

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Fibrous dysplasia (FD) is an uncommon mosaic disorder falling along a broad clinical spectrum. It arises from post-zygotic mutations in GNAS, resulting in constitutive activation of the cAMP pathway-associated G-protein, Gsα, and proliferation of undifferentiated skeletal progenitor cells. FD may occur in isolation, or in association with skin pigmentation and hyperfunctioning endocrinopathies, termed McCune-Albright syndrome (MAS). Disease may involve any part or combination of the skeleton, ranging from an isolated, asymptomatic monostotic lesion, to severe polyostotic disease resulting in fractures, deformity, functional impairment, and progressive scoliosis. FD may be diagnosed clinically in patients with polyostotic disease and/or extraskeletal features of MAS; however, biopsy is typically required to diagnose monostotic disease. Management is focused on treating endocrinopathies, preventing fractures, optimizing function, and treating pain. All patients should be evaluated and treated for extraskeletal features of MAS at the time of diagnosis. In particular control of growth hormone excess is important to prevent craniofacial FD expansion, and control of FGF23-mediated hypophosphatemia is important to prevent fracture, deformity, and bone pain. A mainstay of FD treatment is surgical, and practitioners should be aware that techniques and procedures used in other skeletal disorders, such as bone grafting and prophylactic optic nerve decompression, are frequently ineffective in FD. There are currently no medical therapies capable of altering the disease course in FD. Bisphosphonates may be effective in treating FD-related bone pain but are unlikely to impact bone quality or lesion expansion. There is a critical need to develop novel therapies capable of altering the disease activity of FD lesions. Ongoing efforts include developing drugs to target the mutant Gsα, and devising strategies for targeting mutant skeletal progenitor cells. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Fibrous dysplasia of bone (FD) (OMIM#1174800) is an uncommon skeletal disorder resulting in fractures, pain, and functional impairment. Disease is mosaic and may occur in a single bone (monostotic) or multiple bones (polyostotic). FD thus falls along a broad clinical spectrum, ranging from an incidental, asymptomatic lesion to severe disabling disease. Lesions may occur in isolation or in association with extraskeletal features, most commonly café-au-lait skin macules and hyperfunctioning endocrinopathies including precocious puberty, hyperthyroidism, growth hormone excess, hypercortisolism, and fibroblast growth factor-23 (FGF23)-mediated hypophosphatemia. The combination of FD and/or any of these extraskeletal features is termed McCune-Albright syndrome (MAS).
PATHOGENESIS
FD/MAS results from post zygotic somatic activating (gain-of-function) mutations in GNAS, which encodes the cyclic AMP pathway-associated G-protein, Gsα. GNAS mutations in FD/MAS are known to occur at one of two amino acid residues: Arg201 (>95% of reported cases) [1] or Gln227 (<5%) [2]. These mutations disrupt the intrinsic GTPase activity of Gsα, causing constitutive activation of adenylyl cyclase and inappropriate cyclic AMP signaling. Involvement of the skin, bone, and endocrine systems is consistent with a mutational event early in embryogenesis, occurring prior to derivation of the 3 germ layers. The phenotype in individuals with FD/MAS is thus the result of the distribution of tissues containing the GNAS mutation, and the role played by Gsα signaling in those tissues. This mosaicism is reflected in the clinical features of FD/MAS, including the distinctive appearance of café-au-lait macules which follow developmental lines of Blaschko (Fig 1A), the patchy bone disease (Fig 1B) with extreme variability between individuals, and the lack of any reported cases of vertical transmission [3].
The pathogenesis of FD results from replacement of normal bone and bone marrow by fibro-osseous lesions. Histologically lesions are comprised of skeletal progenitor cells, which proliferate in the multipotent state without the expected differentiation into osteoblast, adipocyte, and hematopoietic-supporting cell lines. Skeletal progenitor cell proliferation and abnormal bone formation result in the characteristic histologic features of FD, including marrow fibrosis, abnormally-shaped trabeculae, and abnormal skeletal matrix formation [4] (Fig 2). Also characteristic is skeletal undermineralization with prominent osteoid, due in part to increased production of FGF23 by FD cells [5, 6].
