

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Severe obesity represents a major risk factor for the development of type 2 diabetes mellitus (T2DM). Due to the strong association of obesity and diabetes, the term “diabesity” was coined, suggesting a causal pathophysiological link between both phenomena. The majority of individuals with T2DM are obese, highlighting the pivotal role of increased adiposity as a risk factor for diabetes. However, only a relatively small fraction of obese individuals will develop T2DM. On a population level, the link between obesity and its secondary complications is well described. However, the molecular mechanisms underlying these complications are still poorly understood. Three main hypotheses have been developed in recent years to bridge the gap between epidemiology and pathobiochemistry: (1) The “inflammation hypothesis” asserts that obesity represents a state of chronic inflammation where inflammatory molecules produced by infiltrating macrophages in adipose tissue exert pathological changes in insulin-sensitive tissues and β-cells. (2) The “lipid overflow hypothesis” predicts that obesity may result in increased ectopic lipid stores due to the limited capacity of adipose tissue to properly store fat in obese subjects. Potentially harmful lipid components and metabolites may exert cytotoxic effects on peripheral cells. (3) The “adipokine hypothesis” refers to the principal feature of white adipose cells to function as an endocrine organ, and to secrete a variety of hormones with auto- and paracrine function. Expanding fat stores can cause dysfunctional secretion of such endocrine factors, thereby resulting in metabolic impairment of insulin target tissues and eventually failure of insulin producing β-cells. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Severe obesity represents a major risk factor for the development of type 2 diabetes mellitus (T2DM), a disease characterized by insulin resistance, insulin hyposecretion and hyperglycaemia.1-3 According to statistics, already 415 million people worldwide were affected by diabetes in 2015 (estimated) and this number is expected to rise to 642 million by 2040.4 Due to the strong association of obesity and diabetes, the term “diabesity” was coined, suggesting a causal pathophysiological link between both phenomena.5,6
SIZE MATTERS (NOT ONLY)!
The majority (~80%) of individuals with T2DM are obese, highlighting the pivotal role of increased adiposity as a risk factor for diabetes. However, only a relatively small fraction of obese individuals develops T2DM.7 In fact, most obese, insulin-resistant individuals do not develop hyperglycemia, indicating that their pancreatic β-cells still produce and secrete sufficient amounts of insulin in order to compensate for the reduced efficiency of insulin action in the periphery.1,8,9 Thus, in addition to an increased adipose mass, additional factors are likely to determine the risk for β-cell dysfunction and the susceptibility for β-cell destruction and diabetes. Nevertheless, despite recent advances in the understanding of body weight regulation and insulin action, the risk factors that determine which obese, non-diabetic individuals will eventually develop diabetes still remain unknown.
ROLE OF FAT DISTRIBUTION
Obesity results from a period of a positive energy balance during which adipocytes store excess triglycerides, resulting in cell hypertrophy and hyperplasia. However, fat depots do not expand uniformly as they accumulate lipids, and the adverse effects of excess fat storage have been frequently attributed to intra-abdominal (i.e. visceral) fat tissue. Using a variety of measures (oral glucose tolerance test, intravenous glucose tolerance test, euglycemic hyperinsulinemic clamps), selective excess of visceral adipose tissue (visceral adiposity) has been linked to insulin resistance in humans.10-19 Interestingly, no relationship between visceral fat and impaired glucose metabolism has been observed in studies with non-obese individuals.20,21 On the other hand, other studies found that abdominal subcutaneous fat correlates with insulin sensitivity as well as visceral fat in euglycemic clamps, thus challenging a unique role for the visceral fat depot in modulating insulin sensitivity.22,23 However, in another study, Klein and coworkers reported that large-volume abdominal liposuction of subcutaneous fat did not improve insulin sensitivity of liver, skeletal muscle, and adipose tissue (as assessed by euglycemic-hyperinsulinemic clamps), at least not within 12 weeks post surgery.24 In accordance to these results, removal of visceral fat has been found to improve insulin sensitivity in humans,25,26 supportive of a causal role of intra-abdominal fat for the insulin resistance in obese individuals. However, the relationship between the amount of visceral fat and insulin sensitivity has been controversially discussed throughout recent years and a number of clinical studies show that surgical removal of this fat depot (e.g. via omentectomy) did not lead to improved whole-body glycemia or even BMI of the patients.27-30 Interestingly, a number of adipose-tissue related sub-phenotypes have been identified, one of these the so-called “TOFI” (“thin on the outside, fat on the inside”) subjects. These patients present a normal BMI (< 25 kg/m2) but increased abdominal obesity and therefore exhibit an increased risk to develop insulin resistance and T2DM.31,32 In contrast to TOFI, the so-called “fat-fit” subjects show no gross impairments of glucose metabolism despite their elevated body adiposity (BMI ≥ 30 kg/m2).32,33
Visceral fat is defined as adipose tissue located inside the peritoneal cavity—within the parietal peritoneum and transversalis fascia, excluding the spine and paraspinal muscles. As such, appropriate techniques for precise measurements of visceral fat, however, have been controversial. In humans, the amount of abdominal visceral fat is determined by a number of different techniques, including anthropomorphic measurements (waist-hip-ratio, waist circumference, abdominal sagittal diameter), computed tomography (CT), magnetic resonance imaging (MRI), and ultrasound. Comparative measurements using CT and MRI have revealed a fairly high correlation between both methods.34-36 In the recent years, Dual-energy X-ray absorptiometry (DXA) has emerged as reliable technique to measure body composition with high-precision, low X-ray exposure, and short-scanning time.37,38 In studies that included these imaging techniques and common clinical measurements, the abdominal sagittal diameter was found to be the most specific predictor of visceral adipose volume, but measurements of waist circumference and sagittal diameter are also highly correlated. Even though waist circumference varies considerably between sexes and among different ethnic groups, it has been proposed as a crude, but efficient, anthropomorphic readout for abdominal adiposity.39,40 And lastly, the amount of visceral fat is correlated to total body fat even though considerable variation in individual fat distribution has been reported,41-44 Thus, despite the lack of a ”gold standard” for the quantitative assessment of regional fat distribution in humans, most of the evidence suggests a specific association between visceral fat with an increased risk for insulin resistance and diabetes.
TO BE (FAT) OR NOT TO BE
Interestingly, deficiency of fat tissue (lipodystrophy) predisposes to similar metabolic complications as an excess of fat in obesity, such as insulin resistance, T2DM, and hepatic steatosis (reviewed in Refs 45 and 4645,46). Moreover, fat transplantation into lipodystrophic mice ameliorated the diabetic phenotype of the animals either partially or completely, implicating that the failure to properly store lipids in depots is causal for lipodystrophic diabetes.47 Also in normal lean animals, fat transplantation has been shown to result in beneficial metabolic effects.48,49 Thus, adipose tissue may have both beneficial and adverse effects on whole-body metabolism, depending on where it accumulates.
OBESITY-INDUCED DIABETES: FACTORS AND MECHANISMS
On a population level, the link between obesity and its secondary complications is well described. However, the molecular mechanisms underlying these complications are still poorly understood.50 Even though the evidence indicates a detrimental role of visceral fat in terms of insulin sensitivity, relatively little is known about distinct physiological and biochemical properties of fat tissue derived from different anatomic locations. On the one hand, transplantation experiments with fat tissue from different adipose depots in mice have not conclusively revealed intrinsic differences between subcutaneous and visceral fat.47,49 On the other hand, factors that have been attributed to confer differential metabolic effects of subcutaneous versus visceral fat include increased portal release of FFA and glycerol from omental/mesenteric fat directly to the liver, and also differences in endocrine and metabolic functions of fat depots.
Three main hypotheses have been developed in recent years to bridge the gap between epidemiology and pathobiochemistry:
(1) The “inflammation hypothesis” asserts that obesity represents a state of chronic inflammation where inflammatory molecules produced by infiltrating macrophages in adipose tissue exert pathological changes in insulin-sensitive tissues and β-cells.
(2) The “lipid overflow hypothesis”, also known as “Adipose Tissue Expandability Hypothesis” predicts that obesity may result in increased ‘ectopic’ lipid stores (lipid that accumulates outside the normal depots, such as in the organ tissue of the liver, muscle, and pancreas) due to the limited capacity of adipose tissue to properly store fat in obese subjects. Potentially harmful lipid components and metabolites may exert cytotoxic effects on peripheral cells, including liver and β-cells, thereby impairing function, survival, and regeneration.
(3) The “adipokine hypothesis” refers to the principal feature of white adipose cells to function as an endocrine organ, and to secrete a variety of hormones with auto- and paracrine function. It has been proposed that expanding fat stores in obesity cause dysfunctional secretion of such endocrine factors, thereby resulting in metabolic impairment of insulin target tissues and eventually failure of insulin producing β-cells.
In the following, these three hypotheses are briefly discussed.
The “Inflammation Hypothesis”
Inflammatory processes are thought to play a key role in the development of obesity-related insulin resistance and type 2-diabetes. In adiposity there are fundamental changes in adipose tissue secretory functions51. An excess of adipose tissue produces a number of pro-inflammatory cytokines leading to a state of chronic subclinical inflammation associated with both insulin resistance and type-2 diabetes.52 The term “metaflammation” describes this low-grade, chronic inflammation orchestrated by metabolic cells in response to excess nutrients and energy.53
How does a spill-over of these inflammatory products into circulation finally lead to insulin resistance? Weisberg et al54 described that macrophages accumulate in adipose tissue of obese subjects and suggested that these macrophages are derived from the circulation. Further studies indicated that adipose tissue macrophages (ATMs) that accumulate during diet-induced obesity (DIO) are not only an important source of adipose tissue inflammation but also mediate insulin resistance in adipocytes.55 The amount of macrophages in adipose tissue correlates positively with two indices of adiposity: Body mass index (BMI) and adipocyte size. The exact mechanisms underlying ATM recruitment and activation are still not fully understood. One factor potentially responsible for obesity-induced inflammation by increasing ATM recruitment is Macrophage Migration Inhibitory Factor (MIF), a chemokine-like inflammatory regulator directly associated with the degree of peripheral insulin resistance.56 Adipose tissue macrophages are considered to be a major reservoir of pro-inflammatory molecules in adipose tissue54. These cytokines exert various functions in the pathogenesis of the disease progression (Figure 1). Some of the most important inflammatory factors are described below.

Figure 1. The “inflammation hypothesis.” Pathophysiology of obesity-induced chronic inflammation and peripheral insulin resistance.
