

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

This chapter is, in part, based on the previous version written by Prof. Rosalind Brown.
ABSTRACT
Thyroid disorders in infancy, childhood and adolescence represent common and usually treatable endocrine disorders. Thyroid hormones are essential for normal development and growth of many target tissues, including the brain and the skeleton. Thyroid hormone action on critical genes for neurodevelopment is limited to specific time window, and even a short period of deficiency of TH can cause irreversible brain damage. During the first trimester of pregnancy fetal brain development is totally dependent on maternal thyroid function. Congenital hypothyroidism is one of the most preventable causes of mental retardation, but early diagnosis is needed in order to prevent irreversible SNC damage. Today more than 70% of the babies worldwide are born in areas without an organized screening program. New insights about genetic causes, screening strategies and treatment of congenital hypothyroidism are reported. Hyperthyroidism in newborns is usually a transient consequence of transplacental passage of TSH receptor stimulating antibodies. Hypothyroidism can be detected in infants born to hyperthyroid mothers, due to transplacental passage of TSH receptor antibodies or hypothalamic-pituitary suppression. In childhood and adolescence autoimmune thyroid disease (AITD) as chronic lymphocytic thyroiditis and Graves’ disease account for the main cause of hypothyroidism and hyperthyroidism, respectively. Incidence of AITD increase from infancy to adolescence. Other autoimmune disorders are frequently associated. An increased risk of thyroid nodules and cancer is suggested. Differentiated thyroid cancer and medullary thyroid carcinoma in childhood and adolescence require specific expertise. Follow up programs are advised for high risk patients as long term survivors of childhood cancer. For complete coverage of this and related areas of Endocrinology, please visit our free online textbook, WWW.ENDOTEXT.ORG.
INTRODUCTION
Thyroid hormone is essential for the growth and maturation of many target tissues, including the brain and skeleton. As a result, abnormalities of thyroid gland function in infancy and childhood result not only in the metabolic consequences of thyroid dysfunction seen in adult patients, but in unique effects on the growth and /or maturation of these thyroid hormone-dependent tissues as well. In most instances, there are critical windows of time for thyroid hormone-dependent development and so the specific clinical consequence of thyroid dysfunction depends on the age of the infant or child. For example, newborn infants with congenital hypothyroidism frequently have hyperbilirubinemia, and delayed skeletal maturation, reflecting immaturity of liver and bone, respectively, and they are at risk of permanent mental retardation if thyroid hormone therapy is delayed or inadequate; their size at birth, however, is normal. In contrast, hypothyroidism that develops after the age of three years (when most thyroid hormone-dependent brain development is complete) is characterized predominantly by a deceleration in linear growth and skeletal maturation but there is no permanent effect on cognitive development. In general, infants with severe defects in thyroid gland development or inborn errors of thyroid hormonogenesis present in infancy whereas babies with less severe defects or acquired abnormalities, particularly autoimmune thyroid disease, present later in childhood and adolescence. In the newborn infant, thyroid function is influenced not only by the neonate ’ s own thyroid gland but by the transplacental passage from the mother of factors that affect the fetal thyroid gland.
In the last several decades, there have been exciting advances in our understanding of fetal and neonatal thyroid physiology, and screening for congenital hypothyroidism has enabled the virtual eradication of the devastating effects of mental retardation due to sporadic congenital hypothyroidism in most developed countries of the world. In addition, advances in molecular biology have led to new insights regarding the early events in thyroid gland embryogenesis and mechanisms of thyroid action in the brain. At the same time, the molecular basis for many of the inborn errors of thyroid hormonogenesis and thyroid hormone action is being unraveled. However, new questions and new challenges arise. In particular, the survival of increasingly small and premature fetuses has resulted in a growing number of neonates with abnormalities in thyroid function and a continuing controversy as to which of these infants require therapy. This chapter will focus on current concepts regarding the ontogenesis of thyroid function in the fetus and will review the major disorders of thyroid gland function in infants and children.
ONTOGENESIS OF THYROID FUNCTION IN THE FETUS AND INFANT
The ontogeny of mature thyroid function involves the organogenesis and maturation of the hypothalamus, pituitary, and thyroid glands as well as the maturation of thyroid hormone metabolism and thyroid hormone action. The placenta also plays a key role in the transfer of hormones and factors other than T4 that impact on thyroid function. In the first half of pregnancy, maternal T4 provides an important source of hormone for the developing fetus. Much of our knowledge derives from work in animal models, particularly sheep and rat. In interpreting these data, it is important to remember potential limitations in these models because of differences both in the structure of the placenta and timing of maturation. For example, the rat thyroid gland is much less mature at birth than its human counterpart and significant maturation of the thyroid gland and of the hypothalamic-pituitary-thyroid axis in this species occurs in the first 2 or 3 weeks after birth in the absence of placental or maternal influence, as compared with the third trimester in human infants.
Thyroid Gland Embryogenesis
Thyroid gland development is extensively reviewed in an earlier chapter and is shown diagrammatically in Figure 1. In brief, the thyroid gland is derived from the fusion of a medial outpouching from the floor of the primitive pharynx, the precursor of the thyroxine (T4)-producing follicular cells, and bilateral evaginations of the fourth pharyngeal pouch, which gives rise to the parafollicular, or calcitonin (C) secreting cells. Commitment towards a thyroid-specific phenotype as well as the growth and descent of the thyroid anlage into the neck results from the coordinate action of a number of transcription factors, including thyroid transcription factor 1 (TTF1, now called NKX2 (1), TTF2 (now called FOXE1) and PAX8 (1,2). Because these transcription factors are also expressed in a limited number of other cell types, it appears to be the specific combination of transcription factors and possibly non-DNA binding cofactors acting coordinately that determine the phenotype of the cell.
Other transcription factors and growth factors that play a role in early thyroid gland organogenesis include HHEX1, HOXA3 (3) and members of the fibroblast growth factor family, i.e., FGF10, but the initial inductive signal is unknown. A role of the neighboring heart primordium in the specification of the thyroid anlage has been postulated. Studies of cadherin expression suggest that the caudal translocation of the thyroid anlage may also arise indirectly, as a result of the growth and expansion of adjacent tissues, including the major blood vessels (4). In late organogenesis, the sonic hedgehog (SHH) gene and its downstream target TBX1 appear to play an important role in the symmetric bilobation of the thyroid (5); SHH also suppresses the ectopic expression of thyroid follicular cells (6).
During caudal migration the pharyngeal region of the thyroid anlage contracts to form a narrow stalk, known as the thyroglossal duct, which subsequently atrophies. Usually no lumen is left in the tract of its descent but, occasionally, an ectopic thyroid and/or persistent thyroglossal duct or cyst form if thyroid descent is abnormal.
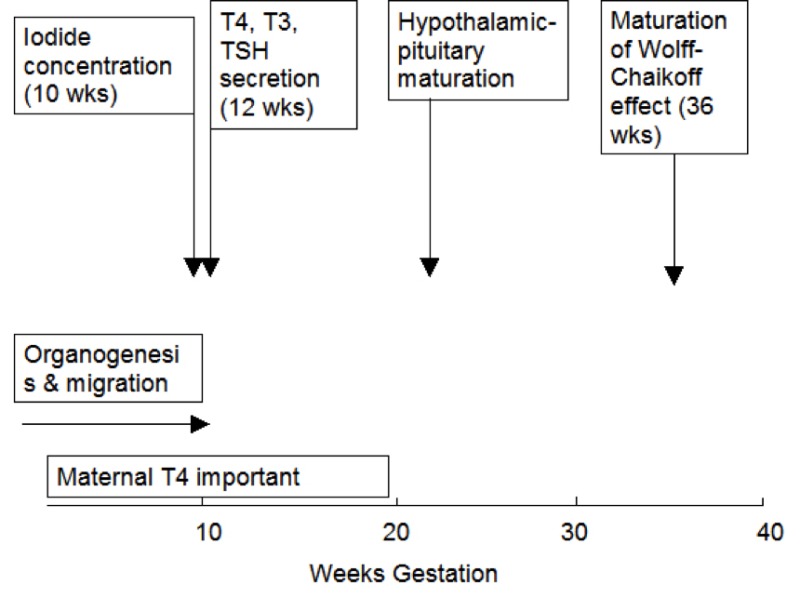
Figure 15- 1Approximate timing of thyroid gland maturation in the human fetus.
In the human, embryogenesis is largely complete by 10 to 12 weeks gestation. At this stage, tiny follicle precursors can be seen, iodine binding can be identified and thyroglobulin (Tg) detected in follicular spaces (7,8) . Thyroid hormones are detectable in fetal serum by gestational age 11 to 12 weeks with both thyroxine (T4) and triiodothyronine (T3) being measurable. However, as discussed in further detail below, it is likely that a fraction of the hormones detectable at this early stage is contributed by the mother through transplacental transfer. Thyroid hormones continue to increase gradually over the entire period of gestation as does serum thyroxine-binding globulin (TBG) (9,10) . TBG is present at levels of 100 nmol/L (5 mg/L) at gestational age 12 weeks and progressively increases up to the time of birth, reaching concentrations of 500 nmol/L (25 mg/L). The serum TBG concentrations are higher in the infant then in adult humans as a consequence of placental estrogen effects on the fetal liver. In addition to the increase in total T4 there is also a progressive increase of the free T4 concentration indicating a maturation of the hypothalamic- pituitary- thyroid axis. The increased total T4 / thyrotropin (TSH) and free T4 /TSH ratios also indicate an increased ability of the thyroid gland to respond to TSH due to upregulation of the TSH receptor (11). Whereas the TBG and total T4 levels rise throughout gestation, the concentrations of free T4, and TSH rise until 31 to 34 weeks, declining thereafter to term (12).
Tg can be identified in the fetal thyroid as early as the 5th week, and is certainly present in follicular spaces by 10 to11 weeks, but maturation of Tg secretion takes much longer and it is not known when circulating Tg first appears in the fetal serum (not shown). By the time of gestational age 27 to 28 weeks, however, Tg levels average approximately 100 mg/L, much higher than in the adult and they remain approximately stable until the time of birth (13,14) . Iodide concentrating capacity can be detected in the thyroid of the 10 to 11 week fetus, but maturation of the Wolff-Chaikoff effect (reduction of iodide trapping in response to excess iodide) does not appear until 36 to 40 weeks gestation. Thus the premature fetus is more sensitive than the full term neonate to the thyroid-suppressive effects of iodine exposure.
The Hypothalamic-pituitary Axis
TSH is detectable at levels of 3 to 4 mU/L at gestational age 12 weeks and increases moderately over the last two trimesters to levels of 6 to 8 mU/L (8,9).The maturation of the negative feedback control of thyroid hormone synthesis is observed by approximately mid-gestation (Figure 1), with elevated serum TSH concentrations being observed in hypothyroid infants as early as 28 weeks (8). When TSH-Releasing Hormone (TRH) is given to mothers, a rise in TSH in the fetal circulation has been noted as early as 25 weeks gestation (15). It is of interest that the fetal TSH increment after TRH is greater than is the paired-maternal response, a consequence either of enhanced TSH release or impaired TSH degradation, perhaps due to immaturity of the hepatic glycoprotein metabolic clearance system. Similarly TSH is reduced in the cord serum of infants with neonatal thyrotoxicosis due to the transplacental passage of thyroid-stimulating antibodies from mothers with Graves’ disease as early as the end of the 2nd trimester.
Serum levels of TRH are higher in the fetal circulation than in maternal blood, the result both of extrahypothalamic TRH production (placenta and pancreas) and the decreased TRH degrading-activity in fetal serum. The physiological significance of these increased levels of TRH in the fetal circulation is not known.
Maturation Of Peripheral Thyroid Hormone Metabolism
As discussed in an earlier chapter, there are three iodothyronine deiodinases involved in the activation and inactivation of thyroid hormone. All three are coordinately regulated during gestation and function to closely regulate the supply of T3 to developing tissues while at the same time protecting the fetus against the effects of excess thyroid hormone. The physiological rationale for the maintenance of reduced circulating T3 concentrations throughout fetal life is still unknown, but it has been suggested that its function may be to avoid tissue thermogenesis and potentiate the anabolic state of the rapidly growing fetus while at the same time permitting highly regulated, tissue- specific maturation in an orderly, temporal sequence.
The seleno-enzyme type 1 iodothyronine deiodinase (D1), an important activating enzyme in adult life, is low throughout gestation. In addition to catalyzing T4 to T3 conversion, D1 catalyzes the inactivation of the sulfated conjugates of T4. As a consequence, circulating T3 concentrations in the fetus are quite low whereas the serum levels of the biologically inactive isomer reverse T3 and of T3 sulfate are increased (10). Unlike D1, both the Type 2 deiodinase (D2), an activating enzyme and D3, an inactivating enzyme are present in fetal brain as early as 7 weeks ’ gestation (16) . D2 converts T4 to T3 while D3 converts T4 to reverse T3. D2 and D3 are the major isoforms present in the fetus and are especially important in defining the level of T3 in the brain and pituitary. The highest concentration of D2 is in brain, pituitary, placenta and brown adipose tissue. D3 is present in many fetal tissues, most prominently the brain, uteroplacental unit, skin, and gastrointestinal tract (17). This is consistent with the key role of D3 in protecting fetal tissues against high maternal T4 concentrations present either in the placenta or in amniotic fluid.
In the presence of hypothyroidism, D2 activity increases while D3 decreases These coordinate activities have been found to be critically important in defending the rat fetus against the effects of fetal hypothyroidism as long as maternal T4 levels are maintained at normal concentrations (18, 19). Despite the low levels of circulating T3, brain T3 levels are 60-80% those of the adult by fetal age 20-26 weeks (20). Thus, whereas the physiological interrelationships between the various deiodinases in the fetus and placenta seem designed to maintain circulating T3 concentrations at a reduced level, specific mechanisms have evolved for maintaining brain T3 concentrations so that normal development can proceed.
Role of the Placenta
Contributions of the maternal thyroid to fetal thyroid economy.
In the human infant under normal circumstances, the placenta has only limited permeability to thyroid hormone and the fetal hypothalamic-pituitary-thyroid system develops relatively independent of maternal influence. Placental thyroid hormone transporters, thyroid hormone binding proteins, iodothyronine deiodinases, sulfotransferases and sulfatases regulate the transport of maternal thyroid hormones to the fetus (20a,20b). The transport of iodine through the placenta is also important as the organ has shown to actively concentrate the anion (20c).
The human placenta expresses iodothyronine Type 2 deiodinase I (D2) (which activates T4 to T3) and Type 3 (D3) (which inactivates T4 and T3). Maternal T4 is metabolized by D3 having 200 times the activity of D2 (20b). Both D2 and D3 activity decrease with advancing gestation (20b). Thus, the relative impermeability of the human placenta to thyroid hormone is due to the presence of D3 which serves to inactivate most of the thyroid hormone presented from the maternal or fetal circulation. The iodide released in this way can then be used for fetal thyroid hormone synthesis. Iodine is actively transported from the maternal circulation to the fetus through the placenta that express placental sodium iodide transporter (NIS) (20c,20d). NIS actively concentrates Iodine. NIS protein levels are significantly correlated with gestational age during early pregnancy and increase with increased placental vascularization (20e).
Interest in the potential role of maternal T4 in the fetal thyroid economy was reawakened with the recognition that in infants with the congenital absence of thyroid peroxidase, the cord serum concentration of T4 is nonetheless between 25 and 50% of normal (21). Since these infants are completely unable to synthesize T4, the measured hormone must be maternal in origin. Similar results are obtained in retrospective studies of cord serum in infants with sporadic congenital athyreosis. This maternal T4 disappears rapidly from the newborn circulation with a half-life of approximately 3 to 4 days.
There is also evidence that maternal-fetal T4 transfer occurs in the first half of pregnancy, when fetal thyroid hormone levels are low (19,22). Low concentrations of T4, presumably of maternal origin, have been detected in human embryonic coelomic fluid as early as 6 weeks gestation and in fetal brain as early 10 weeks gestation prior to the onset of fetal thyroid function indicating its maternal origin (22a-22f). Furthermore, both D2 and D3 activity as well as thyroid hormone receptor (TR) isoforms are present in low concentrations in human fetal brain from the mid first trimester, indicating that the machinery to convert T4 to T3 and to respond to T3 is present. The mechanisms of actions of thyroid hormones in the developing brain are mainly mediated through two ligand activated thyroid hormone receptor isoforms (22b,22c). There is also an important role for the thyroid hormone transporters in one or more of these processes (22g).
Between 6-12 weeks gestation, if maternal total T4 concentration is 100%, the total T4 concentration in the coelomic fluid would be 0.07% and T4 in the amniotic cavity as little as 0.0003-0.0013% of maternal total T4 concentrations. Fetal circulating concentrations of T3 are at least 10 fold lower than T4, whereas by fetal age of 20-26 weeks T3 levels in the fetal brain are 68-80% of the adult brain (20). Unlike adults, the proportion of free unbound T4 is also higher than bound T4 in early gestation. Free T4 levels are determined by the fetal concentrations of the thyroid hormone binding proteins in the circulation and the amount of maternal T4 crossing the placenta (7-9). It seems likely that when fetal thyroid function is normal, the net flux of T4 from mother to fetus is relatively limited. However, when the fetus is hypothyroxinemic, there is significant bulk transfer of T4 to the fetal circulation. This can occur both at the level of the placental maternal capillary interface and via uptake of thyroid hormone from the amniotic fluid through the immature epidermis. T4 uptake by the fetus can also occur via fetal ingestion of amniotic fluid. While the T4 concentrations in amniotic fluid appear modest, the fraction of T4 free in amniotic fluid is approximately ten-fold higher than that of serum and thus the free T4 concentration in amniotic fluid is approximately equal to that in fetal serum at 20 weeks gestation. It has been shown on numerous occasions in both animals and humans that amniotic fluid iodothyronine concentrations reflect those in the maternal circulation (23).
Placenta is permeable to TRH (15) and to immunoglobulins G (IgG) from midgestation. At the time of delivery, cord blood TPOAb correlate with maternal TPOAb concentrations (23a). Maternal passage of TPOAb and TgAb are not associated with thyroid fetal dysfunction. On the contrary, maternal TSH receptor antibodies (both stimulating and blocking) can be dangerous for the fetus and the newborn.
Fetal and neonatal hyperthyroidism can be caused by transplacental passage of TSH receptor antibodies (TRAb), whereas hypothyroidism can be due to transpancental passage of blocking TSH receptor antibodies, from mothers with severe Graves’ disease or severe hypothyroidism due to chronic lymphocytic thyroiditis.( The placenta is also permeable to certain drugs (15). Thus, the administration to the mother of excess iodide, drugs (especially propylthiouracil or methimazole), can affect thyroid function in the fetus and the newborn.
Role of Maternal Thyroid Hormone for fetal brain development and neurocognitive development in the offspring
The essential role that thyroid hormones (TH) play on the fetal brain development starts long before the onset of fetal thyroid function (22-22a). Thus, during the first trimester of pregnancy, fetal brain development is totally dependent on maternal thyroid function. Because the action of TH on critical genes for fundamental neurobiological processes is limited to specific time window, even a short period deficiency of TH may cause permanent brain damage. TH deficiency may affect neuronal cell differentiation and migration, axonal and dendritic outgrowth, myelin formation and synaptogenesis (22b-22f). It is well known that severe Iodine deficiency during pregnancy causes inadequate thyroid hormone production and irreversible brain damage known as cretinism, still endemic in many areas of the world (23b). None of the neurological features of severe endemic cretinism (24) due to iodine deficiency are found in infants with sporadic congenital hypothyroidism whose mothers have normal thyroid function and who receive early and adequate postnatal treatment. Similarly, impaired hearing, when found is much milder and less frequent (25). This would appear to provide unequivocal evidence that the neurological damage sustained by infants with endemic cretinism can be largely prevented by maternal T4. In addition to endemic cretinism, significant developmental delay despite early and adequate postnatal therapy has also been reported in other models of combined maternal-fetal hypothyroidism, such as materno-fetal POU1F1 deficiency (26) and TSH receptor blocking antibody-induced congenital hypothyroidism (27).
In iodine sufficient areas the main cause of maternal thyroid dysfunction (hypothyroidism, subclinical hypothyroidism or hypothyroxinemia) is thyroid autoimmunity, detectable in up to 17% of women (27a). Several studies reported on the consequences of maternal thyroid dysfunction in the progeny. Studies in children born to women with non-iodine deficient hypothyroidism during pregnancy (28,29,29a,29b) as well as in children from hypothyroxinemic mothers (30,30a-30e) have been published. Different parameters and different periods of pregnancy (i.e., increased TSH, low T4, presence or absence of autoimmunitity, prevalent obstetrical or developmental outcome) were analyzed, reporting conflicting results and conclusions.
Impairment in psychomotor development in the offspring of pregnant women with thyroid dysfunction was first reported by Man et al (28). They examined 131 hypothyroxinemic untreated pregnant women and found 36% of their 7-year-old children scored in the dull normal range or below compared to 16% of children of euthyroid mothers (28). Haddow detected a seven point IQ deficit in 7 to 9 year old children whose mothers were retrospectively found to have been hypothyroid at 17 weeks gestation (29). Accordingly, Pop demonstrated that even babies born to women whose free T4 levels were in the lowest 10% of normal at 12 weeks gestation had a measurable impairment in psychomotor development at 2 years of age as compared with the rest of the population, but this effect was not observed if maternal thyroid function was normal at 32 weeks (30). At variance with the aforementioned studies, Liu and more recently, Momotami failed to demonstrate any IQ deficit in babies born to hypothyroid mothers as long as the hypothyroidism was corrected by the end of the second trimester (31a, 31b). Similar results were obtained by Downing et al in 3 children born after severe feto-maternal hypothyroidism due to TSH receptor blocking antibodies (31c). Attention deficit disorder (30f,30g) autistic symptoms in offspring (30h) and schizophrenia in later life (30K) have also been associated with maternal hypothyroxinemia. Attention deficit disorder was previously noted in offspring from mothers with thyroid autoimmunity (30i). Children from mothers with anti-thyroid peroxidase antibodies have been found to have intellectual impairment in early infancy (30j) and a reduced childhood cognitive performance at age 4 and 7 and sensineural hearing loss at both ages (30l). An interesting association study, derived from the Rotterdam cohort, (the population based prospective study from Rotterdam (Generation R) for the first time analyzed the effects of maternal thyroid function on brain morphology of the offspring. In this study 3839 mother-child pairs were included. Maternal serum samples were taken before 18 weeks of gestation (9-18w). MRI were performed in 646 children (mean age 8 years) and IQ determined at mean age 6 years. They found that both high maternal and low FT4 showed an inverted U shaped association with child IQ (-1.4-3.8 points), child grey matter volume and cortex volume (32c). Recently, in the prospective double blind randomized controlled antenatal thyroid screening study (CATS), levothyroxine treatment was started from the 13th week of gestation if serum TSH was >97.5th percentile and/or FT4 was <2.5th percentile. The outcome was the IQ in the offspring at 3 years. No significant differences in IQ values were found between 390 children of treated mothers compared to 404 children of untreated mothers (32).
The incidence of maternal hypothyroidism during pregnancy (3 per 1000 in iodine-sufficient populations (33) is almost ten times that of congenital hypothyroidism for which routine population screening is widespread. Because maternal hypothyroidism has been associated not only with potential adverse effects on fetal brain development but an increased risk of preterm delivery and of miscarriage as well (33a ), some have argued that all pregnant women should be screened for hypothyroidism, a position that has been endorsed by some but not other professional societies.
Updated guidelines for the management of thyroid disease during pregnancy have been recently released from ATA (33b).
THYROID FUNCTION IN THE NEONATE, THE INFANT, AND DURING CHILDHOOD
The Full-term Neonate
Marked changes occur in thyroid physiology at the time of birth in the full term newborn. One of the most dramatic changes is an abrupt rise in the serum TSH which occurs within 30 minutes of delivery, reaching concentrations as high as 60 to 70 mU/L (8). This causes a marked stimulation of the thyroid and an increase in the concentrations of both serum T4 and T3 (34). These consist of an approximate 50% increase in the serum T4 and an increase of three- to four-fold in the concentration of serum T3 to adult levels at 1 to 4 days of life. Serum levels of T4, free T4 and TBG remain elevated over cord levels at 7 days of postnatal life (Figure 2), decreasing thereafter. The T3 concentration rises strikingly at Day 7, and continues to rise for the first 28 days. Opposite effects are noted in the reverse T3 levels and T3 sulfate.
Studies in experimental animals suggest that the increase in TSH is a consequence of the relative hypothermia of the ambient extrauterine environment. However, while a significant portion of the marked increase in T3 from its low basal levels in cord serum can be explained by the abrupt increase in TSH, the simultaneous fall in reverse T3 and T3 sulfate are consistent with an increase in D1 activity occurring at the same time. D2 has been identified in human brown adipose tissue as well as brain and the acute increase in T3 in adipose tissue at birth is required for optimal uncoupling protein synthesis and thermogenesis (35,36).
Premature Infants
Thyroid function in the premature infant reflects, in part, the relative immaturity of the hypothalamic-pituitary-thyroid axis that is found in comparable gestational age infants in utero. Following delivery, there is a surge in T4 and TSH analogous to that observed in term infants, but the magnitude of the increase is less in premature neonates (8). In infants <31 weeks, the circulating T4 concentration may not increase and may even fall in the first 1 to 2 weeks of life (37) (Figure 2). This decrease in the T4 concentration is particularly significant in very premature infants, in whom the serum T4 may occasionally be undetectable. In most cases, the total T4 is more affected than the free T4 (38), a consequence of abnormal protein binding and/or the decreased TBG in these babies with immature liver function.

Figure 15-2
Postnatal changes in of T4, free T4, TBG, T3, rT3 and TSH according to gestational age. Values determined in babies born at gestational age of 23-27, 28-30, 31-34 and 37 weeks or more are reported. Note the increase in T4, free T4 and TBG in the full term infant in the first week of life. T3 also rises strikingly, while rT3 and TSH decline. The increase in T4 and free T4 is blunted in infants
The causes of the decrease in T4 observed postnatally in premature infants are complex. In addition to the clearance of maternal T4 from the neonatal circulation, preterm babies have decreased thyroidal iodide stores (39) (a problem of particular significance in borderline iodine-deficient areas of the world), they are frequently sicker than their more mature counterparts, are less able to regulate iodide balance, and they may be treated by drugs that affect neonatal thyroid function (particularly dopamine and steroids). In addition, since the capacity of the immature thyroid to adapt to exogenous iodide is reduced, there is an increase in sensitivity to the thyroid-suppressive effects of excess iodide found in certain skin antiseptics and drugs to which these babies are frequently exposed (see below).
Despite the reduced total T4 observed in some preterm babies, the TSH concentration is not significantly elevated in most of these infants. In some babies, transient elevations in TSH are seen, the finding of a TSH concentration >40 mU/L being more frequent the greater the degree of prematurity. Frank et al found, for example, that the prevalence of a TSH concentration >40 mU/L in very low birth weight, (<1.5 kg), i.e., very premature, infants was 8-fold higher and in low birth weight, (1.5 kg-2.5 kg) neonates 2-fold higher than the prevalence in term babies (40). Whereas in some cases, an elevated TSH concentration may reflect true primary hypothyroidism, in other instances this increase in TSH may reflect the elevated TSH observed in adults who are recovering from severe illness. Such individuals may develop transient TSH elevations that are associated with still reduced serum T4 and T3 concentrations. These have been interpreted as reflecting a “ re-awakening ” of the illness-induced suppression of the hypothalamic pituitary axis. As the infant recovers from prematurity associated illnesses such as respiratory distress syndrome (RDS), a recovery of the illness-induced suppression of the hypothalamic- pituitary- thyroid axis would also occur.

Figure 15-3
Cord blood levels of T4, free T4, TBG, T3, reverse T3 and TSH in the human infant. Note the low T3 and high reverse T3 concentrations as well as the discrepancy between the total T4 and free T4 levels in very premature babies. (Redrawn from Williams et al (10). See text for details).
Somewhat surprisingly, given the relative immaturity of the thyroid gland, serum Tg concentrations are higher in the premature than in the full term infant (41), particularly in those who are sick with respiratory distress syndrome. In view of the attenuated postnatal TSH rise in the latter babies, it is likely that impaired clearance and/or degradation of this glycoprotein from the circulation rather than increased secretion plays an important role.
Small-for-gestational-age (SGA) Infants
SGA infants have significantly higher TSH and lower total and free T4 values than do infants of normal weight (42). This can be related to the severity of the malnutrition in these infants, as well as to fetal hypoxemia and acidemia. Impaired placental perfusion and chronic starvation may also play a role. This pattern of reduced T4 and elevated TSH differs from the response to starvation in older individuals and healthy adults in whom TSH is reduced. The explanation for the relatively higher TSH in duch infants is not known.
Infants and Children
Following the acute perturbations of the neonatal period there is a slow and progressive decrease in the concentrations of T4, free T4, T3 and TSH during infancy and childhood (43). Younger children tend to have slightly higher serum concentrations of T3 and TSH, so age-specific normative values should always be consulted. The serum concentration of reverse T3 remains unchanged or increases slightly. Serum Tg levels also fall over the first year of life reaching concentrations typical of adults by about 6 months of age. Another important aspect of thyroid physiology in the infant and child is the markedly higher T4 turnover in this age group relative to that in the adult. In infants, T4 production rates are estimated to be on the order of 5 to 6 mcg/kg per day decreasing slowly over the first few years of life to about 2 to 3 mcg/kg/day at ages 3 to 9 years. This is to be contrasted with the production rate of T4 in the adult which is about 1.5 mcg/kg/day. The size of the infant thyroid gland increases quite slowly. The thyroid gland of the newborn weighs approximately 1 gram and increases about 1 gram per year until age 15 when it has achieved its adult size of about 15 to 20 g. In general, the size of the thyroid lobe is comparable to that of the terminal phalanx of the infant or child’s thumb.
THYROID DISEASE IN INFANCY
Congenital hypothyroidism
Non endemic congenital hypothyroidism is one of the commonest treatable causes of mental retardation. The association between goitrous hypothyroidism and mental retardation was first noted more than 400 years ago by Paracelsus in 1527, and Thomas Curling first described sporadic nongoitrous hypothyroidism in 1850. However, despite the demonstration by Murray in 1891 that thyroid extract could ameliorate many of the features of untreated cretinism, it was not until the 1970’ that the importance of early treatment in diminishing the neuro-psychological abnormalities of congenital hypothyroidism was demonstrated convincingly (45). The development by Dussault et al of a sensitive and specific radioimmunoassay for the measurement of T4 in dried whole blood eluated from filter paper (and later tests for T4 and TSH using 1/8 ″ discs) provided the technical means to screen all newborns for congenital hypothyroidism prior to the development of clinical manifestations (46). Thus, as summarized by Delange, congenital hypothyroidism includes all the characteristics of a disease for which screening is justified: 1) it is common (4-5 times more common than phenylketonuria for which screening programs were initially developed); 2) to prevent mental retardation, the diagnosis must be made early, preferably within the first few days of life; 3) at that age, clinical recognition is difficult if not impossible; 4) sensitive, specific screening tests and 5) simple, cheap effective treatment are available; and 6) the benefit-cost ratio is highly favorable (approximately 10/1, a ratio that does not include the loss of tax income that would result from impaired intellectual capacity in the untreated, but non-institutionalized, person) (47). Since the development of the first pilot screening program for the detection of congenital hypothyroidism in Quebec in 1972, newborn screening programs have been introduced throughout the industrialized nations and are under development in many other parts of the world. It has been estimated that as of 1999, some 150 million infants had been screened for congenital hypothyroidism worldwide with 42,000 cases detected (46). Although there continues to be some disagreement as to whether minor neuro-intellectual sequelae remain in the most severely affected infants, accumulating evidence suggests that a normal outcome is possible even in the latter group of babies as long as treatment is started sufficiently early and is adequate (48-50). Certainly, the main objective of screening, the eradication of mental retardation, has been achieved.
National screening programs are well organized in many developed countries. However, it must be emphasized that approximately 71% of babies worldwide are not born in an area with an established national screening program for CH. The economic burden of disability owing to congenital hypothyroidism is still a significant public health challenge (50a).
The prevalence of CH was approximately 1:7000 to 1:10000 in the prescreening era and decreased to1;3000 to 1.4000 in the 1970s and 1980s when the screening programs were applied. Rates ranging from 1:1400 to 1:2800 have been recently reported by screening programs in USA, Canada, Italy, Greece, and New Zealand (50b).
Lower TSH cut off values used in the screening programs and changes in birth population partially explained the higher incidence reported. Lower cutoff values for TSH have been adopted in many countries over the years, leading to the identification of milder forms of CH essentially with eutopic thyroid gland (thyroid in situ). Ford and LaFranchi in 2014 (50a) found that lowering the TSH cutoff value from greater than 20-25 uU/mL to greater than 6-10 approximately doubled the incidence of CH. A study from Italy reported that 21.6% of babies with permanent CH had TSH value at screening less than 15 uU/mL (applied between 2000 and 2006, cutoff TSH value ranged from 15 to 7uU/mL in different regions). The frequency of thyroid dysgenesis in this group was 19.6% and TSH levels at confirmation ranged from 9.9 to 708 uU/mL .It is important to remember that in this study TSH value at screening does not discriminate between transient and permanent forms of CH (50c).
Harris and Pass reported that CH incidence increased from 1:3373 in 1978 to 1:1415 in 2005 (50d). Changes in the demographics of the birth population in New York partially explained the increased incidence of CH. They found a 23% increase with a birth weight < 1500 gr., 50% increase of twin/multiple births, 41% increase in mothers >30 years of age (50d). Also changes in percentage of races or ethnicity of newborns play a role, as shown in the State of California. In this study, the incidence of CH in Asian Indian is reported to be 1:1200 and in Hispanic 1:1600, versus 1:11000 in Non Hispanic Black (50e). A further study from the Italian Study Group, based on data from the Italian National Registry from 1987 to 2008 showed an increased incidence of both permanent and transient CH, in more recent years (50f). The authors investigated trends in the incidence of CH between the period 1987-1998, and 1999-2008. They found an increasing of 38% (from 1:3200 to 1:2320) of the incidence of permanent CH and of 54% (from1:3000 to 1:1940) including the transient forms in the period 1999-2008. The most important factor was the lowering of cutoff TSH values (from greater than 20 to 7/15 uU/ml since 1999. Moreover an increment of 58% of preterm babies with permanent CH was also reported in the second period. Permanent CH due to thyroid dysgenesis had a slight increase, being the great majority of cases presented with normal/hyperplastic thyroid.
