

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Puberty is a biological process that represents the development of secondary sexual characteristics and attainment of reproductive capacity, influenced by genetic, metabolic, environmental, ethnic, geographic, and economic factors. Pubertal onset is characterized by the increased kisspeptin and neurokinin B secretion leading to re-emergence of pulsatile gonadotropin releasing hormone signaling from the hypothalamus which stimulates increased pituitary secretion of luteinizing hormone and follicle stimulating hormone, which in turn stimulate gonadal sex hormone production. Precocious puberty refers to secondary sexual development occurring earlier than the lower end of normal age and delayed puberty refers to secondary sexual development occurring later than the upper end of normal age for the onset of puberty. These changes may represent a serious underlying condition or signify a common variation of normal for which treatment may not be necessary. Clinical evaluation should include a detailed history and physical examination, including anthropometric measurements, calculation of linear growth velocity, and evaluation of secondary sexual characteristics. This chapter summarizes the physiology of pubertal development, variations in pubertal development, and recent developments regarding human puberty. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Puberty is the process through which reproductive competence is achieved (1). Physical characteristics associated with this process include the development of secondary sex characteristics, acceleration in linear growth velocity, and the occurrence of menarche in women and spermatogenesis in men. The sex chromosome karyotype of the embryo, XX or XY, determines the trajectory for differentiation of the gonads and development of the internal and external genital structures. This complex process, beginning in utero, depends on neuroendocrine signaling and gonadal components. Ultimately, integrated communication between the reproductive and metabolic systems is critical for timely pubertal development (2).
Pubertal development and neuroendocrine system maturation involve the ontogeny, activity, and interactions of the gonadotropin releasing hormone (GnRH) neurons. The onset of puberty is accompanied by increased kisspeptin and neurokinin B secretion causing the GnRH neurons to secrete GnRH in a pulsatile manner. Increased GnRH secretion stimulates pulsatile pituitary luteinizing hormone (LH) and follicle stimulating hormone (FSH) secretion (3). LH and FSH stimulate gonadal sex steroid secretion which promotes development of secondary sex characteristics and influences hypothalamic-pituitary function via negative feedback inhibition. This chapter summarizes the physiology of pubertal development, variations in pubertal development, and recent developments regarding human puberty.
CLINICAL FEATURES OF NORMAL PUBERTAL DEVELOPMENT
Children typically demonstrate a predictable sequence of physical changes during pubertal maturation. Within the chronologic age ranges for pubertal development, individual variations regarding age at onset and tempo of pubertal development are expected.
In humans, two physiological processes, gonadarche and adrenarche, govern pubertal transition. Gonadarche reflects the reactivation of the hypothalamic GnRH pulse generator which has been quiescent since late infancy. Increasing pulsatile GnRH secretion stimulates pulsatile gonadotropin secretion which, in turn, stimulates the growth and maturation of the gonads and gonadal sex steroid secretion. Increased estrogen secretion promotes breast development, cornification of the vaginal mucosa, and uterine growth in girls. Increased testosterone secretion promotes penile enlargement. The increased HPG axis activity culminates in folliculogenesis, ovulation, and menses in the female and spermatogenesis in the male.
In addition to gonadal sex steroid secretion, humans experience adrenarche signifying adrenal pubertal maturation. Adrenarche typically begins prior to the first visible physical manifestation of gonadarche, breast development, or testicular enlargement. Pubarche, the physical manifestation of adrenarche, is characterized by the development of pubic hair, axillary hair, apocrine odor, and acne. Adrenarche indicates increased adrenal cortical zona reticularis activity and is accompanied by increased secretion of dehydroepiandrosterone sulfate (DHEAS), dehydroepiandrosterone (DHEA), androstenedione, and 11-hydroxyandrostenedione (4, 5). These so-called “adrenal androgens” are C19 steroids which do not bind directly to the androgen receptor and can be peripherally converted to more potent androgens. Circulating concentrations of two adrenal 11-oxyandrogens, 11-hydroxyandrostenedione and 11-ketotestosterone increase with adrenarche. Whereas 11-hydroxyandrostenedione has minimal androgenic activity, 11-ketotestosterone is almost as potent as testosterone. During adrenarche, 11-ketotestosterone appears to be the major bioactive adrenal C19 steroid and may be responsible for the physical changes associated with pubarche (6).
Gonadarche and adrenarche are dissociated such that the absence of adrenarche does not prevent gonadarche or the attainment of fertility (7). Curiously, the phenomenon of adrenarche appears to be limited to humans and a few species of non-human primates (8, 9). The factors that drive the dynamic changes within a strictly defined zona reticularis within the adrenal cortex, are still poorly defined. How adrenarche and increased adrenal C19 steroids impact brain development during human adolescence is indeterminate (10). Urinary steroid hormone profiling suggest that adrenarche may be a gradual process that likely begins earlier than previously believed (11).
STAGING OF PUBERTY
Tanner and colleagues followed the pubertal development of children living in an orphanage in the UK. Their five-stage classification system continues to be commonly utilized for clinical assessments (12, 13, 14). For girls, Tanner staging is used to describe breast and pubic hair development (See Figure 1). For boys, Tanner staging is used to describe testicular volume, penile development, and pubic hair development (See Figure 2). Tanner and his colleagues also described that the tempo of puberty varies between individuals.

Figure 1.
Tanner Staging for pubertal development in girls. In girls, breast development is rated from 1 (preadolescent) to 5 (mature), and stage 2 (appearance of the breast bud) marks the onset of pubertal development. Pubic hair stages are rated from 1 (preadolescent, no pubic hair) to 5 (adult), and stage 2. Although pubic hair and genital or breast development are represented as synchronous in the illustration, they do not necessarily track together and should be scored separately. Figure 1 Adapted with permission from Carel JC, Léger J. Clinical practice. Precocious puberty. N Engl J Med. 2008;358(22):2367 and Klein DA, Emerick JE, Sylvester JE, Vogt KS. Disorders of Puberty: An Approach to Diagnosis and Management. Am Fam Physician. 2017 Nov 1;96(9):590-599. PMID: 29094880.
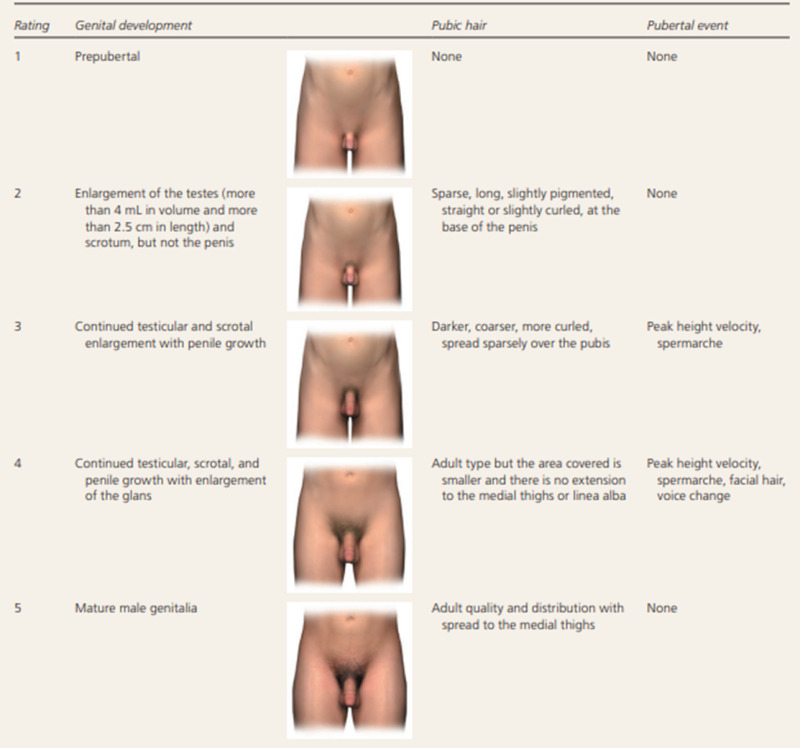
Figure 2.
Tanner Staging for pubertal development in boys. In boys, genital development is rated from 1 (preadolescent) to 5 (adult); stage 2 marks the onset of pubertal development and is characterized by an enlargement of the scrotum and testis and by a change in the texture and a reddening of the scrotal skin. Pubic hair stages are rated from 1 (preadolescent, no pubic hair) to 5 (adult), and stage 2 marks the onset of pubic hair development.Although pubic hair and genital or breast development are represented as synchronous in the illustration, they do not necessarily track together and should be scored separately. In normal boys, stage 2 pubic hair develops at an average of 12 to 20 months after stage 2 genital development. Figure 2 Adapted with permission from Carel JC, Léger J. Clinical practice. Precocious puberty. N Engl J Med. 2008;358(22):2367 and Klein DA, Emerick JE, Sylvester JE, Vogt KS. Disorders of Puberty: An Approach to Diagnosis and Management. Am Fam Physician. 2017 Nov 1;96(9):590-599. PMID: 29094880.
Girls
The typical first clinical sign of puberty in girls is the appearance of breast tissue with elevation of the breast and papilla; this is considered to be Tanner Stage 2 (Figure 1). Initially, breast development (thelarche) may be unilateral. Many girls complain of mild breast tenderness or discomfort during this stage that subsequently resolves. Tanner stage 3 breast development is considered to be additional enlargement of the breast and areola. During Tanner stage 4, the papilla forms a secondary mound above the breast; this stage is often very rapid. Tanner stage 5 represents mature breast development due to recession of the areola to the contour of the breast. Palpation of the breast is obligatory to differentiate breast tissue from lipomastia. In children with obesity without breast development, a palpable depression beneath the nipple in the center of the areola when examined in the supine position gives the impression of a donut and is referred to as the ‘donut’ sign. Breast ultrasound correlates reasonably well with Tanner staging by palpation and can detect breast development slightly earlier than physical exam (15). In most instances, breast development is evident before pubic hair development. Typically, the pubertal growth spurt in girls occurs concurrently with the onset of breast development with only 4-6 cm of linear growth occurring after menarche, however this may be variable.
The appearance of sexual hair including pubic hair (pubarche) signifies the onset of adrenarche. In girls, Tanner stage 2 pubic hair is characterized by sparse, coarse, lightly pigmented hairs along the labia majora. For Tanner stage 3, pubic hair becomes progressively darker, coarser, and spreads over the mons pubis. For Tanner stage 4, pubic hair continues to extend to become an inverse triangle, with spread to the medial aspects of the thighs being considered Tanner stage 5.
With the onset of ovarian estrogen secretion, the vaginal mucosa changes from shiny bright red to pale pink appearance due to cornification of the vaginal mucosa. Increased estrogen secretion promotes uterine growth and causes physiologic leukorrhea, a thin, white, non-foul-smelling vaginal discharge that typically begins 6 to 12 months before menarche. Menarche occurs, on average, 2 to 2.5 years after the onset of breast development (See figure 3A). During the first-year post-menarche, menses are usually irregular and anovulatory. These early years are characterized by inconsistent ovulation and varying lengths of follicular and luteal phases. Ultimately, coordinated maturation of the hypothalamic, pituitary, and ovarian components occurs culminating in cyclic monthly ovulation (16). Although full HPG axis maturation generally occurs over several years, by three years post-menarche, most cycles are between 21-35 days.

Figure 3A and 3B.
Average ages and sequence of pubertal development. Panel A: girls; Panel B: boys.
Boys
For boys, increased testicular volume is the first physical finding indicating onset of gonadarche (See Figure 2 and Figure 3B). Palpation of the testes and use of a Prader orchidometer is essential for accurate assessment. A Prader orchidometer is a collection of 3-D ellipsoids ranging in volume from 1 to 25 mL (See Figure 4). During gonadarche, testicular volume increases, and the penis increases in length and diameter. Flaccid penile length can be measured using a straight edge on the dorsal surface from the pubic ramus to the tip of the glans while compressing the suprapubic fat pad and applying gentle traction to stretch to penis.

Figure 4.
Prader Orchidometer.
Increased testicular volume represents Sertoli cell proliferation, differentiation, and eventually, the initiation of spermatogenesis. The onset of puberty is defined as a testicular volume ≥ 4 ml and a testicular length ≥ 2.5 cm. The volume of mature human testis is approximately 20-25 ml. Spermatozoa (spermaturia) can be found in early morning urine samples beginning during genital stage 3 (16). Nocturnal sperm emissions may also begin around this time.
For boys, Tanner stage 2 pubic hair consists of downy hairs at the base of the penis. During pubic hair stage 3, the hair becomes longer, darker, and extends over the junction of the pubic bones. For pubic hair stage 4, the extent of hair has increased, but has not yet achieved the adult male escutcheon with spread to the medial aspects of the thighs that would be considered Tanner stage 5. Additional features include axillary hair, increased size of the larynx, voice break with deepening of the voice, increased bone mass, and increased muscle strength. Terminal hair develops in androgen-dependent regions on the face and trunk approximately three years after appearance of pubic hair. The distribution and density of beard, chest, abdominal, and back hair varies among individuals.
Peak height velocity is both age and sex-dependent. It occurs earlier in girls, between Tanner breast stages 2 and 3, and later in boys, between Tanner testis stages 3 and 4.
Approximately 50% of boys experience pubertal gynecomastia (17). Typically, pubertal gynecomastia is transient and most prominent in mid-puberty when the ratio of circulating estradiol to testosterone concentrations is relatively higher.
DISCOVERY OF THE HYPOTHALAMIC-PITUITARY-GONADAL (HPG) AXIS
Since ancient times, it was known that castration of animals and humans interfered with development of secondary sex characteristics and fertility (14). In 1935, Ernst Laquer and colleagues isolated testosterone from several tons of steer testes (18). Later that year, Adolf Butenandt, Gunter Hanisch, Leopold Ruzicka, and A. Wettstein published the chemical synthesis of testosterone (19, 20). After showing that follicular fluid obtained from a sow ovary was able to induce cornification of vaginal mucosa, Edgar Allen and Edward Doisy isolated the active substance, estrone (21). Donald MacCorquodale, Stanley Thayer, and Edward Doisy isolated estradiol from 8000 pounds of sow ovaries in 1935 (22). Philip Smith, Bernhard Zondek, Hermann Zondek, H.L. Fevold and colleagues, and Geoffrey Harris established the functional relationships involved in HPG axis function (23, 24, 25, 26). Roger Guillemin and Andrew Schally engaged in a vigorous competition to identify hypothalamic releasing hormones including GnRH (27, 28, 29). Ernst Knobil and his colleagues identified that pulsatile GnRH secretion was essential for sustained pituitary gonadotropin secretion (28, 30). Fred Karsch and Ernst Knobil independently developed the concept of the “GnRH pulse generator” (31). In the 1970s, Melvin Grumbach and colleagues measured circulating gonadotropin concentrations in the human fetus (32). Around the same time, Charles Faiman and Jeremy Winter also reported gonadotropin concentrations in normal and agonadal children (33). Their collective findings led to recognition of early postnatal HPG axis activity followed by quiescence of the HPG axis during childhood until resumption of GnRH pulse generator activity at the onset of puberty.
Ontogeny of GnRH Neurons
Reproductive competence depends on the meticulous development of the GnRH neuron system. In the human fetus, GnRH neurons initially develop in the olfactory placode outside the central nervous system. The olfactory placodes invaginate at approximately 39 days of gestation in the human. Based on the appearance of immunoreactive GnRH protein, the GnRH neuron specification occurs between 39-44 days of gestation (34). The developing GnRH neurons are associated with the embryonic vomeronasal organ. Available data suggest that the GnRH neuron precursors are distinct from those giving rise to the vomeronasal neurons (35).
Subsequently, the GnRH neurons migrate accompanied by olfactory-derived axons, olfactory epithelial sheath cells, and blood vessels towards the cribriform plate (36). Migration of the GnRH neurons seems to pause at the nasal/forebrain junction prior to crossing the cribriform plate (37). During this “pause” phase, multiple tissues, chemokines, growth factors, and neurotransmitters appear to form gradients influencing movement of GnRH neurons. Upon reaching the hypothalamus, the GnRH neurons disperse to their final locations sending projections to the median eminence to release GnRH into the hypophyseal portal vasculature.
The precise origin and particular factors responsible for the specification and differentiation of GnRH neuron precursors remain enigmatic. Inaccessibility of developing human GnRH neurons has led to development of alternative approaches to elucidate the history of GnRH neurons. One approach has involved a protocol to generate GnRH neurons from human pluripotent stem cells (38). With this approach, Yellapragada et al. demonstrated that dose- and time-dependent FGF8 signaling via FGFR1 is indispensable for human GnRH neuron ontogeny (39). Using a differentiation trajectory analysis approach, DLX family of transcription factors have been reported to promote in vitro human GnRH neuron differentiation (40).
Components of the HPG Axis
Gonadotropin-releasing hormone is a decapeptide (pGlu-His-Trp-Ser-Trp-Gly-Leu-Arg-Pro-Gly-NH2) derived from a 92-amino acid precursor, preproGnRH, that was characterized in 1984 (41). LH and FSH are synthesized in the same gonadotroph cell located in the anterior pituitary. LH and FSH are glycoproteins consisting of two subunits. The alpha subunits are identical whereas the beta subunits confer hormone specificity. Each GnRH pulse stimulates an LH pulse.
During human gestation, human chorionic gonadotropin (hCG) drives fetal testicular testosterone secretion in the developing male fetus early during gestation. The pituitary gland begins to secrete gonadotropins with LH and FSH becoming detectable in fetal blood after 14 weeks of gestation (42, 43). Initially, pituitary gonadotropin secretion appears to be GnRH-independent with progressive transition to kisspeptin-GnRH regulation of pituitary gonadotropin secretion during the third trimester (44). Peak gonadotropin concentrations occur around the midpoint of gestation followed by a progressive decline attributed to suppression by placental estrogens (45). In the male fetus, testicular testosterone secretion is essential for normal development of internal and external male genital structures. Comparatively, the fetal ovary is quiescent.
As noted above, GnRH stimulates pituitary LH and FSH secretion. LH and FSH signal through their cognate receptors which are G-protein coupled receptors (46).
GONADS
The gonads synthesize sex steroids from cholesterol. In the testis, LH acting through the LH receptor stimulates conversion of cholesterol to testosterone in the Leydig cell. In specific target tissues such as external genital skin and the prostate, testosterone is converted to dihydrotestosterone by the enzyme, 5α-reductase type 2 encoded by the SRD5A2 gene. Testosterone influences pituitary LH secretion through negative feedback either via direct actions or indirectly after conversion to estradiol. FSH acting through the FSH receptor promotes growth of seminiferous tubules and supports sperm development. Growth of the seminiferous tubules and increasing numbers of germ cells accounts for increasing testicular volume during puberty.
In females, the two cell-two gonadotropin model applies to ovarian steroidogenesis. LH stimulates the theca cell to synthesize androstenedione which diffuses to the granulosa cell where FSH-stimulated aromatase activity stimulates estradiol synthesis. Estradiol has both negative feedback and positive feedback. Estradiol mediated positive feedback is required to elicit the LH surge responsible for ovulation.
Activin and inhibin are heterodimeric glycoproteins secreted by the gonads. Inhibins consist of an alpha subunit and one of two homologous yet distinct beta subunits, βA or βB. Inhibin B is composed of an alpha subunit and a βB subunit whereas inhibin A consists of an alpha subunit and a βA subunit. Inhibins are secreted by Sertoli cells in the testes and granulosa cells in the ovary. Inhibin B influences pituitary FSH secretion by negative feedback. In prepubertal boys, inhibin B concentrations reflect Sertoli cell mass and function. After puberty, inhibin B concentrations reflect spermatogenesis (47). Inhibin B correlates inversely with FSH levels in adult men. Activins are dimers of inhibin β subunits, βA, βB and βC; the best characterized are activin A (βAβA) and activin B (βBβB). Activin A stimulates pituitary FSH secretion(48, 49). Follistatin is a monomeric protein that modulates activin activity and can irreversibly inhibit activin activity.
Leydig cells secrete insulin-like peptide 3 (INSL3), a small peptide that, in utero, acts through the relaxin-like family peptide receptor 2 (RXFP2) to promote trans-abdominal testicular descent. INSL3 concentrations increase in boys during puberty (50).
HYPOTHALAMUS
The hypothalamus serves as the rheostat for many physiological functions especially reproduction and growth. The adult human hypothalamus contains approximately 2000 GnRH neurons with cell bodies diffusely distributed in a rostro-caudal continuum (34). The GnRH neurons send projections to the median eminence that terminate in close association with the capillaries of the primary plexus of the hypophyseal portal circulation. Synchronized activity of the GnRH neurons leads to episodic GnRH release into the median eminence with consequent pulsatile pituitary gonadotropin secretion.
An extrinsic hypothalamic neuronal network, known as the GnRH pulse generator, governs GnRH neuron function. This network is located within the infundibular nucleus (known as the arcuate nucleus in non-human species). In the human, the GnRH pulse generator is responsible for tonic gonadotropin secretion; pulsatile LH and FSH secretion regulate testicular function in men and modulate ovarian function, especially folliculogenesis in women. In women, the developing follicle secretes increasing amounts of estradiol ultimately triggering an LH surge followed by ovulation. In adult men, pulse frequency is relatively constant at approximately one pulse every 90-120 minutes. Among women, pulse frequency varies across the menstrual cycle from approximately one pulse per hour during the follicular phase and one pulse every 180 minutes during the luteal phase.
Among GnRH deficient women, pulsatile GnRH administered at a frequency simulating the follicular phase led to ovulatory menstrual cycles (51). In a preclinical model, administration of pulsatile GnRH to prepubertal rhesus female monkeys initiated pubertal development including ovulatory menstrual cycles (52). Thus, puberty in girls and boys is entirely dependent on resumption of pulsatile GnRH release.
Although the GnRH pulse generator was conceptualized by Fred Karsch and Ernst Knobil, the anatomic location of the pulse generator was indeterminant. Identification of loss of function variants in the kisspeptin receptor (KISS1R) gene in patients with congenital hypogonadotropic hypogonadism launched the investigations establishing kisspeptin, neurokinin B, dynorphin, and their cognate receptors as major components of the pulse generator (53, 54). Kisspeptin signals through its receptor, KISS1R, expressed on GnRH cells. Neurokinin B is a decapeptide encoded by the TAC3 (Tac2 in rodents) gene and its cognate receptor encoded by NK3R gene. Both the kisspeptin and neurokinin B receptors are G-protein coupled receptors. Dynorphin is an opioid peptide that signals through a kappa-opioid receptor which is also a G-protein coupled receptor.
Due to the inaccessibility of human brain, especially the pubertal brain, the contemporary model of the GnRH pulse generator has been delineated by preclinical studies performed in rodents, sheep, and non-human primates (55). This model predicts that reciprocal interactions within a network of kisspeptin neurons in the infundibular nucleus leads to synchronous intermittent activation transmitted to GnRH neurons by kisspeptin fibers that project to the median eminence. These kisspeptin fibers are closely associated with GnRH projections targeting the portal capillaries (56).
Based on the detection of kisspeptin, neurokinin B, and dynorphin in the arcuate kisspeptin neurons of mice and sheep, these neurons have been labeled as KNDy neurons (57). Preclinical data suggest that KNDy neurons serve as the intrinsic GnRH pulse generator (58). Kisspeptin and neurokinin B stimulate GnRH release whereas dynorphin appears to be inhibitory. Coordinated interactions of these neuropeptides within the arcuate kisspeptin neuronal network are ostensibly central to the neurobiology of the GnRH pulse generator resulting in pulsatile kisspeptin output. However, the applicability of these findings to human biology remains to be confirmed.
In humans, the HPG axis is active during gestation and the early neonatal period followed by the quiescent years of childhood until the onset of puberty occurs. This pattern suggests that diverse mechanisms integrate the hierarchical activation and deactivation of various stimulatory and inhibitory neuronal pathways ultimately regulating pubertal onset and progression towards reproductive maturity. Thus, a central inhibition of the axis occurs during childhood. For puberty to occur, increased expression of the key factors, KISS1, NKB3, and GnRH, must begin along with decreased expression of the various inhibitory factors. In other words, during the pubertal transition, the balance between inhibitory and stimulatory factors shifts to favor the re-activation of the HPG axis, onset of pubertal changes, and reproductive competence.