DAG, diacylglycerol; IL-1, interleukine-1; MCP-1, monocyte chemotactic protein-1; TNF, tumor necrosis factor alpha; Toll-like receptor 4, TLR-4; Details in the text.
Tumor Necrosis Factor Alpha:
Tumor necrosis factor alpha (TNF-α) is a pluripotent cytokine primarily produced from macrophages.57 Its expression was shown to be elevated in different mouse and rat models of obesity and diabetes.58 In vitro, TNF-α suppresses the expression of most adipose-specific genes in murine adipocytes, including the enzymes involved in lipogenesis.59 It was also shown that TNF-α induces insulin resistance, in part through its ability to inhibit intracellular signaling from the insulin receptor.60 Moreover, addition of TNF-α to cells in vivo increased the intracellular concentration of ceramides.61 Ceramides can directly induce DNA fragmentation and apoptosis. In skeletal muscle, diacylglycerols and ceramides operate as lipotoxic mediators engaging serine kinases that disrupt the insulin signaling cascade and diminish insulin sensitivity.62 Further, it was discovered that ceramides are able to induce lipoapoptosis in β-cells.63 In addition, TNF-α was shown to induce the formation of reactive oxygen species (ROS).64 Production of ROS increased selectively in adipose tissue of obese mice, causing dysregulated production of adipocytokines (fat-derived hormones), including adiponectin, plasminogen activator inhibitor-1, interleukin-6 (IL-6), and monocyte chemotactic protein-1.65 However, clinical studies with Etanercept, a neutralizing protein for circulating TNF-α failed to demonstrate an improvement of insulin sensitivity in humans,66,67 indicating that acute reduction of systemic TNF-α may not be sufficient to induce metabolic benefits in the periphery.
TNF-Like Weak Inducer of Apoptosis:
TNF-like weak inducer of apoptosis (TWEAK) belongs to the TNF superfamily and was shown to have pro-inflammatory action in adipocytes mediated by the nuclear factor-κB (NFκB) and ERK but not JNK signaling pathways.68 The cytokine promotes the secretion of MCP-1 and RANTES and up-regulates CCl21 and CCL19 expression. Whereas expression levels of membrane-bound TWEAK (mTWEAK) and its receptor Fn14 are increased during obesity, the amount of soluble TWEAK (sTWEAK) was decreased, thereby enhancing the pro-inflammatory activity elicited by TNF-α.69,70
Monocyte Chemotactic Protein-1:
The pro-inflammatory chemokine monocyte chemotactic protein-1 (MCP-1) attracts leukocytes to inflamed sites and is regulated by NFκB. 71 Monocyte chemotactic protein-1 represents the first discovered and most extensively studied human CC chemokine and is also known as CCL2 (Chemokine (C-C motif) ligand 2). CC chemokines are characterized by the conserved position of four cysteine residues responsible for protein stabilization.56 Insulin was found to induce expression and secretion of MCP-1 substantially both in vitro in insulin-resistant adipocytes and in vivo in insulin-resistant obese mice (ob/ob). It was suggested that elevated MCP-1 levels may induce adipocyte dedifferentiation and contribute to pathologic states associated with hyperinsulinemia and obesity, including type 2 diabetes.72 Expression and plasma concentration of MCP-1, however, were shown to be increased both in genetically obese diabetic (db/db) mice and in wildtype mice with high-fat diet-induced obesity, leading to the assumption that increased MCP-1 expression contributes to the macrophage infiltration of adipose tissue and, finally, to the development of insulin resistance.73 Monocyte chemotactic protein-1 has been developed into one of the most important targets for a variety of therapeutic approaches to improve diabetic vascular conditions over the years.74
Interleukin-6:
The role of the cytokine interleukin-6 (IL-6) in the regulation of lipid metabolism is controversial.75 If produced in large amounts by adipose tissue, IL-6 causes insulin resistance in adipocytes and skeletal muscle.76 Contrary to the expectations, IL-6-deficient mice develop obesity. However, excess body weight was only reported in very mature animals.77 Interestingly, chronic exposure of IL-6 produces insulin resistance in skeletal muscle, whereas short-term exposure as consequence of exercise has beneficial effects on insulin sensitivity.78 Thus, despite the evidence of IL-6 as a major player in the regulation of metabolism, the role of this cytokine in the pathogenesis of insulin resistance and diabetes remains incompletely understood.
Interleukin-1:
Interleukin-1 (IL-1) is a cytokine that is also secreted by stimulated macrophages and has many actions that overlap those of TNF-α. For instance, IL-1 increases hepatic triglyceride secretion and serum triglyceride levels.79 Common polymorphisms of the IL-1 gene that influence IL-1 activity are also associated with fat mass in humans.80 Pro-inflammatory pathways in adipose tissue have been shown to be directly activated by free-fatty acids (FFA). In turn, the inflammatory status of macrophages is linked to body fat content. In lean mice, macrophages in WAT are in their active M2 state and produce immunosuppressive factors. However, in obese mice, macrophages are in a pro-inflammatory M1 state (F4/80+, CD11b+, CD11c+), highly responsive to the pro-inflammatory effect of FFA that bind the Toll-Like Receptors (TLRs).81 Increased cytokine release via TLRs as a consequence of FFA binding was proposed as potential pathomechanism causing insulin resistance.82 Interestingly, in a clinical study, blockade of IL-1 receptor with Anakinra, a recombinant IL-1 receptor antagonist, improved HbA1c levels and proinsulin-to-insulin ratio but had no effect on systemic insulin sensitivity.83
Toll-Like Receptor-4:
Toll-like receptors are membrane-spanning, non-catalytic receptors that respond to different microbial antigens, therefore representing an important factor of the innate immunity.84 Toll-like Receptor-4 (TLR-4) is thought to be another important factor in fatty acid-induced insulin resistance. Scherer and coworkers were the first ones that found it expressed on 3T3-L1 adipocytes and activated by lipopolysaccharides (LPS).85 Characterization of TLR-4 as the main endogenous sensor for LPS in adipocytes supports the relevance of fat tissue in immune processes.86 Additionally, TLR-4 was recently shown to be directly activated by dietary saturated fatty acids, thereby promoting inflammatory aspects of the metabolic syndrome and atherosclerosis.87 In addition, stimulation of TLR-4 with activation of the Erk pathway was shown to upregulate IL-6 as well as MCP-1 release in adipose tissue. Therefore, it can be suggested that activation of TLR-4 in adipocytes induces inflammation and, as a consequence, promotes the progression towards diabetes. This mechanism provides new evidence for a coupling of visceral adipose dysfunction with the development insulin resistance and T2DM.88
Summary:
Inflammation is thought to be a major factor in the development of insulin resistance and diabetes. Increased secretion of adipocyte-derived inflammatory cytokines and fatty acids are directly linked to impaired insulin sensitivity in obesity.89 However, inflammatory processes do not account exclusively for the development of insulin resistance since there are studies showing subjects with T2D but without any alterations in inflammatory markers.90 Inflammation alone can therefore not explain how obesity affects insulin sensitivity and certainly not why only a small fraction of obese individuals develop T2DM.
The “Lipid Overflow Hypothesis”
Healthy adipose tissue is characterized by the ability to expand passively to accommodate periods of nutrient excess. In contrast, adipose tissue in polygenic mouse models of obesity-induced diabetes, as well as in obese humans, may fail to fully accommodate excessive nutrient loads.91-93 If the adipose tissue expansion limit is reached, lipids can no longer be stored appropriately in adipose tissue and consequently “overflow” to other peripheral tissues such as skeletal muscle, liver, and pancreas.94,95 Subcutaneous adipose tissue (SAT) represents the largest adipose tissue depot and, in addition, is considered the least metabolically harmful site for lipid storage. The SAT can expand either by increasing the size of the cells (hypertrophic obesity) and/or by recruiting new cells (hyperplastic obesity). In contrast to the hyperplastic response, which seems to be protective against SAT dysfunction, hypertrophic obesity is associated with increased T2D risk.96,97 The storage of this ectopic fat in non-SAT tissues is directly linked to the progression of insulin resistance and type-2-diabetes.98,99 Thus, fat accumulates in tissues that are not adequate for lipid storage, and as a consequence, lipid metabolites might accumulate within those tissues that inhibit insulin signal transduction (Figure 2).
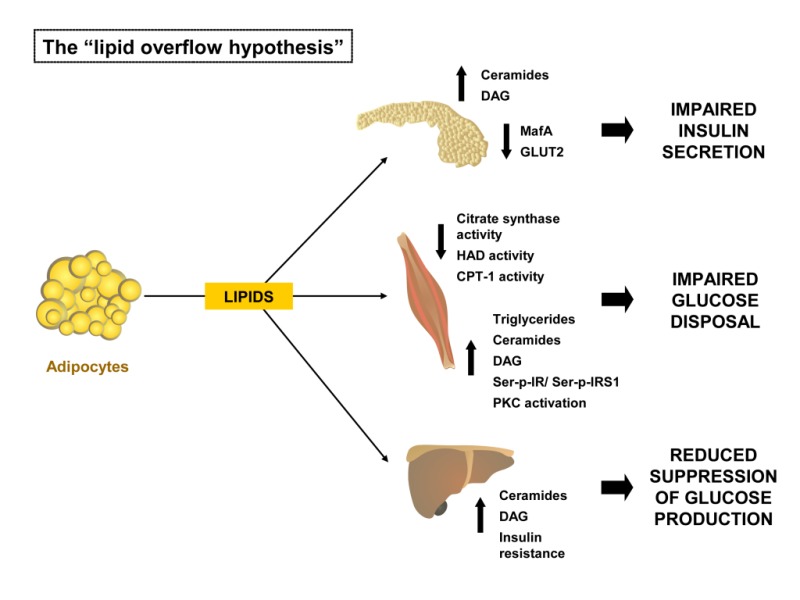
Figure 2: The “lipid overflow hypothesis.” Pathophysiology of obesity-induced ectopic lipid stores that cause peripheral insulin resistance and impaired β-cell function.
CPT-1, carnitine palmitoyltransferase 1; DAG, diacylglycerol; GLUT2, facilitated glucose transporter, member 2 (SLC2A2); HAD, β-hydroxyacyl dehydrogenase; IRS1, insulin receptor substrate 1; MafA, pancreatic beta-cell-specific transcriptional activator MafA; PKC, Protein kinase C; Details in the text.