A national study from France, including 6622 cases of CH identified from 1982 to 2012 showed that the incidence rate CH due to eutopic glands increased by 4.4 fold in this period, regardless of the screening method adopted. Interestingly, also severe eutopic forms of CH increased by 2.1%. The incidence of dysgenesis did not change (50g).
Screening Strategies
Screening for primary CH worldwide should be performed on the basis of national resources. The aim of neonatal screening is the earliest identification of any form of congenital hypothyroidism, but particularly those patients with severe hypothyroidism in whom disability is greatest if not treated. The identification of Central Congenital Hypothyroidism (CCH) by screening programs is under debate. Two screening strategies for the detection of congenital hypothyroidism have evolved. In the primary T4/backup TSH method, still favored in much of North America and the Netherlands, T4 is measured initially while TSH is checked on the same blood spot in those specimens in which the T4 concentration is low. In the primary TSH approach, favored in most parts of Europe and Japan, blood TSH is measured initially.
A primary T4/backup TSH program will detect overt primary hypothyroidism, secondary or tertiary hypothyroidism, babies with a low serum T4 level but delayed rise in the TSH concentration, TBG deficiency and hypothyroxinemia; this approach may, however, miss subclinical hypothyroidism. A primary TSH strategy, on the other hand, will detect both overt and subclinical hypothyroidism, but will miss secondary or tertiary hypothyroidism, a delayed TSH rise, TBG deficiency and hypothyroxinemia. There are fewer false positives with a primary TSH strategy. Both programs will miss the rare infant whose T4 level on initial screening is normal but who later develops low T4 and elevated TSH concentrations. This pattern has been termed “atypical” congenital hypothyroidism or “delayed TSH” and is observed most commonly in premature babies with transient hypothyroidism or infants with less severe forms of permanent disease.
In a few regions, a second routine specimen is collected from all births at 2-4 weeks of age (51). Results from the Northwest Regional Screening program, coordinated in Oregon, (USA), that applied this method, have recently been published (51a). In 2014 the European Society for Pediatric Endocrinology, (ESPE) on behalf of all the scientific societies of pediatric endocrinologists worldwide (ESPE,PES, SLEP, JSPE, APEG, APPES, ISPAE) published updated guidelines about screening, diagnosis, and management of congenital hypothyroidism (51b, 51c).
According to the ESPE guidelines, the most sensitive test for detecting primary CH is the determination of TSH concentration that detects primary CH more effectively than primary T4 screening (51b,51c). Primary T4 screening with confirmatory TSH testing can detect some cases of CCH, but some cases of mild CH can be missed, depending on the cutoff T4 value used.
When available, screening strategies for the identification of CCH are: a) a combination of primary T4 and primary TSH screening, b) a combination of primary T4 screening with secondary TSH testing followed by T4 binding protein determination (TBG). The last one is employed by the Netherlands where, in addition to a primary T4/backup TSH approach, TBG is assessed in those filter paper specimens with the lowest 5% of T4 values (52). The T4/TBG ratio is used as an indirect reflection of the free T4, which is difficult to be measured directly in dried blood spots. This approach has been reported to result in improved sensitivity and specificity in detecting milder cases of primary congenital hypothyroidism that might otherwise be missed. An additional reported advantage was the identification of >90% of infants with central hypothyroidism compared with only 22% with primary T4 screening and none with a primary TSH approach. Since on subsequent testing > 80% of the babies with central hypothyroidism had multiple pituitary hormone deficiencies, a disorder associated with high morbidity and mortality for which effective treatment exists (53,53a), and in view of an apparent frequency (1 in 16,000) similar to that of phenylketonuria (1 in 18,000), the authors have argued that the goals of newborn thyroid screening should be extended to include the detection of babies with central hypothyroidism.
Recently a primary FT4 and TSH strategy was applied in Kanagawa Prefecture in Japan. A different method to determine FT4, based on enzyme-immunometric assays in filter paper blood eluates was used. They found a CCH prevalence of 1:31000 infants (53b,53c).
Measurement of T4 and/or TSH is performed on an eluate of dried whole blood (DBS) collected on filter paper by skin puncture on day 1-4 of life. Primary CH screening has been shown to be effective for the testing of cord blood or the blood collected on filter paper after the age of 24 hours. Blood is applied directly to the filter paper and after drying the card is sent to the laboratory. The best time to collect blood for TSH screening is 48 to 72 hours of age. The practice of early discharge from the hospital of otherwise healthy full term infants has resulted in a greater proportion of babies being tested before this time. For example, it has been estimated that in North America 25% or more of newborns are now discharged within 24 hours of delivery and 40% in the second 24 hours of life (54). Because of the neonatal TSH surge and the dynamic changes in serum T4 and T3 concentrations that occur within the first few days of life, early discharge increases the number of false positive results. It is important that in the screening laboratory the results of TSH are interpreted in relation to time of sampling. Ethnicity seems to play a role in determining mean TSH values at birth (54a).
Physicians caring for infants need to appreciate that there is always the possibility for human error in failing to identify affected infants, whichever screening program is utilized. This can occur due to poor communication, lack of receipt of requested specimens, or the failure to test an infant who is transferred between hospitals during the neonatal period (55). Therefore if the diagnosis of hypothyroidism is suspected clinically, the infant should always be tested (Figure 5).
Similarly, as is obvious from the discussion earlier in the chapter, adult normative values, provided by many general hospital laboratories, differ from those in the newborn period and should never be employed. Normal values according to both gestational and postnatal age for cord blood T4, free T4, TBG, T3, reverse T3, and TSH up to 28 days of life (10) are shown in Figure 2. Normal serum levels of Tg in premature and full-term infants (13,14) and normal serum levels of free T4 and TSH in the first week of life (56) have also been published, though it should be noted that precise values may vary somewhat, depending on the specific assays used.

Figure 15-4
Three month old male infant who was diagnosed clinically when he presented with a history of poor feeding at 3 months of age. The child was born in Puerto Rico prior to the development of newborn screening. Note the dull face, periorbital edema and enlarged tongue.
Screening in special categories of neonates at risk of CH
Special categories of neonates with CH can be missed at screening performed at usual time, particularly preterm babies and neonates with serious illnesses and multiple births. Drugs used in neonatal intensive care (i.e., dopamine, glucocorticoids that suppresses TSH), immaturity of hypothalamic-pituitary thyroid axis, decreased hepatic production of thyroid binding globulin, reduced transfer of maternal T4, reduced intake of iodine or excess iodine exposure, fetal blood mixing in multiple births can affect the first sample, and in many center a second specimen is required to rule out CH. (See section thyroid function in infants for more details).
Preterm babies have a higher incidence of a unique form of hypothyroidism, characterized by a delayed elevation of TSH. These babies can later develop low T4 and elevated TSH concentrations. This pattern has been termed “atypical” congenital hypothyroidism or “delayed TSH”. Preterm babies with a birth weight of less than 1500 gr. have an incidence of congenital hypothyroidism of 1:300. Survival of even extremely premature babies (<28 weeks of gestation) is around 90% in developed countries, and the incidence of prematurity is around 11.5 % in US and 11.8 % worldwide. So, an increasing subpopulation of preterm babies and high risk newborns deserves a special sight about screening and follow up of CH.
In these categories a second specimen 2-6 weeks from the first (ESPE guidelines suggested at about 15 days, or after 15 days from the first) may be indicated: preterm neonates with a gestational age of less than 37 weeks, Low Birth Weight and Very Low Birth Weight neonates and ill and preterm neonates admitted to neonatal intensive care unit, specimen collection within the first 24 hours of life, and multiple births, particularly in the case of same sex twins. The interpretation of the screening results should consider the results of a multiple sampling strategy, the age of sampling and the maturity (GA/birth weight) of the neonate.
Two recent papers (56a,56b) showed that a second screen (using a lower TSH cutoff) is able to detect the delayed elevation of TSH that occurs in these babies. Vigone et al (56a) revaluated the children with a diagnosis of CH detected at second screen and treated with L-thyroxine after 2 years of age and found 24% of cases with permanent congenital hypothyroidism, 52% with transient hypothyroidism and 24% with persistent hypertropinemia. Neither screening nor confirmatory TSH levels were able to predict the thyroid function after 2 years of age in these children.
Timing of normalization of thyroid hormones is critical for brain development (56c) and treatment should be started immediately if DBS TSH concentration is 40 mUI/l or more, after baseline TSH and FT4 serum determination, because this value strongly suggests decompensated hypothyroidism (56d). If TSH is < 40 mUI/l the clinician may postpone treatment, pending the serum results, for 1-2 days. ESPE guidelines (51b,51c) suggest treatment should be started if venous TSH concentration is persistently >20 mUI/l, even if serum FT4 is normal. Overtreatment can be dangerous for neurocognitive outcome and should be avoided, individualizing the dosage.
It is still a matter of debate if treatment can be beneficial in otherwise healthy babies with venous TSH concentration between 6-20 mUI/l and FT4 concentration within the normal limits for age. In these cases, diagnostic imaging is recommended to try to establish a definitive diagnosis. If TSH concentration remains high for more than 3 or 4 weeks, it is possible (in discussion with the family) either starting LT4 supplementation immediately and retesting, off treatment, at a later stage, or retesting two weeks later without treatment. Waiting for larger studies that are able to answer to this question, and given the irreversibility of a possible harm to the child, treating during early childhood and revaluating the thyroid function after myelination of the central nervous system is completed (by 36 to 40 months of age) can be a prudent behavior (56e). LT4 treatment must be started immediately if FT4 or TT4 levels are low, given the known adverse effect of untreated decompensated CH on neurodevelopment and somatic growth.
CH is defined on the basis of serum FT4 levels as severe when FT4 is <5 pmol/l, moderate when FT4 is 5 to 10 pmol/l and mild when FT4 is 10 to 15 pmol/l respectively. Determination of serum thyroglobulin (Tg) is useful, if below the detection threshold, to suggest athyreosis or a complete thyroglobulin synthesis defect. Measurement of Tg is most helpful when a defect in Tg synthesis or secretion is being considered. In the latter condition the serum Tg concentration is low or undetectable despite the presence of a normal or enlarged, eutopic thyroid gland. Serum Tg concentration also reflects the amount of thyroid tissue present and the degree of stimulation. For example, Tg is undetectable in most patients with thyroid agenesis, intermediate in babies with an ectopic thyroid gland and may be elevated in patients with abnormalities of thyroid hormonogenesis not involving Tg synthesis and secretion. Considerable overlap exists, and so, the Tg value needs to be considered in association with the findings on imaging. In patients with inactivating mutations of the TSH receptor discordance between findings on thyroid imaging and the serum Tg concentration has been described in some but not all studies (56f).
Clinical findings are usually difficult to appreciate in the newborn period except in the unusual situation of combined maternal-fetal hypothyroidism. Many of the classic features (large tongue, hoarse cry, facial puffiness, umbilical hernia, hypotonia, mottling, cold hands and feet and lethargy), when present, are subtle and develop only with the passage of time. In addition to the aforementioned findings, nonspecific signs that should suggest the diagnosis of neonatal hypothyroidism include: prolonged, unconjugated hyperbilirubinemia, gestation longer than 42 weeks, feeding difficulties, delayed passage of stools, hypothermia or respiratory distress in an infant weighing over 2.5 kg (57). A large anterior fontanelle and/or a posterior fontanelle > 0.5 cm is frequently present in affected infants but may not be appreciated. In general, the extent of the clinical findings depends on the cause, severity and duration of the hypothyroidism. Babies in whom severe feto-maternal hypothyroidism was present in utero tend to be the most symptomatic at birth. Similarly, babies with athyreosis or a complete block in thyroid hormonogenesis tend to have more signs and symptoms at birth than infants with an ectopic thyroid, the most common cause of congenital hypothyroidism. Unlike acquired hypothyroidism, babies with congenital hypothyroidism are of normal size. However, if diagnosis is delayed, subsequent linear growth is impaired. The finding of palpable thyroid tissue suggests that the hypothyroidism is due to an abnormality in thyroid hormonogenesis or in thyroid hormone action.
Bone maturation reflects the duration and the severity of hypothyroidism. Signs of delayed epiphyseal maturation on knee x-rays, persistence of the posterior fontanelle, a large anterior fontanelle, and a wide sagittal suture all reflect delayed bone maturation. The absence of one or both knee epiphyses has been shown to be related to T4 concentration at diagnosis and to IQ outcome, and is thus a reliable index of intrauterine hypothyroidism.
Imaging Techiniques in CH
Imaging studies are helpful to determine the specific etiology of CH. Both scintigraphy and ultrasound (US) should be considered in neonates with high TSH concentrations. Ideally, the association of US and scintigraphy gives the best information in a child with primary hypothyroidism. Scintigraphy shows the presence/absence (athyreosis), position (ectopic gland, in any point from the foramen caecum at the base of the tongue to the anterior mediastinum) and rough anatomic structure of the thyroid gland.
US, in experienced hands, is a valid tool in defining size and morphology of a eutopic thyroid gland, however, US alone is less effective in detecting ectopic glands. Color Doppler US improves the effectiveness of US (57a).
It is important to remember that an attempt to obtain an imaging of the thyroid in a newborn should never delay the initiation of treatment. Scintigraphy should be carried out within 7 days of starting LT4 treatment. Scintigraphy may be carried out with either 10-20 MBq of technetium-99m (99mTc) or 1-2 MBq of iodine-123 (I123). Tc is more widely available, less expensive, and quicker to use than I 123. Scintigraphy with I123, if available, is usually preferred because of the greater sensitivity and because, I123, unlike of technetium-99 is organified. Therefore, imaging with this isotope allows quantitative uptake measurements and tests for both iodine transport defects and abnormalities in thyroid oxidation. An enrichment of the tracer within the salivary gland can lead to misinterpretation, especially on lateral views, but this can be avoided by allowing the infant to feed before scintigraphy, thus empting the salivary glands and keeping the child calm under the camera. The perchlorate discharge test is considered indicative for a organification defect when a discharge of > 10% of I123 administred dose occurs in a thyroid in normal position (when perchlorate is given at 2 hours).
Excess iodine intake through exposure (i.e from antiseptic preparation), maternal TSH receptor blocking antibodies, inactivating mutation in the TSH receptor and in the sodium/iodide symporter (NIS), and TSH suppression from LT4 treatment can give interfere with the I123 uptake, showing no uptake in the presence of a thyroid in situ (apparent athyreosis).
Thyroid ultrasonography is performed with a high frequency linear array transducer (10-15 MHz) and allows a resolution of 0.7 to 1mm. Thyroid tissue is more echogenic than muscle and less echogenic than fat. In the case of absence of the thyroid fat tissue can be misdiagnosed as dysplastic thyroid gland in situ. Distinguish between thyroid hypoplasia and dysplastic non thyroidal tissue in a newborn requires an enormous experience, and reevaluation at later age can result in a different diagnosis (57a).
Combining scintigraphy and thyroid ultrasound improve diagnostic accuracy, and helps to address further investigations, including molecular genetic studies. Infants found to have a normal sized gland in situ in the absence of a clear diagnosis should undergo further reassessment of the thyroid axis and imaging at a later age.
Therapy
Replacement therapy with L-thyroxine (L-T4) should be begun as soon as the diagnosis of congenital hypothyroidism is confirmed. In babies whose initial results on newborn screening are suggestive of severe hypothyroidism therapy should be begun immediately without waiting for the results of the confirmatory serum. Severe hypothyroidism is defined by T4 <5 mcg/dL (64 nmol/L) and/or TSH >40 mU, or. accordingly with ESPE guidelines(51g,51k), CH is defined on the basis of serum FT4 levels as severe when FT4 is <5 pmol/l, moderate when FT4 is 5 to 10 pmol/l and mild when FT4 is 10 to 15 pmol/l. As noted above, treatment need not be delayed in anticipation of performing thyroid imaging studies as long as the latter are done within 5-7 days of initiating treatment (before suppression of the serum TSH). Parents should be counseled regarding the causes of congenital hypothyroidism, the importance of compliance and the excellent prognosis in most babies if therapy is initiated sufficiently early and is adequate and educational materials should be provided (58). An initial dosage of 10-15 mcg/kg/day of L-T4 is generally recommended to normalize the T4 as soon as possible. The highest dose is indicated in infants with severe disease, and the lower in those with a mild to moderate form. L-T4 Tablets can be crushed and given via a small spoon, with suspension, if necessary in a few milliliters of water or breast milk or formula or juice, but care should be taken that all of the medicine has been swallowed. Thyroid hormone should not be given with substances that interfere with its absorption, such as iron, calcium, soy, or fiber. Drugs such as antacids (aluminium hydroxide) or infantile colic drops (simethicone) can interfere with L-thyroxine absorption. Many babies will swallow the pills whole or will chew the tablets with their gums even before they have teeth. Reliable liquid preparations are not available commercially in the US, although they have been used successfully in Europe. L-T4 can also be administred in liquid form, but only if pharmaceutically produced and licensed L-T4 solutions are available. A brand name rather a generic formulation of L-T4 is recommended because they are not bioequivalent (58a).
The aims of therapy are to normalize the T4 as soon as possible, to avoid hyperthyroidism where possible, and to promote normal growth and development. When an initial dosage of 10-15 mcg/kg is used, the T4 will normalize in most infants within 1 week and the TSH will normalize within 1 month, Subsequent adjustments in the dosage of medication are made according to the results of thyroid function tests and the clinical picture. Often small increments or decrements of L-thyroxine (12.5 mcg) are needed. This can be accomplished by 1/2 tablet changes, by giving an alternating dosage on subsequent days, or by giving an extra tablet once a week.
As stated in ESPE guidelines: “ L-T4 alone is recommended as the medication of choice and should be started as soon as possible, no later than two weeks of life or immediately after confirmatory test results in infants identified in a second routine screening test. L-T4 should be given orally. If intravenous administration is necessary, the dose should be no more than 80% of the oral dose”. Serum or plasma FT4 (or TT4) and TSH concentration should be determined at least 4 hours after the last L-T4 administration. TSH should be maintained in the age-specific reference range and FT4 in the upper half of the age- specific reference range. “The first follow up examination is indicated after 1-2 weeks after the start of LT4 treatment and then every 2 weeks until TSH levels are completely normalized and then every 1- 3 months until 12 months of age. Between the age of one and three years, children should undergo frequent clinical and laboratory evaluations (every 2 to 4 months).” Thereafter, evaluations should be carried out every 3 to 12 months until growth is completed. “More frequent evaluations should be carried out if compliance is questioned or abnormal values are obtained. Any reduction of L-T4 dose should not be based on a single increase of FT4 concentration during treatment. “Measurements should be performed after 4-6 weeks any change in the dosage or in the L-T4 formulation”.
Re-evaluation and Trial Off Therapy
In hypothyroid babies in whom an organic basis was not established at birth and in whom transient disease is suspected, a trial off replacement therapy can be initiated after the age of 3 years when most thyroxine-dependent brain maturation has occurred, as shown by magnetic risonance imaging studies (56e). Re-evaluation is recommended if the treatment was started in a sick child (i.e. preterm), if thyroid antibodies were detectable, if no diagnostic assessment was completed, and in children who have required no increase in L-T4 dosage since infancy. Re-evaluation is recommended also in the case of a eutopic gland with or without goiter, if not enzyme defects have been detected, if any other cause of transient hypothyroidism is suspected.
Re-evaluation is not necessary if venous TSH concentration has risen during the first year of life, due to either LT4 underdosage or poor compliance. To perform a precise diagnosis LT4 treatment is suspended for 4-6 weeks, and biochemical testing and thyroid imaging are carried out. To establish the presence of primary hypothyroidism, without defining the cause, L-T4 dose may be decreased by 20-30% for 2 to 3 weeks. If TSH serum levels rise to > 10 mU/L during this period, the hypothyroidism can be confirmed.
Prognosis
Although all are agreed that the mental retardation associated with untreated congenital hypothyroidism has been largely eradicated by newborn screening, controversy persists as to whether subtle cognitive and behavioral deficits remain, particularly in the most severely affected infants (59-64). Both the initial treatment dose and early onset of treatment (before 2 weeks) are important. Time to normalization of circulating thyroid hormone levels, the initial free T4 concentration, maternal IQ, socioeconomic and ethnic status have also been related to outcome (59,62,63,64). The long term problems for these babies appear to be in the areas of memory, language, fine motor, attention and visual spatial. Inattentiveness can occur both in patients who are overtreated and those in whom treatment was initiated late or was inadequate. In addition to adequate dosage, assurance of compliance and careful long-term monitoring are essential for an optimal developmental outcome. More details about long term follow up are reported in ESPE guidelines (51g,51K). Progressive hearing loss in CH should be recognized and corrected, because strongly influenced the outcome). Recently, extensive reports on long term outcome of congenital hypothyroidism in young adults have been published (64a,64b). In the French cohort of 1202 CH young adults, hearing impairment was found at a mean age of 23.4 years in 9.5% versus 2.5% of general population, and the risk of developing hearing impairment was three times higher in these patients than in general population (64c). Also interesting data about pregnancy outcomes in young women with CH came out from the French cohort (64d).
CAUSES OF PERMANENT CONGENITAL HYPOTHYROIDISM
Permanent congenital thyroidal (primary) hypothyroidism can be the consequence of a disorder in thyroid development and/or migration (thyroid dysgenesis), or due to defects at every step in thyroid hormone synthesis (thyroid dyshormonogenesis). Although congenital hypothyroidism (CH) is in the great majority of cases a sporadic disease, the recent guidelines (51g,51k) for CH recommend genetic counseling in targeted cases. Positive family history for CH, association with cardiac or kidney malformation, midline malformation deafness, neurological sigs (i.e., choreoathetosis, hypotonia, any sign of Albright hereditary osteodystrophy, lung disorders, suggest genetic counseling, in order to assess the risk of recurrence and to provide further information about a possible genetic etiology of CH. Recently a targeted next-generation (NGS) panel, covering all exons of the major CH genes, has been proposed as a useful tool to identify the genetic etiology of CH (64e). Lowering TSH cut off value at screening increases the diagnosis of CH with eutopic thyroid. A targeted next-generation (NGS) panel has been applied to patients with CH and thyroid in situ (64f).
Thyroid Dysgenesis
Unlike in iodine-deficient areas of the world where endemic cretinism continues to be a major health hazard, the majority (85 to 90%) of cases of permanent congenital hypothyroidism in North America, Western Europe and Japan are due to an abnormality of thyroid gland development (thyroid dysgenesis). Thyroid dysgenesis may result in the complete absence of thyroid tissue (agenesis, 20-30%) owing to a defect in survival of the thyroid follicular cells precursors) or it may be partial (hypoplasia); the latter often is accompanied by a failure to descend into the neck (ectopy) mostly located in a sublingual position as a result of a premature arrest of its migratory process. Lowering of cut off TSH values for newborn screening increases the percentage of CH with thyroid in situ. Females are affected twice as often as males. In the United States, thyroid dysgenesis, is less frequent among African Americans and more common among Hispanics and Asians. Babies with congenital hypothyroidism have an increased incidence of cardiac anomalies, particularly atrial and ventricular septal defects (65). An increased prevalence of renal and urinary tract anomalies has also been reported recently (66). Most cases of thyroid dysgenesis are sporadic. Familial cases represent 2%. Discordance between monozigotic twins is inexplained (67). Although both genetic and environmental factors have been implicated in its etiology, in most cases the cause is unknown (67a).
The occasional familial occurrence, the higher prevalence of thyroid dysgenesis in babies of certain ethnic groups and in female versus male infants as well as the increased incidence in babies with Down syndrome (68) all suggest that genetic factors might play a role in some patients. Thyroid transcription factors would appear to be obvious candidate genes in view of their important role in thyroid organogenesis and in thyroid-specific gene expression. To date, however, abnormalities in these genes have been found in only a small proportion of affected patients, usually in association with other developmental abnormalities (68a).
Thyroid transcription factors (TTF) such as NKX2-1 (or formerly TTF1/TITF1), FOXE1 (Forkhread Box E1, formerly TTF2/TITF2), PAX8 (Paired box gene 8), and NKX2-5, are expressed during early phases of thyroid organogenesis (budding and migration), instead thyroid stimulating hormone receptor gene (TSHR) is expressed during the later phases of thyroid development. All these genes are involved in normal thyroid development and in thyroid dysgenesis. Alternately, epigenetic modifications, early somatic mutations or stochastic developmental events may play a role. Five monogenic forms due to mutations in TSHR, NXK2-1, PAX8, FOXE-1. NXK2-5 have been reported. Monogenic forms represent less than 10% in TD (68a).
TABLE 1GENETIC CAUSES OF CONGENITAL HYPOTHYROIDISM
| 1.1 PRIMARY HYPOTHYROIDISM | Gene locus | Inheritance |
| Monogenic forms of thyroid dysgenesis | ||
| · Thyroid stimulating hormone receptor (TSHR) | AR | |
| · NK2 1 (NK2-1, TTF1) brain-lung thyroid syndrome | 14q13 | AD |
| · Paired box gene 8 (PAX8) | 2q11.2 | AD |
| · Forkhead boxE1 (FOXE1, TTF2) (Bambforth-Lazarus syndrome) | 9q22 | AR |
| · NK2 homeobox 5 (NKX2-5) | ||
| New candidates gene | ||
| · Nertrin 1 (NTN-1) | ||
| · JAG1 | 20p.12.2 | |
| Inborn errors of thyroid hormonogenesis | ||
| · Sodium/Iodide symporter (SLC5A5,NIS | 19p13.2 | AR |
| · Thyroid peroxidase (TPO) | 2p25 | AR |
| · Pendred syndrome (SLC26A4,PDS) | 7q31 | AR |
| · Thyroglobulin (TG) | 8q24 | AR |
| · Iodothyrosine deiodinase (IYD,DEHAL1) | 6q24-25 | AR |
| · Dual oxidase 2 (DUOX2) | 15q15.3 | AR/AD |
| · Dual oxidase maturation factor 2 (DUOXA2) | AR/AD | |
| B1.2 CENTRAL HYPOTHYROIDISM | ||
| Isolated TSH deficiency | ||
| · TRHR | 14q31 | AR |
| · TSHB | 1p13 | AR |
| Isolated TSH deficiency or combined pituitary hormone deficiency | ||
| Immunoglobulin superfamily member1 (IGSF1) gene defects | Xq26.1 | X-Linked |
| Combined pituitary hormone deficiency | ||
| · POU1F1 | 3p11 | AR,AD |
| · PROP1 | 5q | AR |
| · HESX1 | 3p21.2-21.2 | AR/AD |
| · LHX3 | 9q.34 | AR |
| · LHX4 | 1q25 | AD |
| · SOX3 | X-linked | |
| · OTX2 | AD |
Monogenic Forms of Thyroid Dysgenesis
Thyroid stimulating hormone receptor resistance (TSHR gene #OMIM 603372)
Described in 1968, is mostly caused by biallelic inactivating mutations in the TSH receptor gene (TSHR). TSH affects follicular thyroid cell proliferation and many cellular processes, including thyroidal iodine uptake, thyroglobulin iodination, and reuptake of iodinated thyroglobulin. Phenotype varies from mild hyperthyrotropinemia with normal thyroid gland to severe CH with thyroid hypoplasia and absence of tracer uptake at scintigraphy (apparent athyreosis).
Inactivating TSHR mutations are the most frequent cause of monogenic TD and non syndromic CH, with prevalence in CH cohorts around 4 % (68b). Clinically a classic and a non-classic TSH resistance form are described, based on different TSHR mutations (68c). Both Gs and Gq proteins are involved Heterozygous non polymorphic TSHR mutations were found in a high frequency (11.8-29%) in children and adolescents with isolated non-autoimmune hyperthyrotropinemia (68d).
NKX2-1 (OMIM 600635)
NKX2-1 (previously TITF-1, TTF-1) gene encodes for a transcription factor of the NK family. It is involved in early development of brain, thyroid and lung. In thyrocytes, NKX2-1 activates the transcription of TG, TPO, TSHR and PDS genes. In the lung is important for the branching of the lobar bronchi and regulates the expression of surfactant proteins in pneumocytes. In the brain, NKX2 is expressed in basal ganglia and forebrain and it is involved in the specification and migration of neurons. Haploinsufficiency of NKX2-1 is responsible for the brain-lung-thyroid (BLT) syndrome (OMIM 610978) characterized by CH, infant respiratory distress syndrome and benign hereditary chorea. NKX2-1 defects occur either as a sporadic cases or as familial cases inherited in an autosomal-dominant manner. The clinical presentation ranges from the complete BLT syndrome (50%) to incomplete forms with brain and thyroid disease (30%) or only benign hereditary chorea (13%), the mildest expression of the syndrome. TD ranges from hypoplasia (about 35%) to normal morphology (>50% of patients) (68e). Recently, a case of BLT syndrome has been reported with thyroid ectopy (68f).
The severity of symptoms varies widely, even in families with the same disease causing mutation. In a detailed study (68g) lung disease, if present at birth, manifests as a surfactant deficiency syndrome and can be fatal. Asthma, recurrent pneumonia in childhood, spontaneous pneumothorax, and interstitial lung disease has also been reported. Neurologic forms present with muscular hypotonia in early infancy and psychomotor delay, which progresses to benign hereditary chorea between 1 and 5 years. Additional non classical features including hypodontia o oligodontia, microcephaly, growth retardation, genitourinary abnormalities, skeletal disorders, and congenital heart defects have been reported in patients with large deletions on chromosome 14, including the NKX2-1 gene and surrounding genes. Interestingly, a more extended phenotype associating hypothalamic symptoms, frequent recurrence of fever without infection, dysrhytmic sleep, and abnormal height in patients with point NKX2-1 mutations was described (68g). So far, 116 NKK2-1 genetic anomalies have been reported worldwide (68h).
PAX8 (OMIM218700)
Paired box gene 8 (PAX8) codes for a TTF of the paired homeodomain transcription factors family. PAX8 is expressed during thyroid organogenesis in the median anlage and in the kidney development. In synergy with NKX2-1, PAX 8 influences the expression of TPO, TG and NIS in thyroid follicular cells. The prevalence of PAX8 mutations in CH patients is about 1%, ranging from 0.3 to 3.4% (68b,68i).Thyroid hypoplasia is the more common phenotype, but athyreosis to normal morphology have also been reported. Thyroid function varies from severe hypothyroidism to mild hypertropinemia, and different phenotypes can be found in the same family. The association with kidney malformations is possible, but remains a facultative sign in CH patients with PAX8 mutations. So far, 29 mutations have been reported (68h).
FOXE1 (OMIM#602617)
The Forkhead Box 1 E1 (FOXE1) gene encodes for a transcription factor of the forkhead/winged-helix transcription factor family. Foxe1 is expressed in the thyroid primordium, in the pharyngeal endoderm derivates such as the palate and the esophagus and in the hair follicoles (68j). Foxe1 interacts with TG and TPO promoters and with regulatory regions of DUOX2 and NIS genes (68k).
The Bamforth-Lazarus syndrome is caused by FOXE1 mutations. It is characterized by CH (usually athyreosis), cleft palate and spinky hair. Bifid epiglottis and choanal atresia can be present. So far, six mutation with loss of function (68h) and 1 mutation with gain of function have been reported in patients with Bamforh-Lazarus syndrome, showing the effect of FOXE1 gene dosage in this disorder (68m).
NKX2-5 (OMIM #600584)
Because an increased prevalence of heart congenital malformations have been reported in CH, genes involved in heart organogenesis as NKX2-5 have been suggested as a cause of CH. NKX2-5, that encodes for a transcription factor with a major role in heart development has been investigated in a cohort of 241 patients with thyroid dysgenesis. Heterozygous missense mutations had been reported in this study in 4 patients with ectopy and athyreosis, and all mutations were transmitted from one of the parents but only 1 patient had minor cardiac phenotype (68n).
A major pathogenetic role of NKX2-5 mutations in thyroid dysgenesis has been questioned: given the absence of TD in carriers of NKX2-5 mutations, and the high number of TD patients without mutations. Better defining the role of NKX2-5 in thyroid organogenesis need further studies (68o).
New Candidates Genes
NTN-1
A new gene Netrin-1 (NTN-1), has been recently identified in a patient with thyroid ectopy and ventricular sept defect, and considered as a possible link between thyroid and heart defects (68p).
JAG1 (20p12.2 OMIM 6019220)
A role for the Notch pathway in thyroid morphogenesis has recently been demonstrated in zebrafish (68q). JAG1 is a gene encoding one single pass transmembrane ligand of the notch receptors. Heterozygous variations of JAG1 are the cause of Alagille syndrome type 1, an autosomal dominant disorder characterized by paucity of intrahepatic bile ducts, cardiac malformations as pulmonary artery stenosis, coarctaction of aorta, atrio-ventricular septal defects and Fallot tetralogy. Many other organs as eye, skeleton, kidney, nervous system can be involved, with a characteristic facial phenotype. A study investigating the role of JAG1 loss of function variations in the pathogenesis of congenital thyroid defects in Alagille syndrome and in patients with congenital hypothyroidism supported the role of this gene as a predisposing factor in congenital hypothyroidism (68r). The authors reported, in a series of 21 patients affected with Alagille syndrome non autommune hypothyroidism in 6 patients (28%), two of them with thyroid hypoplasia. Analyzing 100 patients with congenital hypothyroidism for JAG1 variants they found JAG1 variants in 4. Interestingly, 2 of them had cardiac malformations.
Inborn Errors of Thyroid Hormonogenesis
Inborn errors of thyroid hormonogenesis (thyroid dyshormonogenesis) are responsible for most of the remaining cases (15%) of neonatal thyroidal hypothyroidism. Unlike thyroid dysgenesis, mostly a sporadic condition, these inborn errors of thyroid hormonogenesis are commonly associated with an autosomal recessive form of inheritance, consistent with a single gene abnormality. DUOX2 mutations can be transmitted in autosomal dominant way. Thyroid dysormonogenesis is caused by genetic defects in proteins involved in all steps of thyroid hormone synthesis (68s) often associated with goiter formation. Goiter can be present in utero or at birth.
.A number of different defects have been characterized based on radioiodine uptake and perchlorate test and include:
1) Iodide transport defect (ITD)
(SLC5A5, Sodium/Iodide Symporter NIS), that shows failure to concentrate iodide, with low or absent radioiodine uptake, also in salivary glands and gastric mucosa;
2) Iodide organification defect (IOD)
with normal radioiodine uptake and altered perchlorate discharge test. In these patients, less than 90% of the iodide is organified and remains stored in the follicles. Total IOD is defined as >90% of the given dose back to the blood. Partial IOD is defined as 10-90% of radioiodine washout after perchlorate application. Total IOD is due to Thyroid peroxidase mutations (TPO) and Dual Oxidase 2 (DUOX2), partial IOD is due to DUOX2, Dual Oxidase Maturation Factor 2 mutations (DUOX2A), SLC26A4, pendrin and TPO defects.