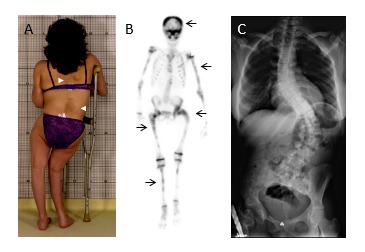
Figure 1.
Clinical and radiographic features of fibrous dysplasia/McCune-Albright syndrome. A. Fibrous dysplasia involving the lower extremities has resulted in a windswept deformity and impaired ambulation. Note the café-au-lait macules (white arrowheads) on the posterior trunk with characteristic jagged borders and location respecting the midline of the body. B. Technetium-99 bone scan shows patchy tracer uptake in affected areas in the skull and limbs (black arrows). C. Radiograph of a patient with axial fibrous dysplasia resulting in severe thoracolumbar scoliosis.
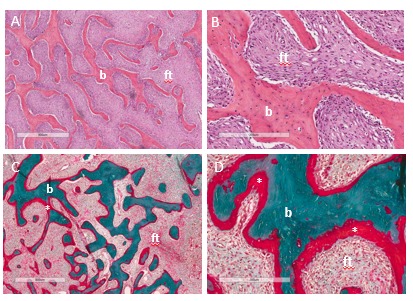
Figure 2.
Histopathologic features of fibrous dysplasia. Hematoxylin-eosin stained sections in low (A) and high power (B) show irregular, discontinuous trabeculae (b) within a fibrous stroma (ft), demonstrating the typical “discontinuous pattern. Goldner’s trichrome stained sections in low (C) and high power (D) reveal osteomalacic changes including excess osteoid (asterisks) and severe undermineralization of the dysplastic bone.
CLINICAL FEATURES
The clinical features in individuals with FD are variable depending upon the location and extent of skeletal lesions. The most commonly involved areas are the proximal femurs and skull base; however, any combination of affected bones is possible. FD in the appendicular skeleton often presents with fractures, limp, and/or pain. Recurrent fractures and FD expansion may lead to deformity with pain and loss of ambulation (Fig 1A). In the craniofacial area FD may present with facial asymmetry or a painless “lump” in the skull. Progressive expansion of craniofacial lesions may rarely lead to functional deficits including vision loss, malocclusion, and obstruction of the nasal and otic canals. Rarely, cranial base deformities may lead to serious neurologic complications [7]. Axial FD commonly leads to scoliosis, which in rare cases may be progressive and potentially fatal [8-10] (Fig 1C).
Radiographic features of FD also vary depending upon location. In the appendicular skeleton lesions appear radiolucent with cortical thinning and a characteristic “ground glass” appearance (Fig 3A). Craniofacial FD may appear sclerotic on radiographs, while computed tomography shows expansile lesions with a homogeneous “ground glass” appearance (Fig 3B). In older individuals craniofacial FD may appear more heterogeneous with focal cystic and sclerotic areas (Fig 3C).
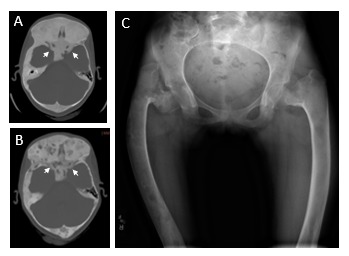
Figure 3.
Radiographic appearance of fibrous dysplasia (FD). A. Extensive FD involvement of the bilateral femurs in an 11-year-old girl demonstrates the characteristic radiographic findings, including the homogeneous “ground glass” appearance, diffuse cortical thinning, and coxa vara (“shepherd’s crook”) deformities (white arrows). B. Computed tomography images of craniofacial FD from a 10-year-old girl demonstrate the typical homogeneous, “ground glass” appearance. White arrows indicate the optic nerves, which are encased in FD. C. Computed tomography images from the same patient at age 19 years demonstrates typical age-related changes, including the development of mixed solid and radiolucent, cystic-appearing lesions. As is typical in FD, this patient has had persistently normal vision despite encasement of the optic nerves.
The natural course of FD is to progress during childhood and adolescence. Patients have grossly normal skeletal development in utero, without obvious features of bone disease at birth. Skeletal lesions appear during the first several years of life and expand during linear growth. The majority of FD lesions are apparent by age 10 years, typically with no new clinically significant lesions occurring after age 15 years [11]. FD lesions typically become less active in adulthood, which is mirrored histologically by age-dependent apoptosis of GNAS mutation-bearing cells in older patients [12], and biochemically by a progressive decline in bone turnover markers over time [13].
