

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

TAKE HOME POINTS
1-The differential diagnosis of HH includes structural, functional and genetic abnormalities affecting the hypothalamic-pituitary-gonadal axis.
2-While our understanding of the genetic basis of HH has expanded considerably in the past decade, mutations have only been identified in approximately 40% of patients to date.
3-The traditional view that congenital HH is a simple monogenic disorder has been challenged by the demonstration of oligogenicity, which helps explain the phenotypic variability underlying this condition.
4-HH is a treatable cause of male infertility with larger testicular size and higher baseline inhibin B levels predicting a favorable response to gonadotropin or GnRH therapy, while negative prognostic indicators include cryptorchidism and possibly prior androgen use.
5-While lifelong androgen replacement is necessary in most men with HH, a brief discontinuation of hormonal therapy should be considered in all cases given that reversibility has been demonstrated in up to 10% of cases.
DEFINITIONS
Pulsatile secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus is required for both the initiation and maintenance of the reproductive axis in the human. Pulsatile GnRH stimulates biosynthesis of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) that in turn initiate both intra-gonadal testosterone production and spermatogenesis as well as systemic testosterone secretion and virilization. Failure of this episodic GnRH secretion or action, or disruption of gonadotropin secretion, results in the clinical syndrome of hypogonadotropic hypogonadism (HH). Disorders causing HH are differentiated from primary testicular disease by the demonstration of low/normal gonadotropin levels in the setting of low testosterone concentrations and sperm counts (Fig. 1). Congenital abnormalities leading to HH are rare but well described and are usually the consequence of deficient GnRH secretion occurring either in isolation (normosmic congenital hypogonadotropic hypogonadism (nCHH)), or in association with anosmia (Kallmann syndrome (KS)). A growing number of loci have been associated with congenital GnRH deficiency and mutations in the GnRH receptor, as well as both LH-b and FSH-b subunits, have been reported. Acquired causes of HH are more common and can be due to any disorder that affects the hypothalamic-pituitary axis.
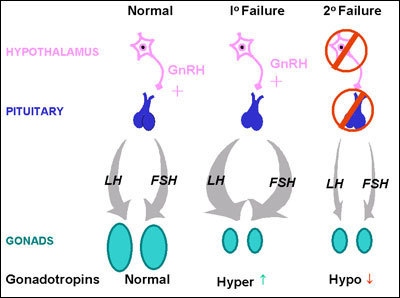
Fig. 1Schematic of the hypothalamic-pituitary-gonadal axis in a normal adult male and in the setting of primary and secondary hypogonadism.
NORMAL GnRH SECRETION ACROSS DEVELOPMENT IN THE MALE
In the human, the pattern of GnRH-induced gonadotropin secretion is constantly changing during sexual development. Therefore, the establishment of a robust normative database is critical for understanding pathologic states like HH.
Fetal/Neonatal Life
GnRH neurons originate outside the central nervous system in the olfactory placode, migrate along the olfactory, terminalis, and vomeronasal nerves up the nasal septum, and through the cribriform plate to the forebrain, ultimately reaching their final destination in the arcuate nucleus of the hypothalamus (1). WhileGnRH neurons have been demonstrated in the fetal hypothalamus by 9 weeks gestation, it is not until 16 weeks that functional connections are established between these neurons and the portal system. From mid-gestation until 6 months of postnatal life, pulsatile secretion of GnRH stimulates gonadotropin biosynthesis and secretion that, in turn, initiates gonadal sex steroid production (2).
Childhood Period
During childhood, the hypothalamic-pituitary axis is not completely quiescent and is characterized by low amplitude GnRH secretion as mirrored by LH secretion using ultrasensitive LH assays (3).
Puberty
The onset of puberty is marked by sleep-entrained reactivation of the reproductive axis characterized by a striking increase in the amplitude of LH pulses with a lesser change in frequency (4). This nocturnal rise of LH secretion stimulates gonadal secretion of both sex steroids and inhibin B, which return to prepubertal levels during the daytime. As puberty progresses, secretion of gonadotropins occurs during both day and night, allowing sexual development to be completed. The precise neuroendocrine trigger to puberty is still unknown. However, it is likely to be a process that removes inhibition of GnRH release rather than one increasing GnRH synthesis as abundant GnRH mRNA is present in the hypothalamic neurons of primates at an equivalent developmental stage (5, 6).
Adulthood
During adulthood, gonadotropins are secreted in a pulsatile fashion. In the adult male, LH is secreted in pulses approximately every 2 hours (Fig. 2A) (7). However, considerable variability is observed in LH pulse patterns and there is a wide range of testosterone secretory patterns. Indeed, in 15% of normal men whose hypothalamic-pituitary-gonadal (HPG) axis was examined using frequent blood sampling, serum testosterone levels as low as 3.5 nmol/L were recorded (to convert to ng/dL, multiply by 28.6) following long inter-pulse intervals of LH secretion, although mean testosterone levels remained within the normal range (Fig. 3). This within-patient variation must be considered when interpreting single LH and testosterone measurements obtained during the evaluation of a male with suspected hypogonadism. This variability is particularly important in middle-aged and older men as up to 30% men that are found to have a low testosterone concentration, will have a normal level on repeat testing (8).
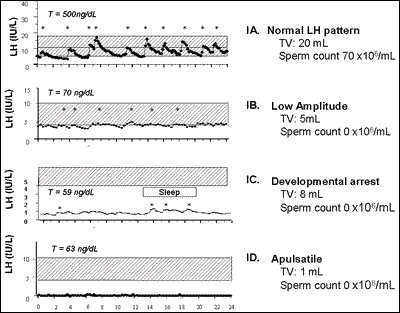
Fig. 2
Spectrum of GnRH-induced LH secretion in men with GnRH deficiency. LH pulsations are indicated by asterisks. A, Normal adult male pattern of GnRH secretion with high amplitude regular LH pulsations and normal serum testosterone (T), testicular volume (TV) and sperm count; B, Disordered amplitude pattern of GnRH secretion in an CHH male, characterized by low amplitude LH pulsations, low serum T, and azoospermia; C, Sleep-entrained or developmental arrest pattern of LH secretion in an CHH male characterized by relatively low amplitude LH pulsations clustered during the night-time hours analogous to the pattern which normally occurs at puberty. Note that the TV is higher than in the subject with the apulsatile pattern; D, Apulsatile pattern of GnRH secretion in an CHH male with complete absence of endogenous LH pulsations, low serum T, prepubertal TV and azoospermia.

Fig. 3
Frequent blood sampling (q 10 min for 24 h) of serum testosterone (T) and LH in a normal male. Note that a T level of 3.2 nmol/L (to convert to ng/dL, multiply by 28.6) which is in the hypogonadal range was recorded after a long interpulse interval of LH secretion.
CLINICAL PRESENTATION
The clinical features of GnRH and/or gonadotropin deficiency are typically first manifested at puberty, a time when there is normally a marked increase in GnRH secretion. The phenotypic presentation of HH varies with age of onset (congenital vs. acquired), severity (complete vs. partial), and duration (functional vs. permanent).
HH may also be diagnosed in the neonatal period. The typical clinical phenotype is that of a male infant with normal sexual differentiation, cryptorchidism and micropenis in whom gonadotropin and sex steroid levels are inappropriately low given the normal activation of the HPG axis during this period. Early in fetal life, the testosterone production required for full sexual differentiation is thought to be stimulated by maternal hCG alone. However, endogenous secretion of GnRH in the late fetal/early neonatal periods appears necessary for inguino-scrotal descent of the testes and full growth of the external genitalia (9, 10). Accordingly, cryptorchidism and microphallus have been reported in up to 50% of patients with IHH and KS in some small series (11, 12) and may represent surrogate markers of failure of activation of GnRH secretion during the neonatal window.
Most often, the diagnosis of HH is delayed until adolescence, when there is a failure to go through puberty. The clinical presentation includes lack of development of secondary sex characteristics, eunuchoidal body proportions (upper/lower body ratio <1 with an arm span 6 cm > standing height), a high-pitched voice, mild anemia, delayed bone age, and pre-pubertal testes (12). However, the syndrome is clinically heterogeneous in that some patients have evidence of partial spontaneous pubertal development reflected by larger gonadal size despite hypogonadal testosterone levels and inappropriately low gonadotropin levels. Gynecomastia is not a typical feature of GnRH deficiency given the hypogonadotropic state and is most commonly seen in patients treated with gonadotropins (13, 14). HH may also present after completion of puberty resulting in a disruption in reproductive function in adulthood characterized by decreased libido, erectile dysfunction and oligo- or azoospermia (15).
DIFFERENTIAL DIAGNOSIS
Hypogonadotropic hypogonadism may be broadly classified into congenital or acquired disorders (Table 1).
Table 1Differential Diagnosis of Hypogonadotropic Hypogonadism (HH)
Congenital HH
Genetic defects of the gonadotropin subunits HH associated with other pituitary hormone deficiencies HH associated with obesity
|
| Acquired HH Structural
Functional
|
Congenital Hypogonadotropic Hypogonadism
CHH is characterized by an isolated defect in GnRH secretion as evidenced by: i) complete or partial absence of GnRH-induced LH pulsations (Fig. 1) (16, 17); ii) normalization of pituitary-gonadal axis function in response to physiological regimens of exogenous GnRH replacement (16-18); iii) otherwise normal hormonal testing of the anterior pituitary including a normal ferritin level and; iv) normal imaging of the hypothalamic-pituitary region.
CHH was first reported in association with anosmia and termed Kallmann syndrome (19). However, it was subsequently appreciated that several patients with CHH lack evidence of an olfactory defect and thus have a normosmic form of CHH. While the majority of CHH patients present with lack of pubertal development, there is considerable clinical heterogeneity. Depending on the degree of prior spontaneous pubertal development, testicular size in men with CHH may range from prepubertal to near-normal adult testes. In addition to anosmia, a variety of other anomalies have been reported to occur in CHH with an increased frequency in the KS subset including cleft lip and palate, synkinesia, sensorineural deafness, cerebellar ataxia, renal agenesis, digital bony abnormalities and dental agenesis (19-21). Some of these clinical features are highly associated with particular genetic causes of KS so that the clinical phenotype can be utilized to prioritize genetic testing (22).
