

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

INTRODUCTION
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare chromaffin cells tumors (PPGLs) characterized by the production, storage, metabolism, and secretion of catecholamines and their metabolites, metanephrines and methoxytyramine. These tumors can raise significant challenges in clinical recognition, diagnosis, and therapy and when undiagnosed can result in severe morbidity as well as mortality, especially due to cardiovascular system toxicity. Despite anatomical dissimilarity – PCC arising from adrenal medulla and PGLs from extra-adrenal sympathetic or parasympathetic paraganglia – these tumors display common embryonic origin, enzymatic milieu, and the ability to produce catecholamines and their metabolites. While once thought as mostly benign and biochemically active, these tumors can show a wide spectrum of cellular and biochemical dedifferentiation, including an aggressive metastatic course and biochemical silence.
The PPGL field has undergone a significant transformation in recent years. We now know that PPGLs represent the highest hereditary-driven endocrine condition with up to 40% of cases related to mutations in 15 well-established driver genes and a growing number of disease-modifying genes (now about 25). We now also appreciate that a significant proportion of what previously was thought to be almost exclusively benign disease is actually malignant and can display biochemical silence and a specific secretory profile with mainly elevated norepinephrine and/or dopamine.
Unfortunately, and despite tremendous advances in our understanding of the biology of PPGLs, the severity of disease-associated morbidity still remains significant since most of these tumors are not well recognized and diagnosis is delayed.
Definition
What would define actual urgency or emergency in PPGL? Is it a biochemical phenotype closely associated with a particular catecholamine secretion or various biomarkers suggestive of dedifferentiation and thus a malignant course, disease symptomatology, or the rapidity of disease progression? Is it an overall basic patient’s health status that can rapidly deteriorate or the expected complications from surgery? Is it the level of comfort and experience of the managing endocrinologist or abilities of the operating surgeon or possible lack of these? Or maybe it is the ability of the patient to follow-up frequently enough for appropriate management or the affordability of tests and medications or lack of those (some due to their price) which would eventually associate with a grim outcome. At the end of the day, it is probably all of the above and even more including some psychological aspects and fear to develop metastatic disease. Any disease that can potentially deteriorate to severely morbid outcomes needs to be seen as urgent and emergent most of the time. Obviously, in the case of a severe hypertensive crisis in the operating room that developed shortly after a previously undiagnosed/misdiagnosed abdominal mass was manipulated, the diagnosis that will drive appropriate therapy will be acute catecholamine crisis and there is a universal awareness of such a situation. Same should be true in case of severe therapy-resistant hypertension which rapidly deteriorates with use of β-adrenoceptor blockers. Unfortunately, there are many other possible scenarios that could start relatively slow and rapidly deteriorate to true medical emergencies. An example is our recent case that shows all aspects of PPGL management, including emergencies. A young female in late pregnancy was admitted for increased BP, thought to be related to noncompliance with BP medications and possible development of pre-eclampsia. Because of resistance to therapy while hospitalized the patient had an assessment of plasma catecholamines, which came back markedly elevated. Her imaging studies showed a 12 cm abdominal PGL with the fetus’ head laying directly on it. The tumor was massively vascularized and engulfed major abdominal vessels. The team that discussed her management could fill a lecture hall and included obstetrics, gynecologic surgery, endocrine surgery, general surgery, vascular surgery, anesthesiology, neonatology ICU, medical ICU, endocrinology, nursing and many more. All of the above worked hard on the day of combined caesarian section delivery and open abdominal surgery, which was complicated, but resulted in full recovery for both mother and baby.
CLINICAL ASPECTS
Clinical course and outcomes of excessive catecholamine secretion by PPGLs closely correlate with multiple factors related to the biochemistry of catecholamine action, secretory profile, acuity, and severity of actual hypercatecholaminemia. This gets very complicated by the fact that clinical symptomatology of hypercatecholaminemia lacks specificity and often presents as much more prevalent conditions, like hypertension, anxiety, or cardiac arrhythmias. If there is a single most important factor to define the overall outcome of the disease, we personally would pick timely suspicion and initiation of appropriate workup. While hypertension – paroxysmal or sustained – usually represents the initial or most common symptom, the overall clinical symptomatology varies widely and is summarized in Table 1.
Table 1.
