

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

CLINICAL RECOGNITION
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders that arise from defective steroidogenesis. The production of cortisol in the zona fasciculata of the adrenal cortex occurs in five major enzyme-mediated steps. CAH results from deficiency in any one of these enzymes. Impaired cortisol synthesis leads to chronic elevations of ACTH via the negative feedback system, causing overstimulation of the adrenal cortex and resulting in hyperplasia and over-secretion of the precursors to the enzymatic defect. The forms of CAH are summarized in Table 1. Impaired enzyme function at each step of adrenal cortisol biosynthesis leads to a unique combination of elevated precursors and deficient products. The most common enzyme deficiency that accounts for more than 90% of all CAH cases is 21-hydroxylase deficiency (21OHD).
Table 1.
Types of Congenital Adrenal Hyperplasia
| Condition Onset Abnormality | Genitalia Mineralocorticoid Effect | Gene Chromosomal Location Typical Features |
|---|---|---|
| Lipoid CAH Congenital StAR Protein | Female, with no sexual development Salt wasting | StAR 8p11.2 All steroid products low |
| Lipoid CAH Congenital P450scc | Female, with no sexual development Salt wasting | CYP11A 15q23-24 All steroid products low |
| 3β-HSD deficiency Congenital 3β-HSD | Females virilized, males hypovirilized Salt wasting | HSD3B2 1p13.1 Elevated DHEA, 17-pregnenolone, low androstenedione, testosterone, elevated K, low Na, CO2 |
| 17α-OH deficiency Congenital P450c17 | Males hypovirilized, Hyperkalemic low-renin hypertension | CYP17 CYP17 10q24.3 Decreased androgens and estrogen, elevated DOC, corticosterone |
| Classic 21-OH deficiency, salt wasting Congenital P450c21 | Females prenatally virilized, males unchanged Salt wasting occurs in ¾ of 21OHD patients | CYP21 6p21.3 Elevated 17-OHP, DHEA, and androstenedione, elevated K, low Na, CO2, low aldosterone, high plasma renin |
| Classic 21-OH deficiency, simple virilizing Congenital P450c21 | Females prenatally virilized, males unchanged No salt wasting | CYP21 6p21.3 Elevated 17-OHP, DHEA, and androstenedione, normal electrolytes |
| Non-classic 21-OH deficiency Postnatal P450c21 | All with normal genitalia at birth, hyperandrogenism postnatally No salt wasting | CYP21 6p21.3 Elevated 17-OHP, DHEA, and androstenedione on ACTH stimulation |
| 11β-OH deficiency Congenital P450c11B1 | Females virilized, males unchanged Low-renin hypertension | CYP11B1 8q24.3 Elevated DOC, 11-deoxycortisol (S); androgens, low K, elevated Na, CO2 |
| P450 Oxidoreductase deficiency (POR), Congenital P450 oxidoreductase | Males undervirilized, females unchanged Variable degree of mineralocorticoid deficiency | P450 Oxidoreductase gene (POR) 7q11.2 Combined and variable enzymatic defects of P450c21, P450c17 and P450aro Wide range of phenotypes: normal to genital ambiguity +/- skeletal abnormalities (Antley Bixler type) |
Classical CAH occurs in 1:13,000 to 1:15,000 live births. It is estimated that 75% of patients have the salt-wasting (SW) phenotype and the rest have simple-virilizing (SV) phenotype. Non-classical 21-OHD CAH (NC-CAH) is more common, and is one of the most common disorders in the Ashkenazi Jewish population with 1 in 27 Jews affected. CAH owing to 11β-hydroxylase deficiency (11β-OHD) is the second most common cause of CAH, accounting for 5-8% of all cases. The other forms of CAH are considered rare diseases and the incidence is unknown in the general population.