3) Forms with normal radioiodine uptake and a normal perchlorate test:
Thyroglobulin TG mutations, iodide recycling defects IYD, Iodothyrosine Deiodinase mutations (DEHAL1).
4) Iodide Transport Defect (OMIM 274400)
ITD is rather a rare form and is due a mutation of the Sodium/Iodide Symporter (NIS). The NIS is expressed at the basolateral membrane of the thyrocite and it is responsible for the active iodide uptake through the membrane into the thyrocite (69). This form of hypothyroidism is characterized by goiter and absence of radioiodine uptake. In contrast with athyreosis, uptake is lacking also in salivary glands and in the stomach (white scintigraphy).
The severity of hypothyroidism depends on the residual function of the mutated NIS protein, ranging with severe to mild forms, often detected in infancy or childhood.
Pendred Syndrome (OMIM274600)
Pendred syndrome is defined by the association of familial profound deafness with multinodular goiter. It is caused by biallelic mutation in the pendrin gene (70-71). Pendred syndrome is the only form of thyroid dyshormonogenesis associated with a malformation. The inner ear presents a characteristic malformation of the cochlea.
Congenital hypothyroidism is present in only 30% of cases, goiter occurs often in childhood. Thyroid phenotype is variable. Perchlorate test shows a partial organification defect. Pendred syndrome is the most frequent etiology of familial deafness. SLC264A mutations (mostly in the heterozygous state) have been also described in isolated enlargement of the vestibular aqueduct, with no thyroid disease (71a). More than 150 mutations have been described. Specific mutation cluster in Asia (H723R), and Europe (L236P, T416P, IVS8, 1-GA) (71b).
Thyroid peroxidase mutations (OMIM #274500)
Thyroid peroxidase (TPO) is a heme peroxidase that regulates two rate-limiting step of thyroid hormones synthesis, first the organification of iodide to iodinated thyrosyl residuates such as MIT and DIT, and then the coupling of MIT and DIT to T3 and T4. TPO action needs hydrogen peroxide as the final electron acceptor. Mutations are mostly in the heme-binding domain of the protein, encoded by exons 7-9 (71c). TPO mutations are a common form of thyroid dyshormonogenesis. Severe congenital hypothyroidism with goiter is present in the great part of patients, with a total IOD. Recently, a few patients with partial IOD have been reported (72,72a).
Dual Oxidase 2 and Dual Oxidase Maturation Factor 2 mutations (OMIM#607200 and 274900)
DUOX2 (formerly THOX2) and DUOXA2 are components of a nicotinamide adenine dinucleotide phosphate oxidase complex that produces hydrogen peroxide indispensable for TPO action.
The first mutation in DUOX2 has been reported in 2002. Heterozygous mutations have been found in a part of the patients, suggesting autosomal dominant and autosomal recessive inheritance both possible in this form (72b). Monoallelic mutations usually cause mild hypothyroidism; biallelic mutations are present in mild to severe hypothyroidism. In some cases, DUOX2 mutations lead to transient congenital hypothyroidism, with normalization of thyroid function at follow up. DUOX2 mutations usually cause partial IOD, but total IOD is also reported (72c). Mutations in DUOXA2 were described in patients detected by neonatal screening with mild CH. Partial IOD was found in these cases (72d).
Thyroglobulin Mutations (OMIM#274700)
Thyroglobulin (Tg) is a glycoprotein synthetized by the thyrocytes that serves as a matrix for thyroid hormones synthesis and storage in the follicles (68t). Tg is also in part released in the blood and it is a useful marker of thyroid tissue.
In CH, Tg serum determination can differentiate between a true and apparent athyreosis, the last with same residual dysgenetic tissue and Tg detectable.
In dyshomonogenesis, Tg levels are low in patients with Tg mutations, but are normal or high in the other defects of hormonogenesis (68t). CH due to Tg mutations is usually severe, with goiter in utero or at birth. Different mechanisms cause hypothyroidism in Tg mutations: a )Tg synthesis defects alter protein synthesis; b) Tg transport defects limit Tg excretion in the follicle; c) a abnormal structure of T impairs coupling of MIT and DIT; d) a large imperfect DNA inversion in Tg gene is a novel cause for CH (72e-72g).
Iodothyrosine Deiodinase Mutations cause Iodide Recycling defects (OMIM#274800)
DEAHAL 1(IYD) is the enzyme that regulates the recycling of iodide from MIT and DIT to the follicle, thus allowing the synthesis of thyroid hormones. Dietary Iodine is scarce in nature and it is the limitating factor for thyroid hormones synthesis. Failure of DEAHL1 cause iodotyrosine deiodinase deficiency, characterized by hypothyroidism, goiter and mental retardation. It is important to stress that these patients are not detected by neonatal screening for CH, probably because the maternal iodine protect for a period the newborn. Diagnosis is reported between 18 month and 16 years with hypothyroidism and mental retardation (72h). The first mutations in DEAHAL1 has been reported in 2008 (72i).
The use of MIT and DIT -as early markers to identify iodotyrosine deiodinase deficiency before mental retardation-is under investigation.
Central Congenital Hypothyroidism (CCH)
Central hypothyroidism (CCH) is caused by an insufficient thyroid hormone biosynthesis due to a defective stimulation by TSH, in the presence of an otherwise normal thyroid. This condition includes all causes of congenital hypothyroidism due to a pituitary or hypothalamic pathology (secondary or tertiary hypothyroidism). CCH was previously considered a very rare disease with a prevalence initially estimated to be 1:100000 in newborns (73). In more recent data, CCH had an incidence that could reach 1:16.000, as shown from results from screening for congenital hypothyroidism applied in the Neetherlands, based on T4/TSH/TBG determination (73a).
Also with this sophisticated method of screening, CCH is sometime not identified at birth, because the limiting step is “how low is a low T4”, low enough to be considered an effective cutoff value and allow the determination of TSH and TBG. Many cases are diagnosed in infancy or childhood, if not later in adulthood (73b). The majority of screening programs are based on TSH determination and a high index of suspicion is needed to identify CCH in the preclinical phase. Delayed diagnosis may result in neurodevelopment delay. More than 50% of children with CCH have moderate or severe hypothyroidism, so, if not treated, the risk of neurodevelopmental delay should not be underestimated (73c).
In the majority of cases identified early, TSH deficiency is a part of combined pituitary hormone deficiency. A timely correction of ACTH and cortisol deficiency, and/or GH deficiency may avoid life threatening emergencies.
CCH can be transient (mostly due to drugs or maternal hyperthyroidism), or permanent.
Genetic Central Hypothyroidism (Table 1)
Isolated Thyroid Stimulating Hormone deficiency
Two forms of non-efficient TSH are known, the first one is very rare and is due to defects in the receptor that regulates the action of TRH on thyrotropes (TRHR), the second form is due to several mutations in the β-subunit of TSH.
a)Thyrotropin-releasing hormone receptor (TRHR ) gene defects. TRHR mediates the correct action of TRH on thyrotropes toward the synthesis, glycosylation and secretion of TSH.
This is a very rare cause of central hypothyroidism. Mutations in TRHR gene have been described so far in 3 patients from 2 families, the first from Canada, the second from Italy, with autosomal recessive inheritance (73d,73e). Index cases were detected at 9 and 11 years for short stature and symptoms related to hypothyroidism. Neonatal hypothyroidism could not be proven because neonatal screening was based on TSH level. No psychomotor delay or intellectual deficit was reported in these children. TSH was in the low normal range with a suspected low bioactivity; T4 or FT4 were low, TRH test showed no response of TSH and PRL.
A compound heterozygosis with 2 different mutations in TRHR gene was found in the Canadian patient. The first mutation in the paternal allele was a premature stop codon R17X that completely inactivated protein function. The second one, on the maternal allele was a complex combination of mutations: 9-nucleotide deletion followed by a point mutation resulting in an in-frame deletion of three aminoacids (Ser115-Thir117) plus a missense change located at the cytoplasmatic end of the transmembrane domain of the receptor (73d).The Italian patient had a homozygous nonsense mutation (pR17X).
A novel homozygous missense mutation (P81R) in TRHR has been published in a female infant presented at age 19 days with prolonged jaundice due to isolated hyperbilirubinemia. Thyroid function showed CCH (TSH 2.2 mU/L (RR 0.4-3.5). FT4 7.9 pmol/L (RR10.7-21.8). She was treated with L-thyroxine and at 4 years of age growth and neurological development are in the normal range. The location of the mutated aminoacid (proline 81) in the second transmembrane helix underlines the functional role of this helix in hormone binding and receptor activation (73f).
b)TSH β Gene defects
TSH is a glycoprotein hormone with an α subunit common with FSH, LH and hCG, and a β-subunit, specific for TSH.
Mutations of the β-subunit of TSH are the cause of the most severe forms of central congenital hypothyroidism. All mutations described so far caused central hypothyroidism, either because truncated protein or alterations in key structural features required for heterodimeric integrity occur (74, 74a, 74b).
Another consequence of mutations of the β-subunit of TSH is the modification of bioactivity and immunoreacivity of the TSH heterodimer. Diagnosis of central hypothyroidism can be complicated because of impaired TSH immunoreactivity and/or bioactivity. For instance, TSH is not detectable when the heterodimer formation between TSHα and TSH β subunit is completely not allowed from mutations (i.e. p.G49, p.32), in other cases some mutant heterodimeric TSH is present and measurable in an immunoassay dependent manner (i. e. p.Q69, c.373 delT). TSH can be measurable but not shows normal bioactivity (74a). Interestingly, a variant (c223A>G, pR75) causing normal bioactive TSH, but with impaired immunoreactivity has been described (74c, 74d). These individuals are euthyroid, but erroneous diagnosis and inappropriate treatment have been reported.
In children affected with CCH due to mutations of the β-subunit of TSH, psychomotor and mental retardation can occur, depending on the time of diagnosis and treatment. Most are clinically diagnosed after 3 months of age because they are not identified by neonatal screening based on increased TSH levels. Hyperplastic pituitary, high levels of serum glycoprotein alfa-subunit and hypoplasic thyroid gland have been reported (74a). Several mutations have been reported, including missense, nonsense and frameshift mutations (74,74a), as well as slice mutations (74b). Recently a homozygous TSHβ mutation was found (74e). A novel missense mutation (c.2T>C) in which a methionin codon, is replaced by a threonine, has been very recently reported in a child with very low levels of TSH (0.45mU/l, (NR 0.4-3.5) and FT4.(<5.1 pmol/l (NR 13.8-22.5). This child was diagnosed at 3.5 months of age because feeding difficulties, somnolence, constipation and severe growth retardation. She was treated with L-thyroxine with a good response in growth, but she has severe neurodevelopmental deficits, with bilateral sensorineural deafness, nistagmus, motor and language development delay at age of 10. She was on autistic/Asperger spectrum and needed special education at school (74f).
Immunoglobulin superfamily member1 (IGSF1) gene defects
IGSF1 (immunoglobulin (Ig) superfamily member1) gene mutations were described in 2012 as a cause of central hypothyroidism, with an incidence of about 1:100.000 (75,75a). IGSF1 gene is located on X chromosome (Xq26.1) and encodes for a plasma membrane glycoprotein that is mainly expressed in the pituitary, brain and testes.
Several pathogenetic mutations in IGSF1 gene have been reported so far (75,75a,75b). An extensive case series, expanding the clinical phenotype has been published very recently (75c,75d). The first patient was diagnosed by neonatal screening in the Neetherlands where a screening program for congenital hypothyroidism that includes T4 determination (T4/TBG/TSH) is applied. Many other cases of central hypothyroidism were identified in this family and in others with an age at diagnosis ranging from 3 weeks to 69.9 years (75). Typical phenotype in adult males includes central hypothyroidism and macroorchidism (>30 ml by Prader orchidometer). Hypoprolactinemia and GH deficiency can be present. GH deficiency is usually transient and detectable in childhood. Body mass index tends to be elevated. Testicular volume is normal in childhood and increases at a normal age in puberty, but the testosterone rise is delayed, as well as the pubertal growth spurt and the appearance of secondary sexual characteristics. Thyroid volume is small, TSH is usually detectable, TSH response to TRH is diminished. No clear correlation genotype-phenotype has been established.
IGSF1 gene is located on X chromosome. Male are affected but 1/3 of females heterozygous carriers shows a milder phenotype, with central hypothyroidism, delayed menarche, mild prolactin deficiency and benign ovarian cysts sometime requiring surgical resection. Recently a familial form of isolated central hypothyroidism with neurological phenotypes due to a novel IGSF1 gene mutation has been reported from Israel (75e).
TSH deficiency in combined pituitary hormone deficiency
Central congenital hypothyroidism can be a component of combined pituitary hormone deficiency. This form represents the majority of cases detected by the neonatal screening when T4 determination is used (73b). Early diagnosis in these cases helps to prevent dangerous hypoglycemic and adrenal crisis due to associated GH and ACTH deficiency.
TSH deficiency can be present at diagnosis or occurs later, as a component of an evolving phenotype. In a minority of patients, mutations of known transcriptor factors (i.e POU1F1 ,PROP1,HESX1,LHX3,LHX4,SOX3 and OTX2) that are involved in pituitary development can be identified (76) (See Table 1).
Mutations in early transcriptor factors cause developmental abnormalities, i.e., septo-optic dysplasia, midline defects, holoprosencephaly, ocular or skeletal defects, intellectual impairment, associated with variable hypopituitarism. Mutations in HESX1, OTX2 and SOX3 have been found in patients with septo-optic dysplasia and TSH deficiency (76).
TSH deficiency in association with other pituitary hormone deficiencies may be associated with abnormal midline facial and brain structures (particularly cleft lip and palate, and absent septum pellucidum and/or corpus callosum) and should be suspected in any male infant with microphallus and persistent hypoglycemia (76a). One of the more common of these syndromes, septo-optic dysplasia, has been related in some cases to a mutation in the HESX 1 homeobox gene in some cases (76b). Other genetic causes of congenital hypopituitarism include molecular defects in the genes for the transcription factors LHX3 (76c), LHX4, POU1F 1 (76d) or PROP 1 (76d). POU1F 1 (Pit-1 in mice) is essential for the differentiation of thyrotrophs, lactotrophs and somatotrophs while PROP 1, a homeodomain protein that is expressed briefly in the embryonic pituitary, is necessary for POU1F 1 expression.
Defects of Thyroid Hormone Transport in Serum
For complete coverage of this and related areas visit the chapter entitled: “Defects of thyroid hormone transport in serum” by Samuel Refetoff, MD in this book.
Inherited abnormalities of the iodothyronine-binding serum proteins include TBG deficiency (partial or complete), TBG excess, transrethyretin (TTR) (prealbumin) variants and familial dysalbuminemic hyperthyroxinemia (FDH). In these conditions the concentration of free hormones is unaltered, but the abnormal total thyroxine concentrations can be misleading during neonatal screening and in the evaluation of thyroid function.
Impaired Sensitivity to Thyroid Hormone
For complete coverage of this and related areas visit the chapter entitled: “Impaired sensitivity to thyroid hormone: defects of transport, metabolism and action” by.Alexandra M. Dumitrescu, MD and Samuel Refetoff, MD, in this book.
Impaired sensitivity to thyroid hormone, previously known as “reduced sensitivity to thyroid hormone”, include defects in thyroid hormone action, transport and metabolism. They are classified in a)Thyroid hormone cell membrane transport defect (THCMTD),b) thyroid hormone metabolism defect (THMD) and c) thyroid hormone action defect that include Resistance to thyroid hormone (RTH) (77).The first defect, recognized almost 50 years ago, produces reduced sensitivity to TH and was given the acronym RTH, for resistance to thyroid hormone (77a, 77b). Its major cause, found in more than 3,000 individuals, is mutations in the TH receptor ß (THRB) gene. More recently mutations in the THRA gene were found to produce a different phenotype owing to the distinct tissue distribution of this TH receptor (77c, 77d). Two other gene mutations, affecting TH action, but acting at different sites have been identified in the last 10 years. One of them, caused by mutations in the TH cell-membrane transporter MCT8, with decreased T4 uptake into brain cells produces severe psychomotor defects (77e,77f). In this syndrome, first described as Allan Herdon Dudley syndrome, (77g) mutations in the monocarboxylate transporter 8 (MCT 8 gene, located on the X-chromosome), have been associated with male- limited hypothyroidism and severe neurological abnormalities, including global developmental delay, dystonia, central hypotonia, spastic quadriplegia, rotary nystagmus and impaired gaze and hearing (77e, 77f). Heterozygous females had a milder thyroid phenotype and no neurological defects. A defect of the intracellular metabolism of TH, identified in 11 members from 9 families, is caused by mutations in the SECISBP2 gene required for the synthesis of selenoproteins, including TH deiodinases (77h). Knowledge of the molecular mechanisms involved in mediation of TH action allows the recognition of the phenotypes caused by genetic defects in the involved pathways. While these defects have opened the avenue for novel insights into thyroid physiology, they continue to pose therapeutic challenges.
CAUSES OF TRANSIENT NEONATAL HYPOTHYROIDISM
Transient neonatal hypothyroidism should be distinguished from a ‘false positive’ result in which the screening result is abnormal but the confirmatory serum sample is normal. Causes of transient abnormalities of thyroid function in the newborn period are listed in Table 2. While iodine deficiency, iodine excess, drugs and maternal TSH receptor blocking antibodies are the most common causes of transient hypothyroidism, in some cases the cause is unknown.
TABLE 2CAUSES OF TRANSIENT HYPOTHYROIDISM IN THE NEWBORN
| · | 2.1 PRIMARY HYPOTHYROIDISM |
| · | · Prenatal or postnatal iodine deficiency or excess |
| · | · Maternal antithyroid medication |
| · | · Maternal TSH receptor blocking antibodies |
| · | · Mild gene mutations (i.e. DUOX2, TSH-R ) |
| · | · Maternal hypothyroidism |
| · | · Prematurity, VLBW |
| · | · Drugs, (i.e. Dopamine, steroids) |
| · Hypothyroxinemia (low T4, normal TSH) | |
| · | 2.2 CENTRAL HYPOTHYROIDISM |
| · | · Prenatal exposure to maternal hyperthyroidism |
| · | · Prematurity (particularly <27 weeks gestation) |
| · | · Drugs |
Iodine Deficiency or Excess
In addition to iodine deficiency, both the fetus and newborn infant are sensitive to the thyroid-suppressive effects of excess iodine, whether administered to the mother during pregnancy, lactation or directly to the baby (78). This occurs, in part because, as noted earlier, recovery from the thyroid-suppressive effect of iodine does not mature before 36 weeks gestation; however, other factors, including increased skin absorption are also likely to play a role. Reported sources of iodine have included drugs (e.g., potassium iodide, amiodarone), radiocontrast agents and antiseptic solutions (e.g., povidone-iodine) used for skin cleansing or vaginal douches. In contrast to Europe, iodine-induced transient hypothyroidism has not been documented frequently in North America (79). For other information see the chapter “Iodine deficiency disorders” in this book.
Maternal Antithyroid Medication
Transient neonatal hypothyroidism may develop in babies whose mothers are being treated with antithyroid medication (either propylthiouracil, PTU or methimazole, MMI) for the treatment of Graves ’ disease. Even maternal PTU doses of 200 mg or less have been associated with an effect on neonatal thyroid function, illustrating the increased fetal sensitivity to these drugs (80). Babies with PTU- or MMI-induced hypothyroidism characteristically develop an enlarged thyroid gland and if the dose is sufficiently large, respiratory embarrassment may occur. Both the hypothyroidism and goiter resolve spontaneously with clearance of the drug from the baby’s circulation. Usually replacement therapy is not required.
Maternal TSH Receptor Antibodies
Maternal TSH receptor blocking antibodies, a population of antibodies closely related to the TSH receptor stimulating antibodies in Graves’ disease, (81) may be transmitted to the fetus in sufficient titer to cause transient neonatal hypothyroidism. The incidence of this disorder has been estimated to be 1 in 180,000 (81a,81b). TSH receptor blocking antibodies most often are found in mothers who have been treated previously for Graves’ disease or who have the non goitrous form of chronic lymphocytic thyroiditis (primary myxedema). Occasionally these mothers are not aware that they are hypothyroid and the diagnosis is made in them only after congenital hypothyroidism has been recognized in their infants (81b). Unlike TSH receptor stimulating antibodies that mimic the action of TSH, TSH receptor blocking antibodies inhibit both the binding and action of TSH (see below). Because TSH-induced growth is blocked, these babies do not have a goiter. Similarly, inhibition of TSH-induced radioactive iodine uptake may result in a misdiagnosis of thyroid agenesis (81c). Usually the hypothyroidism resolves in 3 or 4 months. Babies with TSH receptor blocking-antibody induced hypothyroidism are difficult to distinguish at birth from the more common thyroid dysgenesis but they differ from the latter in a number of important ways (Table 3). They do not require lifelong therapy, and there is a high recurrence rate in subsequent offspring due to the tendency of these antibodies to persist for many years in the maternal circulation. Unlike babies with thyroid dysgenesis in whom a normal cognitive outcome is found if postnatal therapy is early and adequate, babies with maternal blocking-antibody induced hypothyroidism may have a permanent deficit in intellectual development if feto-maternal hypothyroidism was present in utero (27).
TABLE 3. CLINICAL FEATURES OF THYROID DYSGENESIS VERSUS TSH RECEPTOR
- BLOCKING ANTIBODY INDUCED CONGENITAL HYPOTHYROIDISM
| Dysgenesis | Blocking Ab | |
| Severity of CH | + to ++++ | + to ++++ |
| Palpable thyroid | No | No |
| 123I uptake | None to low | None to normal |
| Clinical Course | Permanent | Transient |
| Familial risk | No | Yes |
| TPO Abs | Variable eele | Variable |
| TSH Receptor Abs | Absent | Potent |
Transient Central Hypothyroidism Due to Maternal Hyperthyroidism
Occasionally, babies born to mothers who were hyperthyroid during pregnancy develop transient hypothalamic-pituitary suppression (81,81a,81b,81c). This hypothyroxinemia is usually self-limited, but in some cases it may last for years and require replacement therapy (82). In general the titer of TSH receptor stimulating antibodies in this population of infants is lower than in those who develop transient neonatal hyperthyroidism (see below).
Prematurity
Hypothyroxinemia in the presence of a ‘ normal ’ TSH occurs most commonly in premature infants in whom it is found in 50% of babies of less than 30 weeks gestation. Often the free T4 when measured by equilibrium dialysis is less affected than the total T4 (83). The reasons for the hypothyroxinemia of prematurity are complex. As well as hypothalamic-pituitary immaturity mentioned earlier, premature infants frequently have TBG deficiency due to both immature liver function and undernutrition, and they may have “sick euthyroid syndrome”. They may also be treated with drugs that suppress the hypothalamic-pituitary-thyroid axis. Hypothyroxinemia of prematurity may be associated with adverse neurodevelopmental outcomes. L-T4 treatment overall has no proven benefit and can be harmful (83a). Long term outcome evaluation in young adults did not find association between transient hypothyroxinemia of prematurity and neurodevelopmental outcome (83b). Whether or not premature infants with hypothyroxinemia should be treated remains controversial at the present time (83c,83d,83e). Although several retrospective, cohort studies have documented a relationship between severe hypothyroxinemia and both developmental delay and disabling cerebral palsy in preterm infants <32 weeks gestation a causal relationship could not be determined since the serum T4 in premature infants, as in adults, has been shown to reflect the severity of illness and risk of death (83c).
Drugs
Drugs that suppress the hypothalamic-pituitary axis include known agents such as steroids and dopamine, but also aminophylline, caffeine and diamorphine, other commonly used in sick premature infants (84).
Other causes of hypothyroidism in infancy
Chronic lymphocytic thyroiditis
Chronic lymphocytic thyroiditis (CLT) is a rare disease in infancy, but if not recognized and treated, can cause severe hypothyroidism in a short time with permanent brain damage (85). CLT can be associated with other autoimmune disease as type 1 diabetes or a manifestation of IPEX syndrome (85a). In the cases described by Foley, no goitrous was found. Clinical manifestations and biochemical hypothyroidism (TSH ranged from >42 to 928 mU/L) were severe and very high levels of antibodies were detectable.
Hepatic hemangiomas: consumptive hypothyroidism
Hepatic emangioendothelioma is a rare tumor typically presenting in infancy. Hypothyroidism is caused by a production of type 3 deiodinase by the vascular tumor (85c). D3 deoidinase increases inactivation of T4 and T3 to reverse T3 andT2 and large amount of LT4 (up to 94/ µg/kg/day) are needed to compensate this inactivation (85d). Frequent monitoring is required, adapting the LT4 treatment to the growing proliferative phase of the tumor. Today hemangioendotheliomas in infancy may successfully being treated with steroids and propranolol and may undergo spontaneous regression. Some babies underwent liver transplantation.
HYPERTHYROIDISM
Transient Neonatal Hyperthyroidism
Unlike congenital hypothyroidism which usually is permanent, neonatal hyperthyroidism almost always is transient and results from the transplacental passage of maternal TSH receptor antibodies. Hyperthyroidism develops only in babies born to mothers with the most potent stimulatory activity in serum (86,87,87a). This corresponds to 1-2% of mothers with Graves’ disease, or 1 in 50,000 newborns, an incidence that is approximately four times higher than is that for transient neonatal hypothyroidism due to maternal TSH receptor blocking antibodies (81a). Some mothers have mixtures of stimulating and blocking antibodies in their circulation, the relative proportion of which may change over time. Not surprisingly, the clinical picture in the fetus and neonate of these mothers is more complex and depends not only on the relative proportion of each activity in the maternal circulation at any one time but on the rate of their clearance from the neonatal circulation postpartum. Thus, one affected mother gave birth, in turn, to a normal infant, a baby with transient hyperthyroidism, and one with transient hypothyroidism (87b). In another neonate, the onset of hyperthyroidism did not become apparent until 1-2 months postpartum when the higher affinity blocking antibodies had been cleared from the neonatal circulation (87c). In the latter case, multiple TSH receptor stimulating and blocking antibodies were isolated from the maternal peripheral lymphocytes. Each monoclonal antibody recognized different antigenic determinants (“epitopes”) on the receptor and had different functional properties (87d).
Occasionally, neonatal hyperthyroidism may even occur in infants born to hypothyroid mothers. A prospective study showed that 40% of patients treated for Graves’ disease with radioactive iodine had TRAb detectable after 5 years (87e). In these situations, the maternal thyroid has been destroyed either by prior radioablation, surgery or by coincident destructive autoimmune processes so that potent thyroid stimulating antibodies, present in the maternal circulation, are silent in contrast to the neonate whose thyroid gland is normal (87d). Fetal/neonatal thyrotoxicosis can occur also in newborn from hypothyroid mothers with chronic lymphocytic thyroiditis (87f).
Clinical manifestations
Maternal TSH receptor antibody-mediated hyperthyroidism may present in utero. Fetal hyperthyroidism is suspected in the presence of fetal tachycardia (pulse greater than 160/min) especially if there is evidence of failure to thrive. Obstetric complications are common. Fetal goiter can by monitored by ultrasound. In the neonate infant most often the onset is during the first one-two weeks of life but can occur by 45 days. This is due both to the clearance of maternally-administered antithyroid drug (propylthiouracil, PTU, methimazole or carbimazole) from the infant ’s circulation and to the increased conversion of T4 to the more metabolically active T3 after birth. Rarely, as noted earlier, the onset of neonatal hyperthyroidism may be delayed until later if higher affinity blocking antibodies are also present. In the newborn infant, characteristic signs and symptoms include tachycardia, irritability, poor weight gain, and prominent eyes (Figure 5). Goiter, when present, may be related to maternal antithyroid drug treatment as well as to the neonatal Graves’ disease itself.

Figure 15.5. A
baby with neonatal hyperthyroidism secondary to maternal Graves ‘disease. Note the prominent eyes in the baby and mother in whom Graves’ disease developed after radioiodine therapy for Hodgkin’s disease. In contrast, the father was unaffected.
Rarely, infants with neonatal Graves’ disease present with thrombocytopenia, jaundice, hepatosplenomegaly, and hypoprothrombinemia, a picture that may be confused with congenital infections such as toxoplasmosis, rubella, or cytomegalovirus (87g). In addition, arrhythmias and cardiac failure may develop and may cause death, particularly if treatment is delayed or inadequate. In addition to a significant mortality rate that approximates 20% in some older series, untreated fetal and neonatal hyperthyroidism is associated with deleterious long-term consequences, including premature closure of the cranial sutures (cranial synostosis), failure to thrive, and developmental delay (87h). The half-life of TSH receptor antibodies is 1 to 2 weeks. The duration of neonatal hyperthyroidism, a function of antibody potency and the rate of their metabolic clearance, is usually 2 to 3 months but may be longer.
Laboratory Evaluation
TSH receptor antibodies (TRAb) are Immunoglobulin of G class and freely cross the placenta. Different type of TRAb can be found: TRAb that bind to the TSH receptor and stimulates the production of thyroid hormones, (TSH receptor stimulating antibodies, TSI), TRAb that bind to the TSH receptor, do not stimulate the production of thyroid hormones and can block the binding of TSH (TSH receptor blocking antibodies TBI) .TSH receptor neutral antibodies have also been identified which do not block TSH binding and are unable to stimulate cAMP production (88)..
The receptor binding assays usually used to measure TRAb are not able to distinguish between TSH-receptor stimulating and blocking or neutral antibodies. Bioassays that measure TSI activity based on cAMP on cultured cells can be useful if TRAb are not detectable (88a,88b). The recent guidelines for management of hyperthyroidism (88c) and the updated guidelines for the management of thyroid disease during pregnancy released from the American Thyroid Association ATA (33b) both suggest to anticipate the determination of TRAb in pregnant women with Graves’ disease at 18-22 weeks instead of 20-24 weeks of gestation because a severe case of fetal Graves’ disease has occurred at 18 weeks of pregnancy (88d).
Because of the importance of early diagnosis and treatment, infants at risk for neonatal hyperthyroidism should undergo both clinical and biochemical assessment as soon as possible.
All neonates born from a woman with TRAb positivity in pregnancy should undergo determination of TRAb from cord blood at delivery. If TRAb are negative, the risk to neonatal hyperthyroidism is negligible (Sensitivity is around 100%). FT3, FT4 and TSH determination from cord blood did not predict neonatal hyperthyroidism. Determination of FT4 increase on day 3 to 5 seems to better indicate the onset of hyperthyroidism (88e) Situations that should prompt consideration of neonatal hyperthyroidism are listed in Table 4. A high index of suspicion is necessary in babies of women who have had thyroid ablation because in them a high titer of TSH receptor antibodies would not be evident clinically. Similarly, women with persistently elevated TSH receptor antibodies and with a high requirement for antithyroid medication are at an increased risk of having an affected child. The diagnosis of hyperthyroidism is confirmed by the demonstration of an increased concentration of circulating T4 (and free T4, and T3, if possible) accompanied by a suppressed TSH level in neonatal or fetal blood. The latter can be obtained by cordocentesis if someone experienced in this technique is available. Results should be compared with normal values during gestation. Fetal ultrasonography may be helpful in detecting the presence of a fetal goiter and in monitoring fetal growth. Demonstration in the baby or mother of a high titer of TSH receptor antibodies will confirm the etiology of the hyperthyroidism and, in babies whose thyroid function testing is normal initially, indicate the degree to which the baby is at risk.
TABLE 4SITUATIONS THAT SHOULD PROMPT CONSIDERATION OF NEONATAL HYPERTHYROIDISM
| · Unexplained tachycardia, goiter or stare |
| · Unexplained petechiae, hyperbilirubinemia, or hepatosplenomegaly |
| · History of persistently high TSH receptor antibody titer in mother during pregnancy |
| · History of persistently high requirement for antithyroid medication in mother during pregnancy |
| · History of thyroid ablation for hyperthyroidism in mother |
| · History of previously affected sibling |
As noted in the case of TSH receptor blocking antibody-induced congenital hypothyroidism, the receptor binding assays are a cost-effective, rapid and technically feasible approach. In general, babies likely to become hyperthyroid have the highest TSH receptor antibody titer whereas if TSH receptor antibodies are not detectable, the baby is most unlikely to become hyperthyroid (87g, 89,89a). In the latter case, it can be anticipated that the baby will be euthyroid, have transient hypothalamic-pituitary suppression or have a transiently elevated TSH, depending on the relative contribution of maternal hyperthyroidism versus the effects of maternal antithyroid medication, respectively (89). Close follow up of all babies with abnormal thyroid function tests or detectable TSH receptor antibodies is mandatory.
Therapy
In the fetus, treatment is accomplished by maternal administration of antithyroid medication. Until recently PTU was the preferred drug for pregnant women in North America, but current recommendations suggest the use of MMI rather than PTU after the first trimester because of concerns about potential PTU-induced hepatotoxicity (123) (discussed under Graves’ disease, below). The goals of therapy are to utilize the minimal dosage necessary to normalize the fetal heart rate and render the mother euthyroid or slightly hyperthyroid.
In the neonate MMI (0.5 to 1.0 mg/kg/day) has been used initially in 3 divided doses. If the hyperthyroidism is severe, a strong iodine solution (Lugol’ s solution or SSKI, 1 drop every 8 hours) is added to block the release of thyroid hormone immediately. Often the effect of MMI is not as delayed in infants as it is in older children or adults, a consequence of decreased intrathyroidal thyroid hormone storage. Therapy with both antithyroid drug and iodine is adjusted subsequently, depending on the response. Propranolol (2 mg/kg/day in 2 or 3 divided doses) is added if sympathetic overstimulation is severe, particularly in the presence of pronounced tachycardia. If cardiac failure develops, treatment with digoxin should be initiated, and propranolol should be discontinued. Rarely, prednisone (2 mg/kg/day) is added for immediate inhibition of thyroid hormone secretion. Measurement of TSH receptor antibodies in treated babies may be helpful in predicting when antithyroid medication can be safely discontinued (87). Lactating mothers on antithyroid medication can continue nursing as long as the dosage of PTU or MMI does not exceed 400 mg or 40 mg, respectively. The milk/serum ratio of PTU is 1/10 that of MMI, a consequence of pH differences and increased protein binding, so one might anticipate less transmission to the infant, but concerns about potential PTU toxicity need to be considered. At higher dosages of antithyroid medication, close supervision of the infant is advisable.