Malignant transformation occurs rarely in FD, consistent with the slight increased oncogenic potential associated with GNAS mutations [14]. Malignancies appear to be more common in patients exposed to ionizing radiation, including those receiving pituitary radiation for treatment of growth hormone excess [15, 16]. Clinicians should be alerted to the possibility of malignant transformation in lesions that are rapidly expanding or show signs of cortical disruption on radiographs. Another potential complication of FD is aneurysmal bone cysts, which typically present with acute onset of expansion and localized pain. These lesions may progress rapidly, and frequently require urgent surgical treatment.
EVALUATION AND MANAGEMENT
FD is typically diagnosed based on clinical and radiologic evaluation. All patients with FD should undergo a complete skeletal evaluation at the time of diagnosis to determine the extent of the disease. This is best accomplished with technetium-99 scintigraphy or 18F-NaF PET/CT imaging to identify the areas of involvement, followed by radiographs of affected areas to better visualize lesions anatomically.
The differential diagnosis in patients with FD/MAS is variable depending upon the clinical presentation. Patients with prominent skin findings may be misdiagnosed with neurofibromatosis type 1 (NF-1), which also presents with café-au-lait macules and skeletal abnormalities. The location and distribution of skin lesions in NF-1 includes multiple smooth-bordered macules, which are generally distinct from the jagged bordered lesions in MAS that typically respect the midline of the body. Skeletal findings in NF-1 are less common and include tibial pseudoarthroses and kyphoscoliosis. There are multiple types of fibro-osseous lesions with similar radiographic and histologic features to FD, including ossifying fibromas, osteofibrous dysplasia, and giant cell tumors of bone [17, 18]. These lesions are generally solitary and not associated with extraskeletal features.
The utility of molecular testing for diagnostic purposes should be considered on a case-by-case basis. GNAS mutation detection is highly variable depending upon the level of mosaicism and the sensitivity of the sequencing technique [1, 19]. In patients with monostotic FD and no additional features of MAS, mutation testing may be helpful in distinguishing FD from other fibro-osseous skeletal lesions. In patients with polyostotic disease and/or typical extraskeletal features of MAS, mutation testing is not required to establish the diagnosis, and is unlikely to inform clinical management.
Management in FD is focused on treating endocrinopathies, preventing fractures, optimizing function, and treating pain. There are currently no medical therapies which are capable of altering the disease course. Orthopedic surgery is an important component of management however, practitioners should be aware that techniques used in other skeletal disorders may be unsuccessful in FD. In particular bone grafting, curettage, and external fixation are commonly used techniques that were previously considered standard in FD, but are now known to be frequently ineffective [20]. Diagnosis and treatment of scoliosis is extremely important, as it may be rapidly progressive and lead to fatal respiratory compromise. Surgical fusion has been shown to be effective in treating FD patients with progressive scoliosis [13].
Surgical management of craniofacial FD lesions should similarly be approached cautiously, as complete resection is typically not possible, and partially resected lesions frequently regrow post-operatively [21]. Optic canal involvement is common but only rarely leads to vision loss (Fig 3). Conservative management of optic canal FD has been shown to be superior in asymptomatic patients; prophylactic decompression without objective evidence of optic neuropathy may lead to vision loss and is contraindicated [22]. All patients with craniofacial FD should undergo baseline and yearly ophthalmologic, otolaryngologic, and audiologic evaluations. Patients with FD involving the skull base should also be undergo screening imaging for cranial base deformities around skull age, and should be evaluated regularly for associated neurologic symptoms [7].
Evaluation and treatment for extraskeletal features of MAS is an important component of clinical management in FD. Patients should undergo a staging workup at the time of diagnosis, including complete history and physical examination, review of growth curves, biochemical testing for endocrinopathies, and ultrasonography of the thyroid and testes [23].