This hypothesis is supported by several rodent models of lipodystrophy. These animals are extremely lean but often suffer from marked insulin resistance, diabetes, hypertriglyceridemia, hepatosteatosis, and low HDL (high-density lipoprotein)-cholesterol levels – a metabolic profile similar to that observed in obesity-related metabolic syndrome.100,101 Moreover, recent studies demonstrate a lipodystrophy-like phenotype also in the general human population since subjects who are of normal weight but metabolically unhealthy (∼20% of the normal weight adult population) have a greater than 3-fold higher risk of all-cause mortality and/or cardiovascular events.46 Leptin replacement in patients with generalized lipodystrophy can serve as efficient therapy to improve insulin sensitivity by reducing ectopic fat accumulation, especially in the liver.102-104
Thus, an increased fatty acid flux from normal fat depots towards non-adipose tissues (NAT), e.g. skeletal muscle, heart, liver, and pancreatic β-cells appears to be a critical factor in mediating lipotoxicity. Among the substances known to impair insulin signaling, the most prominent examples include diacylglycerols (DAGs) and ceramides, which have both been shown to impair insulin action in a number of peripheral tissues.105-107
Lipotoxicity- Skeletal Muscle and Adipocytes:
Skeletal muscle insulin resistance is associated with high levels of stored lipids in skeletal muscle cells.108 A high lipid accumulation and/or lower triglyceride turnover can induce lipotoxicity within the skeletal muscle cell.109 Lipid infusion can induce peripheral and hepatic insulin resistance in rats and humans.110,111 There are multiple regulatory sites controlling the complex process of fatty acid (FA) metabolism in skeletal muscle. Long-chain FA (LCFA) oxidation involves lipolysis and LCFA release from the adipose tissue, delivery of FFA to the skeletal muscle, transport across the plasma membrane, lipolysis of intramuscular triacylglycerol (IMTG), activation with addition of a coenzyme A thioester (LCFA-CoA), transport across the mitochondrial membranes and ultimately oxidation.112 Obese individuals display a disturbed lipid oxidation in skeletal muscle. This leads to accumulation of fatty acids and therefore to enhanced levels of triglycerides, fatty acyl CoA, diacylglycerols, and ceramides.113-115 Accumulation of these metabolites may be able to impair insulin signaling through different mechanisms, such as increased serine phosphorylation of the insulin receptor and insulin receptor substrate 1 by Protein kinase C (PKC) β and reduced serine phosphorylation of AKT.116,117 Besides disturbances in the insulin signaling cascade, several other factors could be involved in the direction of LCFA or LCA-CoA towards esterification rather than oxidation in obesity and type-2 diabetes. It has long been debated whether reduced mitochondrial function is the cause of, or secondary to, insulin resistance and T2D. Numerous studies, however, have shown that the activity of the key enzymes of fatty acid oxidation, citrate synthase (CS) and β-hydroxyacyl dehydrogenase (HAD) are significantly reduced in skeletal muscle in obesity and type 2 diabetes.118-121 Additionally, it has also been shown that the activity of carnitine palmitoyltransferase 1 (CPT1) in muscle was also reduced in association with obesity,122 and that mitochondrial oxidative capacity is low in insulin-resistant subjects.123 Carnitine palmitoyltransferase 1 converts acyl-CoA molecules to their acyl carnitine derivatives prior transport of the mitochondrial inner membrane.124
Plasma non-esterified free fatty acids (NEFAs) are suggested to contribute to the development of insulin resistance, since they have been shown to activate the inflammatory nuclear factor kappa-B (NFκB) pathway in human muscle biopsies.125,126 In humans, it was demonstrated that free fatty acids induce insulin resistance by inhibition of glucose transport.127 In addition to the negative impact on insulin sensitivity, there is very recent evidence that lipid droplet (LD) formation is also impaired by an overflow of lipids. It has been shown that LD formation requires some of the same components of the machinery involved in regulated fusion of vesicles including the two soluble N-ethylmaleimide-sensitive-factor attachment protein receptor (SNARE) proteins SNAP23 and syntaxin-5. SNAP23 has been shown to be an essential factor for trafficking of GLUT4-containing vesicles to the plasma membrane, and a more recent study found that SNAP23 is also involved in LD formation in adipocytes.128 Interestingly, the study reported that excessive LD formation inhibited GLUT4 translocation by competing for SNAP23 and that overexpression of Snap23 in these cells restored insulin sensitivity. Thus, SNAP23 might constitute a link between glucose and lipid metabolism, respectively.
Lipotoxicity- Pancreatic Beta Cell:
The development of type 2 diabetes is caused by a combination of insulin resistance and impaired pancreatic β-cell secretion.129 With progression from euglycemia to type 2 diabetes, β-cells progressively fail to compensate for the increase insulin demand in peripheral tissues. The pathogenesis is thereby characterized by different stages, leading from compensatory insulin resistance to decompensated hyperglycemia.130 In manifest type 2 diabetes, β-cells are exposed to both high doses of glucose (glucotoxicity) and lipids (lipotoxicity), respectively.131 Lipotoxicity, manifests as incorporation of large amounts of triglycerides in pancreatic islets, leading to β-cell death.132
While rodents are often preferred models to study disease progression, polygenic mouse models more closely resemble human physiology and preferred over the monogenic models such as in defective leptin signaling (db, ob).133,134 New Zealand Obese (NZO) mice develop a polygenic disease pattern of obesity, insulin resistance, and type 2 diabetes.134,135 The onset of hyperglycemia is characterized by an elevated proliferation rate and hypertrophy of the β-cells136 leading to β-cell failure in most of the male animals.137 The disease progression is characterized by a gradual loss of glucose transporter 2138 and the transcription factor v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian) (MafA).139 Interestingly, NZO mice fed a carbohydrate-free high fat diet become obese and insulin resistant but are protected from β-cell failure.139-141 In contrast mice fed a diet rich in both carbohydrates and fat rapidly develop diabetes, indicating that the additive toxicity of an overflow of carbohydrates and lipids is important for the progression of β-cell failure.139,140,142,143
Summary:
Inability to store fat (lipodystrophy) or overflow of excess lipids from normal fat depots contributes to “ectopic” deposition of lipids and their metabolites in organs important for glucose metabolism, including muscle, liver, and the pancreas. Numerous studies have demonstrated involvement of these lipid metabolites in the development of insulin resistance and diabetes. Moreover, recent evidence indicates that hyperglycemia is a critical factor contributing to lipid-induced beta cell failure and diabetes. Thus, many leading scientists consider type 2 diabetes, a disorder with manifestations of abnormal glucose metabolism, to be at its most fundamental molecular level a disorder of lipid metabolism.
The “Adipokine Hypothesis”
Adipose tissue is not only a storage compartment for triglycerides but also a major endocrine and secretory organ, which releases a wide range of factors (adipokines) that signal through paracrine and hormonal mechanisms.144 Some of these secreted molecules are involved in inflammatory processes, such as TNF-α, IL-1β, IL-6 and MCP-1 as described above. The expanding volume of adipose tissue during obesity raises circulating levels of these inflammatory markers and is therefore thought to contribute to insulin resistance145 and the development of T2DM (Figure 3).

Figure 3: The “adipokine hypothesis.” Pathophysiology of obesity-induced dysfunction of adipokines in adipose cells contributing to peripheral insulin resistance.
AdipoR2, adiponectin receptor 2; AMPK, AMP-activated protein kinase; PEPCK, phosphoenolpyruvate carboxykinase; RBP4, retinol binding protein 4; Details in the text.
More than 100 different factors secreted by adipocytes have been identified over the past years, and it seems likely that this number will increase further due to the progress in analytical chemistry.146 Some of the prominent members of hormones produced by the adipose tissue are described below.