Clinical Syndromes Related to PPGL
| Organ | Syndrome | Mechanism | Receptor action |
|---|---|---|---|
| Heart | Angina Heart attack Cardiomyopathies Myocarditis Arrythmias Heart failure | Coronary spasm Positive inotropy Positive chronotropy Unmatched O2 demand Hypoperfusion | Coronary α1, β2 Conducting system β1, β2 Conducting β1, β2 |
| Brain | Stroke Encephalopathy | Vasoconstriction Unmatched O2 demand Hypoperfusion | Cerebral arterioles α1 Effect of systemic HTN |
| Vascular | Shock Postural hypotension Aortic dissection Organ ischemia Limb ischemia | Arteriolar vasoconstriction Arteriolar vasodilation Vasodilation Unmatched O2 demand Hypoperfusion | Vascular α1, α2, β2 |
| Kidneys | ARF Hematuria | Vasoconstriction Vasodilation Increased renin secretion Unmatched O2 demand Hypoperfusion | Vascular α1, α2, β1, β2 |
| Lungs | Pulmonary edema ARDS Fibrosis Pulmonary HTN | Vasoconstriction Vasodilation Bronchodilation | Vascular α1, α2, β2 |
| GI | Intestinal ischemia | Vasoconstriction Unmatched O2 demand Hypoperfusion | Visceral arterioles α1, β2 |
Physiology of Catecholamine Action
Catecholamine production takes place in both adrenals, as well as sympathetic paraganglia. The synthetic pathway is specific for the abovementioned organs, defined by the unique set of intracellular enzymes able to convert the amino acid tyrosine to end product epinephrine or norepinephrine, dependent on the site of the synthesis. The actual catecholamine is related to the site/type of the cell, as well as degree or differentiation or lack of such (Figure 1). The transport of tyrosine through the cell membrane is active process and carried out by a member of the amino acid transporter family – large neutral amino acid transporters of the L family, mostly LAT1. These transporters can be induced and show overexpression, especially in some cancers. Dedifferentiated/malignant PPGL can produce a phenomenon of massive or predominantly norepinephrine production/secretion profile driven by the both overexpressed LAT1 and the lack of phenylethanolamine N-methyltransferase (PNMT) – the enzyme that converts norepinephrine to epinephrine.

Figure 1.
Catecholamine Synthesis
Although currently we manage both conditions as part of a single syndrome, the physiology of catecholamine production and secretion from both systems is relatively distinct. Adrenals – the mastermind of the fight and flight response – are designed to produce significant amounts of epinephrine/adrenaline, to be secreted in response to stress. The secretion pattern is episodic/paroxysmal and relatively short lived. Epinephrine, the end-product of the synthetic pathway, is stored in secretory granules and secreted on an as-needed basis. Norepinephrine in this case is a co-secretory catecholamine, somewhat different in affinity to adrenergic receptors – through which both substances are signaling (Figure 2 and 3). In cases of catecholamine-producing tumors, the pattern of secretion can vary between paroxysmal or sustained, while the ratio between two catecholamines relates to some degree to the level of differentiation of the adenomatous tissue. Sympathetic paraganglia, on the other hand, is mostly involved in production of norepinephrine as a major sympathetic neurotransmitter rather than as a systemic hormone. The product is mostly secreted into the synaptic space, which than spills over into the systemic circulation. In the physiologic state, a significant amount of norepinephrine re-uptake back into the pre-synapse for repeated use occurs. In the case of PGLs, the increased amount of product reaches the systemic circulation to produce symptoms and signs indistinguishable from adrenally secreted norepinephric clinical picture of adrenergic overactivity.

Figure 2.
Adrenergic Receptors and Ligands
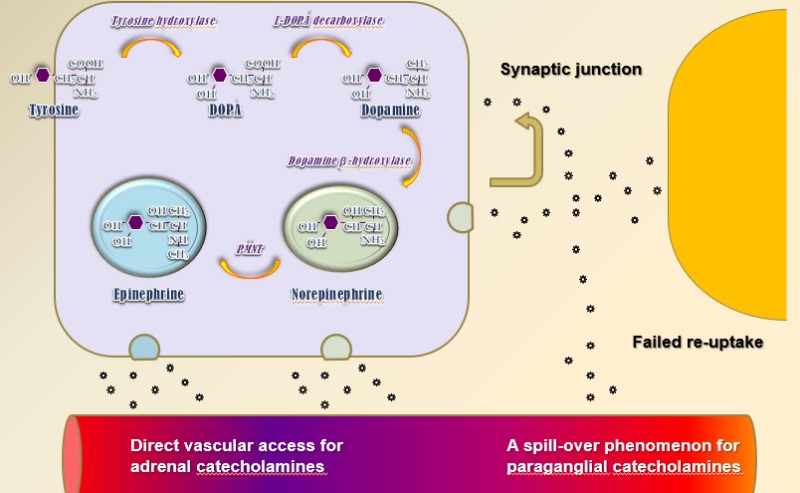
Figure 3.
