetes and mild to moderate HTG despite significantly lowering triglyceride levels (218). Thus, addition of a fibrate to a statin for cardiovascular risk reduction cannot be recommended at this time.
Omega-3 fish oil: Omega-3 (n-3) fatty acids are polyunsaturated fatty acids that lower TGs. The two main n-3 fatty acids are eicosapentanoic acid (EPA) and docosahexanoic acid (DHA) which can lower VLDL secretion and are agonists of PPARa. However, their role in ASCVD prevention also has been controversial as several RCTs of various dosages of n-3 mixtures failed to demonstrate CV benefit in mild to moderate HTG (219). The Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial (REDUCE-IT), evaluated the addition of high dose icosapent ethyl (highly purified eicosapentanoic acid or EPA) compared to placebo in high-risk patients with mild to moderate HTG on statin therapy, demonstrated a surprising 25% lower CV risk in subjects. Notably this effect was independent of baseline TG levels and TG reduction. (220). Subsequently the STRENGTH (Statin Residual Risk Reduction With Epanova in High Cardiovascular Risk Patients with Hypertriglyceridemia) trial of n–3 fatty acid mixtures (EPA + DHA) failed to demonstrate cardiovascular benefit and was terminated early for futility (221). Similarly, use of an EPA/DHA mixture (1.8 g/d) in patients with a recent myocardial infarction in the OMega-3 fatty acids in Elderly with Myocardial Infarction (OMEMI) trial (222) also failed to meet its primary endpoint. Tissue EPA levels may be a contributor to the positive results in the REDUCE-IT study, as there is evidence that EPA inhibits inflammation, causes membrane stabilization, and decreases plaque volume. The REDUCE-IT trial has generated controversy due to use of mineral oil in the control group which resulted in an increase in both LDL-C and C-reactive protein (223, 224). Nonetheless, several current guidelines suggest addition of icosapent ethyl in addition to a statin for residual hypertriglyceridemia in high-risk individuals (those with known ASCVD or diabetes with additional risk factors).
Niacin: Niacin effectively lowers triglycerides and LDL-C while raising HDL-C. Niacin inhibits lipolysis in adipocytes, therefore decreasing the available fatty acids for VLDL synthesis. However, it has fallen out of favor for ASCVD risk reduction. Two RCTs, Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) and Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) demonstrated no benefit to the addition of niacin to statins for decreasing cardiovascular risk. However, it is important to note that neither of these trials were confined to subjects with high TG levels. Therefore, due to the lack of efficacy and potential for increasing insulin resistance, niacin is not recommended for treatment of HTG.
Newer Therapies for HTG
Apo C-III inhibitors: ApoC-III is an endogenous inhibitor of LPL, by displacing ApoC-II an activator of LPL. This leads to inhibition of lipolysis and elevations in TG levels. Apo C-III can also increase TGs by LPL independent mechanisms. Currently, ApoC-III inhibitors include volanesorsen (a second-generation antisense oligonucleotide (ASO)), olezarsen (a third generation ASO), and ARO-APOC3 (a small interfering RNA), all of which are directed at APOC3 gene (225). Volanesorsen has been studied in individuals with FCS and MFCS and has been found to decrease TG levels by up to 70% from baseline and potentially prevent pancreatitis. However, thrombocytopenia and injection site reactions are common. This drug is therefore not approved for use by the FDA due to concerns of bleeding but is approved for use by the European Medicines Agency.
Olezarsen is a third generation ASO for which early studies have been completed.
ANGPTL-3 inhibitors: ANGPTL3 is a key regulator of lipoprotein metabolism and is able to repress LPL and endothelial lipase activity, with resultant increase in TGs and TG rich lipoprotein levels (226). Homozygous loss of function variants in ANGPTL3 result in combined hypolipidemia with low TG and LDL-C levels. ANGPTL3 inhibition results in decreased LDL-C, and TGs. ANGPTL3 inhibitors include evinacumab, a monoclonal antibody, vupanorsen, an antisense oligonucleotide (ASO), and ARO-ANG3, a small-interfering ribonucleic acid (siRNA). Evinacumab has been studied in patients with sHTG. A phase 2 study of evinacumab in patients with sHTG, demonstrated significant reductions in TGs only in patients with MFCS with and without LPL mutations, but not in patients with FCS, suggesting that ANGPTL3 inhibition is dependent on presence of some LPL activity (227). In the cohort with polygenic sHTG and MCS, evinacumab 15 mg/kg IV every 4 weeks resulted in an 81.7% reduction in TGs. It should be noted that evinacumab is currently only approved for homozygous familial hypercholesterolemia.
Management of Severe HTG-Induced Pancreatitis
Because of the low frequency of severe HTG in the general population, and because only some patients with severe HTG develop pancreatitis, large random controlled clinical trials are difficult to perform and unlikely to be undertaken in the foreseeable future. Therefore, therapeutic decisions need to be based on less stringent criteria than might otherwise be desirable. However, keeping TG levels <500 mg/dL should prevent the onset of TG-induced pancreatitis (187, 228, 229).
ACUTE MANAGEMENT
The clinical presentation of HTG-induced pancreatitis is similar to that from other causes of acute pancreatitis and can be preceded by episodic nausea, epigastric pain radiating through to the back, and increasing heart-burn. Individuals with recurrent acute pancreatitis may present without severe elevations in pancreatic enzymes (230). The immediate goal is to lower TGs in hospitalized patients. Management is similar to the management of non-TG induced pancreatitis, which includes cessation of all oral intake for pancreatic rest, fluid resuscitation, pain management, and management of metabolic abnormalities. TGs fall rapidly with discontinuation of oral intake, often to under 1000mg/dL with cessation of oral intake. With clinical improvement, oral diet advancement should be done slowly and cautiously in the hospital as this can result in rebound TG elevations. Supportive care as needed should be instituted for organ failure. Lipid emulsions for parenteral feeding should be avoided since their use will further delay clearance and exacerbate the HTG. If long term nutrition is required for very ill individuals who cannot eat, total parental nutrition without lipids should be utilized.
Heparin: Heparin will liberate LPL into plasma from its endothelial binding sites and hence rapidly lowers TGs (231). However, it also can cause rebound HTG due to rapid degradation of released LPL (232) and increases the risk of hemorrhagic pancreatitis. Therefore, the use of heparin is not recommended (233).
Insulin: The rationale for the use of an IV insulin infusion of regular insulin (in conjunction with IV glucose administration as needed) is that it can activate LPL and enhance clearance of TG-rich lipoproteins (234). Intravenous insulin can be beneficial in individuals with diabetes needing glycemic control. Its use in TG--induced pancreatitis without diabetes has been reported in several case reports (235-239), and has become widespread but it is unclear whether similar changes would have occurred simply by restricting oral intake without the use of insulin. Regular insulin at 0.1-0.2 units/kg/hour with a separate iv dextrose infusion to prevent hypoglycemia in individuals without diabetes is often used. IV insulin can be stopped when TG drops to below 1000mg/dL. However, TGs will increase when the individual consumes an oral diet; therefore, caution should be exercised with slow advancement of the diet. In a study of chylomicronemia with uncontrolled diabetes, insulin infusion lowered TGs more rapidly than plasmapheresis (240).
Plasmapheresis: The use of plasmapheresis to acutely lower TGs is controversial. Plasmapheresis is extracorporeal therapy where plasma is removed and replaced; plasma is separated from the blood and discarded removing chylomicrons. Substitute fluid is replaced to maintain blood volume. The procedure is highly effective in rapidly decreasing TG levels by 85% after 1 session. However, the effect is not persistent, without evidence of long-term efficacy or mortality and morbidity benefit demonstrated. Although recommended by some (241, 242), the current evidence for the benefit of use of plasmapheresis is limited to small uncontrolled anecdotal series (243) from which no firm conclusion can be made regarding its use in acute TG-induced pancreatitis (244). A recent retrospective analysis demonstrated no benefit on length of hospital stay or mortality when therapeutic plasma exchange was added to medical management of severely elevated plasma TGs (245). TG levels fall rapidly with cessation of oral intake and use of non-lipid-containing intravenous fluids. Plasmapheresis requires a specialized center, needs central venous access, and transient anticoagulation; it only temporarily improves TG levels without addressing the underlying cause (83). Risks include line sepsis, deep vein thrombosis, and bleeding. Therefore, we do not recommend its routine use in this situation unless clinical circumstances necessitate plasmapheresis such as severe acute necrotizing pancreatitis (246), shock, or pregnancy (247).
LONG-TERM MANAGEMENT TO PREVENT PANCREATITIS
After TG lowering in the setting of acute pancreatitis, it is essential to determine both the primary and secondary causes of the severe HTG that precipitated the acute pancreatitis. Continued management of any secondary form of HTG, as well as lifestyle and drug therapy to maintain low TG levels is required to prevent recurrent pancreatitis. If fasting plasma TG levels remain above 1000 mg/dL after treating or removing the precipitating causes of the severe HTG, life-long therapy with fibrates or n-3 fatty acids, as described earlier, might be considered for these patients. Limited evidence suggests that orlistat, a gastrointestinal lipase inhibitor that decreases absorption of ingested fat, thereby reducing intestinal chylomicron synthesis, may be of benefit in reducing TG levels when used in conjunction with fibrate therapy (248, 249). TG and glucose control can be particularly challenging in individuals with familial partial lipodystrophy.