A review about management of neonates born to mothers with Graves’ disease has been recently published (89b).
Permanent neonatal hyperthyroidism
Rarely, neonatal hyperthyroidism is permanent and is due to a germline mutation in the TSH receptor (TSH-R) resulting in its constitutive activation (90,90a,90b,90c). A gain of function mutation of the TSH-R should be suspected if persistent neonatal hyperthyroidism occurs in the absence of detectable TSH-R antibodies in the maternal circulation. Prematurity, low birth weight and advanced bone age are common. Most cases result from a mutation in exon 10 which encodes the transmembrane domain and intracytoplasmic tail of the TSH-R, a member of the G protein coupled receptor superfamily (90,90a,90b,90c). Less frequently, a mutation encoding the extracellular domain has been described (90d). An autosomal dominant inheritance has been noted in many of these infants; other cases have been sporadic, arising from a de novo mutation.
Early recognition is important because the thyroid function of affected infants is frequently difficult to manage medically (90a-90c), and, when diagnosis and therapy is delayed, irreversible sequelae, such as cranial synostosis and developmental delay may result (90c). For this reason early, aggressive therapy with either thyroidectomy or even radioablation has been recommended (90c).
Two clinical forms were described: the first one is the “familial non-autoimmune autosomal dominant hyperthyroidism” (FNAH). High variable age of manifestation from neonatal period to 60 years, with. variability also within the same family is reported. Goiter is present in children, with nodules in older age.
The second one is “Persistent sporadic congenital non autoimmune hyperthyroidism” (PSNAH) includes forms with sporadic (de novo) germline mutations in the TSH-R.
PSNAH is characterized by fetal-neonatal onset or within 11 months and more severe hyperthyroidism requiring early aggressive therapy. Guidelines about this rare condition have recently been published (90e).
McCune Albright syndrome
McCune Albright is a syndrome due to somatic activating mutations in Gsα gene, can rarely presents with neonatal hyperthyroidism (90f).
THYROID DISEASE IN CHILDHOOD AND ADOLESCENCE
Hypothyroidism and Thyroiditis
Chronic lymphocytic thyroiditis is the more common cause of acquired hypothyroidism in children and adolescents. Occasionally, patients with disorders classified as congenital hypothyroidism, i.e thyroid dysgenesis, inborn error of thyroid hormonogenesis, central hypothyroidism may be recognized later in childhood and adolescence.
Causes of hypothyroidism in children and adolescents are listed in table 5.
TABLE 5CAUSES OF HYPOTHYROIDISM IN CHILDHOOD AND ADOLESCENCE
| PRIMARY HYPOTHYROIDISM |
| A) Congenital |
| Thyroid dysgenesis |
| Inborn error of thyroid hormonogenesis |
| Thyroidal Gsα protein abnormalities (pseudohypoparathyroidism 1B) |
| B) Acquired |
| Autoimmune |
| Chronic Lymphocytic Thyroiditis |
| Reversible autoimmune hypothyroidism (silent and postpartum thyroiditis, cytokine-induced thyroiditis |
| Infiltrative: Cystinosis, Hemocromathosis, Thalassemia,-Langerhans Cell Histiocytosis |
| Infective: acute, subacute thyroiditis |
| Post ablative Surgery |
| Thyroiditis due to I 131, external irradiation of non-thyroidal tumors (i.e. lymphomas, brain tumors, TBI |
| Iodine deficiency and iodine excess |
| Drugs: antithyroid agents, lithium, natural and synthetic goitrogenic chemicals, tyrosine kinase inhibitors, lithium, thionamides, aminosalicylic acid, aminoglutethimide |
| Goitrogens (cassava, water pollutants, cabbage, sweet potatoes, cauliflower, broccoli, soya beans) |
| CENTRAL HYPOTHYROIDISM |
| A)Congenital |
| Pituitary hypoplasia, septo-optic dysplasia, basal encephalocele |
| Functional defects in TSH biosynthesis and release |
| Mutations in genes encoding for TRH receptor, TSHß, pituitary transcription factors (Pit-1, PROP1, LHX3, LHX4, HESX1), or LEPr, IGSF1 |
| B)Acquired |
| Tumors (pituitary adenoma, craniopharyngioma, meningioma, dysgerminoma, glioma, metastases) |
| Trauma surgery, irradiation, head injury |
| Infections |
| Vascular damage ischemic necrosis, hemorrhage, stalk interrruption, |
| Hypotalamic disorders |
| Drugs: dopamine; glucocorticoids; bexarotene; L-T4 withdrawal |
| “Peripheral” (extrathyroidal) hypothyroidism |
| Consumptive hypothyroidism (massive infantile hemangioma) |
| Mutations in genes encoding for MCT8, SECISBP2, TRα or TR β (impaired sensitivity to thyroid hormones) |
Chronic Lymphocytic Thyroiditis
Autoimmune thyroid diseases (AITD) are defined by the lymphocytic infiltration of the thyroid (91). Usually antibodies against thyroid antigens as thyroperoxidase (TPOAb), thyroglobulin (TgAb), and anti-TSH receptor (TRAb) are detectable in serum. Thyroid antibodies in serum correlate with the presence of lymphocytic infiltrate in the thyroid gland. The clinical spectrum of AITD ranges from hypothyroidism to hyperthyroidism and include chronic lymphocytic thyroiditis (CLT) and Graves’ disease. CLT is the most common cause of hypothyroidism in children and adolescents (91,91a).
Graves’ disease and CLT are closely associated and in fact overlapping syndromes .Patients can move from one to the other category, depending upon the stage of their illness. For example, an individual might first be observed with thyroid enlargement and positive antibody tests for anti-thyroglobulin or anti-TPO antibodies, and thus qualify as having CLT. At a later stage, this individual might become hyperthyroid (Hashitoxicosis) and fit in the category of Graves’ disease. Or, the patient with hyperthyroidism might have progressive destruction of the thyroid, or develops blocking antibodies, and become hypothyroid or ultimately develop myxedema.
Incidence
The prevalence of CLT in children and adolescents was reported to be 1.2% by Rallison in 1975 (91b). In this 6 year-survey study 5179 school children were examined in Arizona. Goiter was evaluated by palpation (91b). More recently, a study from Sardinia in 8040 children and adolescents aged 6-15 years reported TPOAb detectable in 2.9% (91c). Similar results were found in Berlin with a prevalence of TPOAb of 3.4% (mean age 11 years) (91d) and in Greece after correction of iodine deficiency. In this study examining 440 children and adolescents aged 5-18 years a prevalence of TPOAb and TgAb was reported to be 4.6% and 4.3% respectively. The prevalence of CLT, confirmed by ultrasound was 2.5% (91e).
In childhood the most common age at presentation is adolescence, but the disease may occur at any age, even infancy. CLT in infancy is rare, but can cause in a short time severe hypothyroidism and permanent damage to CNS if not recognized and treated (85). The female/male ratio in AITD is up to 6:1, but In prepubertal age the female/male ratio is lower than reported in adolescents and adults.
Etiology and Pathogenesis
CLT is thought to be caused by a combination of genetic susceptibility and environmental factors. Both thyroid-specific genes and genes involved in immune recognition and/or response have been identified (91f, 91g). (See chapter Autoimmunity, by A Weetman for an exhaustive information). Some genes are common to both disorders and some tend to predominate only in Graves’ disease. AITD has a striking predilection for females, but in prepubertal age the female/male ratio is lower. A family history of autoimmune thyroid disease (both chronic lymphocytic thyroiditis and Graves’ disease) is found in 30% to 40% of patients. A study about familial clustering of juvenile AITD found thyroid antibodies detectable in 56% of mothers and 25% of fathers. Interestingly, HLA DQ alleles and antibody status in fathers influenced the susceptibility to AITD in children (91h). Siblings recurrence in childhood is 20-30% (91i). AITD are often associated with other autoimmune disorders. The plethora of associations and their familial occurrence indicates that a defect in the immune system may be more likely than primary defects in each organ, as these diseases often share similar genetic associations, including HLA, CTLA-4, PTPN22 and CD25 gene polymorphisms. It is also clear however that there is a difference in the kind of clustering of other autoimmune disease in CLT and Graves’ disease, presumably related to differences between these two types of thyroid disease in genetic predisposition (91j,91k). There is also an increased incidence of CLT in patients with certain chromosomal abnormalities as Down syndrome (91l) Turner syndrome (91m), Klinefelter syndrome (91n) as well as in patients with Noonan syndrome (91o).
Environmental factors as infection, environmental toxins, substances as iodine, selenium, stress, smoking, estrogens, drugs (amiodarone, interferon alfa, lithium) have been suggested as precipitating factors for CLT (91p). The precise environmental trigger has not been yet established. An epigenetic mechanism may be implicated (91q).
Clinical Manifestations
Both goitrous (Hashimoto’s thyroiditis) and nongoitrous (atrophic thyroiditis, also called primary myxedema) as variants of chronic lymphocytic thyroiditis have been distinguished. The term “Hashimoto’s thyroiditis” is often used as a synonymous of CLT, not necessary linked to the presence of goiter (91, 91a). Goiter, present in approximately two-thirds of children with CLT is caused by lymphocytic infiltration that may be extensive, with lymphoid germinal centers, TSH stimulation, or production of antibodies that stimulate thyroid growth (92). Progressive thyroid cell damage, with cell mediated cytotoxicity and follicular cell apoptosis, can change the apparent clinical picture from goitrous hypothyroidism to that of “atrophic” thyroiditis. Atrophic thyroiditis, or primary hypothyroidism/mixedema, is considered to be the end stage of CLT (91).
Children with chronic lymphocytic thyroiditis may be euthyroid, or may have subclinical or overt hypothyroidism. Occasionally, children may experience an initial thyrotoxic phase due to the discharge of preformed T4 and T3 from the damaged gland. Alternatively, as indicated above, thyrotoxicosis may be due to concomitant thyroid stimulation by TSH receptor stimulatory antibodies (Hashitoxicosis).
The onset of hypothyroidism in childhood is insidious. Affected children often are recognized either because of the detection of a goiter on routine examination or because of a poor interval growth rate present for several years prior to diagnosis (92a). Because the deceleration in linear growth tends to be more affected than weight gain, these children can be relatively overweight for their height, although they rarely are significantly obese (Figure 6). If the hypothyroidism is severe and longstanding, immature facies with an underdeveloped nasal bridge and immature body proportions (increased upper-lower body ratio) may be noted. Dental and skeletal maturation are delayed, the latter often significantly. Patients with central hypothyroidism tend to be even less symptomatic than are those with primary hypothyroidism.

Figure 15-6
Sequential changes in physical appearance in a young girl who presented at 15 years of age with amenorrhea and hyperprolactinemia secondary to severe hypothyroidism. Note her poor linear growth since at least 11 years of age.
The classical clinical manifestations of hypothyroidism can be elicited on careful evaluation, though they often are not the presenting complaints. These include sluggishness, lethargy, cold intolerance, constipation, dry skin or hair texture, and periorbital edema. Bradycardia and delayed deep tendon reflexes can be present. In severe, long-standing hypothyroid children pericardial and pleural effusions may occur. School performance is not usually affected, in contrast to the severe irreversible neuro-intellectual sequelae that occur frequently in inadequately treated babies with congenital hypothyroidism. Causes of hypothyroidism associated with a goiter (CLT, inborn errors of thyroid hormonogenesis, thyroid hormone resistance) should be distinguished from non goitrous causes (primary myxedema, thyroid dysgenesis, central hypothyroidism). The typical thyroid gland in a longstanding chronic lymphocytic thyroiditis is diffusely enlarged and has a rubbery consistency. Although the surface is classically described as ’pebbly’ or bosselated, occasionally asymmetric enlargement occurs and must be distinguished from thyroid neoplasia. Alternatively, the thyroid may be normal in size and consistency or not palpable at all. A palpable lymph node superior to the isthmus (“Delphian node”) is often found and may be confused with a thyroid nodule. The thyroid gland, in thyroid hormone synthetic defects, on the other hand, tends to be softer and diffusely enlarged.
In patients with severe hypothyroidism of longstanding duration, the sella turcica may be enlarged due to thyrotrope hyperplasia. There is an increased incidence of slipped femoral capital epiphyses in hypothyroid children. The combination of severe hypothyroidism and muscular hypertrophy which gives the child a “Herculean” appearance is known as the Kocher-Debre-Semelaigne syndrome (92b).
Puberty tends to be delayed in hypothyroid children in proportion to the retardation in the bone age, although in longstanding severe hypothyroidism, sexual precocity has been described. Females with sexual precocity have menstruation, and breast development but relatively little sexual hair. Multicystic ovaries, the etiology of which is unknown, may be demonstrated on ultrasonography. In other cases, galactorrhea or severe menses have been the presenting features. In boys, testicular enlargement may be found (92c). An elevated serum prolactin, the latter possibly due to raised TRH which is known to stimulate prolactin as well as TSH, has been described in some cases, but gonadotropin levels are not consistently elevated. It has been hypothesized that this syndrome of pseudopuberty in hypothyroid patients is due to cross- interaction of the extremely elevated serum TSH with the FSH receptor (92d). Consistent with the latter hypothesis, there is little increase in serum testosterone as might be expected if the FSH, but not luteinizing hormone (LH) receptor is involved and serum gonadotropins are frequently not increased.
Long term follow up studies of children with chronic lymphocytic thyroiditis have suggested that while most children who are hypothyroid initially remain hypothyroid, spontaneous recovery of thyroid function may occur, particularly in those with initial compensated hypothyroidism (93,93a, 93b). A recent five-years prospective study in children and adolescents affected with CLT showed that thyroid dysfunction increased from 27.3% to 47.4% (93c). Therefore, close follow up is necessary.
Although chronic inflammation, leading to neoplastic transformation, is a well-established clinical phenomenon, if CLT can increase the risk for thyroid nodules and thyroid cancer remains controversial. In the past autoimmune thyroiditis has been thought to be protective from thyroid cancer, but several studies both in adults and in children suggested the opposite. Thyroid nodules in healthy children in iodine replete regions are detected in up to 1.6% (94). High prevalence of thyroid nodules, ranging from 13% to 31%, has been reported in children and adolescents with CLT. In a multicentric pediatric retrospective study from Italy, nodules were found in 115/365 patients with CLT (31.5%), and papillary thyroid carcinoma in 11/115 (9.5%) (94a). In a recent study from Turkey, thyroid nodules were detected in 39/300 (13%) of cases of pediatric CLT and papillary thyroid carcinoma was diagnosed in 2 of the 12 cases that underwent FNAB (94b). Recently, in. a retrospective study from United States examining ultrasound characteristic of the thyroid in 154 children and adolescents with goiter, nodules were reported in 20/154 (13%) and PTC in 4/154 (2.5% ) of children. In this study, the same prevalence of nodules (17%) was found in TPOAb positive and TPOAb negative children. Interestingly, one case of PTC was first classified at ultrasound as pseudonodule. Only 15 % of nodules and none of the papillary thyroid carcinoma in these series (PTC) were palpable, although PTC has a diameter ranging from 1.2 to 2.6 cm (94c). A rare variant of PTC, the diffuse sclerosing variant, has also been reported in children with CLT(94d).
Associated Disease
AITD are frequently associated with other common autoimmune disorders as type 1 diabetes (94e,94f) and celiac disease. AITD can be also the first manifestation of autoimmune polyendocrine syndromes (APS1 and 2) (95,95a,95b). A pletora of other autoimmune conditions, organ or non-organ specific disease, can be associated with AITD in childhood and adolescence. CLT is more frequently associated with adrenal and β cell autoimmunity than Graves’ disease (91j). Early identification and treatment of these disorders may be critical and even preserve children from life-threatening events. Long term surveillance is required, because a second autoimmune disorder may occur any time.
Type 1 diabetes. CLT is the most common associated autoimmune disease in type 1 diabetes. In a ten-years observational study of children and adolescents with type 1 diabetes (mean age at diagnosis 10 years), the prevalence of TPOAb and TgAb at diagnosis was 15.4% and 14.4% respectively. The cumulative incidence increased especially in females in mid puberty. In this study about 14% of patients required treatment with L-thyroxine (95c). Children with AITD had islet cell antibodies in 2.3% (95d). Screening for AITD is suggested at diagnosis and every 2-3 years if negative (ADA and ISPAD recommendation). Thyroid function should be checked every year or more frequently if needed, because thyroid dysfunction (both hypothyroidism and hyperthyroidism) affects metabolic control. In overt hypothyroidism hypoglycemia can occur because glucose absorption may be slow and the sensitivity and rate of degradation of insulin is increased. Hepatic gluconeogenesis and peripheral glucose utilization are also reduced. Long term dyslipidemia may affect cardiovascular risk in these patients.
Hyperthyroidism in type 1 diabetic children can precipitate acute complications. In a study on 60456 children and adolescents with type 1 diabetes, hyperthyroidism was diagnosed in 276 (0.46%). Hyperthyroid state was associated with diabetic ketoacidosis, hypoglycemia and hypertension (95e). Life-long surveillance is required.
Celiac disease. Another strong association is with celiac disease, which is found 3 times more commonly in patients with AITD. Intriguingly the autoantibodies which are the hallmark of celiac disease, directed against transglutaminase, can bind to thyroid cells and thus could be implicated directly in thyroid dysfunction (95f). A recent meta-analysis showed that the prevalence of celiac disease in AITD patients in higher in children (6.2%) than in adults (1.2%) (95g). Prevalence of AITD in celiac patients is about 20%. The presence of celiac disease in type 1 diabetes seems to increase the risk for AITD (95h). Undiagnosed celiac disease causes malabsorption with or without gastrointestinal symptoms. Delayed linear growth may be the first manifestation as unexplained change in L-T4 requirement (95i).
Addison’s disease. Addison’s disease is also associated with AITD. In an old report, the prevalence of adrenal antibodies in children with AITD, was found to be 2.3%, while the great majority of children affected with Addison’s disease presented with CLT (95d). Addison’ disease is more often a component of autoimmune polyendocrine syndromes (APS1 and 2) (95,95a,95b). Addison's disease and/or type 1 diabetes mellitus and AITD occasionally co-exist and form the classical autoimmune polyendocrine syndrome type2 (Schmidt syndrome). Undiagnosed adrenal insufficiency, is a life-threatening condition and can be exacerbated by L-thyroxine therapy, because L-thyroxine increases cortisol clearance. Moreover, symptoms of overt hypothyroidism can overlap with adrenal insufficiency manifestations. Adrenal insufficiency is a rare but non-obvious diagnosis in childhood and should be considered in when autoimmune disorders are diagnosed.
Autoimmune gastritis. Autoimmune gastritis was first described in association with AITD as thyrogastric syndrome. Common clinical manifestations in adults are iron deficient or pernicious anemia (95j). Perhaps 45% of patients with autoimmune thyroiditis have circulating gastric parietal cell antibodies. Also in children with AITD, early manifestations of gastric autoimmunity has been reported, with a prevalence of gastric parietal cell antibodies of 30%. In this series, 45% of PCA positive children presented with increased gastrin plasma levels (a marker of atrophic body gastritis) (95l).
Autoimmune polyendocrine syndromes (APS1 and 2)
CLT can be the first manifestation of autoimmune polyendocrine syndromes (APS1 and 2) (95,95a,95b). APS1 tends to present in early childhood and is characterized primarily by mucocutaneous candidiasis, hypoparathyroidism and adrenal deficiency. APS1 is also defined as autoimmune polyendocrinopaty-candidiasis-ectodermal dystrophy (APECED). APS1 results from mutations in the AIRE (autoimmune regulator) gene. It is a rare autosomal dominant disorder with incomplete penetrance (96). In APS1, chronic lymphocytic thyroiditis is found in approximately 10% of patients. APS1 was originally described in Europe. Recently, a report from USA described different clinical features and diagnostic criteria in APECED patients from Western Hemisphere i.e., as initial signs urticarial eruptions, intestinal dysfunction, enamel dysplasia. Classical triad presentation (mucocutaneous candidiasis, hypoparathyroidism and adrenal deficiency) was delayed. Life threatening endocrine complications can be prevented by an early diagnosis (96a). APS2, tends to occur later in childhood or in the adult with a polygenic predisposition. APS2 can be clustering in families with heterogeneous clinical phenotypes. Other disorders, including vitiligo, celiac disease, myasthenia gravis, premature ovarian failure and chronic active hepatitis may be present (95,95a). An extensive review of these associations has been published (96b) and large population data bases have clarified the strength of the various associations in adults (95k,96c).
Many other autoimmune conditions, organ or non-organ specific disease, can be associated with AITD in childhood and adolescence. Increased prevalence of CLT has been found in juvenile idiopathic arthritis (96d), non-segmental vitiligo (96e) and alopecia areata (96f). CLT may be associated with chronic uriticaria (96g) and rarely with immune-complex glomerulonephritis (96h). High prevalence of antinuclear antibodies (ANA) has recently been reported in a series of 93 children (86 with CLT and 8 with GD) without overt rheumatic disorders. In this series ANA positivity was found in 71% of children, ENA positivity in 4% and anti-DNA antibodies in 1% (96i). Growth hormone deficiency on an autoimmune basis has been suggested in a small number of children, of whom 43% have CLT (92j-96k). Nevertheless this is a rare association, hypothetically a growth disorder in a child with CLT can be due to other causes than hypothyroidism as celiac disease or GH deficiency. It is important that clinicians are cognizant of these associations in order to maintain a high index of vigilance.
Laboratory Evaluation
Measurement of TSH is the best initial screening test for the presence of primary hypothyroidism. If the TSH is elevated, then evaluation of the free T4 will distinguish whether the child has subclinical (normal free T4) or overt (low free T4) hypothyroidism. Measurement of TSH, on the other hand, is not helpful in central hypothyroidism. In these cases hypothyroidism is demonstrated by the presence of a low free T4 accompanied by an “inappropriate ‘’ TSH. In the past TRH testing (TRH 7 mcg/kg) was sometimes utilized to distinguish a hypothalamic versus pituitary origin of the hypothyroidism; in hypothalamic hypothyroidism there tends to be a delayed peak in TSH secretion (60-90 minutes versus the normal maximal response at 15-30 minutes) whereas in hypopituitarism there usually is little or no TSH response. However TRH is no longer available in the USA. Furthermore, the reliability of this test in the pediatric range has been questioned (97). Occasionally mild TSH elevation is seen in individuals with hypothalamic hypothyroidism, a consequence of the secretion of a TSH molecule with impaired bioactivity but normal immunoreactivity. Thyroid hormone resistance is characterized by elevated levels of T4 and T3 and an inappropriately normal or elevated TSH concentration. Antibodies to Tg and TPO, the thyroid antibodies measured in routine clinical practice, are detectable in over 90% of patients with chronic lymphocytic thyroiditis. Therefore, they are useful as markers of underlying autoimmune thyroid damage, TPO antibodies being more sensitive. TSH receptor antibodies also are found in a small proportion of patients with chronic lymphocytic thyroiditis. When stimulatory TSH receptor antibodies are present, they may give rise to a clinical picture of hyperthyroidism, the coexistence of chronic lymphocytic thyroiditis and Graves’ disease is known as Hashitoxicosis. In one study, TSH receptor blocking antibodies were found in <10% of children and adolescents with chronic lymphocytic thyroiditis, patients overall, but in 17.8% of those with severe hypothyroidism (defined as a serum TSH concentration >20 mU/L). Unlike in adults, they were found in goitrous as well as nongoitrous patients, and, when present at a high concentration, appeared to persist indefinitely, suggesting that the presence of potent TSH receptor blocking antibodies in adolescent females might identify patients at risk of having babies with transient congenital hypothyroidism in the future (97a,97b).
Imaging studies (thyroid ultrasonography and/or thyroid uptake and scan) may be performed if thyroid antibody tests are negative or if a nodule is palpable. If no goiter is present, imaging studies are helpful in identifying the presence and location of thyroid tissue, and therefore, of distinguishing primary myxedema from thyroid dysgenesis. Inborn errors of thyroid hormonogenesis beyond a trapping defect are usually suspected by an increased radioiodine uptake, and a large gland on scan. Other etiologies of hypothyroidism usually are evident on history. Ultrasound (US) is useful to define size, anatomy, echogenity of the thyroid. Occasionally the finding of heterogeneous echogenicity on ultrasound examination has been described prior to the appearance of antibodies. Diffuse reduction in echogenity, (hypoechoic), and pseudonodules are common findings (98). Moreover, US can be useful in detecting unsuspected thyroid nodules and cancer. In a study examinating US characteristic of the thyroid in 154 children and adolescents with goiter, nodules were reported in 20/154 (13%) and PTC in 4/154 (2.5%) of children. None of the papillary thyroid carcinoma in these series (PTC) was palpable, although a PTC diameter ranging from 1.2 to 2.6 cm was found. Interestingly, one case of PTC was first classified as pseudonodule (94c). The diffuse sclerosing variant of PTC, with a typical US appearance, has also been reported in children with CLT (94d). If there is a cost-effective benefit in performing US in all cases of children with CLT and/or goiter deserves sufficiently powered prospective studies.
Therapy
The typical replacement dose of L-thyroxine (derived from congenital hypothyroidism) in overt severe hypothyroidism is about 4 to 6 mcg/kg/day for children 1 to 5 years of age, 3 to 4 mcg/kg/day for these ages 6 to10 years and 2-3mcg/kg/day for these 11 ages and older. Lower dose as 1 to 3 mcg/kg/day may be sufficient in less severe cases. The dose should be individually titrated as the lowest useful to keep TSH in the normal range and FT4 or T4 in the upper half of the reference range. L-thyroxine should be given once daily preferably half to 1 hour before meal. Somministration of preparates (i.e., calcium, soya), or drugs that can interfere with absorption should be avoided. T4 and TSH should be measured after the child has received the recommended dosage for at least 6-8 weeks. Once a euthyroid state has been achieved, patients should be monitored every 6 to 12 months. In patients with a goiter a somewhat higher L-thyroxine dosage is used so as to keep the TSH in the low normal range, and thereby minimize its goitrogenic effect.
Close attention is paid to interval growth and bone age as well as to the maintenance of a euthyroid state. Thyroid hormone replacement is not associated with significant weight loss in overweight children, unless the hypothyroidism is severe (99b). Some children with severe, long standing hypothyroidism at diagnosis may not achieve their adult height potential even with optimal therapy (99c), emphasizing the importance of early diagnosis and treatment. Treatment is usually continued indefinitely.
Treatment of children and adolescents with subclinical hypothyroidism (normal free T4, elevated TSH) is controversial (100). Because normalization of TSH is also possible if the patient is not symptomatic, a reasonable option is to reassess thyroid function in 3- 6 months prior to initiating therapy because of the possibility that the thyroid abnormality will be transient.
In adults in whom the risk of progression to overt hypothyroidism is significant, treatment has been recommended whenever the serum TSH concentration is >10 mU/L ; if the TSH is 6-10 mU/L treatment on a case by case basis is suggested (100a). In children and adolescents, a recent five-years prospective study showed that in patients with CLT thyroid dysfunction increased from 27.3% to 47.4% (93c). Some considerations about “to treat or not to treat” “subclinical hypothyroidism” in children with a known cause of thyroid failure as CLT may be useful.
Well designed and adequately powered trials needed to establish the advantages of treating “subclinical hypothyroidism” are not available in adults and seem to be very difficult in children, also because some of the clinical consequences- i.e cardiovascular events- of untreated mild hypothyroidism may hypothetically occur later in adult life.
In adults, variations in thyroid function within the reference range may be associated with adverse health outcome (100b), and some data suggesting clinical consequences of subclinical hypothyroidism are also available in children and adolescents. A positive relationship was found for TSH levels and systolic and diastolic blood pressure (100c), atherogenic lipid profiles, (100d) and other risk factors for cardiovascular diseases (100e).
Moreover, adult patients with a thyroid nodule and TSH in the upper tertiles of the reference range may be at increased risk of malignancy (100f). Given an individual set point for TSH, more than an absolute TSH value (i.e more or less 6 7-, 8 UI/mL) the decision about treatment may consider the temporal trend of TSH in a patient. An increase of TSH value over the time suggests the progression of the grade of hypothyroidism. Both the decisions to “wait and see” or “to treat” require monitoring the thyroid function and clinical follow up. A careful discussion about “the state of the art” must be taken with the child and the family.
Guidelines about the management of subclinical hypothyroidism in pregnancy and in children have been published by the European Thyroid Association in 2014 (100g), but it as an evolving and open field (100).
Thyroid Dysgenesis and Inborn Errors of Thyroid Hormonogenesis
Occasionally, patients with thyroid dysgenesis will escape detection by newborn screening and present later in childhood with non goitrous hypothyroidism or with an enlarging mass at the base of the tongue or along the course of the thyroglossal duct. Similarly, children with inborn errors of thyroid hormonogesis may only be recognized later in childhood because of the detection of a goiter.
Drugs or Goitrogens
In addition to antithyroid medication, a number of drugs used in childhood may affect thyroid function, including certain anticonvulsants, lithium, amiodarone, aminosalicylic acid, aminoglutethimide and sertraline (101-101a). Similarly, a large number of naturally occurring goitrogens (broccoli, cabbage, sweet potatoes, cauliflower, soya beans, cassava and water pollutants) have been identified. Both radioiodine therapy and thyroidectomy, occasionally used in childhood for the definitive treatment of Graves’ disease, frequently cause permanent hypothyroidism.
Worldwide, iodine deficiency continues to be an important cause of hypothyroidism, affecting at least 800 million people living largely in developing countries. In addition, even in certain parts of Europe, an estimated 100-120 million individuals are thought to have borderline iodine deficiency (101b). Although one rarely sees iodine deficiency in North America, an iodine sufficient area, a 6 year old boy with goitrous hypothyroidism has been described in whom iodine deficiency was due to multiple food allergies and severe dietary restriction (101c). In addition, the child consumed a large intake of thiocyanate-containing foods that blocked organification of iodine.
Miscellaneous Causes of Acquired Hypothyroidism
Rarely, the thyroid gland may be involved in generalized infiltrative or infectious disease processes that are of sufficient severity to result in a disturbance in thyroid function (i.e., (Langerhans cell histiocytosis) (101 d). Alternatively, hypothyroidism may be a long term complication of mantle irradiation for Hodgkin’s disease or lymphoma. External irradiation of brain tumors in the posterior fossa of the brain may be associated with both primary and secondary hypothyroidism because of the inclusion of the neck in the radiation field. Rarely, hypothyroidism has been reported in infants with large hemangiomas (85b,85c). In these cases, the hypothyroidism was shown to be due to increased inactivation of T4 by the D3 activity of these tumors.
Central Hypothyroidism
Secondary or tertiary hypothyroidism in less severely affected children with the congenital abnormalities noted earlier in this chapter, may be recognized only later in childhood. Alternatively, secondary or tertiary hypothyroidism may develop as a result of acquired damage to the pituitary or hypothalamus, i.e., by tumors (particularly craniopharyngioma), granulomatous disease, head irradiation, infection (meningitis), surgery or trauma. Usually other trophic hormones are affected, particularly growth hormone.
Impaired Sensitivity to Thyroid Hormone (Thyroid Hormone Resistance)
In contrast to the neonatal period, children with thyroid hormone resistance usually come to attention when thyroid function tests are performed because of poor growth, hyperactivity, a learning disability or other nonspecific signs or symptoms. A small goiter may be appreciated. Affected patients have a high incidence of attention deficit hyperactivity disorder (102). Thyroid hormone resistance has also been described in patients with cystinosis (102a).
Other causes of goiter: Colloid or Simple (Nontoxic) Goiter
Colloid goiter is the second most common cause of euthyroid thyroid enlargement in childhood after CLT. The etiology of colloid goiter is unknown. Not infrequently there is a family history both of goiter, chronic lymphocytic thyroiditis and Graves’ disease, leading to the suggestion that colloid goiter, too, might be an autoimmune disease. Immunoglobulins that stimulated thyroid growth in vitro have been identified in a proportion of patients with simple goiter, but their etiological role is controversial (103). It is important to distinguish patients with colloid goiter from chronic lymphocytic thyroiditis because of the risk of developing hypothyroidism in patients with chronic lymphocytic thyroiditis, but not colloid goiter. Whereas many colloid goiters regress spontaneously, others appear to undergo periods of growth and regression, resulting ultimately in the large nodular thyroid glands later in life.
Clinical Manifestations and Laboratory Investigation
Evaluation of thyroid function by measurement of the serum TSH concentration is the initial approach to diagnosis. In euthyroid patients, the most common situation, chronic lymphocytic thyroiditis should be distinguished from colloid goiter. Clinical examination in both instances reveals a diffusely enlarged thyroid gland. Therefore, the distinction is dependent upon the presence of elevated titers of TPO and Tg antibodies in chronic lymphocytic thyroiditis but not colloid goiter. All patients with negative thyroid antibodies initially should have repeat examinations because some children with chronic lymphocytic thyroiditis will develop positive titers with time.
Therapy
Thyroid suppression in children with a euthyroid goiter is controversial (103a). A significant decrease in goiter size in patients with chronic lymphocytic thyroiditis as assessed by standard deviation score on ultrasonography has been demonstrated recently in patients treated for 3 years (103b). However, the absolute difference quantitatively was not reported and so, whether or not this difference was significant clinically remains unclear. Given the variability in response in different patients, it would be reasonable to attempt a therapeutic trial in patients whose goiter is large.
Painful thyroid: Acute suppurative thyroiditis, subacute thyroiditis
Painful thyroid enlargement is rare in pediatrics and suggests the probability of either acute (suppurative) (106) or subacute thyroiditis (106a). Rarely chronic lymphocytic thyroiditis may be associated with intermittent pain and be confused with the latter disorders. In acute suppurative thyroiditis, progression to abscess formation with the potential of rupture may occur rapidly so prompt recognition and antibiotic therapy are essential (106b). Acute suppurative thyroiditis is a potentially life-threatening endocrine emergency. It is often preceded by an upper respiratory infection, and can be initially misdiagnosed in a young child presenting with high fever, sore throat, and severe dysphagia. A tender very painful swelling in the region of the thyroid gland is present and the abscess can progress to the surrounding tissues and to the skin. Recurrent attacks and involvement of the left lobe suggest a pyriform sinus fistula between the oropharynx and the thyroid as the route of infection (106c). In the latter case, surgical extirpation of the pyriform sinus will frequently prevent further attacks. The management of this condition has recently been reviewed (106,106b). Subacute viral thyroiditis (or de Quervain or granulomatous thyroiditis) it is rarely reported in childhood and adolescence, but cases at 2-3 years of age are known (106a). Usually subacute thyroiditis presents with sore throat, fever and firm, painful tender enlargement of the thyroid. Mild signs of hyperthyroidism can be overlooked. Subacute thyroiditis may occur as a acute, subacute or rarely chronic disorder. A painless variant has been described also in children. (106d). Therapy is usually symptomatic, because the disease is self-limiting. Sometimes treatment with prednisone (0.5-1mg/kg/die) for a short period (i.e. one week) can be useful.
