ABSTRACT
The main function of the thyroid gland is to make hormones, T4 and T3, which are essential for the regulation of metabolic processes throughout the body. As at any factory, effective production depends on three key components - adequate raw material, efficient machinery, and appropriate controls. Iodine is the critical raw material, because 65% of T4 weight is iodine. Ingested iodine is absorbed and carried in the circulation as iodide. The thyroid actively concentrates the iodide across the basolateral plasma membrane of thyrocytes by the sodium/iodide symporter, NIS. Intracellular iodide is then transported in the lumen of thyroid follicles. Meanwhile, the thyrocyte endoplasmic reticulum synthesizes two key proteins, TPO and Tg. Tg is a 660kDa glycoprotein secreted into the lumen of follicles, whose tyrosyls serve as substrate for iodination and hormone formation. TPO sits at the apical plasma membrane, where it reduces H2O2, elevating the oxidation state of iodide to an iodinating species, and attaches the iodine to tyrosyls in Tg. H2O2 is generated at the apex of the thyrocyte by Duox, a NADPH oxydase. Initial iodination of Tg produces MIT and DIT. Further iodination couples two residues of DIT, both still in peptide linkage, to produce T4, principally at residues 5 in the Tg polypeptide chain. When thyroid hormone is needed, Tg is internalized at the apical pole of thyrocytes, conveyed to endosomes and lysosomes and digested by proteases, particularly the endopeptidases cathepsins B, L, D and exopeptidases. After Tg digestion, T4 and T3 are released into the circulation. Nonhormonal iodine, about 70% of Tg iodine, is retrieved intrathyroidally by DEHAL1, an iodotyrosine deiodinase and made available for recycling within the gland. TSH is the stimulator that affects virtually every stage of thyroid hormone synthesis and release. Early control involves the direct activation of the cellular and enzymatic machineries while delayed and chronic controls are on gene expression of key proteins. Iodine supply, either too much or too little, impairs adequate synthesis. Antithyroid drugs act by interfering with iodide oxidation. Genetic abnormalities in any of the key proteins, particularly NIS, TPO, Duox and Tg, can produce goiter and hypothyroidism. For complete coverage of this and related areas in Endocrinology, please visit our free web-book, www.endotext.org.
INTRODUCTION
The thyroid contains two hormones, L-thyroxine (tetraiodothyronine, T4) and L-triiodothyronine (T3) (Figure 2-1, below). Iodine is an indispensable component of the thyroid hormones, comprising 65% of T4's weight, and 58% of T3's. The thyroid hormones are the only iodine-containing compounds with established physiologic significance in vertebrates.

Fig. 2-1Structural formula of thyroid hormones and precursor compounds
The term "iodine" occasionally causes confusion because it may refer to the iodine atom itself but also to molecular iodine (I2). In this chapter "iodine" refers to the element in general, and "molecular iodine" refers to I2. "Iodide" refers specifically to the ion I-.
Ingested iodine is absorbed through the small intestine and transported in the plasma to the thyroid, where it is concentrated, oxidized, and then incorporated into thyroglobulin (Tg) to form MIT and DIT and later T4 and T3 (Figure 2-2). After a variable period of storage in thyroid follicles, Tg is subjected to proteolysis and the released hormones are secreted into the circulation, where specific binding proteins carry them to target tissues. This chapter discusses these broad steps as: (a) iodine availability and absorption; (b) uptake of iodide by the thyroid; (c) oxidation of iodide, which involves the thyroperoxidase (TPO), H2O2, and H2O2 generation; (d) Tg, whose iodination leads to hormone formation; (e) storage of thyroid hormones in a Tg-bound form; (f) Tg breakdown and hormone release; (g) control of synthesis and secretion by iodine supply and TSH; and (h) effects of drugs and other external agents on the process.

The production of thyroid hormones is based on the organization of thyroid epithelial cells in functional units, the thyroid follicles. A single layer of polarized cells (Fig. 2-4A) forms the enveloppe of a spherical structure with an internal compartment, the follicle lumen. Thyroid hormone synthesis is dependent on the cell polarity that conditions the targeting of specific membrane protein, either on the external side of the follicle (facing the blood capillaries) or on the internal side (at the cell-lumen boundary) and on the tightness of the follicle lumen that allows the gathering of substrates and the storage of products of the reactions.Thyroid hormone secretion relies on the existence of stores of pre-synthetized hormones in the follicle lumen and cell polarity-dependent transport and handling processes leading to the delivery of hormones into the blood stream.
IODINE AVAILABILITY AND TRANSPORT
The daily iodine intake of adult humans varies from less than 10 µg in areas of extreme deficiency to several hundred milligrams for some persons receiving medicinal iodine. Milk, meat, vitamin preparations, medicines, radiocontrast material, and skin antiseptics are important sources (Table 2-1) (1;2). In the United States, the average intake in 1960 was about 100-150 µg/day, then rose to 200-500 µg/day in the following decade. It is currently about 150 µg/day (3). The use of iodate as a bread conditioner in the baking industry greatly increased average iodine consumption; this additive has been replaced more recently by other conditioners that do not contain iodine. Iodophors as sterilizing agents in the milk industry also added much iodine to the food chain, but this source may also be diminishing. In the USA and elsewhere, most consumers are unaware of the amount of iodine they ingest. Commerce and manufacturing technology rather than health dictate the presence of iodine in most products. The amounts of iodine are usually unrevealed, and changes in them unannounced.
In the USA, where iodized salt use is optional, about 70% of the population consumes table salt containing approximately 76 ppm iodine (76 mg I/kg salt). Most prepared food in the USA and Europe uses uniodized salt (Switzerland and Macedonia are exceptions) and only about 15% of the daily salt intake is added at the table, so iodized salt in these areas makes only a modest contribution to daily iodine intake (4). The National Health and Nutrition Examination Surveys (NHANES) showed that the median national urinary iodine excretion in the USA in samples collected between 1988 and 1994 was 145 µg/L, a marked decrease from the 321 µg/L in a similar survey two decades before (5;6). Estimates from the NHANES (2001) are about 160 µg/L. The Total Diet Study of the U.S. Food and Drug Administration reported a parallel decrease in iodine consumption between 1970 and 1990 (7) . These fluctuations in iodine intake result from changes in societal and commercial practices that are largely unrecognized and unregulated. Canada mandates that all salt for human consumption contain KI at 100 ppm (76 ppm as iodine). Calculations of the representative Canadian diet in 1986 estimated slightly over 1 mg iodine/person/day, of which iodized salt contributed over half (8). Urinary iodine excretion in a group of men in Ottawa in 1990 was less than 50% of that in the Canadian national survey of 1975 (9), suggesting a decrease in dietary intake there as well as in the USA. Some countries have areas with very high iodine intake (10), from dietary custom (e.g., seaweeds in Japan) or from iodine-rich soil and water (e.g., a few places in China). But many countries have had some degree of iodine deficiency (11) in at least part of their territory. This has been corrected by the widespread programs of iodine prophylaxis promoted by ICCIDD (12).
Too much iodine increases the incidence of iodine-induced hyperthyroidism, autoimmune thyroid disease and perhaps thyroid cancer. Too little causes goiter, hypothyroidism and their consequences i.e.features of the so-called iodine deficiency disorders (5). The global push to eliminate iodine deficiency in the current decades has put both excess and deficiency of iodine in the spotlight. Some countries have already moved rapidly from severe iodine deficiency to iodine excess, while others are only now recognizing iodine deficiency as a problem (5;12). Their experience, as well as that in the USA and Canada, emphasizes the need for continued monitoring to assess trends in iodine intake.
Medicinal sources can provide iodine in amounts much larger than those consumed in an average diet (Table 2-1). For example, 200 mg of amiodarone contains 75 mg of iodine. Radiographic contrast materials typically contain grams of iodine in covalent linkage, and significant amounts (milligrams) may be liberated in the body. Skin disinfectants (e.g., povidone iodine) and iodine-based water purification systems can greatly augment iodine intake. At the other end, some individuals with little consumption of dairy products and of iodized salt have low iodine intakes.
Table 2-1
Some common sources of iodine in adults USA (1,2).
Most dietary iodine is reduced to iodide before absorption throughout the gut, principally in the small intestine. Absorption is virtually complete. Iodinated amino acids, including T4 and T3, are transported intact across the intestinal wall. Short-chain iodopeptides may also be absorbed without cleavage of peptide bonds (13). Iodinated dyes used in radiography are absorbed intact, but some deiodination occurs later. Except in the postabsorptive state, the concentration of iodide in the plasma is usually less than 10 µg/L. Absorbed iodide has a volume of distribution numerically equal to about 38% of body weight (in kilograms) (14), mostly extracellular, but small amounts are found in red cells and bones.
The thyroid and kidneys remove most iodide from the plasma. The renal clearance of iodide is 30-50 mL plasma/min (14-16) and appears largely independent of the load of iodide or other anions. In certain species, such as the rat, large chloride loads can depress iodide clearance. In humans, renal iodide clearance depends principally on glomerular filtration, without evidence of tubular secretion or of active transport with a transfer maximum (17). Reabsorption is partial, passive, and depressed by an extreme osmotic diuresis. Hypothyroidism may decrease and hyperthyroidism may increase renal iodide clearance, but the changes are not marked (14;18).
On iodine diets of about 150 µg/day, the thyroid clears iodide from 10-25 mL of serum (average, 17 mL) per minute (14). The total effective clearance rate in humans is thus 45-60 mL/min, corresponding to a decrease in plasma iodide of about 12%/hr. Thyroidal iodide clearance may reach over 100 mL/min in iodine deficiency, or as low as 3 or 4 mL/min after chronic iodine ingestion of 500-600 µg/day.
The salivary glands and the stomach also clear iodide and small but detectable amounts appear in sweat and in expired air. Breast milk contains large amounts of iodide, mainly during the first 24 hours after ingestion (19). Its content is directly proportional to dietary iodine. For example, in one part of the USA with community adult iodine intake of about 300 µg daily per person, breast milk contained about 18 µg iodine/dL, while in an area of Germany consuming 15 µg iodine per capita daily, the breast milk iodine concentration was only 1.2 µg/dL (20). Milk is the source of virtually all the newborn's iodine, so milk substitutes need to provide adequate amounts.
UPTAKE OF IODINE BY THE THYROID
Thyroid cells extract and concentrate iodide from plasma (21;22). As shown in Fig. 2-3, shortly after administration, radioiodide is taken up from the blood and accumulates within thyroid follicular cells. About 20% of the iodide perfusing the thyroid is removed at each passage through the gland (23). The normal thyroid maintains a concentration of free iodide 20 to 50 times higher than that of plasma, depending on the amount of available iodine and the activity of the gland (24). This concentration gradient may be more than 100:1 in the hyperactive thyroid of patients with Graves' disease. The thyroid can also concentrate other ions, including bromide, astatide, pertechnetate, rhenate, and chlorate, but not fluoride (25;26).
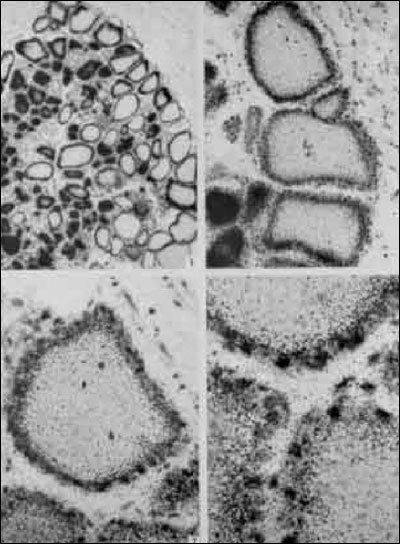
Fig. 2-3
Radioautographs of rat thyroid sections. Animals received iodide shortly before sacrifice, and radioautographs of thyroid sections were coated with emulsion after being stained by the usual methods. The radioautographs indicated the presence of iodide primarily over the cells at these early time intervals. (From Pitt-Rivers, R.J., S.F. Niven, and M.R. Young, in Biochemistry, 90:205, 1964, with permission of the author and publisher.)
The protein responsible for iodide transport, the so-called sodium/iodide symporter or NIS, is located at the basolateral plasma membrane of thyrocytes (Fig. 2-4.). NIS-mediated I- accumulation is a Na+-dependent active transport process that couples the energy released by the inward translocation of Na+ down to its electrochemical gradient to the simultaneous inward translocation of I- against its electrochemical gradient. The maintenance of the Na+ gradient acting as the driving force is insured by Na+-K+-ATPase. NIS belongs to the sodium/glucose cotransport family as the SLC5A5 member. Iodide transport is energy-dependent and requires O2. Ouabain, digitoxin, and other cardiac glycosides block transport in vitro (27;28). Iodide uptake by thyroid cells is dependent on membrane ATPase. During gland hyperplasia, iodide transport usually varies concordantly with plasma membrane Na+-K+-activated, ouabain-sensitive ATPase activity (29).
NIS cDNA was first cloned in rat FRTL-5 cells by Dai et al. (30). The rat NIS gene gives rise to a 3kb transcript with an open reading frame of 1,854 nucleotides encoding a polypeptide chain of 618 amino acids. The mature protein is a glycoprotein with an apparent molecular mass of 85kDa (31;32).It has 13 membrane spanning domains, with the carboxy terminus in the cytoplasm and the amino terminus located outside the cells (33). In the model of Levy et al. (34), a Na+ ion first binds to the transporter which, in the presence of iodide, forms a complex that then transfers iodide and two Na+ ions to the cell interior.
The human NIS gene located on chromosome 19 (35) codes for a protein of 643 amino acids that is 84% homologous with rat NIS (36). The mouse NIS polypeptide chain (37) has the same size (618 amino acids) as the rat NIS. At variance with other species, three different transcripts are generated from the porcine NIS gene by alternative splicing (38); the main form encodes a polypeptide of 643 amino acids as human NIS.
Functional studies clearly show that NIS is responsible for most of the events previously described for iodide concentration by the thyroid. TSH stimulates NIS expression (39;40) and iodide transport (31;32). TSH exerts its regulatory action at the level of transcription through a thyroid-specific far-upstream enhancer denominated NUE (NIS Upstream Enhancer) that contains binding sites for the transcription factor Pax8 and a cAMP response element-like sequence. This original demonstration made on the rat NIS gene (41) has now been extended to human (42) and mouse (43) NIS genes. It has been suggested that TSH could also regulate NIS expression at post-transcriptional level (44). Data from TSH receptor-null mice (44-46) clearly show that TSH is required for expression of NIS. Moderate doses of iodide in the TSH-stimulated dog thyroid inhibit expression of the mRNAs for NIS and TPO, while not affecting that for Tg and TSH receptor (40) . The decrease in thyroid iodide transport resulting from excess iodide administration (escape from the Wolff-Chaikoff effect, see further) is related to a decrease in NIS expression (40;47). Both NIS mRNA and NIS protein are suppressed by TGFb, which also inhibits iodide uptake (48;49). Reviews focus on NIS and its functional importance (50;51).
Several mutations in the NIS gene causing defective iodide transport have been reported in humans (52-59). The most commonly found mutation corresponds to a single base alteration T354P in the ninth putative transmembrane domain of NIS (55). Site directed mutagenesis of rat NIS cDNA to substitute for threonine at residue 354 and transfection into COS cells lead to loss of iodide transport activity (60). Other mutations lead to truncated NIS (58) or to alterations of membrane targeting of the NIS protein (59) . NIS expression is increased in Grave’s disease and hyperactive nodules (61-63) and decreased in adenomas and carcinomas (64;65) appearing as cold nodules at scintigraphy.In hypofuctioning benign or malignant tumors, the impairment of iodide transport would result from both transcriptional and post-transcriptional alterations of NIS expression (66).Other tissues that concentrate iodide also show NIS expression, including salivary glands (67) and mammary glands (68;69).
Iodide supply of follicular lumen involves a two-step transport process: the active transport across the basolateral plasma membrane of thyrocytes by NIS and a passive transport across the apical plasma membrane. The protein(s) insuring the second step is (are) not yet identified. A potential iodide transporter has been proposed: pendrin (70;71). Pendrin, encoded by the PDS gene (72) and composed of 780 amino acids, is expressed in different organs including kidney, inner ear and thyroid. In the thyroid, pendrin is a 110kDA membrane glycoprotein (73), selectively located at the apical plasma membrane (74). Its activity as transporter of anions including iodide has been demonstrated in different experimental systems (71;75-77). Pendrin belongs to the SLC family under the reference SLC26A4. However, the implication of pendrin in thyroid iodide transport remains uncertain for several reasons. First, there is still no direct demonstration of a pendrin-mediated efflux of iodide from thyrocytes to the follicular lumen. Second, the genetic alterations of the PDS gene found in patients with the Pendred syndrome, which lead to a loss of the anion transport activity of pendrin and to a constant and severe hearing loss, only have a moderate impact on the thyroid functioning, generally a euthyroid goiter (78). Third, PDS knock-out mice (79) do not show any thyroid dysfunction. In summary, contrary to NIS for which the anion selectivity (25) corresponds to what was expected, the ion selectivity of thyroid pendrin remains to be elucidated. In the thyroid as in the kidney, pendrin could act primarily as a chloride/bicarbonate anion exchanger. Rather than pendrin, anoctamin-1/TEM 16A, a calcium-activated chloride channel, seems to be responsible for most of the iodide efflux accross the apical membrane of the thyrocytes (80).

Fig. 2-4: NIS-mediated transport of iodide. A, immunolocalization of the human NIS protein at the basolateral plasma membrane of thyrocytes in their typical follicle organization. B, schematic representation of the membrane topology of the NIS polypeptide chain deduced from secondary structure prediction analyses (33). C, transport of iodide from the extracellular fluid (or plasma) to the thyroid follicle lumen. The uptake of iodide at the basolateral plasma membrane of thyrocytes must be active; it operates against an electrical gradient (0 - 50 mV) and a concentration gradient, [ I- ]c being higher than extracellular [ I- ]. The transport of iodide from the cytoplasm to the follicle lumen should be a passive process, the electrical and concentration gradients being favorable.