Management of Specific Syndromes that Accompany Severe HTG
FAMILIAL CHYLOMICRONEMIA SYNDROME (FCS)
Treatment of FCS includes management of an acute crisis (pancreatitis) and long-term management of HTG. Management of acute HTG-induced pancreatitis is described in the previous section. Long-term management of individuals with FCS involves patient education and maintaining a very low-fat diet. Consumption of even small amounts of fat can lead to severe HTG in FCS due to the absence of functional LPL. Infants with FCS presenting with abdominal pain or failure to thrive require discontinuation of breast feeding with replacement by very low-fat formula feeding to decrease TG levels and symptoms. In children and adults with FCS, dietary fat calories should be severely restricted to control the severe HTG and abdominal pain. This translates to about 5% to 10% of total daily calories from fat, which is a major burden for these patients (250). Medium-chain TGs, which are taken up directly by the liver after absorption and do not enter plasma as chylomicrons via the thoracic duct, are a potential alternate fat source for these patients. n-3 fatty acids can aggravate the severe HTG of FCS and therefore are contraindicated in these individuals (251, 252). Fibrates are not efficacious in FCS (253). There are limited studies showing that orlistat might be beneficial in patients with FCS (254, 255). Alcohol, oral contraceptives, and other TG-elevating drugs (see Table 3) can exacerbate severe HTG and precipitate acute pancreatitis in FCS. Successful pregnancies in patients with FCS have become more common of late (256, 257).
Alipogene tiparvovec, an adeno-associated virus LPL gene therapy that was developed and resulted in significant improvement in postprandial chylomicron metabolism in patients with FCS has been abandoned and no longer available (258), Antisense oligonucleotide inhibitors of ApoC-III (volanesorsen is approved in Europe but no in the US) and of ANGPLT3 are in development for FCS (96).
MULTIFACTORIAL CHYLOMICRONEMIA SYNDROME (MFCS)
Management of acute HTG-induced pancreatitis is described in the previous section.
Long term management: To prevent acute HTG-induced pancreatitis in MFCS, the goal is to maintain TG levels below the threshold for pancreatitis, preferably <500 mg/dL. This requires instituting lifestyle adjustments, reversal of any secondary causes of HTG, such as treatment of suboptimally managed or undiagnosed diabetes, treating hypertension with lipid neutral agents such as ACE inhibitors, ARBs, calcium channel inhibitors, or alpha blockers rather than beta-adrenergic blockers and diuretics, and discontinuing other TG-raising drugs (table 3) where possible. Alcohol intake should be limited or eliminated, since even small amounts of alcohol can substantially raise TG levels in individuals with baseline HTG. Attention should be paid to avoid rebound weight gain that commonly occurs after successful weight loss. Oral estrogens should be substituted by transdermal or vaginal preparations, which raise plasma TGs to a lesser extent than oral estrogens (259, 260). Residual HTG should be treated with fibrates (229), which together with management of the secondary disorder or disorders, can reduce TG levels to below the threshold for developing pancreatitis. Other agents that can be used to lower TGs alone or in combination with fibrates, include n-3 fatty acids, and high-dose statins. We do not recommend using niacin due to risk of worsening insulin resistance and lack of clinical trial data for benefit. Lifestyle measures and weight loss are important, but patients should be educated on risks of rapid weight regain after successful weight loss can be associated with rebound severe HTG. Bariatric surgery also has been used to reduce severe HTG in refractory HTG (261). Inhibition of ApoC-III or ANGPTL3, which have been shown to lower TGs in patients with severe HTG (262), may have a role to play in the future the management of severe HTG in patients with MFCS.
REFERENCES
- 1.
- Feingold, K.R., Introduction to Lipids and Lipoproteins, in Endotext, K.R. Feingold, et al., Editors. 2021: South Dartmouth (MA).
- 2.
- Fisher, E.A., The degradation of Apolipoprotein B100: multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim Biophys Acta, 2012. 1821(5): p. 778-81. [PMC free article: PMC3593638] [PubMed: 22342675]
- 3.
- Sundaram, M. and Z. Yao, Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond), 2010. 7: p. 35. [PMC free article: PMC2873297] [PubMed: 20423497]
- 4.
- Yao, Z., Human Apolipoprotein C-III - a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovasc Hematol Disord Drug Targets, 2012. 12(2): p. 133-40. [PubMed: 23030451]
- 5.
- Schneeman, B.O., et al., Relationships between the responses of triglyceride-rich lipoproteins in blood plasma containing Apolipoproteins B-48 and B-100 to a fat-containing meal in normolipidemic humans. Proc Natl Acad Sci U S A, 1993. 90(5): p. 2069-73. [PMC free article: PMC46022] [PubMed: 8446630]
- 6.
- Kindel, T., D.M. Lee, and P. Tso, The mechanism of the formation and secretion of chylomicrons. Atheroscler Suppl, 2010. 11(1): p. 11-6. [PubMed: 20493784]
- 7.
- Kersten, S., Physiological regulation of lipoprotein lipase. Biochim Biophys Acta, 2014. 1841(7): p. 919-33. [PubMed: 24721265]
- 8.
- Gordts, P.L., et al., ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest, 2016. 126(8): p. 2855-66. [PMC free article: PMC4966320] [PubMed: 27400128]
- 9.
- Goldberg, I.J., et al., Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by Apolipoprotein A-IV. J Biol Chem, 1990. 265(8): p. 4266-72. [PubMed: 2307668]
- 10.
- Nilsson, S.K., et al., Apolipoprotein A-V; a potent triglyceride reducer. Atherosclerosis, 2011. 219(1): p. 15-21. [PubMed: 21831376]
- 11.
- Priore Oliva, C., et al., Inherited Apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler Thromb Vasc Biol, 2005. 25(2): p. 411-7. [PubMed: 15591215]
- 12.
- Gonzales, J.C., et al., Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J Clin Invest, 2013. 123(6): p. 2742-51. [PMC free article: PMC3668842] [PubMed: 23676495]
- 13.
- Kroupa, O., et al., Linking nutritional regulation of Angptl4, Gpihbp1, and Lmf1 to lipoprotein lipase activity in rodent adipose tissue. BMC Physiol, 2012. 12: p. 13. [PMC free article: PMC3562520] [PubMed: 23176178]
- 14.
- Lamiquiz-Moneo, I., et al., (Identification of variants in LMF1 gene associated with primary hypertriglyceridemia). Clin Investig Arterioscler, 2015. [PubMed: 25817768]
- 15.
- Inukai, K., et al., ANGPTL3 is increased in both insulin-deficient and -resistant diabetic states. Biochem Biophys Res Commun, 2004. 317(4): p. 1075-9. [PubMed: 15094378]
- 16.
- Shimamura, M., et al., Leptin and insulin down-regulate angiopoietin-like protein 3, a plasma triglyceride-increasing factor. Biochem Biophys Res Commun, 2004. 322(3): p. 1080-5. [PubMed: 15336575]
- 17.
- Koster, A., et al., Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology, 2005. 146(11): p. 4943-50. [PubMed: 16081640]
- 18.
- Foley, E.M., et al., Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol, 2013. 33(9): p. 2065-74. [PMC free article: PMC3821931] [PubMed: 23846497]
- 19.
- Crawford, S.E. and J. Borensztajn, Plasma clearance and liver uptake of chylomicron remnants generated by hepatic lipase lipolysis: evidence for a lactoferrin-sensitive and Apolipoprotein E-independent pathway. J Lipid Res, 1999. 40(5): p. 797-805. [PubMed: 10224148]
- 20.
- Dichek, H.L., et al., Hepatic lipase overexpression lowers remnant and LDL levels by a noncatalytic mechanism in LDL receptor-deficient mice. J Lipid Res, 2001. 42(2): p. 201-10. [PubMed: 11181749]
- 21.
- Brunzell, J.D., et al., Evidence for a common saturable triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. Journal of Clinical Investigation, 1973. 52: p. 1578-1585. [PMC free article: PMC302428] [PubMed: 4352459]
- 22.
- Hegele, R.A., et al., The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol, 2014. 2(8): p. 655-66. [PMC free article: PMC4201123] [PubMed: 24731657]
- 23.
- Berglund, L., et al., Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab, 2012. 97(9): p. 2969-89. [PMC free article: PMC3431581] [PubMed: 22962670]
- 24.
- Subramanian, S., Approach to the Patient With Moderate Hypertriglyceridemia. J Clin Endocrinol Metab, 2022. 107(6): p. 1686-1697. [PubMed: 35184196]
- 25.
- Dron, J.S. and R.A. Hegele, Genetics of Hypertriglyceridemia. Front Endocrinol (Lausanne), 2020. 11: p. 455. [PMC free article: PMC7393009] [PubMed: 32793115]
- 26.