THYROTOXICOSIS AND HYPERTHYROIDISM
Thyrotoxicosis is defined as the clinical syndrome of hypermetabolism resulting from increased free thyroxine (T4) and/or free triiodothyronine (T3) serum levels (107)). The term thyrotoxicosis is not synonymous with hyperthyroidism, the elevation in thyroid hormone levels caused by an increase in their biosynthesis and secretion by the thyroid gland (Table 6). For example, thyrotoxicosis can result from the destruction of thyroid follicles and thyrocytes in the various forms of thyroiditis, or it can be caused by an excessive intake of exogenous thyroid hormone. It should also be noted that the elevation of free thyroid hormone levels does not always result in thyrotoxicosis in all tissues. In the syndrome of Resistance to Thyroid Hormone (RTH), dominant negative mutations in the thyroid hormone receptor β ( TR β) result in decreased thyroid hormone action in tissues where TRβ is the predominant receptor, for example in the liver and the pituitary, whereas other tissues such as the heart, which express mainly TR α, show signs of increased thyroid hormone action. The determination of the etiology of thyrotoxicosis is of importance in order to establish a rational therapy.
TABLE 6CAUSES OF THYROTOXICOSIS IN CHILDHOOD AND ADOLESCENCE
| THYROTOXICOSIS DUE TO HYPERTHYROIDISM (increased production of T3, T4) |
| Autoimmune hyperthyroidism |
| · Graves’ disease |
| · Hashitoxicosis |
| Congenital non autoimmune hyperthyroidism |
| · Sporadic (de novo Persistent sporadic congenital non autoimmune hyperthyroidism (PSNAH) |
| · Hereditary familial non-autoimmune autosomal dominant hyperthyroidism (FNAH) |
| Autonomous functioning nodules |
| · Toxic adenoma |
| · Hyperfunctioning papillary or follicular carcinoma |
| · Toxic multinodular goiter |
| · McCune Albright disease |
| TSH-induced hyperthyroidism |
| · TSH-producing pituitary adenoma |
| · Thyroid Hormone resistance Tumors |
| · Hydatiform mole, choriocarcinoma |
| · Struma ovari, teratoma (autonomous function of thyroid tissue in ovarian) |
| THYROTOXICOSIS WITHOUT HYPERTHYROIDISM- |
| Transient thyrotoxicosis (Release of stored hormones) |
| · Chronic lymphocytic thyroiditis |
| · Subacute thyroiditis |
| · Silent thyroiditis |
| · Drug-induced thyroiditis |
| · Exogenous causes |
| · Thyroid hormone ingestion |
| · Iodine -induced hyperthyroidism (iodine, radiocontrast agents, amiodarone) |
Graves’ Disease
Autoimmune thyroid disease (AITD), including Chronic lymphocytic thyroiditis and Graves’ disease share immunological abnormalities, histological changes in the thyroid, and genetic predisposition and associated diseases. (See chronic lymphocytic thyroiditis section). The clinical spectrum of AITD ranges from hypothyroidism to hyperthyroidism.
More than 95% of cases of hyperthyroidism in children and adolescents are due to Graves’ disease, (107a) an autoimmune disorder characterized by hyperthyroidism, goiter and a particular opthalmopathy. TSH receptor antibodies that mimic the action of TSH (TRAb), causing increased thyroid hormonogenesis and growth are specific of Graves’ disease, but other autoantibodies, as AbTPO and AbTg, are detectable.
Incidence
Graves’ disease is rare in children and adolescents. However, incidence rates of thyrotoxicosis below 15 years of age are increased in the last years. A study from Denmark reported an incidence of 1.58/100.000 person-years in the period 1998-2012 versus 0.79/100.000 person-years in 1982-1988 (107b). There is a strong female predisposition, the female:male ratio being 6 to 8:1. Although it can occur at any age, it is most common in adolescence. Prepubertal children tend to have more severe disease, to require longer medical therapy and to achieve a lower rate of remission as compared with pubertal children (107c). This appears to be particularly true in children who present at <5 years of age (107d). Graves’ disease has been described in children with other autoimmune diseases, both endocrine and non endocrine. These include diabetes mellitus, Addison’s disease, vitiligo, systemic lupus erythematosis, rheumatoid arthritis, myasthenia gravis, periodic paralysis, idiopathic thrombocytopenia purpura and pernicious anemia. (See also associated diseases in CLT). There is an increased risk of Graves’ disease in children with Down syndrome (trisomy 21) (107e).
Pathogenesis
The cause of Graves’ disease is unclear. For unknown reasons the immune system produces TSH receptor antibodies that mimic the action of TSH. Binding of ligand results in stimulation of adenyl cyclase and thyroid hormonogenesis and growth (107f, 107g). Presumably a complex interaction between genetic susceptibility (i.e., HLA, CTLA4, PTN22 genes) and environmental factors contribute. A familial history of AITD is often present, as well as for other autoimmune diseases.
Unlike chronic lymphocytic thyroiditis in which thyrocyte damage is predominant, the major clinical manifestations of Graves’ disease are hyperthyroidism and goiter. As noted earlier, TSH receptor blocking antibodies, in contrast, inhibit TSH-induced stimulation of adenyl cyclase. Both stimulatory and blocking TSH receptor antibodies bind to the extracellular domain of the receptor and appear to recognize apparently discrete linear epitopes in the context of a three-dimensional structure (107g). A number of different monoclonal stimulating Abs including one derived from a patient with Graves’ disease have now been generated (107h) and the crystal structure of the human monoclonal stimulating TSH receptor Ab complexed with a portion of the TSH receptor ectodomain has been accomplished (107i). Taken together, a picture has emerged of distinct but overlapping binding sites of both stimulating and blocking TSH receptor Abs and of TSH to the leucine rich TSH receptor ectodomain (107j). Current evidence suggests that it is the shed A subunit rather than the intact, holoreceptor that induces TSH receptor Abs leading to hyperthyroidism (107j). Studies employing monoclonal TSH receptor antibodies cloned from patients and recombinant mutant TSH receptor have demonstrated that there exist multiple TSH receptor antibodies each with different specificities and functional activities. There is evidence that stimulatory antibodies are mostly lambda and of the IgG1 subclass, strongly suggesting that they are monoclonal or pauciclonal (107k). Blocking antibodies, on the other hand, are not similarly restricted.
Clinical Manifestations
The major clinical manifestations of Graves’ disease are hyperthyroidism and goiter. Opthalmopathy is usually mild, pretibial myxedema and acropachy are not described in children and adolescents.
The onset of Graves’ disease in often insidious and a delay in diagnosis of several months is common, especially in prepubertal children (107c). In children below 4 years of age, the prolonged hyperthyroidism can be dangerous to the CNS (107d). Shortened attention span, and emotional lability may lead to behavioral and school difficulties. Sleep disturbances, and nightmares can occur. Some patients complain of polyuria and of nocturia, the result of an increased glomerular filtration rate. All but a few children with Graves’ disease present with some degree of thyroid enlargement, and most have symptoms and signs of excessive thyroid activity, such as tremors, inability to fall asleep, weight loss despite an increased appetite, diarrhea, proximal muscle weakness, heat intolerance and tachycardia. Acceleration in linear growth may occur, often accompanied by advancement in skeletal maturation (bone age). Adult height is not affected. In the adolescent child, puberty may be delayed. If menarche has occurred, secondary amenorrhea is a common concomitant. If sleep is disturbed, the patient may complain of fatigue. Clinical findings are usually related to hyperthyroid state and disappear with restoration to the euthyroid state (108).
Graves’ opthalmopathy in children and adolescents is reported in up to half of the children and is usually less severe than in adults. Eyelid retraction and “stare” are common and linked to hyperthyroid state (108a). Proptosis is subtle and often overlooked. Normal references for children should be used (108b). Some cases of prominent progressive proptosis requiring treatment have been reported (108c). Surgical therapy is infrequently necessary. In a series of 35 children with Graves’ opthalmopathy from Mayo Clinic 3 patients (8.6%) underwent transantral orbital decomprenssion for proptosis that caused discomfort and exposure keratitis and 1 patient (2.9%) required eyelid surgery. No compressive optic neuropathy was found (108d).
Laboratory Evaluation
The clinical diagnosis of hyperthyroidism is confirmed by the finding of increased concentrations of circulating thyroid hormones (T4 or, preferably, free T4 and T3 or FT3) and low- undetectable TSH. In hyperthyroidism, the circulating T3 concentration frequently is elevated out of proportion to the T4 because, like TSH, TSH receptor antibodies stimulate increased T4 to T3 conversion. Some patients may have at diagnosis high FT3 and normal FT4, a condition known as T3 thyrotoxicosis (109). Children with T3 thyrotoxicosis seem to be younger, with higher levels of TRAb and larger goiter than classical GD. The timing of T3 thyrotoxicosis onset is variable and can require higher doses of ATD to control hyperthyroidism (109a). Demonstration of a suppressed TSH excludes much rarer causes of thyrotoxicosis, such as TSH-induced hyperthyroidism and thyroid hormone resistance in which the TSH is inappropriately “normal” or slightly elevated. If the latter diseases are suspected, free α-subunit should be measured. Alternatively, an elevated T4 level in association with an inappropriately “normal” TSH may be due to an excess of thyroxine-binding globulins (either familial or acquired, for example a result of oral contraceptive use) or rarer binding protein abnormalities (for example, familial dysalbuminemic hyperthyroxinemia) (109b). In the latter cases, serum TBG concentration or electrophoresis of T4 binding proteins, respectively, should be measured. If pregnancy or an hCG-secreting tumor are suspected, serum or urinary hCG concentration can be measured. A low serum Tg can be demonstrated if thyrotoxicosis factitia is suspected (109c).
The diagnosis of Graves’ disease is confirmed by the demonstration of TSH receptor antibodies (TRAb) in serum. TRAb are disease specific antibodies and have a pathogenetic role in Graves’ disease. TRAb are usually determined by binding assays. Bioassays that measure Thyroid Stimulating Immunoglobulins activity based on cAMP on cultured cells can be useful if TRAb are not detectable by binding assays (88a,88b,88c). About 95% of children with GD have TRAb detectable. There is a positive correlation between severity of Graves’ disease and TR antibodies level. Higher levels of TRAb and thyroid hormones at presentation are associated with a need of prolonged ATD treatment (109d). Measurement of TSH receptor antibodies may be useful in distinguishing the toxic phase of chronic lymphocytic thyroiditis (TSH receptor antibody negative) from Graves’ disease. Tg and TPO antibodies are positive in 70% of children and adolescents with Graves’ disease but their measurement is not as sensitive or specific as measurement of TSH receptor antibodies. In contrast to adults, radioactive iodine uptake and scan are used to confirm the diagnosis of Graves’ disease only in atypical cases: for example, if measurement of TSH receptor antibodies is negative, or if a functioning thyroid nodule is suspected.
Therapy
The care of children with Graves’ disease can be complicated and requires physicians with expertise in this area. Treatment guidelines developed for adults guidelines cannot be simply applied to children. For instance, TRAb may be detectable in serum for several years, making the terms “remission” and “recidive” inapplicable in 1-2 years periods for the majority of children and adolescents.
The choice of which of the three therapeutic options (medical therapy, surgery radioactive iodine, or radioactive iodine) to use, should be individualized and discussed with the patient and his/her family. Each approach has its advantages and disadvantages with respect to efficacy, both short and long term complications, the time required to control the hyperthyroidism, and the requirement for compliance. In general, medical therapy with methimazole (MMI) is the initial choice of most pediatricians although radioiodine is gaining increasing acceptance, particularly in noncompliant adolescents, in children who are developmentally delayed, and in those about to leave home (for example, to go to college). Concern about the potential long term induction of cancer by RAI given to children is the discussed later. Alternately, surgery, the oldest form of therapy, may be the initial choice in specific cases if an experienced thyroid surgeon is available. ATA guidelines for hyperthyroidism including a pediatric section have recently been released (88c).
Medical therapy
The thiouracil compounds PTU, MMI and carbimazole (converted to MMI) exert their antithyroid effect by inhibiting the organification of iodine and the coupling of iodotyrosine residues on the Tg molecule to T3 and T4.
The aim of therapy with antithyroid drugs is to control hyperthyroidism for a period sufficient to go to spontaneous remission or until the child is old enough to afford definitive therapy as surgery or RAI. Remission is defined as a state of biochemically euthyroidism or hypothyroidism for one year or more after discontinuation of ATD and occurs in a minority of cases (see later).
Some important considerations arose in the last years: MMI is the drug that should be used, unless special conditions, because of the inacceptable risk of liver failure and transplantation (FDA Propilthyuracil warning) in patients using PTU. PTU can cause fulminant hepatic necrosis and death. The risk was estimated to be 1:2000 in children (110,110a,110b). Propylthiouracil and methimazole have for years been considered effectively interchangeable, and liver damage was considered a very rare event. Recently a commission appointed by the FDA reevaluated this problem, and concluded that the rare but severe complications of liver failure needing transplantation, and death, were sufficient to contraindicate the use of PTU as the normal first-line drug (110c). PTU can be used only in pediatric patients who are allergic to MMI, for a short term, and in whom permanent forms of therapy are not possible. MMI should be used alone, titrating the dosage at the lowest useful to maintain euthyroidism. The “block.and replace therapy”, adding L-thyroxine to MMI should be avoided, because it requires a higher dose of MMI, and the majority of side effect of MMI are dose dependent.
The initial dosage of MMI is 0.5 mg/kg/day (up to 1mg/kg/die, maximal dose 30 mg/die) given every 12 hours. The plasma half-life of methimazole in children is only 3-6 hours, but the drug is concentrated in the thyroid and maintains higher levels there for up to 24 hours after a dose (110d). The initial dosage of PTU is 5 mg/kg/day given every 8 hours. In severe cases, a beta-adrenergic blocker (atenolol, 25 to 50 mg daily or twice daily) can be added to control the cardiovascular overactivity until a euthyroid state is obtained.
Before FDA warning for PTU MMI was generally preferred over PTU because for an equivalent dose it requires taking fewer tablets, it has a longer half-life (and so, requires less frequent medication) and because it has a more favorable safety profile. PTU use has also been advocated in the first trimester of pregnancy. PTU but not MMI inhibits the conversion of T4 to the more active isomer T3.
Patients treated with MMI should be followed every 4 to 6 weeks until the serum concentration of T4 (or free T4 and total T3) normalizes. It should be noted that the TSH concentration may not return to normal until several months later. Therefore, measurement of TSH is useful as a guide to therapy only after it has normalized but not initially. Once the T4 and T3 have normalized, one can decrease the dosage of thioamide drug by 30% to 50%. Maintenance doses of MMI may be administered once daily. PTU may be given twice daily. Usually patients can be followed every 1-4 months once thyroid function has normalized.
As suggested by the ATA guidelines (88c), before starting therapy with ATD, a baseline complete blood count, including WBC with differential, and a liver including bilirubin, transaminases and alkaline phosphatase can be useful. This is because hyperthyroidism itself can determine low WBC count, and premorbid liver disease (i.e autoimmune hepatitis reported in 1% of GD) can exist (110e). Baseline information may help in a correct interpretation of side effects of MMI.
In most children and adolescents, circulating thyroid hormone levels can be normalized readily with antithyroid medication as long as compliance is not a problem. The optimal duration of therapy is controversial. There is no doubt that most children and adolescents, particularly prepubertal ones, require a longer course of therapy than adults. Therefore treatment guidelines developed for older individuals should not be applied to the young. In one retrospective study, TSH receptor Abs disappeared from the circulation in <20% of patients after 13-24 months of medical therapy (110f) in contrast to adults in most of whom TSH receptor Abs normalize by 6 to 12 months (110g,110h, 110i). In another study, approximately 25% of children remitted with every 2 years of therapy up to 6 years of treatment (110j). Equivalent results have been obtained by others (107c). In a recent prospective trial of 154 children with newly diagnosed Graves’ disease treated with carbimazole, 20% of children remitted after 4 years of therapy, 37% after 6 years and 45% after 8 years (110k). The median duration of therapy in most studies is 3 to 4 years, but therapy should be individualized. In patients treated with antithyroid drugs alone, a low drug requirement, small goiter, and lack of orbitopathy are favorable indicators that drug therapy can be tapered gradually and withdrawn. Lower initial degree of hyperthyroxinemia (T4<20 mcg/dL (257.4 nmol/L); T3:T4 ratio <20), lower initial TSH receptor Ab concentration (>4X upper limit of normal (111e) and postpubertal age are favorable prognostic indicators. Persistence of TSH receptor antibodies, on the other hand, indicates a high likelihood of relapse. Initial studies suggesting that combined therapy (i.e., antithyroid drug plus L-thyroxine) might be associated with an improved rate of remission (110l) have not been confirmed (110m).
Side effects
Side effects of drugs were reported in 20-30% of children treated both with PTU and MMI and major side effect are thought to be due to PTU. Cumulative data from more than 500 children (111) with Graves’ disease reported mild increase of liver enzymes in 28%, mild leucopenia in 26% skin reactions in 9%, arthritis in 2.4%, nausea in 1.1%, agranulocytosis and hepatitis in 0.4%. Rare complications can be loss of taste, hypothrombinemia, thrombocytopenia, aplastic anemia, nephrotic syndrome and death (111). Side effects of MMI occur in up to 19% of children. Urticaria, arthralgias, gastrointestinal problems and neutropenia (<1500 granulocytes/mm3) are the most common, myalgias (3%), and cholestatic liver injury (1%) were also reported in a series of 100 children with Graves’ disease exclusively treated with MMI. Side effects usually occur in the first 6 months of therapy (111a) but can occur any time.
Major side effects as Stevens-Johnson syndrome and vasculitis occur rarely (111). Vasculitis can be related with the development of anti-neutrophil-cytoplasmic antibodies (ANCA). ANCA positivity has been reported with MMI and PTU therapy and may develop after many years of therapy (111b). Manifestations of vasculitis typically are polyarthritis and purpuric skin lesions. Pulmonary and renal involvement are also described. In severe cases, glucocorticoids or other immunosuppressive therapy may be needed. Guma et al reported ANCA positivity in 67% of patients with Graves’ disease before medical treatment, suggesting an association with Graves’ disease, rather than a complication of antithyroid drugs (111c).
Rarely, more severe sequelae such as hepatitis, a lupus like syndrome, thrombocytopenia, and agranulocytosis may occur. Most reactions are mild and do not contraindicate continued use. The risk of agranulocytosis (<500 granulocytes/mm3) appears to be greatest within the first 3 months of therapy but it can occur at any time. There is some evidence that close monitoring of the white blood cell count during this initial time period may be useful in identifying agranulocytosis prior to the development of a fever and infection (111d), but most authors do not consider the low risk to be worth the cost of close monitoring. It is important to caution all patients to stop their medication immediately and consult their physician should they develop unexplained fever, sore throat, or gingival sores or jaundice. Unlike PTU, MMI is rarely associated with hepatocellular injury.
Children treated with PTU and MMI tends to excessive weight gain during the first 6 months of therapy and nutrition consultation should be considered if needed (111e). Approximately 10% of children treated medically will develop long term hypothyroidism, a consequence of coincident cell and cytokine-mediated destruction.
Patients with Graves’ disease showed a higher risk of thyroid cancer (111). The Collaborative Thyrotoxicosis Study Group found the incidence of thyroid carcinomas over 10-20 years of follow up 5 fold higher in adults with Graves’ disease treated with thionamides than in patients treated with definitive therapy (111g). Long term stimulation of TSAb can play a role. Patients treated for years with thionamides should be carefully monitored for the detection of thyroid nodules.
Surgery
Surgery, the second therapeutic modality, is performed less frequently now than in the past. The main argument favoring surgery is that it may correct the thyrotoxicosis with surety and speed, and result in less disruption of normal life and development that is associated with long-term administration of antithyroid drugs and the attendant constant medical supervision.
The most important limiting factor is the availably of a high-volume thyroid surgeon to reduce potential complications (112,112a,112b). Near-total thyroidectomy is the procedure of choice in order to minimize the risk of recurrence. Surgery usually is reserved for patients who have failed medical management, who have a markedly enlarged thyroid, who refuse radioactive iodine therapy, and for the rare patient with significant ophthalmopathy in whom radioactive iodine therapy is contraindicated. Often adolescents are unable to maintain the careful dosage schedule needed for control of the disease .and can choice a definitive treatment. Surgical complication rates are higher in younger children (112c). The most common potential complication is transient hypocalcemia which occurs in approximately 10% of patients. Starting therapy with calcitriol 3 days before surgery (0.25 to 0.5 µg twice a day), can reduce the need for calcium infusion and the length of stay (112c). Other, less common potential complications are keloid formation (2.8%), recurrent laryngeal nerve paralysis (2%), hypoparathyroidism (2%) and, rarely (0.08%) death (111). There are fewer complications with an experienced surgeon and when modern methods of anesthesia and pain control are used (112). Prior to surgery, it is important to treat with antithyroid medication in order to render the child euthyroid and prevent thyroid storm. Iodides (Lugols solution, 5 to 10 drops tid or potassium iodide, 2 to 10 drops daily or Na ipodate, 0.5-1 gm every 3 days) are added for 7 to 14 days prior to surgery in order to decrease the vascularity of the gland. L-thyroxine replacement therapy should be given within days of surgery. Following surgical thyroid ablation most patients become hypothyroid and require lifelong thyroid replacement therapy. On the other hand, if therapy is inadequate, hyperthyroidism may recur. Therefore long-term follow-up is mandatory.
131-I Therapy
Definitive therapy with either radioactive iodine or surgical thyroid ablation is usually reserved for patients who have failed drug therapy, developed a toxic drug reaction, or are noncompliant. In recent years, however, radioactive iodine is being favored increasingly, even as the initial approach to therapy (111). The advantages are the relative ease of administration, the reduced need for medical follow up and the lack of demonstrable long term adverse effects (111). The aim of RAI is to ablate completely the thyroid gland and thereby reduce the risk of future neoplasia. RAI should be administrated in a single dose.
Although a dose of 50 to 200 ïCi of 131I/estimated gram of thyroid tissue has been used, the higher dosage is recommended, particularly in younger children, in order to completely ablate the thyroid gland and thereby reduce the risk of future neoplasia. The size of the thyroid gland is estimated, based on the assumption that the normal gland is 0.5-1.0 gms/year of age, maximum 15-20 gms. The formula used is: Estimated thyroid weight in grams X 50-200 mcCi 131 -I/fractional 131I 24 hour uptake Thyroid size can be assessed by ultrasound because underestimation and consequent insufficient RAI treatment is frequent. Surgery may be indicated for goiters larger than 80 gr. Radioactive iodine therapy should be used with caution in children <10 years of age and particularly in those <5 years of age because of the increased susceptibility of the thyroid gland in the young to the proliferative effects of ionizing radiation (113). Pretreatment with antithyroid drugs prior to RAI therapy is advisable if the hyperthyroidism is severe. Thyroid hormone concentrations may rise transiently 4 to 10 days after RAI administration due to the release of preformed hormone from the damaged gland. Beta blockers may be useful during this time period. Similarly, analgesics may be employed if there is mild discomfort due to radiation thyroiditis. Other acute complications of RAI therapy (nausea, significant neck swelling) are rare. One usually sees a therapeutic effect within 6 weeks to 3 months. Worsening of ophthalmopathy, described in adults after RAI, does not appear to be common in childhood. However, if significant ophthalmopathy is present RAI therapy should be used with caution and pretreatment with steroids may be effective. Alternately, another permanent treatment modality (surgery) should be considered.
The question of an age limit below which RAI should not be used frequently arises. With lengthening experience these limits have been lowered. Several studies with average follow-up periods of 12 – 15 years attest to the safety of 131-I therapy in adults (111g,113a,113b). In two studies treated persons showed no tendency to develop thyroid cancer, leukemia, or reproductive abnormalities, and their children had no increase in congenital defects or evidence of thyroid damage (113c,113d). Franklyn and co workers (113e) reported on a population based study of 7417 patients treated with 131-I for thyrotoxicosis in England. They found an overall decrease in incidence of cancer mortality, but a specific increase in mortality from cancer of the small bowel (7 fold) and of the thyroid (3.25) fold 9 (113e). The absolute risk remains very low, and it is not possible to determine whether the association is related to the basic disease, or to radioiodine treatment.
There are less data about long term effects of RAI therapy in pediatric Graves’ disease. In an early report, 73 children and adolescents were so treated. Hypothyroidism developed in 43. Subsequent growth and development were normal (113f). In another group of 23 children treated with 131-I, there were 4 recurrences, at least 5 became hypothyroid, and one was found to have a papillary thyroid cancer 20 months after the second dose (113g). Safa et al. (113a) reviewed 87 children treated over 24 years and found no adverse effects except the well-known occurrence of hypothyroidism. Hamburger (115c) has examined therapy in 262 children ages 3 – 18 and concluded 131-I therapy to be the best initial treatment. Read et al (113h) reviewed experience with 131-I over a 36 year period, including six children under age 6, and 11 between 6 and 11 years. No adverse effects on the patients or their offsprings were found, and they advocate 131-I as a safe and effective treatment. In a review including approximately 1000 children with Graves’ disease treated with RAI and followed for <5 to >20 years to date, (111) there does not appear to be any increased rate of congenital anomalies in offspring nor in thyroid cancer. However, long term follow up data in a larger cohort are still lacking. The epidemic of thyroid cancer apparently induced by radioactive iodine isotopes in infants and children living around Chernobyl suggests caution in use of 131-I in younger children.
Since the possibility of a general induction of cancer by 131-I is of central concern, it is interesting to calculate the risk in children using the data presented by Rivkees et al (113i) who are proponents of use of RAI for therapy in young children. The risk of death from any cancer due specifically to radiation exposure is noted by these authors to be 0.16%/rem for children, and the whole body radiation exposure from RAI treatment at age 10 to be 1.45 rem/mCi administered. Rivkees et al advise treatment with doses of RAI greater than 160 uCi/gram thyroid, to achieve a thyroidal radiation dose of at least 150Gy (about 15000 rads). Assuming a reasonable RAIU of 50% and gland size of 40 gm, the administered dose would thus be 40(gm) x 160uCi/gm x 2 (to account for 50% uptake) =12.8 mCi. Thus the long term cancer death risk would be 12.8 (mCi) x 1.45 rem (per mCi) x 0.16% (per rem) = 3%. For a dose of 15mCi the theoretical incremental risk of a later radiation-induced cancer mortality would be 4% at age 5, 2% at age 10, and 1% at age 15.
Whether or not accepting a specific 2-4% risk of death from any cancer because of this treatment is of course a matter of judgment by the physician and family. However, this would seem to many persons to constitute a significant risk that might be avoided. We note that this is a theoretical risk, based on known effects of ionizing radiation to induce malignancies, but not so far proven in this setting.
Long term studies focused to establish an increased risk of non-thyroid malignancies in children treated with RAI for Graves’ diseases would require about 10.000 children treated below 10 years of age, thus today the decision should be taken on an individual base with the patient and the family. The choice between surgery and RAI therapy in Graves’ disease in children is one of the major long standing controversies in pediatric endocrinology. Most physicians remain concerned about the risks of carcinogenesis, and the experience of Chernobyl has accentuated this concern. Others believe that the risks of surgery and problems with antithyroid drug administration outweigh the potential risk of 131-I therapy. This problem was critically reviewed by Rivkees et al (113j). They point out the significant risks of reaction to antithyroid drugs, and of surgery. Surgery may have a mortality rate in hospital in children of about one per thousand operations, although this may have decreased in recent years. Among problems with radioactive iodide therapy, they note the whole body radiation, possibly worsening of eye disease, and the apparent lack of significant thyroid cancer risk so far reported among children treated with I-131 for Graves’ disease. They assumed that risk would be lower in children after age five, and especially after age ten, and if all thyroid cells were destroyed. They advise using higher doses of radioiodine to minimize residual thyroid tissue, and avoiding treatment of children under age five, but they believe that RAI is a convenient, effective, and useful therapy in children with Graves’ disease. However, as noted above in the section on risks related to use od 131-I, Rivkees own data indicate that treatment of children with conventional doses of RAI may induce a lifetime risk of any fatal cancer of over 2%, a very serious consideration (113i). Many physicians remain reluctant to use 131-I in children under age 15-18 as a first line therapy. Following thyroid ablation most patients become hypothyroid and require lifelong thyroid replacement therapy. On the other hand, if therapy is inadequate, hyperthyroidism may recur. Therefore longterm follow-up is mandatory.
Other Causes of Hyperthyroidism
Non autoimmune hyperthyroidism
Non autoimmune hyperthyroidism is caused by constitutive activation of the TSH receptor (TSHR) (Table 6). Two clinical forms including “familial non-autoimmune autosomal dominant hyperthyroidism (FNAH)” and “persistent sporadic congenital non autoimmune hyperthyroidism (PSNAH)” are described. FNAH is characterized by autosomal dominant inheritance and high variable age of manifestation from neonatal period to 60 years. Variability is present also within the same family. Goiter is present in children, with nodules in older age. PSNAH includes forms with sporadic (de novo) germline mutations in the TSHR. Guidelines about this rare condition have recently been published (90e).
Hyperfunctioning nodules
Hyperthyroidism may be caused by a functioning thyroid adenoma, or functioning thyroid carcinoma. Hyperfunctioning nodules are a rare cause of overt or subclinical hyperthyroidism. Somatic activating mutations within the genes encoding the TSH receptor or the Gs-alpha subunit can be detected (90f). Scintigraphy with Tc 99 or I 123 show hypercaptating nodule and absence of uptake of the surrounding thyroid parenchima. Hyperthyroidism can be controlled with methimazole. Autonomous nodules can be single or a part of multinodular goiter. A recent retrospective study on 31 pediatric cases from US indicated that 45% were overt hyperthyroid at diagnosis and 42% presented with multinodular goiter. Mean age at diagnosis was 15 years, with a range 3-18 yrs. Mean size of the autonomous nodule was 39 mm. In this series of 31 patients, only one patient developed a follicular carcinoma in the controlateral lobe seven years after lobectomy for a benign adenomatoid nodule (114). However, the risk of cancer has been reported up to one third of patients in a series of 31 patients from an iodine- deficient area (114a).
ATA Guidelines for pediatric thyroid nodules and cancer indicate surgery as treatment of children with overt hyperthyroidism due to hyperfunctioning nodules, and surgery is indicated in any nodule >4 cm, because of the decreased sensitivity of FNA to detect malignancy (114b).
Hyperthyroidism may be seen as part of the McCune Albright syndrome (90f) (Table 6). McCune Albright syndrome is due to somatic mutations in Gsα gene that can occur in different tissues as, skin, bones thyroid, adrenal glands.
TSH induced hyperthyroidism
Hyperthyroidism may be due to the inappropriate secretion of TSH by a pituitary adenoma, but thyroid hormone resistance should be excluded.
The syndrome of “inappropriate secretion of TSH” was described in 1975 to indicate two forms of central hyperthyroidism, characterized by high levels of FT3 and FT4 and non suppressed TSH levels(114c). TSH secreting pituitary adenomas are extremely rare in pediatric patients. Guidelines from the ETA has been recently released for these tumors (114d). It is important to consider that a pituitary tumor can be a manifestation of Multiple endocrine neoplasia type 1 and rarely of familial forms of isolated pituitary adenomas with AIP mutations (114e).
In thyroid hormone resistance (RTH) due to mutations of the β isoform of the thyroid hormone receptor hyperthyrodism TSH driven can occur. (See chapter entitled Impaired sensitivity to thyroid hormone. Defects of transport, metabolism and action. .Alexandra M. Dumitrescu, MD and Samuel Refetoff, MD, in this book for a detailed description of this condition).
Tumors secreting chorionic gonadotropin
Recently an adolescent female was described in whom hyperthyroidism resulted from an hCG-secreting hydatidiform mole (114f). Chorioncarcinoma, metastatic embryonal carcinoma of the testis can cause hyperthyroidism (114g).
Transient thyrotoxicosis
Thyrotoxicosis is caused by damage of thyroid cells and release of thyroid hormones stored in the gland. The duration of toxic phase (usually one to three months) depends on the amounts of the thyroid hormones released and the rate of metabolic clearance. Thyroid cell breakdown causes abrupt onset and short duration of symptoms.
Principal causes of transient thyrotoxicosis include:
- Autoimmune thyroiditis (silent thyroiditis): no local symptoms of local inflammation are present.
- Subacute Viral thyroiditis (or de Quervain or granulomatous thyroiditis) it is rarely reported in children and adolescents (106b). Usually presents with sore throat, fever and firm, painful tender enlargement of the thyroid. Mild signs of hyperthyroidism can be overlooked.
- Acute bacterial thyroiditis is rarely a cause of transient thyrotoxicosis.
- Drug-induced thyroiditis (amiodarone and thyrosine kinase inhibitors)
THYROID NODULES AND CANCER
For exhaustive information see also chapters “Thyroid nodules” and “Thyroid cancer “ By F. Pacini and Leslie de Groot in this book.
Recently, the first guidelines specifically elaborated for children with thyroid nodules and differentiated thyroid cancer have been published (114b). Hereditary syndromes (i.e. PTEN related sydromes, DICER1 syndrome, Carney complex, Familial adenomatous polyposis) associated with thyroid cancer in childhood are also been detailed (114b). Medullary thyroid carcinoma guidelines have also been revised, including genetic counseling and modified risk class for children with hereditary MTC (115a).
Thyroid nodules are rare in the first 2 decades of life, but when found, they are more likely to be carcinomatous than are similar masses in adults (115b). Follicular adenomas and colloid cysts account for the majority of benign nodules. Other causes of nodular enlargement include chronic lymphocytic thyroiditis and embryological defects, such as intrathyroidal thyroglossal duct cysts or unilateral thyroid agenesis. Like in adults, the most common form of thyroid cancer in childhood and adolescence is papillary thyroid carcinoma, but other histological types found in the adult may also occur (115c).