Hyperthyroidism may have a deleterious effect on bone density, increasing the risk of skeletal deformities in patients with FD [7, 10]. Hyperthyroidism is treated with anti-thyroidal drugs and thyroidectomy [23]. Peripheral precocious puberty results in increased linear growth and premature epiphyseal maturation, which may compromise final adult height. Symptoms are generally well-controlled with the aromatase inhibitor letrozole in girls, which may be used in combination with a testosterone blocker in boys, as well as leuprolide in children with co-existent central precocious puberty [24-26]. Growth hormone excess has been shown to increase FD expansion, particularly in the craniofacial region, leading to an increased risk of macrocephaly and vision loss [27, 28]. This risk may be partially ameliorated with early diagnosis and treatment [29]. Therapeutic options include somatostatin analogues and pegvisomant, with total hypophysectomy generally reserved for refractory cases [30].
FGF23-mediated hypophosphatemia may have a significant impact on disease severity in FD. Although urinary phosphate wasting is common in FD patients, frank hypophosphatemia occurs less frequently due to a compensatory increase in cleavage of FGF23 into its inactive fragments [31]. The degree of FGF23 overproduction appears to be related to overall disease burden, with frank hypophosphatemia occurring more frequently in individuals with greater amounts of FD. The course of FGF23-mediated hypophosphatemia in any individual may therefore be variable, with low phosphate levels “unmasked” during periods of rapid growth or disease progression. Hypophosphatemia may also potentially normalize as growth and disease activity wanes. In addition to the classic rachitic findings of growth deceleration and long bone deformities, FD patients with uncontrolled hypophosphatemia are also at higher risk for fractures [32], deformities [7, 10] and bone pain [33]. Treatment is similar to other disorders of FGF23 excess, and includes oral phosphorus and calcitriol.
Bone pain is a common feature of FD, and pain management is an important component of clinical care. The pathophysiology of FD-related bone pain is not well-understood [34], and there does not appear to be a clear relationship between pain and overall FD burden [33]. The prevalence and severity of pain is generally greater in adults, however pain is frequently unrecognized and untreated in children [33]. In patients who present with pain, it is important to exclude acute injury, impending fracture, or other causes that may require orthopedic intervention. Patients should also be evaluated and treated for hypophosphatemia, in which pain is frequently the presenting symptom. In the absence of structural or metabolic causes, a reasonable management strategy for bone pain is to use a step-wise approach, starting with conservative measures such as non-steroidal anti-inflammatory medications, rest, and application of heat or cold packs. In pain not responsive to conservative measures, intravenous bisphosphonates such as zoledronic acid or pamidronate may be helpful. In general, these should be administered based on symptoms rather than a set schedule, using the lowest effective dose and interval.
A number of reports have investigated the efficacy of bisphosphonates, medications which increase bone density by inhibiting bone-resorbing osteoclasts, as a potential treatment for FD. Early uncontrolled case series reported subjective improvements in pain and variable effects on the radiographic appearance of FD lesions [35-37]. A placebo-controlled trial of the oral bisphosphonate alendronate showed no effects on pain or FD lesion appearance [38]. Retrospective analyses have demonstrated that bisphosphonates do not appear to prevent progression of FD lesions in children [13], and do not appear to prevent progression of spinal deformity in patients with scoliosis [10]. Concerningly, osteonecrosis of the jaw has been reported in patients with FD, with a prevalence of 5.4% in one series [39]. The role of bisphosphonates in FD management has not been fully elucidated; however, at present there is little evidence to support any effect of bisphosphonates on FD quality or lesion expansion, and the author recommends their use be limited to treatment of bone pain.
FUTURE DIRECTIONS
Current therapeutic options for treatment of FD are inadequate, and there is a critical need to develop medical therapies capable of altering the disease course. Ongoing efforts include developing drugs to target activity of the mutant Gsα [40], and devising strategies for targeting mutant skeletal progenitor cells [41]. Recent evidence suggests a possible role for denosumab, a monoclonal inhibitor of receptor activator of nuclear kappa-B ligand, for treatment of FD. This medication has been approved for treatment of giant cell tumors [42], and showed preliminary positive effects on bone turnover and pain in case reports of FD [43-45]. Of concern are severe, life-threatening metabolic derangements associated with denosumab treatment in one FD patient [44]. Currently there is insufficient evidence to support safety or efficacy of denosumab treatment for FD, and its use should be limited to research protocols.
ACKOWLEDGEMENTS
This research was supported by the Intramural Research Program of the NIH, NIDCR.
REFERENCES
- 1.