Identifying the proximate factors and specific interactions responsible for the on-off-on pattern of HPG axis activity in humans has been a longstanding enigma. Starting with clinical findings, the availability of more sophisticated tools and preclinical models have begun to identify pieces of the puzzle to elucidate the fine details of HPG axis functioning. One factor involved in the suppression of puberty was identified in families with paternally inherited GnRH-dependent/central precocious puberty (CPP). Exome sequencing analyses in multiple families with CPP identified loss of function variants in the makorin 3 (MKRN3) gene (59). This gene, mapped to the Prader Willi region at chromosome 15q11.2, is exclusively expressed from the paternal allele. Consistent with the hypothesis that MKRN3 suppresses the GnRH pulse generator, circulating MKRN3 concentrations decline during puberty (60, 61, 62).
The MKRN3 protein is an E3 ubiquitin ligase consisting of 507 amino acids. It is expressed in KNDy neurons. The protein has five zinc finger domains. Regarding its function, the protein can ubiquitinate substrates and can undergo auto-ubiquitination (63). MKRN3 ubiquitinates methyl-CpG-DNA binding protein 3 (MBP3) interfering with GnRH1 transcription (64). Available preclinical data suggest that MKRN3 functions as a brake on neuronal GnRH release (65). One potential factor influencing MKRN3 expression is microRNA (miRNA) miR-30. Using a rat model, hypothalamic miR-30 expression increased while Mkrn3 expression decreased during puberty. In addition, treatment with agents that interfered with the binding of miR-30 to Mkrn3 were associated with delayed puberty in female rats (66). Using proteomics, MKRN3 targets include insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) and several members of the polyadenylate-binding protein family (67). The decline of hypothalamic Mkrn3 expression in mice and serum MKRN3 protein levels in females prior to the onset of puberty support the hypothesis that MKRN3 suppresses pubertal initiation possibly through effects on prepubertal hypothalamic development and plasticity (61, 67)
Preclinical studies have provided persuasive evidence regarding the regulatory role of epigenetic modifications in pubertal maturation. Epigenetics refers to changes in gene expression and/or activity independent of changes in the primary nucleotide sequence (68). Epigenetic changes include DNA modifications such as methylation/demethylation and histone post-translational modifications such as acetylation/deacetylation. Other post-translational protein modifications such as ubiquitination may also influence protein function. Ubiquitination involves the transfer of ubiquitin to a protein altering its function typically by interfering with protein actions or by promoting protein degradation. As noted above, the MKRN3 protein can function as a ubiquitin ligase. Noncoding RNAs such as miRNAs provide yet another regulatory mechanism.
Another example of epigenetic regulation of pubertal maturation involves two mutually antagonistic histone methylating complexes, the Poly-comb and Trithorax groups. The Poly-comb group represses gene transcription while the Trithorax group appears to function as a gene activator. Preclinical studies performed in rats showed that the Poly-comb group effectively silenced Kiss1 expression until the onset of puberty when increased methylation of the Eed and Cbx7 genes occurred leading to decreased Eed and Cbx7 expression and increased Kiss1 expression (69). Recruitment of the Trithorax activity group further enhanced. Kiss1 expression (70, 71). Genome wide association studies have implicated zinc finger (ZNF) genes. In nonhuman primates, expression of two ZNFs, GATAD1 and ZNF573, decreases upon pubertal reactivation of the GnRH pulse generator (71).
Clinically, it has long been recognized that extremes of body energy status such as chronic malnutrition or severe obesity influence the HPG axis especially in girls and women. The hypothalamic kisspeptin neurons integrate various peripheral and central metabolic signals reflecting energy intake, energy expenditure, and environmental circumstances. Signal coordination between reproductive and metabolic neurons can be direct or indirect. For example, leptin does not directly regulate kisspeptin neurons yet acts as a permissive factor for the onset of puberty (72). Cellular energy and metabolic sensors include mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and sirtuin 1 (SIRT1) (73). Depending on energy status, mTOR and AMPK promote or repress puberty, respectively, by activating or inhibiting Kiss 1 neurons in the arcuate nucleus. Other factors such as melanocortin and agouti-related peptides also interact with kisspeptin pathway (74). In the hypothalamus, neuronal nitric oxide (NO) appears to act on GnRH neurons to integrate metabolic and gonadal information (75, 76). Detailed reviews regarding the neurobiology of the GnRH pulse generator are beyond the scope of this chapter and are available elsewhere (77, 78, 79, 80, 81, 82).
MINI-PUBERTY
Facilitated by the availability of more sensitive hormone assays, Forest and her colleagues described a transient period of increased HPG axis activity in early infancy (83, 84). Following the low gonadotropin concentrations at birth, gonadotropin concentrations were found to rise in both boys and girls within weeks of birth (85). This period of transient gonadotropin secretion has been designated as “minipuberty”. Gonadotropin concentrations in the immediate neonatal period are likely low due to in-utero suppression by placental estrogen. With removal of the placental estrogen suppression, the HPG axis is functional. Relevantly, physical findings typical of pubertal sex steroid secretion are absent with the rare exception of vaginal bleeding attributed to decreased exposure to placental estrogen.
Over the first few years of life, sexual dimorphism in gonadotropin concentrations occurs (86) Boys have higher LH concentrations which peak between 2-10 weeks of age and decline by 4-6 months of age. Girls have higher FSH concentrations which may remain elevated until 2-4 years of age.
In boys, LH stimulates testicular testosterone secretion with testosterone concentrations typically peaking around 1 month of age followed by a decline to prepubertal concentrations by 7-12 months of age. During this phase, the number of germ cells and Sertoli cells increase and penile size increases (87, 88). The proliferation of Sertoli cells leads to a transient increase in testicular volume (89). Sertoli cells secrete Anti-Mullerian Hormone (AMH) and inhibin B. Since Sertoli cells do not express androgen receptors during this stage, spermatogenesis does not occur and AMH secretion remains high (90, 91). A temporary increase in the number of Leydig cells also occurs, but subsequent fetal Leydig cell apoptosis reduces fetal Leydig cell number (92). Longitudinal data obtained from healthy boys suggests a temporal dissociation of Leydig and Sertoli cell activity during minipuberty (93). These data suggest that single blood sample may be insufficient to assess HPG axis activity during early infancy and that obtaining several consecutive samples may be more informative. Curiously, gonadotropin and testosterone concentrations are higher among preterm boys. In addition, increases in testicular volume and penile length are greater in preterm boys compared to full term boys essentially enabling catch-up for testicular volume and penile length (94). Some small studies have documented an exaggerated physiologic hormonal response in extremely premature infants (95).
In girls, the gonadotropins promote granulosa cell proliferation and ovarian estrogen and AMH secretion (96). As would be anticipated, AMH concentrations remain much lower in girls compared to boys (97). A longitudinal study involving healthy full-term infant girls demonstrated two gonadotropin peaks in early infancy with one peak occurring around days 15 to 27 and a later peak occurring at days 164-165 (98). Again, collecting several consecutive samples may be more informative than a single blood sample to assess for minipuberty in infancy.
This transient time period of an active HPG axis, provides an opportunity to diagnose individuals with differences/variants of sex development (DSD/VSD). In a series including both healthy infants and infants with DSD, testosterone measured by LC-MS/MS, AMH concentration, and LH/FSH ratio provided the best discrimination between sexes. The cut-point for LH/FSH ratio was 0.32. Inhibin B and AMH levels were higher in boys with minimal overlap in girls (99). Infants with Turner Syndrome usually have elevated FSH concentrations. Surprisingly, gonadotropin concentrations are typically not elevated in patients with complete androgen insensitivity.
This brief interval of HPG axis activity can also help diagnose congenital hypogonadotropic hypogonadism in boys who present with micropenis accompanied by low gonadotropin and testosterone concentrations (100). Testosterone, LH, FSH, AMH, and inhibin B concentrations may provide information regarding the functionality of testicular tissue in infant boys (101).
As noted above, the human HPG axis displays an “on-off-on” pattern. The biological basis and rationale for transient post-natal HPG axis activity during the first few months of life are enigmatic. At birth, the brain is still plastic with ongoing development. Most axon and synapse formations are completed during the first year of life. Does this transient HPG axis activity imprint specific areas in the brain? Does minipuberty influence future patterns for female and male reproductive function with cyclic gonadotropin patterns in females and not in males? Are gonadal hormones during infancy able to affect future fertility, gender identity, sexual orientation, behaviors, and risk for autism spectrum dysfunction? Data are accruing regarding patterns of hormone secretion during the first six months of life. However, the factors that initiate and terminate this transient period of HPG axis activity and maintain the quiescence of the HPG axis until the onset of puberty are still unknown.
SECULAR TRENDS REGARDING PUBERTY
Over the past few decades, several studies have observed that puberty is beginning at a younger age. Clinical studies examining ages of the onset of puberty depend on the criteria used to denote puberty. Onset of breast development and age at menarche are the conventional indicators of puberty in girls. Prospective observations and retrospective questioning of parents and young girls through in-person questioning has been used to record age at menarche; shorter recall intervals provide the greatest accuracy regarding the details of menarche (102, 103). For boys, age at voice change has been used as a surrogate marker because accurate ascertainment of pubertal onset in boys requires testicular exams using an orchidometer, thus, effectively excluding large-scale epidemiologic clinical studies (104).
During medieval times, available evidence suggests that puberty began around 10-12 years of age. However, the tempo of puberty was slow with menarche occurring closer to 15 years in rural areas and 17 years in London (105). Presumably, undernutrition, increased infections, and greater physical exertion impacted both the timing and tempo of puberty during medieval times (106). The age of menarche declined from 16 to 17 years in the early 19th century to 13 years of age in the late 20th century in Europe and North America. Similarly, the age at menarche has declined in the Yunnan Province in China (107). This decline has been attributed to the improvement in socioeconomic conditions. Currently, the dialogue continues as to whether the trend towards earlier puberty is persisting and, if so, what are the factors driving this process.
Data regarding pubertal milestones in American girls were obtained through the cross-sectional Third National Health and Nutrition Examination Survey (NHANES III) between 1988 and 1994. Among these American girls, mean ages in years for breast development, pubic hair development, and menarche were 9.5, 9.5, and 12.1 for non-Hispanic black girls; 9.8, 10.3, and 12.2 for Mexican-American girls; and 10.3, 10.5, and 12.7 years for non-Hispanic white girls, respectively (108). In 1997, the Pediatric Research in Office Settings (PROS) study reported earlier onset of thelarche with the caveat that breast palpation was not performed (109). The Copenhagen Puberty Study reported that mean age at breast development was lower in the 2006 cohort compared to the 1991 cohort whereas mean age at menarche was similar in both cohorts. Independent of BMI, gonadotropin concentrations were comparable between these cohorts while estradiol concentrations were lower in the 2006 cohort (110).
Beginning in 2004, the Breast Cancer and Environment Research Program (BCERP) prospectively recruited three cohorts of girls aged 6-8 years. This program recruited non-Hispanic white, Hispanic, non-Hispanic black girls, and Asian girls living in New York, Ohio, and California. The overall median age at menarche was 12.25 years with ethnic background median ages as follows: Hispanic girls 11.6 years, black girls at 11.8 years, white girls at 12.5 years, and Asian girls at 12.0 years (111). This cohort differed from the NHANES III study because Hispanic girls experienced menarche earlier than the black girls. These studies, all performed in the United States, report race and ethnicity-related differences in onset of pubertal milestones. Detailed assessment of the potential impact of socio-economic factors was not performed. Notably, differences noted in pubertal timing are smaller than the overall variation among individuals in the population. Most importantly, clinical decision-making should reflect an individual patient's characteristics and family history with less dependence on racial or ethnic backgrounds.
Comparable studies from Spain and Greece have also reported earlier onset of breast development and slower pubertal tempo (112, 113). Thus, available data including a systemic review of international studies largely confirm the ongoing trend for earlier breast development with minimal decline in age at menarche (114).
Several questions regarding this earlier onset of puberty, predominantly earlier thelarche, need to be considered. Does this earlier breast development reflect earlier resumption of GnRH pulse generator activity, extragonadal estrogen production, or environmental exposures? What, if any, is the relationship of BMI to puberty? Another consideration is that race/ethnicity are socio-political constructs and are not fully representative of biology. While genetic ancestry likely influences the onset of puberty, nutritional factors and environmental exposures play important roles. Hence, should cut-off points based primarily on race/ethnicity continue to be utilized?
Based on single unstimulated gonadotropin concentrations, data from the Copenhagen puberty in girls study suggest that gonadotropin concentrations are not obviously increased in girls with early thelarche. Thus, the phenomenon of early thelarche appears to be independent of gonadotropin secretion and may not signify early resumption of GnRH pulse generator activity (115).
The possibility that exposure to endocrine-disrupting chemicals (EDCs) can induce early thelarche has been questioned. EDCs are defined as exogenous chemicals that interfere with hormone action. EDCs include phthalates, phenols, phytoestrogens, organochlorine pesticides, polybrominated flame retardants, diphenyl ethers, heavy metals, and perfluorochemicals. In addition to pesticides, these chemicals can be found in common household products such as hair products, soaps, toothpaste, perfumes, plastics, essential oils, and cleaning products (116). Valid assessment of the consequences of EDCs on puberty is problematic because exposure may occur in utero and generally involves a mixture of assorted EDCs with differing half-lives and activities. Differences in the duration and route of the exposure(s), methodology to detect EDCs, and potential sample contamination further confound analyses. One potential example regarding EDCs involved transient past exposure to organochlorine pesticides among internationally adopted girls in Belgium who subsequently developed precocious puberty (117). Animal models suggest that EDCs can affect puberty through epigenetic mechanisms (118). Nevertheless, most data available regarding the consequences of EDCs on human puberty are inconclusive (119).
Relationship with BMI
Observational data has shown a relationship between BMI and age at puberty in girls (120, 121). The BCERP study found that girls who were overweight or obese at baseline experienced menarche 0.3 years earlier with age at thelarche being inversely correlated with BMI. The BCERP also concluded that BMI had a greater effect than ethnic background on age at menarche (111). Limited data exist regarding the relationship of BMI to pubertal onset in boys. The Puberty Cohort of the Danish National Birth Cohort reported that increased BMI was associated with earlier onset of puberty in boys and girls (122). Among boys, pubertal milestones, testicular enlargement, voice break, and testosterone concentrations showed inverse correlation with BMI (104). Hence, available evidence strongly indicates an inverse relationship between BMI and the onset of puberty in both boys and girls.
Yet, investigating the relationship between puberty and BMI is confounded by potential hormonal and genetic influences (123). Obesity may be associated with hyperinsulinemia and lower sex hormone binding globulin concentrations with consequent higher free sex steroid concentrations. In addition, some genes influence both BMI and pubertal timing (124, 125). The pro-opiomelanocortin (POMC) and central melanocortin systems provide one example of the intricate interrelationships between nutrient signaling and reproductive function. Neurons expressing POMC, producing α-MSH (melanocyte-stimulating hormone), have been suggested to stimulate puberty onset and gonadotropin secretion via modulation of arcuate Kiss1 neurons (126, 127).
Genetic Factors
Genetic factors influence pubertal timing as evidenced by twin studies demonstrating > 50% hereditability for menarche (128). Skeletal maturation, age at pubertal growth spurt, and Tanner staging also show greater concordance between monozygotic twins compared to dizygotic twins emphasizing the relevance of genetic variation in the timing of puberty. Thus, 50-80% of variation in the timing of puberty onset may reflect genetic variation (129). Parental self-reports regarding pubertal timing are associated with timing of specific pubertal milestones in offspring of the concordant sex (130, 131). Genome-wide association studies (GWAS) have detected loci associated with age at menarche (132). Some loci appear to be common and independent of ancestry. A large-scale trans-ethnic GWAS, involving 38,546 women of diverse and predominantly non-European ancestry or ethnicity, identified a novel locus in chromosome 10p15 that is associated with early menarche. This region maps to intron 7 of the aldo-keto reductase Family 1, member C4 (AKR1C4) gene, a member of family of enzymes involved in steroid metabolism and action (133).
To summarize, the secular trends suggesting an earlier onset of puberty appear to be persistent although the age at menarche appears to be relatively static. Likely contributing factors include the rising prevalence of obesity, exposure to potential EDCs, specific dietary influences, and decreased physical activity.
VARIATIONS IN PUBERTAL DEVELOPMENT
Timing of the onset of puberty reflects complex interactions between hormonal and neuronal signals with genetic, metabolic, and environmental factors. These interactions presumably begin early in development and ultimately lead to the re-activation of the HPG axis concomitant with the onset of puberty. Multiple factors, both known and unknown, influence the reactivation of the GnRH pulse generator modulating pubertal onset. As noted above, familial patterns of pubertal development and twin studies highlight the role of genetic factors. Studies of families with either delayed or precocious puberty led to discovery of genes relevant to pubertal onset. In addition, genetic factors including single nucleotide polymorphisms (SNPs) have been associated with pubertal timing in both sexes and across ethnic groups. Epigenetic mechanisms have been suggested to affect the development and function of the GnRH neuronal network ultimately influencing HPG axis function. How confounders such as socioeconomic, environmental, and nutritional status influence pubertal development is unclear. These factors can influence puberty timing, HPG axis function, and fertility.
Precocious puberty is defined as the development of puberty prior to age 8 in girls, and age 9 in boys (134, 135). In girls, delayed puberty is defined as the absence of breast development by age 13 years, absence of menarche by age 15 or lack of menses after 3 years since breast development. In boys, delayed puberty is defined as absence of pubertal development by age 14 (136). Evaluation of a child with abnormal timing of puberty entails thorough knowledge of normal pubertal development, typical variations of normal pubertal development, and causes of abnormal pubertal development. The next section focuses on the evaluation of a patient presenting with a variation in pubertal development.
PRECOCIOUS PUBERTY
Traditionally, the diagnosis of precocious puberty is considered when signs of puberty develop prior to 8 years of age in girls and 9 years in boys (137). These ages are based on Tanner’s original observations on English children regarding typical ages at specific pubertal stages. However, these age criteria should be used as guidelines to complement the evaluation of individual patients. Precocious puberty can be categorized as central or gonadotropin-dependent precocious puberty (CPP) or non-gonadotropin-dependent or peripheral precocious puberty (PPP). Additionally precocious puberty can be further classified as familial or sporadic and syndromic or non-syndromic. The specific etiologies and management differ between the two broad categories of CPP or PPP. Potential consequences of early puberty and menarche in girls include increased risks for breast cancer and diabetes as adults (138, 139).
CENTRAL PRECOCIOUS PUBERTY OR GONADOTROPHIN DEPENDENT PRECOCIOUS PUBERTY
Central precocious puberty (CPP) is associated with early maturation of the HPG with premature reactivation of the GnRH pulse generator and sequential maturation of breasts and pubic hair in females. In males, sequential maturation of testicular volume, penile enlargement, and pubic hair is observed. Typically, the pubertal characteristics are appropriate for the child's sex (isosexual). Despite the earlier onset of puberty, the sequence of pubertal events is usually normal. CPP is due to organic lesions in approximately 40-100 percent of boys whereas idiopathic precocious puberty is the most common diagnosis in girls (69-98%) (140). These children have accelerated linear growth for age, advanced bone age, and pubertal levels of LH and FSH. A Spanish observational report described an annual incidence of CPP ranging between 0.02 and 1.07 new cases per 100,000 (141) while a Korean study reported an incidence of 15.3 per 100,000 girls, and 0.6 per 100,000 boys (142). Distinguishing among CPP, isolated premature thelarche, and premature adrenarche is important because the pathophysiology and therapeutic interventions differ.
CNS LESIONS/INSULTS
CPP can be associated with central nervous system lesions. Hamartomas of the tuber cinereum are congenital benign lesions comprised of heterotopic gray matter, neurons, and glial cells. The prevalence is approximately 1 in 200,000 children (143). Hamartomas are the most commonly recognized CNS lesions associated with CPP in very young children. Hamartomas can be categorized as para-hypothalamic, attached or suspended from the floor of the third ventricle, or as intrahypothalamic, in which the mass is enveloped by the hypothalamus and distorts the third ventricle. The lesions do not grow over time, do not metastasize, and do not produce β-human chorionic gonadotropin-(β-hCG). In some instances, hamartomas are associated with gelastic (laughing or crying) seizures. Yet, most patients with hypothalamic hamartomas do not display neurological symptoms (144, 145). Most hypothalamic hamartomas are sporadic and appear to be idiopathic. Hypothalamic hamartomas can also occur in Pallister-Hall Syndrome (PHS) and oral-facial-digital syndrome (OFD) types I and VI (146). Genetic variants in the sonic hedgehog pathway have been associated with hypothalamic hamartoma (147, 148). The mechanism(s) through which hypothalamic hamartomas lead to CPP is unknown. Hamartoma located close to the infundibulum or tuber cinereum are often associated with CPP whereas those functionally connected to the mammillary bodies and limbic circuit are typically associated with epilepsy without CPP (149, 150). As discussed below, medical treatment is usually indicated for hypothalamic hamartomas associated with CPP. Surgical treatment should be limited to large hamartomas complicated by severe refractory drug-resistant epilepsy (151).
CNS tumors such as astrocytomas, ependymomas, and pinealomas have rarely been associated with CPP. Among girls, factors associated with CNS lesions include: (1) age younger than 6 years; (2) absence of pubic hair; and (3) estradiol concentrations greater than 30 pg/ml (110 pmol/L) (152, 153). As noted above, suspicion for CNS lesions is higher for boys than for girls.
Neurofibromatosis type 1 (NF1) is an autosomal dominant multi-system neurocutaneous disorder due to loss-of-function variants in the neurofibromin-1 (NF1) gene located at chromosome 17q11.2. NF1 is often associated with CPP typically due to optic glioma. The glioma is usually a benign pilocytic astrocytoma that can occur anywhere along the optic tract; the most common locations are within the optic nerve or chiasm. CPP has also been described in NF1 in the absence of optic glioma (154). Children with meningomyelocele and spina bifida also have an increased incidence of CPP. Although the precise mechanism responsible for CPP in these children is unclear, associated factors may include increased perinatal intracranial pressure and brainstem malformations such as Chiari II malformations (155). The mechanistic link between CPP and Rathke cleft cysts, Chiari malformation, and pineal and arachnoid cysts is unclear.
Septo-optic dysplasia (SOD) is a heterogeneous congenital condition characterized by presence of at least two features of the classic triad which include optic nerve hypoplasia, anterior pituitary hormone deficiencies, and midline brain anomalies. SOD is associated with genetic variants in HESX1, SOX2, SOX3, and OTX2 genes. Although SOD is typically associated with delayed puberty, CPP can occur (156, 157).
CNS tumors may be treated with CNS irradiation (158). In some instances, CNS irradiation is associated with acquired CPP (159). In this situation, concurrent growth hormone (GH) deficiency may be present. The linear growth spurt of CPP may mask the decreased linear growth velocity due to GH deficiency. Hence, in this setting, consideration should be given to evaluating the GH axis by provocative GH testing. If testing shows GH deficiency, the patient may benefit from treatment with GH combined with GnRH agonist therapy. Rarely, CPP occurs following head trauma and can develop many years after the injury(160, 161).
SECONDARY CPP
Some children exposed to elevated circulating high sex steroid concentrations occurring in other disorders such as McCune-Albright syndrome, congenital adrenal hyperplasia, and virilizing adrenocortical tumors may develop a secondary CPP (163). These individuals typically have accelerated bone age maturation. The precise mechanism responsible for development of the secondary CPP is unclear. The secondary CPP may represent a priming effect of sex steroids on the hypothalamus or potentially as the consequence of the acute decrease in sex steroid concentrations with treatment of the underlying etiology (164) (165).
NON-SYNDROMIC CPP
Specific genetic variants have been associated with non-syndromic CPP (See Table 1) (166). Loss of function MKRN3 variants are the most reported genetic cause of familial CPP. Paternally inherited loss-of-function MKRN3 variants have been reported in up to 33-46 percent of familial cases of CPP and nearly 0-20% percent of sporadic cases (167) . To date, at least 70 deleterious MKRN3 variants have been identified in patients with CPP. These variants lead to diminished inhibition of puberty results in early onset of puberty. Differing ubiquitination patterns suggests that MKRN3 has multiple molecular mechanisms associated with CPP (168). Curiously, a GWAS study investigating parental effects on pubertal development reported that the paternal allele of a specific SNP (rs12148769, G>A) in MKRN3 was associated with age at menarche in healthy girls suggesting that variants in this region affect pubertal timing within the normal range (132). Although circulating MKRN3 concentrations decrease with onset of puberty, peripheral blood MKRN3 concentrations are not adequately sensitive to distinguish CPP (169).