Iodide that enters the thyroid remains in the free state only briefly before it is further metabolized and bound to tyrosyl residues in Tg. A significant proportion of intrathyroidal iodide is free for about 10-20 minutes after administration of a radioactive tracer (81), but in the steady state, iodide contributes less than 1% of the thyroid total iodine. A major fraction of the intrathyroidal free iodide pool comes from deiodination of MIT and DIT; this iodide is either recycled within the thyroid or leaked into the circulation. Some data suggest that iodide entering the gland by active transport segregates from that generated by deiodination of Tg within the gland (82;83). Once in the thyroid, iodide is organically bound at a rate of 50 to 100% of the pool each minute (24;84). The proportion of an iodide load that is bound varies little, despite wide shifts in daily intake. In contrast, NIS activity is sensitive to both iodine availability and TSH stimulation, and transport rather than intrathyroidal binding is the controlling factor in making iodide available for hormonogenesis.
IODINE IN OTHER TISSUES
The thyroid is not the only organ to concentrate iodine; the others endowed with this capacity are salivary glands, gastric mucosa, mammary glands, and choroid plexus. Ductal cells of the salivary glands express NIS (67) . The plasma membrane of the mammary gland epithelium contains a NIS protein with a molecular mass different from that of thyroid NIS (~75 kDa vs ~90 kDa). In the mammary gland, NIS is processed differently after translation and subjected to regulation by lactogenic stimuli (68). It has been reported that over 80% of human breast cancer samples express this symporter. As it is absent in normal non-lactating tissue, NIS may represent a marker for breast malignancy and even a possible target for radioiodine therapy (69). The thyroid, salivary glands, and gastric mucosa share a common embryologic derivation from the primitive alimentary tract and, in each of these tissues; iodide transport is inhibited by thiocyanate, perchlorate, and cardiac glycosides. TSH stimulates transport only in the thyroid. An active transport for iodide in the gastric mucosa has an obvious value because it provides iodine to the circulation for use in the thyroid. Active concentration by the breast helps transfer iodide to milk. Iodide concentration by the choroid plexus and salivary glands does not have any obvious physiologic benefit, but needs to be remembered for possible insights into pathways as yet undiscovered.
Iodine, particularly in the form of I2, may enter additional metabolic pathways outside the thyroid. Rats administered I2 orally showed much less circulating free iodide and much more iodine bound to proteins and lipids than did animals given iodide (85). In another comparison of I2 versus iodide, administration of iodide to iodine-deficient rats eliminated thyroid hyperplasia much more efficiently than did I2. Additionally, I2 decreased lobular hyperplasia and periductal fibrosis in the mammary glands, while iodide increased the former and had no effect on the latter (86).
THYROPEROXIDASE (TPO)
After concentrating iodide, the thyroid rapidly oxidizes it and binds it to tyrosyl residues in Tg, followed by coupling of iodotyrosines to form T4 and T3. The process requires the presence of iodide, a peroxidase (TPO), a supply of H2O2, and an iodine acceptor protein (Tg).
Thyroperoxidase oxidizes iodide in the presence of H2O2. In crude thyroid homogenates, enzyme activity is associated to cell membranes. It can be solubilized using detergents such as deoxycholate or digitonin. The enzyme activity is dependent on the association with a heme, the ferriprotoporphyrin IX or a closely related porphyrin (87;88). Chemical removal of the prosthetic group inactivates the enzyme, and recombination with the heme protein restores activity (89). The apoprotein from human thyroid is not always fully saturated with its prosthetic group (90). Some congenitally goitrous children have poor peroxidase function because the apoprotein has weak binding for the heme group (90).
Antibodies directed against the thyroid "microsomal antigen," which are present in the serum of patients with autoimmune thyroid disease (AITD), led to identification of TPO. These antibodies were found to react with proteins of 101-107 kDa and to immunoprecipitate thyroid peroxidase (TPO), thus identifying microsomal antigen as TPO (91-95). A monoclonal antibody to purified microsomal antigen or antibodies directed againt thyroperoxidase were then used to clone human TPO (96-98). Different laboratories then cloned TPO from various species: pig (99), rat (100), and mouse (101). Kimura et al. (96) cloned two different cDNAs of humanTPO.TPO1 coded for a protein of 933 residues and TPO2 was identical to TPO1 except that it lacked exon 10 and was composed of 876 residues. Both forms occur in normal and abnormal human thyroid tissue. The C-terminal portion of the proteins exhibits a hydrophobic segment (residues 847-871), likely corresponding to a transmembrane domain; thus, TPO has a short intracellular domain and most of the polypeptide chain is extracellular (Fig. 2-5A). TPO1 is active, but TPO2 appears enzymatically inactive because it does not bind heme, degrades rapidly, and fails to reach the cell surface in transfected cell lines (102). Different degradative pathways exist for the two forms (103). Several other TPO variants resulting from exon skipping have been identified; they appear either active or inactive (104). Pig TPO contains 926 amino acids (99) ; mannose-rich oligosaccharide units occupy four of its five glycosylation sites (105).
Human TPO, which has 46% nucleotide and 44% amino acid sequence homology with human myeloperoxidase, clearly belongs to the same protein family. The TPO gene resides on chromosome 2p13, spans over 150 kbp, and has 17 exons (106). As NIS, Tg, and the TSH receptor (TSHr), TPO expression is controlled by the TSH cAMP pathway (107) through thyroid-specific transcription factors. These include TTF-1/NKx2.1, TTF-2/FOXE1, and Pax-8 (108;109). Tg and TPO genes have the same binding sites for TTF-1/NKx2.1, TTF-2/FOXE1, and Pax-8 in their promoters, and the genes for both have TTF-1/NKx2.1 sites in enhancer regions.
Inactivating mutations in the TPO gene are responsible for a subtype of congenital hypothyroidism characterized by thyroid dyshormonogenesis due to iodide organification defect. More than 60 annotated mutations have been reported; most of them result in total iodide organification defect with severe and permanent hypothyroidism (110;111).
TPO synthesized on polysomes is inserted in the membrane of the endoplasmic reticulum and undergoes core glycosylation. TPO is then transported to the Golgi where it is subjected to terminal glycosylation and packaged into transport vesicles along with Tg (112) (Fig. 2-6). These vesicles fuse with the apical plasma membrane in a process stimulated by TSH. TPO delivered at the apical pole of thyrocytes exposes its catalytic site with the attached heme in the thyroid follicular lumen (113). TPO activity is restricted to the apical membrane, but most of the thyroid TPO is intracellular, being located in the perinuclear part of the endoplasmic reticulum (114;115). Most of this intracellular protein is incompletely or improperly folded; it contains only high mannose-type carbohydrate units, while the membrane TPO has complex carbohydrate units. Glycosylation is essential for enzymatic activity (115). Chronic TSH stimulation increases the amount of TPO and its targetting at the apical membrane (116).
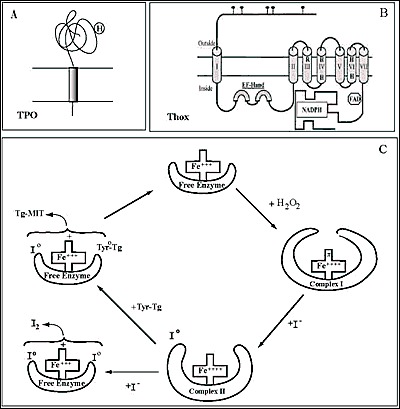
Fig. 2-5
Schematic representation of the membrane topology of Thyroperoxidase, TPO (A) and NADPH thyroid oxidase, ThOX (Duox) (B) at the apical plasma membrane of thyrocytes. C, hypothetical reaction scheme for TPO. H2O2 is presumed to oxidize the free enzyme with a loss of two electrons leading to the formation of complex I. Iodide binds to complex I, is oxidized and form complex II, which then reacts with a tyrosyl residue of Tg, Tyr-Tg.The newly-formed I0 and Tyr0-Tg free radicals interact to form MIT-Tg and the enzyme returns to its free state. I2 may be generated from two cxidized iodine atoms
H 2 O 2 GENERATING SYSTEM
By definition, a peroxidase requires H2O2 for its oxidative function. A large body of older work (reviewed in (117)) investigated possible sources using various in vitro models (117-120). It was already suggested in 1971 that H2O2 would be produced at the apical plasma membrane of the thyrocyte by an enzyme that requires calcium and NADPH originating from the stimulation of the pentose phosphate pathway (121). Further biochemical studies showed that the enzymatic complex producing H2O2 for TPO is a membrane-bound NADPH-dependent flavoprotein (122-126). H2O2 produced by this NADPH-dependent protein is the limiting step of protein iodination and therefore of thyroid hormone synthesis when iodide supply is sufficient (127-129). In human thyroid, the H2O2 production and iodination process are stimulated by the calcium-phosphatidylinositol pathway (129). The quantity of H2O2 produced is important especially in stimulated thyrocytes; it is comparable to the ROS production of activated leukocytes. While the activated leukocyte lives a few hours, the life of an adult thyrocyte is 7 yr (130;131). Thus thyroid cells may be exposed to high doses of H2O2 and have to adapt to it by developing highly regulated generator and efficient protective systems.
More than twenty years passed between the initial biochemical studies and the cloning of Duox as the catalytic enzymatic core of the H2O2 thyroid generating system. By two independent molecular strategies Duox enzymes were uncovered from the thyroid. Starting from a purified fraction of pig thyroid membrane bound NADPH flavoprotein, the team of C. Dupuy isolated p138 Tox which turned out to be Duox2 lacking the first 338 residues (132). Simultaneously, De Deken et al cloned two cDNAs encoding NADPH oxidases using the strategy based on the functional similarities between H2O2 generation in the leukocytes and the thyroid according to the hypothesis that one of the components of the thyroid system would belong to the known gp91phox gene family and display sequence similarities with gp91phox, now called NOX2. Screenings of two cDNA libraries at low stringency with a NOX2 probe enabled the isolation of two sequences coding for two NADPH oxidases of 1551 and 1548 amino acids respectively initially named Thox1 and Thox2 (133). The encoded polypeptides display 83% sequence similarity and are clearly related to gp91phox (53 and 47% similarity over 569 amino acids of the C-terminal end) . The whole protein is composed of : a N-terminus ecto-sequence of 500 amino acids showing a similarity of 43% with thyroperoxidase (hence named Dual oxidase-Duox in the present terminology); a first transmembrane segment preceding a large cytosolic domain which contains two calcium binding EF-hand motifs; the C-terminal portion componed of six transmembrane segments, harbouring the four His and two Arg characteristic of the Nox family protein heme binding site and the conserved FAD- and NADPH-binding sites at the extreme C-terminal cytolic portion (Fig. 2-5B). Duox proteins are localized like TPO at the apical plasma membrane of the thyrocyte as fully glycosylated forms (~190kDa) and in the endoplasmic reticulum as high mannose glycosylated forms (~180kDa) (Fig. 2-6C).
Duox1 and Duox2 genes are co-localized on chromosome 15q15.3, span 75kb, have opposite transcriptional orientations and are separated by a ~16kb region (Fig. 2-6A). Duox1 gene is more telomeric, spans 36 kb and is composed of 35 exons; two first of them are non-coding. Duox2 spans 21.5 kb and is composed of 34 exons; the first being non-coding (134).
In addition to thyroid, Duox expression is reported in several tissues: Duox1 is expressed in lung epithelia, in oocytes (135-137) and Duox2 in gastrointestinal mucosa and salivary glands (138;139). Multiple functions are attributed to Duox enzymes: airway fuid acidification (140), mucin secretion (141), wound healing (142;143) and innate hoste defense (144-147) .
Most of the time Duox activity is associated to a peculiar peroxidase activity like in oocyte with the ovoperoxidase involved in the fertilization process or with the lactoperoxidase in lung epithelia or in the gut (144;145;148;149) . Beside these killing mechanisms, Duox and H2O2 are certainly also involved in the interaction between host mucosa and bacteria to maintain mucosal homeostasis e.g. in bronchi and intestine (146;150). In the thyroid, the specificity of the thyroid hormone machinery using Duox lays on TPO. Thus colocalization of Duox and TPO and their probable association at the apex of the thyrocyte would increase the efficiency of H2O2 producer-consumer system (151-153).
Onset of Duox expression study in thyroid embryonic development pointed Duox as a thyroid differentiation marker. The proteins involved in the synthesis of thyroid hormones are expressed just after the thyroid precursor cells have completed their migration from the primitive pharynx and reached their final location around the trachea (154;155). This final morphological maturation begins in mouse with the expression of Tg at embryonic day 14 followed one day later (E15) by the expression of TPO, NIS, TSH receptor and Duox concomitant with the apparition of iodinared Tg (46;156).
Until 2006, the major obstacle for molecular studies of Duox was the lack of a suitable heterologous cell system for Duox correctly expressed at the plasma membrane in its active state. Several cell lines transfected with Duox1 and/or Duox2 showed Duox expression completely retained in the endoplasmic reticulum in their immature form without displaying any production of H2O2 (157). HEK293 cells transfected with Duox2 generate rather small quantities of superoxide anions in a calcium-depnedent manner (158). The reconstitution of a Duox-based functional H2O2 generating system requires a maturation factor called DuoxA. The two human DuoxA paralogs were initially identified as thyroid specific expressed genes by in silico screenings of multiple parallel signature sequencing data bases (159). The two genes are located on chromosome 15 in the Duox1/Duox2 intergenic region in a tail to tail orientation, DuoxA1 facing Duox1 and DuoxA2 facing Duox2 (Fig. 2-6A.). DuoxA2 ORF spans 6 exons and encodes a 320 amino acid protein predicted to compose five transmembrane segments, a large external loop presenting N-glycosylation sites between the second and third transmembrane helices and a C-terminal cytoplamic region (Fig. 2-6B). DuoxA1 gene was initially annotated “homolog of Drosophila Numb-interacting protein: NIP” (160). Four alternatively spliced DuoxA1 variants have been identified (161). One of the most expressed transcript, DuoxA1α, is the closest homolog of DuoxA2 and encodes a 343 amino acid protein (58% identity of sequence with DuoxA2) adopting the same predicted structure.
In heterologous systems DuoxA proteins in the absence of Duox are mainly retained in the endoplasmic reticulum. When co-transfected with Duox they cotransported with Duox to the plasma membrane where they probably form complexes. Only the Duox1/DuoxA1 and Duox2/DuoxA2 pairs produce the highest levels of H2O2 as they undergo the glycosylation steps through the Golgi. Duox2/DuoxA1 pair does not produce H2O2 but rather superoxide anions and Duox1/DuoxA2 is unable to produce any ROS. In addition it has been shown that the type of Duox-dependent ROS poduction is dictated by defined sequences in DuoxA (162). This means that the Duox activators promote Duox maturation but also are parts of the H2O2 generating complex (163;164). Mice deficient in DuoxA maturation factors present a maturation defect of Duox, lacking the N-glycan processing, and a loss of H2O2 production. These mice develop severe goitrous congenital hypothyroidism with undetectable serum T4 and high serum TSH levels (165).
The reconstitution of this functional H2O2 producing system has been useful to measure and compare the intrinsic enzymatic activities of Duox1 and Duox2 in relationship with their expression at the plasma membrane under stimulation of the major signalling pathways active in the thyroid. It has been shown that the basal activity of both isoenzymes is totally depending on calcium and functional EF-hands calcium binding motifs. However, the two oxidase enzymatic activities are differently regulated after activation of the two main signalling cascades in the thyroid. Duox1 but not Duox2 activity is stimulated by the cAMP dependent cascade triggered by forskolin (EC50=0.1µM) via protein kinase A-mediated phosphorylation on serine 955 of Duox1. In contrast, phorbol esters, at low concentrations, induce Duox2 phosphorylation via protein kinase C activation associated with high H2O2 generation (EC50= 0.8nM) (166). These results suggest that both Duox proteins could be involved in thyroid hormone synthesis by feeding H2O2 to TPO to oxidize iodide and couple iodotyrosines.
From in vitro and in vivo data it has been concluded that Duox-DuoxA constitutes the major if not the unique component of the hormonogenic thyroid H2O2 generating system. The bidirectional promoter allows the coexpression of Duox and DuoxA in the same tissue but the mechanisms regulating their transcription are not well and definitely characterized (167;168). It has been recently shown that Th2 cytokines, IL4 and IL13, up-regulate Duox2 and DuoxA2 genes in human thyrocytes through an activation of Jak-Stat pathway opening new perspectives for a better understanding of the eventual role of Duox in autoimmune diseases (169).
Defects in Duox and/or DuoxA were rapidly recognized possible causes of congenital hypothyroidism (CH) due to thyroid dyshormonogenesis in patients born with a hyperplastic thyroid or developing a goiter postnatally when T4 treatment is delayed after birth.
The first screening of mutations in Duox genes in 2002 was performed on 9 patients who had idiopathic congenital hypothyroidism with positive ClO4- discharge (>10%), one with permanent and 8 with transient hypothyroidism (170). They were identified in the Netherlands by neonatal screening and followed up to determine the evolution of CH with the time. One of the patients with total organification defect (TIOD) presented a permanent hypothyroidism and the 8 others presented a transient hypothyroidism with a partial organification defect (PIOD). Of these last 8 patients 3 harboured heterozygous nonsense or frameshift mutations (Q686X, R701X, S965fsX994) meaning that a single defective Duox2 allele can cause haploinsufficency resulting in mild transient CH. It is noteworthy that this hypothyroid status was limited to the neonatal period, when thyroid hormone requirement is the highest, and was not detectable in adulthood since adult heterozygotes in these families presented normal TSH serum levels. The only case with severe permanent CH was homozygous for a nonsense mutation (R434X= protein devoid of the catalytic core) leading to the conclusion at this time of a complete inability to synthesize thyroid hormone in absence of Duox2. No mutation was detected in Duox1.