- Lumbroso S., Paris F., Sultan C. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study. J Clin Endocrinol Metab. 2004;89(5):2107–13. [PubMed: 15126527]
- 2.
- Idowu B.D., et al. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: the first report of a codon 227 mutation in bone. Histopathology. 2007;50(6):691–704. [PubMed: 17493233]
- 3.
- Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clin Genet. 1986;29(4):321–4. [PubMed: 3720010]
- 4.
- Riminucci M., et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187(2):249–58. [PubMed: 10365102]
- 5.
- Riminucci M., et al. Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL-6 expression. Bone. 2003;33(3):434–42. [PubMed: 13678786]
- 6.
- Riminucci M., et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–92. [PMC free article: PMC182207] [PubMed: 12952917]
- 7.
- Pan K.S., et al. Chiari I Malformation and Basilar Invagination in Fibrous Dysplasia: Prevalence, Mechanisms, and Clinical Implications. J Bone Miner Res. 2018;33(11):1990–1998. [PMC free article: PMC6218312] [PubMed: 29924878]
- 8.
- Leet A.I., et al. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86-A(3):531–7. [PubMed: 14996879]
- 9.
- Mancini F., et al. Scoliosis and spine involvement in fibrous dysplasia of bone. Eur Spine J. 2009;18(2):196–202. [PMC free article: PMC2899336] [PubMed: 19130098]
- 10.
- Berglund J.A., et al. Scoliosis in Fibrous Dysplasia/McCune-Albright Syndrome: Factors Associated with Curve Progression and Effects of Bisphosphonates. J Bone Miner Res. 2018 [PubMed: 29669167]
- 11.
- Hart E.S., et al. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22(9):1468–74. [PubMed: 17501668]
- 12.
- Kuznetsov S.A., et al. Age-dependent demise of GNAS-mutated skeletal stem cells and "normalization" of fibrous dysplasia of bone. J Bone Miner Res. 2008;23(11):1731–40. [PMC free article: PMC2585500] [PubMed: 18597624]
- 13.
- Florenzano P., et al. Age-Related Changes and Effects of Bisphosphonates on Bone Turnover and Disease Progression in Fibrous Dysplasia of Bone. J Bone Miner Res. 2019 [PMC free article: PMC6983318] [PubMed: 30645769]
- 14.
- Ruggieri P., et al. Malignancies in fibrous dysplasia. Cancer. 1994;73(5):1411–24. [PubMed: 8111708]
- 15.
- Hansen M.R., Moffat J.C. Osteosarcoma of the Skull Base after Radiation Therapy in a Patient with McCune-Albright Syndrome: Case Report. Skull Base. 2003;13(2):79–83. [PMC free article: PMC1131834] [PubMed: 15912163]
- 16.
- Liu F., et al. A case of McCune-Albright syndrome associated with pituitary GH adenoma: therapeutic process and autopsy. J Pediatr Endocrinol Metab. 2011;24(5-6):283–7. [PubMed: 21823524]
- 17.
- Kumar K.A., et al. Management and Treatment Outcomes of Maxillofacial Fibro-osseous Lesions: A Retrospective Study. J Maxillofac Oral Surg. 2015;14(3):728–34. [PMC free article: PMC4511885] [PubMed: 26225069]
- 18.
- Steffner R. Benign bone tumors. Cancer Treat Res. 2014;162:31–63. [PubMed: 25070230]
- 19.
- Narumi S., et al. Quantitative and sensitive detection of GNAS mutations causing mccune-albright syndrome with next generation sequencing. PLoS One. 2013;8(3):e60525. [PMC free article: PMC3607597] [PubMed: 23536913]
- 20.
- Stanton RP, I.E., Springfield D, Lindaman L, Wientroub S, Leet A., The surgical management of fibrous dysplasia of bone. Orphanet J Rare Dis, 2012. 24 (7): p. Suppl 1:S1. [PMC free article: PMC3359959] [PubMed: 22640754]
- 21.
- Boyce A.M., et al. Surgical Management of Polyostotic Craniofacial Fibrous Dysplasia: Long-Term Outcomes and Predictors for Postoperative Regrowth. Plast Reconstr Surg. 2016;137(6):1833–9. [PMC free article: PMC5653228] [PubMed: 27219238]
- 22.
- Amit M., et al. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia--a meta-analysis. PLoS One. 2011;6(9):e25179. [PMC free article: PMC3179490] [PubMed: 21966448]
- 23.