PATHOPHYSIOLOGY
Adrenal steroidogenesis occurs in three major pathways: glucocorticoids, mineralocorticoids, and sex steroids as shown in Figure 1. Glucocorticoids (particularly cortisol), androgens, and estrogens are synthesized in the zona fasciculata and reticularis; and aldosterone in the zona glomerulosa. The HPA feedback system is mediated through the circulating level of plasma cortisol by negative feedback of cortisol on CRF and ACTH secretion. Therefore, a decrease in cortisol secretion leads to increased ACTH production, which in turn stimulates (1) excessive synthesis of adrenal products in those pathways unimpaired by the enzyme deficiency and (2) an increase of precursor molecules in pathways blocked by the enzyme deficiency.

Figure 1.
Pathways of Adrenal Steroidogenesis: Five enzymatic steps necessary for cortisol production are shown in numbers. 1= 20, 22 desmolase, 2= 17 hydroxylase (17-OH), 3=3ß-hydroxysteroid dehydrogenase (3ß HSD), 4=21 hydroxylase (21-OHD), 5=11ß hydroxylase (11-OH) In the first step of adrenal steroidogenesis, cholesterol enters mitochondria via a carrier protein called StAR. ACTH stimulates cholesterol cleavage, the rate limiting step of adrenal steroidogenesis.
The clinical symptoms of the five different forms of CAH result from the particular hormones that are deficient and those that are produced in excess as outlined in Table 1. In 21 OHD-CAH, there is an accumulation of 17-hydroxyprogesterone (17-OHP), a precursor to the 21-hydroxylation step, which is then shunted into the intact androgen pathway, where the 17,20-lyase enzyme converts the 17-OHP to D4-androstenedione, which is converted into androgens. Mineralocorticoid deficiency is a feature of SW-CAH, the most severe form of CAH. The enzyme defect in NC-CAH is only partial and salt wasting in this mild form of the disease does not occur. The analogy of all other enzyme deficiencies in terms of precursor retention and product deficiencies are shown in Table 1.
CLINICAL FEATURES
Genitalia
Females with Classical 21-OHD and 11β-hydroxylase deficiency CAH present at birth with virilization of their genitalia. Adrenocortical function begins around the 7th week of gestation; thus, a female fetus with classical CAH is exposed to adrenal androgens at the critical time of sexual differentiation (approximately 9 to 15 weeks gestational age). This leads to clitoral enlargement, fusion and scrotalization of the labial folds, and rostral migration of the urethral/vaginal perineal orifice, placing the phallus in the male position. Degrees of genital virilization are classified into five Prader stages (see Figure 2).
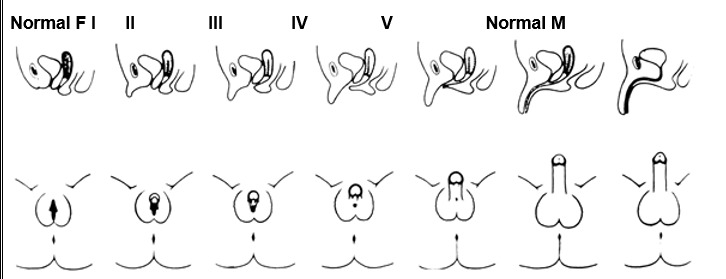
Figure 2.
Different degrees of virilization according to the scale developed by Prader
Stage I: clitoromegaly without labial fusion
Stage II: clitoromegaly and posterior labial fusion
Stage III: greater degree of clitoromegaly, single perineal urogenital orifice, and almost complete labial fusion
Stage IV: increasingly phallic clitoris, urethra-like urogenital sinus at base of clitoris, and complete labial fusion
Stage V: penile clitoris, urethral meatus at tip of phallus, and scrotum-like labia (appear like males without palpable gonads)
Prader, A. Helv Paediatr Acta, 1954. 9:230-248.
Internal female genitalia, such as the uterus, fallopian tubes and ovaries, develop normally. Females with classical CAH maintain the internal genitalia potential for fertility.