Catecholamine Secretion
Catecholamines, both epinephrine and norepinephrine act through activation of the G protein coupled adrenergic receptors (GPCRs), both α1 and 2 and β1 and 2 with minor difference in the fact that norepinephrine has lower affinity to β2-adrenoceptors and thus norepinephric hypercatecholaminemia lack a mild component of peripheral vasodilation and could have slightly different clinical appearance compared to purely epinephric hypercatecholaminemia Table 1 and Figure 2). As with other GPCRs, adrenoceptors can undergo desensitization, which could explain the different clinical presentations in relatively mild long-standing disease compared to more rapidly developing hypercatecholaminemia. One also needs to remember that in massive biochemical hypercatecholaminemia, competitive α- and β-adrenoceptor blockers could be overwhelmed by the concentration of the ligand and safe preoperative adrenoceptor blockade can take longer to achieve and can be partial rather than complete.
Clinical Features
Hypercatecholaminemia-related endocrine emergencies define rare, but truly severe and potentially deadly end of the clinical spectrum of the PPGL syndrome. While it is called a great masquerader, this is misleading because it is not that the disease that masquerades, but rather because of the fact that clinical symptomology is completely non-specific and lacks any definitive symptom or signs that would point towards PPGL as a sole contender. It rather presents with symptoms and signs of much more prevalent conditions – like hypertension, benign cardiac arrhythmias, anxiety – and thus, progresses towards acute or chronic complications without being suspected. Needless to say, that unless the disease is severe or acute, it could be treated as a mainstay symptom-driven state – like hypertension – with at least some success. Clinical emergencies, related to the PPGL represent a completely different scenario – these are usually either unsuspected or only partially treated cases with severe short-term morbidity and significant mortality. In these cases, clinical suspicion is an absolute cornerstone of the management and the delay in diagnosis is adversely proportional to the overall outcome. Clinical scenarios with resultant PPGL-related emergencies usually include unrelated surgeries, where overall stress or tumor manipulation results in massive and acute hypercatecholaminemia with fully sensitized adrenergic receptors and lack of any adrenoceptor blockade, which precipitates acute and severe hypertensive crisis and potentially multiorgan failure. Less dramatic, but still a potentially severe condition includes treating progressive hypertension in the general or obstetric population with a medication that predisposes to unopposed α-adrenoceptor stimulation and thus precipitates severe peripheral vasoconstriction and either worsening of hypertension or heart failure. Obviously, patients with pre-existing heart or renal failure will be much more susceptible to severe outcomes. Because of the fact that some tumors express slow biochemical progression, we need to keep a high index of suspicion not only for patients with resistant HTN, familial HTN, or young age of onset, but for any patient who might have potential to have this disease.
Acute vs chronic hypercatecholaminemia
While an acute increase in catecholamine levels is directly responsible for precipitation of a hypertensive crisis through vascular vasoconstriction and positive inotropy, a long-lasting increase in catecholamine levels, especially of relatively mild degree, can be completely asymptomatic. This can probably be explained to some degree by several physiologic processes, including desensitization of the adrenergic receptors. Slowly progressive disease will mask, at least partially, clinical symptomatology, as well as allow sometime for the patient to try antihypertensive, antiarrhythmic, or antianxiety therapies as part of the therapy for aforementioned nonspecific conditions, as well as clinically desensitize the patient to mild hypertensive symptoms.
As mentioned above, clinical scenarios will mostly associate with unrelated surgeries, obstetric conditions like delivery and pre-eclampsia, as well as sudden or rapidly progressive deterioration of a previously stable person with significant conditions that would be sensitive to rapid increases in BP, pulse rate, or overall oxygen requirement. Currently, there is a well-accepted awareness, especially in the operating/delivery rooms, that sudden and rapid increases in systolic BP must be treated immediately by medications capable to act in the hypercatecholaminemic state. There is also a sufficient awareness of predominant β-adrenoceptor activity of labetalol, which could provide only partial α-blockage and be insufficient alone in full hypercatecholaminemic crisis. On the other hand, IV phentolamine is not a readily available operating room medication and this leaves nitroprusside as a medication mostly available in the operating room settings. Its use for prolonged and complicated surgery or delivery could possibly be associated with generation of methemoglobin and thiocyanite in the patient or the newborn. Because of the fact that majority of acute and severe hypercatecholaminemic states will have either mixed adrenergic/noradrenergic or noradrenergic biochemical phenotype, there is less expectation of β2 -adrenoceptor driven vasodilation and orthostasis.