- Noubiap, J.J., et al., Prevalence of dyslipidaemia among adults in Africa: a systematic review and meta-analysis. Lancet Glob Health, 2018. 6(9): p. e998-e1007. [PubMed: 30103999]
- 27.
- Laufs, U., et al., Clinical review on triglycerides. Eur Heart J, 2020. 41(1): p. 99-109c. [PMC free article: PMC6938588] [PubMed: 31764986]
- 28.
- Pedersen, S.B., A. Langsted, and B.G. Nordestgaard, Nonfasting Mild-to-Moderate Hypertriglyceridemia and Risk of Acute Pancreatitis. JAMA Intern Med, 2016. 176(12): p. 1834-1842. [PubMed: 27820614]
- 29.
- Grundy, S.M., et al., 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation, 2019. 139(25): p. e1082-e1143. [PMC free article: PMC7403606] [PubMed: 30586774]
- 30.
- Pearson, G.J., et al., 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol, 2021. 37(8): p. 1129-1150. [PubMed: 33781847]
- 31.
- Mach, F., et al., 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J, 2020. 41(1): p. 111-188. [PubMed: 31504418]
- 32.
- Expert Panel on Detection, E. and A. Treatment of High Blood Cholesterol in, Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA, 2001. 285(19): p. 2486-97. [PubMed: 11368702]
- 33.
- Miller, M., et al., Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation, 2011. 123(20): p. 2292-333. [PubMed: 21502576]
- 34.
- Jacobson, T.A., et al., National lipid association recommendations for patient-centered management of dyslipidemia: part 1--full report. J Clin Lipidol, 2015. 9(2): p. 129-69. [PubMed: 25911072]
- 35.
- Berglund, L., et al., Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab, 2012. 97(9): p. 2969-89. [PMC free article: PMC3431581] [PubMed: 22962670]
- 36.
- Authors/Task Force, M., et al., 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) Developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis, 2016. 253: p. 281-344. [PubMed: 27594540]
- 37.
- Fredrickson, D., R. Levy, and R. Lees, Fat transport and lipoproteins - an integrated approach to mechanisms and disorders. N Engl J Med, 1967. 276: p. 32,94,148,215,273. [PubMed: 5334042]
- 38.
- Goldstein, J.L., et al., Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest, 1973. 52(7): p. 1544-68. [PMC free article: PMC302426] [PubMed: 4718953]
- 39.
- Nikkila, E.A. and A. Aro, Family study of serum lipids and lipoproteins in coronary heart-disease. Lancet, 1973. 1(7810): p. 954-9. [PubMed: 4121585]
- 40.
- Brunzell, J.D., Clinical practice. Hypertriglyceridemia. N Engl J Med, 2007. 357(10): p. 1009-17. [PubMed: 17804845]
- 41.
- Chait, A., J.J. Albers, and J.D. Brunzell, Very low density lipoprotein overproduction in genetic forms of hypertriglyceridemia. European Journal of Clinical Investigation, 1980. 10: p. 17-22. [PubMed: 6768562]
- 42.
- Brunzell, J.D., et al., Plasma lipoproteins in familial combined hyperlipidemia and monogenic familial hypertriglyceridemia. Journal of Lipid Research, 1983. 24: p. 147-155. [PubMed: 6403642]
- 43.
- Brunzell, J.D., et al., Myocardial infarction in the familial forms of hypertriglyceridemia. Metabolism: Clinical and Experimental, 1976. 25: p. 313-320. [PubMed: 1250165]
- 44.
- Austin, M.A., et al., Cardiovascular disease mortality in familial forms of hypertriglyceridemia: A 20-year prospective study. Circulation, 2000. 101: p. 2777-2782. [PubMed: 10859281]
- 45.
- Hokanson, J.E. and M.A. Austin, Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta- analysis of population-based prospective studies. J Cardiovasc Risk, 1996. 3(2): p. 213-9. [PubMed: 8836866]
- 46.
- McNeely, M., et al., Lipoprotein and Apolipoprotein abnormalities in familial combined hyperlipidemia: a 20-year prospective study. Atherosclerosis, 2001. 159: p. 417-481. [PubMed: 11730829]
- 47.
- Lewis, G.F., C. Xiao, and R.A. Hegele, Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev, 2015. 36(1): p. 131-47. [PubMed: 25554923]
- 48.
- Dron, J.S., et al., Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol, 2019. 13(1): p. 80-88. [PubMed: 30466821]
- 49.
- Eaton, R.P., R.C. Allen, and D.S. Schade, Overproduction of a kinetic subclass of VLDL-ApoB, and direct catabolism of VLDL-ApoB in human endogenous hypertriglyceridemia: an analytical model solution of tracer data. J Lipid Res, 1983. 24(10): p. 1291-303. [PubMed: 6644179]
- 50.
- Grundy, S.M., et al., Transport of very low density lipoprotein triglycerides in varying degrees of obesity and hypertriglyceridemia. Journal of Clinical Investigation, 1979. 63: p. 1274-1283. [PMC free article: PMC372076] [PubMed: 221538]
- 51.
- Beil, U., et al., Triglyceride and cholesterol metabolism in primary hypertriglyceridemia. Arteriosclerosis, 1982. 2(1): p. 44-57. [PubMed: 7059323]
- 52.
- Sigurdsson, G., A. Nicoll, and B. Lewis, Metabolism of very low density lipoproteins in hyperlipidaemia: studies of Apolipoprotein B kinetics in man. Eur J Clin Invest, 1976. 6(2): p. 167-77. [PubMed: 177296]
- 53.
- Ayyobi, A.F. and J.D. Brunzell, Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. Am J Cardiol, 2003. 92(4A): p. 27J-33J. [PubMed: 12957324]
- 54.
- Reynisdottir, S., et al., Impaired activation of adipocyte lipolysis in familial combined hyperlipidemia. J Clin Invest, 1995. 95(5): p. 2161-9. [PMC free article: PMC295819] [PubMed: 7738184]
- 55.
- Reynisdottir, S., et al., Adipose tissue lipoprotein lipase and hormone-sensitive lipase. Contrasting findings in familial combined hyperlipidemia and insulin resistance syndrome. Arterioscler Thromb Vasc Biol, 1997. 17(10): p. 2287-92. [PubMed: 9351402]
- 56.
- Venkatesan, S., et al., Stable isotopes show a direct relation between VLDL ApoB overproduction and serum triglyceride levels and indicate a metabolically and biochemically coherent basis for familial combined hyperlipidemia. Arteriosclerosis and Thrombosis, 1993. 13: p. 1110-1118. [PubMed: 8318511]
- 57.
- Aitman, T., et al., Defects of insulin action on fatty acid and carbohydrate metabolism in familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol, 1997. 17: p. 748-754. [PubMed: 9108790]
- 58.
- Janus, E.D., et al., Kinetic bases of the primary hyperlipidemias: Studies of Apolipoprotein B turnover in genetically defined subjects. European Journal of Clinical Investigation, 1980. 10: p. 161-172. [PubMed: 6780364]
- 59.
- Berneis, K.K. and R.M. Krauss, Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res, 2002. 43(9): p. 1363-79. [PubMed: 12235168]
- 60.
- Cabezas, M.C., et al., Impaired chylomicron remnant clearance in familial combined hyperlipidemia. Arterioscler Thromb, 1993. 13(6): p. 804-14. [PubMed: 8499400]
- 61.
- Hokanson, J.E., et al., Plasma triglyceride and LDL heterogeneity in familial combined hyperlipidemia. Arteriosclerosis and Thrombosis, 1993. 13: p. 427-434. [PubMed: 8443147]
- 62.
- Hokanson, J.E., et al., LDL physical and chemical properties in familial combined hyperlipidemia. Arteriosclerosis, Thrombosis, and Vascular Biology, 1995. 15: p. 452-459. [PubMed: 7749856]
- 63.
- Cruz-Bautista, I., et al., Determinants of VLDL composition and Apo B-containing particles in familial combined hyperlipidemia. Clin Chim Acta, 2015. 438: p. 160-5. [PubMed: 25172037]
- 64.
- Brouwers, M.C. and M.M. van Greevenbroek, Lipid metabolism: the significance of plasma proprotein convertase subtilisin kexin type 9 in the elucidation of complex lipid disorders. Curr Opin Lipidol, 2011. 22(4): p. 317-8. [PubMed: 21743308]
- 65.
- Hopkins, P.N., et al., Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation, 2003. 108(5): p. 519-23. [PubMed: 12847072]
- 66.
- Purnell, J.Q., et al., Relationship of insulin sensitivity and ApoB levels to intra-abdominal fat in subjects with familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol, 2001. 21(4): p. 567-72. [PubMed: 11304474]
- 67.
- Ascaso, J., et al., Insulin resistance in patients wtih familial combined hyperlipidemia and coronary artery disease. Am J Cardiol, 1997. 80: p. 1481-1487. [PubMed: 9399729]
- 68.