Leptin:
Leptin was the first adipokine discovered to influence body fat mass. It is predominantly secreted from white adipose tissue and exerts its main function by repressing food intake and promoting energy expenditure through sites of action in the central nervous system.147 The leptin receptor is expressed in the arcuate, ventromedial, dorsomedial, and lateral hypothalamic nuclei, which are known to regulate food intake.148 Mutation of both the leptin gene (ob) as well as the leptin receptor gene (db) leads to severe obesity, hyperphagia and insulin resistance in mice.149 The ob mutation was first hypothesized in 1950, when animal caretakers of the Jackson Laboratory observed the spontaneous occurrence of an obese phenotype in a mouse.150 but was not described as a non-sense mutation in the leptin gene until more than 40 years later.151
Expression and secretion of leptin is correlated with the amount of body fat and adipocyte size.152 Humans with mutations in both alleles of either leptin or the leptin receptor are obese, but these homozygous mutations are extremely rare.153 To the contrary, the vast majority of obese individuals display high plasma leptin levels in proportion to their increased body fat. Consequently, attempts to treat obesity by leptin administration have been mostly unsuccessful due to an apparent leptin resistance of these patients.154 Nevertheless, leptin improves insulin sensitivity by several mechanisms. In the liver and in skeletal muscle, leptin enhances glucose homeostasis by decreasing intracellular lipid accumulation155 and, in skeletal muscle, by direct activation of AMP-activated protein kinase (AMPK)156. In addition, leptin is able to inhibit insulin secretion by both, a direct effect on pancreatic β-cells, and an indirect mechanism via activation of the SNS (sympathetic nervous system) by the CNS (central nervous system).157-160
Adiponectin:
Adiponectin represents another important adipokine that has to be considered in the pathogenesis of insulin resistance and type 2 diabetes. Up-regulation of this collagen-like plasma protein secreted by adipocytes or its receptor is known to improve insulin sensitivity and endothelial function.52,161,162 Adiponectin has been closely linked to diseases such as obesity, the metabolic syndrome, type 2 diabetes mellitus, dyslipidemia and essential hypertension through its anti-inflammatory effects.163,164 In obesity and diabetes, adiponectin biosynthesis is impaired, and in vitro studies demonstrate suppression of adiponectin expression by various inflammatory and oxidative stress factors.165,166
Adiponectin regulates glucose and lipid metabolism by targeting the liver and skeletal muscle through two transmembrane receptors (AdipoR1 and AdipoR2). While AdipoR1 is most abundant in skeletal muscle, AdipoR2 is predominantly expressed in the liver.167 Improvement of insulin sensitivity is reached through activation of AMPK as well as increased expression of PPARα target genes.168 Adiponectin also has a key role in differentiation of subcutaneous preadipocytes and in the central regulation of energy homeostasis.161,169
Resistin:
Resistin expression and secretion differs between humans and rodents. In rodents, resistin is predominantly secreted from mature adipocytes with some weak expression in pancreatic islets and hypothalamus. In contrast, humans express resistin primarily in macrophages where it is thought to be involved in the recruitment of other immune cells, and in the secretion of pro-inflammatory factors.170 Because of these interspecies differences, it may have a less important role in humans during the pathogenesis of insulin resistance and diabetes. However, insulin-resistant mice display increased resistin levels and treatment with a thiazolidinedione, which activates adipocyte PPAR receptors and improves insulin sensitivity, lowers plasma resistin levels.52,171 In addition, some studies describe a role for resistin in the regulation of hepatic glucose production.172
Opposed to adiponectin, resistin decreases AMPK phosphorylation in liver, which leads to suppression of fatty acid oxidation and stimulation of glucose production.173 In vitro data from cultured adipocytes demonstrated a decreased insulin-stimulated glucose transport and disturbed adipocyte differentiation after resistin treatment.174,175 In humans, resistin is thought to impair insulin signaling by upregulating expression of the lipid phosphatase PTEN.170
Retinol Binding Protein 4:
Retinol binding protein 4 (RBP4) is predominantly expressed in adipose tissue and the liver and was first linked to the pathogenesis of insulin resistance when Abel and coworkers described that RBP4 was highly expressed in adipocytes of insulin resistant GLUT4-knockout mice.176 In addition, injection or overexpression of RBP4 in mice led to impaired insulin sensitivity. On the molecular level, RBP4 was shown to induce hepatic expression of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase (PEPCK) and to inhibit insulin signaling in skeletal muscle.177 Thus, at least in rodents, increased serum RBP4 leads to impaired glucose uptake in skeletal muscle with concomitant increase of hepatic glucose production.178
In humans, RBP4 influence on glucose homeostasis is less clear. Retinol binding protein 4 levels are elevated in plasma from obese and diabetic subjects.179 However, in larger groups a definitive correlation between RBP4 and measures of insulin sensitivity could not be demonstrated.180
Visfatin:
Visfatin, also known as nicotinamide phosphoribosyltransferase (NAMPT) and pre-B-cell colony enhancing factor 1 (PBEF1), is predominantly expressed in visceral adipose tissue, from which the name visfatin was derived. As an adipokine, the protein had been also found in the bloodstream where it has been shown to exert insulin-like functions. In mice, administration of visfatin was shown to lower blood glucose levels, whereas mice with a mutation in visfatin had increased levels of circulating glucose.181 However, subsequent studies have produced conflicting results regarding the association between visceral fat mass and plasma visfatin in humans.182,183 and the initial study was, in part, retracted.184 Despite these inconsistencies, a positive correlation between visfatin gene expression in visceral adipose tissue and BMI was seen in some human studies, as well as a negative correlation between BMI and visfatin gene expression in subcutaneous fat.182,185 In summary, the provided evidence of a direct link between visfatin action and human type 2 diabetes mellitus is still weak and its role in obesity and insulin resistance remains to be elucidated.52
Vaspin:
Visceral adipose tissue-derived serpin or serpinA12 (Vaspin) was originally identified as an adipokine predominantly secreted from visceral adipose tissue. In humans, obesity and T2DM are associated with elevated vaspin serum concentrations and expression levels in adipose tissue, suggesting a compensatory role in response to diminished insulin signaling in obesity. In obese mice, Vaspin administration improves glucose tolerance, insulin sensitivity, and reduces food intake.186,187 The exact cellular mechanisms of Vaspin action have yet to be elucidated, but a recent study demonstrated that Vaspin inhibited TNF-α- and IL-1-mediated activation of NF-κB and its downstream signaling molecules in a concentration-dependent manner and thereby protected endothelial cells from inflammation caused by pro-inflammatory cytokines.188 Moreover, a single-nucleotide polymorphism (rs2236242) was described to be positively associated with type 2 diabetes in 2759 participants in the KORA F3 study bearing an increased risk of diabetes independent of obesity, suggesting a link between vaspin and glucose metabolism.189
Omentin-1:
Omentin was described as a novel adipokine which is mainly produced by visceral adipose tissue and exhibits insulin-sensitizing action. Circulating levels of omentin are reduced in the obese state and in patients with T2DM. The beneficial effects of omentin are thought to be caused by a vasodilatation of blood vessels and attenuation of C-reactive protein-induced angiogenesis, potentially via the nuclear factor B signaling pathway, a potent pro-inflammatory signaling pathway.190 In addition, omentin has been shown to block TNFα-induced JNK and NF-κB activation.191 As with adiponectin, circulating levels of omentin are lower in obesity and also inversely correlated with measures of insulin resistance (HOMA-IR) and lower serum omentin concentrations were found in individuals with impaired glucose tolerance and type 2 diabetes compared to healthy individuals.192 However, it has to be elucidated whether the association of circulating omentin levels with the risk of T2DM is independent of BMI or can entirely be explained by obesity. In addition, further studies are needed to distinguish entirely between adiponectin and omentin action and whether omentin also shows a counter-regulatory increase in pro-inflammatory conditions.193
Apelin:
The peptide apelin is expressed among several tissues and secreted by adipocytes. Apelin gene expression levels are increased in adipose tissue from mouse models of obesity and hyperinsulinemia. Moreover, obese and hyperinsulinemic patients demonstrate elevated plasma levels of apelin. However, apelin plasma levels depend on several factors, including blood glucose levels and plasma triglyceride concentration.194 Currently, apelin is being considered as a biomarker and drug target, but its role in the development of both obesity and type 2 diabetes needs to be clarified with convincing clinical studies.195,196
Cardiotrophin-1:
Cardiotrophin-1 (Ctf1) is expressed in different tissues and secreted as an adipokine. Targeted disruption of cardiotrophin-1 in mice leads to obesity and insulin resistance.197 However, studies in humans have yielded contradictory results regarding cardiotrophin-1 levels and its association with obesity.198 The role of cardiothrophin-1 in the regulation of metabolic circadian rhythms is the focus of current research.199
WNT1-Inducible Signaling Pathway Protein-1:
Wnt1-inducible signaling pathway protein-1 (Wisp1) was recently described as a new adipokine. Its expression and secretion are increased in the course of differentiation of human adipocytes. Changes in body weight regulate both expression of Wisp-1 in adipose tissue and plasma levels of secreted Wisp-1.200 Interestingly, Wisp-1 serum levels are elevated in obese patients affected with polycystic ovary syndrome (PCOS) and in patients with gestational diabetes mellitus.201,202
Micro RNA (miRNA)- Containing Exosomes:
Micro RNA’s are small, noncoding sequences of RNAs that control a multiplicity of gene expression processes in diverse organs. In adipose tissue, miRNA’s are important regulators of cellular metabolism such as cell differentiation and lipid storage. Expression of miRNAs is altered in patients with obesity and type 2 diabetes.203,204 Interestingly, the action of miRNAs is not restricted to the cells of their original expression. Adipocyte-specific targeted disruption of the miRNA-processing enzyme Dicer in mice decreased the number of circulating exosomal miRNAs. Dicer (-/-) mice manifest glucose intolerance and insulin resistance, presumably mediated via increased fibroblast growth factor 21 (FGF21) plasma levels. There is evidence for a direct effect of circulating miRNAs derived from adipose tissue on FGF21 translation in the liver.205 In addition, a recent study showed that adipose tissue macrophages secrete miRNA-containing exosomes. Transfer of exosomes from obese to lean mice led to increased glucose intolerance and insulin resistance, and vice versa. The authors identified miRNA-155 as a potential target acting via the peroxisome proliferator-activated receptor gamma.206
Fatty Acid Esters Of Hydroxy Fatty Acids:
Recently, a novel family of lipids, the so-called fatty acid esters of hydroxy fatty acids (FAHFAs), has been identified. These branched fatty acid esters can be found in a variety of tissues with highest amounts in adipose tissues. A specific group of FAHFAs, the PAHSAs (Palmitic acid esters of hydroxy-stearic acids), have been shown to have beneficial metabolic effects. Circulating PAHSA levels are reduced in insulin resistant people, and serum levels correlate highly with insulin sensitivity. Moreover, treatment of obese mice with PAHSAs leads to improved glucose tolerance and increased insulin secretion. In adipocytes, PAHSAs signal through the omega-3 fatty acid receptor GPR120 to enhance insulin-stimulated glucose uptake.207 The production of FAHFAs in adipose tissue is tightly linked to the abundance of the insulin-responsive glucose transporter 4 (GLUT4) and the ability of adipocytes to transport glucose into the cell. Increased glucose uptake activates the nuclear transcription factor carbohydrate response element binding protein (CREBP), thereby enhancing lipogenesis and the synthesis of FAHFA’s.208 In addition, PAHSAs have been demonstrated to exert anti-inflammatory effects by repressing macrophage-induced tissue inflammation.209
Summary:
Adipocyte-derived factors such as adipokines and cytokines may provide direct links between obesity and the onset and progression of type 2 diabetes. Recent advancements in analytical technologies, in particular mass spectroscopy methods, may lead to further future discoveries of novel adipokines and cytokines that play roles in regulating intra-organ cross talk and metabolism.