Given that it is a rare disease, data on the incidence of CHH is limited with estimates varying from 1/10,000 to 1/86,000 (23, 24). Isolated GnRH deficiency occurs more commonly in men than in women. Based on our review of 250 consecutive cases seen at the Massachusetts GeneralHospital, the male:female ratio is 4:1.
Variant or Partial Forms of GnRH Deficiency
i) Adult onset IHH
An acquired form of isolated GnRH deficiency termed adult-onset idiopathic HH was first reported in 1997 (15). In this group of patients, puberty occurs normally and is followed years later by a decrease in libido, sexual function and fertility. The biochemical profile of these patients is indistinguishable from subjects with congenital GnRH deficiency in that they have an apulsatile pattern of LH secretion associated with low serum testosterone levels. In addition, more than 90% of cases have normal restoration of the pituitary-gonadal axis when treated with physiologic GnRH replacement regimens supporting a hypothalamic defect as the origin of the disorder. Unlike patients with functional GnRH deficiency, no factors known to impair GnRH secretion transiently such as stress, exercise or weight loss have been identified in this population. In addition, longitudinal follow-up of these cases of adult-onset IHH for over a decade suggests that the neuroendocrine defect is permanent in that all remained frankly hypogonadal (25).
ii) Fertile Eunuch Syndrome
In 1950, McCullagh et al provided the first description of a patient with the fertile eunuch syndrome characterized by eunuchoidal proportions and lack of secondary sexual characteristics in the presence of normal size testes and preserved spermatogenesis (26). In this disorder, enfeebled endogenous GnRH secretion appears sufficient to achieve the intra-gonadal testosterone levels needed to support spermatogenesis and testicular growth, but is insufficient to induce virilization. The clinical picture of the fertile eunuch is rather similar to that of mid-pubertal boys; indeed, frequent blood sampling in two men with the fertile eunuch syndrome demonstrated a nocturnal rise of LH and testosterone secretion synchronous with sleep, analogous to the pattern seen in mid-puberty (27). In contrast, we described a patient with the fertile eunuch syndrome displaying a detectable but apulsatile pattern of LH secretion, who was found to harbor a partially inactivating mutation of the GnRH receptor (GnRH-R) (28). The clinical presentation of the fertile eunuch syndrome also reveals some similarity to adult onset IHH in that both are characterized by GnRH deficiency in association with normal or near-normal testicular size. However, “fertile eunuchs” are distinguished by the preservation of spermatogenesis and the achievement of fertility with testosterone or hCG therapy alone (29, 30).
iii) Delayed puberty
Frequently, there is a history of delayed, but otherwise normal, puberty among the families of patients with CHH (21). While the incidence of delayed puberty in the general population is less than 1% (31), rates of up to 12% have been observed in families with CHH (21). These data suggest that delay in initiating, but subsequent normal progression through, puberty may represent the mildest end of the phenotypic spectrum of CHH.
Acquired Hypogonadotropic Hypogonadism Functional
Functional forms of HH are characterized by a transient defect in GnRH secretion. This type of presentation occurs most commonly in female hypogonadotropic subjects with hypothalamic amenorrhea (HA). In susceptible individuals, HA may be precipitated by factors such as significant weight loss, exercise or stress (32-34). In addition, a genetic predisposition to HA has also been identified with the demonstration of rare variants in genes associated with CHH in some women with this disorder (35). Typically, GnRH secretion will resume after correcting the underlying abnormality and menstruation will be restored. While in women the presence or absence of menses acts as a useful clinical marker of the functioning of the HPG axis, there is no comparable clinical marker in the male. Moderate to severe dietary restriction in otherwise healthy men has been shown to decrease testosterone levels by impairing secretion of GnRH (36, 37). In addition, some (38, 39) but not all (40) studies have shown that strenuous physical exercise may adversely affect testosterone concentrations. However to date, a clinical syndrome of functional GnRH deficiency in men that is analogous to HA has not been definitively established.
Drug-Induced GnRH Deficiency
Use of anabolic steroids may result in a functional form of HH manifested by decreased concentrations of both testosterone and dihydrotestosterone and a marked impairment of spermatogenesis (41, 42). While the suppression of the HPG axis induced by anabolic steroids is reversible, rate of recovery following cessation of steroid use is variable and can range from 4-12 months (41, 43). Chronic treatment with glucocorticoids may also lead to hypogonadism. In one study, 16 men with chronic pulmonary disease who received high-dose glucocorticoids for at least one month had a mean serum testosterone of 6.9 nmol/L, compared to 15.6 nmol/L in 11 men matched for age and disease (44). Given that serum LH levels did not increase in this study, these data suggest a predominantly central mechanism for glucocorticoid-induced hypogonadism. Chronic use of oral and intrathecal narcotic analgesics may also suppress LH secretion and result in reversible HH (45). A common side-effect of psychotropic medications such as phenothiazines or risperidone is hyperprolactinemia, which inhibits endogenous GnRH release resulting in HH (46).
Critical Illness
Any period of severe chronic (47), or acute illness such as surgery (48), myocardial infarction (49), burn injury (50), and renal disease (51) may result in low testosterone levels (52). Acute injury is accompanied by a prompt and direct suppression of Leydig cell function (53). When severe stress becomes prolonged, hypogonadotropism ensues largely due to attenuation of pulsatile LH release (53). Endogenous dopamine or opiates may be involved in the pathogenesis of HH induced by critical illness (52).
Structural
Structural lesions of the hypothalamus and pituitary can interfere with the normal pattern of GnRH and/or gonadotropin secretion. The majority of patients with HH secondary to such tumors have multiple pituitary hormone deficiencies in addition to that of gonadotropins (54, 55). However, a mass lesion in the pituitary or hypothalamus is more likely to disrupt the secretion of gonadotropins than that of ACTH or TSH. Thus, patients may present with hypogonadism in the absence of adrenal or thyroid hormone deficiency.
In children, craniopharyngioma is the most common tumor resulting in HH, and is often associated with growth retardation, visual field defects and diabetes insipidus. In adults, prolactinomas are the most frequent cause of HH and may do so by either interfering with GnRH secretion, or in the case of macroadenomas, by local destruction and compression of the gonadotropes. Hyperprolactinemia results in altered dopaminergic function, which has been shown to reduce GnRH mRNA levels and decrease serum levels of LH, FSH and testosterone (56, 57). Although men with hyperprolactinemia may develop galactorrhea, it occurs much less frequently than in women, due to lack of prior stimulation by estrogen and progesterone.
Rarer causes of HH include infiltrative disorders of the hypothalamus or pituitary such as hemochromatosis, sarcoidosis, lymphocytic hypophysitis and histiocytosis, in which case the presence of systemic signs and symptoms frequently leads one to the diagnosis (58-59). Cranial irradiation for the treatment of CNS tumors or leukemia may also result in the gradual onset of hypothalamic-pituitary failure (60). The degree of impairment depends upon both the dose and type of radiation employed and typically reflects hypothalamic dysfunction since the hypothalamus is significantly more radiosensitive than the pituitary. In general, the younger the patient the greater the susceptibility to endocrine dysfunction following radiation therapy (60). Sudden and severe hemorrhage into the pituitary can also results in permanent impairment of pituitary function including hypogonadism (61).
PATHOPHYSIOLOGY
The combined use of both frequent blood sampling and genetic studies has contributed to our understanding of the pathophysiology of isolated GnRH deficiency. Due to its confinement within the hypophyseal-portal blood supply, direct sampling of GnRH is not feasible in the human. In addition, measurements of GnRH in the peripheral circulation do not accurately reflect its secretion due to its rapid half-life of 2-4 min (62, 63). Consequently, inferential approaches must be used to study GnRH secretion in the human.
Traditionally, LH has been used as a surrogate marker of GnRH activity (64, 65). More recently, the pulsatile component of free alpha subunit (FAS) secretion has been shown to be tightly correlated with that of LH (66)and to be driven by GnRH based on its eradication by GnRH receptor blockade (67). Given its half-life of 12-15 min, FAS is useful in tracking GnRH secretion at fast pulse frequencies (68).
Patterns of GnRH Secretion in Subjects with GnRH Deficiency
A range of abnormalities in the neuroendocrine pattern of GnRH secretion mirrors the clinical spectrum of CHH. We examined pulsatile gonadotropin secretion in 50 men with isolated GnRH deficiency during 10-min blood sampling for up to 24 h (16, 17). The largest subset of patients (84%) had no detectable LH pulses (apulsatile pattern, Fig. 2D) and were found to have neither historical nor physical evidence of puberty, thus representing the most severe form of GnRH deficiency. A second pattern of GnRH secretion was observed in which LH pulses occurred at a normal frequency but were of diminished amplitude compared to those of normal men (decreased amplitude pattern, Fig. 2B). This decreased pulse amplitude pattern is suggestive of either enfeebled hypothalamic release of GnRH or a state of GnRH resistance as may be seen with a partial defect at the level of the GnRH-R. A third group of subjects demonstrated predominantly nocturnal LH pulsations (developmental arrest pattern, Fig. 2C); these patients had some testicular growth and a history consistent with an arrest of puberty. Finally, pulsatile LH activity appeared normal by RIA in one patient; however, LH bioactivity was absent when tested in the dispersed rat Leydig cell assay (69). Thus in CHH men, the spectrum of abnormalities in GnRH secretion or action is likely to contribute to the clinical and biochemical heterogeneity of this disorder.
Genetics of CHH
Considerable genetic heterogeneity has also been found to underlie CHH. Most cases of CHH are sporadic (80%), suggesting that either the frequency of spontaneous mutations in this disorder is high or that the etiology of many cases is not genetic. In the last decade considerable advances have been made in unraveling the genetic basis for CHH and to date, mutations have been identified in approximately 40% of cases (70). Unique genetic mechanisms have been described for both KS and nCHH. However, some probands with KS have family members with hypogonadism but normal olfaction (Fig. 4) and the same mutations have been shown to be associated with both KS and nCHH suggesting that they should no longer be viewed as distinct diagnostic subsets (71).
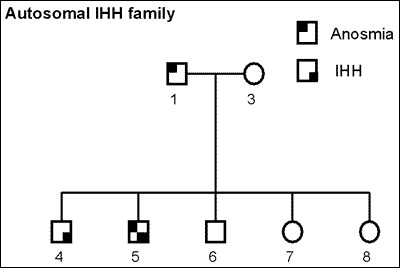
Fig. 4. A
family with GnRH deficiency and multiple affected members in two generations. The pedigree is compatible with autosomal dominant transmission with incomplete penetrance. Note the presence of GnRH deficiency both with and without anosmia in the same family.