With the increasing number of reported Duox2 mutations in CH, it becomes more and more difficult to make the correlation between genotype and phenotype as initially described.
Indeed, subsequent studies have shown a link between biallelic Duox2 defects and PIOD. Patients with compound heterozygous missense (R376W) and a nonsense mutation (R842X), leading to a presumed non functional protein showed PIOD with mild and persistent hyperthyrotropinemia. This suggests that Duox1 can compensate at least partially for the defect in Duox2 (171). Varela et al.. described also two cases of permanent CH with compound heterozygous missense and nonsense or splicing mutations (Q36H and S965fsX994; G418fsX482 and g.IVS19-2A>C conducting to inactive proteins) responsible for congenital goiter with a PIOD (172).
The phenotype-genotype correlation suggested by the work of Moreno et al. is no longer clear. Maruo et al. described a series of transient CH characterized by biallelic defects in Duox2: in one family, four siblings were compound heterozygous for early frameshift mutations (L479SfsX2 and K628RfsX10) resulting in a presumed complete loss of Duox2 activity (not tested at this time). Three of them had low free T4 at birth, mild thyroid enlargement. The thyroid hormone replacement therapy ceased to be necessary by 9yr of age (173). A French-Canadian patient with a transient CH initially detected by neonatal screening presented a compound heterozygozity for a hemizygous missense mutation (G1518S) inherited from the father and a deletion removing the part of the gene coding for the catalytic core of Duox2 inherited from the mother. In vitro test proved that the missense mutant protein was totally inactive (174). This case and others reported later provide further evidence that permanent or transient nature of CH is not directly related to the number of inactivated Duox2 alleles (175-177).
The first homozygous nonsense mutation in DuoxA2 (Y246X) that resulted in a non-functional protein tested in vitro has been found to be responsible of a permanent mild CH in a Chinese patient with a dyshormonogenic goiter (164;178). The mild phenotype can be explained by a partial maintenance of H2O2 production by Duox2/DuoxA1 as demonstrated in vitro. A high level of functional redundancy in Duox/DuoxA system could also explained the mild transient hypothyroidism in a patient with a novel biallelic DuoxA2 mutation and one allele of Duox2 and DuoxA1(179).
The variety of observerd phenotypes associated with Duox2 and now DuoxA2 mutations suggest that the manifestation of Duox2 defects could likely be influenced by the environmental factors like iodine intake or by the activation of Duox1 or DuoxA1 in peculiar circumstances.

Fig 2-6: A, Localization of Duox and DuoxA genes on chromosomes 15q15.3. B, Schematic representation of the predicted structure of DuoxA (from (156). C, Immmunolocalization of human Duox and TPO at the apical membrane of the thyrocyte (upper: Duox immunostaining, middle:preimmune serum, lower:TPO immunostaining) (130).
THYROGLOBULIN (Tg)
Thyroglobulin is the most abundant protein in the thyroid gland; its concentration within the follicular lumen can reach 200-300 g/L. Its main function is to provide the polypeptide backbone for synthesis and storage of thyroid hormones (180). It also offers a convenient depot for iodine storage and retrieval when external iodine availability is scarce or uneven. Neosynthesised Tg polypeptide chains entering the lumen of the rough endoplasmic reticulum (RER) are subjected to core glycosylation, dimerise and are transferred to the Golgi where they undergo terminal glycosylation (Fig. 2-7). Iodination and hormone formation of Tg occur at the apical plasma membrane-lumen boundary and the mature hormone-containing molecules are stored in the follicular lumen, where they make up the bulk of the thyroid follicle colloid content.
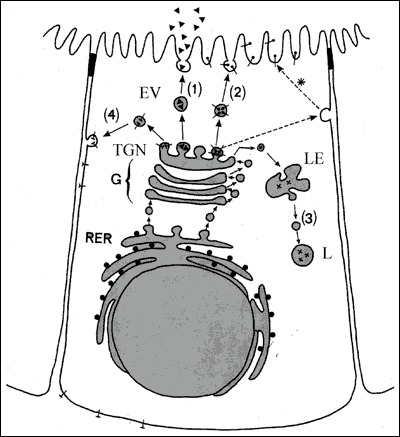
Fig. 2-7
A polarized thyroid epithelial cell synthesizing soluble proteins, Tg (▲) and lysosomal enzymes (X) and membrane proteins, NIS (┴) and TPO (°). The polypeptide chain(s) generated by RER membrane-bound polysomes, enter the lumen of RER for the former and remain inserted into the RER membrane for the latter. Inside the lumen of RER, newly-synthesized proteins undergo core glycosylation and by interacting with chaperones acquire their conformation. Proteins are then transported to the Golgi apparatus (G), where terminal glycosylation and other post-translational reactions take place. In the Trans-Golgi network (TGN), mature proteins undergo sorting processes and are packed into transport vesicles. The vesicles carrying soluble proteins (inside the vesicle) and membrane proteins (as integral vesicle membrane protein) deliver them at the appropriate plasma membrane domain: the apical domain (1) and (2) or the basolateral domain (4). Vesicles carrying lysosomal enzymes (3) conveyed their content to prelysosomes or late endosomes (LE) and lysosomes (L). Apical plasma membrane proteins may reach their final destination by an alternative route involving a transient transfer to and then a retrieval and transport (*) from the basolateral membrane domain to the apical domain.
The Tg peptide chain derives from a gene of more than 200 kbp located on chromosome 8 in humans. The human Tg gene consisting of 48 exons (181) gives rise to a 8.5kb transcript that translates a 2,749 residue peptide (in addition to a 19-residue signal peptide) (182;183). The primary structure deduced from cDNA is also known for bovine, rat, and mouse (184-186). The biochemical traits of human Tg have been reviewed in (187). The N-terminal part of Tg has regions of highly conserved internal homology (10 motives of about 60 amino acids) which appears in several other proteins and are referred to as ‘thyroglobulin type-1 domains’. Such domains have been found to be potent inhibitors of cysteine proteases (188). This finding might be of importance, because these proteases are active in Tg proteolysis (see below). It has been suggested that this region of the Tg molecule may modulate its own degradation and hormone release (189). In the Tg-type 1 repeats, cysteine and proline residues are found in constant position; they may have an important role in the tridimensional structure of the protein. The proximal region of the C-terminal half portion of Tg contains five repeats of another type of cysteine-rich motives. The presence of a high number of cysteine residues in Tg, involved for most of them in disulfide bonds, probably gives rise to peculiar structural constraints. The C-terminal portion of Tg is homologous with acetylcholinesterases (190). Because binding to cell membranes is one feature of acetylcholinesterases, perhaps Tg C-terminus has a similar role. It was reported that the acetylcholinesterase-homology region of Tg could function as a dimerization domain (178;191-193).Furthermore, three highly conserved thioredoxin boxes have been identified in mammalian Tg between residues 1,440 and 1,474; these boxes might be involved in disulfide bond formation leading to intermolecular cross-linking of Tg molecules inside the follicle lumen (194) . Tg gene expression is controlled by the same main thyroid-specific transcription factors that regulate synthesis of TPO (108):TTF-1/NKx2.1, TTF-2/FOXE1, and Pax-8 that bind at the same sites in Tg as they do in TPO. Hydrogen peroxide might be a regulatory factor of Tg expression, based on experimental work showing increased Tg promoter activity with reduced Pax-8 and TTF-1 (195-198). If substantiated, this proposal offers another point of integration between H2O2 generation and transcription of NIS, Tg and TPO genes, all of which being regulated by TSH.
Maturation of the Tg polypeptide chain begins while still on the RER. It undergoes core glycosylation and then monomers fold into stable dimers. Arvan and co-workers (199-204) have mapped this process and emphasize the role of molecular chaperones. The latter are essential for folding the new Tg molecules, and those that are folded improperly are not allowed to proceed further. The principal molecular chaperones are BiP, GRP 94, ERP 72, and calnexin. Only Tg molecules that pass this quality control system unscathed can proceed towards the secretory pathway. Glycosylation is a key event in Tg maturation. Carbohydrates comprise about 10% of Tg weight (205). Human Tg may contain four different types of carbohydrate units. The "polymannose" units consist only of mannose and N-acetylglucosamine. The "complex unit" has a core of three mannose residues with several chains of N-acetylglucosamine, galactose, and fucose or sialic acid extending from them. Both these types of unit are common in glycoproteins and are linked to peptide through an asparagine-N-acetylglucosamine bond. About three quarters of the potential N-glycosylation sites in human Tg are occupied, mostly with the complex unit (206). Two additional units have been found in human Tg; one contains galactosamine and is linked to the hydroxyl group of serine, the other is a chondroitin sulfate unit containing galactosamine and glucuronic acid (207) .
Failure in Tg folding can lead to disease as in the cog/cog mouse; these animals have a large thyroid with a distended ER and sparse Tg storage in follicles (208). Their Tg shows abnormal folding and decreased export from the ER in association with increased levels of several molecular chaperones. In the Tg cDNA of cog/cog mouse, Kim et al.(185) identified a single base substitution that changes leucine to proline at position 2,263. Correction of this defect by site-directed mutagenesis returned Tg export to normal in transfected cells. The cog/cog mouse is an example of endoplasmic reticulum storage disease (209). Other examples are cystic fibrosis, osteogenesis imperfecta, familial neurohypophyseal diabetes insipidus, insulin receptor defect, growth hormone receptor defect, and a variety of lipid disorders (210). In each situation, the underlying defect appears to be a mutation in the coding sequence of exportable proteins. The ER retains the abnormal proteins, which cannot then proceed for further maturation. Several reports describe a similar pathogenesis for cases of congenital goiter and hypothyroidism in humans, although these are not as well characterized. Ohyama et al. (211) investigated a five-year-old euthyroid goitrous boy with high thyroidal radioiodine uptake, a positive perchlorate discharge test, apparently normal H2O2 generation and peroxidase activity in gland tissue, and low amounts of Tg in thyroid tissue overall, but large amounts in the RER. In another report, two hypothyroid goitrous sibs had a 138 bp segment missing between positions 5,590-5,727 in hTg mRNA, translating into a Tg polypeptide chain that lacked 46 residues (212). A third example described four subjects with congenital hypothyroid goiter from two unrelated families (213). Their thyroid tissue showed accumulation of Tg intracellularly with distension of the ER and large increases in activity of specific molecular chaperones, but with failure of Tg to reach the Golgi or the follicular lumen; this case was put forward as an ER storage disease similar to the cog/cog mouse (213).
Tg also contains sulphur and phosphorus. The former is present in the chondroitin sulfate and the complex carbohydrate units, although its form and role are not known (214). Several studies have reported phosphate in Tg, up to 12 mol. per mol Tg. Of this, about half is in the complex carbohydrate units, the remainder is present as phosphoserine and phosphotyrosine (215-217). This may relate to protein kinase A activity (218).
THYROGLOBULIN IODINATION AND HORMONE SYNTHESIS
The step preliminary to thyroid hormone formation is the attachment of iodine to tyrosyl residues in Tg to produce MIT and DIT. This process occurs at the apical plasma membrane-follicle lumen boundary and involves H2O2, iodide, TPO, and glycosylated Tg. All rendezvous at the apical membrane to achieve Tg iodination (Fig. 2-8).
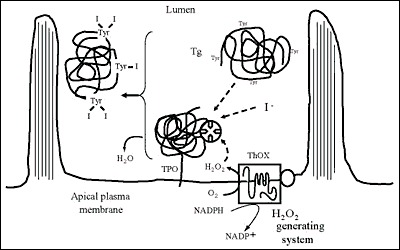
Fig. 2-8
Iodination of Tg at the apical plasma membrane-follicle lumen boundary.The scheme does not account for the relative size of the intervening molecules
First, iodide must be oxidized to an iodinating form. An extensive literature has sought to identify the iodinating species, but the issue is still not resolved (see (219) for a detailed review). One scheme proposes that oxidation produces free radicals of iodine and tyrosine, while both are bound to TPO to form MIT which then separates from the enzyme (Fig.2-5C). Further reaction between free radicals of iodine and MIT gives DIT. Experimental studies by Taurog (219) and others suggest that the TPO reduction occurs directly in a two electron reaction. A second proposal, based on studies of rapid spectral absorption changes (88;220;221), is that TPO-I+ is the iodination intermediate and that the preferred route is oxidation of TPO by H2O2 followed by two electron oxidation of I- to I+, which then reacts within a tyrosine. As a third possibility, Taurog (219) proposed a reaction between oxidized TPO and I- to produce hypoiodite (OI-), which also involves a two electron reaction. Whatever the precise nature of the iodinating species, it is clear that iodide is oxidized by H2O2 and TPO, and transferred to the tyrosyl groups of Tg. All tyrosine residues of Tg are not equally accessible to iodination. The molecule has about 132 tyrosyl residues among its two identical chains; at most, only about 1/3 of the tyrosyls are iodinated. As isolated from the thyroid, Tg rarely contains more than 1% iodine or about 52 iodine atoms.
The final step in hormone synthesis is the coupling of two neighbouring iodotyrosyl residues to form iodothyronine (Fig. 2-9). Two DIT form T4; one DIT and one MIT form T3. Coupling takes place while both acceptor and donor iodotyrosyl are in peptide linkage within the Tg molecule.The reaction is catalyzed by TPO, requires H2O2 (222-225) and is stringently dependent on Tg structure (226).The generation of the iodothyronine residue involves the formation of an ether bond between the iodophenol part of a donor tyrosyl and the hydroxyl group of the acceptor tyrosyl (Fig 2-10). After the cleavage reaction that gives the iodophenol, the alanine side chain of the donor tyrosyl remains in the Tg polypeptide chain as dehydroalanine (227-229). Observations both in vivo and in vitro show an appreciable delay in coupling after initial formation of iodotyrosines. A typical distribution for a Tg containing 0.5% iodine (a normal amount for iodine-sufficient individuals) is 5 residues MIT, 5 of DIT, 2.5 of T4 and 0.7 of T3 (180). More iodine increases the ratios of DIT/MIT and T4/T3, while iodine deficiency decreases them.
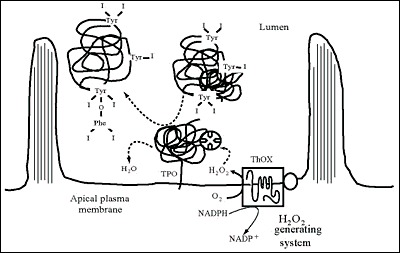
Fig. 2-9
Synthesis of hormone residues (coupling of iodotyrosines) in Tg at the apical plasma membrane-follicle lumen boundary. The scheme does not account for the relative size of the intervening molecules
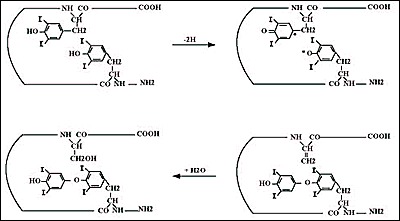
Fig. 2-10
Possible coupling reaction sequence. Oxidation of iodotyrosines may produce iodotyrosyl radicals. The free radicals could combine to generate the iodothyronine residue (at the tyrosine acceptor site) and a dehydroalanine residue (at the tyrosine donor site), which in the presence of H2O converts into a serine
The distribution of hormone among several sites in the Tg molecule has been studied in a number of species (180;230-233). The most important is at tyrosyl 5, quite close to Tg N-terminus. It usually contains about 40% of Tg total T4. The second most important site is at tyrosyl 2554, which may contain for 20-25% of total T4. A third important site is at tyrosyl 2747, which appears favored for T3 synthesis in some species. Tyrosyl 1291 is prominent in T4 formation in guinea pigs and rabbits and very responsive to TSH stimulation. Incremental iodination of low iodine hTg in vitro, with lactoperoxidase as surrogate for TPO, led to the identification of the favored sites for iodination (234). Small increments of iodine go first to tyrosyl residues 2554, 130, 685, 847, 1448, and 5, in that order. Further addition increases the degree of iodination at these sites, iodinates some new tyrosyls, and results in thyroid hormone formation at residues 5, 2554, 2747, and 685, with a trace found at 1291, in that quantitative order. These data identified the most important hormonogenic sites in hTg, and also the favored sites for early iodination. The same work recognized three consensus sequences associated with iodination and hormone formation: i) Asp/Glu-Tyr at three of the four most important sites for hormone synthesis, ii) Ser/Thr-Tyr-Ser associated with hormone formation, including the C-terminal hormonogenic site that favors T3 in some species and iii)Glu-X-Tyr favoring early iodination, although usually not with hormone formation (Fig. 2-11).
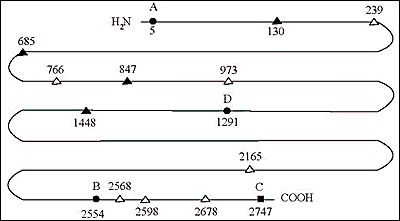
Fig. 2-11Diagram of the human Tg polypeptide chain; residue numbers refer to the human cDNA sequence; (a) sites forming T4 (sites A,B,D) (solid circles) and/or T3 (site C) (solid square); (b) early iodinated sites (solid triangles); (c) other iodinated sites (open triangles).