GENETIC SUSCEPTIBILITY FOR OBESITY AND INSULIN RESISTANCE
Genetic Factors
Genetics clearly plays an important role in conferring the risk for the development of metabolic diseases. Variant genes determine the individual susceptibility towards known risk factors and may explain why only a fraction of obese individuals develop T2DM whereas the majority of diabetics are obese. In recent genome-wide association studies (GWAS’s), numerous variant genes were identified that predispose to diabetes or obesity.210,211 However, due to the relatively small contribution of the individual single nuclear polymorphisms (SNPs) to the overall disease risk, the predictive value of the gene variants is relatively small, and the pathophysiological relevance of many of these SNPs remains to be clarified. When combined, the genes identified so-far by GWAS explain only 15-20% of the heritable variance of metabolic diseases.212,213 An example of contradictory results of GWAS’s versus functional in vivo data represents the fat mass and obesity associated (FTO) gene. In different GWAS’s, SNPs located in the first intron of the FTO gene were associated with an altered body mass index,214,215 whereas Fto knockout mice develop postnatal growth retardation and exhibit a reduced body length.216 In contrast, no association of FTO was detected for height in humans.214,217 Although new in vivo data exist that reflect the human pathophysiology more precisely, the discrepancies of the GWAS’s and functional approaches remain apparent.218
Thus, even though many studies have confirmed FTO and TCF7L2 as two major genes implicated in obesity and diabetes in humans, respectively, GWAS’s have provided only limited mechanistic insights into the pathophysiology of these diseases.214,219-221 Novel approaches combining classical familial linkage analysis methods with whole-genome sequencing (WGS) are currently emerging as an important and powerful analysis method, especially since rare variants, which are not well interrogated by GWAS’s, could be responsible for a substantial proportion of complex human diseases.222,223
Perspectives: Positional Cloning to Identify Novel Genes In (and Out) of the Adipocyte
Polygenic mouse models have proven to be important tools to investigate molecular mechanisms that link obesity and T2DM. Despite novel advancements in the sequencing technology, the most successful strategy to identify and characterize new risk alleles is represented by a positional cloning approach.224,225 This approach capitalizes on a combination of breeding of multiple recombinant congenic mouse lines and of expression profiling of critical genomic regions that confer the phenotype. Using this approach, nine gene variants were identified as candidates for type 2 diabetes and/ or obesity during the last years. Sorcs1 encodes for a protein of largely unknown function that binds to a transcription factor responsible for islet vascularization.226 Lisch-like factor was described to be responsible for reducing β-cell mass and β-cell replication rates.227 Zfp69, a zinc-finger transcription factor, was described as causal gene for the diabetogenic Nidd1 quantitative trait locus (QTL) derived from the lean SJL (Swiss Jim Lambert) mouse strain and responsible for the distribution of lipids between different organs. Recently, it was shown that Zfp69 modulates hepatic insulin sensitivity in mice.93,228,229 Ifi202b, a member of the Ifi200 family of interferon inducible transcriptional modulators modulates fat accumulation through expression of adipogenic genes such as 11β-HSD1.230,231 Syntaxin-binding protein 5-like (Stxbp5l) or tomosyn-2 was identified in an F2 intercross from the BTBR T (+) tf (BTBR) Lep(ob/ob) and C57BL/6 (B6) Lep(ob/ob) mouse strains as a key negative regulator of insulin secretion.232 The same crossbreeding approach yielded Tsc2 as a gene underlying a QTL for nonalcoholic fatty liver disease (NAFLD) on chromosome 17. It was demonstrated that Tsc2(+/-) mice exhibited an increase in lipogenic gene expression levels in the liver in an insulin-dependent manner.233 The gene encoding the bile acid transporter Slco1a6 has been presented as a candidate gene for altered transport of taurocholic acid (TCA), resulting in broad gene regulation in pancreatic islets.234 In an NZO-based crossbreeding approach, one of the components of the KATP channel in pancreatic β-cells, Abcc8, was identified as causative factor in early-phase glucose-mediated insulin secretion.235 Lastly, Tbc1d1, a Rab-GAP protein that is presumably involved in GLUT4 vesicle sorting in skeletal muscle was identified as causal variant for the Nob1 obesity QTL derived from a crossbreeding of lean SJL with obese NZO (New Zealand obese) mice.91,236,237 Interestingly, both QTL, Nidd1 and Nob1 exhibit strong epistatic interaction as well as interaction with dietary fat in an outcross model of NZO and lean SJL mice.91,93,238 and both Zfp69 and Tbc1d1 genes are directly responsible for fat storage and fatty acid oxidation, respectively. This underscores the importance of altered lipid partitioning as a common denominator in the pathogenesis of obesity-driven diabetes.
All nine positionally cloned genes were located within consensus QTL regions, i.e. loci that have been linked to diabetes-related traits in multiple crossbreeding experiments. In fact, our meta-analyses of 77 published genome-wide linkage scans with hundreds of QTL strongly indicated the presence of consensus regions for metabolic traits in the mouse genome, and these hotspots could provide guidance for identifying novel gene variants involved in the development of the disease.239,240 Nevertheless, generation and refinement of novel polygenic mouse models is important since complex genetics seems to contribute significantly to the pathogenesis of the human disease 241 Moreover, diverse genetic tools such as the generation of Chromosome Substitution Strains (CSSs) and combination of classical breeding approaches with high-throughput genotyping, sequencing and genetic engineering technologies, and information repositories highlight the power of the mouse for genetic, functional, and systems studies of complex traits and disease models.242
summary
Although immune system, ectopic fat, and macro/micronutrients all contribute in part to the susceptibility for diabetes in the obese state, most of the underlying molecular mechanisms are still poorly understood. The identification of susceptibility genes mediating the progression of type 2 diabetes is crucial to prevent the massive epidemics of the disease. Future research will be focused not only on gene-gene interactions but also on the interplay of genetic and environmental risk factors.
References
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. Aug 2000;106(4):473-481.
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. Dec 14 2006;444(7121):840-846.
- Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. Jan 1 2003;289(1):76-79.
- Ogurtsova K, da Rocha Fernandes JD, Huang Y, et al. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes research and clinical practice. Jun 2017;128:40-50.
- Farag YM, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. Jan 2011;26(1):28-35.
- Kalra S. Diabesity. JPMA. The Journal of the Pakistan Medical Association. Apr 2013;63(4):532-534.
- Abbasi F, Brown BW, Jr., Lamendola C, McLaughlin T, Reaven GM. Relationship between obesity, insulin resistance, and coronary heart disease risk. J Am Coll Cardiol. Sep 4 2002;40(5):937-943.
- Kahn SE. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. Sep 2001;86(9):4047-4058.
- Fujioka S, Matsuzawa Y, Tokunaga K, Tarui S. Contribution of intra-abdominal fat accumulation to the impairment of glucose and lipid metabolism in human obesity. Metabolism. Jan 1987;36(1):54-59.
- Despres JP, Nadeau A, Tremblay A, et al. Role of deep abdominal fat in the association between regional adipose tissue distribution and glucose tolerance in obese women. Diabetes. Mar 1989;38(3):304-309.
- Pouliot MC, Despres JP, Nadeau A, et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes. Jul 1992;41(7):826-834.
- Park KS, Rhee BD, Lee KU, et al. Intra-abdominal fat is associated with decreased insulin sensitivity in healthy young men. Metabolism. 1991;40(6):600-603.
- Marin P, Andersson B, Ottosson M, et al. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism. Nov 1992;41(11):1242-1248.
- Despres JP, Moorjani S, Lupien PJ, Tremblay A, Nadeau A, Bouchard C. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis. Jul-Aug 1990;10(4):497-511.
- Carey DG, Nguyen TV, Campbell LV, Chisholm DJ, Kelly P. Genetic influences on central abdominal fat: a twin study. Int J Obes Relat Metab Disord. Aug 1996;20(8):722-726.
- Lillioja S, Young AA, Culter CL, et al. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest. Aug 1987;80(2):415-424.
- Solini A, Bonora E, Bonadonna R, Castellino P, DeFronzo RA. Protein metabolism in human obesity: relationship with glucose and lipid metabolism and with visceral adipose tissue. J Clin Endocrinol Metab. Aug 1997;82(8):2552-2558.
- Weiss R, Dufour S, Taksali SE, et al. Prediabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet. Sep 20 2003;362(9388):951-957.
- Bonora E, Del Prato S, Bonadonna RC, et al. Total body fat content and fat topography are associated differently with in vivo glucose metabolism in nonobese and obese nondiabetic women. Diabetes. Sep 1992;41(9):1151-1159.
- Landin K, Lonnroth P, Krotkiewski M, Holm G, Smith U. Increased insulin resistance and fat cell lipolysis in obese but not lean women with a high waist/hip ratio. Eur J Clin Invest. Oct 1990;20(5):530-535.
- Abate N, Garg A, Peshock RM, Stray-Gundersen J, Grundy SM. Relationships of generalized and regional adiposity to insulin sensitivity in men. J Clin Invest. Jul 1995;96(1):88-98.
- Goodpaster BH, Thaete FL, Simoneau JA, Kelley DE. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes. Oct 1997;46(10):1579-1585.
- Klein S, Fontana L, Young VL, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. Jun 17 2004;350(25):2549-2557.
- Barzilai N, She L, Liu BQ, et al. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes. Jan 1999;48(1):94-98.
- Thorne A, Lonnqvist F, Apelman J, Hellers G, Arner P. A pilot study of long-term effects of a novel obesity treatment: omentectomy in connection with adjustable gastric banding. Int J Obes Relat Metab Disord. Feb 2002;26(2):193-199.
- Csendes A, Maluenda F, Burgos AM. A prospective randomized study comparing patients with morbid obesity submitted to laparotomic gastric bypass with or without omentectomy. Obesity surgery. Apr 2009;19(4):490-494.
- Herrera MF, Pantoja JP, Velazquez-Fernandez D, et al. Potential additional effect of omentectomy on metabolic syndrome, acute-phase reactants, and inflammatory mediators in grade III obese patients undergoing laparoscopic Roux-en-Y gastric bypass: a randomized trial. Diabetes care. Jul 2010;33(7):1413-1418.
- Lima MM, Pareja JC, Alegre SM, et al. Acute effect of roux-en-y gastric bypass on whole-body insulin sensitivity: a study with the euglycemic-hyperinsulinemic clamp. J Clin Endocrinol Metab. Aug 2010;95(8):3871-3875.
- Fabbrini E, Tamboli RA, Magkos F, et al. Surgical removal of omental fat does not improve insulin sensitivity and cardiovascular risk factors in obese adults. Gastroenterology. Aug 2010;139(2):448-455.
- Thomas EL, Frost G, Taylor-Robinson SD, Bell JD. Excess body fat in obese and normal-weight subjects. Nutrition research reviews. Jun 2012;25(1):150-161.
- Thomas EL, Parkinson JR, Frost GS, et al. The missing risk: MRI and MRS phenotyping of abdominal adiposity and ectopic fat. Obesity (Silver Spring). Jan 2012;20(1):76-87.
- O'Donovan G, Thomas EL, McCarthy JP, et al. Fat distribution in men of different waist girth, fitness level and exercise habit. Int J Obes (Lond). Dec 2009;33(12):1356-1362.
- van der Kooy K, Seidell JC. Techniques for the measurement of visceral fat: a practical guide. Int J Obes Relat Metab Disord. Apr 1993;17(4):187-196.
- Abate N, Burns D, Peshock RM, Garg A, Grundy SM. Estimation of adipose tissue mass by magnetic resonance imaging: validation against dissection in human cadavers. J Lipid Res. Aug 1994;35(8):1490-1496.