TABLE 1.
| Gene (Reference/s) | Protein encoded | Genetic locus | Comments |
|---|---|---|---|
| MKRN3 (59, 63, 167, 462) | Makorin ring finger protein 3 | 15q11-q13 | Loss-of-function mutation |
| KISS1R (previously named GPR54) (463, 464, 465) | Kisspeptin receptor | 19p13.3 | Gain-of-function mutation |
| KISS1 (465) | Kisspeptin | 1q32 | Gain-of-function mutation |
| DLK1 (466, 467, 468) | Delta-like homolog 1 | 14q32 | -Loss-of-function mutation -Metabolic abnormalities (obesity, type 2 diabetes, hyperlipidemia) |
| ESR1 (469, 470) | Estrogen receptor 1 | 6q25.1-q25.2 | Mutations/polymorphisms, epigenetic change |
| CYP19A1 (471) | Aromatase | 15q21 | (TTTA)n polymorphism, epigenetic change |
Evaluation of another family with CPP led to identification of a loss of function variant in the delta-like 1 homologue (DLK1) gene. DLK1, also known as preadipocyte factor 1, plays a role in the Notch signaling pathway. DLK1 is a paternally expressed gene located at chromosome 14q32.2. Two differentially methylated regions influence the DLK1 imprinting pattern. DLK is located within the genetic locus associated with Temple syndrome. Temple syndrome is characterized by prenatal growth retardation, hypotonia in infancy, motor delay, small hands, CPP, and short stature. In addition to DLK1 loss, two other genes from the paternally inherited chromosome, RTL1 and DIO3, results in Temple Syndrome. Genetic findings associated with Temple syndrome include maternal uniparental disomy, paternal deletion, or loss of differential methylation at the DLK1/MEG3 region on chromosome 14 (170). Women with DLK1 variants also have a metabolic phenotype characterized by overweight/obesity and insulin resistance (171).
Gain-of-function variants in the kisspeptin 1 gene (KISS1) and its cognate receptor, KISS1R, gene have been identified in children with CPP. A heterozygous variant in the KISS1 gene, p.Pro74Ser, was identified in a boy who developed CPP at one year of age; in vitro studies suggested that this variant was more stable than the normal protein leading to a prolonged duration of action (172). A girl with precocious puberty was found to have a variant in the KISS1R gene; in vitro studies of this p.Arg386Pro variant showed prolonged activation of the intracellular signaling pathways following kisspeptin stimulation (173, 174).
Among a series of 586 children with familial CPP, both maternal and paternal inheritance patterns were found. Variants in MKRN3 were the most common cause in paternally inherited CPP. Among the maternally inherited cases, genetic analysis detected rare variants of unknown significance (175).
SYNDROMIC CPP
In addition to genetic and idiopathic CPP, CPP can occur as a feature in specific syndromes. Pallister-Hall and Temple Syndrome are described above. Other syndromes associated with CPP include Cowden and Cowden-like cancer predisposition syndromes associated with PTEN, SDHB-D and KLLN gene variants. These disorders are characterized by multiple multisystemic hamartomas which may be associated with CPP when the skull base, infundibulum, or hypothalamus are affected. Although Prader-Willi syndrome is typically associated with delayed puberty, CPP has also been reported (176). Other genetic syndromes associated with CPP include tuberous sclerosis and Williams-Beuren (See Table 2). Williams-Beuren is associated with genetic variant at chromosome 7q11.23 (177). Rare cases of precocious puberty have also been reported in Russell Silver syndrome (178).
Table 2.
Syndromic Causes of Central Precocious Puberty Without CNS Lesions (CPP)
| Gene (Reference/s) | Genetic locus | Comments |
|---|---|---|
| MECP2 methyl-CpG-binding protein 2 (472) | Xq28 | Rare forms of Rett syndrome |
| X-linked dead-box helicase 3 (461) | Xp11.4 | Neurodevelopmental delay |
| Xp22.33 deletion, SHOX region (473) | Xp22.33 | Body disproportion, short stature, Madelung deformity |
| Xp11.23-p.11.22 duplication (474) | Xp11.23-p11.22 | Intellectual disability, speech delay, electroencephalogram abnormalities, excessive weight, skeletal anomalies |
| Temple syndrome -DLK1 Maternal uniparental disomy or paternal deletion (170, 473) | 14q32.2 | Imprinting defect, act via DLK1, Prenatal and postnatal growth failure, hypotonia, small hands and/or feet, obesity, motor delay |
| Prader-Willi syndrome - MKRN3 Paternal deletion or maternal uniparental disomy of chromosome 15q11-q13 (475) | 15q11-q13 | Changes to the imprinted MKRN3 and/or MAGEL2genes Hypotonia, obesity, growth failure, cognitive disabilities, hypogonadism |
| Silver-Russell syndrome Hypomethylation of chromosome 11p15 or maternal uniparental disomy of chromosome 7 (476) | 11p15.5 | Possible imprinted or recessive factors, not well elucidated, Prenatal and postnatal growth retardation, relative macrocephaly, prominent forehead, body asymmetry, feeding difficulties |
| Williams-Beuren (177, 477, 478) | 7q11.23 | Distinct face, cardiovascular disease, short stature, intellectual disability, hyper-sociability |
| Kabuki syndrome (479) | 12q13.12 | Downregulation of estrogen receptor activation Neurodevelopmental phenotypes, typical distinct face, short stature |
| Mucopolysaccharidosis type IIIA or Sanfilippo disease (480) | 17q25.3 | Severe neurologic deterioration, visceromegaly, skeletal abnormalities |
NONPROGRESSIVE PRECOCIOUS GONADARCHE
Some children experience a nonprogressive (or slowly progressing) CPP (179). Typically, basal gonadotropin concentrations are prepubertal. In general, children with nonprogressive CPP show no or minimal pubertal responsiveness to GnRH stimulation. Height potential is generally unaffected. Typically, these individuals do not usually benefit from GnRH-Ra therapy. Physical findings alone cannot distinguish between progressive and nonprogressive CPP. Presumably this early pubertal development reflects a transient premature activation of the GnRH pulse generator. Longitudinal follow-up to assure that puberty is not progressive is the most appropriate management.
GONADOTROPH ADENOMA
The anterior pituitary gland consists of highly differentiated ectoderm-derived cells expressing specific hormones such as LH, FSH, GH, prolactin, and ACTH. LH and FSH are secreted by gonadotrophs which are derived from the steroidogenesis factor 1(SF-1) lineage. Gonadotroph adenomas, a type of pituitary adenoma, account for approximately 40% of pituitary adenomas (180, 181) in adults. In children, gonadotroph adenomas can very rarely cause central precocious puberty (182). Though, most gonadotroph adenomas are nonfunctional and benign, rare cases of functional adenomas have been reported. Hormone profiles of functioning adenomas most commonly show elevated FSH concentrations with or without increase in LH concentrations. Elevated TSH secretion resulting in hyperthyroidism may occur concurrently (180, 181).
GUT MICROBIOME AND PUBERTY
Microbiota interact with a variety of metabolic and endocrine pathways of the host through genetic expression of more than 100 times the human genome. The gut microbiome variety, composition and impact on health depend on a vast number of variables, both internal, such as age, genetic factors, gender, and endocrine and immune systems, as well as external factors, such as diet, environment, drugs, and pathogens. The relationship between sex hormones and gut microbiome is complex. Sex steroids may directly or indirectly influence the sex-specific gut microbiome that develops during puberty (183). One study reported several gut microbiome alterations in girls with CPP including Ruminococcus bromii, Ruminococcus callidus, Roseburia inulinivorans, Coprococcus eutactus, Clostridium sporosphaeroides, Clostridium lactatifermentans, Alistipes, Klebsiella and Sutterella (176). Although the evidence of the interaction between microbiota and sex hormones remains limited, evidence of diversity of the gut microbiota at different pubertal stages and that alterations may occur in girls with CPP represents an area for potential future development in the prediction and prevention of precocious puberty (184).
Treatment of central precocious puberty
GONADOTROPIN-RELEASING HORMONE ANALOGS
Long-acting Gonadotropin-releasing hormone analogs (GnRHa) have been the standard treatment of CPP since the mid-1980s (185, 186). The GnRHa are super-agonists that bind to the pituitary GnRH receptor downregulating the endogenous pituitary GnRH receptor resulting in decreased gonadotropin and sex steroid secretion. These medications are modified preparations of the native GnRH decapeptide engineered to increase potency and duration of action by substituting a D-isomer amino acid for the naturally occurring L-glycine at position 6. In some analogs, the tenth amino acid is deleted with modification of the naturally occurring L-proline at position 9 (14).
Several distinct GnRHa preparations are available differing in route of administration and duration of action (See Table 3) (28). The choice of a specific GnRHa depends on patient, caregiver, and physician preference and on insurance coverage/payment/authorization. Treatment with GnRHa leads to regression or stabilization of pubertal symptoms, deceleration of linear growth velocity, and slowing of skeletal maturation. Some girls experience estrogen withdrawal bleeding about 2-3 weeks following the first injection. Parents and the patient should be counseled to expect this episode of vaginal bleeding (187).
Table 3.
Currently Available GnRHa Therapeutic Options
| GnRHa Preparations | Dose | Frequency | Route |
|---|---|---|---|
| Goserelin | 3.6mg | Once a month | intramuscular |
| Leuprolide | 7.5mg | Once a month | intramuscular |
| 11.25mg | Once a month | intramuscular | |
| 15mg | Once a month | intramuscular | |
| 11.25mg | Every 3 months | intramuscular | |
| 30mg | Every 3 months | intramuscular | |
| 45mg | Every 6 months | intramuscular | |
| Leuprolide | 45mg | Every 6 months | subcutaneous |
| Triptorelin | 22.5mg | Every 6 months | intramuscular |
| Nafarelin | 800mcg | Twice daily | intranasal |
| Histrelin | 50mg | Annually * | Subdermal implant |
- *
May be used up to 2 years (481).
Adverse Effects
In general, GnRHas are safe and effective. Adverse events include injection site reactions and sterile abscesses at the site of the injection or implant (188, 189, 190) which may result in loss of efficacy. Minor reported side effects include headaches, hot flashes, vaginal withdrawal bleeding, and mood swings (191). Extremely rare side effects include hypersensitivity reactions, seizures, slipped capital femoral epiphysis, idiopathic intracranial hypertension, and anaphylaxis. One concern regarding the histrelin implant is possible device fracture during extraction; ultrasound-guided removal of the remaining fragments may be necessary (192).
GnRHas, specifically only leuprolide and degarelix, have been associated with prolonged QT interval. A prolonged QT interval increases the risk of developing torsades de pointes (TdP) which is a ventricular arrhythmia associated with sudden cardiac death. Low serum potassium or magnesium may exacerbate the risk for prolonged QT interval. Individuals also taking anti-psychotics (typical and atypical), anxiolytics, and anti-depressants may have an increased risk for prolonged QT intervals when taking leuprolide. Hence, providers should inquire regarding other medications, history of congenital heart disease, and family history of Long QT Syndrome or sudden death. If positive, the provider should obtain screening and follow-up EKGs.
Studies conflict regarding how GnRHa treatment impacts weight gain and BMI. Some studies have reported weight gain during treatment (193, 194, 195, 196) whereas others have not found any significant change in weight or BMI (197, 198). As with all patients, counseling patients regarding the pre-treatment weight trajectory and healthy lifestyle is beneficial. Women with a history of CPP have been reported to have similar adult weight to the general population (199).
Bone mineral density is typically elevated at diagnosis with deceleration in bone mineral accrual during treatment. However, follow-up several years after treatment shows normal bone mineral density compared to population norms (200). Available outcome data suggest that fertility is not compromised for women or men with histories of CPP (192, 201, 202, 203, 204).
Despite suggestions that CPP is associated with subsequent development of PCOS, available data are inconsistent. Prospective longitudinal studies are needed to adequately address this concern (205, 206).
Who to Treat?
For patients less than 7 years of age with a confirmed diagnosis of CPP, the benefit of GnRHa treatment is generally unequivocal. However, the value of GnRHa treatment may be unclear for the peripubertal child (typically a girl) with onset of puberty between 7-9 years of age especially when treatment is unlikely to improve the predicted adult height (PAH) (207). Some girls and their families are comfortable with early pubertal onset and early menarche. In contrast, some girls and their families are distraught when even contemplating early puberty and premature menarche. Consistent evidence-based data regarding negative psychosocial consequences in children with CPP are lacking (208). Further, it may be challenging to justify the medical benefits of GnRHa therapy for early puberty due to the accompanying burdens of increased physician office visits and financial impact. Shared decision-making involving the patient, parents, and medical staff is indispensable to address the benefits and risks of GnRHa in the individual patient (209) .
Goals of Treatment
Goals of GnRHa treatment include prevention of pubertal progression and height preservation (210). Growth velocity can significantly decline in some children during GnRHa treatment particularly in those with a markedly advanced bone age (211). The use of other height augmenting medications including recombinant human growth hormone (GH) (212, 213, 214, 215), stanozolol (216, 217), and oxandrolone (218) have been explored but none are recommended for sole use or as an adjunct to GnRHa therapy (219, 220).
Increasing adult height must be judged considering the financial and psychological burdens of this intensive treatment regimen (221). Several recent studies have recommended treatment beyond a bone age of 12 years, however more rigorous studies are needed before such treatment is endorsed (222, 223).
Another goal of CPP treatment is to mitigate psychosocial distress and prevent adverse mental health outcomes. One epidemiological study of over 7000 women showed that adolescents with early age of menarche had higher rates of depression and antisocial behavior, which persisted into adulthood (224). Adverse psychosocial experiences reported in girls with early age at menarche include increased likelihood of teenage pregnancy and childbearing, sexual and physical assault, and reduced likelihood of high school graduation (225). However, studies thus far do not show that GnRHa therapy can mitigate these effects. One small study of 36 girls with CPP treated with GnRHas evaluated behavioral health diagnosis and health-related quality of life and found no abnormalities in psychological functioning (226). In a small study of 15 girls with CPP treated with GnRHa and 15 age-matched controls, comprehensive test batteries revealed similar scores in cognitive performance, behavioral, and psychosocial problems (227). A review of 15 studies evaluating the psychosocial impact of CPP showed an increased psychosocial and health-related quality of life burdens with CPP compared with controls (228). The same study showed qualitative data demonstrating emotional lability in patients with CPP and that physical differences associated with sexual precocity could increase feelings of shame and embarrassment which further increase isolation and social withdrawal (228). Again, larger studies are needed to better establish if and how GnRHas influences the psychosocial issues associated with CPP.
Monitoring of Treatment
Treatment efficacy can be monitored by repeat clinical exams assessing pubertal progression, ultrasensitive LH, FSH and sex hormone concentrations (estradiol in girls, testosterone in boys), rate of progression of bone maturation, estimates of PAH and change in PAH, and patient satisfaction. No uniform consensus exists regarding the optimal strategy for monitoring treatment efficacy in children with CPP. Progression of breast or testicular development may indicate poor adherence, treatment failure, or incorrect diagnosis (188).
Random basal LH concentrations to confirm treatment efficacy may be unhelpful because random LH levels often fail to revert to a prepubertal range even when the HPG axis is fully suppressed (229, 230). Therefore, random LH concentrations cannot be used to indicate treatment failure. To confirm gonadotropin suppression, a GnRH stimulation test with short-acting GnRH or, alternatively, a single LH sample 30–120 min after long-acting GnRH analog administration may be performed (231, 232) and different protocols exist regarding the specific timing and number of LH and FSH measurements (233). Some clinicians prefer to utilize clinical indices particularly in areas where hormone determinations are costly.
During treatment, breast tissue usually becomes softer with variable changes in size. The rate of bone maturation typically slows with adequate treatment resulting in a decline in BA/CA or a change in BA divided by time. Recent data show that the decline in BA/CA is non-linear and that larger declines are seen in the first 18 months of treatment (222). Thereafter, a slower rate of decrease suggests maintenance of suppression rather than treatment failure.
Height velocity is typically rapid prior to treatment and decreases on treatment. The height deceleration is most apparent during the first 18 months of treatment, similar to the deceleration in skeletal maturation. Subsequently, a prepubertal growth rate is often evident (222). Ideally, the rate of bone maturation decelerates resulting in a net gain in height potential. Therefore, calculating PAH during treatment helps assess efficacy. It is also important to understand that mid-parental height (MPH) influences height outcome. GnRHa treatment for CPP may restore genetic potential but rarely causes PAH to surpass genetic potential. Therefore, treatment efficacy by PAH assessment is always in comparison to MPH.
Discontinuation of Therapy
The decision to discontinue GnRHa treatment needs to be tailored to meet the patient’s specific needs. Factors influencing the decision-making process include synchronizing pubertal progression with peers, patient readiness for resumption of puberty, recent linear growth velocity, bone age X-ray results, and adult height prediction (234). Specific considerations for the developmentally delayed child may be reviewed with the caregivers (137, 234, 235). Pubertal manifestations generally reappear within months of discontinuation of GnRHa treatment; the mean time to menarche is approximately 16 months (217, 218). Several studies have reported that ovulatory function and menstrual cycles are normal once they resume (137, 236).
PERIPHERAL PRECOCIOUS PUBERTY OR GONADOTROPIN-INDEPENDENT PRECOCIOUS PUBERTY
Peripheral precocious puberty (PPP) is due to either excessive endogenous gonadal or adrenal sex steroid secretion (estrogens or androgens) or from exogenous exposure to sex steroids. Ectopic gonadotropin secretion typically from a germ-cell tumor often located in the CNS can also lead to PPP. PPP may be appropriate for the child's sex (isosexual) or inappropriate, with virilization of females and feminization of males (heterosexual). In most instances, pubertal development is incomplete, and fertility is not attained. Etiologies of PPP include:
MCCUNE-ALBRIGHT SYNDROME
McCune-Albright syndrome (MAS) is an uncommon disorder characterized by the triad of gonadotropin-independent precocious puberty, irregular café-au-lait skin pigmentation and fibrous dysplasia of bone (237, 238). It has been recognized more recently that MAS may exist as a “form fruste” with only one or two features (239). MAS affects both boys and girls. Importantly, precocious puberty is not observed in all affected individuals and tends to be more common among girls.
MAS is due to a somatic cell (post-zygotic) variant arising early during embryogenesis in the GNAS1 gene which is located at chromosome 20q13.3. This gene encodes the Gsα protein coupled to the G-protein membrane receptors for glycoprotein hormones. Vertical transmission has not been reported suggesting that germline variants are embryonic lethal. Variability in post-zygotic expression of the deleterious variant results in a mosaic pattern of tissue expression and inconsistent clinical manifestations between affected individuals (237).
Two missense variants, Arg201His and Arg201Cys, are the most frequently identified variants. These variants lead to loss of the α-subunit’s intrinsic GTPase activity resulting in inappropriate cyclic AMP production and constitutive receptor activation. The net result is autonomous ligand-independent signaling by LH, FSH, TSH, GHRH, and ACTH receptors leading to the associated hyperfunctioning endocrinopathies(237).
The café-au-lait lesions are generally large with irregular “coast of Maine” borders and typically do not cross the midline. The café-au-lait lesions result from increased tyrosinase gene expression and melanin production in affected melanocytes (240).
Bone manifestations are characterized by dysplastic lesions with abnormal bone turnover and inadequate mineralization. These lesions can be associated with pain, malformations, fractures, or nerve compression. The somatic cell gain-of-function variants alter the differentiation of multi-potent skeletal stem cells resulting in the replacement of normal bone and marrow with immature woven bone and fibrotic stroma. The dysplastic tissue is characterized by abundant osteoclast-like cells. Although the somatic Gsα skeletal variants arise during embryogenesis, bone development appears to be normal in utero.
Bony lesions become apparent during early childhood typically reaching the maximal burden in young adulthood. The variability in the somatic cell expression accounts for the variability in the location and extent of the fibrous dysplasia. To date, an accurate ascertainment of risk to develop bone disease is unavailable. However, younger age and higher skeletal burden score derived from scintigraphic bone scans appear to predict longitudinal progression of bone disease. Importantly, evolution of bony lesions is not associated with the extent of endocrine manifestations (241).
Overproduction of fibroblast growth factor 23 (FGF23) by skeletal cells bearing the GNAS1 variant can lead to increased urinary phosphate excretion and decreased renal 1-α-hydroxylase activity (242). Although overt hypophosphatemic rickets is uncommon due to compensatory mechanisms, affected individuals often manifest increased serum FGF23 levels and renal phosphate wasting (243).
In the gonads, these variants induce ligand independent activation of gonadotropin receptors resulting in subsequent autonomous ovarian estrogen and testicular testosterone secretion in affected prepubertal girls and boys, respectively.
Girls may develop recurrent estrogen-secreting cysts accompanied by breast development and linear growth acceleration. Spontaneous resolution of a cyst decreases the estrogen concentration resulting in withdrawal vaginal bleeding. The sequence of pubertal development may be atypical with vaginal bleeding preceding breast development. Hence, MAS should be considered in females with recurrent ovarian cysts and vaginal withdrawal bleeding. Ovarian torsion rarely occurs. Bloodwork may reveal elevated estradiol concentrations with suppressed gonadotropin concentrations. Pelvic ultrasound typically shows one or more ovarian cysts and uterine enlargement. Nevertheless, serum estradiol concentrations and pelvic ultrasound results may be unremarkable following spontaneous involution of an ovarian cyst. Estrogen exposure may lead to accelerated skeletal maturation with adverse consequences on final adult height. In some instances, a secondary gonadotropin-dependent precocious puberty develops. In adult women, the persistent autonomous ovarian activity can lead to abnormal uterine bleeding, menometrorrhagia, which may be so severe as to require blood transfusion. Spontaneous pregnancies can occur, but relative infertility is common (244).
Among boys, autonomous GNAS1 activation in the testes leads to Leydig and Sertoli cell hyperplasia which can be associated with macro-orchidism. Scrotal ultrasound may show focal masses, diffuse heterogeneity, and microlithiasis. Differing from typical pubertal progression, testicular volume in boys with MAS does not accurately indicate pubertal status. Substantial autonomous testosterone production is uncommon. Approximately 15% of boys manifest clinical signs of excessive androgen secretion (239). Leydig cell hyperplasia, the most common histologic finding of the testes, carries a low risk of malignant transformation. Thus, conservative management with periodic scrotal ultrasound imaging is appropriate for follow-up of testicular masses detected in boys with MAS (245).
Other features associated with MAS include thyrotoxicosis, growth hormone excess (gigantism or acromegaly), and Cushing syndrome. Hypercortisolism is uncommon, typically occurs during the first year of life, and is associated with higher mortality attributed to secondary infections (246). Specific laboratory evaluation and treatment for associated endocrine features should be obtained. Genetic variants can be found in other nonendocrine organs (liver, intestines, and heart) resulting in cholestasis and/or hepatitis, intestinal polyps, and cardiac arrhythmias, respectively (247, 248). Since GNAS1 variants are considered to be weak oncogenes, the risk for malignant transformation is slightly higher than for the general population (239). In addition, women with MAS have an increased risk for breast cancer attributed to earlier estrogen exposure (249).
The diagnosis of MAS is usually based on the characteristic clinical features. Due to GNAS1 variant mosaicism, only 20-30% of peripheral blood lymphocytes are positive for the variant using traditional PCR-based testing. However, variant detection is greater than 80% in the affected tissues (250). Newer circulating cell free DNA testing offers a potential methodology to assess for MAS variants (251) . Importantly, negative testing, especially of peripheral blood lymphocytes, does not exclude the diagnosis of MAS.
Therapeutic goals focus on treating specific clinical manifestations. For manifestations related to puberty, current medications either inhibit sex steroid biosynthesis or block their actions at the level of end organs. Minimal evidence-based data are available because of the low prevalence of MAS. Ketoconazole, an anti-fungal medication, has been used because it inhibits the steroidogenic cytochrome P450 enzymes decreasing adrenal and gonadal steroidogenesis (252). However, ketoconazole may interfere with cortisol synthesis; patients need to be monitored for possible adrenal insufficiency and may benefit from use of stress dose hydrocortisone treatment. Rarely hepatic toxicity can occur.