Human GnRH deficiency has traditionally been viewed as a monogenic disorder that could be inherited as an autosomal dominant, autosomal recessive, or X-linked trait. However, the long appreciated phenotypic variability within and across families with single gene defects combined with the recent demonstration that patients with CHH may harbor a mutation in more than one gene (72, 73) suggest a more complex mode of inheritance. Indeed, a systematic study of nearly 400 patients with isolated GnRH deficiency who were screened for 8 known loci revealed 17% of patients harbor multiple mutations consistent with an oligogenic rather than a monogenic mode of inheritance (74).
Kallmann Syndrome Gene (KAL1 )
Mutations in the KAL1 gene were the first characterized genetic defect reported to cause GnRH deficiency (75). Anosmin, the protein encoded by the KAL gene, shares homology with molecules involved in neural development and contains 4 contiguous fibronectin type III repeats found in neural cell adhesion molecules (76). Confirmation of the causative role of the KAL gene in KS was provided by a study of a KS fetus with a deletion from Xp22.31 to Xpter, i.e. including the entire gene. Histologic examination of the brain of this fetus demonstrated that migration of the GnRH and olfactory neurons was arrested just below the telencephalon at the cribriform plate (77). Therefore, mutations in KAL1 appear to cause premature termination of migration of both the olfactory and GnRH neurons to the brain resulting in anosmia and IHH. This profound defect of GnRH neuronal migration results in the complete failure of activation of the HPG axis in patients with X-linked KS (X-KS). Indeed, X-KS patients typically present with more severe forms of CHH characterized by a high incidence of microphallus, cryptorchidism, complete absence of puberty with small testes, and very low inhibin B levels (78, 79). Further, this subset of men with CHH has the poorest response to therapy to induce spermatogenesis (80).
Mutations in KAL1 are distributed throughout the gene, although most point mutations cluster in the four fibronectin type III repeat domains (81-82). Most X-KS mutations cause alteration of splicing, frameshift or stop codons and result in synthesis of a truncated anosmin protein (83). Missense mutations have rarely been described (83, 84). There appears to be some variability in the phenotypic expression of mutations both within and between families (84, 85). No correlation has been demonstrated between phenotype and location of the mutation described (86).
Autosomal Genes
Despite the biology revealed by the X-linked cases, several lines of evidence suggest that autosomal genes account for the majority of familial cases. Of 36 familial cases with GnRH deficiency studied by Waldstreicher and colleagues, only 21% could be attributed to X-linkage (21). When the analysis was extended to include surrogate markers of IHH (isolated congenital anosmia and delayed puberty), the X-linked pedigrees comprised only 11%, autosomal recessive 25% and autosomal dominant 64%. These data suggest that in familial cases, the X-linked form of GnRH deficiency is the least common.
GnRH1 and GNRHR Mutations
Although an obvious candidate gene, mutations of GNRH1, the gene encoding the preprohormone that is processed to GnRH, remained elusive for many years and it was not until 2009 that mutations in this gene were identified by two independent groups (87, 88). In the first report a homozygous GNRH1 frameshift mutation was identified in a brother and sister that completely deleted the GnRH decapeptide sequence resulting in severe GnRH deficiency manifested by complete absence of puberty (87). Exogenous GnRH administration was shown to restore normal LH pulsatility indicating normal pituitary responsiveness.
While mutations in GNRH1 are a rare cause of CHH, loss-of-function mutations in the GnRH receptor are much more common with 22 novel mutations having been reported to date (89). In this autosomal recessive form of GnRH deficiency, affected patients are homozygous for one mutation or compound heterozygous (with two mutations, each on one allele), and in rare sporadic nIHH cases, heterozygous mutations have been identified (74). Patients harboring GNRHR mutations present with a wide phenotypic spectrum ranging from the fertile eunuch syndrome (28) to partial CHH (90, 91) to the most complete form of GnRH deficiency characterized by cryptorchidism, microphallus, undetectable gonadotropins, and absence of pubertal development (92-93). Patients with hypogonadism caused by inactivating mutations of the GnRH receptor tend to have a poor response to pulsatile GnRH administration (94). However, successful ovulation and conception have been described in a patient with an inactivating receptor mutation receiving high doses of pulsatile GnRH, thus indicating that many GnRH receptor mutations create a relative rather than an absolute resistance to pulsatile GnRH administration (95).
Kisspeptin 1 receptor (KISS1R: formerly GPR54) and its ligand KISS1
Linkage analysis of a large consanguineous family with HH identified this locus for autosomal recessive CHH on chromosome 19p13 (96). Shortly thereafter Seminara et al confirmed KISS1R as an important regulator of puberty in a different consanguineous family (97). Murine studies demonstrated a hypogonadal phenotype similar to that seen in the human that was responsive to exogenous GnRH (97). While mutations in KISS1R appear to be an uncommon cause of GnRH deficiency in the human accounting for <5% of cases, these discoveries opened a new area of exploration in relation to the genetics of puberty and GnRH biology upstream of the hypothalamus. Recently, heterozygous variants in KISS1 have been noted in association with CHH (98).
Fibroblast Growth Factors
The study of two patients affected by different contiguous gene syndromes, both of which included KS, led to the discovery of fibroblast growth factor receptor-1 (FGFR1) as a gene associated with congenital GnRH deficiency (99). Interestingly, mutations in FGFR1 can cause both KS and nCHH (100, 101) and have been identified in as many as 10% of CHH cases. Patients harboring FGFR1 mutations exhibit a wide range of both reproductive and non-reproductive phenotypes including cleft palate, agenesis of the corpus callosum, dental agenesis, unilateral hearing loss, and skeletal anomalies (100-103). The variable expressivity of phenotypes among patients and family members harboring identical FGFR1 mutations is consistent with a more complex, oligogenic model for GnRH deficiency (74, 103).
In 2008, a critical ligand for FGFR-1, FGF8, was found to be involved in GnRH neuron ontogeny (104). Subjects harboring FGF8 mutations also display a broad spectrum of pubertal development ranging from absent, to partial, to complete puberty (in a male with adult onset HH). Additionally, the non-reproductive associated phenotypes are wide ranging and include hearing loss and a variety of skeletal anomalies (high arched palate, cleft lip/palate, severe osteoporosis, camplodactyly, and hyperlaxity of the digits) (105). Further, the variable expressivity observed in FGFR1 families is also evident in family members harboring the same FGF8 mutation (103, 105).
Recently, the key developmental role of FGF signaling in reproduction has been highlighted by the identification of mutations in genes encoding a broader range of modulators of the FGFR1 pathway including FGF17 and other members of the so-called FGF8 synexpression group (106). Mutations in these genes are associated with complex modes of inheritance and they are thought to act primarily as contributors to an oligogenic genetic architecture underlying CHH.
Prokineticins (PmROKR2 and PROK2).
Similar to what has been seen with FGFR1, mutations in the prokineticin system (PROKR2 and PROK2) have been associated with both KS and nCHH (107-110). It is unusual for homozygous mutations to occur and mutations in both ligand and receptor are typically heterozygous. In a review of a large cohort of KS patients harboring mutations in PROK2/PROKR2, Sarfati and colleagues noted that those harboring biallelic mutations exhibit more severe and less variable reproductive phenotypes than those with monoallelic mutations (111).
Neurokinins (TAC3 and TACR3)
The strategy of studying consanguineous families with CHH, which led to the discovery of KISS1R, was also successful in identifying mutations in other autosomal genes (TAC3 and TACR3) with a hitherto unrecognized role in reproduction (112). Patients harboring mutations in TAC3 and TACR3 have a particularly high incidence of microphallus suggesting that this pathway may be important in regulating the “mini-puberty” of infancy (113). A second striking phenotype associated with mutations in this pathway is that recovery of the hypothalamic-pituitary-gonadal axis has been observed in a significant number of patients (113). Further, careful characterization of patients harboring TAC3/TAC3R mutations reveals these patients exhibit low frequency GnRH pulses and a relatively elevated FSH/LH ratio, a biochemical clue for selective gene screening (114).
Genetic Defects of the Gonadotropins
Genetic defects in the genes encoding LH-b subunit and FSH-b are rare and have been summarized in a recent review (115). The initial case report of a male who failed to go through puberty identified a homozygous mutation in amino acid 54 of the LH-b subunit that rendered LH incapable of binding to its receptor (116). The mutant hormone had no biologic activity but had normal immunoreactivity. Testosterone levels were reduced in association with elevated LH and FSH levels. This clinical picture could easily be confused with primary hypogonadism had the patient not been shown to have a normal serum testosterone response to hCG administration. The first reported male with an FSH-b gene mutation (Cys82Arg missense) appeared normally virilized and had normal testosterone levels, but small testes, azoospermia, a high LH and a low FSH level (117). The second reported case of a man with an FSH-b gene mutation (Val61X) had very small testes (1-2 mL) and an unexpected absence of pubertal development; he was found to harbor an additional defect in Leydig cell function as evidenced by high LH and low testosterone levels (118).
Other rare genetic syndromes associated with hypogonadotropic hypogonadism
HH also occurs as one of a constellation of features in a number of rare syndromes including X-linked adrenal hypoplasia congenita (AHC) caused by defects in the DAX1 (NROB1) gene (Xp21) (119, 120), CHARGE syndrome (coloboma, heart defects, atresia of the choanae, retardation of growth and development, genital anomaly, ear abnormalities) resulting from CHD7 (8q12.1) defects (121, 122), leptin deficiency (123, 124), and prohormone convertase 1/3 deficiency (5q15-21) (125).
In addition, combined pituitary hormone deficiency has been linked with rare abnormalities in genes encoding transcription factors necessary for pituitary development. Mutations in the gene, PROP1 (Prophet of Pit-1) appear to be the most common cause of both familial and sporadic congenital combined pituitary hormone deficiency (126). Most patients with inactivating mutations in PROP1 exhibit low gonadotropin levels and fail to enter puberty as well as having additional deficiencies in GH, PRL, and TSH (126). However, there is a report of two sibships with a homozygous mutation, Arg120Cys in PROP1 in which the affected children entered puberty spontaneously and then developed gonadotropin deficiency over time (127). The same sequence of events was observed in several children with mutations causing a complete loss of function raising questions as to whether this should more correctly be viewed as a model of acquired rather than congenital gonadotropin deficiency (128).