Postnatal Effects, Growth and Puberty
Lack of appropriate postnatal treatment in boys and girls results in continued exposure to excessive androgens, causing progressive penile or clitoral enlargement, the development of premature pubic hair, axillary hair and acne. Advanced somatic and epiphyseal development occurs with exaggerated growth and is usually accompanied by premature epiphyseal maturation and closure, resulting in a final adult height that is typically significantly below that expected from parental heights. Excess glucocorticoid treatment can also lead to poor growth. The mean age at onset of puberty in both males and females is slightly younger than the general population. In those who are inadequately treated, central precocious puberty can occur. Following the onset of puberty, in a majority of successfully treated patients, the milestones of further development of secondary sex characteristics in general appear to be normal. In female adolescents and adults, signs of hyperandrogenism may include male-pattern alopecia (temporal balding), acne, hirsutism, menstrual irregularities, secondary PCOS and impaired fertility. Although the expected age of menarche may be delayed in females with classical CAH, when adequately treated many have regular menses after menarche. In males, short stature and impaired fertility are observed.
Gender Role Behavior and Cognition
Prenatal androgen exposure in females affected with classical forms of CAH not only has a masculinizing effect on the development of the external genitalia, but also on childhood behavior. Both physical and behavioral masculinization are related to each other and to genotype, indicating that behavioral masculinization in childhood is a consequence of prenatal androgen exposure. The majority of genetic females with CAH retain the female gender identity even in the setting of prenatal androgen exposure and postnatal hyperandrogenism.
Fertility
Difficulty with fertility in females with CAH may be due to anovulation, secondary polycystic ovarian syndrome, irregular menses, non-suppressible serum progesterone levels, or an inadequate introitus. Fertility is reduced in SW-CAH with rare reports of pregnancy. Non-classical CAH is an important and frequently unrecognized form of infertility. Males with CAH, particularly if poorly treated, may have reduced sperm counts and low testosterone as a result of high androstenedione concentrations which suppress gonadotropins and testicular adrenal rest tumors. Testicular adrenal rest tumors (TART) are thought to arise from aberrant adrenal cells in the testes; TARTs are always benign and mostly bilateral. Microscopic examination shows that adrenal rest cells are present in the testicles of all male patients with CAH and often detected radiographically in those with longstanding poorly controlled disease. Regular testicular examination and periodic testicular ultrasonography are recommended for early detection of adrenal rest tumors of the testes. However, MRI studies have been increasingly used to diagnose TARTs.
Salt-Wasting 21-Hydroxylase Deficiency
When the loss of 21-hydroxylase function is severe, adrenal aldosterone secretion is not sufficient for sodium reabsorption by the distal renal tubules, and individuals suffer from salt wasting as well as cortisol deficiency and androgen excess. Infants with renal salt wasting have poor feeding, weight loss, failure to thrive, vomiting, dehydration, hypotension, hyponatremia, and hyperkalemic metabolic acidosis progressing to adrenal crisis (azotemia, vascular collapse, shock, and death). Adrenal crisis can occur as early as age one to four weeks. Affected males who are not detected in a newborn screening program are at high risk for a salt-wasting adrenal crisis because their normal male genitalia do not alert medical professionals to their condition. It is important to recognize that the extent of genital virilization may not differ among SV-CAH and SW-CAH.
Simple-Virilizing 21-Hydroxylase Deficiency
The salient features of classical SV-CAH are prenatal virilization and progressive postnatal masculinization with rapid somatic growth and advanced epiphyseal maturation leading to early epiphyseal closure and likely short stature. There is no evidence of mineralocorticoid deficiency in this disorder and serum electrolyte concentrations are normal. Diagnosis at birth of a female with SV-CAH is usually made immediately because of the apparent genital ambiguity. Since the external genitalia are not affected in newborn males, hyperpigmentation may be the only clue suggesting increased ACTH secretion and cortisol deficiency. Diagnosis at birth in males thus rests on prenatal or newborn screening.