Severe vs mild hypercatecholaminemia
It is accepted that patients with mild hypercatecholaminemia can be relatively asymptomatic or mildly symptomatic with some response to the usual antihypertensive therapy, thus disease can be present for a significant length of time undiagnosed. Severe hypercatecholaminemia, on the other hand, is markedly symptomatic and should be suspected right away. Clinical problems arise in cases when this happens during unrelated surgeries, as stated above, especially when an unrecognized abdominal mass, which in this case will be a PGL, is manipulated and releases massive amounts of pre-synthesized catecholamines. These cases are rare and close to impossible to predict, but in cases of severe intra-operative or intra-labor hypertension, should be immediately suspected and treated. Another scenario represents rapidly progressive disease in a younger patient – these are usually familial paragangliomas that can rapidly progress and metastasize. In this case, younger patients present with what suggest anxiety, especially in patients with an episodic secretory profile. Appropriate diagnosis can be significantly delayed when these patients enter the “outpatient workup mode” with infrequent appointments to assess the efficacy of anti-anxiety medications. This delay in diagnosis can associate with development of significant complications in patients with other pre-existing conditions. Obviously, acute concomitant illnesses will precipitate acute hypertensive crisis. Although over-suspicion could result in significant number of questionably necessary tests, it seems reasonable to test keeping in mind potentially morbid outcomes of severe untreated hypercatecholaminemia.
Episodic vs continuous secretion
Both episodic and sustained secretion of catecholamines can produce hypertension as well as an acute crisis. One can argue that the episodic form is more symptomatic owing to the nature of an on and off symptomatology that can be easier to detect for both the patient and the physician. We are not aware of differential adrenoceptor desensitization of episodic hypercatecholaminemia when compared to a persistent secretory state. Both forms are capable of rapid secretion of massive amounts of catecholamines in case of stress or manipulation, so the actual presentation or management of acute hypertensive emergency will not differ.
Large vs small mass
Historically, the size of the mass was thought to be proportional to the biochemical activity of PCC, with the exception of larger tumors, which were thought to overgrow their vascular supply, become necrotic and decrease the ability to be significantly active biochemically. Current knowledge complicates this to a significant degree because of several added details. The degree of differentiation of PPGL can massively affect both the actual profile of the secreted catecholamines (the higher the differentiation, the more probable the synthetic catecholamine pathway leads to epinephrine), as well as the amount of secreted catecholamines, where lesser differentiation could associate with a significant decrease in the amount of the synthetic catecholamine.
One should also remember that larger intra-abdominal masses can also result in local tissue invasion, including large or multiple vessels, adjacent organs etc. In this case, knowing the anatomical relationship between the tumor and the adjacent tissues can help avoid a potentially prolonged and complicated surgery.
We are also historically aware of the fact that the actual size of the adrenal tumor correlates with possible metastatic/malignant state/course. In PPGL this postulate is also relative, making genetic milieu more important factor for the prediction of malignancy (like SDHB mutation or younger age), then the actual size of the initial adrenal mass. It worth mentioning that multiple masses and PGLs per se will have a higher predisposition to malignancy as compared to a single adrenal pheochromocytoma.
In any case, finding a small PPGL and assuming that there would be no significant hypercatecholaminemia during stress or surgery is as wrong as finding a large mass and assuming that it had outgrown the vascular supply and thus is necrotic and incapable of acute delivery of massive hypercatecholaminemia.
Pheochromocytoma vs paraganglioma
The division of PPGL tumors into PCC and PGL is mostly anatomical rather than functional. The only major difference is that PCCs express significantly higher content of PNMT and thus higher probability of predominantly the adrenergic or mixed biochemical phenotype, as compared to predominantly noradrenergic phenotype of PGLs. With that said, the actual profile will strongly depend on the degree of tumor differentiation, as well as possibility of mixed PPGL cases.
Another possible cause of differences in the acute conditions associated with different PPGL tumors is the fact that adrenal incidentalomas are readily diagnosed on unrelated imaging studies, especially in recent years when both chest and abdominal CT scans, which both image adrenal glands, are done for progressively increasing number of conditions. PGL are frequently missed, especially in cases where clinical symptomatology is less severe or the patient is young and is otherwise seen as “healthy”. Acute and severe hypercatecholaminemic crisis can occur when a previously unknown abdominal or chest mass is seen during unrelated surgery or invasive procedure and is manipulated, causing release of massive amount of pre-synthesized catecholamines. In these cases, surgical awareness of uncommon locations and anesthesiology readiness for appropriate therapy of potentially life-threatening crisis is the true cornerstone of the management of this endocrine emergency.
Single tumor vs metastatic disease
The main difference in the approach to the possibility of metastatic disease is based on the expectation that rapid postoperative withdrawal of adrenoceptor blockade will associate with rebound hypertensive crisis. In addition to this, possibility of distant metastatic disease with significant morbidity associated with involvement of affected organs needs to be kept in mind.