- Castro Cabezas, M., et al., Impaired fatty acid metabolism in familial combined hyperlipidemia: a mechanism associating hepatic Apolipoprotein B overproduction and insulin resistance. J Clin Invest, 1993. 92: p. 160-168. [PMC free article: PMC293556] [PubMed: 8100834]
- 69.
- van der Kallen, C., et al., Evidence of insulin resistant lipid metabolism in adipose tissue in familial combined hyperlipidemia, but not type 2 diabetes mellitus. Atherosclerosis, 2002. 164: p. 337-346. [PubMed: 12204806]
- 70.
- Arner, P., et al., Dynamics of human adipose lipid turnover in health and metabolic disease. Nature, 2011. 478(7367): p. 110-3. [PMC free article: PMC3773935] [PubMed: 21947005]
- 71.
- Brouwers, M.C., et al., Fatty liver is an integral feature of familial combined hyperlipidaemia: relationship with fat distribution and plasma lipids. Clin Sci (Lond), 2007. 112(2): p. 123-30. [PubMed: 16958621]
- 72.
- Brouwers, M.C., et al., Increased arterial stiffness in familial combined hyperlipidemia. J Hypertens, 2009. 27(5): p. 1009-16. [PubMed: 19402225]
- 73.
- Keulen, E.T., et al., Increased intima-media thickness in familial combined hyperlipidemia associated with Apolipoprotein B. Arterioscler Thromb Vasc Biol, 2002. 22(2): p. 283-8. [PubMed: 11834529]
- 74.
- Chait, A. and J.D. Brunzell, Severe hypertriglyceridemia: Role of familial and acquired disorders. Metabolism: Clinical and Experimental, 1983. 32: p. 209-214. [PubMed: 6827992]
- 75.
- Herink, M. and M.K. Ito, Medication Induced Changes in Lipid and Lipoproteins, in Endotext, K.R. Feingold, et al., Editors. 2018: South Dartmouth (MA).
- 76.
- Feingold, K.R., The Effect of Inflammation and Infection on Lipids and Lipoproteins, in Endotext, K.R., Feingold, et al., Editors. 2022: South Dartmouth (MA).
- 77.
- Feingold, K.R., Obesity and Dyslipidemia, in Endotext, K.R. Feingold, et al., Editors. 2023: South Dartmouth (MA).
- 78.
- Feingold, K.R., The Effect of Endocrine Disorders on Lipids and Lipoproteins, in Endotext, K.R. Feingold, et al., Editors. 2023: South Dartmouth (MA).
- 79.
- Chait, A., Secondary hyperlipidemia. Journal of Clinical Pathology, 1973. 26(suppl 5): p. 68-71. [PMC free article: PMC1436099] [PubMed: 4582172]
- 80.
- Chait, A. and J.D. Brunzell, Acquired hyperlipidemia (secondary dyslipoproteinemia). Endocrinology and Metabolism Clinics of North America, 1990. 19: p. 259-278. [PubMed: 2192873]
- 81.
- Brunzell, J.D. and E.L. Bierman, Chylomicronemia syndrome. Interaction of genetic and acquired hypertriglyceridemia. Medical Clinics of North America, 1982. 66: p. 455-468. [PubMed: 7040847]
- 82.
- Chait, A., H.T. Robertson, and J.D. Brunzell, Chylomicronemia syndrome in diabetes mellitus. Diabetes Care, 1981. 4: p. 343-348. [PubMed: 7344882]
- 83.
- Brahm, A.J. and R.A. Hegele, Chylomicronaemia-current diagnosis and future therapies. Nat Rev Endocrinol, 2015. [PubMed: 25732519]
- 84.
- Brunzell, J. and S. Deeb, Familial lipoprotein lipase deficiency, Apo CII deficiency, and hepatic lipase deficiency, in The Metabolic and Molecular Basis of Inherited Disease, C. Scriver, et al., Editors. 2001, McGraw-Hill Book Co.: New York. p. 2789-2816.
- 85.
- Rahalkar, A.R., et al., Novel LPL mutations associated with lipoprotein lipase deficiency: two case reports and a literature review. Can J Physiol Pharmacol, 2009. 87(3): p. 151-60. [PubMed: 19295657]
- 86.
- Martin-Campos, J.M., et al., Molecular analysis of chylomicronemia in a clinical laboratory setting: diagnosis of 13 cases of lipoprotein lipase deficiency. Clin Chim Acta, 2014. 429: p. 61-8. [PubMed: 24291057]
- 87.
- Blom, D.J., et al., Characterizing familial chylomicronemia syndrome: Baseline data of the APPROACH study. J Clin Lipidol, 2018. 12(5): p. 1234-1243 e5. [PubMed: 30318066]
- 88.
- Peterson, J., et al., Structural and functional consequences of missense mutations in exon 5 of the lipoprotein lipase gene. J Lipid Res, 2002. 43(3): p. 398-406. [PubMed: 11893776]
- 89.
- Nickerson, D.A., et al., DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet, 1998. 19(3): p. 233-40. [PubMed: 9662394]
- 90.
- Breckenridge, W.C., et al., Hypertriglyceridemia associated with deficiency of Apolipoprotein C-II. New England Journal of Medicine, 1978. 298: p. 1265. [PubMed: 565877]
- 91.
- Rabacchi, C., et al., Spectrum of mutations of the LPL gene identified in Italy in patients with severe hypertriglyceridemia. Atherosclerosis, 2015. 241(1): p. 79-86. [PubMed: 25966443]
- 92.
- Surendran, R.P., et al., Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med, 2012. 272(2): p. 185-96. [PMC free article: PMC3940136] [PubMed: 22239554]
- 93.
- Kristensen, K.K., et al., A disordered acidic domain in GPIHBP1 harboring a sulfated tyrosine regulates lipoprotein lipase. Proc Natl Acad Sci U S A, 2018. 115(26): p. E6020-E6029. [PMC free article: PMC6042107] [PubMed: 29899144]
- 94.
- Calandra, S., et al., APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr Opin Lipidol, 2006. 17(2): p. 122-7. [PubMed: 16531747]
- 95.
- Peterfy, M., Lipase maturation factor 1: a lipase chaperone involved in lipid metabolism. Biochim Biophys Acta, 2012. 1821(5): p. 790-4. [PMC free article: PMC3288453] [PubMed: 22063272]
- 96.
- Baass, A., et al., Familial chylomicronemia syndrome: an under-recognized cause of severe hypertriglyceridaemia. J Intern Med, 2020. 287(4): p. 340-348. [PubMed: 31840878]
- 97.
- Dron, J.S. and R.A. Hegele, The evolution of genetic-based risk scores for lipids and cardiovascular disease. Curr Opin Lipidol, 2019. 30(2): p. 71-81. [PubMed: 30676533]
- 98.
- Parsons, S.K., et al., Asparaginase-associated lipid abnormalities in children with acute lymphoblastic leukemia. Blood, 1997. 89(6): p. 1886-95. [PubMed: 9058708]
- 99.
- Tozuka, M., et al., Characterization of hypertriglyceridemia induced by L-asparaginase therapy for acute lymphoblastic leukemia and malignant lymphoma. Ann Clin Lab Sci, 1997. 27(5): p. 351-7. [PubMed: 9303174]
- 100.
- Yadav, D. and C.S. Pitchumoni, Issues in hyperlipidemic pancreatitis. J Clin Gastroenterol, 2003. 36(1): p. 54-62. [PubMed: 12488710]
- 101.
- Brunzell, J.D. and H.G. Schrott, The interaction of familial and secondary causes of hypertriglyceridemia: Role in pancreatitis. Transactions of the Association of American Physicians, 1973. 86: p. 245-254. [PubMed: 4788799]
- 102.
- Brunzell, J.D. and H.G. Schrott, The interaction of familial and secondary causes of hypertriglyceridemia: role in pancreatitis. J Clin Lipidol, 2012. 6(5): p. 409-12. [PubMed: 23009776]
- 103.
- Johansen, C.T., et al., An increased burden of common and rare lipid-associated risk alleles contributes to the phenotypic spectrum of hypertriglyceridemia. Arterioscler Thromb Vasc Biol, 2011. 31(8): p. 1916-26. [PMC free article: PMC3562702] [PubMed: 21597005]
- 104.
- Schneider, W.J., et al., Familial dysbetalipoproteinemia. Abnormal binding of mutant Apoprotein E to low density lipoprotein receptors of human fibroblasts and membranes from liver and adrenal of rats, rabbits, and cows. J Clin Invest, 1981. 68(4): p. 1075-85. [PMC free article: PMC370895] [PubMed: 6270194]
- 105.
- Vedhachalam, C., et al., The C-terminal lipid-binding domain of Apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry, 2007. 46(10): p. 2583-93. [PubMed: 17305370]
- 106.
- Siest, G., et al., Apolipoprotein E: an important gene and protein to follow in laboratory medicine. Clin Chem, 1995. 41(8 Pt 1): p. 1068-86. [PubMed: 7628082]
- 107.
- Weisgraber, K.H., S.C. Rall, Jr., and R.W. Mahley, Human E Apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the Apo-E isoforms. J Biol Chem, 1981. 256(17): p. 9077-83. [PubMed: 7263700]
- 108.