Non-Classical 21-Hydroxylase Deficiency
Individuals with the non-classical (NC) form of 21-OHD have only mild to moderate enzyme deficiency and present postnatally, eventually developing signs of hyperandrogenism. Females with NC-CAH do not have virilized genitalia at birth. NC-CAH may present at any age after birth with a variety of hyperandrogenic symptoms. While serum cortisol concentration is typically low in patients with the classic form of the disease, it is usually normal in patients with NC 21-OHD. Similar to classical CAH, NC-CAH may cause premature development of pubic hair, acne, secondary PCOS, advanced bone age with accelerated linear growth velocity, and short stature. In adult males, early balding, acne, infertility or short stature may prompt the diagnosis of NC-CAH.
DIAGNOSIS
Diagnosis of CAH must be suspected in infants born with ambiguous genitalia. The physician is obliged to make the diagnosis as quickly as possible to initiate therapy. The diagnosis and rational decision of sex assignment must rely on the determination of genetic sex, the hormonal determination of the specific deficient enzyme, genotype, and an assessment of the patient's potential for future sexual activity and fertility. As indicated in Table 1, each form of CAH has its own unique hormonal profile, consisting of elevated levels of precursors and elevated or diminished levels of adrenal steroid products. Diagnosis of the 21-OHD CAH can also be confirmed biochemically by a hormonal evaluation. In a randomly timed blood sample, a very high concentration of 17-hydroxyprogesterone (17-OHP), the precursor of the defective enzyme, is diagnostic of classical 21-OHD. Such testing is the basis of the newborn-screening program developed to identify classically affected patients who are at risk for salt wasting crisis. False-positive results are, however, common with premature infants. Appropriate references based on weight and gestational age are therefore in place in many screening programs. False negative results may occur if samples are drawn late in the afternoon as adrenal hormones exhibit diurnal variation. The gold standard for hormonal diagnosis is the corticotropin stimulation test (250 μg cosyntropin intravenously), measuring levels of 17-OHP and Δ4 androstenedione at baseline and 60 min. These values can then be plotted in the published nomogram to ascertain disease severity. The corticotropin stimulation test should not be performed during the initial 24 hours of life as samples from this period are typically elevated in all infants and may yield false-positive results. Establishing a genetic diagnosis is not only important for the genotype-phenotype correlation, but also for genetic counseling for future pregnancies and for genetic counseling for the patient and his/her reproductive future.
For 21-OHD CAH, genetic analysis of the CYP21A2 gene may provide more clues to predict phenotypic severity. In about 50% of the causative genotypes, genotype-phenotype correlation can be found, although certain mutations can lead to variable phenotypes in different population groups especially in the simple virilizer group. Sequencing of the entire gene should be performed to detect rare mutations when genotype–phenotype non-concordance is observed in patients with CAH.
Newborn screening for CAH, which utilizes 17 hydroxyprogesterone levels, is a useful tool for early detection of CAH prior to the development of adrenal crisis in the affected neonate. However, screening is associated with a high rate of false positive results as levels are affected by prematurity and birth weight. Molecular genetics, especially genotyping of the CYP21A2 gene should be considered as a second-tier screening test in the new born screening program.
Prenatal testing for CAH in utero has historically utilized invasive techniques like amniocentesis and chorionic villus sampling which cannot be done prior to 14 weeks of gestation. Prenatal dexamethasone treatment must begin prior to genital formation occurring at approximately 9 weeks, in order to avoid genital ambiguity in the affected female fetus. Massive parallel sequencing using hybridization probes on cell-free fetal DNA in maternal plasma indicated that the fetal CAH status was correctly deduced as early as 5 weeks 6 days of gestation. This is a noninvasive technique that accurately diagnoses CAH before the ninth week of gestation.