Spontaneous vs familial/syndromal case
Recent years have tremendously changed many aspects of our understanding of the biology and management of the PPGL particularly the progress in understanding the genetics of the disease. While possibility of a genetically driven condition should be increased in younger patients or ones with a positive family history, finding a predominantly noradrenergic biochemical phenotype, multiple masses on imaging studies, or additional clinical findings – thyroid nodules happen to be medullary thyroid cancer (MTC), renal tumors etc. – should strongly suggest a genetic condition. The opposite is even more important – like sending patient with thyroid nodule of unclear pathology to surgery and ending up with it being MTC and patient having a hypertensive crisis during the surgery. Possible syndromal association with SDHB mutation should prompt assessment of multiple tumors, as well as early recurrence and metastatic disease to prevent early post-operative discontinuation of medical therapy and rebound hypertension or discontinuation of long term follow up. In addition, the head and neck PGLs, rarely seen by endocrinologists in the past, are associated with a SDHD gene mutation and can metastasize and locally invade, while being secretory silent. Establishment of a genetic disorder requires institution of testing and biochemical screening of relatives.
Dopamine vs norepinephrine vs epinephrine secreting tumors
Based on the differences in the affinity of epinephrine and norepinephrine to adrenoceptors, with norepinephrine having lesser action on the β2-adrenoceptor, one can expect a pure “vasoconstrictive” clinical presentation in cases with pure norepinephric secretory profile, while with epinephrine and dopamine-secreting tumors, orthostatic or episodic hypotension will be much more frequent.
PPGL in pregnancy
PPGL during pregnancy is a rare clinical entity. In the case of pregnancy, there are 2 patients at the same time, both the mother and the fetus. Both can be severely affected by the disease, although in a somewhat different manner. PPGL is difficult to suspect during pregnancy because of pre-eclampsia-driven management attitude. Diagnosis can be significantly delayed causing fetal morbidity and affecting both the pregnancy and delivery. Several physiologic phenomena drive the unique behavior of PPGL in pregnancy. These include high placental expression of catechol-O-methyltransferase (COMT) and monoamine oxidase (MAO) and lack of autoregulation in uteroplacental circulation. While both enzymes are responsible for production of inactive catecholamine metabolites, they provide some kind of “fetal barrier”, shielding the fetus from exposure to increased catecholamine levels. Lack of uteroplacental vascular autoregulation, on the other hand, directly affects placental blood flow and fetal blood supply in the hypertensive vasoconstricted mother and can associate with rapid development of uteroplacental insufficiency. As far as management – MRI will be the preferred imaging modality, medical therapy will be started with same medications as in non-pregnant patients, and the management of acute severe hypercatecholaminemia will be similar to non-pregnant cases, with exception for the need to avoid methyldopa and more prevalent use of intravenous magnesium sulfate, which will be effective in both PPGL and pre-eclampsia. Surgery still remains the treatment of choice and there is continuous debate about the sequence of delivery versus surgery.
PPGL in pediatric population
Hypertension in the pediatric population is mostly secondary and is mostly related to renal disease with endocrine causes happening much less frequently. With this said, the possibility of both genetically-driven as well as a malignant course is much higher and needs to be assessed in every pediatric case. Overall management is similar to adult PPGL. On the other hand, the patient will need an extended follow up to assure that any possibility of recurrence is monitored.
Co-secretory substances
The unique enzymatic machinery of PPGL cells provides a series of steps that transforms an amino acid to an amine. In this case, the amino acid is tyramine and the end product are catecholamines. One should appreciate that PPGLs can co-secrete multiple active substances, most clinically relevant of which will probably be ACTH/CRH, which can cause frank and at times severe Cushing syndrome. This needs to be kept in mind, especially when the patient presents with suspicious symptoms or biochemical findings. PPGL as part of MEN2 will associate with overproduction of calcitonin and disseminated metastatic disease, which needs to be diagnosed, hopefully prior to PPGL surgery.
Symptomatic vs silent
While we had discussed in length symptomatic PPGL, parasympathetic PGL could associate with silent tumors, which could be associated with SDHB/D mutation and might have a malignant/metastatic course with local involvement of carotid sinus, as well as major neck vessels, in times associated with different – “silent/local” urgencies/emergencies.
Treatment-associated severe hypercatecholaminemia
Inoperable or recurrent metastatic disease can be treated through multiple modalities, which usually cause different degrees of tumor destruction. These include older therapies (radiofrequency ablation, cryotherapy, external beam radiation, transarterial chemoembolization, ethanol injection), as well as newly rediscovered 131I-MIBG and somatostatin receptor-driven peptide receptor radionuclide therapy (PRRT) with 90Y-DOTATOC/DOTATATE and 177Lu-DOTATATE. This tumor destruction is associated with the potential of massive release of the pre-synthesized catecholamines and could generate severe hypercatecholaminemia for a prolonged period of time. In preparation for therapy, patients need to undergo a protocol, identical to surgical preparation and their biochemical response needs to be followed for weeks after therapy. Overtly secreting or very large tumors should probably generate post-procedural admission for closer monitoring to make sure that the patient will not develop a hypertensive emergency. As we had discussed above, pre-treatment with a competitive α-adrenoceptor antagonist must be used in almost all patients but may provide insufficient α-adrenoceptor blockade with massive hypercatecholaminemia. On the other hand, use of phenoxybenzamine in a full dose, can potentially result in prolonged hypotension but is less problematic than a severe hypertensive crisis and its consequences.