- Boyce, A.M. and M.T. Collins, Fibrous Dysplasia/McCune-Albright Syndrome, in GeneReviews(R), R.A. Pagon, et al., Editors. 1993, University of Washington, Seattle: Seattle WA. [PMC free article: PMC274564] [PubMed: 25719192]
- 24.
- Feuillan P., et al. Letrozole treatment of precocious puberty in girls with the McCune-Albright syndrome: a pilot study. J Clin Endocrinol Metab. 2007;92(6):2100–6. [PubMed: 17405850]
- 25.
- Boyce A.M., et al. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab. 2012;97(9):E1782–90. [PMC free article: PMC3431566] [PubMed: 22745241]
- 26.
- Estrada A., et al. Long-term outcomes of letrozole treatment for precocious puberty in girls with McCune-Albright syndrome. Eur J Endocrinol. 2016;175(5):477–483. [PMC free article: PMC5066167] [PubMed: 27562402]
- 27.
- Roszko K.L., Collins M.T., Boyce A.M. Mosaic Effects of Growth Hormone on Fibrous Dysplasia of Bone. N Engl J Med. 2018;379(20):1964–1965. [PubMed: 30428295]
- 28.
- Akintoye S.O., et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab. 2002;87(11):5104–12. [PubMed: 12414879]
- 29.
- Boyce A.M., et al. Optic Neuropathy in McCune-Albright Syndrome: Effects of Early Diagnosis and Treatment of Growth Hormone Excess. J Clin Endocrinol Metab. 2012 [PMC free article: PMC3537097] [PubMed: 23093488]
- 30.
- Salenave S., et al. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014:jc20133826. [PMC free article: PMC4037730] [PubMed: 24517150]
- 31.
- Bhattacharyya N., et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27(5):1132–41. [PMC free article: PMC7448291] [PubMed: 22247037]
- 32.
- Leet A.I., et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res. 2004;19(4):571–7. [PubMed: 15005844]
- 33.
- Kelly M.H., Brillante B., Collins M.T. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19(1):57–63. [PubMed: 17622477]
- 34.
- Chapurlat R.D., et al. Pathophysiology and medical treatment of pain in fibrous dysplasia of bone. Orphanet J Rare Dis. 2012;7 Suppl 1:S3. [PMC free article: PMC3359957] [PubMed: 22640953]
- 35.
- Liens D., Delmas P.D., Meunier P.J. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. Lancet. 1994;343(8903):953–4. [PubMed: 7909013]
- 36.
- Chapurlat R.D., et al. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long-term effectiveness and evaluation of predictors of response to treatment. Bone. 2004;35(1):235–42. [PubMed: 15207763]
- 37.
- Plotkin H., et al. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88(10):4569–75. [PubMed: 14557424]
- 38.
- Boyce A.M., et al. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014:jc20141371. [PMC free article: PMC4223439] [PubMed: 25033066]
- 39.
- Metwally T., et al. Fibrous Dysplasia and Medication-Related Osteonecrosis of the Jaw. J Oral Maxillofac Surg. 2016;74(10):1983–99. [PMC free article: PMC5039058] [PubMed: 27137436]
- 40.
- Bhattacharyya N., et al. A high throughput screening assay system for the identification of small molecule inhibitors of gsp. PLoS One. 2014;9(3):e90766. [PMC free article: PMC3965391] [PubMed: 24667240]
- 41.
- Piersanti S., et al. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. 2010;25(5):1103–16. [PMC free article: PMC5524372] [PubMed: 19874199]
- 42.
- Singh A.S., Chawla N.S., Chawla S.P. Giant-cell tumor of bone: treatment options and role of denosumab. Biologics. 2015;9:69–74. [PMC free article: PMC4507456] [PubMed: 26203221]
- 43.
- Benhamou J., Gensburger D., Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine. 2014;81(6):549–50. [PubMed: 24962974]
- 44.
- Boyce A.M., et al. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012;27(7):1462–70. [PMC free article: PMC3377825] [PubMed: 22431375]
- 45.
- Ganda K., Seibel M.J. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. 2014;25(2):777–82. [PubMed: 24311113]
- Fibrous Dysplasia - EndotextFibrous Dysplasia - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...