- Smelt, A.H. and F. de Beer, Apolipoprotein E and familial dysbetalipoproteinemia: clinical, biochemical, and genetic aspects. Semin Vasc Med, 2004. 4(3): p. 249-57. [PubMed: 15630634]
- 109.
- Dong, L.M., et al., The carboxyl terminus in Apolipoprotein E2 and the seven amino acid repeat in Apolipoprotein E-Leiden: role in receptor-binding activity. J Lipid Res, 1998. 39(6): p. 1173-80. [PubMed: 9643348]
- 110.
- Mahley, R.W., Y. Huang, and S.C. Rall, Jr., Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res, 1999. 40(11): p. 1933-49. [PubMed: 10552997]
- 111.
- Koopal, C., A.D. Marais, and F.L. Visseren, Familial dysbetalipoproteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes, 2017. 24(2): p. 133-139. [PubMed: 28098593]
- 112.
- Koopal, C., et al., Autosomal dominant familial dysbetalipoproteinemia: A pathophysiological framework and practical approach to diagnosis and therapy. J Clin Lipidol, 2017. 11(1): p. 12-23 e1. [PubMed: 28391878]
- 113.
- Cenarro, A., et al., The p.Leu167del Mutation in APOE Gene Causes Autosomal Dominant Hypercholesterolemia by Down-regulation of LDL Receptor Expression in Hepatocytes. J Clin Endocrinol Metab, 2016. 101(5): p. 2113-21. [PubMed: 27014949]
- 114.
- Bea, A.M., et al., Contribution of APOE Genetic Variants to Dyslipidemia. Arterioscler Thromb Vasc Biol, 2023. 43(6): p. 1066-1077. [PubMed: 37051929]
- 115.
- Paquette, M., S. Bernard, and A. Baass, Diagnosis of remnant hyperlipidaemia. Curr Opin Lipidol, 2022. 33(4): p. 227-230. [PubMed: 35942808]
- 116.
- Pallazola, V.A., et al., Modern prevalence of dysbetalipoproteinemia (Fredrickson-Levy-Lees type III hyperlipoproteinemia). Arch Med Sci, 2020. 16(5): p. 993-1003. [PMC free article: PMC7444722] [PubMed: 32863987]
- 117.
- Mahley, R.W. and S.C. Rall, Jr., Type III hyperlipoproteinemia (dysbetalipoproteinemia): The role of Apolipoprotein E in normal and abnormal lipoprotein metabolism, in The Metabolic Basis of Inherited Disease, C.R. Scriver, et al., Editors. 1989, McGraw-Hill: New York. p. 1195.
- 118.
- Brummer, D., et al., Expression of type III hyperlipoproteinemia in patients homozygous for Apolipoprotein E-2 is modulated by lipoprotein lipase and postprandial hyperinsulinemia. J Mol Med (Berl), 1998. 76(5): p. 355-64. [PubMed: 9587070]
- 119.
- Feussner, G. and R. Ziegler, Expression of type III hyperlipoproteinaemia in a subject with secondary hypothyroidism bearing the Apolipoprotein E2/2 phenotype. J Intern Med, 1991. 230(2): p. 183-6. [PubMed: 1865171]
- 120.
- Breslow, J.L., et al., Studies of familial type III hyperlipoproteinemia using as a genetic marker the ApoE phenotype E2/2. J Lipid Res, 1982. 23(8): p. 1224-35. [PubMed: 7175379]
- 121.
- Chait, A., et al., Type-III Hyperlipoproteinaemia ("remnant removal disease"). Insight into the pathogenetic mechanism. Lancet, 1977. 1(8023): p. 1176-8. [PubMed: 68276]
- 122.
- Chait, A., et al., Impaired very low density lipoprotein and triglyceride removal in broad beta disease: comparison with endogenous hypertriglyceridemia. Metabolism, 1978. 27(9): p. 1055-66. [PubMed: 210351]
- 123.
- Rothschild, M., et al., Pathognomonic Palmar Crease Xanthomas of Apolipoprotein E2 Homozygosity-Familial Dysbetalipoproteinemia. JAMA Dermatol, 2016. 152(11): p. 1275-1276. [PubMed: 27603268]
- 124.
- Albers, J.J., G.R. Warnick, and W.R. Hazzard, Type III hyperlipoproteinemia: a comparative study of current diagnostic techniques. Clin Chim Acta, 1977. 75(2): p. 193-204. [PubMed: 191218]
- 125.
- Blom, D.J., F.H. O'Neill, and A.D. Marais, Screening for dysbetalipoproteinemia by plasma cholesterol and Apolipoprotein B concentrations. Clin Chem, 2005. 51(5): p. 904-7. [PubMed: 15855667]
- 126.
- Paquette, M., et al., A simplified diagnosis algorithm for dysbetalipoproteinemia. J Clin Lipidol, 2020. 14(4): p. 431-437. [PubMed: 32631794]
- 127.
- Morganroth, J., R.I. Levy, and D.S. Fredrickson, The biochemical, clinical, and genetic features of type III hyperlipoproteinemia. Ann Intern Med, 1975. 82(2): p. 158-74. [PubMed: 163608]
- 128.
- Havel, R.J. and J.P. Kane, Primary dysbetalipoproteinemia: Predominancy of a specific Apoprotein species in triglyceride-rich lipoproteins. Proceedings of the National Academy of Sciences of the USA, 1973. 70: p. 2015. [PMC free article: PMC433655] [PubMed: 4352966]
- 129.
- Koopal, C., et al., Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: a European cross-sectional study. Atherosclerosis, 2015. 240(1): p. 90-7. [PubMed: 25768710]
- 130.
- Mahley, R. and S. Rall, Type III Hyperlipoproteinemia (Dysbetalipoproteinemia): The Role of Apolipoprotein E in Normal and Abnormal Lipoprotein Metabolism, in The Metabolic & Molecular Bases of Inherited Disease, C. Scriver, et al., Editors. 2001, McGraw-Hill: New York. p. 2835-2862.
- 131.
- Koopal, C., et al., The relation between Apolipoprotein E (APOE) genotype and peripheral artery disease in patients at high risk for cardiovascular disease. Atherosclerosis, 2016. 246: p. 187-92. [PubMed: 26800308]
- 132.
- Paquette, M., S. Bernard, and A. Baass, Dysbetalipoproteinemia Is Associated With Increased Risk of Coronary and Peripheral Vascular Disease. J Clin Endocrinol Metab, 2022. 108(1): p. 184-190. [PubMed: 36056815]
- 133.
- Garg, A., Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab, 2011. 96(11): p. 3313-25. [PMC free article: PMC7673254] [PubMed: 21865368]
- 134.
- Akinci, B., M. Sahinoz, and E. Oral, Lipodystrophy Syndromes: Presentation and Treatment, in Endotext, K.R. Feingold, et al., Editors. 2000: South Dartmouth (MA).
- 135.
- Simha, V. and A. Garg, Inherited lipodystrophies and hypertriglyceridemia. Curr Opin Lipidol, 2009. 20(4): p. 300-8. [PubMed: 19494770]
- 136.
- Lightbourne, M. and R.J. Brown, Genetics of Lipodystrophy. Endocrinol Metab Clin North Am, 2017. 46(2): p. 539-554. [PMC free article: PMC5424609] [PubMed: 28476236]
- 137.
- Lotta, L.A., et al., Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet, 2017. 49(1): p. 17-26. [PMC free article: PMC5774584] [PubMed: 27841877]
- 138.
- Guillin-Amarelle, C., et al., Type 1 familial partial lipodystrophy: understanding the Kobberling syndrome. Endocrine, 2016. 54(2): p. 411-421. [PubMed: 27473102]
- 139.
- Agarwal, A.K. and A. Garg, A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab, 2002. 87(1): p. 408-11. [PubMed: 11788685]
- 140.
- Ajluni, N., et al., Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf), 2017. 86(5): p. 698-707. [PMC free article: PMC5395301] [PubMed: 28199729]
- 141.
- Subramanyam, L., V. Simha, and A. Garg, Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin Genet, 2010. 78(1): p. 66-73. [PMC free article: PMC3150739] [PubMed: 20041886]
- 142.
- Hussain, I. and A. Garg, Lipodystrophy Syndromes. Endocrinol Metab Clin North Am, 2016. 45(4): p. 783-797. [PMC free article: PMC7590232] [PubMed: 27823605]
- 143.
- Hussain, I., N. Patni, and A. Garg, Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology, 2019. 51(2): p. 202-212. [PMC free article: PMC6402807] [PubMed: 30595509]
- 144.
- Jacob, K.N., et al., Phenotypic heterogeneity in body fat distribution in patients with atypical Werner's syndrome due to heterozygous Arg133Leu lamin A/C mutation. J Clin Endocrinol Metab, 2005. 90(12): p. 6699-706. [PubMed: 16174718]
- 145.
- Garg, A., et al., Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab, 2009. 94(12): p. 4971-83. [PMC free article: PMC2795646] [PubMed: 19875478]
- 146.