Identifying the donor tyrosyls has attracted considerable investigational interest over the past several decades. The fact that some tyrosyls are iodinated early but do not go on to provide the acceptor ring of T4 makes them potential donor candidates (234). On the basis of in vitro iodination of an N-terminal cyanogen bromide Tg peptide, Marriq et al. (235) concluded that residue 130 was a donor tyrosine for the major hormonogenic site at Tyr5. This conclusion was challenged by Xiao et al. (236) in a similar in vitro system. A baculovirus system expressing the 1-198 fragment of Tg, either normal or mutated on tyrosyl residues, showed that iodination of a fragment containing tyrosyls only at residue 5, 107 and 130 formed T4 as did the intact normal peptide, but this fragment could also form T4 with substitutions at residue 5 or 130 (237). Dunn et al.(238) who incorporated 14C-Tyr into beef thyroid slices followed by in vitro iodination and trypsin digestion of the N-terminal portion of Tg localized pyruvate (as a derivative of dehydroalanine) to residue 130 by mass spectrometry. They proposed that Tyr130 was the donor tyrosine for the most important hormonogenic site at Tyr5. Gentile et al. (239) used mass spectrometry to identify a peptide containing dehydroalanine at tyrosine 1375 of bTg and proposed this tyrosine as the donor for the hormonogenic site at residue 1291. Donors for the other major hormonogenic sites have not yet been identified.
In addition to its role as component of the iodoamino acids, iodine is associated with cleavage of peptide bonds of Tg, at least in vitro (180). This has been attributed to generation of free radicals during oxidation (240). Exposure of Tg to reducing agents yields an N-terminal peptide of about 20-26kDa, depending on the animal species, that contains the major hormonogenic site of Tg (241). This peptide appears in parallel with iodination or may slightly precede it (242). Further addition of iodine cleaves the 26kDa further, to produce an 18kDa (on human Tg), an event that also occurs with TSH stimulation (242). Thus, iodination-associated cleavage appears to be part of the maturation of the Tg molecule. These discrete N-terminal peptides have been found in all vertebrate Tg examined so far (231).
The amount of iodine has important effects on thyroid hormone production (243). The initial reaction between TPO and H2O2 produces the so-called "compound I," which oxidizes iodide and iodinates Tg. Next, the two reactants form compound II, which is necessary for the coupling reaction to make thyroid hormones. However, if excessive iodine is present, conversion to compound II does not take place, and hormone synthesis is impaired. (Fig. 2-12) Other iodinated compounds occasionally inhibit the thyroid. Thyroalbumin excited considerable interest several decades ago. This is an iodinated albumin, shown to be serum albumin that is iodinated in the thyroid (244). Occasionally, large amounts are found in certain thyroid diseases, including Hashimoto's thyroiditis (245), congenital metabolic defects (246), thyrotoxicosis (247) and thyroid carcinoma (248). In all these cases, there are abnormalities in thyroid structure which might explain the access of serum albumin to intrathyroidal iodination sites. However, in physiological conditions, serum albumin can reach thyroid follicle lumina by transcytosis i.e. basolateral endocytosis and vesicular transport to the apical plasma membrane (249). The thyroid also iodinates lipids and many different iodolipids have been described after high doses of iodide in vitro (250;251). Of particular interest is 2-iodohexadecanal (252;253). It occurs in the thyroid of several species following administration of KI, and its amount increases linearly with additional iodine, in contrast to iodination of Tg which eventually is inhibited by excess iodide. This compound inhibits the action of NADPH oxidase, which is responsible for H2O2 production (254;255). These findings suggested that iodination of lipids impairs H2O2 production and, therefore, decreases further Tg iodination. This is the most probable mechanism for the Wolff-Chaikoff effect (128).
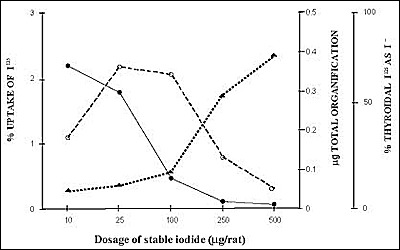
Fig. 2-12
Demonstration of the Wolff-Chaikoff block induced by iodide in the rat. Animals were given increasing doses of stable iodide. There was at first an increase in total organification, but then, as the dose was increased further, a depression of organification of iodide and an increase in the free iodide present in the thyroid gland occurred.
HORMONE STORAGE
Tg molecules vectorially delivered to the follicule lumen by exocytosis accumulates to reach uncommun concentrations i.e. 0.3-0.5 mM.The mechanism operating such a “packaging” is unknown. Water and ion extraction from the follicle lumen might represent an active process leading toTg concentration. As the follicle lumen is a site of Ca++ accumulation (256;257), the high degree of compaction of lumenal Tg might depend on electrostatic interactions between Ca++ and anionic residues of Tg, which is an acidic protein. Stored Tg molecules undergo iodination and hormone formation reactions at the apical plasma membrane-lumen boundary (257-259), where TPO and H2O2 generating system reside. The mature Tg molecules, now containing MIT, DIT, T4 and T3, remains extracellular in the lumen of thyroid follicles. Turnover of intrafollicular material or so-called colloid varies greatly with gland activity. For normal humans, the organic iodine pool (largely in intrafollicular material), turns over at a rate of about 1% per day (14). When the turnover increases, less Tg is stored, and with extreme hyperplasia, none is evident and the entire organic iodine content may be renewed daily (14). In this situation, secretion of Tg and resorption of Tg (see below) probably occur at similar rates and only tiny amounts of intrafollicular material are present at any time.
Thyroglobulin as usually isolated from the thyroid is chiefly the 19S 660kDa dimer that has been glycosylated and iodinated. Iodination and hormone formation of Tg is more complex than generally thought because of the slow diffusion of molecules that are in a colloidal state in the follicle lumen. It has been reported that TSH alters the hydrodynamic properties of intrafollicular Tg molecules (260;261). The diffusion coefficient of Tg which is about 26mm2 / sec in water would only be in the order of 10-100mm2 / hour in the thyroid follicle lumen. There is evidence for the presence of insoluble Tg in the form of globules of 20-120 microns, at a protein concentration of almost 600 mg/mL, in the lumen of thyroid follicles of different animal species (262). In human, about 34% of the gland Tg would be in this form (263). In pig, insoluble Tg contains more iodine than did the 660kDa Tg, and had virtually no thyroid hormone (264). Insoluble Tg has many internal crosslinks through disulfide bonds, dityrosine, and glutamyl-lysine bonds, the latter generated by transglutaminase (265). The formation of Tg multimers that probably results from oxidative processes might be limited by the presence of molecular chaperones such as the protein disulfide isomerase (PDI) and BiP in the follicle lumen (266).
THYROGLOBULIN ENDOCYTOSIS
To be useful, thyroid hormones must be released from Tg and delivered to the circulation for action at their distant target tissues. Depending on numerous factors including - the supply of iodide as substrate, the activity of enzymes catalyzing hormone formation, the concentration and physico-chemical state of Tg - the hormone content of lumenal Tg molecules varies to a rather large extent. Tg molecules newly arrived in the follicle lumen with no or a low hormone content would co-exist with “older” Tg exhibiting up to 6-8 hormone residues. The downstream processes responsible for the production of free thyroid hormones from these prohormonal molecules must therefore adequately manage the use of these lumenal heterogeneous Tg stores to provide appropriate amounts of hormones for peripheral utilization. One would expect to find i) control systems preventing excess hormone production that would result from the processing of excessive amounts of prohormonal Tg molecules and ii) checking systems avoiding the use of Tg molecules with no or a low hormone content.
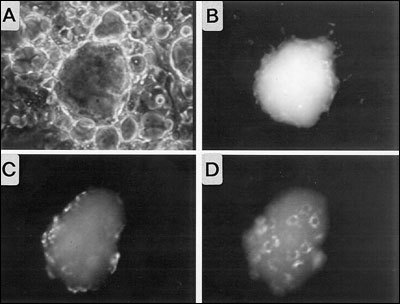
Fig. 2- 13
Visualization of Tg endocytosis by in vitro reconstituted thyroid follicles obtained from porcine thyrocytes in primary culture. Purified porcine Tg molecules labeled by covalent coupling of fluorescein were microinjected into the lumen of a follicle. A and B, phase contrast and fluorescence images taken at the time of microinjection. C and D, fluorescence images of the top (C) and the bottom (D) of the follicle after 2hr of incubation. Fluorescently-labeled Tg is present inside thyrocytes.
The way the thyroid follicle proceeds to generate free hormones from stored hormone containing Tg molecules has been known for a long time. Tg molecules are first taken up by polarized thyrocytes (Fig. 2-13) and then conveyed to lysosomal compartments for proteolytic cleavage that release T4 and T3 from their peptide linkages. The first step represents the limiting point in the thyroid hormone secretory pathway. Over the last decade, there has been substantial improvement in the knowledge of the cellular and molecular mechanisms governing the internalization or endocytosis and intracellular transport of the prohormone, Tg. The evolution has first been to consider that it could proceed via a mechanism different from phagocytosis, also named macropinocytosis, evidenced in rats under acute TSH stimulation (reviewed in (267)). Results obtained in rats and dogs have been for a long time extrapolated to the different animal species including human. There is now a number of experimental data indicating that in the thyroid of different species under physiological circumstances, basal internalization of Tg, mainly if not exclusively, occurs via vesicle-mediated endocytosis or micropinocytosis (reviewed in (268)), while macropinocytosis results from acute stimulation (Fig. 2-14) (269;270).
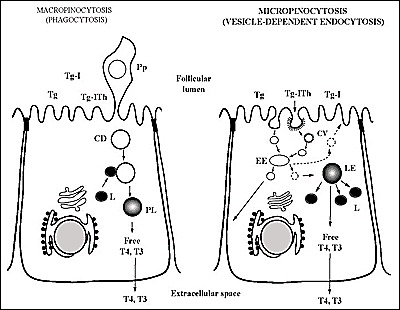
Fig. 2-14
Schematic representation of the two modes of internalization of Tg; Micropinocytosis (on the right) and Macropinocytosis or phagocytosis (on the left). Intralumenal Tg stores potentially subjected to endocytosis are composed of (recently secreted) non-iodinated Tg, iodinated Tg (Tg-I) and iodinated Tg containing iodothyronine residues (Tg-Ith).Abbreviations are: CV, Coated Vesicle; EE, Early Endosome; LE, Late Endosome; L, Lysosome; Pp, Pseudopod; CD, Colloid Droplet; PL, Phagolysosome. The scheme on the right indicates the three possible routes of transport of internalized Tg molecules reaching the EE: transport to LE, recycling towards the follicle lumen and transcytosis i.e.transport towards the basolateral plasma membrane.
The internalization process starts with the organization of microdomains at the apical plasma membrane of thyrocytes; these microdomains or pits, resulting from the recruitment and assembly of proteins (clathrin, adaptins…) on the cytoplasmic side of the membrane, invaginate to finally generate coated vesicles after membrane fission. Lumenal Tg molecules, either free or associated to membrane proteins acting as Tg receptors, enter the pits and are then sequestrated into the newly-formed vesicles (267-269). Tg internalization via vesicle-mediated endocytosis is regulated by TSH (268). The vesicles lose their coat and, through a complex fusion process, deliver their content into a first type of endocytic compartments, the early apical endosomes (270) (Fig 2-15). In these compartments, Tg molecules probably undergo sorting on the basis of recognition of different physico-chemical parameters either linked or independent such as the hormone content, exposed carbohydrates, conformation of peptide domains… A step of sorting appears as a prerequisite for subsequent differential cellular handling of Tg molecules. It has been shown that internalized Tg molecules can follow different intracellular pathways. Part of Tg molecules are conveyed via a vesicle transport system to the second type of endocytic compartments, late endosomes or prelysosomes. This route ending to lysosomes corresponds to the Tg degradation pathway for the generation of free thyroid hormones. It is reasonable to think that Tg molecules following this route are the more mature molecules (with a high hormone content) but, this has not been firmly demonstrated. The other Tg molecules with no or a low hormone content, present in early apical endosomes, enter either of the two following routes; they are recycled back into the follicle lumen through a direct vesicular transport towards the apical plasma membrane (271) or via a two-step vesicular transport to the Golgi apparatus and then to the apical plasma membrane (272). Alternately, Tg molecules are transported and released at the basolateral membrane domain of thyrocytes via transcytotic vesicles (262;273); a process accounting for the presence of Tg in plasma. The orientation of Tg molecules towards one or the other of these three routes requires the presence of receptors. However, one route could simply convey Tg molecules that are not selected for entering the other pathways.
Receptors involved in Tg endocytosis may operate at the apical plasma membrane for Tg internalization and downstream in apical early endosomes for Tg sorting. The requirement and/or the involvement of apical cell surface receptors has long been debated. Most investigators now recognize that receptors are not needed for internalization since Tg is present at a high concentration at the site of vesicle formation. So, Tg molecules are most likely internalized by fluid-phase endocytosis and not by receptor-mediated endocytosis. On the contrary, if apical membrane Tg receptors exist, their function would be to prevent the internalization of sub-classes of Tg molecules (274;275). As it is not conceivable that internalized Tg molecules could enter the different intracellular routes, described above, at random, Tg receptors must exist in early apical endosomes. A detailed review on potential Tg receptors has been made by Marino and Mc Cluskey (276).
The first candidate receptor, initially described by Consiglio et al.(277;278) was later identified as the asialoglycoprotein receptor composed of three subunits (RLH1,2 and 3). This receptor binds Tg at acidic pH and recognizes both sugar moities and peptide determinants on Tg (279). As low-iodinated Tg molecules are known to have a low sialic acid content, this receptor could be involved in sorting immature Tg molecules for recycling to the follicle lumen. A second receptor, still not identified, named N-acetylglucosamine receptor (280;281), presumably located in sub-apical compartments, interacts with Tg at acidic pH; it could also act as a receptor for recycling immature Tg molecules back to the follicle lumen. A third receptor; megalin, has more recently been discovered in the thyroid and has been the subject of extensive studies yielding convincing data (276;282-285). Megalin is an ubiquitous membrane protein belonging to the LDL receptor family. It is located in the apical region of thyrocytes and its expression is regulated by TSH. Megalin, that binds multiple unrelated ligands, interacts with Tg with a high affinity.In vitro and in vivo data indicate that Megalin is involved in the transcellular transport or transcytosis of Tg molecules, possibly with a low hormone content (286).
From the properties and subcellular location of these receptors, one can propose an integrated view of the sorting processes that would operate in early apical endosomes. The asialoglycoprotein receptor and/or the less defined N-acetylglucosamine receptor would recognize immature Tg for recycling and megalin would interact with Tg subjected to apical to basolateral transcytosis. The remaining Tg molecules would enter, without sorting, the functionally important pathway i.e. the prelysosome-lysosome route.
Under TSH stimulation, macropinocytosis would be triggered and would become operative in Tg internalization. Pseudopods representing extensions of the apical plasma membrane project into the follicle lumen and pinch off to form a resorption vacuole known as colloid droplet (287) .The colloid droplets then deliver their content to lysosomes. Pseudopod formation is one of the earliest effects of TSH on the gland, evident within several minutes after administration (288;289). In most species but perhaps not in rat, TSH stimulates macropinocytosis through the activation of the cyclic AMP cascade (290;291).
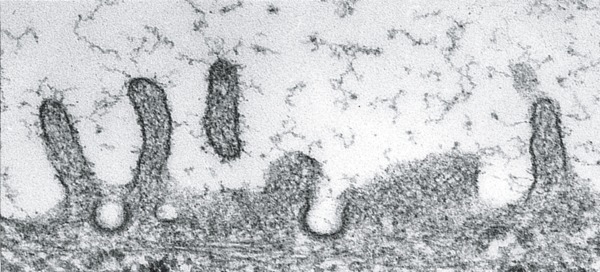

Fig. 2-15
Transmission electron microscope observations of apical endocytic structures in thyrocytes. Top: coated pits at the apical plasma membrane. Bottom: an early endosome located in the apical region. Bars, 200 nm.
PROTEOLYTIC CLEAVAGE OF THYROGLOBULIN
Internalized Tg molecules that are conveyed to lysosome compartments are subjected to diverse hydrolytic reactions leading to the generation of free thyroid hormones and to complete degradation of the protein. Given its composition, Tg is likely the substrate for the different lysosomal enzymes: proteases, glycohydrolases, phosphatases, sulfatases.... Efforts have been made to identify proteases involved in the release of hormonal residues from their peptide linkage in Tg. Endopeptidases such as cathepsin D, H and L (292-299) are capable of cleaving Tg.
Initial cleavage would bring into play endopeptidases and resulting products would be further processed by exopeptidases. Dunn et al. (295) showed that cathepsin B has exopeptidase activity as well as an endopeptidase action (295;297). These investigators tested the activities of human enzyme preparations against the 20kDa N-terminal peptide from rabbit Tg, which contains the dominant T4 site at residue 5. Extended cathepsin B incubation produced the dipeptide T4-Gln, corresponding to residues 5 and 6 of Tg. The combination of cathepsin B with the exopeptidase dipeptidase I released T4 from this dipeptide, although lysosomal dipeptidase I alone was not effective. Thus, the combination of cathepsin B and lysosomal dipeptidase I was sufficient to release free thyroid hormone from its major site at residue 5. The exopeptidase lysosomal dipeptidase II may also be involved in release of free T4, but from a site in Tg other than residue 5 (297). Thus, Tg probably undergoes selective cleavage reactions at its N- and C- terminal ends to release iodothyronines that are located nearby (297;300). Starting from highly purified preparations of thyroid lysosomes, Rousset et al. (301-303) have identified intralysosomal Tg molecules with very limited structural alterations but devoid of hormone residue. One may think that proteolysis of Tg occurs in two sequential steps; i) early and selective cleavages to release T3 and T4 residues and ii) delayed and complete proteolysis. The reduction of the very high number of disulfide bonds might be the limiting reaction between the two steps. The nature and the origin of the reducing compounds acting on Tg are not known. Noteworthy, the possibility of proteolytic cleavage of Tg inside the follicle lumen, before internalization, has been proposed (304-307) but not yet confirmed by other groups.