Aromatase inhibitors prevent conversion of androgens to estrogens. Initial reports for testolactone, fadrozole, and anastrozole were disheartening because no enduring beneficial effects on skeletal growth and bone maturation were observed. Letrozole has been used and showed sustained beneficial effects on skeletal maturation and predicted final height in one small series (253).
Selective estrogen receptor modulators such as tamoxifen and fulvestrant have been used. Tamoxifen has both agonist and antagonist activity at the estrogen receptor. Despite reports regarding the efficacy of tamoxifen to reduce vaginal bleeding accompanied by positive effects on bone, this medication has been reported to increase risk of endometrial disease in adult women (254). In view of potential risks for endometrial cancer, tamoxifen should be used with great caution in women with MAS (255).
Fulvestrant is a pure estrogen receptor blocker administered by intramuscular injections at monthly intervals. In one small series, vaginal bleeding was reduced with complete cessation of vaginal bleeding in only 8/25 girls. The rate of skeletal maturation decreased without any significant change in linear growth velocity or predicted adult height. Fulvestrant was reported to be well tolerated; additional studies are needed to supplement these initial findings (256).
In the past, surgery cystectomy or oophorectomy had been performed in girls with MAS (257). Since cyst recurrence is common, cystectomy should be avoided if possible. Women with MAS have the potential for fertility and spontaneous pregnancy; hence, oophorectomy should be avoided (258).
For boys with MAS associated precocious puberty, therapeutic interventions include androgen receptor blockers, aromatase inhibitors, and ketoconazole to interfere with testosterone synthesis (258). Combination therapy with bicalutamide and anastrozole was successfully utilized in one boy with PPP due to MAS (259). Bicalutamide is a potent nonsteroidal antiandrogen that binds to and inhibits the androgen receptor and increases the receptor’s degradation. Surgical intervention should only be considered for rapidly enlarging palpable testicular masses due to the risk of malignancy (245).
PREMATURE MENARCHE AND OVARIAN CYSTS
Functioning ovarian follicular cysts can secrete estradiol resulting in isolated premature vaginal bleeding or peripheral precocious puberty (260, 261). Additional signs of puberty may be absent in girls with isolated premature menarche. Although some girls may present with slight breast development followed by vaginal bleeding. The bleeding typically lasts only a few days and is usually attributed to spontaneous resolution/regression of an estrogen-secreting ovarian cyst. By the time a pelvic ultrasound can be obtained, the cyst has resolved, and the ultrasound shows no abnormalities. Isolated premature menarche may be limited to a single episode or may be recurrent. In most instances, linear growth velocity, onset of cyclic menstrual cycles, and final adult height are unaltered.
Differential diagnosis includes sexual abuse, vaginal foreign body, vaginal infections, MAS, or primary hypothyroidism (262). Due to the intermittent nature of these cysts, conservative medical management is usually appropriate (263). Large cysts may predispose to ovarian torsion (264, 265, 266, 267). Patients with ovarian torsion usually present with short duration of pain and systemic symptoms such as vomiting. Given the low frequency of malignancy in such an ovarian, detorsion with or without cystectomy is generally preferred (268). Gonadectomy should be avoided to preserve fertility. Rarely, rhabdomyosarcoma or sclerosing stromal tumors can present with vaginal bleeding.
OVARIAN TUMORS
Estrogen-secreting ovarian tumors are a rare cause of peripheral precocious puberty. Specific types of tumors include granulosa cell, gonadal stromal cell, ovarian sex cord stromal, and theca cell tumors.
Juvenile granulosa cell tumors (JGCT) are the most common ovarian tumors. Typically, these tumors present with rapidly progressive isosexual precocity (269). Most JGCT are large enough to be palpated during an examination and are typically limited to the ovary at the time of diagnosis. Circulating estradiol concentrations may be extremely elevated with suppressed gonadotropin concentrations. Circulating tumor markers including α-fetoprotein (AFP), lactate dehydrogenase (LDH), β-human chorionic gonadotropin (β-hCG), cancer antigen 125 (CA-125), and inhibin B can be identified. Genetic somatic variants have been identified in juvenile granulosa cell tumors. Over 60% of JGCT carry in frame duplications in the AKT1 gene; this gene codes for a kinase involved in ovarian mitogenic signaling (270). Other identified variants include KMT2C-truncating and the ribonuclease III domain of DICER1 variants. In contrast to adult granulosa cell tumors of the ovary, variants in the FOXL2 gene are generally not found in JGCT. Ollier and Maffucci syndromes, rare disorders associated with benign cartilaginous enchondroma, have been associated with JGCT (271). Surgical excision with peritoneal cytology for staging is the primary treatment.
Rarely, other tumors including gonadoblastoma, lipid tumors, cystadenomas, and ovarian carcinomas can secrete sex steroids. Finding elevated serum inhibin and AMH concentrations suggest that the tumor cells are derived from granulosa or Sertoli cells.
Sex cord tumors with annular tubules can occur in patients with Peutz-Jeghers Syndrome. Peutz-Jeghers Syndrome is an autosomal dominant disorder associated with mucocutaneous pigmentation, gastrointestinal polyposis, and genetic variants in the STK11 gene located at chromosome 19p13.3. (272) The gonadal tumors can be multi-focal, bilateral, and can differentiate into granulosa cell or large cell calcifying Sertoli cell tumors with the potential to secrete estrogen. Thus, girls may present with precocious puberty whereas boys may present with gynecomastia.
Sertoli-Leydig cell tumors are rare ovarian tumors often associated with somatic or germline DICER1 variants (273). Most are unilateral, but bilateral tumors have been described. These tumors contain testicular structures, Sertoli and Leydig cells, and can rarely secrete androgens. Hence, girls can virilize with pubic hair development (274, 275). Girls known to carry germline DICER1 variants should undergo regular pelvic ultrasounds to screen for ovarian tumors (276).
LEYDIG CELL TUMORS
Leydig cell tumors are a subtype of testicular stromal tumors that arise from testosterone producing Leydig cells. In prepubertal boys, presenting features include penile enlargement, acne, development of pubic and axillary hair, and accelerated linear growth velocity. Examination of the testes typically show asymmetric testicular volume due to a unilateral testicular tumor. Leydig cell tumors are usually benign. Bloodwork shows elevated circulating testosterone concentrations and suppressed gonadotropin concentrations. Ultrasound is useful to assess testicular volume and morphology.
Treatment involves surgical removal of the tumor. When possible, testis-sparing enucleation is preferred to radical orchiectomy to preserve testicular function and fertility. The surgical approach is dictated by the intraoperative assessment of tumor size, location, and the amount of remaining normal testicular parenchyma (277, 278).
HUMAN CHORIONIC GONADOTROPIN SECRETING GERM CELL TUMORS
During early gestation, primordial germ cells migrate from the hindgut to the gonads. In some instances, the germ cells can migrate to locations outside of the gonad, fail to undergo apoptosis, proliferate in these atypical locations, and ultimately become hCG-secreting germ cell tumors (279). Common locations for germ cell tumors include the CNS, lung, or liver (280). Boys and men with Klinefelter syndrome are at higher risk to develop extra-gonadal GCTs particularly in the mediastinum (281). In addition, hepatoblastoma secreting hCG and α-fetoprotein can also present with precocious puberty (282). Due to the similarity between hCG and LH which have identical α-subunits and related β-subunits, tumor-derived hCG stimulates testicular LH receptors resulting in testosterone secretion. In the prepubertal boy, the aberrant hormone exposure can result in precocious puberty (27, 28). Testicular volume may not increase since seminiferous tubule growth does not occur in the absence of FSH stimulation. Bloodwork shows elevated hCG and testosterone concentrations with suppressed/variable LH and FSH concentrations.
Prepubertal girls generally do not develop isosexual precocious puberty with hCG-secreting germ cell tumors because in the absence of FSH, the granulosa cells do not express aromatase and are unable to synthesize estradiol.
GERM CELL TUMORS
Chromosomal aneuploidy or genetic variants can interfere with gonadal development resulting in dysgenetic gonads. In this situation, the appropriate microenvironment for normal germ cell maturation is absent, thereby disrupting the normal maturational progression for germ cells. This situation may result in the development of gonadal germ cell tumors. Precursor lesions of germ cell tumors include germ cell neoplasia in situ (GCNIS, formerly termed carcinoma in situ – CIS) and gonadoblastoma (283). Subsequently, dysgerminoma, seminoma, or non-seminoma may develop. Usually, such germ cell tumors do not secrete significant amounts of sex steroids.
FAMILIAL MALE LIMITED PRECOCIOUS PUBERTY (FMPP)
Familial male-limited precocious puberty (also known as testotoxicosis) is due to an autosomal dominant activating germline variant in the LH/choriogonadotropin receptor (LHCGR) gene located at chromosome 2p21. The LH/CG receptor is a G-protein coupled receptor (284). The variant is associated with autonomous ligand-independent receptor signaling leading to Leydig cell hyperplasia and premature testosterone secretion in prepubertal boys. Pathogenic missense variants associated with FMPP tend to congregate in an apparent hot spot located in the 6th transmembrane segment and in the 3rd intracellular loop (285).
Affected males typically present between two to six years of age with penile enlargement, linear growth acceleration, advanced skeletal maturation, acne, and pubarche. The testes are usually symmetrically enlarged due to the Leydig cell hyperplasia, but the size is disproportionately smaller compared to the testosterone levels (286, 287). A large portion of the testicular volume is formed by Sertoli cells which are not stimulated in this condition (286, 287). Circulating testosterone concentrations are elevated with suppressed gonadotropin concentrations. Although adult height is generally compromised, fertility has been reported (288). Longitudinal follow-up with testicular self-examination and scrotal ultrasound is recommended because malignant testicular germ cell tumors have been described in a few individuals (289).
Therapeutic goals include decreasing autonomous testicular testosterone secretion and slowing epiphyseal maturation. To date, several medications including ketoconazole, spironolactone, bicalutamide, and aromatase inhibitors have been used with varying efficacy (286). To date, the most effective therapy is combination treatment with an anti-androgen and an aromatase inhibitor (290, 291). If secondary GnRH-dependent precocious puberty develops, GnRH agonist therapy can be added to the therapeutic regimen. Abiraterone, a selective CYP17A1 inhibitor, was utilized in a young boy with bilateral Leydig cell tumors and resistance to the usual combination regimen of an anti-androgen and aromatase inhibitor. He required glucocorticoid replacement therapy and monitoring for possible excessive mineralocorticoid action because of abiraterone treatment-associated iatrogenic 17-hydroxylase/17,20-lyase deficiency (292).
Although inherited as an autosomal dominant disorder, girls do not develop precocious puberty (293). Since only the LHCGR gene is affected, the presumably minimally increased theca cell androgens cannot be aromatized to estradiol in the absence of FSH stimulation. Importantly, asymptomatic women can transmit the affected allele to their sons.
PRIMARY HYPOTHYROIDISM
Children with profound chronic primary hypothyroidism may present with precocious puberty. Van Wyk and Grumbach described this association in 1960 (294). Clinical features in girls include early breast development, vaginal bleeding, and galactorrhea. Boys present with testicular enlargement. Pubic and axillary hair are absent. Typical features associated with hypothyroidism such as short stature, impaired linear growth, puffy face, dry skin, constipation, and delayed skeletal maturation (despite pubertal changes) are usually evident. Pituitary imaging shows an enlarged pituitary gland. Abdominal ultrasound may show ovarian enlargement with or without ovarian cysts. Labs show mild elevation in FSH levels but LH levels usually remain prepubertal.
Levothyroxine therapy induces regression of pubertal symptoms, stops vaginal bleeding, and decreases pituitary volume. However, final height may often be compromised due to accelerated skeletal maturation upon initiation of thyroxine treatment. One potential mechanism for the precocious puberty is cross-reactivity of TSH at the ovarian FSH receptor. TSH and FSH share a common α-subunit with hormone specificity due to the differing β-subunits (295). This mechanism was tested using recombinant human TSH in an in vitro bioassay which, at high concentrations, was able to stimulate human FSH receptors (296, 297, 298, 299).
VIRILIIZING CONGENITAL ADRENAL HYPERPLASIAS
The virilizing congenital adrenal hyperplasias (CAHs) are autosomal recessive disorders associated with impaired adrenal steroidogenesis due to genetic variants in steroidogenic enzyme genes. The most common is 21-hydroxylase deficiency due to variants in the 21-hydroxylase gene (CYP21A2) located at chromosome 6p21.33. Clinically, congenital adrenal hyperplasias reflect a phenotypic spectrum ranging from presentation in neonatal period with classic salt-losing CAH to presentation during infancy/todder age with classic simple virilizing CAH to later presentations with non-classic CAH. Milder or non-classic forms have been described for 11-β-hydroxylase deficiency and 3β-hydroxysteroid dehydrogenase deficiency (300).
Children with non-classic CAH typically present with premature pubarche characterized by pubic/axillary hair development, acne, accelerated linear growth velocity, and advanced skeletal maturation. Girls may have clitoromegaly whereas boys may have phallic enlargement with prepubertal testicular volume. Adult women with non-classic CAH usually present with irregular menses, hirsutism, and infertility.
The diagnostic test for 21-hydroxylase deficiency is an elevated 17-hydroxyprogesterone (17-OHP) concentration. Early morning basal 17-OHP values have been suggested as an effective screening test with reports of 100% sensitivity and 99% specificity with a threshold value of 200 ng/dl (6 nmol/L) to diagnose NCAH in children who present with premature pubarche (301). If the diagnosis is highly suspected despite relatively normal 17-OHP concentrations, an ACTH stimulation test may be indicated to exclude the diagnosis of 21-hydroxylase deficiency. For an ACTH stimulation test, following collection of a basal blood sample, 0.25 mg synthetic ACTH (Cortrosyn) is administered by intravenous or intramuscular routes; a second blood sample is collected at 30 and/or 60 minutes. Physician preference governs the timing of the ACTH-stimulated 17-OHP concentration. In the future, 21-deoxycortisol and 11-oxyandrogens may be increasingly utilized in the diagnosis and management of 21-hydroxylase deficiency (302, 303). The reader is referred to more extensive discussion of the virilizing CAH (304, 305).
ADRENOCORTICAL TUMORS
Androgen-secreting adrenocortical tumors are extremely rare causes of PPP accounting for less than 1% of all childhood malignancies. Most tumors occur in children younger than 4 years of age with a second smaller peak in adolescents. Pediatric adrenocortical tumors are categorized as adenomas or carcinomas based on histological features. However, histopathologic differentiation may be challenging, and biologic behavior of the tumor may help with this categorization (306).
Pediatric adrenocortical carcinoma is more common in girls than boys and has a bimodal pattern with peaks under age 5 and over 10 years of age (307). Adrenocortical tumors are associated with several genetic syndromes such as Li-Fraumeni syndrome and Beckwith-Wiedemann syndrome (BWS). Li-Fraumeni Syndrome is an autosomal dominant familial cancer syndrome associated with germline p53 gene variants. The p53 gene (or TP53 gene) is a tumor suppressor gene located at chromosome 17p13.1, and codes for the protein p53. Malignancies associated with Li-Fraumeni syndrome include adrenocortical carcinomas, breast cancer, brain tumors, and sarcoma. The incidence of adrenocortical tumors is 10-15 times higher in southern Brazil; this has been attributed to the higher prevalence of the R337H variant of the TP53 gene (308).
Beckwith-Wiedemann syndrome is characterized by macroglossia, macrosomia, organomegaly, neonatal hypoglycemia due to hyperinsulinism, and abdominal wall defects. This disorder is associated with uniparental disomy in the 11p15 chromosomal region leading to IGF2 growth factor overexpression. Although only 1% of children with Beckwith-Wiedemann Syndrome will develop adrenocortical carcinomas, these adrenal tumors account for approximately 20% of the neoplasms in children with this disorder (309). Other disorders associated with adrenal tumors include Multiple Endocrine Neoplasia Syndrome Type 1 (MEN1) and Carney complex (310).
The next section reviews variants of puberty associated with early pubertal changes and are important differentials to consider in the evaluation of CPP.
PREMATURE THELARCHE
Premature thelarche is the premature development of glandular breast development. The breast development may be unilateral or bilateral. Typically, premature thelarche develops in otherwise healthy girls between 12-24 months of age and is self-limited. No other pubertal changes are evident; linear growth velocity is normal and pubic/axillary hair are absent. On physical examination, the areolae and vaginal mucosa are prepubertal. The diagnosis can usually be made on a clinical basis without bloodwork or bone age X-rays (311). Pelvic ultrasound showed increased prevalence of ovarian microcysts in girls with premature thelarche compared to age-matched controls; no correlation between ovarian cysts, gonadotropin concentrations, and estradiol concentrations has been found (312). Longitudinal follow-up is appropriate to confirm the diagnosis and assess for the unlikely possibility of progression to CPP.
PREMATURE ADRENARCHE
Pubarche refers to the appearance of pubic/axillary hair, increased apocrine odor, and acne due to the onset of adrenarche. Adrenarche refers to the pubertal maturation of the adrenal zona reticularis. Adrenarche which normally occurs in children between 6-8 years of age and is characterized by increased secretion of the adrenal androgen precursors DHEA, DHEAS, and androstenedione.
Premature adrenarche is characterized by premature pubarche, which is defined as the development of pubic or axillary hair before 8 years in girls or 9 years in boys. There is no breast development in females and no testicular enlargement in males. Bone age is usually not advanced. Premature adrenarche is a diagnosis of exclusion. Thus, exclusion of other disorders such as CAH, androgen-secreting tumors, exogenous androgen exposures, and other rare genetic disorders such as apparent cortisone reductase and PAPS synthase 2 (PAPSS2) deficiencies is essential (313).
Children with premature adrenarche and early androgen excess may be at a higher risk to develop the metabolic syndrome. Waist circumference (WC), waist/hip ratio, and total and truncal fat mass increase are detected in premature adrenarche. Increases in systolic and diastolic blood pressure (BP), total cholesterol (TC), very low-density lipoprotein (VLDL), TC/high density lipoprotein (HDL), low density lipoprotein (LDL)/HDL ratio, and atherogenic index (AI) have been reported. Increased insulin concentrations starting from prepubertal ages may occur suggesting that premature adrenarche may be one of the first symptom of insulin resistance (IR) in childhood (314, 315). T2DM may occur in a subset of these cases. Ovarian hyperandrogenism, hirsutism, ovulatory dysfunction, and polycystic ovaries may be more frequent in girls with premature adrenarche during post pubertal ages than normal population. Although, early retrospective data in a homogenous population suggests an association between premature adrenarche and adolescent hyperandrogenism (316), more recent longitudinal data suggests that premature adrenarche was not associated with adolescent ovarian dysfunction and was only associated with lower SHBG concentrations (317).
EXPOSURE TO EXOGENOUS SEX STERIODS
Feminization, including gynecomastia in males, has been attributed to excess estrogen exposure from creams, ointments, and sprays. Other possible sources of estrogen exposure include contamination of food with hormones, phytoestrogens (e.g., in soy), and over-the-counter remedies such as lavender oil and tea tree oil (318, 319, 320, 321). Similarly, virilization of young children has been described following inadvertent exposure to androgen-containing creams/gels (322).
Endocrine-Disrupting Chemicals
Various endocrine-disrupting chemicals (EDCs) are found in the environment (323, 324, 325, 326). Most EDCs have chemical structures similar to those of endogenous sex steroids. These chemicals can disrupt steroid hormone receptor binding and hormone metabolism altering hormone concentrations or changing hormone synthesis/degradation (327). EDCs can act beyond steroid hormone receptors by affecting transcriptional modulators and direct effects on genes. Some EDCs have mixed activities, and most EDCs include several different chemicals. The patient’s age and duration of exposure modulate the consequences of EDC exposure. In addition, EDCs can be classified as persistent (long-lasting) or non-persistent (short half-lives). Environmental EDC exposures can be transgenerational such that future generations could be affected (328). Mechanisms for EDC exposures include ingestion, topical use, inhalation, and transfer across the placenta (329).
The consequences of mixed “cocktail” EDC exposures on pubertal development are indeterminate. A systematic review with a stringent meta-analysis found no consistent association between xenobiotic EDCs and pubertal timing apart from an insinuation that, in girls, postnatal exposure to phthalates could be associated with earlier thelarche and later pubarche, consistent with their anti-androgenic properties. Methodological heterogeneity, limited number of studies, and variability in statistical analyses constrained the conclusions of this systematic review. Hence, future longitudinal epidemiologic studies to clarify the specific EDCs, age at exposure, and duration of exposure will be valuable (327, 330).
DELAYED PUBERTY
Gonadarche associated with the reactivation of the GnRH pulse generator, is signified by breast development in girls and testicular enlargement in boys. Delayed puberty is defined as absence or delayed onset of gonadarche at a chronologic age >2 standard deviations later than the population mean. In girls, delayed puberty is defined as absence of breast development by age 13 years or lack of menarche by age 15 years (331) or 3 years from onset of thelarche (332). In boys, delayed puberty is defined as the lack of testicular enlargement to a volume >= 4 ml by age 14 years (333). Delayed puberty is more common in boys than in girls.
Four main categories of delayed puberty have been described (See Table 4).
- transient hypogonadotropic hypogonadism associated with delayed maturation of the HPG axis also known as constitutional delay of growth and puberty (CDGP)
- hypergonadotropic hypogonadism characterized by primary gonadal dysfunction with consequent elevated LH and FSH concentrations.
- hypogonadotropic hypogonadism with low LH and FSH concentrations due to congenital (CHH) or acquired causes
- functional hypogonadotropic hypogonadism (FHH), as seen in chronic health disorders such as cystic fibrosis, renal failure, inflammatory bowel disease, restrictive eating disorders etc.
Table 4.
Etiologies of Delayed Puberty (458)
| Condition | Etiology |
|---|---|
| Constitutional Delay of Growth and Puberty | Genetic basis has infrequently been described in HS6ST1, FTO, IGSF10, EAP1 genes |
| Hypergonadotropic Hypogonadism | Congenital: -Klinefelter’s syndrome -Turner syndrome -Gonadal dysgenesis -Anorchia -Primary ovarian insufficiency -Testicular regression syndrome -Genetic causes: FMR1, STAG3, NR0B1, NR5A1, FOXL2, WT1 and others -Galactosemia |
| Acquired: -Infectious (mumps) -Autoimmune (polyglandular syndromes) -Surgery (torsion, trauma) -Chemotherapy (alkylating agents) -Radiation -Gonadal tumor | |
| Hypogonadotropic Hypogonadism | Congenital: -Isolated HH: Over 50 genes have been identified; notable are ANOS1, FGFR1, FGF8, PROK2, CHD7, KISS1, KISS1R, GNRH, GNRHR and others -Prader Willi -CHARGE syndrome -Noonan -Bardet-Biedl - Panhypopituitarism associated with genetic variants in PROP1, HESX1, LHX, LHB, FSHB and others. |
| Acquired: -Central nervous system tumors (e.g., craniopharyngiomas, germinomas), cysts, -Cranial surgeries, -Cranial radiation therapy greater than 30 Gy -Other inflammatory, autoimmune (hypophysitis), and infiltrative (Langerhans cell histiocytosis) diseases of the pituitary gland | |
| Functional Hypogonadotropic Hypogonadism | -intense physical stress (competitive gymnastics, ballerina syndrome) -emotional stress (elevated glucocorticoids) -caloric deficit (anorexia nervosa) -chronic systemic illness (celiac, inflammatory bowel disease, CF, renal disease) -endocrinopathies (hypothyroidism, excess glucocorticoids, hyperprolactinemia) -medication adverse effects -pituitary iron deposits in chronic transfusion dependent children |
Transient Hypogonadotropic Hypogonadism Associated with Delayed Maturation of the HPG Axis/ Constitutional Delay of Growth and Puberty (CDGP)
Transient hypogonadotropic hypogonadism also known as constitutional delay in growth and puberty (CDGP) is the most common etiology of delayed puberty occurring in 70% of boys and 32% of girls with delayed puberty (334). In both sexes, CDGP is self-limited and is considered to represent a variant of normal pubertal timing. CDGP has a strong genetic component, with a positive family history of delayed puberty reported in 50% to 75% of cases (335).