- Calvo, M. and E. Martinez, Update on metabolic issues in HIV patients. Curr Opin HIV AIDS, 2014. 9(4): p. 332-9. [PubMed: 24824886]
- 147.
- Castelli, W.P., The triglyceride issue: a view from Framingham. Am Heart J, 1986. 112(2): p. 432-7. [PubMed: 3739899]
- 148.
- Harchaoui, K.E., et al., Triglycerides and cardiovascular risk. Curr Cardiol Rev, 2009. 5(3): p. 216-22. [PMC free article: PMC2822144] [PubMed: 20676280]
- 149.
- Langsted, A., et al., Nonfasting cholesterol and triglycerides and association with risk of myocardial infarction and total mortality: the Copenhagen City Heart Study with 31 years of follow-up. J Intern Med, 2011. 270(1): p. 65-75. [PubMed: 21198993]
- 150.
- Nordestgaard, B.G. and A. Varbo, Triglycerides and cardiovascular disease. Lancet, 2014. 384(9943): p. 626-35. [PubMed: 25131982]
- 151.
- Hokanson, J.E. and M.A. Austin, Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk, 1996. 3(2): p. 213-9. [PubMed: 8836866]
- 152.
- Zilversmit, D.B., Atherogenesis: A postprandial phenomenon. Circulation, 1979. 60: p. 473-485. [PubMed: 222498]
- 153.
- Zilversmit, D.B., Atherogenic nature of triglycerides, postprandial lipidemia, and triglyceride-rich remnant lipoproteins. Clin Chem, 1995. 41(1): p. 153-8. [PubMed: 7813071]
- 154.
- Nordestgaard, B.G. and D.B. Zilversmit, Large lipoproteins are excluded from the arterial wall in diabetic cholesterol-fed rabbits. J Lipid Res, 1988. 29(11): p. 1491-500. [PubMed: 3241125]
- 155.
- Nordestgaard, B.G., S. Stender, and K. Kjeldsen, Reduced atherogenesis in cholesterol-fed diabetic rabbits. Giant lipoproteins do not enter the arterial wall. Arteriosclerosis, 1988. 8(4): p. 421-8. [PubMed: 3395278]
- 156.
- Williams, K.J. and I. Tabas, The response-to-retention hypothesis of early atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 1995. 15: p. 551-561. [PMC free article: PMC2924812] [PubMed: 7749869]
- 157.
- Williams, K.J. and I. Tabas, The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol, 1998. 9(5): p. 471-4. [PubMed: 9812202]
- 158.
- Krauss, R.M., Dense low density lipoproteins and coronary artery disease. Am J Cardiol, 1995. 75(6): p. 53B-57B. [PubMed: 7863975]
- 159.
- Olin-Lewis, K., et al., ApoC-III content of ApoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res, 2002. 43(11): p. 1969-77. [PubMed: 12401896]
- 160.
- Chait, A., et al., Susceptibility of small, dense low density lipoproteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern B. American Journal of Medicine, 1993. 94: p. 350-356. [PubMed: 8475928]
- 161.
- Tribble, D.L., et al., Oxidative susceptibility of LDL density subfractions is related to their ubiquinol-10 and à-tocopherol content. Proceedings of the National Academy of Sciences of the USA, 1994. 91: p. 1183-1187. [PMC free article: PMC521478] [PubMed: 8302851]
- 162.
- Cornier, M.A., et al., The metabolic syndrome. Endocr Rev, 2008. 29(7): p. 777-822. [PMC free article: PMC5393149] [PubMed: 18971485]
- 163.
- Rosenson, R.S., et al., Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol, 2014. 64(23): p. 2525-40. [PubMed: 25500239]
- 164.
- Do, R., et al., Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet, 2013. 45(11): p. 1345-52. [PMC free article: PMC3904346] [PubMed: 24097064]
- 165.
- Waterworth, D.M., et al., Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol, 2010. 30(11): p. 2264-76. [PMC free article: PMC3891568] [PubMed: 20864672]
- 166.
- Teslovich, T.M., et al., Biological, clinical and population relevance of 95 loci for blood lipids. Nature, 2010. 466(7307): p. 707-13. [PMC free article: PMC3039276] [PubMed: 20686565]
- 167.
- Rip, J., et al., Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol, 2006. 26(6): p. 1236-45. [PubMed: 16574898]
- 168.
- Ference, B.A., et al., Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA, 2019. 321(4): p. 364-373. [PMC free article: PMC6439767] [PubMed: 30694319]
- 169.
- Pimstone, S.N., et al., Mutations in the gene for lipoprotein lipase. A cause for low HDL cholesterol levels in individuals heterozygous for familial hypercholesterolemia. Arterioscler Thromb Vasc Biol, 1995. 15(10): p. 1704-12. [PubMed: 7583547]
- 170.
- Burkhardt, R., et al., Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J Clin Invest, 2010. 120(12): p. 4410-4. [PMC free article: PMC2993600] [PubMed: 21084752]
- 171.
- Douvris, A., et al., Functional analysis of the TRIB1 associated locus linked to plasma triglycerides and coronary artery disease. J Am Heart Assoc, 2014. 3(3): p. e000884. [PMC free article: PMC4309087] [PubMed: 24895164]
- 172.
- Tg, et al., Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med, 2014. 371(1): p. 22-31. [PMC free article: PMC4180269] [PubMed: 24941081]
- 173.
- Jorgensen, A.B., et al., Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med, 2014. 371(1): p. 32-41. [PubMed: 24941082]
- 174.
- Soufi, M., et al., Mutation screening of the APOA5 gene in subjects with coronary artery disease. J Investig Med, 2012. 60(7): p. 1015-9. [PubMed: 22914599]
- 175.
- Do, R., et al., Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature, 2015. 518(7537): p. 102-6. [PMC free article: PMC4319990] [PubMed: 25487149]
- 176.
- Dewey, F.E., et al., Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med, 2016. 374(12): p. 1123-33. [PMC free article: PMC4900689] [PubMed: 26933753]
- 177.
- Myocardial Infarction, G., et al., Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med, 2016. 374(12): p. 1134-44. [PMC free article: PMC4850838] [PubMed: 26934567]
- 178.
- Nordestgaard, B.G., R. Wootton, and B. Lewis, Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo. Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler Thromb Vasc Biol, 1995. 15(4): p. 534-42. [PubMed: 7749867]
- 179.
- Norata, G.D., et al., Post-prandial endothelial dysfunction in hypertriglyceridemic subjects: molecular mechanisms and gene expression studies. Atherosclerosis, 2007. 193(2): p. 321-7. [PubMed: 17055512]
- 180.
- Malloy, M.J. and J.P. Kane, A risk factor for atherosclerosis: triglyceride-rich lipoproteins. Adv Intern Med, 2001. 47: p. 111-36. [PubMed: 11795072]
- 181.
- Mamo, J.C., S.D. Proctor, and D. Smith, Retention of chylomicron remnants by arterial tissue; importance of an efficient clearance mechanism from plasma. Atherosclerosis, 1998. 141 Suppl 1: p. S63-9. [PubMed: 9888645]
- 182.
- Havel, R.J. and R.S. Gordon, Jr., Idiopathic hyperlipemia: metabolic studies in an affected family. J Clin Invest, 1960. 39: p. 1777-90. [PMC free article: PMC441903] [PubMed: 13712364]
- 183.
- Benlian, P., et al., Premature atherosclerosis in patients with familial chylomicronemia caused by mutations in the lipoprotein lipase gene. N Engl J Med, 1996. 335(12): p. 848-54. [PubMed: 8778602]
- 184.
- Zafrir, B., et al., Clinical features and outcomes of severe, very severe, and extreme hypertriglyceridemia in a regional health service. J Clin Lipidol, 2018. 12(4): p. 928-936. [PubMed: 29685592]
- 185.
- Austin, M.A. and J.E. Hokanson, Epidemiology of triglycerides, small dense low-density lipoprotein, and lipoprotein(a) as risk factors for coronary heart disease. Med Clin North Am, 1994. 78(1): p. 99-115. [PubMed: 8283937]
- 186.
- Goldberg, I.J., R.H. Eckel, and R. McPherson, Triglycerides and heart disease: still a hypothesis? Arterioscler Thromb Vasc Biol, 2011. 31(8): p. 1716-25. [PMC free article: PMC3141088] [PubMed: 21527746]
- 187.
- Brown, W.V., et al., Severe hypertriglyceridemia. J Clin Lipidol, 2012. 6(5): p. 397-408. [PubMed: 23009775]
- 188.
- Goldberg, A.S. and R.A. Hegele, Severe hypertriglyceridemia in pregnancy. J Clin Endocrinol Metab, 2012. 97(8): p. 2589-96. [PubMed: 22639290]
- 189.
- Brunzell, J.D., Lipoprotein lipase deficiency and other causes of the chylomicronemia syndrome, in The Metabolic Basis of Inherited Disease, C.R. Scriver, et al., Editors. 1989, McGraw-Hill: New York. p. 1165-1180.
- 190.
- Tremblay, K., et al., Etiology and risk of lactescent plasma and severe hypertriglyceridemia. J Clin Lipidol, 2011. 5(1): p. 37-44. [PubMed: 21262505]
- 191.