After Tg digestion, T4 and T3 must go from the lysosomal compartments to the cytoplasm and from the cytoplasm out of the cell to enter the circulation. It has been postulated for decades that thyroid hormones are released from thyrocytes by simple diffusion. There are many objections to this view (308). One of these comes from the chemical nature of iodothyronines; T4 and T3, which are generally considered as lipophylic compounds possess charges on both their proximal (amino acid side chain) and distal (phenolate) parts. As now known for the entry of thyroid hormones in peripheral target cells, the exit of thyroid hormones from thyrocytes probably involves membrane transporter(s). Details of hormone transport across the lysosomal membrane and then across the basolateral plasma membrane are unknown, including whether it is an active or passive process. At present, only a lysosomal membrane transporter for iodotyrosines has been reported (309;310). Nevertheless the role of newly cloned peripheral tissue thyroid transporters (311;312) in this process remains to be defined.
The type I and type II iodothyronine 5'-deiodinase is present in the thyroid (63;313;314) and deiodinate about 10% of T4 to T3. The extent of this intrathyroidal deiodination is increased when the thyroid is stimulated by TSH (315;316). Estimates of average normal secretion for euthyroid humans are 94-110 µg T4 and 10-22 µg T3 daily (317). The thyroid may also convert some T4 to 3,3'5'-T3 (reverse T3) within the thyroid. About 70% of the Tg iodine content is in the form of DIT and MIT, so this represents an important part of the intrathyroid iodine pool. Rather than lose it to the circulation, the thyroid deiodinates MIT and DIT and returns most of iodide to the intrathyroidal iodide pool. The responsible enzyme i.e. the iodotyrosine deiodinase is an NADPH-dependent flavoprotein with a estimated molecular weight of about 42kDa (318) and recently identified as DEHAL1(319-322). About 3-5 times more iodide is formed inside the gland each day by this deiodinase than enters the cell from the serum (14). The importance of the internal recycling of iodide is demontrated by congenitally goitrous subjects who harbour mutations in DEHAL gene (322) and cannot deiodinate iodotyrosines. These patients are successfully treated with large amounts of iodide (323). Some iodine is lost from the gland through inefficiency of its recycling by the iodotyrosine deiodinase (14;87;317;324). This leak may increase as the thyroid adapts to a high daily iodine intake (325), possibly as an autoregulatory process to prevent excessive Tg iodination. Much more iodide can be lost from diseased glands. Ohtaki et al. (87) found that some iodide leaks from all glands, including normal ones, but that the amount increases markedly with gland iodine content, presumably reflecting a dependence on dietary iodine intake. Fisher et al. (317) reported that about 38 µg iodide was released when the mean T4 secretion was 53 µg/day.
Among other products which are released or leak out from the thyroid, there is Tg (326;327). The secretion of Tg is clinically important. Its presence in serum can be detected by a routine assay and provides a sensitive (although not always specific) marker for increased thyroid activity. Attempts have been made to determine the biochemical characteristics of circulating Tg molecules in terms of iodine content (328), structural integrity (329) and hormone content (330). Serum levels are elevated in patients with hyperplastic thyroid or thyroid nodules including differentiated thyroid cancer. Tg measurement can identify congenital hyperplastic goiter, endemic goiter, and many benign multinodular goiters, but its greatest application is in the follow-up of differentiated thyroid cancer (331). Most papillary and follicular cancers retain some of the metabolic functions of the normal thyrocyte, including the ability to synthesize and secrete Tg. Subjects who have differentiated thyroid cancer treated by surgery and radioiodine should not have normal thyroid tissue left, and therefore, should not secrete Tg. If Tg is found in their serum, it reflects the continuing presence either of normal tissue, unlikely after its previous ablation, or of thyroid cancer. The depolarized cancer cells presumably secrete Tg directly in intercellular space. Tracking serum Tg levels is probably the most sensitive and practical means for the follow-up of such patients. It is more sensitive when the subject is stimulated by TSH. Until recently, this could only be done by withdrawal of thyroid hormone and consequent symptomatic hypothyroidism, but now recombinant human TSH can be administered to enhance the sensitivity of the serum Tg and thyroid scan (332-337).
CONTROL OF HORMONE SYNTHESIS
The most important controlling factors are iodine availability and TSH. Inadequate amounts of iodine lead to inadequate thyroid hormone production, increased TSH secretion and thyroid stimulation, and goiter in an attempt to compensate. Excess iodide acutely inhibits thyroid hormone synthesis, the Wolff-Chaikoff effect (243), apparently by inhibiting H2O2 generation, and therefore, blocking Tg iodination (127). A proposed mechanism is that the excess iodide leads to the formation of 2-iodohexadecanal (255), which is endowed with an inhibitory action on H2O2 generation.
TSH influences virtually every step in thyroid hormone synthesis and release. In humans the effects on secretion appear to be mediated through the cAMP cascade (see chapter 1) while the effects on synthesis are mediated by the Gq/phospholipase C cascade (338). Elsewhere in this chapter, we have mentioned instances of TSH regulation. To summarize, TSH stimulates the expression of NIS, TPO, Tg and the generation of H2O2 , increases formation of T3 relative to T4, alters the priority of iodination and hormonogenesis among tyrosyls and promotes the rapid internalization of Tg by thyrocytes. These several steps are interrelated and have the net effects of increasing the amount of iodine available to the cells and of making and releasing a larger amount and a more effective type of thyroid hormone (T3).
Anti-thyroid drugs are external compounds influencing thyroid hormone synthesis. The major inhibitory drugs are the thionamides: propylthiouracil and methimazole. In the thyroid, they appear to act by competing with tyrosyl residues of Tg for oxidized iodine, at least in the rat (219). Iodotyrosyl coupling is also inhibited by these drugs and appears more sensitive to their effects than does tyrosyl iodination.
Reference List
- Pennington JA, Young BE. Total diet study nutritional elements, 1982-1989. J Am Diet Assoc 1991; 91(2):179-183.
- Dunn JT. Sources of dietary iodine in industrialized countries. In: Delange F, Dunn JT, Glinoer D, editors. Iodine Deficiency in Europe. A Continuing Concern. New-York: Plenum Press, 1993: 17-21.
- Soldin OP, Soldin SJ, Pezzullo JC. Urinary iodine percentile ranges in the United States. Clin Chim Acta 2003; 328(1-2):185-190.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87(2):486-488.
- Dunn JT. What's happening to our iodine? J Clin Endocrinol Metab 1998; 83(10):3398-3400.
- Hollowell JG, Staehling NW, Hannon WH et al. Iodine nutrition in the United States. Trends and public health implications: iodine excretion data from National Health and Nutrition Examination Surveys I and III (1971-1974 and 1988-1994). J Clin Endocrinol Metab 1998; 83(10):3401-3408.
- Pennington JAT, Schoen SA, Salmon GD, Young B, Johnson RD, Marts R.W. Composition of core foods of the U.S. food supply,1982-1991. J Food Comp Anal 1995; 8:171-217.
- Fischer PWF, Giroux A. Iodine content of a representative Canadian diet. J Can Diet Assoc 1987; 48:24-27.
- Fischer PWF, Giroux A. Urinary iodine excretion by a selected sample of healthy Canadian adults. J Can Diet Assoc 1990; 51:337-340.
- Takamura N, Hamada A, Yamaguchi N et al. Urinary iodine kinetics after oral loading of potassium iodine. Endocr J 2003; 50(5):589-593.
- Vitti P, Delange F, Pinchera A, Zimmermann M, Dunn JT. Europe is iodine deficient. Lancet 2003; 361(9364):1226.
- International Council for Control of Iodine Deficiency Disorders. Current Iodine Deficiency Disorders Status Database. http://www.iccidd.org . 2010.
- Wynn JO. Components of the serum protein-bound iodine following administration of I-131-labeled hog thyroglobulin. J Clin Endocrinol Metab 1961; 21:1572-1578.
- De Groot. LJ. Kinetic analysis of iodine metabolism. J Clin Endocrinol Metab 1966; 26:149-173.
- McConahey WM, Keating FRJ, Power. MH. An estimation of the renal and extrarenal clearance of radioiodide in man. J Clin Invest 1951; 30(7):778-780.
- Bricker NS, Hlad CJJ. Observations on the mechanism of the renal clearance of I131. J Clin Invest 1955; 34(7, Part 1):1057-1072.
- Perry WF, Hughes. JF. The urinary excretion and thyroid uptake of iodine in renal disease. J Clin Invest 1952; 31(5):457-463.
- Berson SA, Yalow RS, Sorrentino. J, Roswit. B. The determination of thyroidal and renal plasma I131 clearance rates as a routine diagnostic test of thyroid dysfunction. J Clin Invest 1952; 31(2):141-158.
- Weaver. JC, Kamm. ML, Dobson. RL. Excretion of radioline in human milk. J Am Med Assoc 1960; 173:872-875.
- Delange F. Requirements of iodine in humans. In: Delange F, Dunn JT, Glinoer D, editors. Iodine Deficiency in Europe. A Continuing Concern. New-York: Plenum Press, 1993: 5-13.
- Andros G, Wollman SH. Autoradiographic localization of radioiodide in the thyroid gland of the mouse. Am J Physiol 1967; 213(1):198-208.
- Pitt-Rivers R, Trotter. WR. The site of accumulation of iodide in the thyroid of rats treated with thiouracil. Lancet 1953; 265(6792):918-919.
- Pochin EE. Investigation of thyroid function and disease with radioactive iodine. Lancet 1950; 2(6620):84-91.
- Berson SA, Yalow RS. The iodide trapping and binding functions of the thyroid. J Clin Invest 1955; 34(2):186-204.
- Van Sande J., Massart C, Beauwens R et al. Anion selectivity by the sodium iodide symporter. Endocrinology 2003; 144(1):247-252.
- Wolff J. Transport of iodide and other anions in the thyroid gland. Physiol Rev 1964; 44:45-90.
- Tyler DD, Gonze J, Lamy F, Dumont JE. Influence of mitochondrial inhibitors on the respiration and energy-dependent uptake of iodide by thyroid slices. Biochem J 1968; 106(1):123-133.
- Wolff J. Thyroidal iodine transport. I. Cardiac glycosides and the role of potassium. Biochim Biophys Acta 1960; 38:316-324.
- Brunberg JA, Halmi NS. The role of ouabain-sensitive adenosine triphosphatase in the stimulating effect of thyrotropin on the iodide pump of the rat thyroid. Endocrinology 1966; 79(4):801-807.
- Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature 1996; 379(6564):458-460.
- Levy O, Dai G, Riedel C et al. Characterization of the thyroid Na+/I- symporter with an anti-COOH terminus antibody. Proc Natl Acad Sci U S A 1997; 94(11):5568-5573.
- Paire A, Bernier-Valentin F, Selmi-Ruby S, Rousset B. Characterization of the rat thyroid iodide transporter using anti-peptide antibodies. Relationship between its expression and activity. J Biol Chem 1997; 272(29):18245-18249.
- Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N. N-linked glycosylation of the thyroid Na+/I- symporter (NIS). Implications for its secondary structure model. J Biol Chem 1998; 273(35):22657-22663.
- Eskandari S, Loo DD, Dai G, Levy O, Wright EM, Carrasco N. Thyroid Na+/I- symporter. Mechanism, stoichiometry, and specificity. J Biol Chem 1997; 272(43):27230-27238.
- Smanik PA, Ryu KY, Theil KS, Mazzaferri EL, Jhiang SM. Expression, exon-intron organization, and chromosome mapping of the human sodium iodide symporter. Endocrinology 1997; 138(8):3555-3558.
- Smanik PA, Liu Q, Furminger TL et al. Cloning of the human sodium lodide symporter. Biochem Biophys Res Commun 1996; 226(2):339-345.
- Perron B, Rodriguez AM, Leblanc G, Pourcher T. Cloning of the mouse sodium iodide symporter and its expression in the mammary gland and other tissues. J Endocrinol 2001; 170(1):185-196.
- Selmi-Ruby S, Watrin C, Trouttet-Masson S et al. The porcine sodium/iodide symporter gene exhibits an uncommon expression pattern related to the use of alternative splice sites not present in the human or murine species. Endocrinology 2003; 144(3):1074-1085.
- Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology 1997; 138(6):2227-2232.
- Uyttersprot N, Pelgrims N, Carrasco N et al. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I- symporter mRNAs in dog thyroid. Mol Cell Endocrinol 1997; 131(2):195-203.
- Ohno M, Zannini M, Levy O, Carrasco N, Di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol 1999; 19(3):2051-2060.
- Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3',5'-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol 2002; 16(10):2266-2282.
- Lin X, Ryu KY, Jhiang SM. Cloning of the 5'-flanking region of mouse sodium/iodide symporter and identification of a thyroid-specific and TSH-responsive enhancer. Thyroid 2004; 14(1):19-27.
- Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem 2001; 276(24):21458-21463.
- Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A 2002; 99(24):15776-15781.
- Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A 2002; 99(24):15462-15467.
- Eng PH, Cardona GR, Fang SL et al. Escape from the acute Wolff-Chaikoff effect is associated with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid and protein. Endocrinology 1999; 140(8):3404-3410.
- Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem 2004; 279(5):3439-3446.
- Pekary AE, Hershman JM. Tumor necrosis factor, ceramide, transforming growth factor-beta1, and aging reduce Na+/I- symporter messenger ribonucleic acid levels in FRTL-5 cells. Endocrinology 1998; 139(2):703-712.
- Dohan O, De la Vieja A, Paroder V et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 2003; 24(1):48-77.
- Riedel C, Dohan O, De la Vieja A, Ginter CS, Carrasco N. Journey of the iodide transporter NIS: from its molecular identification to its clinical role in cancer. Trends Biochem Sci 2001; 26(8):490-496.
- Fujiwara H, Tatsumi K, Miki K et al. Congenital hypothyroidism caused by a mutation in the Na+/I- symporter. Nat Genet 1997; 16(2):124-125.
- Fujiwara H, Tatsumi K, Miki K et al. Recurrent T354P mutation of the Na+/I- symporter in patients with iodide transport defect. J Clin Endocrinol Metab 1998; 83(8):2940-2943.
- Kosugi S, Inoue S, Matsuda A, Jhiang SM. Novel, missense and loss-of-function mutations in the sodium/iodide symporter gene causing iodide transport defect in three Japanese patients. J Clin Endocrinol Metab 1998; 83(9):3373-3376.
- Kosugi S, Sato Y, Matsuda A et al. High prevalence of T354P sodium/iodide symporter gene mutation in Japanese patients with iodide transport defect who have heterogeneous clinical pictures. J Clin Endocrinol Metab 1998; 83(11):4123-4129.
- Kosugi S, Bhayana S, Dean HJ. A novel mutation in the sodium/iodide symporter gene in the largest family with iodide transport defect. J Clin Endocrinol Metab 1999; 84(9):3248-3253.
- Matsuda A, Kosugi S. A homozygous missense mutation of the sodium/iodide symporter gene causing iodide transport defect. J Clin Endocrinol Metab 1997; 82(12):3966-3971.
- Pohlenz J, Rosenthal IM, Weiss RE, Jhiang SM, Burant C, Refetoff S. Congenital hypothyroidism due to mutations in the sodium/iodide symporter. Identification of a nonsense mutation producing a downstream cryptic 3' splice site. J Clin Invest 1998; 101(5):1028-1035.
- Pohlenz J, Duprez L, Weiss RE, Vassart G, Refetoff S, Costagliola S. Failure of membrane targeting causes the functional defect of two mutant sodium iodide symporters. J Clin Endocrinol Metab 2000; 85(7):2366-2369.
- Levy O, Ginter CS, De la Vieja A, Levy D, Carrasco N. Identification of a structural requirement for thyroid Na+/I- symporter (NIS) function from analysis of a mutation that causes human congenital hypothyroidism. FEBS Lett 1998; 429(1):36-40.
- Saito T, Endo T, Kawaguchi A et al. Increased expression of the Na+/I- symporter in cultured human thyroid cells exposed to thyrotropin and in Graves' thyroid tissue. J Clin Endocrinol Metab 1997; 82(10):3331-3336.
- van Staveren WC, Solis DW, Delys L et al. Gene expression in human thyrocytes and autonomous adenomas reveals suppression of negative feedbacks in tumorigenesis. Proc Natl Acad Sci U S A 2006; 103(2):413-418.
- Wattel S, Mircescu H, Venet D et al. Gene expression in thyroid autonomous adenomas provides insight into their physiopathology. Oncogene 2005; 24(46):6902-6916.
- Delys L, Detours V, Franc B et al. Gene expression and the biological phenotype of papillary thyroid carcinomas. Oncogene 2007; 26(57):7894-7903.
- Lazar V, Bidart JM, Caillou B et al. Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab 1999; 84(9):3228-3234.
- Trouttet-Masson S, Selmi-Ruby S, Bernier-Valentin F et al. Evidence for transcriptional and posttranscriptional alterations of the sodium/iodide symporter expression in hypofunctioning benign and malignant thyroid tumors. Am J Pathol 2004; 165(1):25-34.
- Jhiang SM, Cho JY, Ryu KY et al. An immunohistochemical study of Na+/I- symporter in human thyroid tissues and salivary gland tissues. Endocrinology 1998; 139(10):4416-4419.
- Cho JY, Leveille R, Kao R et al. Hormonal regulation of radioiodide uptake activity and Na+/I- symporter expression in mammary glands. J Clin Endocrinol Metab 2000; 85(8):2936-2943.
- Tazebay UH, Wapnir IL, Levy O et al. The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat Med 2000; 6(8):871-878.
- Royaux IE, Suzuki K, Mori A et al. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 2000; 141(2):839-845.