A high index of suspicion is necessary if the nodule is painless, of firm or hard consistency, if it is fixed to surrounding tissues or if there is a family history of thyroid cancer. Other worrisome findings include a history of rapid increase in size, associated cervical adenopathy, hoarseness or dysphagia. Even the findings of a cystic component or a functioning nodule, commonly used as favorable signs in adult patients, do not exclude the possibility of neoplasia (115c). Occasionally, thyroid cancer presents in childhood as unexplained cervical adenopathy, or neoplasia is found in patients who also have chronic lymphocytic thyroiditis (115c). The possibility of a rare medullary thyroid carcinoma should be considered if there is a family history of thyroid cancer or pheochromocytoma or if the child has multiple mucosal neuromas and a marfanoid habitus, findings suggestive of multiple endocrine neoplasia (MEN) types 2A and/or 2B (115d).
Children exposed previously to thyroid irradiation comprise a high-risk group. The increased risk of thyroid cancer in adults exposed during childhood to low levels of thyroid irradiation for benign conditions of the head and neck is well known (115e). The increased incidence of both benign and carcinomatous nodules in patients with Hodgkin disease who had received radiotherapy to the neck during childhood is also being documented increasingly (115f, 115g). Thyroid cancer is now known to be the most common second malignancy in childhood survivors of Hodgkin’s and is also seen with increased frequency in leukemia survivors (115h). Similarly, children exposed to high levels of radioactive iodine in the first decade of life or in utero, a consequence of the Chernobyl disaster, are at a markedly increased risk of developing papillary thyroid cancer (113). The risk of thyroid cancer is related to the dose of external irradiation and, unlike the 19 year average latency after low dose irradiation, the average latent period in survivors of Hodgkin disease appears to be only 9 years (115g). In Chernobyl victims, the latency was only 4 years (113). As compared with adults, there appears to be a higher prevalence of gene rearrangements in children with differentiated thyroid cancer, the clinical significance of which is unclear (115h).
Initial investigation of a thyroid nodule includes evaluation of thyroid function and TPO and Tg antibodies. A suppressed serum TSH concentration accompanied by an elevation in the circulating T4 and/or T3 suggests the possibility of a functioning nodule, which can be confirmed with a radionuclide scan. The finding of positive antibodies, on the other hand, usually indicates the presence of underlying chronic lymphocytic thyroiditis, but in some cases, positive antibodies may simply constitute evidence of an immune response to the presence of neoplastic cells. Ultrasonography provides information about whether the nodule is solid or cystic, and whether it is single or multifocal. Fine-needle aspiration biopsy, popular in the investigation of thyroid carcinoma in adults, is gaining increasing acceptance and is now considered to be the procedure of choice in the evaluation of nodules >0.5 cm (115k).
There is an increased incidence of both cervical node involvement and of pulmonary metastases at the time of diagnosis in children with thyroid carcinoma (115c). Nonetheless, the long term cancer specific mortality rate is no greater in children than in adults <40 years of age (115i). Guidelines specifically elaborated for management of children with thyroid nodules differentiated thyroid cancer have been published (114b). Excision of the tumor or lobe is the appropriate treatment for benign tumors and cysts, whereas total thyroidectomy with preservation of the parathyroid glands and recurrent laryngeal nerves is the initial therapy for malignant thyroid tumors. The latter procedure is followed by radioablation if there is evidence of residual gland or tumor after surgery. The issue of prophylactic lymph node dissection is controversial (115h). After radioiodine therapy, the dose of thyroxine is adjusted to keep the serum TSH concentration suppressed (between 0.05 mU/L and 0.1 mU/L in a sensitive assay). Measurement of serum Tg, a thyroid follicular cell-specific protein, is used to detect evidence of metastatic disease in differentiated forms of thyroid cancer, such as papillary or follicular carcinoma. This is best performed after a period (usually 6 weeks) of thyroxine withdrawal or after the exogenous administration of recombinant TSH (115). Measurement of circulating calcitonin is used as a tumor marker for medullary thyroid cancer (MTC), a C-cell derived malignancy (115m). Mutations of the RET protooncogene, detectable in nearly all familial forms of MTC, is of value in screening family members (115e, 115m). In families affected with multiple endocrine neoplasia type 2, screening of children as young as 5 years followed by total thyroidectomy has been successful in curing patients with microscopic MTC, an otherwise highly malignant neoplasm with a poor prognosis (115e). See Medullary Thyroid Carcinoma guidelines for updated genetic counseling and modified risk class for children with hereditary MTC (115a).
Optimal monitoring of patients with a history of thyroid irradiation during childhood remains controversial. Because of the insensitivity of clinical palpation, regular assessment of thyroid function (TSH and, as necessary free T4) as well as ultrasound examinations should be performed. There is evidence that thyroid suppression is associated with a reduction in the development of new nodules after partial surgical resection of an irradiated thyroid gland (115q) but whether it plays any role if the TSH is not elevated or in preventing neoplasia is unknown. Recently, a study that followed a cohort of 4338 5- years survivors of pediatric solid cancer suggested that chemotherapy (nitrosureas), splenectomy, and radiation dose to pituitary gland also play a role in predicting thyroid cancer risk (115r).
A retrospective study on the effects of total body irradiation (TBI) preceding hemopoietic cell transplation in childhood suggested short term and life-long monitoring for thyroid nodules and thyroid cancer in these patients (115s). Although it was a small size, retrospective study they found the time from TBI to thyroid carcinoma detection ranged from 2.2 years to 15.3 years. Follow up programs are advised for long term survivors of childhood cancer.
REFERENCES
- Missero C, Cobellis G, De Felice M, Di Lauro R. Molecular events involved in differentiation of thyroid follicular cells. Mol Cell Endocrinol. 1998;140:37-43.
- De Felice M, Di Lauro R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev. 2004;25:722-46.
- Manley NR, Capecchi MR. The role of Hoxa-3 in mouse thymus and thyroid development. Development. 1995;121;1989-2003.
- Fagman H, Grande M, Edsbagge J, Semb H, Nilsson M. Expression of classical cadherins in thyroid development: maintenance of an epithelial phenotype throughout organogenesis. Endocrinology. 2003;144:3618-24.
- Fagman H, Liao J, Westerlund J, Andersson L, Morrow BE, Nilsson M. The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Hum Mol Genet. 2007;16:276-85.
- Fagman H, Grande M, Gritli-Linde A, Nilsson M. Genetic deletion of sonic hedgehog causes hemiagenesis and ectopic development of the thyroid in mouse. Am J Pathol. 2004;164:1865-72.
- Burrow GN, Fisher DA, Larsen PR. Maternal and fetal thyroid function. N Engl J Med. 1994;331:1072-8.
- Fisher DA, Klein AH. Thyroid development and disorders of thyroid function in the newborn. N Engl J Med. 1981;304:702-12.
- Thorpe-Beeston JG, Nicolaides KH, McGregor AM. Fetal thyroid function. Thyroid.1992;2:207-17.
- Williams FL, Simpson J, Delahunty C, et al. Developmental trends in cord and postpartum serum thyroid hormones in preterm infants. J Clin Endocrinol Metab. 2004;89:5314-20.
- Brown RS, Shalhoub V, Coulter S, et al. Developmental regulation of thyrotropin receptor gene expression in the fetal and neonatal rat thyroid: relation to thyroid morphology and to thyroid-specific gene expression. Endocrinology. 2000;141:340-5.
- Hume R, Simpson J, Delahunty C, et al. Human fetal and cord serum thyroid hormones: developmental trends and interrelationships. J Clin Endocrinol Metab. 2004;89:4097-103.
- De Nayer P, Cornette C, Vanderschueren M, et al. Serum thyroglobulin levels in preterm neonates. Clin Endocrinol. 1984;21:149-53.
- Sobrero G, Munoz L, Bazzara L, et al. Thyroglobulin reference values in a pediatric infant population. Thyroid. 2007;17:1049-54.
- Roti E, Gnudi A, Braverman LE. The placental transport, synthesis and metabolism of hormones and drugs which affect thyroid function. Endocr Rev.1983;4:131-49.
- Kester MH, Kaptein E, Van Dijk CH, et al. Characterization of iodothyronine sulfatase activities in human and rat liver and placenta. Endocrinology. 2002;143:814-9.
- Van der Geyten S, Segers I, Gereben B, et al. Transcriptional regulation of iodothyronine deiodinases during embryonic development. Mol Cell Endocrinol. 2001;183:1-9.
- Ruiz de Ona C, Obregon MJ, Escobar del Rey F, Morreale de Escobar G. Developmental changes in rat brain 5′-deiodinase and thyroid hormones during the fetal period: the effects of fetal hypothyroidism and maternal thyroid hormones. Pediatr Res. 1988;24:588-94.
- Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Is neuropsychological development related to maternal hypothyroidism or to maternal hypothyroxinemia? J Clin Endocrinol Metab. 2000;85:3975-87.
- Ferreiro B, Bernal J, Goodyer CG, Branchard CL. Estimation of nuclear thyroid hormone receptor saturation in human fetal brain and lung during early gestation. J Clin Endocrinol Metab. 1988;67:853-6.
20a. Chan SY, Vasilopoulou E, Kilby MD. The role of the placenta in thyroid hormone delivery to the fetus. Nat Clin Pract Endocrinol Metab. 2009; 5:45-54.
20b. Patel J.Li H, Mortimer RH, Richard K. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol Metab. 2011;22:164-70.
20c. Burns R, O’Herlihy C, Smyth PP. The Placenta as a Compensatory Iodine Storage Organ. Thyroid. 2011; 21: 541-46.
20d. Burns R, O’Herlihy C, Smyth PP. Regulation of iodide uptake in placental primary cultures. Eur Thyroid J. 2013;2:243-51.
20e. Li H, Patel J, Mortimer RH, Richard K. Ontogenic changes in human placental sodium iodide symporter expression. Placenta. 2012;33:946-8.
- 21.
Vulsma T, Gons MH, de Vijlder JJ. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N Engl J Med. 1989;321:13-6.
- 22.
Calvo RM, Jauniaux E, Gulbis B, et al. Fetal tissues are exposed to biologically relevant free thyroxine concentrations during early phases of development. J Clin Endocrinol Metab. 2002;87:1768-77.
22a. Contempre B, Jauniaux E, Calvo R, Jurkovic D, Campbell S, de Escobar GM. Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J Clin Endocrinol Metab.1993; 77:1719-22.
22b. Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Practice. 2007; 3:249-259.
22c. Rovet JF. The role of thyroid hormones for brain development and cognitive function. Endocr Dev. 2014;26:26-43.
22d. Morreale de Escobar G, Obregon MJ, Escobar del Rey F: Maternal thyroid hormones early in pregnancy and fetal brain development. In: Best Practice & Research in Clinical Endocrinology and Metabolism: The Thyroid and Pregnancy. Editor: Glinoer D 2004; 18:225.
22e. Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. Neuroendocrinol. 2008;20:784-94.
22f. Schroeder AC, Privalsky ML. Thyroid hormones, T3 and T4, in the brain. Front Endocrinol. 2014;5:40. doi: 10.3389/fendo.2014.00040. eCollection 2014.
22g. Bernal J, Guadaño-Ferraz A, Morte B. Thyroid hormone transporters-functions and clinical implications. Nat Rev Endocrinol. 2015;11:406-17.
- 23.
Meinhold H, Dudenhausen JW, Wenzel KW, Saling E. Amniotic fluid concentrations of 3,3′,5′-tri-iodothyronine (reverse T3), 3,3′-di-iodothyronine, 3,5,3′-tri-iodothyronine (T3) and thyroxine (T4) in normal and complicated pregnancy. Clin Endocrinol.1979;10:355-65.
23a. Seror R, Amand G, Guibourdenche J, Ceccaldi PF, Luton D, Anti-TPO antibodies diffusion through the placental barrier during pregnancy. PloS One. 2014, 9:e84647.
23b. Zimmermann MB. The effects of iodine deficiency in pregnancy and infancy. Paediatr Perinat Epidemiol. 2012; 26 (Supp1):108-117.
- 24.
Boyages SC. Clinical review 49: Iodine deficiency disorders. J Clin Endocrinol Metab. 1993;77:587-91.
- 25.
Rovet J, Walker W, Bliss B, Buchanan L, Ehrlich R. Long-term sequelae of hearing impairment in congenital hypothyroidism. J Pediatr. 1996;128:776-83.
- 26.
de Zegher F, Pernasetti F, Vanhole C, Devlieger H, Van den Berghe G, Martial JA. The prenatal role of thyroid hormone evidenced by fetomaternal Pit-1 deficiency. J Clin Endocrinol Metab. 1995;80:3127-30.
- 27.
Matsuura N, Konishi J. Transient hypothyroidism in infants born to mothers with chronic thyroiditis–a nationwide study of twenty-three cases. The Transient Hypothyroidism Study Group. Endocrinol Jpn. 1990;37:369-79.
27a. Hallowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, Braveman LE. Serum T4 and thyroid antibodies in the United States population (1988-1994): National Health and Nutrition Examination Survay (NHANES III). J Clin Endocrinol Metab. 2002; 489-499.
- 28.
Man EB, Jones WS, Holden RH, Mellits ED. Thyroid function in human pregnancy. 8. Retardation of progeny aged 7 years; relationships to maternal age and maternal thyroid function. Am J Obstet Gynecol. 1971;111:905-16.
- 29.
Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O’Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, Faix JD, Klein RZ. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med.1999;341:549-55.
29a. Smit BJ, Kok JH, Vulsma T, Briët JM, Boer K, Wiersinga WM. Neurologic development of the newborn and young child in relation to maternal thyroid function. Acta Paediatr. 2000;89:291-5.
29b. Li Y, Shan Z, Teng W, Yu X, Li Y, Fan C, Teng X, Guo R, Wang H, Li J, Chen Y, Wang W, Chawinga M, Zhang L, Yang L, Zhao Y, Hua T. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months. Clin Endocrinol. 2010;72:825-9.
- 30.
Pop VJ, Kuijpens JL, van Baar AL, Verkerk G, van Son MM, de Vijlder JJ, Vulsma T, Wiersinga WM, Drexhage HA, Vader HL. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol. 1999;50:149-55.
30a. Pop VJ, Brouwers EP, Vader H, Vulsma T, van Baar AL, de Vijlder JJ. Maternal hypothyroxinaemia during early pregnancy and subsequent child development: a 3-year follow-up study. Clin Endocrinol. 2003;59:282-8.
30b. Henrichs J, Bongers-Schokking JJ, Schenk JJ, Ghassabian A, Schmidt HG, Visser TJ, Hooijkaas H, de Muinck Keizer-Schrama SM, Hofman A, Jaddoe VV, Visser W, Steegers EA, Verhulst FC, de Rijke YB, Tiemeier H. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab. 2010;95:4227-34.
30c. Suárez-Rodríguez M, Azcona-San Julián C, Alzina de Aguilar V. Hypothyroxinemia during pregnancy: the effect on neurodevelopment in the child. Int J Dev Neurosci. 2012;30:435-8.
30d. Finken MJ, van Eijsden M, Loomans EM, Vrijkotte TG, Rotteveel J. Maternal hypothyroxinemia in early pregnancy predicts reduced performance in reaction time tests in 5- to 6-year-old offspring. J Clin Endocrinol Metab. 2013;98:1417-26.
30e. Julvez J, Alvarez-Pedrerol M, Rebagliato M, Murcia M, Forns J, Garcia-Esteban R, Lertxundi N, Espada M, Tardón A, Riaño Galán I, Sunyer J. Thyroxine levels during pregnancy in healthy women and early child neurodevelopment. Epidemiology. 2013;24:150-7.
30f. Vermiglio F, Lo Presti VP, Moleti M, Sidoti M, Tortorella G,Scaffidi G, Castagna MG, Mattina F, Violi MA, Crisà A, Artemisia A, Trimarchi F. Attention deficit and hyperactivity disorders in the offspring of mothers exposed to mild-moderate iodine deficiency:a possible novel iodine deficiency disorder in developed countries. J Clin Endocrinol Metab. 2004;89:6054-60.
30g. Modesto T, Tiemeier H, Peeters RP, Jaddoe VW, HofmanA, Verhulst FC, Ghassabian A. Maternal Mild Thyroid Hormone Insufficiency in Early Pregnancy and Attention-Deficit/Hyperactivity Disorder Symptoms in Children. JAMA Pediatr. 2015;169:838-45.
30h. Román GC, Ghassabian A, Bongers-Schokking JJ, Jaddoe VW,Hofman A, de Rijke YB, Verhulst FC, Tiemeier H. Association of gestational maternal hypothyroxinemia and increased autism risk. Ann Neurol. 2013;74:733-42.
30k. Gyllenberg D, Sourander A, Surcel HM, Hinkka-Yli-SalomäkiS, McKeague IW, Brown AS. Hypothyroxinemia During Gestation and Offspring Schizophrenia in a National Birth Cohort. Biol Psychiatry. 2015 19. pii: S0006-3223(15)00520-X.
30i. Ghassabian A, Bongers-Schokking JJ, de Rijke YB, van Mil N, Jaddoe VW, de Muinck Keizer-Schrama SM, Hooijkaas H, Hofman A, Visser W, Roman GC, Visser TJ, Verhulst FC, Tiemeier H. Maternal thyroid autoimmunity during pregnancy and the risk of attention deficit/hyperactivity problems in children: the Generation R Study.Thyroid. 2012; 22:178-86.
30j. Pop VJ, de Vries E, van Baar AL, Waelkens JJ, de Rooy HA, Horsten M, Donkers MM, Komproe IH, van Son MM,Vader HL. Maternal thyroid peroxidase antibodies during pregnancy: a marker of impaired child development? J Clin Endocrinol Metab. 1995;80:3561-6.
30l. Wasserman EE, Pillion JP, Duggan A, Nelson K, Rohde C, Seaberg EC, Talor MV, Yolken RH, Rose NR. Childhood IQ, hearing loss, and maternal thyroid autoimmunity in the Baltimore Collaborative Perinatal Project. Pediatr Res. 2012;72:525-30.
30m. Kooistra L, Crawford S, van Baar AL, Brouwers EP, Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy. Pediatrics. 2006;117:161-7.
30n. Mirabella G, Westall CA, Asztalos E, Perlman K, Koren G, Rovet J. Development of contrast sensitivity in infants with prenatal and neonatal thyroid hormone insufficiencies. Pediatr Res. 2005;57:902-7.
30o. Ghassabian A, Bongers-Schokking JJ, Henrichs J, Jaddoe VW, Visser TJ, Visser W, de Muinck Keizer-Schrama SM, Hooijkaas H, Steegers EA, Hofman A, Verhulst FC, van der Ende J, de Rijke YB, Tiemeier H. Maternal thyroid function during pregnancy and behavioral problems in the offspring: the generation R study. Pediatr Res. 2011;69:454-9.
- 31.
Momotani N, Iwama S, Momotani K. Neurodevelopment in children born to hypothyroid mothers restored to normal thyroxine (T₄) concentration by late pregnancy in Japan: no apparent influence of maternal T₄ deficiency. J Clin Endocrinol Metab. 2012;97:1104-8.
31a. Liu H, Momotani N, Noh JY, Ishikawa N, Takebe K, Ito K. Maternal hypothyroidism during early pregnancy and intellectual development of the progeny. Arch Intern Med. 1994;154:785-7.
31b. Downing S, Halpern L, Carswell J, Brown RS Severe maternal hypothyroidism corrected prior to the third trimester is associated with normal cognitive outcome in the offspring. Thyroid. 2012; 22:625-30.
- 32.
Lazarus JH, Bestwick JP, Channon S et al. Antenatal thyroid screening and childhood cognitive function. N Engl J Med. 2012; 366:493-501.
32a. Casey B. Effect of treatment of maternal subclinical hypothyroidism or hypothyroxinemia on IQ in offspring. American Journal of Obstetrics & Gynecology. Supplement to JANUARY 2016 Abstr 2 S2.
32b. Su PY, Huang K, Hao JH, Xu YQ, Yan SQ, Li T, Xu YH, Tao FB. Maternal thyroid function in the first twenty weeks of pregnancy and subsequent fetal and infant development: a prospective population-based cohort study in China. J Clin Endocrinol Metab. 2011;96:3234-41.
32c. Korevaar TI, Muetzel R, Medici M, Chaker L, Jaddoe VW, de Rijke YB, Steegers EA, Visser TJ, White T, Tiemeier H, Peeters RP. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol. 2016; 4:35-43.
32d. Samadi A, Skocic J, Rovet JF. Children born to women treated for hypothyroidism during pregnancy show abnormal corpus callosum development.Thyroid. 2015; 25:494-502.
32e. Lischinsky JE, Skocic J, Clairman H, Rovet J. Preliminary Findings Show Maternal Hypothyroidism May Contribute to Abnormal Cortical Morphology in Offspring. Front Endocrino . 2016;25:7-16.
32f. Williams FL, Watson J, Ogston SA, Visser TJ, Hume R, Willatts P. Maternal and umbilical cord levels of T4, FT4, TSH, TPOAb, and TgAb in term infants and neurodevelopmental outcome at 5.5 years. J Clin Endocrinol Metab. 2013;98: 829-38.
- 33.
Klein RZ, Haddow JE, Faix JD, et al. Prevalence of thyroid deficiency in pregnant women. Clin Endocrinol. 1991;35:41-6.
33a. Thangaratinam S, Tan A, Knox E, Kiliby MD, Franklyn J, Coomarasamy A. Association between thyroid autoantibodies and miscarriage and preterm birth: meta-analysis of evidence. BMJ. 2011; 342:d2616.
33b. Alexander EK, Pearce EN, Brent GA, Brown RS, Chen H, Dosiou C, grobman W, Laurberg P, Lazarus JK, Mandel SJ, Peeters R, Sullivan S. 2016 guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid 2017, Jan 6. doi: 10.1089/thy.2016.0457. [Epub ahead of print]
- 34.
Abuid J, Stinson DA, Larsen PR. Serum triiodothyronine and thyroxine in the neonate and the acute increases in these hormones following delivery. J Clin Invest..1973;52:1195-9.
- 35.
Bianco AC, Silva JE. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. J Clin Invest. 1987;79:295-300.
- 36.
Houstek J, Vizek K, Pavelka S, et al. Type II iodothyronine 5 ′ -deiodinase and uncoupling protein in brown adipose tissue of human newborns. J Clin Endocrinol Metab. 1993;77:382-7.
- 37.
Mercado M, Yu VY, Francis I, Szymonowicz W, Gold H. Thyroid function in very preterm infants. Early Hum Dev. 1988;16:131-41.
- 38.
Nelson JC, Weiss RM, Wilcox RB. Underestimates of serum free thyroxine (T4) concentrations by free T4 immunoassays. J Clin Endocrinol Metab. 1994;79:76-9.
- 39.
Ares S, Escobar-Morreale HF, Quero J, et al. Neonatal hypothyroxinemia: effects of iodine intake and premature birth. J Clin Endocrinol Metab. 1997;82:1704-12.
- 40.
Frank JE, Faix JE, Hermos RJ, et al. Thyroid function in very low birth weight infants: effects on neonatal hypothyroidism screening. J Pediatr. 1996;128:548-54.
- 41.
Kok JH, Tegelaers WH, de Vijlder JJ. Serum thyroglobulin levels in preterm infants with and without the respiratory distress syndrome. I. Cord blood study. Pediatr Res. 1986;20:996-1000.
- 42.
Thorpe-Beeston JG, Nicolaides KH, Snijders RJ, Felton CV, McGregor AM. Thyroid function in small for gestational age fetuses. Obstet Gynecol. 1991;77:701-6.
- 43.
Zurakowski D, Di Canzio J, Majzoub JA. Pediatric reference intervals for serum thyroxine, triiodothyronine, thyrotropin, and free thyroxine. Clin Chem. 1999;45:1087-91.
- 44.
Fisher D. Next generation newborn screening for congenital hypothyroidism? J Clin Endocrinol Metab. 2005;90:3797-9.
44b. Marvin L. Mitchell, Ho-Wen Hsu, Inderneel Sahai and the Massachusetts Pediatric Endocrine Work Group. The increased incidence of congenital hypothyroidism: fact or fancy? Clin Endocrinol. 2011;75:806-10.
- 45.
Klein AH, Meltzer S, Kenny FM. Improved prognosis in congenital hypothyroidism treated before age three months. J Pediatr. 1972;81:912-5.
- 46.
Dussault JH. The anedoctal history of screening for congenital hypothyroidism. J Clin Endocrinol Metab. 1999;84:4332-4.
- 47.
Delange F. Neonatal screening for congenital hypothyroidism: results and perspectives. Horm Res.1997;48:51-61.
- 48.
Rovet J, Daneman D. Congenital hypothyroidism: a review of current diagnostic and treatment practices in relation to neuropsychologic outcome. Paediatr Drugs. 2003;5:141-9.
50a. Ford G, LaFranchi SH. Screening for congenital hypothyroidism: a worldwide view of strategies. Best Practice & Research Clinical Endocrinology & Metabolism. 2014;28:175-187.
50b. Wassner AJ, Brown RS. Congenital Hypothyroidism: recent advances. Curr Opin Endocrinol Diabetes Obes. 2015, 22:407-412.
50c. Olivieri A, Corbetta C, Weber G, Vigone MC, Fazzini C,Medda E, and The Italian Study Group for Congenital Hypothyroidism. Congenital hypothyroidism due to defects of thyroid development and mild increase of TSH at screening: data from the Italian registry of infants with congenital hypothyroidism. J Clin Endocrinol Metab. 2013; 98:140:3-1408.
50d. Harris KB and Pass KA. Increase of congenital hypothyroidism in New York State and in The United States. Molecular Genetics and Metabolism. 2007; 91:268-277.
50e. Hinton CF, Harris KB, Borgfeld L et al. trends in incidence of congenital hypothyroidism related to select demographic factors: data from the United States , California, Massachussets, New York and Texas. Pediatrics. 2010;125:S37-S47.
50f. Olivieri A, Fazzini C, Medda E. Multiple factors influencing the incidence of congenital hypothyroidism detected by neonatal screening. Horm Res Pediatr. 2015 83:86-93.
50g. Barry Y, Bonaldi C, Goulet V, Coutant R, Leger J, Paty AC, Delmas D, Cheillan D, Roussey M. Increased incidence of congenital hypotiroidism in France from 1982 to 2012: a nationwide multicenter analysis. Ann Epidemiol. 2016;26:100-105.
- 51.
LaFranchi SH, Hanna CE, Krainz PL, Skeels MR, Miyahira RS, Sesser DE. Screening for congenital hypothyroidism with specimen collection at two time periods: results of the Northwest Regional Screening Program. Pediatrics. 1985;76:734-40.
51a. Ford GA, Denniston S, Sessere D, Skeels MR, LaFranchi SH. Transient versus permanent congenital hypothyroidism after the age of 3 years in infants, detected on the first versus second newborn screening test in Oregon, USA. Horm Res Paediatr. 201616;86(3):169-177
51b. Leger J, Olivieri A, Donaldson M, Torresani T,Krude H, van Vliet G, Polak M, Butler G on behalf of ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE, and the Congenital Hypothyroidism Consensus Conference Group. European Society for Pediatric Endocrinology Consensus Guidelines on Screening, Diagnosis, and management of congenital hypothyroidism. Horm Res Pediatr. 2014;81:80-103.
51c. Leger J, Olivieri A, Donaldson M, Torresani T, Krude H, van Vliet G, Polak M, Butler G on behalf of ESPE-PES-SLEP-JSPE-APEG-APPE-ISPAE, and the Congenital Hypothyroidism Consensus Conference Group. European Society for Pediatric Endocrinology Consensus Guidelines on Screening, Diagnosis, and management of congenital hypothyroidism. J Clin Endocrinol Metab. 2014; 99:363-384.
- 52.
Lanting CI, van Tijn DA, Loeber JG, Vulsma T, de Vijlder JJ, Verkerk PH. Clinical effectiveness and cost-effectiveness of the use of the thyroxine/thyroxine-binding globulin ratio to detect congenital hypothyroidism of thyroidal and central origin in a neonatal screening program. Pediatrics. 2005;116:168-73.
- 53.
van Tijn DA, de Vijlder JJ, Verbeeten B, Jr., Verkerk PH, Vulsma T. Neonatal detection of congenital hypothyroidism of central origin. J Clin Endocrinol Metab. 2005; 90:3350-9.
53a. Nesbesio TD, McKenna MP, Nabhan ZM et al. Newborn screening results in children with central hypothyroidism. J Pediatr. 2010;156:990-3.
53b. Adachi M, Soneda A, Asakura Y, Muroya K, Yamagami Y, Hirahara F. Mass screening of newborns for congenital hypothyroidism of central origin by free thyroxine measurement of blood samples on filter paper. Eur J Endocrinol. 2012; 166:829-838.
53c. Soneda, A, Adachi M, Muroya K, Asakura Y, Yamagami Y, Hirahara F. Overall usefulness of newborn screening for congenital hypothyroidism by using free thyroxine measurements. Endocr J 2014; 61:025-1-30.
- 54.
American Academy of Pediatrics AAP Section on Endocrinology and Committee on Genetics, and American Thyroid Association Committee on Public Health: Newborn screening for congenital hypothyroidism: recommended guidelines. Pediatrics. 1993;91:1203-9.
54a. Peters C, Brooke I, Ifederu A, Langham S, Hindmarsh P, Cole TJ. Defining the newborn blood spot screening reference interval for TSH: impact of ethnicity. J Clin Endocrinol Metab. 2016; 101:3445-3449.
- 55.
55. Fisher DA. Effectiveness of newborn screening programs for congenital hypothyroidism: prevalence of missed cases. Pediatr Clin North Am. 1987;34:881-90.
- 56.
Adams LM, Emery JR, Clark SJ, Carlton EI, Nelson JC. Reference ranges for newer thyroid function tests in premature infants. J Pediatr. 1995;126:122-7.
56a. Vigone MC, Caiulo S, Di Frenna M et al. Evolution of thyroid function in preterm infants detected by screening for congenital hypothyroidism. J Pediatrics. 2014;164: 1296-1302.
56b. LaFranchi SH. Screening preterm infants for congenital hypothyroidism: better the second time around. J Pediatr. 2014; 164:1259-1261.
56c. Zoller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observation and experimental findings. J Neuroendocrinol. 2004:16:809-818.
56d. Spencer CA, LoPresti JS, Patel A et al. Application of a new chemiluminometric thyrotropin assay to subnormal measurement. J Clin Endocrinol Metab. 1990;70:453-460.
56e. Parazzini C, Baldoli C, Scotti G, Triulzi F. Terminal zones of myelination: MR evaluation of children aged 20-40 months. Am J Neuroradiol. 2002:23:1669-1673.
56f. Gagne N, Parma J, Deal C, Vassart G, Van Vliet G. Apparent congenital athyreosis contrasting with normal plasma thyroglobulin levels and associated with inactivating mutations in the thyrotropin receptor gene: are athyreosis and ectopic thyroid distinct entities? J Clin Endocrinol Metab. 1998;83:1771-5.
- 57.
Smith DW, Klein AM, Henderson JR, Myrianthopoulos NC. Congenital hypothyroidism–signs and symptoms in the newborn period. J Pediatr. 1975;87(6 Pt 1):958-62.
57a. Karakoc-Aydiner E, Turan S, Akpinar I, Dede F, Isguven P, Adal E, Guran T, Akcay T, Bereket A. Pitfalls in the diagnosis of thyroid disgenesis by thyroid untrasonography and scintigraphy. Eur J Endocrinol. 2012; 166:43 -48.
- 58.
Brown RS, LaFranchi S, Rose SR. Patient information page from the hormone foundation. Congenital hypothyroidism. J Clin Endocrinol Metab. 2009;94:1835-6.
58a. Carswell JM, Gordon JH, Popovsky E, Hale A, Brown RS. Generic and brand-name L-thyroxine are not bioequivalent for children with severe congenital hypothyroidism. J Clin Endocrinol Metab. 2013;98: 610-617.
- 59.
Bongers-Schokking JJ, de Muinck Keizer-Schrama SM. Influence of timing and dose of thyroid hormone replacement on mental, psychomotor, and behavioral development in children with congenital hypothyroidism. J Pediatr. 2005;147:768-74.
- 60.
Heyerdahl S, Oerbeck B. Congenital hypothyroidism: developmental outcome in relation to levothyroxine treatment variables. Thyroid. 2003;13:1029-38.
- 61.
Simoneau-Roy J, Marti S, Deal C, Huot C, Robaey P, Van Vliet G. Cognition and behavior at school entry in children with congenital hypothyroidism treated early with high-dose levothyroxine. J Pediatr. 2004;144:747-52.
- 62.
Rovet JF. In search of the optimal therapy for congenital hypothyroidism. J Pediatr. 2004;144:698-700.
- 63.
Dimitropoulos A, Molinari L, Etter K, et al. Children with congenital hypothyroidism: long-term intellectual outcome after early high-dose treatment. Pediatr Res. 2009;65:242-8.
- 64.
Fisher DA. The importance of early management in optimizing IQ in infants with congenital hypothyroidism. J Pediatr. 2000;136:273-4.
64a. Leger J. Congenital hypothyroidism: a clinical update of long-term outcome in young adults. Eur J Endocrinol. 2015;172, R67-R77.
64b. Leger J, Ecosse E, Roussey M, Lanoe JL, Larroque B. French Congenital hypothyroidism study group. Subtle health impairment and socioeducational attainment in young adult patients with congenital hypothyroidism diagnosed by neonatal screening: a longitudinal population-based cohort study. J Clin Endocrinol Metab. 2011;96:1771-82.
64c. Lichtenberer-Geslin L, Dos Santos S, Hassani Y, Ecosse E, Van Den Abbele T, Leger J. Factors associated with hearing impairment in patient with congenital hypothyroidism treated since the neonatal period: a national population-based study. J Clin Endocrinol Metab. 2013; 98:3644-3652.
64d. Leger J, dos Santos S, larroque B, Ecosse E. Pregnancy outcomes and relationship to treatment adequacy in women treated early for congenital hypothyroidism: a longitudinal population-based study. J Clin Endocrinol Metab. 2015; 100: 800-809.
64e. Lof C, Patyra K, Kuulasmaa T, Vangipurapu J, Undeutsch H, Jaeschke H, Pajunen T, Kero A, Krude H, Biebermann, Kleinau G, Kuhnen P, Rantakari K, Miettinen P, Kirjavainen T, Pursiheimo JP, Mustila T, Jaaskelainen J, Ojaniemi M, Toppari J, Ignatius J, Laakso M, Kero J. Detection of novel gene variants associated with congenital hypothyroidism in a finnish patient cohort. Thyroid. 2016; 26:1215-1224.