CLINICAL SYNDROMES ASSOCIATED WITH PPGL
Multisystem Failure
This is by far most feared complication because of the high morbidity and mortality associated with a rapid and at times unexpected and unpredicted development, resembling an avalanche starting small but rapidly leaping into a clinical disaster. While it could be preceded by a hypertensive crisis, patients who are sicker and fragile at baseline can develop it with little or no warning symptoms. The blood pressure pattern can show either hypertension or hypotension in case of progressive shock and cardiac failure. It can associate with fever, encephalopathy, as well as renal failure, pulmonary edema and even disseminated intravascular coagulation. Clinical outcomes mostly depend on delays in diagnosis and initiation of appropriate therapy.
Cardiovascular Emergencies
Hypertensive crisis
While hypertension in patients with PPGL can be both paroxysmal and sustained, a severe hypertensive crisis is usually precipitated by stress, postural changes, food containing large amounts of catecholamine precursors, as well as local manipulation of an unsuspected tumor. Medications can also induce hypertensive crisis through direct stimulation of release of stored catecholamines – which could be of a massive quantity. These medications include ACTH, tricyclic antidepressants, phenothiazine, nasal decongestants containing sympathomimetic or histamine, and metoclopramide. Treatment needs to be initiated immediately, intravenously and one needs to remember that α-adrenoceptor blockade is the drug of choice, as well as the fact that β-adrenoceptor medication can both cause and precipitate acute deterioration of the hypertensive crisis.
Hypotensive shock
Hypotension in PPGL is usually perceived as exclusively related to dopamine or epinephrine-secreting tumors. While this is true, hypovolemia and acute heart failure due to an acute coronary event, myocarditis, or pulmonary edema can produce profound hypotension and shock in norepinephrine-secreting tumors too.
Cardiac arrhythmias
Tachyarrhythmias are frequently associated with PPGL and are related to β-adrenergic stimulation-driven positive inotropy. These are mostly supraventricular including atrial fibrillation and flutter, as well as wide complex ventricular tachycardia. One needs to remember that in case of myocarditis, cardiomyopathy, or an acute coronary event, myocardial susceptibility to any type of rhythm disturbances is significantly increased and can manifest with bradyarrhythmia’s.
Myocarditis and cardiomyopathy
Development of myocarditis and cardiomyopathy in PPGL is well known and described, but still remains poorly understood as far as the actual mechanistic process. It could relate to direct myocardial toxicity of significant and prolonged hypercatecholaminemia, as well as prolonged hypertension or a coronary event. It could be of any type – either hypertrophic or dilated – as well as asymmetric (tako-tsubo type). It could improve to some degree after successful treatment.
Acute myocardial or peripheral ischemia
Both could be caused by prolonged hypertension, resulting in intimal hypertrophy, as well as local spasm in the naïve or already sclerotic vessel. It can also result from increased and uncompensated oxygen demand.
Pulmonary Emergencies
Pulmonary edema can be both cardiac and non-cardiac of origin. The first is discussed above, while the last is mostly related to increased capillary pressure together with vasoconstriction related stasis and an increase in vascular permeability.
Gastrointestinal Emergencies
Clinically, acute GI emergencies are usually associated with abdominal pain and vomiting. These could be related to mesenteric ischemia, which consequently can result in bowel perforation, ileus, and GI bleeding. Ileus in PPGL can be both paralytic and pseudo-obstructive and can also associate with megacolon. Although rarely thought to be related to PPGL, these disorders need to be diagnosed early and treated to prevent rapid deterioration and the need for urgent surgery. Severe hypertension can also associate with an aneurysm of the aorta that can undergo dissection with a hypertensive spike.
Renal Emergencies
Acute vasoconstriction of the renal arteries can result in acute renal failure, while prolonged hypertension can cause progressive deterioration of renal function over a relatively short period of time, especially in patients with underlying hypertension or susceptibility to significant vascular changes.
Neurologic Emergencies
Strokes are known to occur with both paroxysmal and sustained hypertension, but other neurologic emergencies could include hypertensive encephalopathy, subarachnoid hemorrhage, and seizures. Neurologic deficiencies related to brain or spinal metastases, as well as local neurologic deficits caused by paragangliomas are also seen.