- Thomas EL, Fitzpatrick JA, Malik SJ, Taylor-Robinson SD, Bell JD. Whole body fat: content and distribution. Progress in nuclear magnetic resonance spectroscopy. Aug 2013;73:56-80.
- Xia Y, Ergun DL, Wacker WK, Wang X, Davis CE, Kaul S. Relationship between dual-energy X-ray absorptiometry volumetric assessment and X-ray computed tomography-derived single-slice measurement of visceral fat. Journal of clinical densitometry : the official journal of the International Society for Clinical Densitometry. Jan-Mar 2014;17(1):78-83.
- Kaul S, Rothney MP, Peters DM, et al. Dual-energy X-ray absorptiometry for quantification of visceral fat. Obesity (Silver Spring). Jun 2012;20(6):1313-1318.
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. Jul 1998;15(7):539-553.
- Alberti KG, Zimmet P, Shaw J. The metabolic syndrome--a new worldwide definition. Lancet. Sep 24-30 2005;366(9491):1059-1062.
- Seidell JC, Oosterlee A, Deurenberg P, Hautvast JG, Ruijs JH. Abdominal fat depots measured with computed tomography: effects of degree of obesity, sex, and age. Eur J Clin Nutr. Sep 1988;42(9):805-815.
- Ferland M, Despres JP, Tremblay A, et al. Assessment of adipose tissue distribution by computed axial tomography in obese women: association with body density and anthropometric measurements. Br J Nutr. Mar 1989;61(2):139-148.
- Despres JP, Prud'homme D, Pouliot MC, Tremblay A, Bouchard C. Estimation of deep abdominal adipose-tissue accumulation from simple anthropometric measurements in men. Am J Clin Nutr. Sep 1991;54(3):471-477.
- Bouchard C, Despres JP, Mauriege P. Genetic and nongenetic determinants of regional fat distribution. Endocr Rev. Feb 1993;14(1):72-93.
- Hegele RA, Joy TR, Al-Attar SA, Rutt BK. Thematic review series: Adipocyte Biology. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res. Jul 2007;48(7):1433-1444.
- Stefan N, Schick F, Haring HU. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. Aug 01 2017;26(2):292-300.
- Gavrilova O, Marcus-Samuels B, Graham D, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. Feb 2000;105(3):271-278.
- Konrad D, Rudich A, Schoenle EJ. Improved glucose tolerance in mice receiving intraperitoneal transplantation of normal fat tissue. Diabetologia. Apr 2007;50(4):833-839.
- Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. May 2008;7(5):410-420.
- Virtue S, Vidal-Puig A. It's not how fat you are, it's what you do with it that counts. PLoS Biol. Sep 23 2008;6(9):e237.
- Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. Dec 2003;112(12):1821-1830.
- Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. Nov 2008;93(11 Suppl 1):S64-73.
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415-445.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. Dec 2003;112(12):1796-1808.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. Dec 14 2006;444(7121):860-867.
- Van Coillie E, Van Damme J, Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine & growth factor reviews. Mar 1999;10(1):61-86.
- Old LJ. Tumor necrosis factor (TNF). Science. Nov 8 1985;230(4726):630-632.
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. Jan 1 1993;259(5091):87-91.
- Torti FM, Dieckmann B, Beutler B, Cerami A, Ringold GM. A macrophage factor inhibits adipocyte gene expression: an in vitro model of cachexia. Science. Aug 30 1985;229(4716):867-869.
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem. Dec 15 1993;268(35):26055-26058.
- Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. Mar 19 1993;259(5102):1769-1771.
- Muoio DM. Intramuscular triacylglycerol and insulin resistance: Guilty as charged or wrongly accused? Biochim Biophys Acta. Dec 1 2009.
- Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. Dec 30 2002;1585(2-3):202-212.
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. Mar 11 2005;120(5):649-661.
- Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. Dec 2004;114(12):1752-1761.
- Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res. Nov-Dec 2005;42(6):517-525.
- Martinez-Abundis E, Reynoso-von Drateln C, Hernandez-Salazar E, Gonzalez-Ortiz M. Effect of etanercept on insulin secretion and insulin sensitivity in a randomized trial with psoriatic patients at risk for developing type 2 diabetes mellitus. Arch Dermatol Res. Nov 2007;299(9):461-465.
- Vendrell J, Maymo-Masip E, Tinahones F, et al. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: up-regulation in severe obesity is mediated by inflammation but not hypoxia. J Clin Endocrinol Metab. Jun 2010;95(6):2983-2992.
- Chacon MR, Richart C, Gomez JM, et al. Expression of TWEAK and its receptor Fn14 in human subcutaneous adipose tissue. Relationship with other inflammatory cytokines in obesity. Cytokine. Feb 7 2006;33(3):129-137.
- Maymo-Masip E, Fernandez-Veledo S, Garcia Espana A, et al. The rise of soluble TWEAK levels in severely obese subjects after bariatric surgery may affect adipocyte-cytokine production induced by TNFalpha. J Clin Endocrinol Metab. Aug 2013;98(8):E1323-1333.
- Aljada A, Ghanim H, Saadeh R, Dandona P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J Clin Endocrinol Metab. Jan 2001;86(1):450-453.
- Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A. Jun 10 2003;100(12):7265-7270.
- Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. Jun 2006;116(6):1494-1505.
- Panee J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine. Oct 2012;60(1):1-12.
- Krook A. IL-6 and metabolism-new evidence and new questions. Diabetologia. Jul 2008;51(7):1097-1099.
- Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. Nov 14 2003;278(46):45777-45784.
- Wallenius V, Wallenius K, Ahren B, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. Jan 2002;8(1):75-79.
- Nieto-Vazquez I, Fernandez-Veledo S, de Alvaro C, Lorenzo M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes. Dec 2008;57(12):3211-3221.
- Feingold KR, Soued M, Adi S, et al. Effect of interleukin-1 on lipid metabolism in the rat. Similarities to and differences from tumor necrosis factor. Arterioscler Thromb. May-Jun 1991;11(3):495-500.
- Strandberg L, Lorentzon M, Hellqvist A, et al. Interleukin-1 system gene polymorphisms are associated with fat mass in young men. J Clin Endocrinol Metab. Jul 2006;91(7):2749-2754.
- Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. Nov 30 2007;282(48):35279-35292.
- Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. Oct 2005;25(10):2062-2068.
- Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. Apr 12 2007;356(15):1517-1526.
- Netea MG, van der Graaf C, Van der Meer JW, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. May 2004;75(5):749-755.
- Lin Y, Lee H, Berg AH, Lisanti MP, Shapiro L, Scherer PE. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem. Aug 11 2000;275(32):24255-24263.
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. Jul 8 2004;430(6996):257-263.
- Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol. Oct 2009;20(5):379-385.
- Kopp A, Buechler C, Bala M, Neumeier M, Scholmerich J, Schaffler A. Toll-like receptor ligands cause proinflammatory and prodiabetic activation of adipocytes via phosphorylation of extracellular signal-regulated kinase and c-Jun N-terminal kinase but not interferon regulatory factor-3. Endocrinology. Mar 2010;151(3):1097-1108.
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. Jul 2006;116(7):1793-1801.
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. Feb 12 2004;350(7):664-671.
- Chadt A, Leicht K, Deshmukh A, et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet. Nov 2008;40(11):1354-1359.
- Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res. Jun 2007;48(6):1253-1262.
- Scherneck S, Nestler M, Vogel H, et al. Positional cloning of zinc finger domain transcription factor Zfp69, a candidate gene for obesity-associated diabetes contributed by mouse locus Nidd/SJL. PLoS Genet. Jul 2009;5(7):e1000541.
- Tan CY, Vidal-Puig A. Adipose tissue expandability: the metabolic problems of obesity may arise from the inability to become more obese. Biochem Soc Trans. Oct 2008;36(Pt 5):935-940.
- Smith U. Abdominal obesity: a marker of ectopic fat accumulation. J Clin Invest. May 2015;125(5):1790-1792.
- Krotkiewski M, Bjorntorp P, Sjostrom L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest. Sep 1983;72(3):1150-1162.
- Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. Jan 2010;24(1):326-331.
- Perry RJ, Camporez JP, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. Feb 12 2015;160(4):745-758.
- Smith U, Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. Journal of internal medicine. Nov 2016;280(5):465-475.
- Barak Y, Nelson MC, Ong ES, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. Oct 1999;4(4):585-595.
- Moitra J, Mason MM, Olive M, et al. Life without white fat: a transgenic mouse. Genes Dev. Oct 15 1998;12(20):3168-3181.
- Simha V, Szczepaniak LS, Wagner AJ, DePaoli AM, Garg A. Effect of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes care. Jan 2003;26(1):30-35.
- Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. May 2002;109(10):1345-1350.
- Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. Feb 21 2002;346(8):570-578.
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. Jan 2006;45(1):42-72.
- van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. May 23 2008;94(2):231-241.
- Petersen MC, Shulman GI. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends in pharmacological sciences. Jul 2017;38(7):649-665.
- Turcotte LP, Fisher JS. Skeletal muscle insulin resistance: roles of fatty acid metabolism and exercise. Phys Ther. Nov 2008;88(11):1279-1296.
- Moro C, Bajpeyi S, Smith SR. Determinants of intramyocellular triglyceride turnover: implications for insulin sensitivity. Am J Physiol Endocrinol Metab. Feb 2008;294(2):E203-213.
- Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. Jul 2001;50(7):1612-1617.
- Li L, Yang G, Li Q, Tang Y, Li K. High-fat- and lipid-induced insulin resistance in rats: the comparison of glucose metabolism, plasma resistin and adiponectin levels. Ann Nutr Metab. 2006;50(6):499-505.
- Holloway GP, Bonen A, Spriet LL. Regulation of skeletal muscle mitochondrial fatty acid metabolism in lean and obese individuals. Am J Clin Nutr. Jan 2009;89(1):455S-462S.
- Adams JM, 2nd, Pratipanawatr T, Berria R, et al. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. Jan 2004;53(1):25-31.
- Goodpaster BH, Kelley DE. Skeletal muscle triglyceride: marker or mediator of obesity-induced insulin resistance in type 2 diabetes mellitus? Curr Diab Rep. Jun 2002;2(3):216-222.
- Krssak M, Falk Petersen K, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. Jan 1999;42(1):113-116.
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. Dec 2006;55 Suppl 2:S9-S15.
- Cooney GJ, Thompson AL, Furler SM, Ye J, Kraegen EW. Muscle long-chain acyl CoA esters and insulin resistance. Ann N Y Acad Sci. Jun 2002;967:196-207.