- Lloret Linares, C., et al., Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas, 2008. 37(1): p. 13-2. [PubMed: 18580438]
- 192.
- Cameron, J.L., et al., Acute pancreatitis with hyperlipidemia: The incidence of lipid abnormalities in acute pancreatitis. Annals of Surgery, 1973. 177: p. 483-489. [PMC free article: PMC1355660] [PubMed: 4691868]
- 193.
- Farmer, R.G., et al., Hyperlipoproteinemia and pancreatitis. American Journal of Medicine, 1973. 54: p. 161-165. [PubMed: 4685843]
- 194.
- Wang, Q., et al., Elevated Serum Triglycerides in the Prognostic Assessment of Acute Pancreatitis: A Systematic Review and Meta-Analysis of Observational Studies. J Clin Gastroenterol, 2017. 51(7): p. 586-593. [PubMed: 28682990]
- 195.
- Mirtallo, J.M., et al., State of the art review: Intravenous fat emulsions: Current applications, safety profile, and clinical implications. Ann Pharmacother, 2010. 44(4): p. 688-700. [PubMed: 20332339]
- 196.
- Devaud, J.C., et al., Hypertriglyceridemia: a potential side effect of propofol sedation in critical illness. Intensive Care Med, 2012. 38(12): p. 1990-8. [PubMed: 23052949]
- 197.
- Havel, R.J., Pathogenesis, differentiation and management of hypertriglyceridemia. Adv Intern Med, 1969. 15: p. 117-54. [PubMed: 4908616]
- 198.
- Yang, F., et al., The role of free fatty acids, pancreatic lipase and Ca+ signalling in injury of isolated acinar cells and pancreatitis model in lipoprotein lipase-deficient mice. Acta Physiol (Oxf), 2009. 195(1): p. 13-28. [PubMed: 18983441]
- 199.
- Tsuang, W., et al., Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol, 2009. 104(4): p. 984-91. [PubMed: 19293788]
- 200.
- Valdivielso, P., A. Ramirez-Bueno, and N. Ewald, Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med, 2014. 25(8): p. 689-94. [PubMed: 25269432]
- 201.
- Saharia, P., et al., Acute pancreatitis with hyperlipemia: studies with an isolated perfused canine pancreas. Surgery, 1977. 82(1): p. 60-7. [PubMed: 877857]
- 202.
- Seplowitz, A.H., S. Chien, and F.R. Smith, Effects of lipoproteins on plasma viscosity. Atherosclerosis, 1981. 38(1-2): p. 89-95. [PubMed: 7470209]
- 203.
- Chang, Y.T., et al., Association of cystic fibrosis transmembrane conductance regulator (CFTR) mutation/variant/haplotype and tumor necrosis factor (TNF) promoter polymorphism in hyperlipidemic pancreatitis. Clin Chem, 2008. 54(1): p. 131-8. [PubMed: 17981921]
- 204.
- Ivanova, R., et al., Triglyceride levels and Apolipoprotein E polymorphism in patients with acute pancreatitis. Hepatobiliary Pancreat Dis Int, 2012. 11(1): p. 96-101. [PubMed: 22251476]
- 205.
- Durrington, P., Dyslipidaemia. Lancet, 2003. 362(9385): p. 717-31. [PubMed: 12957096]
- 206.
- Parker, F., et al., Evidence for the chylomicron origin of lipids accumulating in diabetic eruptive xanthomas: A correlative lipid biochemical, histochemical and electron microscopic study. Journal of Clinical Investigation, 1970. 49: p. 2172-2187. [PMC free article: PMC322718] [PubMed: 5480845]
- 207.
- Rosenson, R.S., et al., Hypertriglyceridemia and other factors associated with plasma viscosity. Am J Med, 2001. 110(6): p. 488-92. [PubMed: 11331062]
- 208.
- Inokuchi, R., et al., Hypertriglyceridemia as a possible cause of coma: a case report. J Med Case Rep, 2012. 6: p. 412. [PMC free article: PMC3520695] [PubMed: 23198781]
- 209.
- Feingold, K.R., Triglyceride Lowering Drugs, in Endotext, K.R. Feingold, et al., Editors. 2021: South Dartmouth (MA).
- 210.
- Feingold K.R., The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels, in Endotext, K.R. Feingold, et al., Editors. 2021: South Dartmouth (MA).
- 211.
- Scandinavian Simvastatin Survival Study Group, Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet, 1994. 344: p. 1383-1389. [PubMed: 7968073]
- 212.
- Cannon, C.P., et al., Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med, 2015. [PubMed: 26039521]
- 213.
- Koskinen, P., et al., Coronary heart disease incidence in NIDDM patients in the Helsinki Heart Study. Diab Care, 1992. 15: p. 825-829. [PubMed: 1516498]
- 214.
- ACCORD Study Group, et al., Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med, 2010. 362(17): p. 1563-74. [PMC free article: PMC2879499] [PubMed: 20228404]
- 215.
- Keech, A., et al., Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet, 2005. 366(9500): p. 1849-61. [PubMed: 16310551]
- 216.
- Bezafibrate Infarction Prevention, s., Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation, 2000. 102(1): p. 21-7. [PubMed: 10880410]
- 217.
- Camejo, G., Phase 2 clinical trials with K-877 (pemafibrate): A promising selective PPAR-alpha modulator for treatment of combined dyslipidemia. Atherosclerosis, 2017. 261: p. 163-164. [PubMed: 28434555]
- 218.
- Das Pradhan, A., et al., Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med, 2022. 387(21): p. 1923-1934. [PubMed: 36342113]
- 219.
- Aung, T., et al., Associations of Omega-3 Fatty Acid Supplement Use With Cardiovascular Disease Risks: Meta-analysis of 10 Trials Involving 77917 Individuals. JAMA Cardiol, 2018. 3(3): p. 225-234. [PMC free article: PMC5885893] [PubMed: 29387889]
- 220.
- Bhatt, D.L., et al., Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med, 2018. [PubMed: 30415628]
- 221.
- Nicholls, S.J., et al., Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA, 2020. 324(22): p. 2268-2280. [PMC free article: PMC7667577] [PubMed: 33190147]
- 222.
- Kalstad, A.A., et al., Effects of n-3 Fatty Acid Supplements in Elderly Patients After Myocardial Infarction: A Randomized, Controlled Trial. Circulation, 2021. 143(6): p. 528-539. [PubMed: 33191772]
- 223.
- Goff, Z.D. and S.E. Nissen, N-3 polyunsaturated fatty acids for cardiovascular risk. Curr Opin Cardiol, 2022. 37(4): p. 356-363. [PubMed: 35275889]
- 224.
- Mason, R.P., S.C.R. Sherratt, and R.H. Eckel, Omega-3-fatty acids: Do they prevent cardiovascular disease? Best Pract Res Clin Endocrinol Metab, 2023. 37(3): p. 101681. [PubMed: 35739003]
- 225.
- Zambon, A., et al., New and Emerging Therapies for Dyslipidemia. Endocrinol Metab Clin North Am, 2022. 51(3): p. 635-653. [PubMed: 35963633]
- 226.
- Haller, J.F., et al., ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J Lipid Res, 2017. 58(6): p. 1166-1173. [PMC free article: PMC5454515] [PubMed: 28413163]
- 227.
- Rosenson, R.S., et al., Evinacumab in severe hypertriglyceridemia with or without lipoprotein lipase pathway mutations: a phase 2 randomized trial. Nat Med, 2023. 29(3): p. 729-737. [PMC free article: PMC10033404] [PubMed: 36879129]
- 228.
- Chaudhry, R., A. Viljoen, and A.S. Wierzbicki, Pharmacological treatment options for severe hypertriglyceridemia and familial chylomicronemia syndrome. Expert Rev Clin Pharmacol, 2018. 11(6): p. 589-598. [PubMed: 29842811]
- 229.
- Capell, W.H. and R.H. Eckel, Treatment of hypertriglyceridemia. Curr Diab Rep, 2006. 6(3): p. 230-40. [PubMed: 16898578]
- 230.
- Yuan, G., K.Z. Al-Shali, and R.A. Hegele, Hypertriglyceridemia: its etiology, effects and treatment. CMAJ, 2007. 176(8): p. 1113-20. [PMC free article: PMC1839776] [PubMed: 17420495]
- 231.
- Whayne, T.F., Jr. and J.M. Felts, Activation of lipoprotein lipase. Effects of rat serum lipoprotein fractions and heparin. Circ Res, 1970. 27(6): p. 941-51. [PubMed: 5487079]
- 232.
- Weintraub, M., et al., Continuous intravenous heparin administration in humans causes a decrease in serum lipolytic activity and accumulation of chylomicrons in circulation. J Lipid Res, 1994. 35(2): p. 229-38. [PubMed: 8169526]
- 233.
- Whayne, T.F., Jr., Concerns about heparin therapy for hypertriglyceridemia. Arch Intern Med, 2010. 170(1): p. 108-9; author reply 109. [PubMed: 20065209]
- 234.