TREATMENT
Routine Treatment
The goal of therapy in CAH is to both correct the deficiency in cortisol secretion and to suppress ACTH overproduction. Proper treatment with glucocorticoid reduces stimulation of the androgen pathway, thus preventing further virilization and allowing normal growth and development. The usual requirement of hydrocortisone (or its equivalent) for the treatment of classical 21-OHD form of CAH is about 10-15 mg/m2/day divided into 2 or 3 doses per day and for non-classical 21-OHD 5-8 mg/m2/day divided into 2 or 3 doses per day. Hydrocortisone is the glucocorticoid of choice in the pediatric age group. Prednisolone and dexamethasone are not used in growing children given growth suppressive effects. A small dose of dexamethasone at bedtime (0.25 to 0.5 mg) is usually adequate for androgen suppression in non-classical adult patients. Adequate biochemical control is assessed by measuring serum levels 17-OHP and androstenedione; serum testosterone can be used in females and prepubertal males (but not newborn males). We recommend that hormone levels are measured at a consistent time in relation to medication dosing, usually 1-2 hours after the morning corticosteroid. Titration of the dose should be aimed at maintaining 17-OHP concentrations below 1000 ng/dL and androstenedione concentrations below 200 ng/dl. Over-treatment should be avoided because it can lead to Cushing syndrome. Patients with salt wasting CAH have elevated plasma renin in response to the sodium-deficient state, and they require treatment with the salt-retaining 9α-fludrocortisone acetate. The average dose is 0.1 mg daily (0.05-0.2 mg daily). Infants should also be started on salt supplementation, as sodium chloride, at 1-2 g daily, divided into several feedings. Measurements of plasma renin and aldosterone are used to monitor the efficacy of mineralocorticoid therapy. Advancement of bone age is monitored by bone age x-rays. Growth hormone therapy, in conjunction with a GnRH analogue, has been shown to be effective in improving final adult height. Patients may also experience peripheral precocious puberty, which requires treatment with gonadotropin-releasing hormone analogues. Aromatase inhibitors and growth hormone therapy should only be used in patients with a very short predicted final stature or in clinical trials. Use of aromatase inhibitors in CAH has been shown decrease bone maturation rates and some increase in adult height but the differences were not statistically significant.
Treatment During Illness and Emergency
Adrenal crisis can present as hypotension or shock and serum electrolyte abnormalities (hypoglycemia, hyponatremia, hyperkalemia, acidosis). During adrenal crisis, an immediate bolus of hydrocortisone 50-100 mg can be given intravenously or intramuscularly followed by hydrocortisone 100 mg/m2/day given as either continuous infusion or divided at least every 6 hours. Rehydration can be started with 20ml/kg isotonic saline with D5 as rapid bolus followed by repeat boluses or continuous infusion guided by level of dehydration. Hypoglycemia may require dextrose bolus and an initial bolus of 0.5-1 gram/kg of dextrose can be given intravenously at 2-3 ml per minute. If hyperkalemia is present, cardiac monitoring should be done to monitor for EKG changes. If changes are present, hyperkalemia should be treated using insulin with glucose infusion with or without other measures.
In non-life-threatening periods of illness or physiologic stress, the corticosteroid dose should be increased to 2 or 3 times the maintenance dose for the duration of that period, divided into 3 daily doses. Each family should be given injection kits of hydrocortisone, i.e. Solu-Cortef, for emergency use, and all family members should be trained in its intramuscular administration. The injectable dose of hydrocortisone in an emergency is 25 mg for infants, 50 mg for children under 40 kg, and 100 mg for children over 40 kg and for adults. In the event of a surgical procedure, 5-10 times the daily maintenance dose of hydrocortisone is needed, with 25-100 mg hydrocortisone IM/IV administered before and during a surgical procedure (as per infant, child, adult recommendations above), followed by high doses of hydrocortisone during the first 24-48 post-operative hours; the dose can then be tapered over the following days to the normal preoperative schedule. Stress doses of dexamethasone should not be given because of the delayed onset of action. It is not necessary for increased mineralocorticoid doses during these periods of stress. It is imperative for all patients who are receiving corticosteroid replacement therapy, such as patients with CAH, to wear a Medical Alert bracelet or medallion that will enable correct and appropriate therapy in case of emergencies. It is also crucial to re-educate parents at regular intervals on the life-threatening nature of this emergency.