Distinguishing between CDGP and congenital hypogonadotropic hypogonadism (CHH) may be challenging because these conditions share clinical features, hormone levels, and radiological findings. Inhibin B and LH levels tend to be lower in boys with CDGP, but the overlap in values precludes the use of these hormones to distinguish between CDGP and CHH.
For boys, 3-4 months of steroid priming with testosterone followed by 3-4 months of observation is commonly used to discriminate CHH from CDGP (336). It has been suggested that this sex steroid exposure stimulates resumption of the HPG axis activity leading to secondary sex characteristics typical of male puberty (337). Individuals who show no pubertal progression during the observation period should be evaluated for CHH or another disorder affecting the HPG axis. Estradiol priming has been used similarly in girls to distinguish CHH from CDGP (338). Due to the differences in the long-term outcomes, accurate diagnosis is essential, with CDGP being largely a diagnosis of exclusion (339).
CDGP occurs more commonly in family members of individuals with CHH compared to the general population (340). Individuals with CDGP appear to have higher prevalence of pathogenic variants compared to unaffected family members or controls (341). Some genetic variants have been detected in both individuals with CDGP and CHH; these genes include HS6ST1, PROKR2, TAC3, TAC3R, and IL17RD (342). Genetic variants associated primarily with CDGP include IGFS10, EAP1, and FTO (338).
Hypergonadotropic Hypogonadism
Pubertal delay associated with hypergonadotropic hypogonadism is usually associated with disorders affecting gonadal function, specifically gonadal steroidogenesis. With the onset of gonadarche and increased GnRH and gonadotropin secretion, inadequate gonadal steroid secretion and lack of negative feedback leads to increasing gonadotropin secretion. These conditions may be present at birth or acquired.
TURNER SYNDROME
Turner Syndrome refers to deletions or structural rearrangements of the X chromosome. The reported incidence is around 1 in 2500 liveborn female births (343). The initial in utero process of ovarian differentiation proceeds normally with migration of the primordial germ cells into the developing ovary during the fourth week of gestation. By 18 weeks of gestation, premature degeneration of ovarian follicles has begun. The ovarian follicles are typically replaced by connective tissue resulting in the characteristic streak gonad. This accelerated follicular atresia usually leads to premature ovarian insufficiency. Girls with Turner syndrome have gonadal dysgenesis or “streak gonads” in 85% of cases at birth. However, because adrenal androgen secretion is not impaired, the onset of pubarche usually occurs at a normal time. Typical clinical features of girls with Turner syndrome include short stature, short/webbed neck, shield shaped chest with the appearance of widely spaced nipples, cubitus valgus, and Madelung deformity of the forearm and wrist, shortened fourth metacarpals/metatarsals, horseshoe kidneys, coarctation of the aorta, increased risk for autoimmune conditions, and aberrant development of the lymphatic system. Many girls with Turner syndrome may remain undiagnosed until later in childhood or adolescence when they present with short stature and/or delayed puberty. With increased utilization of noninvasive prenatal screening (NIPS), many girls with Turner Syndrome are detected prenatally. The reader is referred to other publications for more extensive discussion regarding the features and approach to multidisciplinary health care management (344, 345, 346).
KLINEFELTER SYNDROME
Klinefelter Syndrome is a chromosomal aneuploidy characterized by 47, XXY karyotype and premature testicular insufficiency. Increased NIPS utilization has led to detection of many boys in utero and is estimated to occur in 1 in 667 males based on prenatal cytogenetic analysis (347). However, many men remain underdiagnosed, with less than 10% of patients being diagnosed prior to puberty. Men with Klinefelter syndrome typically present with tall stature, incomplete puberty, or gynecomastia. Generally, the onset of puberty is not delayed. Klinefelter syndrome is associated with small firm testes, Sertoli cell dysgenesis, impaired spermatogenesis, and variable degrees of testosterone deficiency (348). Learning disabilities, language and visuospatial processing defects, and neuropsychiatric conditions such as attention-deficit/hyperactivity disorder and depression are common (349). If a tumor is found in the anterior mediastinum, a karyotype should be performed to evaluate for Klinefelter syndrome because of its association with mediastinal germinoma (350). Despite normal BMI, the body fat percentage, and the ratio between android fat percentage and gynoid fat percentage are significantly higher than normal. They may also have an impaired bone metabolism starting during childhood and adolescence. Systematic studies are needed to evaluate whether testosterone replacement therapy during puberty will improve these parameters (351). The reader is referred to other publications for more extensive discussion regarding the features and approach to multidisciplinary health care management (352, 353, 354, 355, 356).
DIFFERENCES OF SEX DEVELOPMENT
Differences of sex development (DSDs) are a group of conditions where external genital development is atypical. These disorders are associated with chromosomal anomalies, genetic variants, and environmental influences (357). Gonadal function is impaired in some types of DSDs resulting in primary gonadal failure and hypergonadotropic hypogonadism. Detailed review of DSDs is beyond the scope of this chapter. The interested reader is referred to other Endotext chapters for more extensive review of DSDs.
GENETIC CAUSES OF PREMATURE OVARIAN INSUFFICIENCY (POI)
POI can present with primary or secondary amenorrhea. Fragile X-associated premature ovarian insufficiency is among a family of disorders caused by the expansion of a CGG trinucleotide repeat sequence located in the 5′ untranslated region (UTR) of the fragile X messenger ribonucleoprotein 1 (FMR1) gene on the X chromosome. One etiology is premutation of the FMR1 gene associated with 55-200 CGG repeats without abnormal methylation of the neighboring CpG island and promoter, responsible for both fragile X associated premature ovarian insufficiency in females and fragile X associated tremor ataxia syndrome in males and females where patients may present with mild to moderate intellectual disability, intentional tremor and cerebellar ataxia, peripheral neuropathy, Parkinsonism, and urinary and bowel incontinence.
The X chromosome carries many genes that govern follicular maturation and overall ovarian function, and numerical and structural changes in this chromosome, as in Turner syndrome or triple X syndrome, are associated with POI.
Multiple genes are involved in ovarian differentiation, oocyte development, and, ultimately, folliculogenesis and variants in these genes may be associated with premature ovarian insufficiency (358, 359). The clinical phenotype ranges from delayed puberty to secondary amenorrhea (360).
GALACTOSEMIA
Galactosemia is a rare cause of delayed puberty. Classic galactosemia is a rare inborn error of galactose metabolism due to a defect in the gene encoding the galactose-1-phospate uridyltransferase enzyme (GALT). The prevalence is approximately 1/30,000–60,000 (361). Early manifestations include lactose intolerance, jaundice, failure to thrive, lethargy, hepatocellular damage, renal tubular disease, and cataracts. A galactose-free diet can reverse the neonatal symptoms. However, some long-term complications such as developmental delay, intellectual disability, epilepsy, osteoporosis, and premature ovarian insufficiency may still develop. In females, hypergonadotropic hypogonadism resulting in delayed puberty, primary or secondary amenorrhea, and infertility may occur (362, 363). Available data from patients with classic galactosemia suggest that the primary ovarian insufficiency is due to dysregulation of pathways essential for folliculogenesis culminating in premature ovarian insufficiency (364). Several previous cohort studies in males showed delayed puberty and below-target final height (365, 366, 367), however a recent study with 47 males showed that puberty and fertility were normal and in contrast to earlier reports, AMH, testosterone and Inhibin B levels were normal (361).
Hypogonadotropic Hypogonadism
Pubertal delay associated with hypogonadotropic hypogonadism is usually associated with disorders affecting the neurons that secrete GnRH or the pituitary gonadotrophs that secrete the FSH and LH. These conditions may be present at birth or acquired as described below.
CONGENITAL HYPOGONADOTROPIC HYPOGONADISM
The initiation and maintenance of reproductive capacity in humans depends on pulsatile GnRH secretion. Congenital hypogonadotropic hypogonadism (CHH) results from the absence of the normal pulsatile GnRH secretion or deficient pituitary gonadotropin secretion leading to delayed puberty and infertility. The number of genetic loci associated with CHH continues to expand (Table 4). CHH may be associated with variants in genes involved in the development or migration of GnRH neurons as well as genes involved in the secretion or action of GnRH (368). Autosomal recessive, autosomal dominant, X-linked, and oligogenic inheritance have been described (369, 370). Additional genetic influences include epigenetic factors (371). Clinical heterogeneity has been described between and within families (372).
Given the developmental origins of GnRH neurons in the olfactory placode, CHH can be associated with anosmia or hyposmia. The association of CHH and anosmia is known as Kallmann syndrome. Classic Kallmann syndrome is associated with variants in the ANOS1 gene which is mapped to the X chromosome. Other features of Kallmann syndrome due to ANOS1 variants include unilateral renal agenesis, sensorineural hearing loss, dental agenesis, synkinesia (alternating mirror movements), and cleft lip/palate (373).
In syndromic CHH, associated clinical features may help identify the possible gene(s). For example, clinical features associated with FGFR1 variants include anosmia/hyposmia, cleft lip/cleft palate, dental agenesis, and skeletal anomalies. CHH can also occur in the CHARGE syndrome, which is characterized by coloboma, congenital heart disease, choanal atresia, genital anomalies, ear anomalies, and development delay. CHH can occur with impaired pituitary development associated with PROP1, HESX1, or LHX variants. CHH is also associated with variants in the GnRH, GnRHR, LHB, and FSHB genes (See Table 5). Although some genetic loci are common to both CDGP and CHH, the genetic architectures of these two conditions are largely distinct (374).
Table 5.
| Gene (Reference/s) | Protein encoded | Genetic locus | Associated features/syndromes |
|---|---|---|---|
| SYNDROMIC CAUSES | |||
|
FGFR1/FGF8
(484, 485) | Fibroblast Growth Factor Receptor 1/fibroblast growth factor 8 | 8p11.23 | Hartsfield syndrome |
| LEPR/LEP (486, 487, 488) | Leptin receptor and Leptin | 1p31.3 | Severe obesity syndromes |
| PCSK1 (489) | Prohormone convertase 1 gene | 5q15 | Obesity, ACTH deficiency, diabetes |
| DMXL2 (490) | DmX-like protein 2 | 15q21 | Polyendocrinopathy, Polyneuropathy syndrome |
|
RNF216/
OTUD4 (491) | Ring finger protein 216/ OTU domain-containing protein 4 | 4q31.21 | Gordon Holmes |
| PNPLA6 (492, 493) | Patatin-like phospholipase domain-containing protein 6 | 19p13.2 | Gordon Holmes, Oliver McFarlane, Lawrence Moon, Boucher-Neuhauser syndrome |
| SOX10 (494) | Sex determining region Y-Box transcription factor 10 | 22q13.1 | Wardenburg syndrome |
|
SOX2
(495) | Sex determining region Y-Box transcription factor 2 | 3q26.33 | Optic nerve hypoplasia, CNS abnormalities |
|
SOX3
(496) | Sex determining region Y-Box transcription factor 3 | Xq27.1 | Intellectual disability, craniofacial abnormalities, multiple pituitary hormone deficiencies |
|
IGSF1
(497, 498) | Immunoglobulin superfamily member 1 | Xq26 | Associated with X-linked central hypothyroidism, macro-orchidism |
|
HESX1
(499) | HESX homeobox 1 | 3p14.3 | Hypopituitarism, septo-optic dysplasia |
| CHD7 (500, 501, 502) | Chromodomain helicase DNA binding protein 7 | 8q12.2 | CHARGE syndrome |
|
POLR3A/
POLR3B (503, 504, 505) | RNA polymerase III | 12q23.3 | Hypomyelination, hypodontia |
| NROB1 (DAX-1) (506, 507) | Nuclear Receptor Subfamily 0 Group B Member 1/ dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 | Xp21 | Adrenal hypoplasia |
|
REV3L/
PLXND1 (508) | Catalytic subunit of DNA polymerase zeta | 6q21 | Möbius syndrome |
| PWS (509, 510) | 15q11.2 | Prader-Willi syndrome | |
|
BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10,
TRIM32, BBS12 (511, 512) | Encoded protein may play a role in eye, limb, cardiac and reproductive system development | 11q13.2, 20p12, 16q21, 15q22.3‐23, 14q32.1 (multiple loci) | Bardet-Biedl syndrome |
|
PHF6
(513) | Plant homeodomain (PHD)-like finger protein 6 | Xp26.2 | Borjeson‐Forssman‐Lehmann syndrome |
|
SMCHD1
(514) | Structural maintenance of chromosomes flexible hinge domain containing 1 | 18p11.32 | Bosma arhinia microphthalmia syndrome |
|
TBC1D20/
RAB18 (459, 515) | TBC1 Domain Family Member 20, GTPase activator proteins of Rab-like small GTPases | 20p13 | Warburg micro syndrome |
|
HDAC8
(516) | Histone deacetylase 8 | Xq13.1 | Cornelia de Lange syndrome |
| NON-SYNDROMIC CAUSES | |||
|
FGF17
(517) | Fibroblast Growth Factor 17 | 8p21.3 | |
|
ANOS1 (KAL1)
(518, 519) | Kallmann syndrome protein, which is now known as Anosmin 1 | Xp22.31 | involved in fibroblast growth factor (FGF) signaling |
|
GNRHR/
GNRH1 (520, 521, 522) | Gonadotropin-releasing hormone receptor/ gonadotropin-releasing hormone 1 | 4q13.2 | |
| KISS1R/ KISS1 (54, 523) | Kisspeptin-1 receptor/ kisspeptin-1 | 19p13.3 | |
| KLB (524) | Klotho Beta | 4p14 | Metabolic defects |
| TAC3/TACR3 (342, 525) | Tachykinin 3, Tachykinin 3 receptor Encodes neurokinin b | 4q24 | |
|
IL17RD
(517) | Interleukin 17 Receptor D | 3p14.3 | |
|
DUSP6
(517) | Dual specificity phosphatase 6 | 12q22–q23 | |
| SEMA3A/ SEMA3E/ SEMA7A (526) | Semaphorin 3A | 7q21.11 | |
| SPRY4 (517) | Sprouty homolog 2 | 5q31.3 | |
| FLRT3 (517) | Fibronectin leucine rich transmembrane protein 3 | 20p11 | |
| PROKR2/ PROK2 (527, 528) | Prokineticin-2 and Prokineticin receptor 2 | 3p13 | |
|
WDR11
(529) | WD repeat domain 11 | 10p26.12 | |
|
CCDC141
(530) | Coiled-Coil Domain Containing 141 | 2q31.2 | |
| FEZF1 (531) | FEZ family zinc finger 1 | 7q31.32 | |
|
LHB
(532) | Luteinizing hormone | 19q13.33 | |
|
FSHB
(533, 534) | Follicle-stimulating hormone | 11p14.1 | |
|
AXL
(458) | AXL receptor tyrosine kinase | 19q13.2 | |
|
EAP1
(535) | Enhanced at puberty 1 | 14q24 | Trans-activates the GnRH promoter |
|
LGR4
(536) | Receptor for R-spondins which, once activated, potentiates the canonical Wnt signaling pathway | 11p14.1 | |
|
TUBB3
(483) | Microtubule protein β-III-tubulin | 16q24.3 | Congenital fibrosis of the extraocular muscles |
|
WDR11, PROP1, PROK2, PROKR2
(529) | Bromodomain and WD repeat-containing protein 2, Homeobox protein prophet of PIT-1, prokinectin 2 | 10q26.12, 5q35.3, 3p13 | Combined pituitary hormone deficiency |
|
FTO
(482, 537) | Fat mass and obesity-associated protein | 16q12.2 | Mice lacking FTO had significantly delay in pubertal onset |
ACQUIRED HYPOGONADOTROPIC HYPOGONADISM
Several conditions are associated with primary gonadal insufficiency. These conditions include auto-immune disorders, trauma, neoplasia, vascular events, and infection. Autoimmune disorders can be associated with premature ovarian and testicular insufficiency. Biallelic mutations in the autoimmune regulator (AIRE) gene are associated with autoimmune polyendocrine syndrome type 1 which is also known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Associated features include mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency (375). The detection of autoantibodies directed against tissue-specific antigens suggests an autoimmune diagnosis.
Antineoplastic chemotherapy with alkylating agents, as well as localized ionizing radiation, may permanently damage germ cells leading to infertility. In males, Sertoli cells are more susceptible to such toxicity than Leydig cells such that testosterone production may remain intact despite Sertoli and germ cell injury. Mumps orchitis should be considered, especially in unvaccinated males. Decreased blood flow to the gonads from surgical injury (e.g., orchiopexy in boys), torsion, or trauma can lead to ischemia and atrophy, with resultant primary testicular insufficiency.
The presence of otherwise normal male external genitalia associated with nonpalpable gonads indicates that the testes were present and functioning at least early in gestation. The “vanishing testes syndrome” also known as testicular regression is associated with atrophy or regression of testicular tissue initially formed during early embryonic development. Potential etiologies of the testicular regression include in utero vascular disruption or testicular torsion. Pathogenic variants of the DEAH-box RNA helicase DHX37 (DHX37) gene have been identified in boys with testicular regression and in association with gonadal dysgenesis (376).
In addition to autoimmune etiologies, premature ovarian insufficiency can be associated with ovarian/pelvic tumors, chemotherapy, especially alkylating agents, and radiation therapy. The location of the pelvic tumor and treatments influence the ovarian reserve and risk for premature ovarian insufficiency. Low or declining serum AMH levels provide an indirect measure of ovarian reserve. However, due to much variability and lack of diagnostic thresholds, measuring AMH values does not accurately predict ovarian insufficiency in cancer survivors (377).
Acquired hypogonadotropic hypogonadism (HH) can be due to central nervous system tumors such as craniopharyngiomas and germ cell tumors. Such tumors can disrupt the hypothalamic-pituitary stalk or can impact pituitary function producing decreased gonadotropin production. Hyperprolactinemia due to prolactin-secreting adenomas can cause acquired HH (378). Other central nervous system disorders associated with acquired HH include hypophysitis, histiocytosis, and hemochromatosis. Intracranial surgeries and/or cranial radiation therapy greater than 30 Gy are known risk factors for HH. Moderate to severe trauma to the brain is associated with injuries to the hypothalamus, stalk (infundibulum), or pituitary gland itself; the consequences of traumatic brain injury may not manifest for many years. Chronic steroid treatment can be associated with acquired HH in boys with Duchenne muscular dystrophy (379, 380). Inflammatory and infiltrative diseases of the pituitary gland are other rare causes of acquired HH.
FUNCTIONAL HYPOGONADOTROPIC HYPOGONADISM
Functional HH is the hypothalamic response to intense physical or emotional stress, caloric deficit, or chronic systemic illness (381). In this situation, the otherwise normal HPG axis fails to function due to the concomitant stress. Puberty can be delayed or stalled until the underlying condition has been adequately addressed. The finding of hypercortisolemia in women with functional HH associated with restrictive eating disorders highlights the relevance of HPA axis function in FHH (382). Importantly, functional HH can have long lasting adverse consequences on bone health (383).
The hypothalamus receives numerous inputs regarding body energy status and subsequently modulates reproductive status based on this information. Hence, nutritional status and energy output influence HPG axis activity in part via leptin signaling which regulates the sensitivity of the pituitary to GnRH (384). Energy deficits may occur due to weight loss, excessive energy expenditure (rigorous physical activity, renal disease, cystic fibrosis, congenital heart disease), decreased caloric intake or malabsorption (disordered eating behaviors, bowel disorders such as celiac, Crohn’s, and ulcerative colitis) are associated with delayed or stalled puberty and functional hypothalamic amenorrhea (385, 386, 387). Elevated circulating levels of cytokines (as seen in some acute or inflammatory conditions) may also inhibit the HPG axis. Elevated prolactin levels, due to prolactinoma or severe primary hypothyroidism may inhibit gonadotropin release.
Some boys with obesity have low gonadotropin and testosterone levels and manifest delayed puberty (388). It is important to recognize that certain medications such as antipsychotics (typical and atypical), certain antidepressants, and opioids can alter menses (364).
Treatment of Delayed Puberty
A variety of therapeutic regimens for pubertal induction have been described for both boys and girls. However, large, randomized trials providing evidence-based data regarding the optimum regimen are lacking (389). Sex steroid replacement therapy remains a mainstay of treatment. The type and route of administration of the sex steroids is dependent on patient preference, insurance coverage, and health care provider practices. Importantly, the specific treatment regimen depends on the underlying etiology of the pubertal delay. Future novel therapies could include kisspeptin and neurokinin B analogs (390).
BOYS
Pulsatile GnRH therapy is the most physiological method and can induce adult secondary sex characteristics, achieving normal adult testosterone concentrations, and spermatogenesis (370) in boys with HH. However, the inconvenience of wearing a mini-pump and conflicting outcome data limits its usefulness. Other approaches include hCG, FSH, hMG, and/or GnRH treatments. Despite much heterogeneity, a systematic study reported that treatment with hCG and FSH induced greater increase in testicular volume and rate of spermatogenesis compared to hCG alone (391)370). Importantly, available limited data suggest that testosterone administration prior to gonadotropin treatments does not interfere with the beneficial effects on testicular growth and spermatogenesis. Based on the physiologic roles of LH and FSH, pubertal induction should begin with FSH to promote testicular maturation followed by combined FSH and hCG treatment. The subsequent hCG treatment will promote testicular testosterone secretion leading to virilization, growth spurt, and psychosocial development.
Still, at the present time, testosterone is the most established treatment for pubertal induction in boys with delayed puberty. Traditionally, IM testosterone esters, primarily testosterone enanthate or testosterone cypionate, have been used. A subcutaneous testosterone enanthate auto-injector has recently been approved, but this approach requires more weekly injections and is more expensive. However, no evidence-based guidelines exist for testosterone-induced pubertal initiation. Potential adverse consequences of testosterone therapy include erythrocytosis, premature epiphyseal closure especially with excessive doses which may result in aggressive behavior, mood swings, and priapism.
Other testosterone formulations include testosterone gels, pills, and pellets. Limitations of testosterone gels include difficulties in accurately titrating low doses, potential testosterone exposure to household members, and the cost. Oral methyltestosterone and its 17α-derivatives have been associated with hepatic dysfunction and should be avoided. Oral testosterone undecanoate was approved by the FDA in 2019 to treat hypogonadal adult men. However, due to its short half-life, multiple daily doses are necessary, and no data are available regarding use for puberty induction. Testosterone pellets require surgical placement every 3-4 months, are expensive, and often spontaneously extrude (392).
For the younger adolescent boy with a strong family history of CDGP, reassurance and continued clinical monitoring may be adequate. However, discerning CDGP from CHH is essential because the treatment, genetics, and psychosocial implications differ. Hence, low dose testosterone for 3-4 months followed by a similar period of observation may be helpful to distinguish CDGP from CHH. Individuals with CHH will show persistently low gonadotropin and sex steroid hormone levels after the 3–4-month period of observation whereas individuals with CDGP will usually show spontaneous pubertal progression. Curiously, testosterone exposure apparently activates GnRH production and secretion leading to “reversal” with onset of HPG axis activity in some boys with CHH. This reversal is associated with specific genetic variants and may be transient (393, 394).
GIRLS
Timely induction of pubertal development is fundamental. Two major goals of estrogen therapy are mimicking typical pubertal progression with breast development and promoting adequate uterine growth (395). Although pulsatile GnRH treatment can be used, this approach has no advantage over estrogen for pubertal induction in girls. Though all therapeutic approaches utilize estrogens, details regarding specific formulations and methods of administration vary. Transdermal estradiol is preferred for replacement therapy because this approach avoids the first pass through the liver and the potential for adverse effects on clotting factors.
Typically, low transdermal estradiol doses are used for the initial phase of pubertal induction. Transdermal estradiol doses of 3-7 mcg/day can be achieved by cutting matrix patches (0.014-0.025 mg/24 h) into quarters or eighths. Subsequently, the dose can be increased approximately every six months until adult replacement dosage is achieved taking about 24-36 months to do so. High initial estrogen doses should be avoided due to increased likelihood for atypical breast development characterized by prominent nipples with little supporting breast tissue. High estrogen doses should also be avoided as premature epiphyseal fusion could impair additional linear growth.