MANAGEMENT
The diagnostic approach and treatment of PPGL are discussed in detail in the PPGL section of Endotext and shown in Table 2, but what we will discuss here is the approach to PPGL-related medical emergencies.
Table 2.
Treatment of PPGL
| Stage | Goal | Primary | Alternative |
|---|---|---|---|
| Initial oral | Normalization of BP Minimal organ effect | In the following order: α-blocker β blocker Metyrosine | Calcium channel blocker Labetalol |
| Pre-operative | Normal BP Normovolemia Optimized cardiac performance | As Initial Fluids to normovolemia | As Initial |
| Intra-operative | Prevention of the following: Severe hypercatecholaminemia Severe hypertension Severe hypotension | Phentolamine IV Nitroprusside IV Aggressive fluid replacement | Labetalol IV |
| Post-operative | Prevention of hypotension Prevention of hypoglycemia | Aggressive fluid replacement Glucose supplementation | |
| Inoperable disease | Maintenance of normal BP Treatment of metastatic disease | Chemotherapy Radiotherapy Debulking | Experimental Therapy |
As with non-urgent PPGL, the main part of successful management and prevention of its deterioration into a medical emergency is timely suspicion and diagnosis. Obviously, this cannot happen in each and every case and we will continue to see acute severe hypertensive crises and poor outcomes that could not be prevented. But otherwise, the overall suspicion should be relatively high, even if it will generate some unnecessary workups, while preventing avoidable death. We also feel that the common flamboyancy of PPGL being a great and friendly masquerader should probably be revised to some degree. It should include a quite real possibility of metamorphosis of this apparent benignity into behavioral tendencies of a Grim Reaper; just to make sure that the reality of severely morbid outcomes is known and respected.
As was discussed above, in case of acute intra-operative hypertensive crisis with or without identifiable mass, therapy should be initiated assuming PPGL-related severe hypercatecholaminemia. Medications are to be administered IV and should include phentolamine or nitroprusside. Nitroprusside is more readily available in ORs compared to phentolamine, which needs to be prepared by the pharmacy. Nitroprusside can cause adverse effects when administered over an extended period during complicated surgery (discussed above). If existence of a PPGL is established prior to surgery, it would definitely be advisable to procure phentolamine to be available in OR/ICU. Phentolamine, an α-adrenoceptor antagonist, is given as an i.v. bolus of 2.5 mg to 5 mg at 1 mg/min, which can be repeated every 5 min for adequate control of hypertension. Alternatively, it can be given as a continuous infusion (100 mg of phentolamine in 500 mL of 5% dextrose in water, not available in USA) with an infusion rate adjusted to the patient’s blood pressure during continuous blood pressure monitoring. Sodium nitroprusside can be administered at 0.5 to 10.0 mcg/kg per minute (stop if no results are seen after 10 minutes). Magnesium sulfate acts as vasodilator and antiarrhythmic and is administered as a 1-2 gm bolus and then continuously at 1 to 3 gm/h. Esmolol, a short acting β1-adrenoceptor antagonist can improve uncontrolled tachycardia. Continuous infusion of Nicardipine, that is usually a very good initial choice, can prevent catecholamine-induced coronary vasospasm, hypertension, and tachycardia and it is given intravenously at 1 to 2.5 mg for 2 min, then at 5 to 15 mg/h. If the patient was not on a α-adrenoceptor blocker prior to the surgery, use of Labetalol could precipitate deterioration in blood pressure because of 1:7 α:β-specific blocking effect. If the patient is on a short-acting competitive α-adrenoceptor blocker, using i.v. Labetalol bolus could be beneficial for better control of blood pressure.
The same approach should be carried out in cases of a severe hypertensive crisis if it happens acutely in a patient with known and insufficiently treated PPGL (recurrence, lack of compliance), acute deterioration of hypertension, or resistant to initial therapy including pre-eclampsia. There will be little time to sufficiently and efficiently pre-load patient with oral therapy, which should be carried out after resolution of the crisis if surgery was not performed.