GUIDELINES
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. [PMC free article: PMC6456929] [PubMed: 30272171]
- J Clin Endocrinol Metab. 2018 Nov 1;103(11):4043–4088. [PMC free article: PMC6456929] [PubMed: 30272171]
- Rodriguez A, Ezquieta B, Labarta JI, Clemente M, Espino R, Rodriguez A, et al. Recommendations for the diagnosis and treatment of classic forms of 21-hydroxylase-deficient congenital adrenal hyperplasia. An Pediatr (Barc). 2017;87(2):116 e1- e10. [PubMed: 28161392]
REFERENCES
- New M, Yau M, Lekarev O, Lin-Su K, Parsa A, Pina C, Yuen T, Khattab A. Congenital Adrenal Hyperplasia. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2017 Mar 15.
- El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 2017 Nov 11;390(10108):2194–2210. [PubMed: 28576284]
- Fluck CE, Miller WL. P450 oxidoreductase deficiency: a new form of congenital adrenal hyperplasia. Curr Opin Pediatr. 2006;18(4):435–41. [PubMed: 16915000]
- Yilmaz R, Sahin D, Aghayev A, Erol OB, Poyrazoglu S, Saka N, et al. Sonography and Magnetic Resonance Imaging Characteristics of Testicular Adrenal Rest Tumors. Pol J Radiol. 2017;82:583–8. [PMC free article: PMC5894055] [PubMed: 29662589]
- New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2013;110(7):2611–6. [PMC free article: PMC3574953] [PubMed: 23359698]
- Balsamo A, Baldazzi L, Menabo S, Cicognani A. Impact of molecular genetics on congenital adrenal hyperplasia management. Sex Dev. 2010;4(4-5):233–48. [PubMed: 20639616]
- New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99(6):E1022–30. [PMC free article: PMC4037720] [PubMed: 24606108]
- Lin-Su K, Harbison MD, Lekarev O, Vogiatzi MG, New MI. Final adult height in children with congenital adrenal hyperplasia treated with growth hormone. J Clin Endocrinol Metab. 2011;96(6):1710–7. [PMC free article: PMC3206397] [PubMed: 21450983]
- Review Congenital adrenal hyperplasia: not really a zebra.[Am Fam Physician. 1999]Review Congenital adrenal hyperplasia: not really a zebra.Deaton MA, Glorioso JE, McLean DB. Am Fam Physician. 1999 Mar 1; 59(5):1190-6, 1172.
- Review The use of liquid chromatography-tandem mass spectrometry in newborn screening for congenital adrenal hyperplasia: improvements and future perspectives.[Front Endocrinol (Lausanne). 2...]Review The use of liquid chromatography-tandem mass spectrometry in newborn screening for congenital adrenal hyperplasia: improvements and future perspectives.de Hora M, Heather N, Webster D, Albert B, Hofman P. Front Endocrinol (Lausanne). 2023; 14:1226284. Epub 2023 Oct 2.
- Congenital adrenal hyperplasia: current surgical management at academic medical centers in the United States.[J Urol. 2015]Congenital adrenal hyperplasia: current surgical management at academic medical centers in the United States.Sturm RM, Durbin-Johnson B, Kurzrock EA. J Urol. 2015 May; 193(5 Suppl):1796-801. Epub 2015 Mar 25.
- Review Management of adolescents with congenital adrenal hyperplasia.[Lancet Diabetes Endocrinol. 2013]Review Management of adolescents with congenital adrenal hyperplasia.Merke DP, Poppas DP. Lancet Diabetes Endocrinol. 2013 Dec; 1(4):341-52. Epub 2013 Nov 15.
- A case of lipoid congenital adrenal hyperplasia presenting with cholestasis.[Iran J Pediatr. 2011]A case of lipoid congenital adrenal hyperplasia presenting with cholestasis.Khodadad A, Modaresi V, Kiani MA, Rabani A, Pakseresht B. Iran J Pediatr. 2011 Dec; 21(4):539-42.
- Congenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment - EndotextCongenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...