Oral micronized 17β-estradiol can be used for those with severe skin irritation or aversion to the use of a patch. Oral preparations containing conjugated equine estrogens or ethinyl estradiol should be avoided for both pubertal induction and maintenance therapy. Most combined oral contraceptives contain ethinyl estradiol at doses higher than appropriate for induction of puberty. Approximately 18-24 months after initiation of unopposed estrogen therapy, progestogens can be added to induce withdrawal bleeding and to reduce the risk for endometrial hyperplasia. Progestogens can be introduced earlier if breakthrough vaginal bleeding occurs. Pelvic ultrasounds before and during pubertal induction can be planned to assess uterine size and shape as well as to evaluate endometrial thickness to ascertain optimal timing to introduce progestins. Progestins vary in potency and can be administered by transdermal, oral, or uterine routes. Although increased potency may have beneficial effects on withdrawal bleeding, greater progestogenic side effects may develop.
No evidence-based data exist, and no single regimen has been demonstrated to be superior. Pubertal induction therapy should be individualized based on clinical response and other auxologic parameters.
Oral contraceptive pills may be used for convenience but should be limited to after completion of pubertal development. Since some girls may experience sporadic ovulation, contraception should be utilized by those at risk of undesired pregnancies.
EVALUATION OF A CHILD WITH A VARIATION IN PUBERTAL DEVELOPMENT
The diagnostic tools are comparable for the evaluation of either precocious or delayed puberty. Detailed medical history and physical examination provide the preliminary information to guide the differential diagnosis for a child with a variation in pubertal development (165, 396) (see Figures 5-8). Laboratory, imaging, and genetic studies are subsequently utilized to ascertain the specific diagnosis. The tools for evaluation of a child with a variation in pubertal development are described below. The tools are comparable, but the interpretation of test results differs for precocious and delayed puberty.

Figure 5.
Algorithm to evaluate a girl presenting with precocious puberty. *follow clinical progression every 3-6 months. FSH: Follicle Stimulating Hormone; LH: Luteinizing Hormone; CAH: Congenital Adrenal Hyperplasia; ACTH: Adrenocorticotrophic Hormone; GnRH: Gonadotropin Releasing Hormone; DHEAS: Dehydroepiandrosterone Sulfate; 17OHP: 17-hydroxy progesterone; NF-1: Neurofibromatosis-1.

Figure 6.
Algorithm to evaluate a boy presenting with precocious puberty. FSH: Follicle Stimulating Hormone; LH: Luteinizing Hormone; MRI: Magnetic Resonance Imaging.

Figure 7.
Algorithm to evaluate a girl presenting with delayed puberty. FSH: Follicle Stimulating Hormone; LH: Luteinizing Hormone; CDGP: Constitutional Delay in Growth and Puberty; CHH: Congenital Hypogonadotropic Hypogonadism; MRI: Magnetic Resonance Imaging.

Figure 8.
Algorithm to evaluate a boy presenting with delayed puberty. FSH: Follicle Stimulating Hormone; LH: Luteinizing Hormone; CDGP: Constitutional Delay in Growth and Puberty; CHH: Congenital Hypogonadotropic Hypogonadism; MRI: Magnetic Resonance Imaging.
History and Physical Examination
The medical history focuses on the timing and sequence of the pubertal changes in the patient as well as parents, grandparents, and siblings. Review of past medical history and medications (including chemotherapy and nutritional supplements) is essential. Inquiry regarding exposures (tea tree/lavender oils, sex steroids, radiation) may help to identify potential environmental factors (162). Obtaining birth history, length and weight, history of SGA (397), prematurity, or CNS insult at birth or later provide relevant information (162).
A history of gelastic seizures may point to a hypothalamic hamartoma. Inquiry regarding use of transdermal testosterone by a family member may identify the cause of premature virilization. Pubertal delay associated with micropenis, anosmia, cryptorchidism, deafness, choanal atresia, hearing loss, and/or digital abnormalities suggests congenital hypogonadotropic hypogonadism (CHH). A family history of anosmia, subfertility, and deafness should be sought for those with pubertal delay. Multiple syndromes are associated with CHH (see Table 3); suggestive features include absent/reduced sense of smell, choanal atresia, hearing loss, morbid obesity, visual impairment. Family history of precocious or delayed puberty in close relatives may be discovered (398). Behavioral difficulties or learning disabilities may be associated with specific syndromes such as Turner or Klinefelter syndromes.
A complete physical examination including height, weight, arm span, and sitting height is essential. Review of the child’s growth curves provides valuable information regarding changes in linear growth and weight gain. Acceleration in linear growth and upward crossing of centiles may be seen in precocious puberty. A gradual downward crossing of centiles may be noted in constitutional delay in growth and puberty (CDGP) as linear growth slows compared to peers who are entering puberty (399). Pubic hair development (pubarche) may also be delayed in CDGP as opposed to CHH where adrenarche occurs at the normal age for population (372).
Physical exam includes ascertainment of the sexual maturity rating for breast, pubic hair, and testicular volume based on the scoring system derived by Tanner and colleagues (Figure 1). Due to challenges in discriminating lipomastia from true glandular breast development, palpation of the breasts is important. Firm glandular tissue under the areolae is indicative of thelarche. Accurate measurement of testicular volume using an orchidometer is essential (see Figure 2). A testicular volume of ≤ 1.1 mL has a reported sensitivity and specificity of 100% and 91%, respectively, for CHH (400).
The physical examination should assess for midline defects, dysmorphic features, visual field abnormalities, and features characteristic for specific syndrome. For example, short stature, cubitus valgus, low hair line, widely spaced nipples, and delayed puberty suggest Turner Syndrome. The physical examination needs to include palpation of the thyroid gland, skin examination for acne or café-au-lait macules (which would suggest neurofibromatosis or McCune-Albright syndrome) and a visual field exam. Melanocytic macules typical of Peutz-Jeghers syndrome could point to the presence of a sex cord tumor causing gonadotropin independent (peripheral) sexual precocity.
Laboratory Evaluation
Laboratory evaluation assists the diagnostic process to identify the etiology of “off-time puberty.” Circulating gonadotropin and sex steroid concentrations reflect HPG axis status (187, 236). Most current gonadotropin assays are sandwich assays specific to the β-subunit. Ultrasensitive FSH and LH assays should be used when available. For LH, samples should preferably be obtained in the morning. The lower limit of detection for most ultrasensitive immunochemiluminescent assays (ICMA) is ≤0.1 mIU/mL (230, 401, 402).
When the clinical concern is precocious puberty, LH concentrations greater than 0.3-0.5 mIU/mL suggest central precocious puberty (CPP) with higher cut-points increasing the sensitivity and specificity of the LH determination (403). Elevated basal LH levels show high sensitivity and specificity for boys when high quality immunochemiluminometric assays (ICMA) is used (404). Different cut-points need to be used to interpret LH concentrations in girls under two years of age because LH concentrations may be elevated at this age leading to misdiagnosis of CPP followed by inappropriate treatment during this phase of development (405). For the child with physical signs of premature puberty, LH concentrations in the prepubertal range are consistent with either peripheral precocity or a benign pubertal variant such as premature thelarche. Typically, LH and FSH concentrations are suppressed in children with peripheral precocious puberty (406).
In the evaluation for delayed puberty, low gonadotropin concentrations suggest a central etiology such as CDGP or hypogonadotropic hypogonadism while elevated gonadotropin concentrations suggest primary gonadal insufficiency. Random gonadotropin concentrations may provide only limited information because gonadotropin secretion is pulsatile. Distinguishing hypogonadotropic hypogonadism from CDGP is often challenging because LH, FSH and sex hormone reference intervals vary widely even in healthy adolescents (407). Similarly, due to significant overlap in hormone reference intervals, GnRH agonist and human chorionic gonadotropin (hCG) stimulated gonadotropin (408) and sex steroid concentrations fail to distinguish youth with CHH from those with CDGP (407, 409).
Due to the small structural differences between steroid molecules, immunoassays are confounded by cross-reactivity issues. Assay issues are amplified in children because commercial immunoassays for estradiol and testosterone are usually designed to measure hormone concentrations within the normal adult reference interval. Hence, most estradiol immunoassays have low sensitivity and specificity to quantify the low concentrations (< 30 pg/ml) typically found in prepubertal children and individuals with hypogonadism. Similar issues occur with testosterone immunoassays. Hence, steroid hormone concentrations should be measured by liquid chromatographic separation followed by mass spectrometry (LC-MS/MS). Serum testosterone is best measured using LC-MS/MS technology to limit cross-reactivity and increase sensitivity and specificity especially when low hormone concentrations might be anticipated. LC-MS/MS is also the optimal technique to measure circulating concentrations of other steroids including 17-hydroxyprogesterone, DHEA, androstenedione, and the 11-oxy androgens. It offers greater sensitivity and specificity and allows simultaneous measurement of multiple hormone concentrations (410).
Sex steroids such as estradiol and testosterone circulate bound to sex hormone binding globulin (SHBG). Tissue availability of the free hormone, presumed to be the active moiety, is regulated by SHBG. Direct free testosterone concentrations should be avoided because direct immunoassays have poor reproducibility and reliability. When free testosterone concentrations need to be determined, equilibrium dialysis should be performed despite known potential limitations including increased expense, reliance on total testosterone accuracy, temperature control, and sample dilution (411).
Another confounding factor is biotin (vitamin B7) which is an over-the counter supplement by itself or as an addition to many preparations used to strengthen nails and hair. Biotin interferes with the technical aspects of immunoassays and can lead to either falsely elevated or falsely low result when streptavidin binding is utilized in the assay detection system. When immunoassay results seem incongruous, use of biotin-containing products should be queried. Biotin does not interfere with LC-MS/MS assays (412).
GnRH or GnRH AGONIST STIMULATION TEST
Historically, the established gold standard to diagnosis CPP was the LH and FSH response to a standard bolus of native GnRH. With decreased availability of native GnRH, most stimulation tests are now performed with the GnRH agonist (GnRHa) leuprolide acetate, a synthetic nonapeptide with much greater potency. The timing and peak values of FSH and LH levels differ between GnRH and leuprolide acetate. Following native GnRH administration, LH levels peak after 20–40 minutes, followed by a decline. With leuprolide acetate, peak LH occurs between 0.5 - 4 hours followed by sustained LH elevation.
The optimal cutoff value of peak stimulated LH for identifying children with CPP is unclear due to assay variability. For most LH assays, a value of 3.3 to 5 mIU/mL defines the upper limit of normal for stimulated LH values in prepubertal children. Stimulated LH concentrations above this range suggest CPP (232). Children with progressive CPP tend to have a high stimulated LH:FSH ratios compared with those with non- or intermittently progressive precocious puberty. Measuring the appropriate sex steroid 24 hours following GnRHa administration can help confirm a CPP diagnosis (413). However, obtaining this second sample may burden the family because of the need for a second venipuncture, expense of another hormone determination, and missed school and work.
As noted above regarding basal LH levels, care must be taken in interpreting the results of GnRH stimulation test in females under the age of two years, as both basal and stimulated LH levels can be elevated as part of the normal hormonal changes associated with mini-puberty (405).
To assess GnRH production by the hypothalamus, kisspeptin-stimulated LH response has been proposed to identify individuals with GnRH deficiency and thus CHH. Kisspeptin stimulates GnRH secretion, thus promoting LH, and to a lesser extent FSH, secretion.
One study found that maximal LH rise after kisspeptin administration was more accurate for diagnosis of men with GnRH deficiency than GnRH stimulation testing (414). A similar study in adolescents with pubertal delay (3 females and 13 males), peak LH post kisspeptin stimulation was demonstrated to be superior to GnRH stimulation testing for predicting capacity to progress through puberty (noting that the LH cut off values were different and an ideal cutoff value still needs to be determined) (346). Further research is required to better define the parameters of using kisspeptin stimulation in clinical practice (404).
In children with precocious pubarche, measurement of adrenal steroids may be necessary to help distinguish between peripheral precocity and benign premature adrenarche. Children with premature adrenarche can have mild elevation in adrenal hormones (415). Since premature adrenarche is a diagnosis of exclusion, further investigation for congenital adrenal hyperplasia and virilizing adrenal tumors may be indicated. In children, an early-morning 17-hydroxyprogesterone (17-OHP) value >200 ng/dL (6 nmol/L) has a high sensitivity and specificity for non-classic congenital adrenal hyperplasia secondary to 21-hydroxylase deficiency. An adrenocorticotropic hormone (ACTH) stimulation test is needed to confirm the diagnosis (313, 416). An ACTH stimulation test involves administration of 0.25 mg synthetic ACTH (1-24) or 15 mcg/kg for children up to 2 years of age with blood samples obtained at baseline and either 30 or 60 minutes after synthetic ACTH administration. Although 21-hydroxylase deficiency is the most common virilizing form of CAH, 17-hydroxypregnenolone, DHEA, and 11-deoxycortisol determinations may be necessary to assess for 3β-hydroxysteroid dehydrogenase or 11β-hydroxylase deficiencies.
Boys with hypogonadotropic hypogonadism tend to have lower inhibin B values compared to boys with CDGP. However, a validated cut-point for inhibin B concentrations remains to be established (417, 418, 419). FSH stimulated inhibin B concentrations < 116 pmol/L have been demonstrated in a study of adolescents with delayed puberty to have more accurate diagnostic discrimination and a promising test for prediction of onset of puberty (414).
Human chorionic gonadotropin concentrations can be measured in males to evaluate for the possibility of an hCG-secreting tumor leading to peripheral precocity (280). A thyroid-stimulating hormone (TSH) concentration should be measured if chronic primary hypothyroidism is suspected as the underlying cause for the sexual precocity, known as the Van-Wyk-Grumbach syndrome (296, 298) .
Targeted diagnostic tests are warranted in some cases to investigate for specific causes of apparent functional hypogonadotropic hypogonadism, such as anti-transglutaminase IgA for celiac disease. Despite promising data, measurement of AMH and INSL3 in addition to testosterone, as endocrine markers to guide the differential diagnosis (418, 420), need additional studies.
The testosterone response to long-term hCG stimulation and peak serum FSH response to GnRH were found to be significantly different in CHH patients (421). However, there are potential long-term drawbacks to prolonged hCG therapy in males who are FSH-naïve regarding premature stimulation of Sertoli and germ cell differentiation prior to FSH exposure (338).
Imaging Studies
BONE AGE
Assessing the skeletal maturation based on a radiograph of the left hand and wrist is an important diagnostic tool in pubertal evaluation. For the commonly utilized Greulich and Pyle method, the patient’s bone age radiograph is compared with an atlas of radiographs from children of known ages (422). For the Tanner-Whitehouse 2 method, 20 different hand and wrist bones are scored. Bone age standards are largely based on hand and wrist radiographs obtained from children of European ancestry between the 1930s to the 60s (423). Despite this limitation, the bone age radiograph is a valuable indicator regarding sex steroid exposure and epiphysial (growth plate) maturation. Additional factors such as other hormones, obesity, genetics, nutritional status, various disease states, and certain medications can influence the rate of epiphyseal maturation (424) (425).
Bone age has been used to predict adult height using the tables of Bayley and Pinneau (426), but reliability is low with a tendency toward overestimation (427). The use of automated measurement systems with artificial intelligence has increased, mitigating previous limitations due to intra- and inter-observer variability (428, 429). Bone age readings within two standard deviations of the chronologic age are considered to be within normal limits. A delayed bone age is usually observed in patients with delayed puberty and an advanced bone age is observed with precocious puberty. One exception is patients with precocious puberty associated with hypothyroidism (Van Wyk Grumbach syndrome) where the bone age is delayed despite pubertal changes. In some instances, monitoring the predicted adult height (PAH) during the course of treatment of pubertal disorders helps to assess treatment efficacy.
ULTRASOUND IMAGING
In females, pelvic ultrasound is a rapid, non-invasive, and relatively low-cost method to ascertain the anatomy of the ovaries and uterus, ovarian volume, and uterine development. This imaging is generally readily accessible and does not require sedation, radiation, or use of contrast material. However, the quality of the device and operator experience influence the analysis.
During puberty, increased gonadotropin secretion promotes ovarian growth, increased estradiol secretion, and increased uterine volume (430). Girls with CPP have increased uterine size and ovarian volumes compared to prepubertal girls or those with premature thelarche. However, the overlap between prepubertal and early pubertal girls for ovarian volume and uterine size confounds interpretation of the ultrasound findings (431) (432). In a prepubertal patient with isolated vaginal bleeding, a normal pelvic ultrasound does not exclude the diagnosis of a functional ovarian cyst because the cyst may have regressed prior to imaging. Pelvic ultrasounds should be obtained in girls with primary amenorrhea who fail progesterone withdrawal to assess for Mullerian duct and renal anomalies. For patients with rapid development of secondary sex characteristics, pelvic ultrasound studies may be needed to assess for gonadal tumors.
The use of Doppler ultrasound to assess utero-ovarian blood flow may also provide helpful information. With increased estradiol secretion and stimulation of the estrogen receptors, vascular resistance of the uterine arteries is reduced. The pulsatility index (PI) is defined as the difference between peak systolic flow and end-diastolic flow divided by the mean flow velocity; the PI reflects impedance to blood flow distal to the sampling point. A review showed that PI is lower among pubertal girls. However, definitive cut-points for PI values have not been established. In addition, testing is operator dependent (433, 434, 435, 436).
Ultrasound examination of the testes, especially if asymmetric in size, should be performed in males with peripheral precocity to evaluate for the possibility of a Leydig cell tumor (437, 438). Testicular ultrasound imaging should be performed regularly to assess for testicular rest tissue in boys with congenital adrenal hyperplasia (439).
MR AND CT IMAGING
Brain MR or CT imaging is performed to define brain and pituitary anatomy. Brain and pituitary MR is helpful to assess for intracranial pathology among those with CNS symptoms. Most studies recommend a contrast-enhanced brain MRI for girls with onset of secondary sexual characteristics before six years of age because of higher rates of CNS abnormalities in these patients (137). In a 2018 meta-analysis (440), the prevalence of intracranial lesions was 3 percent among girls presenting with CPP after six years of age, compared with 25 percent among those presenting before six years. Thus, girls with pubertal onset between six and eight years of age may not need the MRI in the absence of clinical evidence of CNS pathology (441, 442, 443). MRI should be limited to high-risk individuals (younger age, neurologic symptoms) (444). Current guidelines recommend that in otherwise asymptomatic girls with CPP, a discussion occur with the parents regarding the pros and cons of brain imaging and assist in informed decision making (137, 445, 446). While contrast-enhanced brain MRIs are recommended for all boys presenting with CPP (412), one study found that these rates may be overestimated. The prevalence of intracranial lesions among boys who were healthy, did not have neurological symptoms, and were diagnosed with CPP was lower than that previously reported and none of the identified lesions necessitated treatment, suggesting the need to globally reevaluate the prevalence of pathological brain lesions among boys with CPP (447).
For children with delayed puberty, MR imaging of the pituitary gland and olfactory structures can assess for features of CHH such as absence of the olfactory bulbs (448, 449, 450).
Pelvic MRI is helpful to characterize and stage pediatric ovarian masses due to excellent soft tissue contrast. In addition, MR imaging does not involve the use of ionizing radiation and allows better assessment of the abdomen and kidneys. Disadvantages of MRI include that it is time-consuming, expensive, and may require sedation.
In both girls and boys, adrenal tumors can cause peripheral precocious puberty, progressive virilization, and/or markedly elevated serum adrenal androgens (e.g., DHEAS). If diagnoses such as congenital adrenal hyperplasia and exogenous androgen or testosterone exposure have been excluded, such patients should have an imaging study of the adrenal glands (451, 452, 453). CT may be preferable for evaluation, staging, surgical planning for adrenal tumors (454). Despite radiation exposure, CT can be readily performed in emergent situations.
Genetic Testing
A karyotype can help with a diagnosis of Turner or Klinefelter syndrome (455).Newer sequencing technologies along with increased knowledge regarding genes involved in puberty has advanced the usefulness of genetic testing (456). The known genetic causes of CPP and HH have increased exponentially over the past five years. Genetic testing could therefore precede brain MRI, at least in familial CPP cases (167, 457).
Patients with delayed puberty associated with phenotypic features such as anosmia/hyposmia, synkinesia, or hearing loss, the probability of detecting a pathogenic variant on genetic testing for HH is increased (458) (459, 460). Consideration should be given to using genetic testing early in the diagnostic process while recognizing the limitations of genetic testing. Challenges in using genetic testing as a discriminatory test between CHH and CDGP remain, and more research is needed in this area.
SUMMARY
Pubertal development and maturation of the neuroendocrine system involve the ontogeny, activity, and interactions of the GnRH neurons. Pubertal onset is accompanied by an increase in kisspeptin and neurokinin B secretion regulating the pulsatile GnRH secretion that stimulates pulsatile pituitary LH and FSH secretion. LH and FSH stimulate gonadal sex steroid secretion promoting development of secondary sex characteristics and influencing hypothalamic-pituitary function via negative feedback inhibition.
Alterations of gut microbiome at different pubertal stages may present an area for future development in the prediction and prevention of precocious puberty. Use of genetic testing including targeted next generation sequencing and whole exome sequencing may have increasing utility as diagnostic tools early on in the evaluation of pubertal disorders.
Discovery regarding the details of normal reproductive physiology followed by identification of the genetic basis for disorders of pubertal timing established our current knowledge base for the evaluation and management of children with disorders affecting the timing of puberty. Despite the vast expansion of our knowledge, much remains to be learned about the physiology and regulation of the HPG axis from the fetus to the young adult.
REFERENCES
- 1.
- Springfield: Mass, G. & C. Merriam Co; 1961. Webster’s third new international dictionary of the English language, unabridged.
- 2.
- Argente J, Dunkel L, Kaiser UB, Latronico AC, Lomniczi A, Soriano-Guillen L, et al. Molecular basis of normal and pathological puberty: from basic mechanisms to clinical implications. Lancet Diabetes Endocrinol. 2023;11(3):203-16. [PMC free article: PMC10198266] [PubMed: 36620967]
- 3.
- Abreu AP. Unveiling the central regulation of pubertal development. J Clin Endocrinol Metab. 2023. [PubMed: 37589951]
- 4.
- Reiter EO, Fuldauer VG, Root AW. Secretion of the adrenal androgen, dehydroepiandrosterone sulfate, during normal infancy, childhood, and adolescence, in sick infants, and in children with endocrinologic abnormalities. J Pediatr. 1977;90(5):766-70. [PubMed: 140222]
- 5.
- Witchel SF BA, Oberfield SE. Adrenal Androgens, Adrenarche and Adrenopause. 8th Edition ed. Robertson RP GL, Grossman A, Hammer GD, Jensen MD, Kahaly GJ, Swerdloff R, Thakker RV (eds), editor: Elsevier; 2021.
- 6.
- Rege J, Turcu AF, Kasa-Vubu JZ, Lerario AM, Auchus GC, Auchus RJ, et al. 11-Ketotestosterone Is the Dominant Circulating Bioactive Androgen During Normal and Premature Adrenarche. J Clin Endocrinol Metab. 2018;103(12):4589-98. [PMC free article: PMC6226603] [PubMed: 30137510]
- 7.
- Counts DR, Pescovitz OH, Barnes KM, Hench KD, Chrousos GP, Sherins RJ, et al. Dissociation of adrenarche and gonadarche in precocious puberty and in isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1987;64(6):1174-8. [PubMed: 3571422]
- 8.
- Conley AJ, Moeller BC, Nguyen AD, Stanley SD, Plant TM, Abbott DH. Defining adrenarche in the rhesus macaque (Macaca mulatta), a non-human primate model for adrenal androgen secretion. Mol Cell Endocrinol. 2011;336(1-2):110-6. [PMC free article: PMC5881168] [PubMed: 21184803]
- 9.
- Cutler GB, Jr., Glenn M, Bush M, Hodgen GD, Graham CE, Loriaux DL. Adrenarche: a survey of rodents, domestic animals, and primates. Endocrinology. 1978;103(6):2112-8. [PubMed: 155005]
- 10.
- Cumberland AL, Hirst JJ, Badoer E, Wudy SA, Greaves RF, Zacharin M, et al. The Enigma of the Adrenarche: Identifying the Early Life Mechanisms and Possible Role in Postnatal Brain Development. Int J Mol Sci. 2021;22(9). [PMC free article: PMC8122518] [PubMed: 33919014]
- 11.