Recently the critical role of PROK2, PROKR2 and FGF8 in the developing pituitary has been noted in the genetic overlap between KS and developmental disorders including combined pituitary hormone deficiency, septo-optic dysplasia, and holoprosencephaly (129, 130). Mutations in the gene SOX2 have been identified in patients with HH in association with anopthalmia or microphhalmia (131, 132), while SOX10 and IL17RD are key genes to consider when deafness is present in association with CHH (106, 133).
EVALUATION
Hypogonadism is defined as a defect in one of the two major functions of the testes i.e. production of testosterone and spermatogenesis. The presence of hypogonadism can reflect disorders intrinsic to the testes (primary or hypergonadotropic hypogonadism) or disorders of the pituitary or hypothalamus (secondary or hypogonadotropic hypogonadism). These two entities can be distinguished by measuring serum LH and FSH concentrations (Fig. 5). Primary hypogonadism is characterized by a low serum testosterone level and oligo- or azoospermia in the presence of elevated serum LH and FSH concentrations. In contrast, secondary hypogonadism is diagnosed in the setting of a low testosterone level and sperm count in association with low or inappropriately normal serum LH and FSH concentrations.
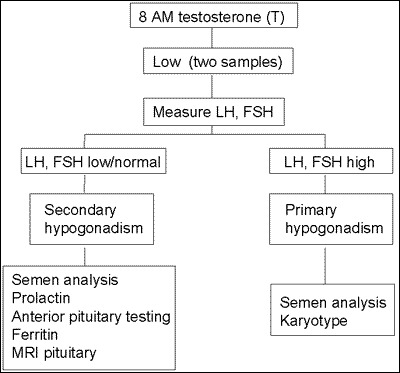
Figure 5Algorithm for evaluation of hypogonadism in a male.
It is best to measure testosterone in a morning sample given the normal diurnal rhythm of testosterone secretion. Repeat measurements should be performed if the initial reading is low. In secondary hypogonadism, measuring the serum LH response to a single bolus of exogenous GnRH is not helpful in distinguishing pituitary from hypothalamic disease, because a subnormal response may occur in both settings. Patients with HH due to congenital absence of hypothalamic GnRH secretion may have had no prior exposure to GnRH; in this setting, repeated administration of GnRH is needed to prime the gonadotropes to elicit a gonadotropin response. Characterization of the pulsatile pattern of LH secretion with frequent blood sampling is useful in refining the diagnosis in a research setting, but is not practical for clinical purposes.
A semen analysis should be obtained to assess sperm production and the count, motility, and morphology determined. Based on WHO criteria, the parameters for a normal semen analysis are a sperm density ³ 15 million sperm/mL of ejaculate with total motility ³ 40% and strictly defined normal morphology of ³ 4% (134). Two normal semen analyses are sufficient to indicate that the sperm density and motility are normal.
In the case of secondary hypogonadism, it is critical to assess the rest of the pituitary axis, including a prolactin level to ensure that the defect is isolated to the HPG axis. Transferrin saturation should be measured to exclude hemochromatosis. Patients with HH typically undergo adrenarche at a normal age and should therefore have normal adult male levels of DHEAS. Radiographic evaluation should include a bone age determination, MRI of the pituitary and hypothalamic area and, in adults, a DEXA scan to assess bone mineral density. Renal ultrasound should be considered to assess for unilateral renal agenesis in patients with KS especially those known to have a KAL1 mutation.
The growing genetic heterogeneity of CHH poses a significant challenge to clinicians in the diagnostic work of patients. While in familial cases the mode of inheritance can be used to guide genetic testing, the majority of CHH cases are actually sporadic. However, a careful clinical evaluation can be helpful in prioritizing genetic testing. In a recent analysis of 219 patients with KS the following clinical features were highly associated with specific gene defects: synkinesia (KAL1), dental agenesis (FGF8/FGFR1), digital bony abnormalities (FGF8/FGFR1), and hearing loss (CHD7) (135). Weaker associations were observed between renal agenesis and KAL1, cleft lip/palate and FGF8/FGFR1, CHD7 and HS6ST1, and male reversal and FGF8/FGFR1 and HS6ST1 (135).
TREATMENT
Choosing the most appropriate treatment for men with HH should be based upon an informed discussion between the patient and his physician. Androgen therapy, whether by exogenous testosterone replacement or induction of endogenous testosterone production by hCG is needed in all HH patients. Androgens play a number of important physiologic roles in the human and are required not only for virilization and normal sexual function, but also for maintenance of both muscle and bone mass, as well as normal mood and cognition.
While testosterone is the primary treatment modality used to induce and maintain secondary sexual characteristics and sexual function in men with hypogonadism, treatment with testosterone does not restore fertility. Therefore, in patients in whom fertility is the treatment goal, induction of gonadotropin secretion by pulsatile GnRH or exogenous gonadotropins is necessary. While hormone therapy is the mainstay of treatment for CHH, cryptorchidism, if present, should be treated surgically with orchidopexy, ideally before the age of 2 years to improve fertility outcomes and reduce the risk of future testicular malignancy (136).
The traditional teaching has been that patients with CHH require life long therapy but the recent demonstration of reversal of hypogonadism suggests that this many not always be the case (113, 137, 138). In a study of a large cohort of men with CHH, 10% of cases demonstrated a sustained reversal after discontinuation of treatment. Reversal occurred across a spectrum of phenotypes including patients with and without anosmia and those with absent or partial spontaneous puberty (137). A key, but sometimes subtle, clinical finding that can predict reversal is spontaneous testicular enlargement on testosterone therapy (137, 138). Reversal has been documented in patients with a variety of genetic defects including GNRHR, FGFR1 and CHD (137, 138) but has been shown to be particularly common in those with defects in the neurokinin pathway (113). While the mechanism of reversal in CHH is not understood it is possible is that exposure to sex steroids may enhance the plasticity of the neuronal network responsible for GnRH secretion. In light of these studies, it seems reasonable to recommend that a brief discontinuation of hormonal therapy be considered in patients with CHH to assess for reversibility. The trial period off treatment will depend on the formulation used but should be approximately 2-3 months for intramuscular testosterone and 4 weeks for those receiving transdermal preparations.
Testosterone Substitution
Testosterone is currently available in a variety of formulations for clinical use including injectable esters, transdermal patches, and gel preparations, each with its own unique pharmacokinetic profile. This topic is discussed in detail in Chapter 2.
Induction of Spermatogenesis
In patients who desire fertility, the options to induce spermatogenesis include exogenous gonadotropins, which are freely available or pulsatile GnRH, use of which is restricted to specialist centers. Conventional therapy uses hCG as an LH substitute in conjunction with FSH in the form of either human menopausal gonadotropins, highly purified urinary FSH preparations or recombinant FSH formulations. Both FSH and LH seem to be required.
The role of FSH in the initiation and maintenance of spermatogenesis in the human is controversial. There is discordance between the phenotype of men with mutations in the FSH receptor gene (FSHR) who are oligospermic (139) and those with mutations of the FSH-b subunit who are azoospermic (117, 118). If one assumes that the FSHR mutations were able to abolish FSH action completely, this report suggests that LH and testosterone alone are sufficient to induce spermatogenesis given that all five reported cases had sperm in the ejaculate and two had fathered children (139). However, the demonstration that the two patients with a mutation in the FSH-ß subunit were azoospermic (117, 118) suggests that the degree of inactivation of the FSH-R may have been incomplete. However, interpretation of this report is confounded by the fact that one of these patients also had hypogonadal testosterone levels so that the azoospermia in this case could not be attributed to lack of FSH alone (118). The importance of FSH in initiating spermatogenesis is further called in to question by the demonstration that in a subset of patients with HH and larger testicular size, spermatogenesis can be stimulated with hCG alone (140). However, this group largely comprises men with the fertile eunuch syndrome (29, 30), who may well have sufficient endogenous FSH secretion to sustain normal spermatogenesis with hCG alone.
Several other lines of evidence support a role for FSH in the maintenance of spermatogenesis. In contraceptive trials using testosterone esters, azoospermia is achieved only when serum FSH levels are rendered undetectable (141, 142). Similarly, there is a case report of a patient with a history of a hypophysectomy for a pituitary adenoma who was found to be fertile when treated with testosterone alone due a concomitant activating mutation of the FSH-R (143). A small Finnish study of boys with very small testes (< 3mL) resulting from severe GnRH deficiency or panhypopituitarism following treatment of intracranial tumors showed that treatment with recombinant human FSH (r-hFSH) alone prior to induction of puberty with hCG and FSH increased testicular size leading the authors to postulate that priming with FSH may have beneficial effects on spermatogenesis later in life (144). We recently expanded these findings in a randomized, open label study of the effects of r-hFSH on both hormonal and histologic outcomes in a cohort of men with CHH and prepubertal testes (145). Seven men were randomized to 4 months of r-hFSH followed by 24 months of GnRH and six men received 24 months of pulsatile GnRH only. Treatment with r-hFSH resulted in a doubling of testicular size, increased inhibin B levels into the normal range, and induced Sertoli cell and spermatagonia proliferation. All men receiving r-hFH pretreatment developed sperm in the ejaculate versus 4/6 of the GnRH only group and trended towards higher maximal sperm counts. However, given the small sample size r-hFSH pretreatment was not shown to be statistically superior to GnRH alone (145).
In addition to FSH, adequate intra-testicular testosterone concentrations are also essential for normal spermatogenesis as illustrated by a study of hypogonadal men in which spermatogenesis could be induced by the combination of hCG and hMG but not by a regimen comprising purified FSH and testosterone (146). In summary, the evidence suggests that qualitatively and quantitatively normal spermatogenesis is best maintained in the presence of both FSH and LH-induced T secretion.