- Yoshida A, Taniguchi S, Hisatome I et al. Pendrin is an iodide-specific apical porter responsible for iodide efflux from thyroid cells. J Clin Endocrinol Metab 2002; 87(7):3356-3361.
- Everett LA, Glaser B, Beck JC et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 1997; 17(4):411-422.
- Porra V, Bernier-Valentin F, Trouttet-Masson S et al. Characterization and semiquantitative analyses of pendrin expressed in normal and tumoral human thyroid tissues. J Clin Endocrinol Metab 2002; 87(4):1700-1707.
- Bidart JM, Mian C, Lazar V et al. Expression of pendrin and the Pendred syndrome (PDS) gene in human thyroid tissues. J Clin Endocrinol Metab 2000; 85(5):2028-2033.
- Gillam MP, Sidhaye AR, Lee EJ, Rutishauser J, Stephan CW, Kopp P. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem 2004; 279(13):13004-13010.
- Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 1999; 21(4):440-443.
- Soleimani M, Greeley T, Petrovic S et al. Pendrin: an apical Cl-/OH-/. Am J Physiol Renal Physiol 2001; 280(2):F356-F364.
- Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab 2002; 87(4):1778-1784.
- Everett LA, Belyantseva IA, Noben-Trauth K et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 2001; 10(2):153-161.
- Twyffels L, Strickaert A, Virreira M et al. Anoctamin-1/TMEM16A is the major apical iodide channel of the thyrocyte. Am J Physiol Cell Physiol 2014; 307(12):C1102-C1112.
- De Groot. LJ, Davis. AM. The early stage of thyroid hormone formation. Studies on rat in vivo. Endocrinology 1961; 69:695-718.
- Hildebrandt JD, Scranton JR, Halmi NS. Intrathyroidally generated iodide: its measurement and origins. Endocrinology 1979; 105(3):618-626.
- Rosenberg IN, Athans JC, Ahr CS, Behar A. Thyrotropin-induced release of iodide from the thyroid. Endocrinology 1961; 69:438-455.
- Ingbar SH. Simultaneous measurement of the iodide-concentrating and protein-binding capacities of the normal and hyperfunctioning human thyroid gland. J Clin Endocrinol Metab 1955; 15(2):238-264.
- Thrall KD, Bull RJ, Sauer RL. Distribution of iodine into blood components of the Sprague-Dawley rat differs with the chemical form administered. J Toxicol Environ Health 1992; 37(3):443-449.
- Eskin BA, Grotkowski CE, Connolly CP, Ghent WR. Different tissue responses for iodine and iodide in rat thyroid and mammary glands. Biol Trace Elem Res 1995; 49(1):9-19.
- Ohtaki S, Moriya S, Suzuki H, Moriuchi Y. Nonhormonal iodine escape from the normal and abnormal thyroid gland. J Clin Endocrinol Metab 1967; 27(5):728-740.
- Ohtaki S, Nakagawa H, Kimura S, Yamazaki I. Analyses of catalytic intermediates of hog thyroid peroxidase during its iodinating reaction. J Biol Chem 1981; 256(2):805-810.
- Krinsky MM, Alexander NM. Thyroid peroxidase. Nature of the heme binding to apoperoxidase. J Biol Chem 1971; 246(15):4755-4758.
- Niepomniszcze H, Degroot LJ, Hagen GA. Abnormal thyroid peroxidase causing iodide organification defect. J Clin Endocrinol Metab 1972; 34(4):607-616.
- Czarnocka B, Ruf J, Ferrand M, Carayon P, Lissitzky S. Purification of the human thyroid peroxidase and its identification as the microsomal antigen involved in autoimmune thyroid diseases. FEBS Lett 1985; 190(1):147-152.
- Hamada N, Portmann L, Degroot LJ. Characterization and isolation of thyroid microsomal antigen. J Clin Invest 1987; 79(3):819-825.
- Kotani T, Umeki K, Matsunaga S, Kato E, Ohtaki S. Detection of autoantibodies to thyroid peroxidase in autoimmune thyroid diseases by micro-ELISA and immunoblotting. J Clin Endocrinol Metab 1986; 62(5):928-933.
- Portmann L, Hamada N, Heinrich G, Degroot LJ. Anti-thyroid peroxidase antibody in patients with autoimmune thyroid disease: possible identity with anti-microsomal antibody. J Clin Endocrinol Metab 1985; 61(5):1001-1003.
- Ruf J, Czarnocka B, De Micco C, Dutoit C, Ferrand M, Carayon P. Thyroid peroxidase is the organ-specific 'microsomal' autoantigen involved in thyroid autoimmunity. Acta Endocrinol Suppl (Copenh) 1987; 281:49-56.
- Kimura S, Kotani T, McBride OW et al. Human thyroid peroxidase: complete cDNA and protein sequence, chromosome mapping, and identification of two alternately spliced mRNAs. Proc Natl Acad Sci U S A 1987; 84(16):5555-5559.
- Libert F, Ruel J, Ludgate M et al. Thyroperoxidase, an auto-antigen with a mosaic structure made of nuclear and mitochondrial gene modules. EMBO J 1987; 6(13):4193-4196.
- Seto P, Hirayu H, Magnusson RP et al. Isolation of a complementary DNA clone for thyroid microsomal antigen. Homology with the gene for thyroid peroxidase. J Clin Invest 1987; 80(4):1205-1208.
- Magnusson RP, Gestautas J, Taurog A, Rapoport B. Molecular cloning of the structural gene for porcine thyroid peroxidase. J Biol Chem 1987; 262(29):13885-13888.
- Derwahl M, Seto P, Rapoport B. Complete nucleotide sequence of the cDNA for thyroid peroxidase in FRTL5 rat thyroid cells. Nucleic Acids Res 1989; 17(20):8380.
- Kotani T, Umeki K, Yamamoto I et al. Nucleotide sequence of the cDNA encoding mouse thyroid peroxidase. Gene 1993; 123(2):289-290.
- Niccoli P, Fayadat L, Panneels V, Lanet J, Franc JL. Human thyroperoxidase in its alternatively spliced form (TPO2) is enzymatically inactive and exhibits changes in intracellular processing and trafficking. J Biol Chem 1997; 272(47):29487-29492.
- Fayadat L, Siffroi-Fernandez S, Lanet J, Franc JL. Degradation of human thyroperoxidase in the endoplasmic reticulum involves two different pathways depending on the folding state of the protein. J Biol Chem 2000; 275(21):15948-15954.
- Ferrand M, Le F, V, Franc JL. Increasing diversity of human thyroperoxidase generated by alternative splicing. Characterized by molecular cloning of new transcripts with single- and multispliced mRNAs. J Biol Chem 2003; 278(6):3793-3800.
- Rawitch AB, Pollock G, Yang SX, Taurog A. Thyroid peroxidase glycosylation: the location and nature of the N-linked oligosaccharide units in porcine thyroid peroxidase. Arch Biochem Biophys 1992; 297(2):321-327.
- de Vijlder JJ, Dinsart C, Libert F et al. Regional localization of the gene for thyroid peroxidase to human chromosome 2pter----p12. Cytogenet Cell Genet 1988; 47(3):170-172.
- Gerard CM, Lefort A, Libert F, Christophe D, Dumont JE, Vassart G. Transcriptional regulation of the thyroperoxydase gene by thyrotropin and forskolin. Mol Cell Endocrinol 1988; 60(2-3):239-242.
- Damante G, Di Lauro R. Thyroid-specific gene expression. Biochim Biophys Acta 1994; 1218(3):255-266.
- Kambe F, Seo H. Thyroid-specific transcription factors. Endocr J 1997; 44(6):775-784.
- Bakker B, Bikker H, Vulsma T, de Randamie JS, Wiedijk BM, de Vijlder JJ. Two decades of screening for congenital hypothyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update). J Clin Endocrinol Metab 2000; 85(10):3708-3712.
- Ris-Stalpers C, Bikker H. Genetics and phenomics of hypothyroidism and goiter due to TPO mutations. Mol Cell Endocrinol 2010; 322(1-2):38-43.
- Ericson LE, Johanson V, Molne J, Nilsson M, Ofverholm T. Intracellular transport and surface cell expression of thyroperoxidase. In: Carayon P, Ruf J, editors. Thyroperoxidase and Thyroid Autoimmunity. London: John Libbey Eurotext, 1990: 107-116.
- Yokoyama N, Taurog A. Porcine thyroid peroxidase: relationship between the native enzyme and an active, highly purified tryptic fragment. Mol Endocrinol 1988; 2(9):838-844.
- Alquier C, Ruf J, thouel-Haon AM, Carayon P. Immunocytochemical study of localization and traffic of thyroid peroxidase/microsomal antigen. Autoimmunity 1989; 3(2):113-123.
- Fayadat L, Niccoli-Sire P, Lanet J, Franc JL. Human thyroperoxidase is largely retained and rapidly degraded in the endoplasmic reticulum. Its N-glycans are required for folding and intracellular trafficking. Endocrinology 1998; 139(10):4277-4285.
- Penel C, Gruffat D, Alquier C, Benoliel AM, Chabaud O. Thyrotropin chronically regulates the pool of thyroperoxidase and its intracellular distribution: a quantitative confocal microscopic study. J Cell Physiol 1998; 174(2):160-169.
- Dupuy C, Virion A, Kaniewski J, Deme D, Pommier J. Thyroid NADPH--dependent H2O2 generating system: mechanism of H2O2 formation and regulation by Ca2+. In: Carayon P, Ruf J, editors. Thyroperoxidase and Thyroid Autoimmunity. London: John Libbey Eurotext, 1990: 95-102.
- Bjorkman U, Ekholm R. Hydrogen peroxide generation and its regulation in FRTL-5 and porcine thyroid cells. Endocrinology 1992; 130(1):393-399.
- Carvalho DP, Dupuy C, Gorin Y et al. The Ca2+- and reduced nicotinamide adenine dinucleotide phosphate-dependent hydrogen peroxide generating system is induced by thyrotropin in porcine thyroid cells. Endocrinology 1996; 137(3):1007-1012.
- Massart C, Hoste C, Virion A, Ruf J, Dumont JE, Van Sande J. Cell biology of H(2)O(2) generation in the thyroid: Investigation of the control of dual oxidases (DUOX) activity in intact ex vivo thyroid tissue and cell lines. Mol Cell Endocrinol 2011; 343(1-2):32-44.
- Dumont JE. The action of thyrotropin on thyroid metabolism. Vitam Horm 1971; 29:287-412.
- Bjorkman U, Ekholm R. Generation of H2O2 in isolated porcine thyroid follicles. Endocrinology 1984; 115(1):392-398.
- Deme D, Virion A, Hammou NA, Pommier J. NADPH-dependent generation of H2O2 in a thyroid particulate fraction requires Ca2+. FEBS Lett 1985; 186(1):107-110.
- Dupuy C, Kaniewski J, Deme D, Pommier J, Virion A. NADPH-dependent H2O2 generation catalyzed by thyroid plasma membranes. Studies with electron scavengers. Eur J Biochem 1989; 185(3):597-603.
- Nakamura Y, Ogihara S, Ohtaki S. Activation by ATP of calcium-dependent NADPH-oxidase generating hydrogen peroxide in thyroid plasma membranes. J Biochem 1987; 102(5):1121-1132.
- Virion A, Michot JL, Deme D, Kaniewski J, Pommier J. NADPH-dependent H2O2 generation and peroxidase activity in thyroid particular fraction. Mol Cell Endocrinol 1984; 36(1-2):95-105.
- Corvilain B, Van Sande J, Dumont JE. Inhibition by iodide of iodide binding to proteins: the "Wolff-Chaikoff" effect is caused by inhibition of H2O2 generation. Biochem Biophys Res Commun 1988; 154(3):1287-1292.
- Corvilain B, Van Sande J, Laurent E, Dumont JE. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology 1991; 128(2):779-785.
- Corvilain B, Laurent E, Lecomte M, Van Sande J, Dumont JE. Role of the cyclic adenosine 3',5'-monophosphate and the phosphatidylinositol-Ca2+ cascades in mediating the effects of thyrotropin and iodide on hormone synthesis and secretion in human thyroid slices. J Clin Endocrinol Metab 1994; 79(1):152-159.
- Coclet J, Foureau F, Ketelbant P, Galand P, Dumont JE. Cell population kinetics in dog and human adult thyroid. Clin Endocrinol (Oxf) 1989; 31(6):655-665.
- Saad AG, Kumar S, Ron E et al. Proliferative activity of human thyroid cells in various age groups and its correlation with the risk of thyroid cancer after radiation exposure. J Clin Endocrinol Metab 2006; 91(7):2672-2677.
- Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J Biol Chem 1999; 274(52):37265-37269.
- De Deken X, Wang D, Many MC et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem 2000; 275(30):23227-23233.
- Pachucki J, Wang D, Christophe D, Miot F. Structural and functional characterization of the two human ThOX/Duox genes and their 5'-flanking regions. Mol Cell Endocrinol 2004; 214(1-2):53-62.
- Fischer H, Gonzales LK, Kolla V et al. Developmental regulation of DUOX1 expression and function in human fetal lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2007; 292(6):L1506-L1514.
- Forteza R, Salathe M, Miot F, Forteza R, Conner GE. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol 2005; 32(5):462-469.
- Wong JL, Creton R, Wessel GM. The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev Cell 2004; 7(6):801-814.
- El Hassani RA, Benfares N, Caillou B et al. Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol 2005; 288(5):G933-G942.
- Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 2003; 17(11):1502-1504.
- Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid Redox Signal 2009; 11(10):2453-2465.
- Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci U S A 2005; 102(3):767-772.
- Boots AW, Hristova M, Kasahara DI, Haenen GR, Bast A, van der Vliet A. ATP-mediated activation of the NADPH oxidase DUOX1 mediates airway epithelial responses to bacterial stimuli. J Biol Chem 2009; 284(26):17858-17867.
- Luxen S, Noack D, Frausto M, Davanture S, Torbett BE, Knaus UG. Heterodimerization controls localization of Duox-DuoxA NADPH oxidases in airway cells. J Cell Sci 2009; 122(Pt 8):1238-1247.
- Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal 2006; 8(9-10):1549-1561.
- Moskwa P, Lorentzen D, Excoffon KJ et al. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med 2007; 175(2):174-183.
- Botteaux A, Hoste C, Dumont JE, Van Sande J, Allaoui A. Potential role of Noxes in the protection of mucosae: H(2)O(2) as a bacterial repellent. Microbes Infect 2009; 11(5):537-544.
- Bae YS, Choi MK, Lee WJ. Dual oxidase in mucosal immunity and host-microbe homeostasis. Trends Immunol 2010; 31(7):278-287.
- Ha EM, Oh CT, Bae YS, Lee WJ. A direct role for dual oxidase in Drosophila gut immunity. Science 2005; 310(5749):847-850.
- Ha EM, Lee KA, Park SH et al. Regulation of DUOX by the Galphaq-phospholipase Cbeta-Ca2+ pathway in Drosophila gut immunity. Dev Cell 2009; 16(3):386-397.
- Allaoui A, Botteaux A, Dumont JE, Hoste C, De Deken X. Dual oxidases and hydrogen peroxide in a complex dialogue between host mucosae and bacteria. Trends Mol Med 2009; 15(12):571-579.
- Song Y, Driessens N, Costa M et al. Roles of hydrogen peroxide in thyroid physiology and disease. J Clin Endocrinol Metab 2007; 92(10):3764-3773.
- Fortunato RS, Lima de Souza EC, Ameziane-ElHassani R et al. Functional Consequences of Dual Oxidase-Thyroperoxidase Interaction at the Plasma Membrane. J Clin Endocrinol Metab 2010;95:5403-5411.
- Song Y, Ruf J, Lothaire P et al. Association of duoxes with thyroid peroxidase and its regulation in thyrocytes. J Clin Endocrinol Metab 2010; 95(1):375-382.
- De Felice M., Di Lauro R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev 2004; 25(5):722-746.
- Lazzaro D, Price M, De Felice M, Di Lauro R. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development 1991; 113(4):1093-1104.
- Milenkovic M, De Deken X, Jin L et al. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. J Endocrinol 2007; 192(3):615-626.
- De Deken X, Wang D, Dumont JE, Miot F. Characterization of ThOX proteins as components of the thyroid H(2)O(2)-generating system. Exp Cell Res 2002; 273(2):187-196.
- El Hassani RA, Benfares N, Caillou B et al. Dual oxidase2 is expressed all along the digestive tract. Am J Physiol Gastrointest Liver Physiol 2005; 288(5):G933-G942.
- Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 2006; 281(27):18269-18272.
- Xie X, Hu J, Liu X et al. NIP/DuoxA is essential for Drosophila embryonic development and regulates oxidative stress response. Int J Biol Sci 2010; 6(3):252-267.
- Morand S, Chaaraoui M, Kaniewski J et al. Effect of iodide on nicotinamide adenine dinucleotide phosphate oxidase activity and Duox2 protein expression in isolated porcine thyroid follicles. Endocrinology 2003; 144(4):1241-1248.
- Hoste C, Dumont JE, Miot F, De D, X. The type of DUOX-dependent ROS production is dictated by defined sequences in DUOXA. Exp Cell Res 2012; 318(18):2353-2364.
- Morand S, Ueyama T, Tsujibe S, Saito N, Korzeniowska A, Leto TL. Duox maturation factors form cell surface complexes with Duox affecting the specificity of reactive oxygen species generation. FASEB J 2009; 23(4):1205-1218.
- Zamproni I, Grasberger H, Cortinovis F et al. Biallelic inactivation of the dual oxidase maturation factor 2 (DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab 2008; 93(2):605-610.
- Grasberger H, De Deken, X, Mayo OB et al. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol Endocrinol 2012; 26(3):481-492.
- Rigutto S, Hoste C, Grasberger H et al. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. J Biol Chem 2009; 284(11):6725-6734.