If, on the other hand, there is time for oral therapy in less urgent situations or while awaiting upcoming surgery, α-adrenoceptor blockade should be initiated as soon as possible and the patient should be clinically evaluated on a frequent basis to adjust therapy as tolerated. The choice of medication should be dictated by several factors. In cases of severe hypercatecholaminemia or relatively recent onset, where one should expect less time available for significant desensitization of adrenoceptors, competitive α-adrenoceptor blockers could provide lesser control of symptoms just by the virtue of pharmacokinetics against massive concentration of catecholamines. In such cases, use of a non-competitive agonist – phenoxybenzamine - will make more sense. Otherwise, competitive adrenoceptor blockers seem to be efficient and safe and could result in shorter hospitalization due to shorter action and lesser postoperative hypotension. Also, doxazosin (as well as others (prazosin and terazosin, which seem to be used less frequently) seems to be both safe and efficient in PPGL management. Physician’s preferences and experience play a major role in the selection of prescribed medication. In addition to this, the cost and availability affects the choice of medication. Endocrinologist need to be comfortable with multiple different classes of medications used for therapy. Calcium channel blockers (nicardipine/amlodipine) proved to be also safe and efficient, but, again, we would suggest avoiding them alone in cases of severe hypercatecholaminemia, especially with concomitant congestive heart failure. A competitive inhibitor of tyrosine hydroxylase - α-methyl-para-tyrosine (metyrosine, Demser) can be used to both compete with tyrosine (the substrate for catecholamine synthesis), as well as a direct inhibitor of tyrosine hydroxylase.
Hypotension is managed by fluid administration and/or vasopressors including phenylephrine. Hypoglycemia can occur after removal of PPGL-related catecholamine excess as a result of rebound release of insulin secretion and is treated with intravenous glucose.
GUIDELINES
- Lenders JW, Duh QY, Eisenhofer G, et al. Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. [PubMed: 24893135]
REFERENCES
- Pacak K, Tella SH. Pheochromocytoma and Paraganglioma. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2018 Jan 4.
- Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocrine Reviews. 2017;38(6):489–515. [PMC free article: PMC5716829] [PubMed: 28938417]
- Fishbein L, Leshchiner I, Walter V, et al. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181–193. [PMC free article: PMC5643159] [PubMed: 28162975]
- Prete A, Paragliola RA, Salvatori R, Salvatore MC. Management of catecholamine-secreting tumors in pregnancy: a review. Endocrine Practice. 2015;22(3):357–70. [PubMed: 26536138]
- Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108–119. [PubMed: 24442145]
- Tischler AS, Pacak K, Eisenhofer G. The Adrenal Medulla and Extra-adrenal Paraganglia: Then and Now. Endocr Pathol. 2013 Dec 24; [PubMed: 24362581]
- Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92(11):4069–79. [PubMed: 17989126]
- Hereditary pheochromocytoma/paraganglioma syndrome with a novel mutation in the succinate dehydrogenase subunit B gene in a Japanese family: two case reports.[J Med Case Rep. 2021]Hereditary pheochromocytoma/paraganglioma syndrome with a novel mutation in the succinate dehydrogenase subunit B gene in a Japanese family: two case reports.Hirose R, Tsurutani Y, Sugisawa C, Inoue K, Suematsu S, Nagata M, Hasegawa N, Kakuta Y, Yonamine M, Takekoshi K, et al. J Med Case Rep. 2021 May 22; 15(1):282. Epub 2021 May 22.
- Challenges in the diagnosis of pheochromocytoma and paraganglioma syndrome.[Neuro Endocrinol Lett. 2014]Challenges in the diagnosis of pheochromocytoma and paraganglioma syndrome.Zdrojowy-Wełna A, Bednarek-Tupikowska G. Neuro Endocrinol Lett. 2014; 35(5):355-8.
- Laparoscopic Surgery for Pheochromocytoma and Paraganglioma Removal: A Retrospective Analysis of Anaesthetic Management.[Curr Hypertens Rev. 2016]Laparoscopic Surgery for Pheochromocytoma and Paraganglioma Removal: A Retrospective Analysis of Anaesthetic Management.Ramachandran R, Rewari V, Sharma A, Kumar R, Trikha A. Curr Hypertens Rev. 2016; 12(3):222-227.
- Review Genetics of pheochromocytomas and paragangliomas.[Best Pract Res Clin Endocrinol...]Review Genetics of pheochromocytomas and paragangliomas.Opocher G, Schiavi F. Best Pract Res Clin Endocrinol Metab. 2010 Dec; 24(6):943-56.
- Review Clinical and Molecular Features of Renal and Pheochromocytoma/Paraganglioma Tumor Association Syndrome (RAPTAS): Case Series and Literature Review.[J Clin Endocrinol Metab. 2017]Review Clinical and Molecular Features of Renal and Pheochromocytoma/Paraganglioma Tumor Association Syndrome (RAPTAS): Case Series and Literature Review.Casey RT, Warren AY, Martin JE, Challis BG, Rattenberry E, Whitworth J, Andrews KA, Roberts T, Clark GR, West H, et al. J Clin Endocrinol Metab. 2017 Nov 1; 102(11):4013-4022.
- Emergencies Related to Pheochromocytoma/Paraganglioma Syndrome - EndotextEmergencies Related to Pheochromocytoma/Paraganglioma Syndrome - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...