1. Exogenous Gonadotropin Therapy
The typical gonadotropin regimen for induction of spermatogenesis in men comprises hCG in combination with FSH. Purified hCG is an effective substitute for LH given the structural homology between these 2 hormones which act through the same receptor on Leydig cells. While a variety of FSH formulations are now available in different countries, there is little to choose between them in terms of therapeutic efficacy. Traditionally, FSH has been administered in the form of human menopausal gonadotropins (hMG) derived from the urine of postmenopausal women. Although hMG has both FSH and LH activity, FSH activity predominates and LH activity is so low that combined administration with hCG is necessary to achieve fertility. Subsequently, highly purified urinary FSH preparations were developed, which have enhanced specific activity in comparison to hMG (10,000 IU/mg of protein vs 150 IU/mg of protein for hMG). In the early 1990s, r-hFSH formulations were developed, which have greater purity and specific activity than any of the urinary preparations and no intrinsic LH activity (147, 148). r-hFSH is produced in genetically engineered Chinese hamster ovary cells, in which the genes encoding the alpha and beta subunits have been introduced using recombinant DNA technology (147). Pharmacokinetic studies of r-hFSH indicate a half-life of 48 ± 5 h and a dose-dependent increase in the serum level of FSH (147).
In our experience, the subcutaneous route of administration is as effective as the intramuscular route for both gonadotropins and significantly increases patient compliance. Therapy is typically initiated with hCG alone at a dose of 1,000 IU on alternate days and the dose titrated based on trough testosterone levels and testicular growth (149). Alternatively, recombinant human hCG can also be administered subcutaneously from a prefilled syringe. In some patients the dose of hCG can be decreased over time as testicular size increases. In the majority of patients with larger testes at baseline, spermatogenesis can be initiated with hCG alone most likely due to residual FSH secretion (140, 150). Once there is a plateau in the response to hCG which typically occurs at around 6 months, therapy with FSH (in one of the three forms described above) should be added at a dose of 75 IU on alternate days. If sperm output and testicular growth remain suboptimal the dose of FSH can be gradually increased to 150 U daily. Continuation of this combined regimen for 12-24 months induces testicular growth in almost all patients, spermatogenesis in approximately 80% and pregnancy rates in the range of 50% (151-153). In an Australian study of 75 men with HH treated with gonadotropins the median time for sperm to appear in the ejaculate was 7.1 months and for conception was just over 28 months (152). Similar data were reported in a compilation of clinical trial data from Asian, European, Australian and American patients (153). Factors predictive of better outcome include larger baseline testicular size and absence of cryptorchidism. The Australian study reported that prior androgen use is also a negative prognostic indicator of response (152). While the study investigators propose that gonadotropin therapy be considered to induce puberty based on their results, confirmation that such an approach is superior to the conventional practice of giving testosterone would require a large clinical trial the logistics of which would be challenging given the rarity of this patient population. Gynecomastia is the most common side effect of gonadotropin therapy and is due to hCG stimulation of aromatase causing increased secretion of estradiol. This undesirable side effect can be prevented by using the lowest dose of hCG capable of maintaining serum testosterone levels towards the lower end of the normal range.
In the majority of HH patients treated with gonadotropins sperm density remains below the normal range. However, failure to achieve a normal sperm density does not preclude fertility. Indeed, the median sperm concentrations reported at conception range from 5-8 million/mL (140, 152). While spermatogenesis can be initiated even in patients with very small testes (18, 140), a longer duration of therapy is typically required and it may take up to 24 months for spermatogenesis to be induced. Accordingly when discussing the issue of fertility with patients, we recommend that they start treatment at least 6 to 12 months prior to the time at which fertility is desired. Once pregnancy is achieved, we advise continuing therapy until at least the second trimester. If the couple plans to have another child in the near future, then hCG monotherapy should be continued. However, if a long interval is expected to elapse before the next pregnancy, it may be more convenient for the patient to resume testosterone therapy. Patients should also be offered the option of storing sperm for subsequent use in intrauterine insemination or intracytoplasmic sperm injection. In patients in whom the combination of hCG and FSH is required to induce spermatogenesis initially, treatment with hCG alone may be sufficient for subsequent pregnancies due to larger testicular size.
In patients with panhypopituitarism who fail to respond to gonadotropin therapy, the addition of recombinant growth hormone (rGH) therapy should be considered. It is thought that a direct effect of growth hormone on Leydig cells may play a role in the delayed puberty encountered commonly in patients with isolated growth hormone deficiency (154, 155). While small non-randomized studies suggest that rGH may enhance the testosterone response to hCG administration (154), larger, randomized studies are needed before a definitive decision about the benefit of rGH in inducing spermatogenesis in men with hypopituitarism can be reached.
2. Pulsatile GnRH Therapy
The alternative to gonadotropin therapy is pulsatile administration of GnRH, which may be administered by a programmable, portable mini-infusion pump (Fig. 6) (156). While intravenous administration produces the most physiologic GnRH pulse contour and ensuing LH response (157), the subcutaneous route is clearly more practical for the long term treatment required to stimulate spermatogenesis. Based on our normative data (7), the frequency of GnRH administration that we employ is every 2 hours. The dose of GnRH is titrated for each individual to ensure normalization of testosterone, LH and FSH and varies from 25 to 600 ng/kg per bolus. Patients on longterm therapy are monitored with serum testosterone and gonadotropin levels at monthly intervals. Once testicular volume reaches 6-8 mL, regular semen analyses are obtained. The majority of patients require treatment for 18-24 months to maximize testicular growth and achieve spermatogenesis, although the time taken to reach these endpoints tends to be shorter in those with a larger initial gonadal size. In our analysis of 76 men with CHH, pulsatile GnRH restored normal testosterone levels in 93% of cases and was successful in inducing spermatogenesis in 77% by 12 months and 82% by 24 months (158). As with gonadotropin therapy, testicular size is an important predictor of successful treatment while negative predictors include a history of cryptorchidism and a pre-treatment inhibin B level <60 pg/mL (158).
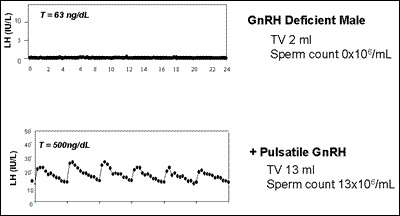
Fig. 6
LH and testosterone (T) at baseline and in response to GnRH therapy in a man with complete GnRH deficiency. Baseline LH secretion pattern is apulsatile with a hypogonadal T level. Exogenous pulsatile GnRH therapy administered every 2 h stimulates normal LH pulses which in turn induce a normal serum T level.
If pulsatile GnRH treatment fails, a mutation of GNRHR should be considered (94) and genetic testing arranged, if available. A second cause of failure of pulsatile GnRH treatment is the appearance of anti-GnRH antibodies, which typically occur in the setting of erratic compliance and are associated with a progressive decrease in T and gonadotropins levels.
Both exogenous gonadotropins and pulsatile GnRH are very effective in stimulating spermatogenesis. Most studies have no shown no advantage of either therapy in terms of testicular growth, onset of spermatogenesis, final sperm counts or pregnancy rates (151, 159).
However, pulsatile GnRH therapy is not approved for induction of spermatogenesis by the Food and Drug Administration in the United States, and its use is thus confined to specialist centers.
Assisted Reproductive Technology
With the advances that have been made in the area of assisted reproductive technology (ART), couples who would previously have been offered donor insemination or adoption are now achieving pregnancies despite persistent severe impairments in sperm number and/or quality after hormonal therapy. While male factor infertility was initially considered a contraindication to in vitro fertilization (IVF), IVF is now considered as an acceptable treatment option for the infertile male. However, for men with severe male factor infertility, IVF is often unsuccessful and more sophisticated techniques to assist fertilization such as intracytoplasmic sperm injection (ICSI) are required. The success rates of ICSI are high with fertilization rates of 50-60% and pregnancy rates of ~30% per cycle. If no sperm are present in the ejaculate, sperm retrieved either from epididymal aspiration or from testicular aspiration/biopsy can be used successfully for ICSI. (See Chapter 7)
The timeframe for referring men who are undergoing therapy to stimulate spermatogenesis for ART has changed in the last decade as both access to ICSI and success rates have improved. In the study by Liu et al of pregnancy outcomes in men treated with gonadotropins, 7/58 pregnancies (12%) resulted from IVF (152). Early referral for ICSI should certainly be considered in couples where there is concern that reduced ovarian reserve due to the age of the female partner may pose an additional barrier to natural conception. However, for other couples lack of insurance coverage may make the procedure prohibitively expensive. In addition, couples may have concerns about the ethics or safety of ART given that follow up of children conceived by ICSI suggests that there is a small increase in the risk of major malformations and chromosomal abnormalities particularly hypospadias and imprinting disorders (160-164). While the evidence is still inconclusive due to the inherent bias associated with observational studies (165), the potential for harm to the offspring underscores the importance of genetic counseling of couples contemplating assisted reproduction.
REFERENCES
1. Sullivan KA, Silverman A-J. 1993 The ontogeny of gonadotropin-releasing hormone neurons in the chick. Neuroendocrinology. 58:597-608.
2. Waldhauser F, Weissenbacher G, Frisch H, Pollak A. 1981 Pulsatile secretion of gonadotropins in early infancy. European Journal of Pediatrics. 137:71-74.
3. Apter D, Cacciatore B, Alfthan H, Stenman UH. 1989 Serum luteinizing hormone concentrations increase 100-fold in females from 7 years to adulthood, as measured by time-resolved immunofluorometric assay. J Clin Endocrinol Metab. 68:53-57.
4. Boyar RM, Rosenfeld RS, Kapen S, et al. 1974 Human puberty. Simultaneous augmented secretion of luteinizing hormone and testosterone during sleep. J Clin Invest. 54:609-18.
5. Wiemann JN, Clifton DK, Steiner RA. 1989 Pubertal changes in gonadotropin-releasing hormone and proopiomelanocortin gene expression in the brain of the male rat. Endocrinology. 124:1760-1767.
6. El Majdoubi M, Sahu A, Ramaswamy S, Plant TM. 2000 Neuropeptide Y: A hypothalamic brake restraining the onset of puberty in primates. Proc Natl Acad Sci USA. 97:6179-6184.
7. Spratt DI, O'Dea LSL, Schoenfeld D, Butler J, Rao PN, Crowley Jr WF. 1988 Neuroendocrine-gonadal axis in men: frequent sampling of LH, FSH, and testosterone. American Journal of Physiology. 254:E658-E666.