- Cardoso-Weide LC, Cardoso-Penha RC, Costa MW, Ferreira AC, Carvalho DP, Santisteban PS. DuOx2 Promoter Regulation by Hormones, Transcriptional Factors and the Coactivator TAZ. Eur Thyroid J 2015; 4(1):6-13.
- Christophe-Hobertus C, Christophe D. Human Thyroid Oxidases genes promoter activity in thyrocytes does not appear to be functionally dependent on Thyroid Transcription Factor-1 or Pax8. Mol Cell Endocrinol 2007; 264(1-2):157-163.
- Raad H, Eskalli Z, Corvilain B, Miot F, De Deken, X. Thyroid hydrogen peroxide production is enhanced by the Th2 cytokines, IL-4 and IL-13, through increased expression of the dual oxidase 2 and its maturation factor DUOXA2. Free Radic Biol Med 2013; 56:216-225.
- Moreno JC, Bikker H, Kempers MJ et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 2002; 347(2):95-102.
- Vigone MC, Fugazzola L, Zamproni I et al. Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Hum Mutat 2005; 26(4):395-402.
- Varela V, Rivolta CM, Esperante SA, Gruneiro-Papendieck L, Chiesa A, Targovnik HM. Three mutations (p.Q36H, p.G418fsX482, and g.IVS19-2A>C) in the dual oxidase 2 gene responsible for congenital goiter and iodide organification defect. Clin Chem 2006; 52(2):182-191.
- Maruo Y, Takahashi H, Soeda I et al. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab 2008; 93(11):4261-4267.
- Hoste C, Rigutto S, Van VG, Miot F, De Deken X. Compound heterozygosity for a novel hemizygous missense mutation and a partial deletion affecting the catalytic core of the H(2)O(2)-generating enzyme DUOX2 associated with transient congenital hypothyroidism. Hum Mutat 2010; 31(4):E1304-E1319.
- Jin HY, Heo SH, Kim YM et al. High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Horm Res Paediatr 2014; 82(4):252-260.
- Muzza M, Rabbiosi S, Vigone MC et al. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab 2014; 99(3):E544-E553.
- Wang F, Lu K, Yang Z et al. Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clin Endocrinol (Oxf) 2014; 81(3):452-457.
- Liu S, Liu L, Niu X, Lu D, Xia H, Yan S. A novel missense mutation (I26M) in DUOXA2 causing congenital goiter hypothyroidism impairs NADPH oxidase activity but not protein expression. J Clin Endocrinol Metab 2015; 100(4):1225-1229.
- Hulur I, Hermanns P, Nestoris C et al. A single copy of the recently identified dual oxidase maturation factor (DUOXA) 1 gene produces only mild transient hypothyroidism in a patient with a novel biallelic DUOXA2 mutation and monoallelic DUOXA1 deletion. J Clin Endocrinol Metab 2011; 96(5):E841-E845.
- Dunn JT, Dunn AD. Thyroglobulin: chemistry, biosynthesis, and proteolysis. In: Braverman LE, Utiger R, editors. The Thyroid. Philadelphia: Lippicott Williams & Wilkins, 2000: 91-104.
- Mendive FM, Rivolta CM, Moya CM, Vassart G, Targovnik HM. Genomic organization of the human thyroglobulin gene: the complete intron-exon structure. Eur J Endocrinol 2001; 145(4):485-496.
- Malthiery Y, Lissitzky S. Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur J Biochem 1987; 165(3):491-498.
- van de Graaf SA, Pauws E, de Vijlder JJ, Ris-Stalpers CR. The revised 8307 base pair coding sequence of human thyroglobulin transiently expressed in eukaryotic cells. Eur J Endocrinol 1997; 136(5):508-515.
- Di Lauro R., Avvedimento EV, Cerillo R, et al. Structure and function of the rat thyroglobulin gene. In: Eggo MC, Burrow GN, editors. Thyroglobulin-The prothyroid hormone. Progress in Endocrine Research and Therapy. New York: Raven Press, 1985: 77-86.
- Kim PS, Hossain SA, Park YN, Lee I, Yoo SE, Arvan P. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc Natl Acad Sci U S A 1998; 95(17):9909-9913.
- Mercken L, Simons MJ, Swillens S, Massaer M, Vassart G. Primary structure of bovine thyroglobulin deduced from the sequence of its 8,431-base complementary DNA. Nature 1985; 316(6029):647-651.
- van de Graaf SA, Ris-Stalpers C, Pauws E, Mendive FM, Targovnik HM, de Vijlder JJ. Up to date with human thyroglobulin. J Endocrinol 2001; 170(2):307-321.
- Lenarcic B, Krishnan G, Borukhovich R, Ruck B, Turk V, Moczydlowski E. Saxiphilin, a saxitoxin-binding protein with two thyroglobulin type 1 domains, is an inhibitor of papain-like cysteine proteinases. J Biol Chem 2000; 275(20):15572-15577.
- Molina F, Pau B, Granier C. The type-1 repeats of thyroglobulin regulate thyroglobulin degradation and T3, T4 release in thyrocytes. FEBS Lett 1996; 391(3):229-231.
- Swillens S, Ludgate M, Mercken L, Dumont JE, Vassart G. Analysis of sequence and structure homologies between thyroglobulin and acetylcholinesterase: possible functional and clinical significance. Biochem Biophys Res Commun 1986; 137(1):142-148.
- Lee J, Wang X, Di Jeso B, Arvan P. The cholinesterase-like domain, essential in thyroglobulin trafficking for thyroid hormone synthesis, is required for protein dimerization. J Biol Chem 2009; 284(19):12752-12761.
- Wang X, Lee J, Di Jeso B et al. Cis and trans actions of the cholinesterase-like domain within the thyroglobulin dimer. J Biol Chem 2010; 285(23):17564-17573.
- Park YN, Arvan P. The acetylcholinesterase homology region is essential for normal conformational maturation and secretion of thyroglobulin. J Biol Chem 2004; 279(17):17085-17089.
- Klein M, Gestmann I, Berndorfer U, Schmitz A, Herzog V. The thioredoxin boxes of thyroglobulin: possible implications for intermolecular disulfide bond formation in the follicle lumen. Biol Chem 2000; 381(7):593-601.
- Cao X, Kambe F, Ohmori S, Seo H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochem Biophys Res Commun 2002; 297(2):288-293.
- Kambe F, Nomura Y, Okamoto T, Seo H. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol Endocrinol 1996; 10(7):801-812.
- Tell G, Scaloni A, Pellizzari L, Formisano S, Pucillo C, Damante G. Redox potential controls the structure and DNA binding activity of the paired domain. J Biol Chem 1998; 273(39):25062-25072.
- Tell G, Pines A, Paron I et al. Redox effector factor-1 regulates the activity of thyroid transcription factor 1 by controlling the redox state of the N transcriptional activation domain. J Biol Chem 2002; 277(17):14564-14574.
- Kim PS, Arvan P. Folding and assembly of newly synthesized thyroglobulin occurs in a pre-Golgi compartment. J Biol Chem 1991; 266(19):12412-12418.
- Kim PS, Bole D, Arvan P. Transient aggregation of nascent thyroglobulin in the endoplasmic reticulum: relationship to the molecular chaperone, BiP. J Cell Biol 1992; 118(3):541-549.
- Kim PS, Arvan P. Calnexin and BiP act as sequential molecular chaperones during thyroglobulin folding in the endoplasmic reticulum. J Cell Biol 1995; 128(1-2):29-38.
- Martin-Belmonte F, Alonso MA, Zhang X, Arvan P. Thyroglobulin is selected as luminal protein cargo for apical transport via detergent-resistant membranes in epithelial cells. J Biol Chem 2000; 275(52):41074-41081.
- Muresan Z, Arvan P. Thyroglobulin transport along the secretory pathway. Investigation of the role of molecular chaperone, GRP94, in protein export from the endoplasmic reticulum. J Biol Chem 1997; 272(42):26095-26102.
- Muresan Z, Arvan P. Enhanced binding to the molecular chaperone BiP slows thyroglobulin export from the endoplasmic reticulum. Mol Endocrinol 1998; 12(3):458-467.
- Arima T, Spiro MJ, Spiro RG. Studies on the carbohydrate units of thyroglobulin. Evaluation of their microheterogeneity in the human and calf proteins. J Biol Chem 1972; 247(6):1825-1835.
- Yang SX, Pollock HG, Rawitch AB. Glycosylation in human thyroglobulin: location of the N-linked oligosaccharide units and comparison with bovine thyroglobulin. Arch Biochem Biophys 1996; 327(1):61-70.
- Spiro MJ. Presence of a glucuronic acid-containing carbohydrate unit in human thyroglobulin. J Biol Chem 1977; 252(15):5424-5430.
- Mayerhofer A, Amador AG, Beamer WG, Bartke A. Ultrastructural aspects of the goiter in cog/cog mice. J Hered 1988; 79(3):200-203.
- Kim PS, Kwon OY, Arvan P. An endoplasmic reticulum storage disease causing congenital goiter with hypothyroidism. J Cell Biol 1996; 133(3):517-527.
- Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev 1998; 19(2):173-202.
- Ohyama Y, Hosoya T, Kameya T et al. Congenital euthyroid goitre with impaired thyroglobulin transport. Clin Endocrinol (Oxf) 1994; 41(1):129-135.
- Targovnik HM, Vono J, Billerbeck AE et al. A 138-nucleotide deletion in the thyroglobulin ribonucleic acid messenger in a congenital goiter with defective thyroglobulin synthesis. J Clin Endocrinol Metab 1995; 80(11):3356-3360.
- Medeiros-Neto G, Kim PS, Yoo SE et al. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J Clin Invest 1996; 98(12):2838-2844.
- Cauvi D, Nlend MC, Venot N, Chabaud O. Sulfate transport in porcine thyroid cells. Effects of thyrotropin and iodide. Am J Physiol Endocrinol Metab 2001; 281(3):E440-E448.
- Consiglio E, Acquaviva AM, Formisano S et al. Characterization of phosphate residues on thyroglobulin. J Biol Chem 1987; 262(21):10304-10314.
- Spiro MJ, Gorski KM. Studies on the posttranslational migration and processing of thyroglobulin: use of inhibitors and evaluation of the role of phosphorylation. Endocrinology 1986; 119(3):1146-1158.
- Yamamoto K, Tsuji T, Tarutani O, Osawa T. Structural changes of carbohydrate chains of human thyroglobulin accompanying malignant transformations of thyroid glands. Eur J Biochem 1984; 143(1):133-144.
- Alvino CG, Acquaviva AM, Catanzano AM, Tassi V. Evidence that thyroglobulin has an associated protein kinase activity correlated with the presence of an adenosine triphosphate binding site. Endocrinology 1995; 136(8):3179-3185.
- Taurog A. Hormone synthesis. In: Braverman LE, Utiger R, editors. The Thyroid. Philadelphia: Lippincott Williams & Wilkins, 2000: 61-85.
- Ohtaki S, Nakagawa H, Nakamura M, Yamazaki I. Reactions of purified hog thyroid peroxidase with H2O2, tyrosine, and methylmercaptoimidazole (goitrogen) in comparison with bovine lactoperoxidase. J Biol Chem 1982; 257(2):761-766.
- Ohtaki S, Nakagawa H, Nakamura M, Yamazaki I. One- and two-electron oxidations of tyrosine, monoiodotyrosine, and diiodotyrosine catalyzed by hog thyroid peroxidase. J Biol Chem 1982; 257(22):13398-13403.
- Cahnmann HJ, Pommier J, Nunez J. Spatial requirement for coupling of iodotyrosine residues to form thyroid hormones. Proc Natl Acad Sci U S A 1977; 74(12):5333-5335.
- Deme D, Pommier J, Nunez J. Specificity of thyroid hormone synthesis. The role of thyroid peroxidase. Biochim Biophys Acta 1978; 540(1):73-82.
- Virion A, Pommier J, Deme D, Nunez J. Kinetics of thyroglobulin iodination and thyroid hormone synthesis catalyzed by peroxidases: the role of H2O2. Eur J Biochem 1981; 117(1):103-109.
- Virion A, Courtin F, Deme D, Michot JL, Kaniewski J, Pommier J. Spectral characteristics and catalytic properties of thyroid peroxidase-H2O2 compounds in the iodination and coupling reactions. Arch Biochem Biophys 1985; 242(1):41-47.
- Lamas L, Taurog A. The importance of thyroglobulin structure in thyroid peroxidase-catalyzed conversion of diiodotyrosine to thyroxine. Endocrinology 1977; 100(4):1129-1136.
- Gavaret JM, Nunez J, Cahnmann HJ. Formation of dehydroalanine residues during thyroid hormone synthesis in thyroglobulin. J Biol Chem 1980; 255(11):5281-5285.
- Gavaret JM, Cahnmann HJ, Nunez J. Thyroid hormone synthesis in thyroglobulin. The mechanism of the coupling reaction. J Biol Chem 1981; 256(17):9167-9173.
- Johnson TB, Tewkesbury LB. The Oxidation of 3,5-Diiodotyrosine to Thyroxine. Proc Natl Acad Sci U S A 1942; 28(3):73-77.
- Fassler CA, Dunn JT, Anderson PC et al. Thyrotropin alters the utilization of thyroglobulin's hormonogenic sites. J Biol Chem 1988; 263(33):17366-17371.
- Kim PS, Dunn JT, Kaiser DL. Similar hormone-rich peptides from thyroglobulins of five vertebrate classes. Endocrinology 1984; 114(2):369-374.
- Roe MT, Anderson PC, Dunn AD, Dunn JT. The hormonogenic sites of turtle thyroglobulin and their homology with those of mammals. Endocrinology 1989; 124(3):1327-1332.
- Xiao S, Dorris ML, Rawitch AB, Taurog A. Selectivity in tyrosyl iodination sites in human thyroglobulin. Arch Biochem Biophys 1996; 334(2):284-294.
- Lamas L, Anderson PC, Fox JW, Dunn JT. Consensus sequences for early iodination and hormonogenesis in human thyroglobulin. J Biol Chem 1989; 264(23):13541-13545.
- Marriq C, Lejeune PJ, Venot N, Vinet L. Hormone formation in the isolated fragment 1-171 of human thyroglobulin involves the couple tyrosine 5 and tyrosine 130. Mol Cell Endocrinol 1991; 81(1-3):155-164.
- Xiao S, Pollock HG, Taurog A, Rawitch AB. Characterization of hormonogenic sites in an N-terminal, cyanogen bromide fragment of human thyroglobulin. Arch Biochem Biophys 1995; 320(1):96-105.
- den Hartog MT, Sijmons CC, Bakker O, Ris-Stalpers C, de Vijlder JJ. Importance of the content and localization of tyrosine residues for thyroxine formation within the N-terminal part of human thyroglobulin. Eur J Endocrinol 1995; 132(5):611-617.
- Dunn AD, Corsi CM, Myers HE, Dunn JT. Tyrosine 130 is an important outer ring donor for thyroxine formation in thyroglobulin. J Biol Chem 1998; 273(39):25223-25229.
- Gentile F, Ferranti P, Mamone G, Malorni A, Salvatore G. Identification of hormonogenic tyrosines in fragment 1218-1591 of bovine thyroglobulin by mass spectrometry. Hormonogenic acceptor TYR-12donor TYR-1375. J Biol Chem 1997; 272(1):639-646.
- Duthoit C, Estienne V, Delom F et al. Production of immunoreactive thyroglobulin C-terminal fragments during thyroid hormone synthesis. Endocrinology 2000; 141(7):2518-2525.
- Dunn JT, Kim PS, Dunn AD. Favored sites for thyroid hormone formation on the peptide chains of human thyroglobulin. J Biol Chem 1982; 257(1):88-94.
- Dunn JT, Kim PS, Dunn AD, Heppner DG, Jr., Moore RC. The role of iodination in the formation of hormone-rich peptides from thyroglobulin. J Biol Chem 1983; 258(15):9093-9099.
- Wolff J, Chaikoff IL. Plasma inorganic iodide as a homeostatic regulator of thyroid function. J Biol Chem 1948; 174(2):555-564.
- de Vijlder JJ, Veenboer GJ, van Dijk JE. Thyroid albumin originates from blood. Endocrinology 1992; 131(2):578-584.
- De Groot. LJ, Hall. R, McDermott WV, Davis. AM. Hashimoto's thyroiditis. A genetically conditioned disease. N Engl J Med 1962; 267:267-273.
- De Groot. LJ, Stanbury JB. The syndrome of congenital goiter with butanol-insoluble serum iodine. Am J Med 1959; 27:586-595.
- Stanbury JB, Janssen. MA. Labeled iodoalbumin in the plasma in thyrotoxicosis after I-125 and I-131. J Clin Endocrinol Metab 1963; 23:1056-1058.
- Robbins J, Wolff J, Rall JE. Iodoproteins in normal and abnormal human thyroid tissue and in normal sheep thyroid. Endocrinology 1959; 64(1):37-52.
- Gire V, Kostrouch Z, Bernier-Valentin F, Rabilloud R, Munari-Silem Y, Rousset B. Endocytosis of albumin and thyroglobulin at the basolateral membrane of thyrocytes organized in follicles. Endocrinology 1996; 137(2):522-532.
- Boeynaems JM, Van Sande J., Dumont JE. Which iodolipids are involved in thyroid autoregulation: iodolactones or iodoaldehydes? Eur J Endocrinol 1995; 132(6):733-734.
- Gartner R, Dugrillon A, Bechtner G. Iodolipids and thyroid function and growth. In: Nauman J, Glinoer D, Braverman LE, Hostalek U, editors. The Thyroid and Iodine. Stutgard,New York: Merck European Thyroid Symposium, 2000: 19-27.