64f. Nicholas AK, Serra EG, Cangui H, Alyaarubi S, Ullah I, Schoenmakes E, Deeb A, Habeb AM, AI Maghamsi M, Peters C, Nathwani N, Aycan Z, Saglam H, Bober E, Dattani M, Shenoy S, Murray PG, Babiker A, Willensen R, Thankarmony A, Lyons G, Irwin R, Padidela R, Tharian K, Davies JH, Puthi V, Park SM, Massoud AF, Gregory JW, Albanese A, Pease-Gevers E, Martin H, Brugger K, Maher ER, Chatterje K, Anderson CA, Schoenmakers N. Comprehensive screening of eight known causative genes in congenital hypothyroidism with gland-in-situ. J Clin Endocrinol Metab. 2016; 12:4521-31.
- 65.
Siebner R, Merlob P, Kaiserman I, Sack J. Congenital anomalies concomitant with persistent primary congenital hypothyroidism. Am J Med Genet. 1992;44:57-60.
- 66.
Kumar J, Gordillo R, Kaskel FJ, Druschel CM, Woroniecki RP. Increased prevalence of renal and urinary tract anomalies in children with congenital hypothyroidism. J Pediatr. 2009;154:263-6.
- 67.
Perry R, Heinrichs C, Bourdoux P, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab. 2002;87:4072-7.
67a. Brown RS, Demmer LA. The etiology of thyroid dysgenesis-still an enigma after all these years. J Clin Endocrinol Metab. 2002;87:4069-71.
- 68.
Fort P, Lifshitz F, Bellisario R, et al. Abnormalities of thyroid function in infants with Down syndrome. J Pediatr. 1984;104:545-9.
68a. Stoupa A, Kariyawasam D, Carre’ A, Polak M. Update of thyroid development genes. Endocrinol Metab Clin N Am. 45:243-246, 2016.
68b. Szinnai G. genetics of normal and abnormal thyroid development in humans. Best Pract Res Clin Endocrinol Metab. 2014; 28:133-50.
68c. Satoshi Narumi and Tobonobu Hasegawa. TSH resistance revised. Endocrine Journal. 2015, 62:393-398.
68d. Calebiro D, Gelmini G, Cordella D et al. Frequent TSH receptor genetic alterations with variable signaling impairment in a large series of children with nonautoimmune isolated hyperthyrotropinemia. J Clin Endocrinol Metab. 2012; 97: E156-160.
68e. Carre A, Szinnai G, Castanet M et al. Five new TTF1/NKX2-1 mutations in brain–lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18:2266-76.
68f. de Filippis T, Marelli F, Vigone MC, Di Frenna M, Weber G, Persani L. Novel NKX2-1 frameshift mutations in patients with athypical phenotypes of the brain-lung-thyroid syndrome. Eu Thyroid J. 2014; 3: 227-233.
68g. Thowarth A, Schittert-Hubener S , Schrumpf P et al. Comprehensive genotyping and clinical characterization reveal 27 novel NKX2-1 mutations and expand the phenotypic spectrum. J Med Genet. 2014;51:375-387.
68h. Stenson PD, Mort M, Ball EV et al. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133: 1-9.
68i. Narumi S, Muroya K, Asakura Y et al. Transcription factor mutations and congenital hypothyroidism: systematic genetic screening of a population-based cohort of Japanese patients. J Clin Endocrinol Metab. 2010: 95: 1981-5.
68j. Fernandez LP, Lopez- Marquez A, Santisteban P. Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol. 2015; 11:29-42.
68k. Fernandez LP, Lopez-Marquez A, Martinez AM, Gomez-Lopez G, Santisteban P l. New insights into FOXE1 functions: identification of direct FOXE1 targets in thyroid cells. PLoS One. 2013, 8:e62849.
68m. Carre A Hamza RT, Kariyawasam D, et al. A novel FOXE1 mutation (R73S) in Bamforth-Lazarus syndrome causing increased thyroid gene expression. Thyroid. 2014:24:649-65.
68n. Dentice M, Cordeddu V, Rosica A et al. Missense mutation in the transcription factor NKX2-5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91:1428-1433.
68o. Opitz R, Hitz MP, Vandernoot I et al. Functional zebrafish studies based on human genotyping point to netrin-1 as a link between aberrant cardiovascular development and thyroid dysgenesis. Endocrinology. 2015;156:377-388.
68q. Porazzi P, Marelli F, Benato F,de Filippis T,Calebiro D, Argenton F, Tiso N, Persani L . Disruptors of global and JAGGED1 mediated notch signaling affect thyroid morfogenesis in the zebrafish. Endocrinology. 2012; 153:5645-5658.
68r. De Filippis T, Marelli F. Nebbia G, Porazzi P, Corbetta S, Fugazzola L, Gastaldi R, Vigone MC, Biffanti R, Frizziero D, Mandara’ L, Prontera P, Salerno M, Maghnie M, Tiso N, Radetti G, Weber G, Persani L. JAG1 loss- of- function variations as a novel predisposing event in the pathogenesis of congenital thyroid defects. J Clin Endocrinol Metab. 2016;101:861-870.
68s. Knobel M, Madeires-Neto G. An outline of inherited disorders of the thyroid hormone generating system. Thyroid. 2003; 13:771-801.
- 69.
Portulano C, Paroder-Belenitsky M, Carrasco N. The Na+/I symporter (NIS): mechanism and medical impact. Endocr Rev. 2014;35 :106-149.
- 70.
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani F, Nassir E, Baxevanis AD, Sheffied VC, Green ED. Pendred syndrome is caused by mutations in putative sulphate transporter gene (PDS). Nat Genet. 1997;17:411-422.
- 71.
71. Kopp P. Pendred’s syndrome: identification of the genetic defect a century after its recognition. Thyroid. 1999;9:65-9.
71a. Ladsous M, Vlaeminck-Guillem V, Dumur V, Vincent C, Dubrulle F, Dhanens CM, Wemeau JL. Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and non syndromic enlargement of the vestibular aqueduct. Thyroid. 2014; 24:639-648.
71b. Bizhanova A, Kopp P. Genetics and phenomics of Pendred syndrome. Mol Cell Endocrinol. 2010;322:83-90.
- 72.
Nascimiento AC, Guendes DR, Santos CS, Knobel M, Rubio IG, Medeiros-Neto G. Thyroperoxidase gene mutations in congenital goitrous hypothyroidism with total and partial iodide organification defect. Thyroid. 2003;13:1145-51.
72a. Delodoey J, Pfarr N, Vuissoz JM, Parma J, Vassart G, Biesterfeld S, Phlenz J, Van Vliet G. Pseudodominant inherence of goitrous congenital hypothyroidism caused byTPO mutations; molecular and in silico studies. J Clin Endocrinol Metab. 2008;93:627-633.
72b. Moreno JC,Bikker H, Kempers MJE, vanTrotsenburg AS, Baas F, deVijlder JJM, Vulsma T, Ris-Stalpers C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med. 2002; 347:95-102.
72c. Muzza M, Rabbiosi S, Vigone MC, Zamproni I, Cirelio V, Maffini M, Maruca K, Schoenmarkers N, Beccaria L, Gallo F, Park SM, Beck-Peccoz P, Persani L, Weber G, Fugazzola L. The clinical and molecular characterization of patients with dyshormonogenetic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab. 2014;99:E544-E553.
72d. Zamproni I, Grasberger H, Cortinovis F, Vigone MC, Chiumello G, Mora S, Onigata K, Fugazzola L, Refetoff S, Persani L, Weber G. Biallelic inactivation of the dual oxidation maturation factor 2 (DUAXA2) gene as a novel cause of congenital hypothyroidism. J Clin Endocrinol Metab. 2008; 93:605-610.
72e. Ieri T, Cochaux P, Targovinik HM, Suzuki M, Shimoda S, Perret J, Vassart G. A 3’splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest..1991:88:1901-1905.
72f. Medeiros-Neto G, Kim PS, Yoo SE, Vono J, Targovnik H, Camargo R, Hossain SA, Arvan P. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J Clin Invest. 1996; 98:2838-2844.
72g. Citterio CE, Rossetti LC, Souchon PF, Morales C, Thouvard-Viprey M, Salmon-Musial AS, Mauran PL, Doco-Fenzy M, Gonzales-Sarmiento R, Rivolta CM, De Brasi CD, Targovink HM. Novel mutational mechanism in the thyroglobulin gene: imperfect DNA inversion as a cause for hereditary hypothyroidism. Mol Cell Endocrinol. 2013; 381:220-229.
72h. Iglesias A, Garcia-Nimo L, Cocho de Juan JA, Moreno JC. Towards the pre-clinical diagnosis of hypothyroidism caused by iodotyrosine deiodinase (DEHAL1) deficts. Best Prac Res Clin Endocrinol Metab. 2014; 28:151-159.
72i. Moreno JC, Klootwijk W, van Toor H, Pinto G, D’Allessandro M, Leger A, Goundie D, Polak M, Gruters A, Visser TJ. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Eng J Med. 2008;358:1856-1859.
- 73.
Fisher DA, Dussault JH, Foley TP, Klein AH, LaFranchi S, Larsen PR, Mitchell ML, Murphey WH, Walfish PG. Screening for congenital hypothyroidism: results of screening one million North American infants. J Pediatr. 1979;94: 700-705.
73a. Lanting CI, van Tijn DA, Loeber JG, Vulsma T, de Vijder JJ, Verkerk PH. Clinical effectiveness and cost-effectiveness of the use of the thyroxine-binding globulin ratio to detect congenital hypothyroidism of thyroidal and central origin in a neonatal screening program. Pediatrics. 2005;116:168-173.
73b. Kempers MJ, Lanting CI, van Heijst AF, van Trotsenburg AS, Wiedijk BM. De Vijder JJ,Vulsma T. Neonatal screening for congenital hypothyroidism based on thyroxine, thyrotropin and thyroxine-binding globulin measurement potentials and pitfalls. J Clin Endocrinol Metab.2006; 91:3370-3376.
73c. Zwaveling-Soonawala N, van Trotsenburg AS, Verkerk PH. The severity of congenital hypothyroidism of central origin should not be underestimated. J Clin Endocrinol Metab. 2015:100:E297-E300.
73d. Collu R, Tang J, Castagne’ J, Legace’ G, Masson N, Huot C, Deal C, Delvin E, Faccenda F, Edne KA, Van Vliet G. A novel mechanism for isolated central hypothyroidism: Inactivating mutations in the thyrotropin releasing-hormone receptor gene. J Clin Endocrinol Metab. 1997:82:1561-1565.
73e. Bonomi M, Busnelli M, Beck-Peccoz P, Costanzo D, Antonica F, Dolci C, Pilotta A, Buzi F, Persani L. A family with complete resistance to thyrotropin-releasing hormone receptor gene. N Engl J Med. 2009;360:731-734.
73f. Koulouri O, Nicholas AK, Shoenmakers E, Mokrosinki J, Lane F, Cole T, Kirk J, Farooqi IS, Chatterjee VK, Gurnell M, Shoenmakers N. A novel thyrotropin-releasing hormone receptor missense mutation (P81R) in central congenital hypothyroidism. J Clin Endocrinol Metab. 2016; 101:847-851.
- 74.
Dacou-Voutekatis C, Feltquate DM, Drakopoulou M, Kourides IA, Dracopoli NC. Familial hypothyroidism caused by a nonsense mutation in the thyroid stimulating hormone beta-subunit gene. Am J Hum Genet. 1990;46:988-993.
74a. Bonomi M, Proverbio MC, Weber G, Chiumello G, Beck-Peccoz P, Persani L. Hyperplastic pituitary gland, high serum glycoprotein hormone alfa-subunit, and variable circulating thyrotropin (TSH) levels as a hallmark of central hypothyroidism due to mutations of the TSH beta gene. J Clin Endocrinol Metab. 2001;86:1600-1604.
74b. Bequadano MS, Ciaccio M, Dujovne N et al. Two novel mutations of the TSH- β-subunit gene underlying congenital central hypothyroidism undetectable in neonatal TSH screening. J Clin Endocrinol Metab. 2010;95:E98-E103.
74c. Drees JC, Stone JA, Reamer CR, Arboleda VF, Huang K, Hrynkow J, Greene DN, Petrie MS, Hoke C, Lorey TS et al. Falsely undetectable TSH in a cohort of Sauth Asian euthyroid patients. J Clin Endocrinol Metab. 2014;99:1171-1179.
74d. PappaT, Johannesen J, Schemberg N, Torrent M, Dumitriescu A, Refetoff S. A TSHB variant with impaired immunoreactivity but intact biological activity and its clinical implications. Thyroid. 2015; 25:869-876.
74e. Hermanns P, Couch R, Leonard N, Klotz C, Pohlenz J. A novel deletion in the thyrotropin β-subunit gene identified by array comparative genomyc hybridization analysis causes central congenital hypothyroidism in a boy originating from Turkey. Horm ResPediatr. 2014;82:201-205.
74f. Nicholas AK, Jaleel S, Kyons E, Shoenmakers E, Dattani MT, Crowne E, Bernhard B, Kirk J, Roche EF, Chatterjee VK, Shoenmakers N. Molecular spectrum of TSHβ subunit gene defects in central hypothyroidism in the UK and Ireland. Clin Endocrinol. 2016; Jun 30 doi:10111/cen. 13149.
- 75.
Sun Y, Bak B, Schoenmaker N, van Trotsenburg AS, Oostdijk W, Voshol P et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012; 44:1375-1381.
75a. Tajima T, Nakamura A, Ishizu K. A novel mutation of IGSF1 in a Japanese patient of congenital central hypothyroidism without macroorchidism. Endocr J. 2013; 60:245-249.
75b. Nakamura A, Bak B, Silander TL, Lam J, Hotsubo T, Yorifuji T, Ishizu K, Bernard DJ, Tajima T. Three novel IGSF1 mutations in four Japanese patients with X-linked congenital central hypothyroidism. J Clin Endocrinol Metab. 2013;98:E1682-E1691.
75c. Joustra SD, Schoenmakers N, Persani L, Campi I, Bonomi M, Radetti G, Beck-Peccoz P, Zhu H, Davis TM, Sun Y, Corssmit EP, Appelman-Dijkstra NM, Heinen CA, Pereira AM, Varewick AJ, Janssen JA, Endert E, Hennekam RC, Lombardi MP, Mannens MM, Bak B, Bernard DI, Breuning MH, Chatterijee K et al. The IGSF1 deficiency syndrome:characteristics of male and females patients. J Clin Endocrinol Metab. 2013; 98:4942-4952.
75d. Joustra SD, Heinen CA, Schoenmakers N, Bonomi M, Ballieux MO, Turgeon D, Bernard E, Fliers E, van Trotsenburg ASP, Losekoot M, Persani L, Wit JM, Biermasz NR, Pereira AM, Oostdijk W, on behalf of the IGSF1 Clinical Care Group. IGSF1 Deficiency: lessons from an extensive case series and recommendations for clinical management. J Clin Endocrinol Metab. 2016; 101:1627-1636.
75e. Tenenbaum-Rakover Y, Turgeon MO, London S,Hermanns P,Pohlenz J, Bernard DJ, Bercovich D. Familial central hypothyroidism caused by a novel IGSF1 gene mutation. Thyroid. 2016 ;26:1693-1700..
- 76.
Alatzoglou KS, Dattani MT. Genetic forms of hypopituitarism and their manifestations in neonatal period. Early Hum Dev. 2009;85:705-712.
76a. Brown RS, Bhatia V, Hayes E. An apparent cluster of congenital hypopituitarism in central Massachusetts: magnetic resonance imaging and hormonal studies. J Clin Endocrinol Metab. 1991;72:12-8.
76b. Dattani ML, Martinez-Barbera J, Thomas PQ, et al. Molecular genetics of septo-optic dysplasia. Horm Res. 2000; 53 Suppl 1:26-33.
76c. Machinis K, Pantel J, Netchine I, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69:961-8.
76d. Parks JS, Brown MR. Transcription factors regulating pituitary development. Growth Horm IGF Res. 1999;9 Suppl B:2-8; discussion -11.
- 77.
Refetoff S, Bassett JH, Beck-Peccoz P et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport and metabolism. Thyroid. 2014; 24:407.
77a. Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993; 14:348-399.
77b. Weiss RE, Balzano S, Scherberg NH, Refetoff S. Neonatal detection of generalized resistance to thyroid hormone. Jama. 1990;264:2245-50.
77c. Bochukova E, Schoenmakers N, Agostini E, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med. 2012;366:243-9.
77d. Moran C, Chatterje K, Resistance to thyroid hormone due to defective thyroid hormone receptor alpha. Best Pract Res Clin Endocrinol Metab. 2015; 29: 647.
77e. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168-75.
77f. Friesema EC, Gruters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004; 364:1435-1437.
77g. Allan W, Erdon CN, Dudley FC. Some exemples of the inheritance of mental deficiency apparently sex-linked idiocy and microcefaly. Am J Ment Defic. 1944, 48:325.
77h. Dumitrescu AM, Liao XH, Abdullah MS, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247-52.
- 78.
l’Allemand D, Gruters A, Beyer P, Weber B. Iodine in contrast agents and skin disinfectants is the major cause for hypothyroidism in premature infants during intensive care. Horm Res. 1987;28:42-9.
- 79.
Brown RS, Bloomfield S, Bednarek FJ, Mitchell ML, Braverman LE. Routine skin cleansing with povidone-iodine is not a common cause of transient neonatal hypothyroidism in North America: a prospective controlled study. Thyroid.1997;7:395-400.
- 80.
Cheron RG, Kaplan MM, Larsen PR, Selenkow HA, Crigler JF, Jr. Neonatal thyroid function after propylthiouracil therapy for maternal Graves’ disease. N Engl J Med. 1981;304:525-8.
- 81.
Mitsuda N, Tamaki H, Amino N, Hosono T, Miyai K, Tanizawa O. Risk factors for developmental disorders in infants born to women with Graves disease. Obstet Gynecol. 1992;80(3 Pt 1):359-64.
81a. Brown RS, Bellisario RL, Botero D, et al. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J Clin Endocrinol Metab. 1996;81:1147-51.
81b. Brown RS, Keating P, Mitchell E. Maternal thyroid-blocking immunoglobulins in congenital hypothyroidism. J Clin Endocrinol Metab. 1990;70:1341-6.
81c. Connors MH, Styne DM. Transient neonatal ‘athyreosis’ resulting from thyrotropin-binding inhibitory immunoglobulins. Pediatrics. 1986;78:287-90.
- 82.
Mandel SH, Hanna CE, LaFranchi SH. Diminished thyroid-stimulating hormone secretion associated with neonatal thyrotoxicosis. J Pediatr. 1986;109:662-5.
- 83.
Deming DD, Rabin CW, Hopper AO, Peverini RL, Vyhmeister NR, Nelson JC. Direct equilibrium dialysis compared with two non-dialysis free T4 methods in premature infants. J Pediatr. 2007;151:404-8.
83a van Wassener-Leemhuis A, Ares S, Golombek S, Kok J, Paneth N, Kase J, LaGamma E. Thyroid hormone supplementation in preterm infants born before 28 weeks of gestational age and neurodevelopmental outcome at age 36 months. Thyroid. 2014; 24:1162-1169.
83b. Hollanders JJ, Israels J, van del Pal SM, Verkerk PH, Rotteveel J, Finken MJ, Duch POPS-19 collaborative study group. No Association Between Transient Hypothyroxinemia of Prematurity and Neurodevelopmental Outcome in Young Adulthood. J Clin Endocrinol Metab. 2015; 100:4648-53.
83c. Williams FL, Visser TJ, Hume R. Transient hypothyroxinaemia in preterm infants. Early Hum Dev. 2006;82:797-802.
83d. van Wassenaer AG, Kok JH, de Vijlder JJ, et al. Effects of thyroxine supplementation on neurologic development in infants born at less than 30 weeks’ gestation. N Engl J Med. 1997;336:21-6.
83e. van Wassenaer AG, Westera J, Houtzager BA, Kok JH. Ten-year follow-up of children born at <30 weeks’ gestational age supplemented with thyroxine in the neonatal period in a randomized, controlled trial. Pediatrics. 2005;116:e613-8.
- 84.
Williams FL, Ogston SA, van Toor H, Visser TJ, Hume R. Serum thyroid hormones in preterm infants: associations with postnatal illnesses and drug usage. J Clin Endocrinol Metab. 2005;90:5954-63.
- 85.
Foley TP, Jr., Abbassi V, Copeland KC, Draznin MB. Brief report: hypothyroidism caused by chronic autoimmune thyroiditis in very young infants. N Engl J Med. 1994;330:466-8.
85a. Verbansky JW, Chatila TA. Immune dysregulation, Poliendocrinopathy, Enteropathy, X linked (IPEX) and IPEX related disorders: an evolving web of heritable autoimmune disease. Curr Opin Pediatr. 2013; 25:708-714.
85b. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Med Genet. 2002;39:537-45.
85c. Huang SA, Tu HM, Harney JW et al. Severe hypothyroidism caused by type 3 iodothyronine dieiodinase in infantile hemangiomas. N Eng J Med. 2000;343: 185-9.
85d. Balatz AE, Athanassaki I, Gunn SK, Tatevian N, Huang SA, Haymond MW, Karaviti L. Rapid resolution of consumptive hypothyroidism in a child with hepatic hemangioendothelioma following liver transplantation. Ann Cli Lab Sc. 2007;37:280-284.
- 86.
Zakarija M, McKenzie JM. Pregnancy-associated changes in the thyroid-stimulating antibody of Graves’ disease and the relationship to neonatal hyperthyroidism. J Clin Endocrinol Metab. 1983;57:1036-40.
- 87.
Skuza KA, Sills IN, Stene M, Rapaport R. Prediction of neonatal hyperthyroidism in infants born to mothers with Graves disease. J Pediatr. 1996;128:264-8.
87a. Abeillon-du Payret, Chikh K, Bossart N, Bretones P, Gaucherand P, Claris O, Charrie’ A, Raverot V, Orgiazzi J, Borzon- Chazot F, Bournaud C. Predictive value of maternal second-generation thyroid-binding inhibitory immunoglobulin assay for neonatal autoimmune hyperthyroidism. Eur J Endocrinol. 2014, 171:451-460.
87b. Fort P, Lifshitz F, Pugliese M, Klein I. Neonatal thyroid disease: differential expression in three successive offspring. J Clin Endocrinol Metab. 1988;66:645-7.
87c. Zakarija M, McKenzie JM, Munro DS. Immunoglobulin G inhibitor of thyroid-stimulating antibody is a cause of delay in the onset of neonatal Graves’ disease. J Clin Invest. 1983;72:1352-6.
87d. Kohn LD, Suzuki K, Hoffman WH, et al. Characterization of monoclonal thyroid-stimulating and thyrotropin binding-inhibiting autoantibodies from a Hashimoto’s patient whose children had intrauterine and neonatal thyroid disease. J Clin Endocrinol Metab. 1997;82:3998-4009.
87e. Launberg P, Wallin G, Tallstedt L, Abraham-Nording M, Lundell G, Terring O. TSH-receptor autoimmunity in Graves’ disease after therapy with antithyroid –drugs, surgery or radioiodine: a 5 year prospective randomized study. Eur J Endocrinol. 2008; 158:69-75.
87f. Kiefier FW, Kierbemass-Screhof K, Steiner M, Worda C, Kaisprian G, Tanja D, Kahaly G, Gess A. Fetal/neonatal thyrotoxicosis in a newborn from a hypothyroid woman with Hashimoto thyroiditis, J Clin Endocrinol Metab. 2017;102:6-9.
87g. Neal PR, Jansen RD, Lemons JA, Mirkin LD, Schreiner RL. Unusual manifestations of neonatal hyperthyroidism. Am J Perinatol. 1985;2:231-5.
87h. Daneman D, Howard NJ. Neonatal thyrotoxicosis: intellectual impairment and craniosynostosis in later years. J Pediatr. 1980;97:257-9.
- 88.
Morshed SA, Ando T, Latif R, Davies TF. Neutral antibodies to the TSH receptor are present in Graves' disease and regulate selective signaling cascades. Endocrinology. 151:5537-49, 2010.
88a. Barbesino G, Tomer Y. Clinical utility of TSH receptor antibodies. J Clin Endocrinol Metab. 2013;98:2247-2255.
88b. Diana T, Brown RS, Bossowski A, Segni M, Niedziela M, Konig J, Bossowka A, Ziora K, Smith J, Pitz S, Kanitz M, Kahaly GJ. Clinical relevance of thyroid-stimulating autoantibodies in pediatric Graves’ disease. A multicenter study. J Clin Endocrinol Metab. 2014;99:1648-1655.
88c. Ross DS, Burch HB, Cooper DS, Greenlee MC, Launberg P, Maia AL, Rivkees S, Samuels M, Sosa JA, Stan MN, Walter MA. 2016 American Thyroid Association Guidelines for Diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis.Thyroid.. 2016;2:1343-1421.
88d. Donnelly MA, Wood C, Casey B, Hobbins J, Barbour LA. Early severe fetal Graves disease in a mother after after thyroid ablation and thyroidectomy. Obstetric Gynecol. 2015; 125:141-152.
88e. Besancon A, Beltrand J, Le Gac I, Luton D, Polak M. Management of neonates born to women with Graves’ disease: a cohort study. Eur J Endocrinol. 2014;170:855-862.
- 89.
Matsuura N, Konishi J, Fujieda K, et al. TSH-receptor antibodies in mothers with Graves’ disease and outcome in their offspring. Lancet. 1988;1:14-7.
89a. Tamaki H, Amino N, Aozasa M, et al. Universal predictive criteria for neonatal overt thyrotoxicosis requiring treatment. Am J Perinatol. 1988;5:152-8.
89b. Van der Kaay D, Wasserman JD, Palmert MR. Management of neonates born to mothers with Graves’ disease. Pediatrics. 2016;137:e20151878.
- 90.
de Roux N, Polak M, Couet J, et al. A new mutation of the thyroid-stimulating hormone receptor in a severe neonatal hyperthyroidism. J Clin Endocrinol Metab. 1996;81:2023-6.
90a. Holzapfel HP, Wonerow P, von Petrykowski W, Henschen M, Scherbaum WA, Paschke R. Sporadic congenital hyperthyroidism due to a spontaneous germline mutation in the thyrotropin receptor gene. J Clin Endocrinol Metab. 1997;82:3879-84.
90b. Schwab KO, Gerlich M, Broecker M, Sohlemann P, Derwahl M, Lohse MJ. Constitutively active germline mutation of the thyrotropin receptor gene as a cause of congenital hyperthyroidism. J Pediatr. 1997;131:899-904.
90c. Kopp P, Muirhead S, Jourdain N, Gu WX, Jameson JL, Rodd C. Congenital hyperthyroidism caused by a solitary toxic adenoma harboring a novel somatic mutation (serine281–>isoleucine) in the extracellular domain of the thyrotropin receptor. J Clin Invest .1997;100:1634-9.
90d. Gruters A, Schoneberg T, Biebermann H, et al. Severe congenital hyperthyroidism caused by a germ-line neo mutation in the extracellular portion of the thyrotropin receptor. J Clin Endocrinol Metab. 1998;83:1431.
90e. Paschke R, Niedzela M, Vaidya B, Persani L, Rapaport B, Leclere J. The management of familial and persistent sporadic non-autoimmune hyperthyroidism caused by thyroid stimulating hormone receptor germline mutations. Eur Thyroid J. 2012;1:142-147.
90f. Yoshimoto M. Nakayama M, Baba T, Uehara y, Nikawa N, Ito M, Tsuji Y. A case of neonatal McCune Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediatr Scand. 1991;80:984-987.
- 91.
Davies TF, Amino N. A new classification for human autoimmune thyroid disease. Thyroid. 1993; 3: 331-333,
91a. Fisher DA, Pandian MR, Carlton E. Autoimmune disease: an expanding spectrum. Pediatr Clin North Am. 1987; 34: 907-18.
91b. Rallison ML, Dobyns FR, Keating FR, Rall JE, Tyler FH, Occurrence and natural history of chronic lymphocytic thyroiditis in childhood. J Pediatr. 1975; 86: 675-82.
91c. Loviselli A, Veluzzi F, Mossa P, Cambosu MA, Secci G, Atzeni F, Taberlet A, Balestrieri A, Martino E, Grasso L, Songini M, Bottazzo GF, Mariotti S. Sardinian Schoolchildren Study Group. The Sardinian Autoimmunity Study: 3. Studies on circulating antithyroid antibodies in Sardinian schoolchildren: relationship to goiter prevalence and thyroid function.Thyroid. 2001;1: 849-57.
91d. Kabelitz M, Liesenkotter KP, Stach B, Willigerodt H, Stablen W, Singendok W, Jager-Roman E, Litzenborger H, EhnertB, Gruters A. The prevalence of anti-thyroid peroxidase antibodies and autoimmune thyroiditis in children and adolescents in an iodine replete area. Eur J Endocrinol. 2003;48:301-307.
91e. Kaloumenou I, Mastorakos G, Alevizaki M, Duntas LH, Mantzou E, Ladopoulos C, Antoniou A, Chiotis D, Popassotiriou I, Chousos GP, Dacou-Voutetakis-C. Thyroid autoimmunity in schoolchildren in an area with long-standing iodine sufficiency: correlation with gender, pubertal stage, and maternal thyroid autoimmunity. Thyroid. 2008;18:747-58.
91f. Davies TF. Really significant genes for autoimmune thyroid disease do not exist–so how can we predict disease? Thyroid. 2007;17:1027-9.
91g. Brown RS. Autoimmune thyroid disease: unlocking a complex puzzle. Curr Opin Pediatr. .2009;21:523-8.
91h .Segni M, Pani MA, Pasquino AM, Badenhoop K. Familial clustering of juvenile thyroid autoimmunity: higher risk is conferred by human leukocyte antigen DR3-DQ2 and thyroid peroxidase antibody status in fathers. J Clin Endocrinol Metab 2002; 87: 3779-82.
91i.Segni M, Wood J, Pucarelli I, Toscano V, Toscano R, Pasquino AM. Clustering of autoimmune thyroid diseases in children and adolescents: a study of 66 families. J Pediatr Endocrinol Metab. 2001, 14 Supp 5:1271-5.
91j. Wiebolt J, Achterbergh R, den Boer A, van der Leij S, Marsch E, Suelmann B, de Vries R, van Haeften TW. Clustering of additional autoimmunity behaves differently in Hashimoto's patients compared with Graves' patients. Eur J Endocrinol. 2011;164:789-94.
91k. Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid. 2002;12:975-986.
91l. Karlsson B, Gustafsson G, Ivarson SA, Anneren G, Thyroid dysfunction in Down’s syndrome: relation to age and thyroid autoimmunity. Arch Dis Child. 1998;79:243-245.
91m. Eisheikh M, Wass JA, Conway GS. Autoimmune thyroid disease in women with Turner ‘s syndrome-the association with karyotype. Clin Endocrinol. 2001; 55:223-226,
91n. Seminog OO, Seminog AB, Yeates D, Goldacre MJ. Associations between Klinefelter’s syndrome and autoimmune diseases: English national recortd linkage studies. Autoimmunity. 2015;48: 125-280.
91o. Quaio CR, Carvalho JF, da Silva CA, Bueno C, Brasil AS, Pereira AC, Jorge AA, Malaquias AC, Kim CA, Bertola DR. Autoimmune disease and multiple autoantibodies in 42 patients with RASopathies. Am J Med Genet. 2012; 158A: 1077-82.
91p. Brent GA. Enviromental exposures and autoimmune thyroid disease. Thyroid. 2010;20:755-761.
91q. Eshler DC, Hasam A, Tomer Y. Cutting edge: the etiology of autoimmune thyroid disease. Clin Rev Allergy Immunol. 2011; 41:190-197.
- 92.
Brown RS. Immunoglobulins affecting thyroid growth: a continuing controversy. J Clin Endocrinol Metab. 1995;80:1506-8.
92a. deVries L, Bulvik S, Philllip M. Chronic autoimmune thyroiditis in children and adolescents at presentation and during long-term follow up. Arch Dis Child. 2009; 94:33-7.
92b. Najjar SS. Muscular hypertrophy in hypothyroid children: the Kocher-Debre-Semelaigne syndrome; a review of 23 cases. J Pediatr. 1974;85:236-9.
92c. Hopwood NJ, Lockhart LH, Bryan GT. Acquired hypothyroidism with muscular hypertrophy and precocious testicular enlargement. J Pediatr. 1974;85:233-6.
92d. Anasti JN, Flack MR, Froehlich J, Nelson LM, Nisula BC. A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J Clin Endocrinol Metab. 1995;80:276-9.
- 93.
Maenpaa J, Raatikka M, Rasanen J, Taskinen E, Wager O. Natural course of juvenile autoimmune thyroiditis. J Pediatr. 1985;107:898-904.
93a. Moore DC. Natural course of ‘subclinical’ hypothyroidism in childhood and adolescence. Arch Pediatr Adolesc Med. 1996;150:293-7.
93b. Lazar L, Frumkin RB, Battat E, Lebenthal Y, Phillip M, Meyerovitch J. Natural history of thyroid function tests over 5 years in a large pediatric cohort. J Clin Endocrinol Metab. 2009;94:1678-82.
93c. Aversa T, Corrias A, Salerno MC, Tessaris D, Di Mase R, Valenzise M, Corica D, De Luca F, Wasniewka M. Five-years prospective evaluation of thyroid function test evolution in children with Hashimoto’s thyroiditis presenting with either euthyroidism or subclinical hypothyroidism. Thyroid. 2016, 26:1450-1456.
- 94.
Hayashida N,Imaizumi M, Shimura H, Okubo N, Asari Y, Nigarawa T, Midorikava Sm Kotani K, Nakaji S, Otssuru A, Akamizu T, Kitaoka M, Suzuki S, Taniguchi N, Yamashita S, Takamura N. Thyroid ultrasound findings in children from three Japanese prefectures: Aomori, Yamanashi and Nagasaki. Plos One. 2013,8:e83220.
94a. Corrias A, Cassio A, Weber G, Mussa A, Wasniewska M, Rapa A, Gastaldi R, Einaudi S, Baronio F, Vigone MC, Messina MF, Bal M, Bona G, De Santis C; for the study for thyroid diseases of the Italian Society for Pediatric Endocrinology and Diabetology. Thyroid nodules and cancer in children and adolescents affected by autoimmune thyroiditis. Arch Pediatr Adolesc Med. 2008; 162:526-531.