8. Brambilla DJ, O’Donnell AB, Matsumoto AM, McKinlay JB. 2007 Intraindividual variation in levels of serum testosterone and other reproductive and adrenal hormones in men. Clin Endocrinol (Oxf) 67:853-862
9. Hutson JM. 1985 A biphasic model for the hormonal control of testicular descent. Lancet. 2:419-21.
10. Berkowitz GS, Lapinski RH, Dolgin SE, Gazella JG, Bodian CA, Holzman IR. 1993 Prevalence and natural history of cryptorchidism. Pediatrics. 92:44-9.
11. Bardin CW, Ross GT, Rifkind AB, Cargille CM, Lipsett MB. 1969 Studies of the pituitary-Leydig cell axis in young men with hypogonadotropic hypogonadism and hyposmia: comparison with normal men, prepubertal boys, and hypopituitary patients. J Clin Invest. 48:2046-2056.
12. Van Dop C, Burstein S, Conte FA, Grumbach MM. 1987 Isolated gonadotropin deficiency in boys: Clinical characteristics and growth. Journal of Pediatrics. 111:684-692.
13. Boyar RM, Finkelstein JW, Witkin M, Kapen S, Weitzman E, Hellman L. 1973 Studies of endocrine function in "isolated" GnRH deficiency. Journal of Clinical Endocrinology and Metabolism. 36:64-72.
14. Lieblich JM, Rogol AD, White BJ, Rosen SW. 1982 Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann syndrome). American Journal of Medicine. 73:506-519.
15. Nachtigall LB, Boepple PA, Pralong FP, Crowley Jr WF. 1997 Adult-onset idiopathic hypogonadotropic hypogonadism-a treatable form of male infertility. New England Journal of Medicine. 336:410-415.
16. Santoro N, Filicori M, Crowley Jr WF. 1986 Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocrine Reviews. 7:11-23.
17. Spratt DI, Carr DB, Merriam GR, Scully RE, Rao PN, Crowley Jr WF. 1987 The spectrum of abnormal patterns of gonadotropin-releasing hormone secretion in men with idiopathic hypogonadotropic hypogonadism: clinical and laboratory correlations. Journal of Clinical Endocrinology and Metabolism. 64:283-291.
18. Whitcomb RW, Crowley Jr WF. 1990 Clinical review 4: diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. Journal of Clinical Endocrinology and Metabolism. 70:3-7.
19. Kallmann FJ, Schoenfeld WA. 1944 The genetic aspects of primary eunuchoidism. American Journal of Mental Deficiency. 158:203-236.
20. Schwankhaus JD, Currie J, Jaffe MJ, Rose SR, Sherins RJ. 1989 Neurologic findings in men with isolated hypogonadotropic hypogonadism. Neurology. 39:223-226.
21. Waldstreicher J, Seminara SB, Jameson JL, et al. 1996 The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. Journal of Clinical Endocrinology and Metabolism. 81:4388-4395.
22. Costa-Barbosa FA, Balasubramanian R, Keefe KW, et al. 2013 Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. Journal of Clinical Endocrinology and Metabolism. 98:E943-953.
23. Filippi G. 1986 Klinefelter's syndrome in Sardinia. Clinical report of 265 hypogonadic males detected at the time of military check-up. Clinical Genetics. 30:276-284.
24. Fromantin M, Gineste J, Didier A, Rouvier J. 1973 Les impuberismes et les hypogonadismes a l'incorporation. Probl Actuels Endocrinol Nutr. 16:179-199.
25. Dwyer AA, Hayes FJ, Plummer L, Pitteloud N, Crowley WF Jr. 2010 The long-term clinical follow-up and natural history of men with adult-onset idiopathic hypogonadotropic hypogonadism. Journal of Clinical Endocrinology and Metabolism. 95:4235-4243.
26. McCullagh EP, Beck JC, Schaffenburg CA. 1953 A syndrome of eunuchoidism with spermatogenesis, normal urinary FSH and low or normal ICSH: "Fertile Eunuchs". Journal of Clinical Endocrinology and Metabolism. 13:489-509.
27. Boyar RM, Wu RH, Kapen S, Hellman L, Weitzman ED, Finkelstein JW. 1976 Clinical and laboratory heterogeneity in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 43:1268-75.
28. Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr, Hayes FJ. 2001 The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the GnRH receptor. J Clin Endocrinol Metab.86: 2470-75.
29. Rogol AD, Mittal KK, White BJ, McGinniss MH, Lieblich JM, Rosen SW. 1980 HLA-compatible paternity in two "fertile eunuchs" with congenital hypogonadotropic hypogonadism and anosmia (the Kallmann syndrome). Journal of Clinical Endocrinology and Metabolism. 51:275-279.
30. Smals AGH, Kloppenborg PWC, van Haelst UJG, Lequin R, Benraad TJ. 1978 Fertile eunuch syndrome versus classic hypogonadotrophic hypogonadism. Acta Endocrinologica. 87:389-399.
31. Styne DM. 1991 Puberty and its disorders in boys. Endocrine and Metabolism Clinics of North America. 20:43-69.
32. Berga SL, Girton LG. 1989 The psychoneuroendocrinology of functional hypothalamic amenorrhea. Psychiatric Clinics of North America. 12:105-116.
33. Laughlin GA, Dominguez CE, Yen SSC. 1998 Nutritional and endocrine-metabolic aberrations in women with functional hypothalamic amenorrhea. Journal of Clinical Endocrinology and Metabolism. 83:25-32.
34. Warren MP. 1980 The effects of exercise on pubertal progression and reproductive function in girls. Journal of Clinical Endocrinology and Metabolism. 51:1150-1157.
35. Caronia LM, Martin C, Welt CK, et al. 2011 A genetic basis for functional hypothalamic amenorrhea. New Engl J Med. 364:215-225.
36. Klibanski A, Beitins IZ, Badger T, Little R, McArthur JW. 1981 Reproductive function during fasting in men. Journal of Clinical Endocrinology and Metabolism. 53:258-263.
37. Veldhuis JD, Iranmanesh A, Evans WS, Lizarralde G, Thorner MO, Vance ML. 1993 Amplitude suppression of the pulsatile mode of immunoradiometric luteinizing hormone release in fasting-induced hypoandrogenemia in normal men. Journal of Clinical Endocrinology and Metabolism. 76:587-593.
38. McColl EM, Wheeler GD, Gomes P, Bhambhani Y, Cumming DC. 1989 The effects of acute exercise on pulsatile LH release in high-mileage male runners. Clin Endocrinol (Oxf). 31:617-21.
39. Wheeler GD, Wall SR, Belcastro AN, Cumming DC. 1984 Reduced serum testosterone and prolactin levels in male distance runners. Journal of the American Medical Association. 252:514-6.
40. Snegovskaya V, Viru A. 1993 Steroid and pituitary hormone responses to rowing: relative significance of exercise intensity and duration and performance level. Eur J Appl Physiol Occup Physiol. 67:59-65.
41. Alen M, Reinila M, Vihko R. 1985 Response of serum hormones to androgen administration in power athletes. Med Sci Sports Exerc. 17:354-9.
42. Ruokonen A, Alen M, Bolton N, Vihko R. 1985 Response of serum testosterone and its precursor steroids, SHBG and CBG to anabolic steroid and testosterone self-administration in man. J Steroid Biochem. 23:33-8.
43. Turek PJ, Williams RH, Gilbaugh JH 3rd, Lipshultz LI. 1995 The reversibility of anabolic steroid-induced azoospermia. J Urol 153:1628-1630.
44. MacAdams MR, White RH, Chipps BE. 1986 Reduction of serum testosterone levels during chronic glucocorticoid therapy. Ann Intern Med. 104:648-51.
45. Abs R, Verhelst J, Maeyaert J, et al. 2000 Endocrine consequences of long-term intrathecal administration of opioids. J Clin Endocrinol Metab. 85:2215-22.
46. Molitch ME. 2005 Medication-induced hyperprolactinemia. Mayo Clinic Proc. 80:1050-1057.
47. Van den Berghe G, de Zegher F, Lauwers P, Veldhuis JD. 1994 Luteinizing hormone secretion and hypoandrogenaemia in critically ill men: effect of dopamine. Clin Endocrinol (Oxf). 41:563-9.
48. Wang C, Chan V, Yeung RT. 1978 Effect of surgical stress on pituitary-testicular function. Clin Endocrinol (Oxf). 9:255-66.
49. Wang C, Chan V, Tse TF, Yeung RT. 1978 Effect of acute myocardial infarction on pituitary-testicular function. Clin Endocrinol (Oxf). 9:249-53.
50. Lephart ED, Baxter CR, Parker CR, Jr. 1987 Effect of burn trauma on adrenal and testicular steroid hormone production. J Clin Endocrinol Metab. 64:842-8.
51. Iglesias P, Carrero JJ, Díez JJ. 2012 Gonadal dysfunction in men with chronic kidney disease: clinical features, prognostic implications and therapeutic options. J Nephrol. 25:31-42.
52. Van den Berghe G, de Zegher F, Bouillon R. 1998 Clinical review 95: Acute and prolonged critical illness as different neuroendocrine paradigms. J Clin Endocrinol Metab. 83:1827-34.
53. Spratt DI, Bigos ST, Beitins I, Cox P, Longcope C, Orav J. 1992 Both hyper- and hypogonadotropic hypogonadism occur transiently in acute illness: bio- and immunoactive gonadotropins. J Clin Endocrinol Metab. 75:1562-70.
54. Bates AS, Van't Hoff W, Jones PJ, Clayton RN. 1996 The effect of hypopituitarism on life expectancy. J Clin Endocrinol Metab. 81:1169-72.
55. Arafah BM. 1986 Reversible hypopituitarism in patients with large nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 62:1173-9.
56. Selmanoff M, Shu C, Petersen SL, Barraclough CA, Zoeller RT. 1991 Single cell levels of hypothalamic messenger ribonucleic acid encoding luteinizing hormone-releasing hormone in intact, castrated, and hyperprolactinemic male rats. Endocrinology. 128:459-66.
57. Carter JN, Tyson JE, Tolis G, Van Vliet S, Faiman C, Friesen HG. 1978 Prolactin-secreting tumors and hypogonadism in 22 men. N Engl J Med. 299:847-52.
58. Charbonnel B, Chupin M, Le Grand A, Guillon J. 1981 Pituitary function in idiopathic haemochromatosis: hormonal study in 36 male patients. Acta Endocrinol (Copenh). 98:178-83.