- Remer T, Boye KR, Hartmann MF, Wudy SA. Urinary markers of adrenarche: reference values in healthy subjects, aged 3-18 years. J Clin Endocrinol Metab. 2005;90(4):2015-21. [PubMed: 15671100]
- 12.
- Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44(235):291-303. [PMC free article: PMC2020314] [PubMed: 5785179]
- 13.
- Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45(239):13-23. [PMC free article: PMC2020414] [PubMed: 5440182]
- 14.
- Fuqua JS, Eugster EA. History of Puberty: Normal and Precocious. Horm Res Paediatr. 2022;95(6):568-78. [PubMed: 36446322]
- 15.
- Bruserud IS, Roelants M, Oehme NHB, Madsen A, Eide GE, Bjerknes R, et al. References for Ultrasound Staging of Breast Maturation, Tanner Breast Staging, Pubic Hair, and Menarche in Norwegian Girls. J Clin Endocrinol Metab. 2020;105(5):1599-607. [PMC free article: PMC7275631] [PubMed: 32140730]
- 16.
- Pedersen JL, Nysom K, Jorgensen M, Nielsen CT, Muller J, Keiding N, et al. Spermaturia and puberty. Arch Dis Child. 1993;69(3):384-7. [PMC free article: PMC1029527] [PubMed: 8215551]
- 17.
- Mieritz MG, Raket LL, Hagen CP, Nielsen JE, Talman ML, Petersen JH, et al. A Longitudinal Study of Growth, Sex Steroids, and IGF-1 in Boys With Physiological Gynecomastia. J Clin Endocrinol Metab. 2015;100(10):3752-9. [PubMed: 26287961]
- 18.
- David K DE, Freud J, Laqueur E. Crystalline male hormone from the testes (Testosterone) is more effective than androsterone derived from urine or cholesterin. Hoppe-Seyler Z Physiol Chem. 1935:233:81–2.
- 19.
- Butenandt A HG. Umwandlung des Dehydroandrosterons in Androstendiol und Testosteron; ein Weg zur Darstellung des Testosterons aus Cholesterin. Hoppe-Seyler Z Physiol Chem. 1935:237:89–98.
- 20.
- Ruzicka L WA. Synthetische Darstellung des Testikelhormons Testosteron (Androsten 3-on-17-ol). Helv Chim Acta. 1935:18:1264–75.
- 21.
- Doisy EA TS, Veler CD The Crystals of the Follicular Ovarian Hormone. Proceedings of the Society for Experimental Biology and Medicine. 1930(27(5):417-419).
- 22.
- Maccorquodale DW, Thayer SA, Doisy EA. The Crystalline Ovarian Follicular Hormone. Proceedings of the Society for Experimental Biology and Medicine. 1935;32:1182 -
- 23.
- Smith PE. Ablation and transplantation of the hypophysis in the rat. Anat Rec. 1926;32(221):15-6.
- 24.
- Zondek B. Uber die hormone des hypophysenvorderlappes. I. Wachstums Hormon, follikelreifungshormon (Prolan A), Luteinisierungshormon (Prolan B) Stoffwechselhormon. Klin Wochschr. 1930(9(6):245–8) .
- 25.
- H. L. Fevold FLH, and S. L. Leonard. THE GONAD STIMULATING AND THE LUTEINIZING HORMONES OF THE ANTERIOR LOBE OF THE HYPOPHESI. The American Journal of Physiology. 1931:97.2.291.
- 26.
- Harris GW. The Induction of Ovulation in the Rabbit, by Electrical Stimulation of the Hypothalamo-hypophysial Mechanism. Proceedings of The Royal Society B: Biological Sciences. 1937;122:374-94.
- 27.
- Schally AV, Arimura A, Kastin AJ, Matsuo H, Baba Y, Redding TW, et al. Gonadotropin-releasing hormone: one polypeptide regulates secretion of luteinizing and follicle-stimulating hormones. Science. 1971;173(4001):1036-8. [PubMed: 4938639]
- 28.
- Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science. 1978;202(4368):631-3. [PubMed: 100883]
- 29.
- Burgus R, Butcher M, Ling N, Monahan M, Rivier J, Fellows R, et al. [Molecular structure of the hypothalamic factor (LRF) of ovine origin monitoring the secretion of pituitary gonadotropic hormone of luteinization (LH)]. C R Acad Hebd Seances Acad Sci D. 1971;273(18):1611-3. [PubMed: 5002981]
- 30.
- Plant TM. Recognition that sustained pituitary gonadotropin secretion requires pulsatile GnRH stimulation: a Pittsburgh Saga. F S Rep. 2023;4(2 Suppl):3-7. [PMC free article: PMC10201287] [PubMed: 37223769]
- 31.
- Plant TM, Steiner RA. The fifty years following the discovery of gonadotropin-releasing hormone. J Neuroendocrinol. 2022;34(5):e13141. [PubMed: 35726373]
- 32.
- Kaplan SL, Grumbach MM. The ontogenesis of human foetal hormones. II. Luteinizing hormone (LH) and follicle stimulating hormone (FSH). Acta Endocrinol (Copenh). 1976;81(4):808-29. [PubMed: 946570]
- 33.
- Winter JS, Faiman C, Hobson WC, Prasad AV, Reyes FI. Pituitary-gonadal relations in infancy. I. Patterns of serum gonadotropin concentrations from birth to four years of age in man and chimpanzee. J Clin Endocrinol Metab. 1975;40(4):545-51. [PubMed: 1127071]
- 34.
- Casoni F, Malone SA, Belle M, Luzzati F, Collier F, Allet C, et al. Development of the neurons controlling fertility in humans: new insights from 3D imaging and transparent fetal brains. Development. 2016;143(21):3969-81. [PubMed: 27803058]
- 35.
- Taroc EZM, Naik AS, Lin JM, Peterson NB, Keefe DL, Jr., Genis E, et al. Gli3 Regulates Vomeronasal Neurogenesis, Olfactory Ensheathing Cell Formation, and GnRH-1 Neuronal Migration. J Neurosci. 2020;40(2):311-26. [PMC free article: PMC6948949] [PubMed: 31767679]
- 36.
- Duittoz AH, Forni PE, Giacobini P, Golan M, Mollard P, Negron AL, et al. Development of the gonadotropin-releasing hormone system. J Neuroendocrinol. 2022;34(5):e13087. [PMC free article: PMC9286803] [PubMed: 35067985]
- 37.
- Fueshko SM, Key S, Wray S. GABA inhibits migration of luteinizing hormone-releasing hormone neurons in embryonic olfactory explants. J Neurosci. 1998;18(7):2560-9. [PMC free article: PMC6793115] [PubMed: 9502815]
- 38.
- Lund C, Pulli K, Yellapragada V, Giacobini P, Lundin K, Vuoristo S, et al. Development of Gonadotropin-Releasing Hormone-Secreting Neurons from Human Pluripotent Stem Cells. Stem Cell Reports. 2016;7(2):149-57. [PMC free article: PMC4982984] [PubMed: 27426041]
- 39.
- Yellapragada V, Eskici N, Wang Y, Madhusudan S, Vaaralahti K, Tuuri T, et al. FGF8-FGFR1 signaling regulates human GnRH neuron differentiation in a time- and dose-dependent manner. Dis Model Mech. 2022;15(8). [PMC free article: PMC9403748] [PubMed: 35833364]
- 40.
- Wang Y, Madhusudan S, Cotellessa L, Kvist J, Eskici N, Yellapragada V, et al. Deciphering the Transcriptional Landscape of Human Pluripotent Stem Cell-Derived GnRH Neurons: The Role of Wnt Signaling in Patterning the Neural Fate. Stem Cells. 2022;40(12):1107-21. [PMC free article: PMC9806769] [PubMed: 36153707]
- 41.
- Seeburg PH, Adelman JP. Characterization of cDNA for precursor of human luteinizing hormone releasing hormone. Nature. 1984;311(5987):666-8. [PubMed: 6090951]
- 42.
- Hagen C, McNeilly AS. The gonadotrophins and their subunits in foetal pituitary glands and circulation. J Steroid Biochem. 1977;8(5):537-44. [PubMed: 599925]
- 43.
- Clements JA, Reyes FI, Winter JS, Faiman C. Studies on human sexual development. III. Fetal pituitary and serum, and amniotic fluid concentrations of LH, CG, and FSH. J Clin Endocrinol Metab. 1976;42(1):9-19. [PubMed: 1249196]
- 44.
- Guimiot F, Chevrier L, Dreux S, Chevenne D, Caraty A, Delezoide AL, et al. Negative fetal FSH/LH regulation in late pregnancy is associated with declined kisspeptin/KISS1R expression in the tuberal hypothalamus. J Clin Endocrinol Metab. 2012;97(12):E2221-9. [PubMed: 23015653]
- 45.
- Kaplan SL, Grumbach MM. Pituitary and placental gonadotrophins and sex steroids in the human and sub-human primate fetus. Clin Endocrinol Metab. 1978;7(3):487-511. [PubMed: 215355]
- 46.
- Althumairy D, Zhang X, Baez N, Barisas G, Roess DA, Bousfield GR, et al. Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors. Diseases. 2020;8(3). [PMC free article: PMC7565105] [PubMed: 32942611]
- 47.
- Andersson AM, Skakkebaek NE. Serum inhibin B levels during male childhood and puberty. Mol Cell Endocrinol. 2001;180(1-2):103-7. [PubMed: 11451578]
- 48.
- Vale W, Rivier J, Vaughan J, McClintock R, Corrigan A, Woo W, et al. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 1986;321(6072):776-9. [PubMed: 3012369]
- 49.
- Wijayarathna R, de Kretser DM. Activins in reproductive biology and beyond. Hum Reprod Update. 2016;22(3):342-57. [PubMed: 26884470]
- 50.
- Abbara A, Koysombat K, Phylactou M, Eng PC, Clarke S, Comninos AN, et al. Insulin-like peptide 3 (INSL3) in congenital hypogonadotrophic hypogonadism (CHH) in boys with delayed puberty and adult men. Front Endocrinol (Lausanne). 2022;13:1076984. [PMC free article: PMC9745113] [PubMed: 36523592]
- 51.
- Crowley WF, Jr., McArthur JW. Simulation of the normal menstrual cycle in Kallman's syndrome by pulsatile administration of luteinizing hormone-releasing hormone (LHRH). J Clin Endocrinol Metab. 1980;51(1):173-5. [PubMed: 6247361]
- 52.
- Wildt L, Marshall G, Knobil E. Experimental induction of puberty in the infantile female rhesus monkey. Science. 1980;207(4437):1373-5. [PubMed: 6986658]
- 53.
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr., Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349(17):1614-27. [PubMed: 14573733]
- 54.
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100(19):10972-6. [PMC free article: PMC196911] [PubMed: 12944565]
- 55.
- Plant TM. The neurobiological mechanism underlying hypothalamic GnRH pulse generation: the role of kisspeptin neurons in the arcuate nucleus. F1000Res. 2019;8. [PMC free article: PMC6600864] [PubMed: 31297186]
- 56.
- Herbison AE. The Gonadotropin-Releasing Hormone Pulse Generator. Endocrinology. 2018;159(11):3723-36. [PubMed: 30272161]
- 57.
- Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology. 2010;151(1):301-11. [PMC free article: PMC2803147] [PubMed: 19880810]
- 58.
- Nagae M, Uenoyama Y, Okamoto S, Tsuchida H, Ikegami K, Goto T, et al. Direct evidence that KNDy neurons maintain gonadotropin pulses and folliculogenesis as the GnRH pulse generator. Proc Natl Acad Sci U S A. 2021;118(5). [PMC free article: PMC7865162] [PubMed: 33500349]
- 59.
- Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. 2013;368(26):2467-75. [PMC free article: PMC3808195] [PubMed: 23738509]
- 60.
- Busch AS, Hagen CP, Almstrup K, Juul A. Circulating MKRN3 Levels Decline During Puberty in Healthy Boys. J Clin Endocrinol Metab. 2016;101(6):2588-93. [PubMed: 27057785]
- 61.
- Hagen CP, Sorensen K, Mieritz MG, Johannsen TH, Almstrup K, Juul A. Circulating MKRN3 levels decline prior to pubertal onset and through puberty: a longitudinal study of healthy girls. J Clin Endocrinol Metab. 2015;100(5):1920-6. [PubMed: 25695892]
- 62.
- Varimo T, Dunkel L, Vaaralahti K, Miettinen PJ, Hero M, Raivio T. Circulating makorin ring finger protein 3 levels decline in boys before the clinical onset of puberty. Eur J Endocrinol. 2016;174(6):785-90. [PubMed: 27025240]
- 63.
- Palumbo S, Cirillo G, Aiello F, Papparella A, Miraglia Del Giudice E, Grandone A. MKRN3 role in regulating pubertal onset: the state of art of functional studies. Front Endocrinol (Lausanne). 2022;13:991322. [PMC free article: PMC9523110] [PubMed: 36187104]
- 64.
- Li C, Lu W, Yang L, Li Z, Zhou X, Guo R, et al. MKRN3 regulates the epigenetic switch of mammalian puberty via ubiquitination of MBD3. Natl Sci Rev. 2020;7(3):671-85. [PMC free article: PMC8288866] [PubMed: 34692086]
- 65.
- Abreu AP, Toro CA, Song YB, Navarro VM, Bosch MA, Eren A, et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. J Clin Invest. 2020;130(8):4486-500. [PMC free article: PMC7410046] [PubMed: 32407292]
- 66.
- Heras V, Sangiao-Alvarellos S, Manfredi-Lozano M, Sanchez-Tapia MJ, Ruiz-Pino F, Roa J, et al. Hypothalamic miR-30 regulates puberty onset via repression of the puberty-suppressing factor, Mkrn3. PLoS Biol. 2019;17(11):e3000532. [PMC free article: PMC6863565] [PubMed: 31697675]
- 67.
- Naule L, Mancini A, Pereira SA, Gassaway BM, Lydeard JR, Magnotto JC, et al. MKRN3 inhibits puberty onset via interaction with IGF2BP1 and regulation of hypothalamic plasticity. JCI Insight. 2023;8(8). [PMC free article: PMC10243807] [PubMed: 37092553]
- 68.
- Lomniczi A, Loche A, Castellano JM, Ronnekleiv OK, Bosch M, Kaidar G, et al. Epigenetic control of female puberty. Nat Neurosci. 2013;16(3):281-9. [PMC free article: PMC3581714] [PubMed: 23354331]
- 69.
- Lomniczi A, Ojeda SR. The Emerging Role of Epigenetics in the Regulation of Female Puberty. Endocr Dev. 2016;29:1-16. [PMC free article: PMC4955615] [PubMed: 26680569]
- 70.
- Toro CA, Wright H, Aylwin CF, Ojeda SR, Lomniczi A. Trithorax dependent changes in chromatin landscape at enhancer and promoter regions drive female puberty. Nat Commun. 2018;9(1):57. [PMC free article: PMC5754362] [PubMed: 29302059]
- 71.
- Lomniczi A, Wright H, Castellano JM, Matagne V, Toro CA, Ramaswamy S, et al. Epigenetic regulation of puberty via Zinc finger protein-mediated transcriptional repression. Nat Commun. 2015;6:10195. [PMC free article: PMC4703871] [PubMed: 26671628]
- 72.
- Barker-Gibb ML, Sahu A, Pohl CR, Plant TM. Elevating circulating leptin in prepubertal male rhesus monkeys (Macaca mulatta) does not elicit precocious gonadotropin-releasing hormone release, assessed indirectly. J Clin Endocrinol Metab. 2002;87(11):4976-83. [PubMed: 12414861]
- 73.
- Vazquez MJ, Toro CA, Castellano JM, Ruiz-Pino F, Roa J, Beiroa D, et al. SIRT1 mediates obesity- and nutrient-dependent perturbation of pubertal timing by epigenetically controlling Kiss1 expression. Nat Commun. 2018;9(1):4194. [PMC free article: PMC6179991] [PubMed: 30305620]
- 74.
- Manfredi-Lozano M, Roa J, Tena-Sempere M. Connecting metabolism and gonadal function: Novel central neuropeptide pathways involved in the metabolic control of puberty and fertility. Front Neuroendocrinol. 2018;48:37-49. [PubMed: 28754629]
- 75.
- Chachlaki K, Messina A, Delli V, Leysen V, Maurnyi C, Huber C, et al. NOS1 mutations cause hypogonadotropic hypogonadism with sensory and cognitive deficits that can be reversed in infantile mice. Sci Transl Med. 2022;14(665):eabh2369. [PMC free article: PMC7613826] [PubMed: 36197968]
- 76.
- Constantin S, Reynolds D, Oh A, Pizano K, Wray S. Nitric oxide resets kisspeptin-excited GnRH neurons via PIP2 replenishment. Proc Natl Acad Sci U S A. 2021;118(1). [PMC free article: PMC7817197] [PubMed: 33443156]
- 77.
- Witchel SF, Plant TM. Neurobiology of puberty and its disorders. Handb Clin Neurol. 2021;181:463-96. [PubMed: 34238478]
- 78.
- Goodman RL, Herbison AE, Lehman MN, Navarro VM. Neuroendocrine control of gonadotropin-releasing hormone: Pulsatile and surge modes of secretion. J Neuroendocrinol. 2022;34(5):e13094. [PMC free article: PMC9948945] [PubMed: 35107859]
- 79.
- Terasawa E. The mechanism underlying the pubertal increase in pulsatile GnRH release in primates. J Neuroendocrinol. 2022;34(5):e13119. [PMC free article: PMC9232993] [PubMed: 35491543]
- 80.
- Avendano MS, Vazquez MJ, Tena-Sempere M. Disentangling puberty: novel neuroendocrine pathways and mechanisms for the control of mammalian puberty. Hum Reprod Update. 2017;23(6):737-63. [PubMed: 28961976]
- 81.
- Aylwin CF, Lomniczi A. Sirtuin (SIRT)-1: At the crossroads of puberty and metabolism. Curr Opin Endocr Metab Res. 2020;14:65-72. [PMC free article: PMC7467505] [PubMed: 32905232]
- 82.
- Lhomme T, Clasadonte J, Imbernon M, Fernandois D, Sauve F, Caron E, et al. Tanycytic networks mediate energy balance by feeding lactate to glucose-insensitive POMC neurons. J Clin Invest. 2021;131(18). [PMC free article: PMC8439611] [PubMed: 34324439]
- 83.
- Forest MG, Cathiard AM. Pattern of plasma testosterone and delta4-androstenedione in normal newborns: Evidence for testicular activity at birth. J Clin Endocrinol Metab. 1975;41(5):977-80. [PubMed: 1184729]
- 84.
- Forest MG, Sizonenko PC, Cathiard AM, Bertrand J. Hypophyso-gonadal function in humans during the first year of life. 1. Evidence for testicular activity in early infancy. J Clin Invest. 1974;53(3):819-28. [PMC free article: PMC333063] [PubMed: 4812441]
- 85.
- Grumbach MM. The neuroendocrinology of human puberty revisited. Horm Res. 2002;57 Suppl 2:2-14. [PubMed: 12065920]
- 86.
- Bizzarri C, Cappa M. Ontogeny of Hypothalamus-Pituitary Gonadal Axis and Minipuberty: An Ongoing Debate? Front Endocrinol (Lausanne). 2020;11:187. [PMC free article: PMC7154076] [PubMed: 32318025]
- 87.
- Cortes D, Muller J, Skakkebaek NE. Proliferation of Sertoli cells during development of the human testis assessed by stereological methods. Int J Androl. 1987;10(4):589-96. [PubMed: 3654012]
- 88.
- Muller J, Skakkebaek NE. Fluctuations in the number of germ cells during the late foetal and early postnatal periods in boys. Acta Endocrinol (Copenh). 1984;105(2):271-4. [PubMed: 6421044]
- 89.
- Kuijper EA, van Kooten J, Verbeke JI, van Rooijen M, Lambalk CB. Ultrasonographically measured testicular volumes in 0- to 6-year-old boys. Hum Reprod. 2008;23(4):792-6. [PubMed: 18281246]
- 90.
- Chemes HE, Rey RA, Nistal M, Regadera J, Musse M, Gonzalez-Peramato P, et al. Physiological androgen insensitivity of the fetal, neonatal, and early infantile testis is explained by the ontogeny of the androgen receptor expression in Sertoli cells. J Clin Endocrinol Metab. 2008;93(11):4408-12. [PubMed: 18713818]
- 91.
- Boukari K, Meduri G, Brailly-Tabard S, Guibourdenche J, Ciampi ML, Massin N, et al. Lack of androgen receptor expression in Sertoli cells accounts for the absence of anti-Mullerian hormone repression during early human testis development. J Clin Endocrinol Metab. 2009;94(5):1818-25. [PMC free article: PMC2699416] [PubMed: 19276236]
- 92.
- Codesal J, Regadera J, Nistal M, Regadera-Sejas J, Paniagua R. Involution of human fetal Leydig cells. An immunohistochemical, ultrastructural and quantitative study. J Anat. 1990;172:103-14. [PMC free article: PMC1257207] [PubMed: 2272896]
- 93.
- Busch AS, Ljubicic ML, Upners EN, Fischer MB, Raket LL, Frederiksen H, et al. Dynamic Changes of Reproductive Hormones in Male Minipuberty: Temporal Dissociation of Leydig and Sertoli Cell Activity. J Clin Endocrinol Metab. 2022;107(6):1560-8. [PubMed: 35225342]
- 94.
- Kuiri-Hanninen T, Seuri R, Tyrvainen E, Turpeinen U, Hamalainen E, Stenman UH, et al. Increased activity of the hypothalamic-pituitary-testicular axis in infancy results in increased androgen action in premature boys. J Clin Endocrinol Metab. 2011;96(1):98-105. [PubMed: 20881260]
- 95.
- Santos MC, Limao S, Ferreira P. Exacerbated mini-puberty of infancy in an ex-extreme preterm girl. BMJ Case Rep. 2020;13(9). [PMC free article: PMC7470511] [PubMed: 32878858]
- 96.
- Kuiri-Hanninen T, Kallio S, Seuri R, Tyrvainen E, Liakka A, Tapanainen J, et al. Postnatal developmental changes in the pituitary-ovarian axis in preterm and term infant girls. J Clin Endocrinol Metab. 2011;96(11):3432-9. [PubMed: 21900380]
- 97.
- Hagen CP, Aksglaede L, Sorensen K, Main KM, Boas M, Cleemann L, et al. Serum levels of anti-Mullerian hormone as a marker of ovarian function in 926 healthy females from birth to adulthood and in 172 Turner syndrome patients. J Clin Endocrinol Metab. 2010;95(11):5003-10. [PubMed: 20719830]
- 98.
- Ljubicic ML, Busch AS, Upners EN, Fischer MB, Petersen JH, Raket LL, et al. A Biphasic Pattern of Reproductive Hormones in Healthy Female Infants: The COPENHAGEN Minipuberty Study. J Clin Endocrinol Metab. 2022;107(9):2598-605. [PubMed: 35704034]
- 99.
- Johannsen TH, Main KM, Ljubicic ML, Jensen TK, Andersen HR, Andersen MS, et al. Sex Differences in Reproductive Hormones During Mini-Puberty in Infants With Normal and Disordered Sex Development. J Clin Endocrinol Metab. 2018;103(8):3028-37. [PubMed: 29917083]
- 100.
- Swee DS, Quinton R. Congenital Hypogonadotrophic Hypogonadism: Minipuberty and the Case for Neonatal Diagnosis. Front Endocrinol (Lausanne). 2019;10:97. [PMC free article: PMC6393341] [PubMed: 30846970]
- 101.
- Jespersen K, Ljubicic ML, Johannsen TH, Christiansen P, Skakkebaek NE, Juul A. Distinguishing between hidden testes and anorchia: the role of endocrine evaluation in infancy and childhood. Eur J Endocrinol. 2020;183(1):107-17. [PubMed: 32422605]
- 102.
- Koo MM, Rohan TE. Accuracy of short-term recall of age at menarche. Ann Hum Biol. 1997;24(1):61-4. [PubMed: 9022907]
- 103.
- Dorn LD, Sontag-Padilla LM, Pabst S, Tissot A, Susman EJ. Longitudinal reliability of self-reported age at menarche in adolescent girls: variability across time and setting. Dev Psychol. 2013;49(6):1187-93. [PMC free article: PMC3514629] [PubMed: 22889389]
- 104.