- Blaak EE, Wagenmakers AJ, Glatz JF, et al. Plasma FFA utilization and fatty acid-binding protein content are diminished in type 2 diabetic muscle. Am J Physiol Endocrinol Metab. Jul 2000;279(1):E146-154.
- Ortenblad N, Mogensen M, Petersen I, et al. Reduced insulin-mediated citrate synthase activity in cultured skeletal muscle cells from patients with type 2 diabetes: evidence for an intrinsic oxidative enzyme defect. Biochim Biophys Acta. Jun 30 2005;1741(1-2):206-214.
- Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. Jul 1997;83(1):166-171.
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. Oct 2002;51(10):2944-2950.
- Hulver MW, Berggren JR, Cortright RN, et al. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. Apr 2003;284(4):E741-747.
- Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. Dec 2001;86(12):5755-5761.
- Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. Jan 2006;86(1):205-243.
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. Jul 2002;51(7):2005-2011.
- Boden G. Free fatty acids-the link between obesity and insulin resistance. Endocr Pract. Jan-Feb 2001;7(1):44-51.
- Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. Jun 15 1996;97(12):2859-2865.
- Bostrom P, Andersson L, Vind B, et al. The SNARE protein SNAP23 and the SNARE-interacting protein Munc18c in human skeletal muscle are implicated in insulin resistance/type 2 diabetes. Diabetes. May 11 2010.
- Kahn BB. Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell. Mar 6 1998;92(5):593-596.
- Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. Dec 2004;53 Suppl 3:S16-21.
- Joost HG. Pathogenesis, Risk Assessment and Prevention of Type 2 Diabetes mellitus. Obesity Facts. JUN 2008;1(3):128-137.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. Dec 2003;144(12):5159-5165.
- Mauvais-Jarvis F, Kahn CR. Understanding the pathogenesis and treatment of insulin resistance and type 2 diabetes mellitus: what can we learn from transgenic and knockout mice? Diabetes Metab. Dec 2000;26(6):433-448.
- Leiter EH, Reifsnyder PC. Differential levels of diabetogenic stress in two new mouse models of obesity and type 2 diabetes. Diabetes. Feb 2004;53 Suppl 1:S4-11.
- Ortlepp JR, Kluge R, Giesen K, et al. A metabolic syndrome of hypertension, hyperinsulinaemia and hypercholesterolaemia in the New Zealand obese mouse. Eur J Clin Invest. Mar 2000;30(3):195-202.
- Lange C, Jeruschke K, Herberg L, Leiter EH, Junger E. The diabetes-prone NZO/Hl strain. Proliferation capacity of beta cells in hyperinsulinemia and hyperglycemia. Arch Physiol Biochem. Feb 2006;112(1):49-58.
- Junger E, Herberg L, Jeruschke K, Leiter EH. The diabetes-prone NZO/Hl strain. II. Pancreatic immunopathology. Lab Invest. Jul 2002;82(7):843-853.
- Chankiewitz E, Peschke D, Herberg L, et al. Did the gradual loss of GLUT2 cause a shift to diabetic disorders in the New Zealand obese mouse (NZO/Hl)? Exp Clin Endocrinol Diabetes. May 2006;114(5):262-269.
- Jurgens HS, Neschen S, Ortmann S, et al. Development of diabetes in obese, insulin-resistant mice: essential role of dietary carbohydrate in beta cell destruction. Diabetologia. Jul 2007;50(7):1481-1489.
- Dreja T, Jovanovic Z, Rasche A, et al. Diet-induced gene expression of isolated pancreatic islets from a polygenic mouse model of the metabolic syndrome. Diabetologia. Feb 2010;53(2):309-320.
- Kluth O, Mirhashemi F, Scherneck S, et al. Dissociation of lipotoxicity and glucotoxicity in a mouse model of obesity associated diabetes: role of forkhead box O1 (FOXO1) in glucose-induced beta cell failure. Diabetologia. Nov 24.
- Leiter EH, Coleman DL, Ingram DK, Reynolds MA. Influence of dietary carbohydrate on the induction of diabetes in C57BL/KsJ-db/db diabetes mice. J Nutr. Jan 1983;113(1):184-195.
- Mirhashemi F, Kluth O, Scherneck S, et al. High-Fat, Carbohydrate-Free Diet Markedly Aggravates Obesity but Prevents beta-Cell Loss and Diabetes in the Obese, Diabetes-Susceptible db/db Strain. Obesity Facts. DEC 2008;1(6):292-297.
- Ahima RS, Osei SY. Adipokines in obesity. Front Horm Res. 2008;36:182-197.
- Trayhurn P, Wood IS. Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans. Nov 2005;33(Pt 5):1078-1081.
- Gnacinska M, Malgorzewicz S, Stojek M, Lysiak-Szydlowska W, Sworczak K. Role of adipokines in complications related to obesity: a review. Adv Med Sci. 2009;54(2):150-157.
- Houseknecht KL, Baile CA, Matteri RL, Spurlock ME. The biology of leptin: a review. J Anim Sci. May 1998;76(5):1405-1420.
- Fei H, Okano HJ, Li C, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. Jun 24 1997;94(13):7001-7005.
- Hamann A, Matthaei S. Regulation of energy balance by leptin. Exp Clin Endocrinol Diabetes. 1996;104(4):293-300.
- Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. Dec 1950;41(12):317-318.
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. Dec 1 1994;372(6505):425-432.
- Houseknecht KL, Flier SN, Frevert EU, Frederich RC, Flier JS, Kahn BB. Leptin secretion correlates with adipocyte size in genetic and dietary obesity. Diabetes. MAY 1996;45:144-144.
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. Dec 14 2006;444(7121):847-853.
- Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. Jama. Oct 27 1999;282(16):1568-1575.
- Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. Sep 25 1997;389(6649):374-377.
- Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. Jan 17 2002;415(6869):339-343.
- Kulkarni RN, Wang ZL, Wang RM, et al. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest. Dec 1 1997;100(11):2729-2736.
- Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. Sep 2000;141(9):3072-3079.
- Meek TH, Morton GJ. Leptin, diabetes, and the brain. Indian journal of endocrinology and metabolism. Dec 2012;16(Suppl 3):S534-542.
- Park S, Ahn IS, Kim DS. Central infusion of leptin improves insulin resistance and suppresses beta-cell function, but not beta-cell mass, primarily through the sympathetic nervous system in a type 2 diabetic rat model. Life sciences. Jun 5 2010;86(23-24):854-862.
- Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. Sep 2007;117(9):2621-2637.
- Kawano J, Arora R. The role of adiponectin in obesity, diabetes, and cardiovascular disease. J Cardiometab Syndr. Winter 2009;4(1):44-49.
- Kazumi T, Kawaguchi A, Hirano T, Yoshino G. Serum adiponectin is associated with high-density lipoprotein cholesterol, triglycerides, and low-density lipoprotein particle size in young healthy men. Metabolism. May 2004;53(5):589-593.
- Weyer C, Funahashi T, Tanaka S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. May 2001;86(5):1930-1935.
- Kamigaki M, Sakaue S, Tsujino I, et al. Oxidative stress provokes atherogenic changes in adipokine gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. Jan 13 2006;339(2):624-632.
- Kim KY, Kim JK, Jeon JH, Yoon SR, Choi I, Yang Y. c-Jun N-terminal kinase is involved in the suppression of adiponectin expression by TNF-alpha in 3T3-L1 adipocytes. Biochem Biophys Res Commun. Feb 11 2005;327(2):460-467.
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. Jul 2006;116(7):1784-1792.
- Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. May 2005;26(3):439-451.
- Kusminski CM, McTernan PG, Schraw T, et al. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia. Mar 2007;50(3):634-642.
- Barnes KM, Miner JL. Role of resistin in insulin sensitivity in rodents and humans. Curr Protein Pept Sci. Feb 2009;10(1):96-107.
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. Jan 2005;1(1):15-25.
- Rajala MW, Obici S, Scherer PE, Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest. Jan 2003;111(2):225-230.
- Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond). Sep 2008;32 Suppl 4:S7-12.
- Kim KH, Lee K, Moon YS, Sul HS. A cysteine-rich adipose tissue-specific secretory factor inhibits adipocyte differentiation. J Biol Chem. Apr 6 2001;276(14):11252-11256.
- Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. Feb 2008;34(1):2-11.
- Abel ED, Peroni O, Kim JK, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. Feb 8 2001;409(6821):729-733.
- Yang Q, Graham TE, Mody N, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. Jul 21 2005;436(7049):356-362.
- Wolf G. Serum retinol-binding protein: a link between obesity, insulin resistance, and type 2 diabetes. Nutr Rev. May 2007;65(5):251-256.
- Graham TE, Yang Q, Bluher M, et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. Jun 15 2006;354(24):2552-2563.
- von Eynatten M, Humpert PM. Retinol-binding protein-4 in experimental and clinical metabolic disease. Expert Rev Mol Diagn. May 2008;8(3):289-299.
- Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. Jan 21 2005;307(5708):426-430.
- Berndt J, Kloting N, Kralisch S, et al. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes. Oct 2005;54(10):2911-2916.
- Arner P. Visfatin--a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. Jan 2006;91(1):28-30.
- Fukuhara A, Matsuda M, Nishizawa M, et al. Retraction. Science. Oct 26 2007;318(5850):565.
- Varma V, Yao-Borengasser A, Rasouli N, et al. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J Clin Endocrinol Metab. Feb 2007;92(2):666-672.
- Bluher M. Vaspin in obesity and diabetes: pathophysiological and clinical significance. Endocrine. Apr 2012;41(2):176-182.
- Heiker JT. Vaspin (serpinA12) in obesity, insulin resistance, and inflammation. Journal of peptide science : an official publication of the European Peptide Society. Mar 5 2014.
- Liu S, Dong Y, Wang T, et al. Vaspin inhibited proinflammatory cytokine induced activation of nuclear factor-kappa B and its downstream molecules in human endothelial EA.hy926 cells. Diabetes research and clinical practice. Dec 25 2013.
- Kempf K, Rose B, Illig T, et al. Vaspin (SERPINA12) genotypes and risk of type 2 diabetes: Results from the MONICA/KORA studies. Exp Clin Endocrinol Diabetes. Mar 2010;118(3):184-189.
- Tan BK, Adya R, Randeva HS. Omentin: a novel link between inflammation, diabesity, and cardiovascular disease. Trends in cardiovascular medicine. Jul 2010;20(5):143-148.