59. Thodou E, Asa SL, Kontogeorgos G, Kovacs K, Horvath E, Ezzat S. 1995 Clinical case seminar: lymphocytic hypophysitis: clinicopathological findings. J Clin Endocrinol Metab. 80:2302-11.
60. Appelman-Dijkstra NM, Kokshoorn NE, Dekkers OM, Neelis KJ, Biermasz NR, Romijn JA, Smit JW, Pereira AM. 2011 Pituitary dysfunction in adult patients after cranial radiotherapy: systematic review and meta-analysis. J Clin Endocrinol Metab. 96: 2330-40.
61. Randeva HS, Schoebel J, Byrne J, Esiri M, Adams CB, Wass JA. 1999 Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol (Oxf). 51:181-8.
62. Arimura A, Kastin AJ, Gonzales-Barcena D, Siller J, Weaver RE, Schally AV. 1974 Disappearance of LH-releasing hormone in man as determined by radioimmunoassay. 3:421-425.
63. Pimstone B, Epstein S, Hamilton SM, LeRoith D, Hendricks S. 1977 Metabolic clearance and plasma half-disappearance time of exogenous gonadotropin-releasing hormone in normal subjects and in patients with liver disease and chronic renal failure. Journal of Clinical Endocrinology and Metabolism. 44:356-360.
64. Clarke IJ, Cummins JT. 1982 The temporal relationship between gonadotropin-releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 111:1737-1739.
65. Levine JN, Pau KYF, Ramirez VD, Jackson GL. 1982 Simultaneous measurement of luteinizing hormone releasing hormone and luteinizing hormone release in unanesthetized, ovariectomized sheep. Endocrinology. 111:1449-1455.
66. Whitcomb RW, O'Dea LSL, Finkelstein JS, Heavern DM, Crowley Jr WF. 1990 Utility of free alpha-subunit as an alternative neuroendocrine marker of gonadotropin-releasing hormone (GnRH) stimulation of the gonadotroph in the human: evidence from normal and GnRH-deficient men. Journal of Clinical Endocrinology and Metabolism. 70:1654-1661.
67. Hall JE, Whitcomb RW, Rivier JE, Vale WW, Crowley Jr WF. 1990 Differential regulation of luteinizing hormone, follicle-stimulating hormone, and free à-subunit from the gonadotrope by gonadotropin-releasing hormone (GnRH): Evidence from the use of two GnRH antagonists. Journal of Clinical Endocrinology and Metabolism. 70:328-335.
68. Hayes FJ, McNicholl DJ, Schoenfeld D, Marsh EE, Hall JE. 1999 Free alpha-subunit is superior to luteinizing hormone as a marker of gonadotropin-releasing hormone despite desensitization at fast pulse frequencies. Journal of Clinical Endocrinology and Metabolism. 1999:1028-1036.
69. Beitins IZ, Dufau ML, O'Loughlin K, Catt KJ, McArthur JW. 1977 Analysis of biological and immunological activities in the two pools of LH released during constant infusion of luteinizing hormone-releasing hormone (LHRH) in men. Journal of Clinical Endocrinology and Metabolism. 45:605-608.
70. Balasubramanian R, Dwyer A, Seminara SB et al. 2010 Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology. 92:81-99.
71. Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, et al. 2012 Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. 97:E136-44
72. Dode C, Teixera L, Fouveaut C, et al. 2006 Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. October; 2(10): e175.
73. Pitteloud N, Quinton R, Pearse S, et al 2007 Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 117:457-463.
74. Sykiotis G, Plummer L, Hughes VA, et al. 2010 Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 107:15140-15144.
75. Bick D, Franco B, Sherins RJ, et al. 1992 Brief report: intragenic deletion of the KALIG-1 gene in Kallmann's syndrome. New England Journal of Medicine. 326:1752-1755.
76. Edelman GM, Crossin KL. 1991 Cell adhesion molecules: implications for a molecular histology. 60:155-190.
77. Schwanzel-Fukuda M, Pfaff D. 1989 Origin of luteinizing hormone-releasing hormone neurons. Nature. 338:161-164.
78. Pitteloud N, Hayes FJ, Boepple PA, DeCruz S, Seminara SB, MacLaughlin DT, Crowley WF Jr. 2002. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 87:152-160
79. Salenave S, Chanson P, Bry H, et al. Kallmann's syndrome: a comparison of the reproductive phenotypes in men carrying KAL1 and FGFR1/KAL2 mutations. J Clin Endocrinol Metab. 93:758-763.
80. Pitteloud N, Hayes FJ, Dwyer A, et al 2002. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 87:4128-4136.
81. Ballabio A, Bardoni B, Carrozzo R, et al. 1989 Continguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proceedings of the National Academy of Sciences of the United States of America. 86:10001-10005.
82. Cohen-Salmon M, Tronche F, del Castillo I, Petit C. 1995 Characterization of the promoter of the human KAL gene, responsible for the X-chromosome-linked Kallmann syndrome. Gene. 164:235-242.
83. Hardelin JP, Levilliers J, Blanchard S, et al. 1993 Heterogeneity in the mutations responsible for X chromosome- linked Kallmann syndrome. Human Molecular Genetics. 2:373-377.
84. Georgopoulos NA, Pralong FP, Seidman CE, Seidman JG, Crowley Jr WF, Vallejo M. 1997 Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. Journal of Clinical Endocrinology and Metabolism. 82:213-217.
85. Parenti G, Rizzolo MG, Ghezzi M, et al. 1995 Variable penetrance of hypogonadism in a sibship with Kallmann syndrome due to a deletion of the KAL gene. American Journal of Medical Genetics. 57:476-478.
86. Quinton R, Duke VM, de Zoysa PA, et al. 1996 The neuroradiology of Kallmann's syndrome: A genotypic and phenotypic analysis. Journal of Clinical Endocrinology and Metabolism. 81:3010-3017.
87. Bouligand J, Ghervan C, Tello JA, et al. 2009 Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 360:2742-2748.
88. Chang Y-M, de Guillebon A, Lang-Muritano M, et al. 2009 GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 106: 11703–11708.
89. Chevrier L, Guimiot F, de Roux N. 2011 GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol. 346:21-28.
90. de Roux N, Young J, Misrahi M, et al. 1997 A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. New England Journal of Medicine. 337:1597-1602.
91. de Roux N, Young J, Brailly-Tabard S, Misrahi M, Milgrom E, Schaison G. 1999 The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. Journal of Clinical Endocrinology and Metabolism. 84:567-572.
92. Pralong FP, Gomez F, Castillo E, et al. 1999 Complete hypogonadotropic hypogonadism associated with a novel inactivating mutation of the gonadotropin-releasing hormone receptor. Journal of Clinical Endocrinology and Metabolism. 84:3811-3816.
93. Layman LC, Cohen DP, Jin M, et al. 1998 Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nature Genetics. 18:14-15.
94. Caron P, Chauvin S, Christin-Maitre S, et al. 1999 Resistance of hypogonadic patients with mutated GnRH receptor genes to pulsatile GnRH administration. Journal of Clinical Endocrinology and Metabolism. 84:990-996.
95. Seminara SB, Beranova M, Oliveira LMB, et al. 2000 Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 85:556-562.
96. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. 2003. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 100(19):10972-10976.
97. Seminara SB, Messager S, Chatzidaki EE, JF, et al. 2003. The GPR54 gene as a regulator of puberty. N Engl J Med. 349:1614-1627.
98. Chan YM, Broder-Fingert S, Paraschos S, et al. 2011 GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J Clin Endocrinol Metab. 96:E1771-1781
99. Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pêcheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. 2003. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 33(4):463-465.
100. Pitteloud N, Acierno JS Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF Jr. 2006. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 103(16):6281-6286.
101. Raivio T, Sidis Y, Plummer L, Chen H, Ma J, Mukherjee A, Jacobson-Dickman E, Quinton R, Van Vliet G, Lavoie H, Hughes VA, Dwyer A, Hayes FJ, Xu S, Sparks S, Kaiser UB, Mohammadi M, Pitteloud N. 2009 Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 94:4380-4390.
102. Pitteloud N, Meysing A, Quinton R, Acierno JS Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, Bouloux P, Mohammadi M, Crowley WF Jr. 2006 Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 254-255:60-69.
103. Miraoui H, Dwyer A, Pitteloud N. 2011. Role of fibroblast growth factor (FGF) signaling in the neuroendocrine control of human reproduction. Mol Cell Endocrinol. 346:37-43.
104. Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. 2008. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and in mice. J Clin Invest. 118(8):2822-2831.
105. Trarbach EB, Abreu AP, Silveira LF, Garmes HM, Baptista MT, Teles MG, Costa EM, Mohammadi M, Pitteloud N, Mendonca BB, Latronico AC. 2010. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab. 95:3491-6.
106. Miraoui H, Dwyer AA, Sykiotis GP, et al. 2013 Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 92(5):725-743.
107. Dode C, Teixeira L, Levilliers J, et al. 2006 Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2:1648–1652
108. Pitteloud N, Zhang C, Pignatelli D, Li JD, Raivio T, Cole LW, Plummer L, Jacobson-Dickman EE, Mellon PL, Zhou QY, Crowley Jr WF 2007 Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 104:17447–17452
109. Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, Garmes HM, Mendonca BB, Latronico AC 2008 Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab 93:4113–4118
110. Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Raivio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley Jr WF, Zhou QY, Pitteloud N 2008 Mutations in prokineticin 2 (PROK2) and PROK2 receptor 2 (PROKR2) in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab 93:3551–3559
111. Sarfati J, Guiochon-Mantel A, Rondard P, et al. 2010. A comparative phenotypic study of Kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab. 95:659-69.
112. Topaloglu AK, Reimann F, Guclu M, et al. 2009. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat Genet. 41(3):354-358.
113. Gianetti E, Tusset C, Noel SD, et al. 2010. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 95(6):2857-2867.
114. Francou B, Bouligand J, Voican A, et al. 2011. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One. 6(10):e25614.
115. Salvi R, Pralong FP. Molecular characterizyation and phenotypic expression of mutations in genes for gonadotropins and their receptors in humans. Front Horm Res. 2010;39:1-12.
116. Weiss J, Axelrod L, Whitcomb RW, Harris PE, Crowley Jr WF, Jameson JL. 1992 Hypogonadism caused by a single amino acid substitution in the beta subunit of luteinizing hormone. N Engl J Med. 326:179-183.