94b. Keskin M, Savas-Erdeve S, Aycan Z. Co-existence of thyroid nodule and thyroid cancer in children and adolescents with Hashimoto thyroiditis: a single-center study. Horm Res Pediatr. 2016; 85:181-187.
94c. Kambalpalli M, Gupta A, Prasad UR, Francis GL. Ultrasound characteristics of the thyroid in children and adolescents with goiter: a single center experience. Thyroid. 2015; 25:176-182.
94d. Jeong SH, Hong HS, Lee FH, Kwak JJ. Papillary thyroid carcinoma arising in children and adolescent Hashimoto’s thyroiditis: ultrasonographic and pathologic findings. Int J Endocrinolol. 2016, 2397690.
94e. Gilani BB, MacGillivray MH, Voorhess ML, Mills BJ, Riley WJ, MacLaren NK. Thyroid hormone abnormalities at diagnosis of insulin-dependent diabetes mellitus in children. J Pediatr. 1984;105:218-22.
94f. Kakleas K, Soldatou A, Karachaliou F, Karavanaki K. Associated autoimmune diseases in children and adolescents with type 1 diabetes. Autoimmunity Reviews. 2015; 14:781-797.
- 95.
Neufeld M, Maclaren N, Blizzard R. Autoimmune polyglandular syndromes. Pediatr Ann. 1980;9:154-62.
95a. Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med. 2004;350:2068-2079.
95b. Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83:1049-55.
95c. Kordonouri O, Harmann R, Deiss D, Wilms M, Gruters-Kieslich A. Natural course of autoimmune thyroiditis in type 1 diabetes: association with gender, age, diabetes duration and and puberty. Arch Dis Child. 2005; 90:411-414.
95d. Bright GM, Blizzard RM, Kaiser DM, Clarke WL. Organ-specific autoantibodies in children with common endocrine disease. J Pediatr. 1982; 100:8-14.
95e. Dost A, Roher TR, Frolich-Reitherer E, Bollow E, Karges B, Bockmann A, Harmann J, Holl RW: DPV initiative and the German Competence Network Diabetes Mellitus. Horm Res Paediatr. 2015; 84: 190-8.
95f. Naiyer AJ, Shah J, Hernandez L, Kim SY, Ciaccio EJ, Cheng J, Manavalan S, Bhagat G, Green PH. Tissue transglutaminase antibodies in individuals with celiac disease bind to thyroid follicles and extracellular matrix and may contribute to thyroid dysfunction. Thyroid. .2008;18:1171-8.
95g. Roy A, Laszowska M, Sundstrom J, Lebwohl B, green PH, Kampe O, Ludvigsoon JF. Prevalence of celiac disease in patients with autoimmune thyroid disease: a meta-analysis. Thyroid. 2016; 26:880-90.
95h. Kurien M, Mollazadegan K, Sanders DS, Ludvigssson JF. Celiac disease increased risk of thyroid disease in patients with type 1 diabetes: a nationwide cohort study. Diab Care. 2016 39:371-375.
95i. Virili C, Bassotti G, Santaguida MG, Iourio R, Del Duca SC, Mercuri V, Pocarelli A, Gargiulo P. Gargano L, Centanni M. Atypical celiac disease as a cause of increased need for thyroxine: a systematic study. J Clin Endocrinol Metab. 2012; 97:E419-22.
95j. Boelaert K, Newby PR, Simmonds MJ, Holder RL, Carr-Smith JD, Heward JM, Manji N, Allahabadia A, Armitage M, Chatterjee KV, Lazarus JH, Pearce SH, Vaidya B, Gough SC, Franklyn JA. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. 2010;123:183.e1-9.
95k Irvine WJ, Davies SH, Teitelbaum S, Delamore IW, Williams AW. The clinical and pathological significance of gastric parietal cell antibody. Ann NY Acad Sci.1965; 124:657-91.
95l. Segni M, Borrelli O, Pucarelli I, Delle Fave G, Pasquino AM, Annibale B. Early manifestations of gastric autoimmunity in patients with juvenile autoimmune thyroid diseases. J Clin Endocrinol Metab. 2004; 4944-8.
- 96.
Scott HS, Heino M, Peterson P, et al. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol Endocrinol.1998;12:1112-9.
96a. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, Browne SK, Hsu AP, Rampertaap S, Swamydas M, Collar AL, Kong HH, Lee CR, Chascsa D, Simcox T, Pham A, Bondici A, Natarajan M, Monsale J, Kleiner DE, Quezado M, Alevizos I, Moutsopoulos NM, Yockey L, Frein C, Soldatos A, Calvo KR, Adjemian J, Similuk MN, Lang DM, Stone KD, Uzel G, Kopp JB, Bishop RJ, Holland SM, Olivier KN, Fleisher TA, Heller T, Winer KK, Lionakis MS. Redifined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI insight. 2016;1(13):e88782.
96b. Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid. 2002; 12:975-986.
96c. Fallahi P, Ferrari SM, Ruffilli I, Elia G, Biricotti M, Vita R, Benvenga S, Antonelli A. The association of other autoimmune diseases in patients with autoimmune thyroiditis: Review of the literature and report of a large series of patients. Autoimm Rev. 2016;15:1125-1128.
96d. Stagi S, Giani T, Simonini G, Falcini F. Thyroid function, autoimmune thyroiditis and coeliac disease in juvenile idiopathic arthritis. Reumatology. 2005; 44:517-20.
96e. Kroon MW, Vrijman C, Chandeck C, Wind BS, Wolkerstofer A, Luiten RM, Bos JD, Geskus RB, van Trosenburg P, van der Veen JP. High prevalence of autoimmune thyroiditis in children and adolents with vitiligo. Horm Res Paediatr. 2013;79:137-44.
96f. Noso S, Park C, Babaya N, Hiromine Y, Harada T, Ito H, Taketomo Y, Kanto K, Oiso N, Kawada A, Suzuki T, Kawabata Y, Ikegami H. Organ specificity in autoimmune diseases: thyroid and islet autoimmunity in alopecia areata. J Clin Endocrinol Metab. 2015; 100:1976-1983.
96g. Leznoff A, Josse RG, Denburg J, Dolovich J. Association of chronic urticaria and angioedema with thyroid autoimmunity. Arch Dermatol. 1983;119:636-40.
96h. O’Regan S, Fong JS, Kaplan BS, Chadarevian JP, Lapointe N, Drummond KN. Thyroid antigen-antibody nephritis. Clin Immunol Immunopathol. 1976;6:341-6.
96i. Segni M, Pucarelli I, Truglia S, Turriziani I, Serafinelli C, Conti F. High prevalence of antinuclear antibodies in children with thyroid autoimmunity. J Immunol Res 2014, 150239.
96j. Manetti L, Lupi I, Morselli L, Albertini S, Cosottini M, Grasso L, Genovesi M, Pinna G, Mariotti S, Bogazzi F, Bartalena L, Martino E. Prevalence and functional significance of antipituitary antibodies in patients with autoimmune and non-autoimmune thyroid diseases. J Clin Endocrinol Metab. 2007.92:2176-81.
96k. De Bellis A, Bellastella G, Maiorino MI, Aitella E, Lucci E, Cozzolino D, Bellastella A, Bizzarro A, Giugliano D, Esposito K, Italian Autoimmune Hypophysitis network group. Longitudinal behavior of autoimmune GH deficiency: from childhood to transition age. Eur J Endocrinol. 2016;174:381-7.
- 97.
Mehta A, Hindmarsh PC, Stanhope RG, Brain CE, Preece MA, Dattani MT. Is the thyrotropin-releasing hormone test necessary in the diagnosis of central hypothyroidism in children?. J Clin Endocrinol Metab. 2003;88:5696-703.
97a. Feingold SB, Smith J, Houtz J, et al. Prevalence and functional significance of thyrotropin (TSH) receptor blocking antibodies in children and adolescents with chronic lymphocytic thyroiditis. J Clin Endocrinol Metab. 2009;94: 4742-8.
97b. Brown RS, Alter CA, Sadeghi-Nejad A. Severe unsuspected maternal hypothyroidism discovered after the diagnosis of thyrotropin receptor-blocking antibody-induced congenital hypothyroidism in the neonate: failure to recognize and implication to the fetus Horm Res Pediatr. 2015; 83:132-8.
- 98.
Pedersen OM, Aadal NP, Larssen TB, Varhuag JE, Myking O, Vik-Mo H. The value of ultrasonography in predicting autoimmune thyroid disease. Thyroid. 2000, 10:251-259.
- 99.
Rovet JF, Daneman D, Bailey JD. Psychologic and psychoeducational consequences of thyroxine therapy for juvenile acquired hypothyroidism. J Pediatr. 1993;122:543-9.
99a. Van Dop C, Conte FA, Koch TK, Clark SJ, Wilson-Davis SL, Grumbach MM. Pseudotumor cerebri associated with initiation of levothyroxine therapy for juvenile hypothyroidism. N Engl J Med.1983;308:1076-80.
99b. Lomenick JP, El-Sayyid M, Smith WJ. Effect of levo-thyroxine treatment on weight and body mass index in children with acquired hypothyroidism. J Pediatr. 2008. 152:96-100.
99c. Rivkees SA, Bode HH, Crawford JD. Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med. 1988;318:599-602.
- 100.
Salerno M, Capalbo D, Cerbone M, De Luca F. Subclinical hypothyroidism in childhood- current knowledge and open issues. Nat Rev Endocrinol. 2016; 734-746.
100a. Surks MI, Ortiz E, Daniels GH, et al. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. Jama. 2004;291:228-38.
100b. Taylor P, Razvi S, Pearce Sh, Colin D. A Review of the clinical consequences of variation in thyroid function whitin the reference range. J Clin Endocrinol Metab. 2013; 98: 3562-3571,
100c. Ittermann T, Thamm M, Wallschofski H, Rettig R, Volzke H. Serum thyroid-stimulating hormone levels are associated with blood pressure in children and adolescents. J Clin Endocrinol Metab. 2012; 97:828-834.
100d. Witte T, Ittermann T, Thamm M, Ribiet NB, Votzke H. Association between serum thyroid stimulating hormone levels and serum lipids in children and adolescents: a population-based study on german youth. J Clin Endocrinol Metab. 2015;100:2090-7.
100e. Nader NS, Bahn RS, Johnson ,MD, Weaver AL, Singh R, Kumar S. Relationships between thyroid function and lipid status or insulin resistance in a pediatric population. Thyroid. 2010;20:1333-1339.
100f. McLeod DS, Watters KF, Carpenter AD, Landenson PW, Cooper DS, Ding EL. Thyrotropin and thyroid cancer diagnosis: a systematic review and dose-response meta-analysis. J Clin Endocrinol Metab. 2012; 97:2682-2692.
100g. Lazarus J, Brown RS, Daumerie C, Hubalewska-Dydejczyk A, Negro R, Vaydya B. European thyroid association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur Thyroid J. 2014; 3: 76-94.
- 101.
Surks MI, Sievert R. Drugs and thyroid function. N Engl J Med. 1995;333:1688-94.
101a. McCowen KC, Garber JR, Spark R. Elevated serum thyrotropin in thyroxine-treated patients with hypothyroidism given sertraline. N Engl J Med. 1997;337:1010-1.
101b. Delange FM. Iodine Deficiency. In: Braverman LE, Utiger,R.D., ed. Werner & Ingbar’s The Thyroid. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2000.
101c. Pacaud D, Van Vliet G, Delvin E, et al. A Third World endocrine disease in a 6-year-old North American boy. J Clin Endocrinol Metab. 1995;80:2574-6.
101d. Donadieu J, Rolon MA, Thomas C, et al. Endocrine involvement in pediatric-onset Langerhans ’ cell histiocytosis: a population-based study. J Pediatr. 2004;144:344-50.
- 102.
Hauser P, Zametkin AJ, Martinez P, et al. Attention deficit-hyperactivity disorder in people with generalized resistance to thyroid hormone. N Engl J Med. 1993;328:997-1001.
102a. Bercu BB, Orloff S, Schulman JD. Pituitary resistance to thyroid hormone in cystinosis. J Clin Endocrinol Metab. 1980;51:1262-8.
- 103.
van der Gaag RD, Drexhage HA, Wiersinga WM, et al. Further studies on thyroid growth-stimulating immunoglobulins in euthyroid nonendemic goiter. J Clin Endocrinol Metab. 1985;60:972-9.
- 104.
Rother KI, Zimmerman D, Schwenk WF. Effect of thyroid hormone treatment on thyromegaly in children and adolescents with Hashimoto disease. J Pediatr. 1994;124:599-601.
- 105.
Svensson J, Ericsson UB, Nilsson P, Olsson C, Jonsson B, Lindberg B, Ivarsson SA. Levothyroxine treatment reduces thyroid size in children and adolescents with chronic autoimmune thyroiditis. J Clin Endocrinol Metab. 2006. 91:1729-34.
- 106.
Smith SL, Pereira KD Suppurative thyroiditis in children. A management algorithm. Pediatr Emerg Care. 2008; 24:764-767.
106a. Greene JN. Subacute thyroiditis. Am J Med. 1971:51:97.
106b. Paes JE, Burman KD,.Cohen J, Franklyn J, McHemry CR, Shoham S, Kloos RT. Acute bacterial suppurative thyroiditis: a clinical review and expert opinion. Thyroid. 2010; 20:247-255.
106c. Mali VP, Prabhakaran K. Recurrent acute thyroid swellings because of pyriform sinus fistula. J Pediatr Surg. 2008;43:e27-30.
106d. Radfar N,Kenny FM, Larsen PR. Subacute thyroiditis in a lateral thyroid gland:evaluation of the pituitary-thyroid axis during the acute distructive and the recovery phases. J Pediatr. 1975; 87:34.
- 107.
Braverman L, Utiger R: Introduction to thyrotoxicosis. In: Braverman L, Utiger R eds. The Thyroid. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 453-455, 2005.
107a. Graves RJ: Clinical lectures. London Med Surg J (pt 2):516, 1835.
107b. Kjaer H, Andersen SM, Hansen D. Increasing incidence of juvenile thyrotoxicosis in Denmark: a nationwide study, 1998-2012. Horm Res Paediatr. 2015;84: 102-107.
107c. Shulman DI, Muhar I, Jorgensen EV, Diamond FB, Bercu BB, Root AW. Autoimmune hyperthyroidism in prepubertal children and adolescents: comparison of clinical and biochemical features at diagnosis and responses to medical therapy. Thyroid. 1997;7:755-60.
107d. Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM. Special features of Graves ’ disease in early childhood. Thyroid. 1999;9:871-7.
107e. Goday-Arno A, Cerda-Esteva M, Flores-Le-Roux JA, Chillaron-Jordan JJ, Corretger JM, Cano-Perez JF. Hyperthyroidism in a population with Down syndrome (DS). Clin Endocrinol. 2009;71:110-4.
107f. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19:673-716.
107g. Smith BR, Sanders J, Furmaniak J. TSH receptor antibodies. Thyroid. 2007;17:923-38.
107h. Sanders J, Evans M, Premawardhana LD, et al. Human monoclonal thyroid stimulating autoantibody. Lancet. 2003;362:126-8.
107i. Sanders J, Miguel RN, Bolton J, et al. Molecular interactions between the TSH receptor and a thyroid-stimulating monoclonal autoantibody. Thyroid. 2007;17:699-706.
107j. Rapoport B, McLachlan SM. The thyrotropin receptor in Graves’ disease. Thyroid .2007;17:911-22.
107k. Williams RC, Jr., Marshall NJ, Kilpatrick K, et al. Kappa/lambda immunoglobulin distribution in Graves’ thyroid-stimulating antibodies. Simultaneous analysis of C lambda gene polymorphisms. J Clin Invest. 1988;82:1306-12.
- 108.
Segni M, Gorman CA. The aftermath of childhood hyperthyroidism. J Pediatr Endocrinol Metab. 2001; Suppl 5 1277-82.
108a. Goldstein SM, Katowitz WR, Modhang T, Katowitz JA. Pediatric Thyroid-Associated Orbitopathy: The children’s hospital of Philadelphia experience and literature review. Thyroid. 2008;18:997-999.
108b. Nucci P, Brancato R, Bandello F, Alfarano R, Bianchi S. Normal exophtalmometric values in children. Am J Opthal 1989;108:582-584.
108c. Liu GT, Helher KL, Katowitz JA, Kazim M, Moazami G, Moshang T, Teener JW, Sladeky J, Volpe NJ, Galetta SL. Prominent proptosis in childhood thyroid eye disease. Opthalmology. 1996;103:779-784.
108d. Durairaj VD, Bartley GB, Garrity JA. Clinical features and treatment of Graves’ opthalmopathy in pediatric patients. Opthal Plast Reconstr Surg 2006; 22:7-12.
- 109.
Sterling K, Refetoff S, Selenkow HA. T3 thyrotoxicosis due to elevated serum triiodothyronine levels. JAMA
. 1970;213:571-5.
109a. Harvengt J, Boizeau P, Chevenne D, Zenaty D, Paulsen A, Simon D, Crepon SG, Alberti C, Carel JC, Leger J. Triiodothyronine- predominant Graves’ disease in childhood:detection and therapeutic inplications. Eur J Endocrinol. 2015; 172: 715-23.
109b. Ruiz M, Rajatanavin R, Young RA, Taylor C, Brown R, Braverman LE, Ingbar SH. Familial dysalbuminemic hyperthyroxinemia: a syndrome that can be confused with thyrotoxicosis. N Engl J Med. 1982;306:635-9.
109c. Mariotti S, Martino E, Cupini C, et al. Low serum thyroglobulin as a clue to the diagnosis of thyrotoxicosis factitia. N Engl J Med. 1982;307:410-2.
109d. Kaguelidou F, Alberti C, Castanet M, Guitteny MA, Czernichow P, Leger J. Predictors of autoimmune hyperthyroidism relapse in children after discontinuation of anthythyroid drug treatment. J Clin Endocrinol Metab. 2008; 93:3817-3826.
- 110.
Ichiki Y, Akahoshi M, Yamashita N, et al. Propylthiouracil-induced severe hepatitis: a case report and review of the literature. J Gastroenterol. 1998;33:747-50.
110a. Russo MW, Galanko JA, Shrestha R, Fried MW, Watkins P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transpl. 2004;10:1018-23.
110b. Rivkees SA, Mattison DR. Ending propylthiouracil-induced liver failure in children. N Engl J Med. 2009;360:1574-5.
110c. Bahn RS, Burch HS, Cooper DS, Garber JR, Greenlee CM, Klein IL, Laurberg P, McDougall IR, Rivkees SA, Ross D, Sosa JA, Stan MN. The Role of Propylthiouracil in the Management of Graves’ Disease in Adults: report of a meeting jointly sponsored by the American Thyroid Association and the Food and Drug Administration. Thyroid. 2009;19:673-4.
110d. Okuno A, Yano K, Inyaku F, Suzuki Y, Sanae N, Kumai M, Naitoh Y. Pharmacokinetics of methimazole in children and adolescents with Graves’ disease. Acta Endocrinol.1987. 115:112-118.
110e. Rivkees SA. Pediatric Graves’ disease: management in the post-propilthyuracil era .Inter J Pediatr Endocrinolol. 2014; 10:1-10.
110f. Smith J, Brown RS. Persistence of thyrotropin (TSH) receptor antibodies in children and adolescents with Graves’ disease treated using antithyroid medication. Thyroid. 2007;17:1103-7.
110g. Fenzi G, Hashizume K, Roudebush CP, DeGroot LJ. Changes in thyroid-stimulating immunoglobulins during antithyroid therapy. J Clin Endocrinol Metab. 1979;48:572-6.
110h. Teng CS, Yeung RT. Changes in thyroid-stimulating antibody activity in Graves’ disease treated with antithyroid drug and its relationship to relapse: a prospective study. J Clin Endocrinol Metab. 1980;50:144-7.
110i. Bliddal H, Kirkegaard C, Siersbaek-Nielsen K, Friis T. Prognostic value of thyrotrophin binding inhibiting immunoglobulins (TBII) in longterm antithyroid treatment, 131I therapy given in combination with carbimazole and in euthyroid ophthalmopathy. Acta Endocrinol. 1981;98:364-9.
110j. Collen RJ, Landaw EM, Kaplan SA, Lippe BM. Remission rates of children and adolescents with thyrotoxicosis treated with antithyroid drugs. Pediatrics. 1980;65(3):550-6.
110k. Leger J, Gelwane G, Kaguelidou F et al. Positive impact of long-term antithyroid drug treatment on the outcome of children with Graves disease: natural long-term cohort study. J Clin Endocrinol Metab. 2012; 97:110-9.
110l. Hashizume K, Ichikawa K, Sakurai A, et al. Administration of thyroxine in treated Graves’ disease. Effects on the level of antibodies to thyroid-stimulating hormone receptors and on the risk of recurrence of hyperthyroidism. N Engl J Med. 1991;324:947-53.
110m. McIver B, Rae P, Beckett G, Wilkinson E, Gold A, Toft A. Lack of effect of thyroxine in patients with Graves’ hyperthyroidism who are treated with an antithyroid drug. N Engl J Med 1996;334:220-4.
- 111.
Rivkees SA, Sklar C, Freemark M. Clinical review 99: The management of Graves’ disease in children, with special emphasis on radioiodine treatment. J Clin Endocrinol Metab. 1998;83:3767-76.
111a. Rivkees SA, Stephenson K, Dinauer C. Adverse events associated with methimazole therapy of Graves’ disease in children. Int J Pediatr Endocrinol. 2010; 176970.
111b. Wada N, Mukai M, Kohono M, Notoya A, Ito T, Yoshioka N. Prevalence of serum anti-myeloperoxidase antineutrophil cytoplasmatic antibodies (MPO-ANCA) in patients with Graves’ disease trated with prophilthyuracil and thiamazole. Endocr J. 2002; 49: 329-334.
111c. Guma M, Salinas I, Reverter JL, Roca J, Valls-Roc M, Juan M, Olive’ A. Frequency of antineutrophil cytoplasmic antibody in Graves’ disease patients treated with methimazole. J Clin Endocrinol Metab. 2003, 88:2141-6.
111d. Tajiri J, Noguchi S, Murakami T, Murakami N. Antithyroid drug-induced agranulocytosis. The usefulness of routine white blood cell count monitoring. Arch Intern Med. 1990;150:621-4.
111e. van Veenendaal NR, Rivkees SA. Treatment of pediatric Graves’ disease is associated with excessive weight gain. J Clin Endocrinol Metab. 2011; 96:3257-63.
111f. Mazzaferri EL. Thyroid cancer and Graves’ disease. J Clin Endocrinol Metab. 1990; 70:825-829.
111g. Dobyns BM, Sheline GE, Workman JB, Tompkins EA, McConahey WM, Becker DV. Malignant and benign neoplasm of the thyroid in patients treated for hyperthyroidism: a report of the cooperative thyrotoxicosis therapy follow-up study. J Clin Endocrinol Metab. 1974; 38:976-988.
- 112.
Sherman J, Thompson GB, Lteif A, et al. Surgical management of Graves disease in childhood and adolescence: an institutional experience. Surgery. 2006;140:1056-61. Discussion 61-2.
112a.Tuggle CT, Roman SA, Wang TS, Thomas DC, Uldelsman R, Ann Sosa J. Pediatric endocrine surgery: who is operating on our children? Surgery. 2008; 144:869-877, Discussion 877.
112b. Kundel A, Thompson GB, Richards ML, Qiu LX, Cai Y, Schwenk FW, Lief AN, Pittock ST, Kumar S, Tebben PJ Hay ID, Grant CS. Pediatric endocrine surgery: a 20 year experience at the Mayo Clinic. J Clin Endocrinol Metab. 2014. 99: 399-406.
112c. Beuer CK, Solomon D, Donovan P, Rivkees SA, Udelsman R. Effect of patient age on surgical outcome for Graves’ disease: a case control study of 100 consecutive patients at a high volume thyroid surgical center. Int J Pediatr Endocrionol. 2013;1: 9856.
- 113.
Nikiforov Y, Gnepp DR, Fagin JA. Thyroid lesions in children and adolescents after the Chernobyl disaster: implications for the study of radiation tumorigenesis. J Clin Endocrinol Metab. 1996;81:9-14.
113a. Safa AM, Schumacher P, Rodriguez-Antunez A. Long-term follow-up results in children and adolescents treated with radioactive iodine (131-Iodine). N Engl J Med. 1975; 292:167-171.
113b. Holm LE, Dahlqvist I, Israelsson A, Lundell GM. Malignant thyroid tumors after 131-iodine therapy. N Engl J Med. 1980; 303:188-191.
113c. Hamburger JI. Management of hyperthyroidism in children and adolescents. J Clin Endocrinol Metab. 1985; 60:1019.
113d. Hayek A, Chapman E, Crawford JD. Long-term results of treatment of thyrotoxicosis in children and adolescents with radioactive iodine. N Engl J Med .1970; 283:949, 953.
113e. Franklyn JA, Maisonneuve P, Sheppard M, Betteridge PB.Bovie P. Cancer incidence and mortality after radioiodine treatment for hyperthyroidism: a population-based cohort study. Lancet. 1999;353:2111-5
113f. Starr P, Jaffe HL, Oettinger L Jr. Late results of 131-I treatment of hyperthyroidism in seventy-three children and adolescents. J Nucl Med.1964; 5:81-9.
113g. Kogut MD, Kaplan SA, Collipp PJ, Tiamsic T, Boyle D. Treatment of hyperthyroidism in children. N Engl J Med.1965; 272:217-22.
113h. Read CH Jr, Tansey MJ, Menda Y. A 36-year retrospective analysis of the efficacy and safety of radioactive iodine in treating young Graves’ patients. J Clin Endocrinol Metab. 2004;89:4229-33.
113i. Rivkees SA, Dinauer C. An optimal treatment for pediatric Graves’ disease is radioiodine. J Clin Endocrinol Metab. 2007;92:797-800.
113j. Rivkees SA. Controversies in the management of Graves’ disease in children. J Endocrinol Invest. 2016;39:1247-1257.
- 114.
Ly S, Frates MC, Benson CB, Peters HE, Grant FD, Drubach LA, Voss SD, Feldman HA, Smith JR, Barletta J, Hollowell M, Cibas ES, Moore FD, Modi BM, Shamberger RC, Huang SA. Features and outcome of autonomous thyroid nodules in children: 31 consecutive patients seen at a single center. J Clin Endocrinol Metab. 2016;101:3856-3862.
114a. Niedzela M, Breborowicz D, Trejster E, Koeman E. Hot nodules in children and adolents in western Poland from 1996 to 2000: clinical analysis of 31 patients. J Pediatr Endocrinol Metab. 2002;15:823-830.
114b. Francis GL, Waguenspack SG, Bauer AJ, Angelos P, Benvenga S, Cerutti JM, Dinauer CA, Hamilton J, Hay ID, Luster M, Parisi MT, Rachmiel M, Thompson GB, Yamashita S. Management guidelines for children with thyroid nodules and differentiated thyroid cancer. Thyroid. 2015:716-759.
114c. Gershergorn MC, Weintraub BD. Thyrotropin-induced hyperthyroidism caused by selective pituitary resistance to thyroid hormone. A new syndrome of “inappropriate secretion of TSH”. J Clin Invest. 1975;56:633-642.
114d. Beck-Peccoz P, Lania A, Beckers A, Chatterrjee K, Wernau JL. European Thyroid association guidelines for the diagnosis and treatment of thyrotropin-secreting pituitary tumors. Thyroid J. 2013;2-76-82.
114e. Daly AF, Tichomirova MA, Petronians P, Heliovaara E, Jaffrain-Rea ML, Barlus R et al. Clinical characteristics and therapeutic responses.in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. 2010; 95:E373-E383.
114f. Misra M, Levitsky LL, Lee MM. Transient hyperthyroidism in an adolescent with hydatidiform mole. J Pediatr. 2002;140:362-6.
114g. Hershman JM. Human chorionic gonadotropin and the thyroid: hyperemesis gravidarum and throphoblastic tumors. Thyroid. 1999; 9:653-657.
115a. Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robison B, Rosental MS, Santoro M, Schulberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines task force on medullary thyroid carcinoma. Thyroid. 2015; 25:567-610.
115b. Hung W, Anderson KD, Chandra RS, et al. Solitary thyroid nodules in 71 children and adolescents. J Pediatr Surg. 1992;27:1407-9.
115c . Schlumberger M, De Vathaire F, Travagli JP, et al. Differentiated thyroid carcinoma in childhood: long term follow-up of 72 patients. J Clin Endocrinol Metab. 1987;65:1088-94.
115d . Flannery TK, Kirkland JL, Copeland KC, Bertuch AA, Karaviti LP, Brandt ML. Papillary thyroid cancer: a pediatric perspective. Pediatrics 1996;98(3 Pt 1):464-6.
115e. Lairmore TC, Frisella MM, Wells SA, Jr. Genetic testing and early thyroidectomy for inherited medullary thyroid carcinoma. Ann Med. 1996;28:401-6.
115f. Sarne D, Schneider AB. External radiation and thyroid neoplasia. Endocrinol Metab Clin North Am. 1996;25:181-95.
115g. Healy JC, Shafford EA, Reznek RH, et al. Sonographic abnormalities of the thyroid gland following radiotherapy in survivors of childhood Hodgkin’s disease. Br J Radiol. 1996;69:617-23.
115h. Soberman N, Leonidas JC, Cherrick I, Schiff R, Karayalcin G. Sonographic abnormalities of the thyroid gland in longterm survivors of Hodgkin disease. Pediatr Radiol. 1991;21:250-3.
115k. Dinauer CA, Breuer C, Rivkees SA. Differentiated thyroid cancer in children: diagnosis and management. Curr Opin Oncol. 2008;20:59-65.
115i. Waguespack SG, Francis G. Initial management and follow up of differentiated thyroid cancer in children. J Natl Compr Canc Netw. 2010; 8:1289-300.
115l. Gharib H, Goellner JR. Fine-needle aspiration biopsy of thyroid nodules. Endocr Pract .1995;1:410-7.
115m. Zimmerman D, Hay ID, Gough IR, et al. Papillary thyroid carcinoma in children and adults: long-term follow-up of 1039 patients conservatively treated at one institution during three decades. Surgery. 1988;104:1157-66.
115o. Ladenson PW, Braverman LE, Mazzaferri EL, et al. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma. N Engl J Med. 1997;337:888-96.
115p. Wohllk N, Cote GJ, Evans DB, Goepfert H, Ordonez NG, Gagel RF. Application of genetic screening information to the management of medullary thyroid carcinoma and multiple endocrine neoplasia type 2. Endocrinol Metab Clin North Am. 1996;25:1-25.
115q. Fogelfeld L, Wiviott MB, Shore-Freedman E, et al. Recurrence of thyroid nodules after surgical removal in patients irradiated in childhood for benign conditions. N Engl J Med. 1989;320:835-40.
115r. de Vathaire F1, Haddy N1, Allodji RS1, Hawkins M1, Guibout C1, El-Fayech C1, Teinturier C1, Oberlin O1, Pacquement H1, Diop F1, Kalhouche A1, Benadjaoud M1, Winter D1, Jackson A1, Bezin Mai-Quynh G1, Benabdennebi A1, Llanas D1, Veres C1, Munzer M1, Nguyen TD1, Bondiau PY1, Berchery D1, Laprie A1, Deutsch E1, Lefkopoulos D1, Schlumberger M1, Diallo I1, Rubino C. Thyroid Radiation Dose and Other Risk Factors of Thyroid Carcinoma Following Childhood Cancer. J Clin Endocrinol Metab. 2015 ;100:4282-90.
115s. Vivanco M1, Dalle JH, Alberti C, Lescoeur B, Yakouben K, Carel JC, Baruchel A, Léger J. Malignant and benign thyroid nodules after total body irradiation preceding hematopoietic cell transplantation during childhood. Eur J Endocrinol. 2012 ;167):225-233.
- ABSTRACT
- INTRODUCTION
- ONTOGENESIS OF THYROID FUNCTION IN THE FETUS AND INFANT
- THYROID FUNCTION IN THE NEONATE, THE INFANT, AND DURING CHILDHOOD
- THYROID DISEASE IN INFANCY
- CAUSES OF PERMANENT CONGENITAL HYPOTHYROIDISM
- CAUSES OF TRANSIENT NEONATAL HYPOTHYROIDISM
- HYPERTHYROIDISM
- THYROID DISEASE IN CHILDHOOD AND ADOLESCENCE
- THYROTOXICOSIS AND HYPERTHYROIDISM
- THYROID NODULES AND CANCER
- REFERENCES
- Review Foetal and neonatal thyroid disorders.[Minerva Pediatr. 2002]Review Foetal and neonatal thyroid disorders.Radetti G, Zavallone A, Gentili L, Beck-Peccoz P, Bona G. Minerva Pediatr. 2002 Oct; 54(5):383-400.
- Autoimmune thyroid disease in pregnant women and their offspring.[Endocr Pract. 1996]Autoimmune thyroid disease in pregnant women and their offspring.Brown RS. Endocr Pract. 1996 Jan-Feb; 2(1):53-61.
- A Prospective Study to Evaluate the Possible Role of Cholecalciferol Supplementation on Autoimmunity in Hashimoto's Thyroiditis.[J Assoc Physicians India. 2023]A Prospective Study to Evaluate the Possible Role of Cholecalciferol Supplementation on Autoimmunity in Hashimoto's Thyroiditis.Bhakat B, Pal J, Das S, Charaborty SK, SircarMedical NR, Kolkata, RGKar, NorthBengal, Siliguri. J Assoc Physicians India. 2023 Jan; 71(1):1.
- Short-term hyperthyroidism followed by transient pituitary hypothyroidism in a very low birth weight infant born to a mother with uncontrolled Graves' disease.[Pediatrics. 2001]Short-term hyperthyroidism followed by transient pituitary hypothyroidism in a very low birth weight infant born to a mother with uncontrolled Graves' disease.Higuchi R, Kumagai T, Kobayashi M, Minami T, Koyama H, Ishii Y. Pediatrics. 2001 Apr; 107(4):E57.
- Review Genetic Defects in Thyroid Hormone Supply.[Endotext. 2000]Review Genetic Defects in Thyroid Hormone Supply.Nettore IC, Fenzi G, Macchia PE. Endotext. 2000
- Disorders of the Thyroid Gland in Infancy, Childhood and Adolescence - EndotextDisorders of the Thyroid Gland in Infancy, Childhood and Adolescence - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...