- Yamawaki H, Kuramoto J, Kameshima S, Usui T, Okada M, Hara Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem Biophys Res Commun. May 6 2011;408(2):339-343.
- Yan P, Liu D, Long M, Ren Y, Pang J, Li R. Changes of serum omentin levels and relationship between omentin and adiponectin concentrations in type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. Apr 2011;119(4):257-263.
- Herder C, Carstensen M, Ouwens DM. Anti-inflammatory cytokines and risk of type 2 diabetes. Diabetes, obesity & metabolism. Sep 2013;15 Suppl 3:39-50.
- Castan-Laurell I, Dray C, Attane C, Duparc T, Knauf C, Valet P. Apelin, diabetes, and obesity. Endocrine. Aug 2011;40(1):1-9.
- Bertrand C, Valet P, Castan-Laurell I. Apelin and energy metabolism. Frontiers in physiology. 2015;6:115.
- Alipour FG, Ashoori MR, Pilehvar-Soltanahmadi Y, Zarghami N. An overview on biological functions and emerging therapeutic roles of apelin in diabetes mellitus. Diabetes & metabolic syndrome. Dec 2017;11 Suppl 2:S919-S923.
- Moreno-Aliaga MJ, Perez-Echarri N, Marcos-Gomez B, et al. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metab. Aug 03 2011;14(2):242-253.
- Escote X, Gomez-Zorita S, Lopez-Yoldi M, et al. Role of Omentin, Vaspin, Cardiotrophin-1, TWEAK and NOV/CCN3 in Obesity and Diabetes Development. International journal of molecular sciences. Aug 15 2017;18(8).
- Lopez-Yoldi M, Stanhope KL, Garaulet M, et al. Role of cardiotrophin-1 in the regulation of metabolic circadian rhythms and adipose core clock genes in mice and characterization of 24-h circulating CT-1 profiles in normal-weight and overweight/obese subjects. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. Apr 2017;31(4):1639-1649.
- Murahovschi V, Pivovarova O, Ilkavets I, et al. WISP1 is a novel adipokine linked to inflammation in obesity. Diabetes. Mar 2015;64(3):856-866.
- Sahin Ersoy G, Altun Ensari T, Vatansever D, Emirdar V, Cevik O. Novel adipokines WISP1 and betatrophin in PCOS: relationship to AMH levels, atherogenic and metabolic profile. Gynecological endocrinology : the official journal of the International Society of Gynecological Endocrinology. Feb 2017;33(2):119-123.
- Sahin Ersoy G, Altun Ensari T, Subas S, Giray B, Simsek EE, Cevik O. WISP1 is a novel adipokine linked to metabolic parameters in gestational diabetes mellitus. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet. Apr 2017;30(8):942-946.
- Arner P, Kulyte A. MicroRNA regulatory networks in human adipose tissue and obesity. Nature reviews. Endocrinology. May 2015;11(5):276-288.
- Hashimoto N, Tanaka T. Role of miRNAs in the pathogenesis and susceptibility of diabetes mellitus. Journal of human genetics. Feb 2017;62(2):141-150.
- Thomou T, Mori MA, Dreyfuss JM, et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. Feb 23 2017;542(7642):450-455.
- Ying W, Riopel M, Bandyopadhyay G, et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell. Oct 05 2017;171(2):372-384 e312.
- Moraes-Vieira PM, Saghatelian A, Kahn BB. GLUT4 Expression in Adipocytes Regulates De Novo Lipogenesis and Levels of a Novel Class of Lipids With Antidiabetic and Anti-inflammatory Effects. Diabetes. Jul 2016;65(7):1808-1815.
- Yore MM, Syed I, Moraes-Vieira PM, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. Oct 09 2014;159(2):318-332.
- Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. Sep 03 2010;142(5):687-698.
- McCarthy MI, Zeggini E. Genome-wide association studies in type 2 diabetes. Curr Diab Rep. Apr 2009;9(2):164-171.
- Stancakova A, Laakso M. Genetics of metabolic syndrome. Reviews in endocrine & metabolic disorders. Dec 2014;15(4):243-252.
- Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. Sep 2012;44(9):981-990.
- So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genetic epidemiology. Jul 2011;35(5):310-317.
- Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. May 11 2007;316(5826):889-894.
- Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. Jun 2007;39(6):724-726.
- Fischer J, Koch L, Emmerling C, et al. Inactivation of the Fto gene protects from obesity. Nature. Apr 16 2009;458(7240):894-898.
- Hakanen M, Raitakari OT, Lehtimaki T, et al. FTO genotype is associated with body mass index after the age of seven years but not with energy intake or leisure-time physical activity. J Clin Endocrinol Metab. Apr 2009;94(4):1281-1287.
- Church C, Lee S, Bagg EA, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. Aug 2009;5(8):e1000599.
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. Apr 26 2007.
- Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. Feb 22 2007;445(7130):881-885.
- Scott LJ, Mohlke KL, Bonnycastle LL, et al. A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science. Apr 26 2007.
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. Apr 16 2010;141(2):210-217.
- Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nature reviews. Genetics. May 2015;16(5):275-284.
- Schwenk RW, Vogel H, Schurmann A. Genetic and epigenetic control of metabolic health. Molecular metabolism. Sep 25 2013;2(4):337-347.
- Joost HG, Schurmann A. The genetic basis of obesity-associated type 2 diabetes (diabesity) in polygenic mouse models. Mammalian genome : official journal of the International Mammalian Genome Society. Oct 2014;25(9-10):401-412.
- Clee SM, Yandell BS, Schueler KM, et al. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat Genet. Jun 2006;38(6):688-693.
- Dokmanovic-Chouinard M, Chung WK, Chevre JC, et al. Positional cloning of "Lisch-Like", a candidate modifier of susceptibility to type 2 diabetes in mice. PLoS Genet. 2008;4(7):e1000137.
- Chung B, Stadion M, Schulz N, et al. The diabetes gene Zfp69 modulates hepatic insulin sensitivity in mice. Diabetologia. Oct 2015;58(10):2403-2413.
- Scherneck S, Vogel H, Nestler M, Kluge R, Schurmann A, Joost HG. Role of zinc finger transcription factor zfp69 in body fat storage and diabetes susceptibility of mice. Results and problems in cell differentiation. 2010;52:57-68.
- Vogel H, Scherneck S, Kanzleiter T, et al. Loss of function of Ifi202b by a microdeletion on chromosome 1 of C57BL/6J mice suppresses 11beta-hydroxysteroid dehydrogenase type 1 expression and development of obesity. Human molecular genetics. Sep 1 2012;21(17):3845-3857.
- Vogel H, Jahnert M, Stadion M, Matzke D, Scherneck S, Schurmann A. A vast genomic deletion in the C56BL/6 genome affects different genes within the Ifi200 cluster on chromosome 1 and mediates obesity and insulin resistance. BMC genomics. Feb 15 2017;18(1):172.
- Bhatnagar S, Oler AT, Rabaglia ME, et al. Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet. Oct 2011;7(10):e1002323.
- Wang CY, Stapleton DS, Schueler KL, et al. Tsc2, a positional candidate gene underlying a quantitative trait locus for hepatic steatosis. J Lipid Res. Aug 2012;53(8):1493-1501.
- Tian J, Keller MP, Oler AT, et al. Identification of the Bile Acid Transporter Slco1a6 as a Candidate Gene That Broadly Affects Gene Expression in Mouse Pancreatic Islets. Genetics. Nov 2015;201(3):1253-1262.
- Andrikopoulos S, Fam BC, Holdsworth A, et al. Identification of ABCC8 as a contributory gene to impaired early-phase insulin secretion in NZO mice. The Journal of endocrinology. Jan 2016;228(1):61-73.
- Dokas J, Chadt A, Joost HG, Al-Hasani H. Tbc1d1 deletion suppresses obesity in leptin-deficient mice. Int J Obes (Lond). Aug 2016;40(8):1242-1249.
- Chadt A, Immisch A, de Wendt C, et al. "Deletion of both Rab-GTPase-activating proteins TBC1D1 and TBC1D4 in mice eliminates insulin- and AICAR-stimulated glucose transport [corrected]. Diabetes. Mar 2015;64(3):746-759.
- Plum L, Giesen K, Kluge R, et al. Characterisation of the mouse diabetes susceptibilty locus Nidd/SJL: islet cell destruction, interaction with the obesity QTL Nob1, and effect of dietary fat. Diabetologia. Jun 2002;45(6):823-830.
- Wuschke S, Dahm S, Schmidt C, Joost HG, Al-Hasani H. A meta-analysis of quantitative trait loci associated with body weight and adiposity in mice. Int J Obes (Lond). May 2007;31(5):829-841.
- Schmidt C, Gonzaludo NP, Strunk S, et al. A meta-analysis of QTL for diabetes-related traits in rodents. Physiol Genomics. Jun 12 2008;34(1):42-53.
- Leiter EH. Selecting the "right" mouse model for metabolic syndrome and type 2 diabetes research. Methods Mol Biol. 2009;560:1-17.
- Buchner DA, Nadeau JH. Contrasting genetic architectures in different mouse reference populations used for studying complex traits. Genome research. Jun 2015;25(6):775-791.
- Review Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells.[Front Endocrinol (Lausanne). 2...]Review Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells.Murakami T, Inagaki N, Kondoh H. Front Endocrinol (Lausanne). 2022; 13:869414. Epub 2022 Mar 31.
- Review Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus.[Int J Obes Relat Metab Disord....]Review Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus.Heilbronn L, Smith SR, Ravussin E. Int J Obes Relat Metab Disord. 2004 Dec; 28 Suppl 4:S12-21.
- Review Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus.[Ann N Y Acad Sci. 2002]Review Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus.Ravussin E, Smith SR. Ann N Y Acad Sci. 2002 Jun; 967:363-78.
- Review Adipose Tissue: Physiology to Metabolic Dysfunction.[Endotext. 2000]Review Adipose Tissue: Physiology to Metabolic Dysfunction.Richard AJ, White U, Elks CM, Stephens JM. Endotext. 2000
- Feeding the critically ill obese patient: a systematic review protocol.[JBI Database System Rev Implem...]Feeding the critically ill obese patient: a systematic review protocol.Secombe P, Harley S, Chapman M, Aromataris E. JBI Database System Rev Implement Rep. 2015 Oct; 13(10):95-109.
- Molecular links between Obesity and Diabetes: “Diabesity” - EndotextMolecular links between Obesity and Diabetes: “Diabesity” - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...