- Panneels V, Macours P, Van den BH, Braekman JC, Van SJ, Boeynaems JM. Biosynthesis and metabolism of 2-iodohexadecanal in cultured dog thyroid cells. J Biol Chem 1996; 271(38):23006-23014.
- Pereira A, Braekman JC, Dumont JE, Boeynaems JM. Identification of a major iodolipid from the horse thyroid gland as 2-iodohexadecanal. J Biol Chem 1990; 265(28):17018-17025.
- Ohayon R, Boeynaems JM, Braekman JC, Van den Bergen H, Gorin Y, Virion A. Inhibition of thyroid NADPH-oxidase by 2-iodohexadecanal in a cell-free system. Mol Cell Endocrinol 1994; 99(1):133-141.
- Panneels V, Van den Bergen H, Jacoby C et al. Inhibition of H2O2 production by iodoaldehydes in cultured dog thyroid cells. Mol Cell Endocrinol 1994; 102(1-2):167-176.
- Fonlupt P, Audebet C, Gire V, Bernier-Valentin F, Rousset B. Calcium is transported into the lumen of pig thyroid follicles by fluid phase basolateral to apical transcytosis. J Cell Physiol 1997; 171(1):43-51.
- Haeberli A, Millar FK, Wollman SH. Accumulation and localization of radiocalcium in the rat thyroid gland. Endocrinology 1978; 102(5):1511-1519.
- Ekholm R. Iodination of thyroglobulin. An intracellular or extracellular process? Mol Cell Endocrinol 1981; 24(2):141-163.
- Wollman SH, Ekholm R. Site of iodination in hyperplastic thyroid glands deduced from autoradiographs. Endocrinology 1981; 108(6):2082-2085.
- Gerber H, Studer H, von GC. Paradoxical effects of thyrotropin on diffusion of thyroglobulin in the colloid of rat thyroid follicles after long term thyroxine treatment. Endocrinology 1985; 116(1):303-310.
- Ofverholm T, Ericson LE. Intraluminal iodination of thyroglobulin. Endocrinology 1984; 114(3):827-835.
- Herzog V. Transcytosis in thyroid follicle cells. J Cell Biol 1983; 97(3):607-617.
- Berndorfer U, Wilms H, Herzog V. Multimerization of thyroglobulin (TG) during extracellular storage: isolation of highly cross-linked TG from human thyroids. J Clin Endocrinol Metab 1996; 81(5):1918-1926.
- Baudry N, Lejeune PJ, Delom F, Vinet L, Carayon P, Mallet B. Role of multimerized porcine thyroglobulin in iodine storage. Biochem Biophys Res Commun 1998; 242(2):292-296.
- Saber-Lichtenberg Y, Brix K, Schmitz A et al. Covalent cross-linking of secreted bovine thyroglobulin by transglutaminase. FASEB J 2000; 14(7):1005-1014.
- Delom F, Mallet B, Carayon P, Lejeune PJ. Role of extracellular molecular chaperones in the folding of oxidized proteins. Refolding of colloidal thyroglobulin by protein disulfide isomerase and immunoglobulin heavy chain-binding protein. J Biol Chem 2001; 276(24):21337-21342.
- Bernier-Valentin F, Kostrouch Z, Rabilloud R, Munari-Silem Y, Rousset B. Coated vesicles from thyroid cells carry iodinated thyroglobulin molecules. First indication for an internalization of the thyroid prohormone via a mechanism of receptor-mediated endocytosis. J Biol Chem 1990; 265(28):17373-17380.
- Bernier-Valentin F, Kostrouch Z, Rabilloud R, Rousset B. Analysis of the thyroglobulin internalization process using in vitro reconstituted thyroid follicles: evidence for a coated vesicle-dependent endocytic pathway. Endocrinology 1991; 129(4):2194-2201.
- Pierce LR, Zurzolo C, Salvatore G, Edelhoch H. Coated vesicles from the thyroid gland: isolation, characterization, and a search for a possible role in thyroglobulin transport. J Endocrinol Invest 1985; 8(4):303-312.
- Kostrouch Z, Bernier-Valentin F, Munari-Silem Y, Rajas F, Rabilloud R, Rousset B. Thyroglobulin molecules internalized by thyrocytes are sorted in early endosomes and partially recycled back to the follicular lumen. Endocrinology 1993; 132(6):2645-2653.
- Kostrouch Z, Bernier-Valentin F, Munari-Silem Y, Rajas F, Rabilloud R, Rousset B. Thyroglobulin molecules internalized by thyrocytes are sorted in early endosomes and partially recycled back to the follicular lumen. Endocrinology 1993; 132(6):2645-2653.
- Miquelis R, Courageot J, Jacq A, Blanck O, Perrin C, Bastiani P. Intracellular routing of GLcNAc-bearing molecules in thyrocytes: selective recycling through the Golgi apparatus. J Cell Biol 1993; 123(6 Pt 2):1695-1706.
- Romagnoli P, Herzog V. Transcytosis in thyroid follicle cells: regulation and implications for thyroglobulin transport. Exp Cell Res 1991; 194(2):202-209.
- Cortese F, Schneider AB, Salvatore G. Isopycnic centrifugation of thyroid iodoproteins: selectivity of endocytosis. Eur J Biochem 1976; 68(1):121-129.
- van den Hove MF, Couvreur M, de Visscher M, Salvatore G. A new mechanism for the reabsorption of thyroid iodoproteins: selective fluid pinocytosis. Eur J Biochem 1982; 122(2):415-422.
- Marino M, McCluskey RT. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. Am J Physiol Cell Physiol 2000; 279(5):C1295-C1306.
- Consiglio E, Salvatore G, Rall JE, Kohn LD. Thyroglobulin interactions with thyroid plasma membranes. The existence of specific receptors and their potential role. J Biol Chem 1979; 254(12):5065-5076.
- Consiglio E, Shifrin S, Yavin Z et al. Thyroglobulin interactions with thyroid membranes. Relationship between receptor recognition of N-acetylglucosamine residues and the iodine content of thyroglobulin preparations. J Biol Chem 1981; 256(20):10592-10599.
- Montuori N, Pacifico F, Mellone S et al. The rat asialoglycoprotein receptor binds the amino-terminal domain of thyroglobulin. Biochem Biophys Res Commun 2000; 268(1):42-46.
- Mezgrhani H, Mziaut H, Courageot J, Oughideni R, Bastiani P, Miquelis R. Identification of the membrane receptor binding domain of thyroglobulin. Insights into quality control of thyroglobulin biosynthesis. J Biol Chem 1997; 272(37):23340-23346.
- Miquelis R, Alquier C, Monsigny M. The N-acetylglucosamine-specific receptor of the thyroid. Binding characteristics, partial characterization, and potential role. J Biol Chem 1987; 262(31):15291-15298.
- Marino M, Zheng G, McCluskey RT. Megalin (gp330) is an endocytic receptor for thyroglobulin on cultured fisher rat thyroid cells. J Biol Chem 1999; 274(18):12898-12904.
- Marino M, Friedlander JA, McCluskey RT, Andrews D. Identification of a heparin-binding region of rat thyroglobulin involved in megalin binding. J Biol Chem 1999; 274(43):30377-30386.
- Marino M, Zheng G, Chiovato L et al. Role of megalin (gp330) in transcytosis of thyroglobulin by thyroid cells. A novel function in the control of thyroid hormone release. J Biol Chem 2000; 275(10):7125-7137.
- Zheng G, Marino M, Zhao J, McCluskey RT. Megalin (gp330): a putative endocytic receptor for thyroglobulin (Tg). Endocrinology 1998; 139(3):1462-1465.
- Lisi S, Pinchera A, McCluskey RT et al. Preferential megalin-mediated transcytosis of low-hormonogenic thyroglobulin: a control mechanism for thyroid hormone release. Proc Natl Acad Sci U S A 2003; 100(25):14858-14863.
- Neve P, Willems C, Dumont JE. Involvement of the microtubule-microfilament system in thyroid secretion. Exp Cell Res 1970; 63(2):457-460.
- Engstrom G, Ericson LE. Effect of graded doses of thyrotropin on exocytosis and early phase of endocytosis in the rat thyroid. Endocrinology 1981; 108(2):399-405.
- Ericson LE, Engstrom G. Quantitative electron microscopic studies on exocytosis and endocytosis in the thyroid follicle cell. Endocrinology 1978; 103(3):883-892.
- Cochaux P, Van Sande J, Dumont JE. Inhibition of the cyclic AMP-adenylate cyclase system and of secretion by high concentrations of adenosine in the dog thyroid. Biochem Pharmacol 1982; 31(23):3763-3767.
- Johanson V, Ofverholm T, Ericson LE. Forskolin-induced elevation of cyclic AMP does not cause exocytosis and endocytosis in rat thyroid follicle cells in vivo. Mol Cell Endocrinol 1988; 59(1-2):27-34.
- Dunn AD, Dunn JT. Thyroglobulin degradation by thyroidal proteases: action of purified cathepsin D. Endocrinology 1982; 111(1):280-289.
- Dunn AD, Dunn JT. Thyroglobulin degradation by thyroidal proteases: action of thiol endopeptidases in vitro. Endocrinology 1982; 111(1):290-298.
- Dunn AD, Dunn JT. Cysteine proteinases from human thyroids and their actions on thyroglobulin. Endocrinology 1988; 123(2):1089-1097.
- Dunn AD, Crutchfield HE, Dunn JT. Thyroglobulin processing by thyroidal proteases. Major sites of cleavage by cathepsins B, D, and L. J Biol Chem 1991; 266(30):20198-20204.
- Dunn AD, Crutchfield HE, Dunn JT. Proteolytic processing of thyroglobulin by extracts of thyroid lysosomes. Endocrinology 1991; 128(6):3073-3080.
- Dunn AD, Myers HE, Dunn JT. The combined action of two thyroidal proteases releases T4 from the dominant hormone-forming site of thyroglobulin. Endocrinology 1996; 137(8):3279-3285.
- Nakagawa H, Ohtaki S. Partial purification and characterization of two thiol proteases from hog thyroid lysosomes. Endocrinology 1984; 115(1):33-40.
- Yoshinari M, Taurog A. Lysosomal digestion of thyroglobulin: role of cathepsin D and thiol proteases. Endocrinology 1985; 117(4):1621-1631.
- Nakagawa H, Ohtaki S. Thyroxine (T4) release from thyroglobulin and its T4-containing peptide by thyroid thiol proteases. Endocrinology 1985; 116(4):1433-1439.
- Alquier C, Guenin P, Munari-Silem Y, Audebet C, Rousset B. Isolation of pig thyroid lysosomes. Biochemical and morphological characterization. Biochem J 1985; 232(2):529-537.
- Rousset B, Selmi S, Bornet H, Bourgeat P, Rabilloud R, Munari-Silem Y. Thyroid hormone residues are released from thyroglobulin with only limited alteration of the thyroglobulin structure. J Biol Chem 1989; 264(21):12620-12626.
- Selmi S, Rousset B. Identification of two subpopulations of thyroid lysosomes: relation to the thyroglobulin proteolytic pathway. Biochem J 1988; 253(2):523-532.
- Brix K, Linke M, Tepel C, Herzog V. Cysteine proteinases mediate extracellular prohormone processing in the thyroid. Biol Chem 2001; 382(5):717-725.
- Friedrichs B, Tepel C, Reinheckel T et al. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest 2003; 111(11):1733-1745.
- Linke M, Herzog V, Brix K. Trafficking of lysosomal cathepsin B-green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J Cell Sci 2002; 115(Pt 24):4877-4889.
- Tepel C, Bromme D, Herzog V, Brix K. Cathepsin K in thyroid epithelial cells: sequence, localization and possible function in extracellular proteolysis of thyroglobulin. J Cell Sci 2000; 113 Pt 24:4487-4498.
- Rousset B, Mornex R. The thyroid hormone secretory pathway--current dogmas and alternative hypotheses. Mol Cell Endocrinol 1991; 78(1-2):C89-C93.
- Andersson HC, Kohn LD, Bernardini I, Blom HJ, Tietze F, Gahl WA. Characterization of lysosomal monoiodotyrosine transport in rat thyroid cells. Evidence for transport by system h. J Biol Chem 1990; 265(19):10950-10954.
- Tietze F, Kohn LD, Kohn AD et al. Carrier-mediated transport of monoiodotyrosine out of thyroid cell lysosomes. J Biol Chem 1989; 264(9):4762-4765.
- Dumitrescu AM, Refetoff S. Novel biological and clinical aspects of thyroid hormone metabolism. Endocr Dev 2007; 10:127-139.
- van der Deure WM, Peeters RP, Visser TJ. Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters. J Mol Endocrinol 2010; 44(1):1-11.
- Salvatore D, Tu H, Harney JW, Larsen PR. Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest 1996; 98(4):962-968.
- Toyoda N, Nishikawa M, Mori Y et al. Identification of a 27-kilodalton protein with the properties of type I iodothyronine 5'-deiodinase in human thyroid gland. J Clin Endocrinol Metab 1992; 74(3):533-538.
- Laurberg P. T4 and T3 release from the perfused canine thyroid isolated in situ. Acta Endocrinol (Copenh) 1976; 83(1):105-113.
- Toyoda N, Nishikawa M, Mori Y et al. Thyrotropin and triiodothyronine regulate iodothyronine 5'-deiodinase messenger ribonucleic acid levels in FRTL-5 rat thyroid cells. Endocrinology 1992; 131(1):389-394.
- Fisher DA, Oddie TH, Thompson CS. Thyroidal thyronine and non-thyronine iodine secretion in euthyroid subjects. J Clin Endocrinol Metab 1971; 33(4):647-652.
- Rosenberg IN, Goswami A. Purification and characterization of a flavoprotein from bovine thyroid with iodotyrosine deiodinase activity. J Biol Chem 1979; 254(24):12318-12325.
- Gnidehou S, Caillou B, Talbot M et al. Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J 2004; 18(13):1574-1576.
- Gnidehou S, Lacroix L, Sezan A et al. Cloning and characterization of a novel isoform of iodotyrosine dehalogenase 1 (DEHAL1) DEHAL1C from human thyroid: comparisons with DEHAL1 and DEHAL1B. Thyroid 2006; 16(8):715-724.
- Moreno JC. Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res 2003; 60 Suppl 3:96-102.
- Moreno JC, Klootwijk W, van Toor H et al. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med 2008; 358(17):1811-1818.
- Medeiros-Neto G, Stanbury JB. The iodotyrosine deiodinase defect. Inherited Disorders of the Thyroid System. CRC Press, 1994: 139-159.
- Kubota K, Uchimura H, Mitsuhashi T, Chiu SC, Kuzuya N, Nagataki S. Effects of intrathyroidal metabolism of thyroxine on thyroid hormone secretion: increased degradation of thyroxine in mouse thyroids stimulated chronically with thyrotrophin. Acta Endocrinol (Copenh) 1984; 105(1):57-65.
- Eggo MC, Burrow GN. Integrated regulation of growth and of function. Adv Exp Med Biol 1989; 261:327-339.
- Hjort T. Determination of serum-thyroglobulin by a haemagglutination-inhibition test. Lancet 1961; 1(7189):1262-1264.
- Roitt IM, Torrigiani G. Identification and estimation of undegraded thyroglobulin in human serum. Endocrinology 1967; 81(3):421-429.
- Schneider AB, Ikekubo K, Kuma K. Iodine content of serum thyroglobulin in normal individuals and patients with thyroid tumors. J Clin Endocrinol Metab 1983; 57(6):1251-1256.
- Druetta L, Croizet K, Bornet H, Rousset B. Analyses of the molecular forms of serum thyroglobulin from patients with Graves' disease, subacute thyroiditis or differentiated thyroid cancer by velocity sedimentation on sucrose gradient and Western blot. Eur J Endocrinol 1998; 139(5):498-507.
- Druetta L, Bornet H, Sassolas G, Rousset B. Identification of thyroid hormone residues on serum thyroglobulin: a clue to the source of circulating thyroglobulin in thyroid diseases. Eur J Endocrinol 1999; 140(5):457-467.
- Spencer CA. Serum thyroglobulin measurements: clinical utility and technical limitations in the management of patients with differentiated thyroid carcinomas. Endocr Pract 2000; 6(6):481-484.
- Ladenson PW, Braverman LE, Mazzaferri EL et al. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma. N Engl J Med 1997; 337(13):888-896.
- Mazzaferri EL, Robbins RJ, Spencer CA et al. A consensus report of the role of serum thyroglobulin as a monitoring method for low-risk patients with papillary thyroid carcinoma. J Clin Endocrinol Metab 2003; 88(4):1433-1441.
- McDougall IR, Weigel RJ. Recombinant human thyrotropin in the management of thyroid cancer. Curr Opin Oncol 2001; 13(1):39-43.
- Robbins RJ, Tuttle RM, Sharaf RN et al. Preparation by recombinant human thyrotropin or thyroid hormone withdrawal are comparable for the detection of residual differentiated thyroid carcinoma. J Clin Endocrinol Metab 2001; 86(2):619-625.
- Schlumberger M, Ricard M, Pacini F. Clinical use of recombinant human TSH in thyroid cancer patients. Eur J Endocrinol 2000; 143(5):557-563.
- Wartofsky L. Management of low-risk well-differentiated thyroid cancer based only on thyroglobulin measurement after recombinant human thyrotropin. Thyroid 2002; 12(7):583-590.
- Song Y, Massart C, Chico-Galdo V et al. Species specific thyroid signal transduction: conserved physiology, divergent mechanisms. Mol Cell Endocrinol 2010; 319(1-2):56-62.
Publication Details
Author Information and Affiliations
Publication History
Last Update: September 2, 2015.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Rousset B, Dupuy C, Miot F, et al. Chapter 2 Thyroid Hormone Synthesis And Secretion. [Updated 2015 Sep 2]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.