- Goldberg, I.J., Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res, 1996. 37(4): p. 693-707. [PubMed: 8732771]
- 235.
- Aryal, M.R., et al., Acute pancreatitis owing to very high triglyceride levels treated with insulin and heparin infusion. BMJ Case Rep, 2013. 2013. [PMC free article: PMC3645636] [PubMed: 23608843]
- 236.
- Khan, A.S., S.U. Latif, and M.A. Eloubeidi, Controversies in the etiologies of acute pancreatitis. JOP, 2010. 11(6): p. 545-52. [PubMed: 21068485]
- 237.
- Coskun, A., et al., Treatment of hypertriglyceridemia-induced acute pancreatitis with insulin. Prz Gastroenterol, 2015. 10(1): p. 18-22. [PMC free article: PMC4411402] [PubMed: 25960810]
- 238.
- Mikhail, N., et al., Treatment of severe hypertriglyceridemia in nondiabetic patients with insulin. Am J Emerg Med, 2005. 23(3): p. 415-7. [PubMed: 15915436]
- 239.
- Jabbar, M.A., M.I. Zuhri-Yafi, and J. Larrea, Insulin therapy for a non-diabetic patient with severe hypertriglyceridemia. J Am Coll Nutr, 1998. 17(5): p. 458-61. [PubMed: 9791843]
- 240.
- Thuzar, M., et al., Extreme hypertriglyceridemia managed with insulin. J Clin Lipidol, 2014. 8(6): p. 630-4. [PubMed: 25499946]
- 241.
- Szczepiorkowski, Z.M., et al., Guidelines on the use of therapeutic apheresis in clinical practice--evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher, 2010. 25(3): p. 83-177. [PubMed: 20568098]
- 242.
- Stefanutti, C. and U. Julius, Treatment of primary hypertriglyceridemia states - General approach and the role of extracorporeal methods. Atheroscler Suppl, 2015. 18: p. 85-94. [PubMed: 25936310]
- 243.
- Furuya, T., et al., Plasma exchange for hypertriglyceridemic acute necrotizing pancreatitis: report of two cases. Ther Apher, 2002. 6(6): p. 454-8. [PubMed: 12460410]
- 244.
- Click, B., et al., The role of apheresis in hypertriglyceridemia-induced acute pancreatitis: A systematic review. Pancreatology, 2015. 15(4): p. 313-20. [PMC free article: PMC6609092] [PubMed: 25800175]
- 245.
- Webb, C.B., et al., Effect of TPE vs medical management on patient outcomes in the setting of hypertriglyceridemia-induced acute pancreatitis with severely elevated triglycerides. J Clin Apher, 2021. 36(5): p. 719-726. [PubMed: 34228372]
- 246.
- Koutroumpakis, E., et al., Management and outcomes of acute pancreatitis patients over the last decade: A US tertiary-center experience. Pancreatology, 2017. 17(1): p. 32-40. [PubMed: 28341116]
- 247.
- Huang, C., et al., Clinical features and treatment of hypertriglyceridemia-induced acute pancreatitis during pregnancy: A retrospective study. J Clin Apher, 2016. 31(6): p. 571-578. [PubMed: 26946248]
- 248.
- Wierzbicki, A.S., T.M. Reynolds, and M.A. Crook, Usefulness of Orlistat in the treatment of severe hypertriglyceridemia. Am J Cardiol, 2002. 89(2): p. 229-31. [PubMed: 11792350]
- 249.
- Tolentino, M.C., et al., Combination of gemfibrozil and orlistat for treatment of combined hyperlipidemia with predominant hypertriglyceridemia. Endocr Pract, 2002. 8(3): p. 208-12. [PubMed: 12113634]
- 250.
- Davidson, M., et al., The burden of familial chylomicronemia syndrome: interim results from the IN-FOCUS study. Expert Rev Cardiovasc Ther, 2017. 15(5): p. 415-423. [PubMed: 28338353]
- 251.
- Rouis, M., et al., Therapeutic response to medium-chain triglycerides and omega-3 fatty acids in a patient with the familial chylomicronemia syndrome. Arterioscler Thromb Vasc Biol, 1997. 17(7): p. 1400-6. [PubMed: 9261273]
- 252.
- Brunzell, J.D., Familial Lipoprotein Lipase Deficiency, in GeneReviews at GeneTests: Medical Genetics Information Resource. 2011, University of Washington: Seattle. p. 1997-2010.
- 253.
- Brunzell JD, D.S., Familial lipoprotein lipase deficiency, Apo CII deficiency and hepatic lipase deficiency., in The Metabolic and Molecular Basis of Inherited Disease, 8th edition 2001, McGraw-Hill Book Co.: New York. p. 2789-2816.
- 254.
- Patni, N., C. Quittner, and A. Garg, Orlistat Therapy for Children With Type 1 Hyperlipoproteinemia: A Randomized Clinical Trial. J Clin Endocrinol Metab, 2018. 103(6): p. 2403-2407. [PMC free article: PMC6456945] [PubMed: 29659879]
- 255.
- Blackett, P., et al., Lipoprotein abnormalities in compound heterozygous lipoprotein lipase deficiency after treatment with a low-fat diet and orlistat. J Clin Lipidol, 2013. 7(2): p. 132-9. [PubMed: 23415432]
- 256.
- Tsai, E.C., et al., Potential of essential fatty acid deficiency with extremely low fat diet in lipoprotein lipase deficiency during pregnancy: A case report. BMC Pregnancy Childbirth, 2004. 4(1): p. 27. [PMC free article: PMC544881] [PubMed: 15610556]
- 257.
- Al-Shali, K., et al., Successful pregnancy outcome in a patient with severe chylomicronemia due to compound heterozygosity for mutant lipoprotein lipase. Clin Biochem, 2002. 35(2): p. 125-30. [PubMed: 11983347]
- 258.
- Carpentier, A.C., et al., Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab, 2012. 97(5): p. 1635-44. [PubMed: 22438229]
- 259.
- Sanada, M., et al., Substitution of transdermal estradiol during oral estrogen-progestin therapy in postmenopausal women: effects on hypertriglyceridemia. Menopause, 2004. 11(3): p. 331-6. [PubMed: 15167313]
- 260.
- Hemelaar, M., et al., Oral, more than transdermal, estrogen therapy improves lipids and lipoprotein(a) in postmenopausal women: a randomized, placebo-controlled study. Menopause, 2003. 10(6): p. 550-8. [PubMed: 14627865]
- 261.
- Hsu, S.Y., et al., Laparoscopic bariatric surgery for the treatment of severe hypertriglyceridemia. Asian J Surg, 2015. 38(2): p. 96-101. [PubMed: 25161086]
- 262.
- Gaudet, D., et al., Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N Engl J Med, 2015. 373(5): p. 438-47. [PubMed: 26222559]
- Spectrum of mutations of the LPL gene identified in Italy in patients with severe hypertriglyceridemia.[Atherosclerosis. 2015]Spectrum of mutations of the LPL gene identified in Italy in patients with severe hypertriglyceridemia.Rabacchi C, Pisciotta L, Cefalù AB, Noto D, Fresa R, Tarugi P, Averna M, Bertolini S, Calandra S. Atherosclerosis. 2015 Jul; 241(1):79-86. Epub 2015 May 1.
- Review Clinical Management of Hypertriglyceridemia in the Prevention of Cardiovascular Disease and Pancreatitis.[Curr Atheroscler Rep. 2021]Review Clinical Management of Hypertriglyceridemia in the Prevention of Cardiovascular Disease and Pancreatitis.Hernandez P, Passi N, Modarressi T, Kulkarni V, Soni M, Burke F, Bajaj A, Soffer D. Curr Atheroscler Rep. 2021 Sep 13; 23(11):72. Epub 2021 Sep 13.
- The longitudinal triglyceride phenotype in heterozygotes with LPL pathogenic variants.[J Clin Lipidol. 2023]The longitudinal triglyceride phenotype in heterozygotes with LPL pathogenic variants.Perera SD, Wang J, McIntyre AD, Dron JS, Hegele RA. J Clin Lipidol. 2023 Jan-Feb; 17(1):87-93. Epub 2022 Nov 22.
- Review Management of Hospitalized Children with Severe Hypertriglyceridemia.[Endotext. 2000]Review Management of Hospitalized Children with Severe Hypertriglyceridemia.De La Torre A, Hamilton L, Wilson DP. Endotext. 2000
- Review Genetic Disorders Causing Hypertriglyceridemia in Children and Adolescents.[Endotext. 2000]Review Genetic Disorders Causing Hypertriglyceridemia in Children and Adolescents.Shah AS, Wilson DP. Endotext. 2000
- Hypertriglyceridemia: Pathophysiology, Role of Genetics, Consequences, and Treat...Hypertriglyceridemia: Pathophysiology, Role of Genetics, Consequences, and Treatment - Endotext
- Familial Hypertriglyceridemia - StatPearlsFamilial Hypertriglyceridemia - StatPearls
- MPI [Macaca nemestrina]MPI [Macaca nemestrina]Gene ID:105490968Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...