117. Lindstedt G, Nystrom E, Matthews C, Ernest I, Janson PO, Chatterjee K. 1998 Follitropin (FSH) deficiency in an infertile male due to FSH beta gene mutation. A syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin Chem Lab Med. 36:663-5.
118. Phillip M, Arbelle JE, Segev Y, Parvari R. 1998 Male hypogonadism due to a mutation in the gene for the beta subunit of follicle stimulating hormone. New Engl J Med. 338:1729-1732.
119. Muscatelli F, Strom TM, Walker AP, et al. 1994. Mutations in DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotrophic hypogonadism. Nature. 372:672-676.
120. Jadhav U, Harris RM, Jameson JL. 2011. Hypogonadotropic hypogonadism in subjects with DAX1 mutations. Mol Cell Endocrinol. 346:65-73.
121. Kim HG, Kurth I, Lan F, Meliciani I, et al. 2008. Mutations in CHD7, encoding a chromatin remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 83:511-519.
122. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al. 2008. CHD7 mutations in patients initially diagnosed with Kallmann syndrome: the clinical overlap with CHARGE syndrome. Clin Genet. 75(1):65-71.
123. Montague CT, Farooqi IS, Whitehead JP, et al. 1997 Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 387:903-908.
124. Farooqi IS, Wangensteen T, Collins S, et al. 2007. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 356(3):237-47.
125. Farooqi IS, Volders K, Stanhope R, et al. 2007 Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 92(9):3369-73.
126. Parks JS, Brown MR, Hurley DL, Phelps CJ, Wajnrajch MP. 1999 Heritable disorders of pituitary development. J Clin Endocrinol Metab. 84:4362-4370.
127. Fluck C, Deladoey J, Rutishauser K, et al. 1998 Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting an the substitution of ArgàCys at Codon120 (R120C). J Clin Endocrinol Metab 83 :3727-3734.
128. Deladoey J, Fluck C, Buyukgebiz A, et al. 1999 ‘Hot spot’ in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 84:1645-50
129. McCabe MJ, Gaston-Massuet C, Gregory LC, et al. 2013 Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab. 98(3):E547-57.
130. Raivio T, Avbelj M, McCabe MJ, et al. 2012 Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 97:E694-9.
131. Sato N, Kamachi Y, Kondoh H, Shima Y, Morohashi K, Horikawa R, Ogata T. 2007. Hypogonadotropic hypogonadism in an adult female with a heterozygous hypomorphic mutation of SOX2. Eur J Endocrinol 156:167-171
132. Stark Z, Storen R, Bennetts B, Savarirayan R, Jamieson RV. 2011. Isolated hypogonadotropic hypogonadism with SOX2 mutation and anopthalmia/microphtalmia in offspring. Eur J Hum Genet. 19(7):753-6.
133. Pingault V, Bodereau V, Baral V, et al. 2013 Loss-of-Function mutations in SOX10 cause Kallmann Syndrome with deafness. Am J Hum Genet. 92(5):707-24.
134. World Health Organization Laboratory Manual for the evaluation and processing of human semen. 5th Edition (2010) CambridgeUniversity Press, Cambridge.
135. Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, Dwyer AA, Buck CL, Choi JH, Seminara SB, Quinton R, Monies D, Meyer B, Hall JE, Pitteloud N, Crowley WF Jr. 2013 Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. 98(5):E943-953.
136. Canaves F, Mussa A, Manenti M, et al. 2009 Sperm count of young men surgically treated for cryptorchidism in the first and second year of life: fertility is better in children treated at a younger age. Eur J Paedr Surg. 19 :388-391.
137. Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF Jr, Pitteloud N. 2007 Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 357(9):863-873.
138. Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. 2012 Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One. 2012;7(6):e39450.
139. Tapanainen JS, Aittomaki K, Min J, Vaskivuo T, Huhtaniemi IT. 1997 Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nature (London). 15:205-206.
140. Burris AS, Rodbard HW, Winters SJ, Sherins RJ. 1988 Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 66:1144-51.
141. Behre HM, Baus S, Kliesch S, Keck C, Simoni M, Nieschlag E. 1995 Potential of testosterone buciclate for male contraception: endocrine differences between responders and nonresponders. J Clin Endocrinol Metab. 80:2394-2403.
142. O'Donnell L, Narula A, Balourdos G, et al. 2001 Impairment of spermatogonial development and spermiation after testosterone-induced gonadotropin suppression in adult monkeys (Macaca fascicularis). J Clin Endocrinol Metab. 86:1814-1822.
143. Gromoll J, Simoni M, Nieschlag E. 1996 An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab. 81:1367-1370.
144. Raivio T, Wikström AM, Dunkel L. 2007. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol. 156(1):105-111.
145. Dwyer AA, Sykiotis GP, Hayes FJ, Poepple PA, Lee H, Loughlin KR, Dym M, Sluss PM, Crowley WF JR, Pitteloud N. 2013 Trial of recombinant follicle stimulating hormone (rFSH) pre-treatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 98:E1790-1795.
146. Schaison G, Young J, Pholsena M, Nahoul K, Couzinet B. 1993 Failure of combined follicle-stimulating hormone-testosterone administration to initiate and/or maintain spermatogenesis in men with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 77:1545-1549.
147. Recombinant Human FSH Product Development Group 1998 Recombinant follicle stimulating hormone: development of the first biotechnology product for the treatment of infertility. Hum Reprod Update. 4:862-81.
148. Mannaerts B, Fauser B, Lahlou N, et al. 1996 Serum hormone concentrations during treatment with multiple rising doses of recombinant follicle stimulating hormone (Puregon) in men with hypogonadotropic hypogonadism. Fertil Steril. 65:406-410.
149. King TFJ, Hayes FJ. 2012 Long-term outcome of idiopathic hypogonadotropic hypogonadism. Curr Opin Endocrinol Diabetes Obes. 19:204-210.
150. Vicari E, Mongioì A, Calogero AE, Moncada ML, Sidoti G, Polosa P, D'Agata R. 1992. Therapy with human chorionic gonadotrophin alone induces spermatogenesis in men with isolated hypogonadotrophic hypogonadism--long-term follow-up. Int J Androl. 15:320-329.
151. Büchter D, Behre HM, Kliesch S, Nieschlag E. 1998. Pulsatile GnRH or human chorionic gonadotropin/human menopausal gonadotropin as effective treatment for men with hypogonadotropic hypogonadism: a review of 42 cases. Eur J Endocrinol. 139:298-303.
152. Liu PY, Baker HW, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. 2009 Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 94:801-808.
153. Warne DW, Decosterd G, Okada H, Yano Y, Koide N, Howles CM. 2009 A combined analysis of data to identify predictive factors for spermatogenesis in men with hypogonadotropic hypogonadism treated with recombinant human follicle-stimulating hormone and human chorionic gonadotropin. Fertil Steril. 92:594-604.
154. Kulin HE, Samojlik E, Santen R, Santner S. 1981 The effect of growth hormone on the Leydig cell response to chorionic gonadotrophin in boys with hypopituitarism. Clin Endocrinol (Oxf). 15:463-472.
155. Chatelain PG, Sanchez P, Saez JM. 1991 Growth hormone and insulin-like growth factor I treatment increase testicular luteinizing hormone receptors and steroidogenic responsiveness of growth hormone deficient dwarf mice. Endocrinology. 128:1857-62.
156. Hoffman AR, Crowley WF, Jr. 1982. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 307: 1237-1241.
157. Spratt DI, Crowley Jr WF, Butler JP, Hoffman AR, Conn PM, Badger TM. 1985 Pituitary luteinizing hormone responses to intravenous and subcutaneous administration of gonadotropin releasing hormone in men. Journal of Clinical Endocrinology and Metabolism. 61:890-895.
158. Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. 2002. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 87(9):4128-4136.
159. Liu L, Banks SM, Barnes KM, Sherins RJ. 1988 Two-year comparison of testicular responses to pulsatile gonadotropin-releasing hormone and exogenous gonadotropins from the inception of therapy in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 67:1140-1145.
160. Bonduelle M, Wennerholm UB, Loft A, Tarlatzis BC, Peters C, Henriet S, Mau C, Victorin-Cederquist A, Van Steirteghem A, Balaska A, Emberson JR, Sutcliffe AG. A multi-centre cohort study of the physical health of 5-year-old children conceived after intracytoplasmic sperm injection, in vitro fertilization and natural conception. Hum Reprod. 2005;20(2):413.
161. Belva F, Henriet S, Liebaers I, Van Steirteghem A, Celestin-Westreich S, Bonduelle M. Medical outcome of 8-year-old singleton ICSI children (born>or=32 weeks' gestation) and a spontaneously conceived comparison group. Hum Reprod. 2007;22(2):506.
162. Whitelaw N, Bhattacharya S, Hoad G, Horgan GW, Hamilton M, Haggarty P. Epigenetic status in the offspring of spontaneous and assisted conception.Hum Reprod. 2014 Jul;29(7):1452-8
163. Hansen M, Bower C, Milne E, de Klerk N, Kurinczuk JJ. Assisted reproductive technologies and the risk of birth defects—a systematic review. Hum Reprod 2005;20:328-338.
164. Reefhuis J, Honein MA, Schieve LA, Correa A, Hobbs CA, Rasmussen SA, National Birth Defects Prevention Study. 2009. Assisted reproductive technology and major structural birth defects in the United States. Hum Reprod. 24(2):360-366.
165. Bukulmez O. 2009 Does assisted reproductive technology cause birth defects? Curr Opin Obstet Gynecol. 2009 Jun;21(3):260-264.
- TAKE HOME POINTS
- DEFINITIONS
- CLINICAL PRESENTATION
- DIFFERENTIAL DIAGNOSIS
- Congenital Hypogonadotropic Hypogonadism
- Acquired Hypogonadotropic Hypogonadism Functional
- Drug-Induced GnRH Deficiency
- Critical Illness
- Structural
- PATHOPHYSIOLOGY
- Genetic Defects of the Gonadotropins
- Other rare genetic syndromes associated with hypogonadotropic hypogonadism
- EVALUATION
- TREATMENT
- REFERENCES
- Hypogonadotropic Hypogonadism (HH) and Gonadotropin Therapy - EndotextHypogonadotropic Hypogonadism (HH) and Gonadotropin Therapy - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...