- Busch AS, Hollis B, Day FR, Sorensen K, Aksglaede L, Perry JRB, et al. Voice break in boys-temporal relations with other pubertal milestones and likely causal effects of BMI. Hum Reprod. 2019;34(8):1514-22. [PMC free article: PMC6688887] [PubMed: 31348498]
- 105.
- Lewis ME, Shapland F, Watts R. The influence of chronic conditions and the environment on pubertal development. An example from medieval England. Int J Paleopathol. 2016;12:1-10. [PubMed: 29539515]
- 106.
- Arthur NA, Gowland RL, Redfern RC. Coming of age in Roman Britain: Osteological evidence for pubertal timing. Am J Phys Anthropol. 2016;159(4):698-713. [PubMed: 26739513]
- 107.
- Liu W, Yan X, Li C, Shu Q, Chen M, Cai L, et al. A secular trend in age at menarche in Yunnan Province, China: a multiethnic population study of 1,275,000 women. BMC Public Health. 2021;21(1):1890. [PMC free article: PMC8524999] [PubMed: 34666747]
- 108.
- Wu T, Pauline M, Germaine BM. Ethnic differences in the Presence of Secondary Sex Characteristics and Menarche Among US Girls:The Third National Health and Nutrition Examination Survey,1988-1994. Pediatrics. 2002;110(4). [PubMed: 12359790]
- 109.
- Herman-Giddens ME, Slora EJ, Wasserman RC, Bourdony CJ, Bhapkar MV, Koch GG, et al. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics. 1997;99(4):505-12. [PubMed: 9093289]
- 110.
- Aksglaede L, Sorensen K, Petersen JH, Skakkebaek NE, Juul A. Recent decline in age at breast development: the Copenhagen Puberty Study. Pediatrics. 2009;123(5):e932-9. [PubMed: 19403485]
- 111.
- Biro FM, Pajak A, Wolff MS, Pinney SM, Windham GC, Galvez MP, et al. Age of Menarche in a Longitudinal US Cohort. J Pediatr Adolesc Gynecol. 2018;31(4):339-45. [PMC free article: PMC6121217] [PubMed: 29758276]
- 112.
- Marti-Henneberg C, Vizmanos B. The duration of puberty in girls is related to the timing of its onset. J Pediatr. 1997;131(4):618-21. [PubMed: 9386670]
- 113.
- Pantsiotou S, Papadimitriou A, Douros K, Priftis K, Nicolaidou P, Fretzayas A. Maturational tempo differences in relation to the timing of the onset of puberty in girls. Acta Paediatr. 2008;97(2):217-20. [PubMed: 18177444]
- 114.
- Eckert-Lind C, Busch AS, Petersen JH, Biro FM, Butler G, Brauner EV, et al. Worldwide Secular Trends in Age at Pubertal Onset Assessed by Breast Development Among Girls: A Systematic Review and Meta-analysis. JAMA Pediatr. 2020;174(4):e195881. [PMC free article: PMC7042934] [PubMed: 32040143]
- 115.
- Sorensen K, Mouritsen A, Aksglaede L, Hagen C, Mogensen S, Juul A. Recent secular Trends in Pubertal Timing: Implications for Evaluation and Diagnosis of Precocious Puberty. Hormone Research in Paediatrics. 2012;77:137-45. [PubMed: 22508036]
- 116.
- Predieri B, Iughetti L, Bernasconi S, Street ME. Endocrine Disrupting Chemicals' Effects in Children: What We Know and What We Need to Learn? Int J Mol Sci. 2022;23(19). [PMC free article: PMC9570268] [PubMed: 36233201]
- 117.
- Krstevska-Konstantinova M, Charlier C, Craen M, Du Caju M, Heinrichs C, de Beaufort C, et al. Sexual precocity after immigration from developing countries to Belgium: evidence of previous exposure to organochlorine pesticides. Hum Reprod. 2001;16(5):1020-6. [PubMed: 11331654]
- 118.
- Lopez-Rodriguez D, Franssen D, Heger S, Parent AS. Endocrine-disrupting chemicals and their effects on puberty. Best Pract Res Clin Endocrinol Metab. 2021;35(5):101579. [PubMed: 34563408]
- 119.
- Fudvoye J, Lopez-Rodriguez D, Franssen D, Parent AS. Endocrine disrupters and possible contribution to pubertal changes. Best Pract Res Clin Endocrinol Metab. 2019;33(3):101300. [PubMed: 31401055]
- 120.
- Kaplowitz PB, Slora EJ, Wasserman RC, Pedlow SE, Herman-Giddens ME. Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics. 2001;108(2):347-53. [PubMed: 11483799]
- 121.
- Kaplowitz PB. Link between body fat and the timing of puberty. Pediatrics. 2008;121 Suppl 3:S208-17. [PubMed: 18245513]
- 122.
- Brix N, Ernst A, Lauridsen LLB, Parner ET, Arah OA, Olsen J, et al. Childhood overweight and obesity and timing of puberty in boys and girls: cohort and sibling-matched analyses. Int J Epidemiol. 2020;49(3):834-44. [PMC free article: PMC7394964] [PubMed: 32372073]
- 123.
- Ahmed ML, Ong KK, Dunger DB. Childhood obesity and the timing of puberty. Trends Endocrinol Metab. 2009;20(5):237-42. [PubMed: 19541497]
- 124.
- Day FR, Perry JR, Ong KK. Genetic Regulation of Puberty Timing in Humans. Neuroendocrinology. 2015;102(4):247-55. [PMC free article: PMC6309186] [PubMed: 25968239]
- 125.
- Shalitin S, Gat-Yablonski G. Associations of Obesity with Linear Growth and Puberty. Horm Res Paediatr. 2022;95(2):120-36. [PubMed: 34130293]
- 126.
- Reid RL, Ling N, Yen SS. Gonadotropin-releasing activity of alpha-melanocyte-stimulating hormone in normal subjects and in subjects with hypothalamic-pituitary dysfunction. J Clin Endocrinol Metab. 1984;58(5):773-7. [PubMed: 6423658]
- 127.
- Israel DD, Sheffer-Babila S, de Luca C, Jo YH, Liu SM, Xia Q, et al. Effects of leptin and melanocortin signaling interactions on pubertal development and reproduction. Endocrinology. 2012;153(5):2408-19. [PMC free article: PMC3381095] [PubMed: 22408174]
- 128.
- Morris DH, Jones ME, Schoemaker MJ, Ashworth A, Swerdlow AJ. Familial concordance for age at menarche: analyses from the Breakthrough Generations Study. Paediatr Perinat Epidemiol. 2011;25(3):306-11. [PubMed: 21470270]
- 129.
- Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol Genet Metab. 2003;80(1-2):1-10. [PubMed: 14567953]
- 130.
- Busch AS, Hagen CP, Juul A. Heritability of pubertal timing: detailed evaluation of specific milestones in healthy boys and girls. Eur J Endocrinol. 2020;183(1):13-20. [PubMed: 32348954]
- 131.
- Wohlfahrt-Veje C, Mouritsen A, Hagen CP, Tinggaard J, Mieritz MG, Boas M, et al. Pubertal Onset in Boys and Girls Is Influenced by Pubertal Timing of Both Parents. J Clin Endocrinol Metab. 2016;101(7):2667-74. [PubMed: 27014950]
- 132.
- Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. 2017;49(6):834-41. [PMC free article: PMC5841952] [PubMed: 28436984]
- 133.
- Sarnowski C, Cousminer DL, Franceschini N, Raffield LM, Jia G, Fernandez-Rhodes L, et al. Large trans-ethnic meta-analysis identifies AKR1C4 as a novel gene associated with age at menarche. Hum Reprod. 2021;36(7):1999-2010. [PMC free article: PMC8213450] [PubMed: 34021356]
- 134.
- Klein KO. Precocious puberty: who has it? Who should be treated? J Clin Endocrinol Metab. 1999;84(2):411-4. [PubMed: 10022393]
- 135.
- Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. 2003;24(5):668-93. [PubMed: 14570750]
- 136.
- Kaplowitz PB. Delayed puberty. Pediatr Rev. 2010;31(5):189-95. [PubMed: 20435710]
- 137.
- Bangalore Krishna K, Fuqua JS, Rogol AD, Klein KO, Popovic J, Houk CP, et al. Use of Gonadotropin-Releasing Hormone Analogs in Children: Update by an International Consortium. Horm Res Paediatr. 2019;91(6):357-72. [PubMed: 31319416]
- 138.
- Goldberg M, D'Aloisio AA, O'Brien KM, Zhao S, Sandler DP. Pubertal timing and breast cancer risk in the Sister Study cohort. Breast Cancer Res. 2020;22(1):112. [PMC free article: PMC7590599] [PubMed: 33109223]
- 139.
- Mueller NT, Duncan BB, Barreto SM, Chor D, Bessel M, Aquino EML, et al. Earlier age at menarche is associated with higher diabetes risk and cardiometabolic disease risk factors in Brazilian adults: Brazilian Longitudinal Study of Adult Health (ELSA-Brasil). Cardiovasc Diabetol. 2014;13. [PMC free article: PMC3899384] [PubMed: 24438044]
- 140.
- Partsch CJ, Heger S, Sippell WG. Management and outcome of central precocious puberty. Clin Endocrinol (Oxf). 2002;56(2):129-48. [PubMed: 11874402]
- 141.
- Soriano-Guillen L, Corripio R, Labarta JI, Canete R, Castro-Feijoo L, Espino R, et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metab. 2010;95(9):4305-13. [PubMed: 20554707]
- 142.
- Kim SH, Huh K, Won S, Lee KW, Park MJ. A Significant Increase in the Incidence of Central Precocious Puberty among Korean Girls from 2004 to 2010. Plos One. 2015;10(11). [PMC free article: PMC4634943] [PubMed: 26539988]
- 143.
- Brandberg G, Raininko R, Eeg-Olofsson O. Hypothalamic hamartoma with gelastic seizures in Swedish children and adolescents. Eur J Paediatr Neurol. 2004;8(1):35-44. [PubMed: 15023373]
- 144.
- Cukier P, Castro LH, Banaskiwitz N, Teles LR, Ferreira LR, Adda CC, et al. The benign spectrum of hypothalamic hamartomas: infrequent epilepsy and normal cognition in patients presenting with central precocious puberty. Seizure. 2013;22(1):28-32. [PubMed: 23092686]
- 145.
- Harrison VS, Oatman O, Kerrigan JF. Hypothalamic hamartoma with epilepsy: Review of endocrine comorbidity. Epilepsia. 2017;58 Suppl 2(Suppl 2):50-9. [PMC free article: PMC5533614] [PubMed: 28591479]
- 146.
- Yang Y, Shen F, Jing XP, Zhang N, Xu SY, Li DD, et al. Case Report: Whole-Exome Sequencing of Hypothalamic Hamartoma From an Infant With Pallister-Hall Syndrome Revealed Novel de novo Mutation in the GLI3. Front Surg. 2021;8:734757. [PMC free article: PMC8493334] [PubMed: 34631784]
- 147.
- Fujita A, Higashijima T, Shirozu H, Masuda H, Sonoda M, Tohyama J, et al. Pathogenic variants of DYNC2H1, KIAA0556, and PTPN11 associated with hypothalamic hamartoma. Neurology. 2019;93(3):e237-e51. [PubMed: 31197031]
- 148.
- Hildebrand MS, Griffin NG, Damiano JA, Cops EJ, Burgess R, Ozturk E, et al. Mutations of the Sonic Hedgehog Pathway Underlie Hypothalamic Hamartoma with Gelastic Epilepsy. Am J Hum Genet. 2016;99(2):423-9. [PMC free article: PMC4974069] [PubMed: 27453577]
- 149.
- Chan YM, Fenoglio-Simeone KA, Paraschos S, Muhammad L, Troester MM, Ng YT, et al. Central precocious puberty due to hypothalamic hamartomas correlates with anatomic features but not with expression of GnRH, TGFalpha, or KISS1. Horm Res Paediatr. 2010;73(5):312-9. [PMC free article: PMC2868525] [PubMed: 20389100]
- 150.
- Harrison VS, Oatman O, Kerrigan JF. Hypothalamic hamartoma with epilepsy: Review of endocrine comorbidity. Epilepsia. 2017;58:50-9. [PMC free article: PMC5533614] [PubMed: 28591479]
- 151.
- Bourdillon P, Ferrand-Sorbet S, Apra C, Chipaux M, Raffo E, Rosenberg S, et al. Surgical treatment of hypothalamic hamartomas. Neurosurg Rev. 2021;44(2):753-62. [PubMed: 32318922]
- 152.
- Chalumeau M, Chemaitilly W, Trivin C, Adan L, Breart G, Brauner R. Central precocious puberty in girls: an evidence-based diagnosis tree to predict central nervous system abnormalities. Pediatrics. 2002;109(1):61-7. [PubMed: 11773542]
- 153.
- Stephen MD, Zage PE, Waguespack SG. Gonadotropin-dependent precocious puberty: neoplastic causes and endocrine considerations. Int J Pediatr Endocrinol. 2011;2011(1):184502. [PMC free article: PMC3212801] [PubMed: 21603196]
- 154.
- Pinheiro SL, Maciel J, Cavaco D, Figueiredo AA, Damasio IL, Donato S, et al. Precocious and accelerated puberty in children with neurofibromatosis type 1: results from a close follow-up of a cohort of 45 patients. Hormones. 2023;22(1):79-85. [PubMed: 36269545]
- 155.
- Almutlaq N, O'Neil J, Fuqua JS. Central precocious puberty in spina bifida children: Guidelines for the care of people with spina bifida. J Pediatr Rehabil Med. 2020;13(4):557-63. [PMC free article: PMC7838954] [PubMed: 33325409]
- 156.
- Cerbone M, Guemes M, Wade A, Improda N, Dattani M. Endocrine morbidity in midline brain defects: Differences between septo-optic dysplasia and related disorders. EClinicalMedicine. 2020;19:100224. [PMC free article: PMC7046495] [PubMed: 32140665]
- 157.
- Oatman OJ, McClellan DR, Olson ML, Garcia-Filion P. Endocrine and pubertal disturbances in optic nerve hypoplasia, from infancy to adolescence. Int J Pediatr Endocrinol. 2015;2015(1):8. [PMC free article: PMC4397734] [PubMed: 25878671]
- 158.
- Siegel DA, King JB, Lupo PJ, Durbin EB, Tai E, Mills K, et al. Counts, incidence rates, and trends of pediatric cancer in the United States, 2003-2019. J Natl Cancer Inst. 2023. [PMC free article: PMC11018256] [PubMed: 37433078]
- 159.
- Chemaitilly W, Merchant TE, Li Z, Barnes N, Armstrong GT, Ness KK, et al. Central precocious puberty following the diagnosis and treatment of paediatric cancer and central nervous system tumours: presentation and long-term outcomes. Clin Endocrinol (Oxf). 2016;84(3):361-71. [PMC free article: PMC4755813] [PubMed: 26464129]
- 160.
- De Sanctis V, Soliman AT, Elsedfy H, Soliman NA, Elalaily R, El Kholy M. Precocious Puberty Following Traumatic Brain Injury in Early Childhood: A Review of the Literature. Pediatr Endocrinol Rev. 2015;13(1):458-64. [PubMed: 26540762]
- 161.
- Dassa Y, Crosnier H, Chevignard M, Viaud M, Personnier C, Flechtner I, et al. Pituitary deficiency and precocious puberty after childhood severe traumatic brain injury: a long-term follow-up prospective study. Eur J Endocrinol. 2019;180(5):281-90. [PubMed: 30884465]
- 162.
- Partsch CJ, Sippell WG. Pathogenesis and epidemiology of precocious puberty. Effects of exogenous oestrogens. Hum Reprod Update. 2001;7(3):292-302. [PubMed: 11392376]
- 163.
- Stecchini MF, Braid Z, More CB, Aragon DC, Castro M, Moreira AC, et al. Gonadotropin-dependent pubertal disorders are common in patients with virilizing adrenocortical tumors in childhood. Endocr Connect. 2019;8(5):579-89. [PMC free article: PMC6499918] [PubMed: 30959478]
- 164.
- Chen H, Mo CY, Zhong LY. Central precocious puberty secondary to peripheral precocious puberty due to a pineal germ cell tumor: a case and review of literature. BMC Endocr Disord. 2023;23(1):237. [PMC free article: PMC10601200] [PubMed: 37884982]
- 165.
- Maione L, Bouvattier C, Kaiser UB. Central precocious puberty: Recent advances in understanding the aetiology and in the clinical approach. Clin Endocrinol (Oxf). 2021;95(4):542-55. [PMC free article: PMC8586890] [PubMed: 33797780]
- 166.
- Moise-Silverman J, Silverman LA. A review of the genetics and epigenetics of central precocious puberty. Front Endocrinol (Lausanne). 2022;13:1029137. [PMC free article: PMC9757059] [PubMed: 36531492]
- 167.
- Valadares LP, Meireles CG, De Toledo IP, Santarem de Oliveira R, Goncalves de Castro LC, Abreu AP, et al. MKRN3 Mutations in Central Precocious Puberty: A Systematic Review and Meta-Analysis. J Endocr Soc. 2019;3(5):979-95. [PMC free article: PMC6483926] [PubMed: 31041429]
- 168.
- Magnotto JC, Mancini A, Bird K, Montenegro L, Tutunculer F, Pereira SA, et al. Novel MKRN3 Missense Mutations Associated With Central Precocious Puberty Reveal Distinct Effects on Ubiquitination. J Clin Endocrinol Metab. 2023;108(7):1646-56. [PMC free article: PMC10653150] [PubMed: 36916482]
- 169.
- Li M, Chen Y, Liao B, Tang J, Zhong J, Lan D. The role of kisspeptin and MKRN3 in the diagnosis of central precocious puberty in girls. Endocr Connect. 2021;10(9):1147-54. [PMC free article: PMC8494402] [PubMed: 34414898]
- 170.
- Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51(8):495-501. [PubMed: 24891339]
- 171.
- Gomes LG, Cunha-Silva M, Crespo RP, Ramos CO, Montenegro LR, Canton A, et al. DLK1 Is a Novel Link Between Reproduction and Metabolism. J Clin Endocrinol Metab. 2019;104(6):2112-20. [PubMed: 30462238]
- 172.
- Silveira LG, Noel SD, Silveira-Neto AP, Abreu AP, Brito VN, Santos MG, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. 2010;95(5):2276-80. [PMC free article: PMC2869552] [PubMed: 20237166]
- 173.
- Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. 2008;358(7):709-15. [PMC free article: PMC2859966] [PubMed: 18272894]
- 174.
- Bianco SD, Vandepas L, Correa-Medina M, Gereben B, Mukherjee A, Kuohung W, et al. KISS1R intracellular trafficking and degradation: effect of the Arg386Pro disease-associated mutation. Endocrinology. 2011;152(4):1616-26. [PMC free article: PMC3060635] [PubMed: 21285314]
- 175.
- Tinano FR, Canton APM, Montenegro LR, de Castro Leal A, Faria AG, Seraphim CE, et al. Clinical and Genetic Characterization of Familial Central Precocious Puberty. J Clin Endocrinol Metab. 2023;108(7):1758-67. [PubMed: 36611250]
- 176.
- Nicoara DM, Scutca AC, Mang N, Juganaru I, Munteanu AI, Vitan L, et al. Central precocious puberty in Prader-Willi syndrome: a narrative review. Front Endocrinol (Lausanne). 2023;14:1150323. [PMC free article: PMC10214499] [PubMed: 37251677]
- 177.
- Spielmann S, Partsch CJ, Gosch A, Pankau R. Treatment of central precocious puberty and early puberty with GnRH analog in girls with Williams-Beuren syndrome. J Pediatr Endocrinol Metab. 2015;28(11-12):1363-7. [PubMed: 26197460]
- 178.
- Patti G, Malerba F, Calevo MG, Schiavone M, Scaglione M, Casalini E, et al. Pubertal timing in children with Silver Russell syndrome compared to those born small for gestational age. Front Endocrinol (Lausanne). 2022;13:975511. [PMC free article: PMC9451521] [PubMed: 36093089]
- 179.
- Palmert MR, Malin HV, Boepple PA. Unsustained or slowly progressive puberty in young girls: initial presentation and long-term follow-up of 20 untreated patients. J Clin Endocrinol Metab. 1999;84(2):415-23. [PubMed: 10022394]
- 180.
- Cote DJ, Smith TR, Sandler CN, Gupta T, Bale TA, Bi WL, et al. Functional Gonadotroph Adenomas: Case Series and Report of Literature. Neurosurgery. 2016;79(6):823-31. [PMC free article: PMC4912468] [PubMed: 26692108]
- 181.
- Ntali G, Capatina C. Updating the Landscape for Functioning Gonadotroph Tumors. Medicina (Kaunas). 2022;58(8). [PMC free article: PMC9414558] [PubMed: 36013538]
- 182.
- Uhing A, Ahmed A, Salamat S, Chen M. A Rare Case of Precocious Puberty Secondary to an LH-secreting Pituitary Adenoma. JCEM Case Rep. 2023;1(3):luad055. [PMC free article: PMC10580444] [PubMed: 37908585]
- 183.
- Sisk-Hackworth L, Kelley ST, Thackray VG. Sex, puberty, and the gut microbiome. Reproduction. 2023;165(2):R61-R74. [PMC free article: PMC9847487] [PubMed: 36445259]
- 184.
- Calcaterra V, Rossi V, Massini G, Regalbuto C, Hruby C, Panelli S, et al. Precocious puberty and microbiota: The role of the sex hormone-gut microbiome axis. Front Endocrinol (Lausanne). 2022;13:1000919. [PMC free article: PMC9634744] [PubMed: 36339428]
- 185.
- Chaussain JL, Roger M, Couprie C, Lahlou N, Canlorbe P. Treatment of precocious puberty with a long-acting preparation of D-Trp6-LHRH. Horm Res. 1987;28(2-4):155-63. [PubMed: 2969859]
- 186.
- Lahlou N, Roger M, Chaussain JL, Feinstein MC, Sultan C, Toublanc JE, et al. Gonadotropin and alpha-subunit secretion during long term pituitary suppression by D-Trp6-luteinizing hormone-releasing hormone microcapsules as treatment of precocious puberty. J Clin Endocrinol Metab. 1987;65(5):946-53. [PubMed: 2959680]
- 187.
- Carel JC, Léger J. Clinical practice. Precocious puberty. N Engl J Med. 2008;358(22):2366-77. [PubMed: 18509122]
- 188.
- Chen M, Eugster EA. Central Precocious Puberty: Update on Diagnosis and Treatment. Pediatr Drugs. 2015;17(4):273-81. [PMC free article: PMC5870137] [PubMed: 25911294]
- 189.
- Johnson SR, Nolan RC, Grant MT, Price GJ, Siafarikas A, Bint L, et al. Sterile abscess formation associated with depot leuprorelin acetate therapy for central precocious puberty. J Paediatr Child Health. 2012;48(3):E136-9. [PubMed: 21564386]
- 190.
- Miller BS, Shukla AR. Sterile abscess formation in response to two separate branded long-acting gonadotropin-releasing hormone agonists. Clin Ther. 2010;32(10):1749-51. [PubMed: 21194598]
- 191.
- Pienkowski C, Tauber M. Gonadotropin-Releasing Hormone Agonist Treatment in Sexual Precocity. Endocr Dev. 2016;29:214-29. [PubMed: 26680581]
- 192.
- De Sanctis V, Soliman AT, Di Maio S, Soliman N, Elsedfy H. Long-term effects and significant Adverse Drug Reactions (ADRs) associated with the use of Gonadotropin-Releasing Hormone analogs (GnRHa) for central precocious puberty: a brief review of literature. Acta Biomed. 2019;90(3):345-59. [PMC free article: PMC7233750] [PubMed: 31580327]
- 193.
- van der Sluis IM, Boot AM, Krenning EP, Drop SL, de Muinck Keizer-Schrama SM. Longitudinal follow-up of bone density and body composition in children with precocious or early puberty before, during and after cessation of GnRH agonist therapy. J Clin Endocrinol Metab. 2002;87(2):506-12. [PubMed: 11836277]
- 194.
- Paterson WF, McNeill E, Young D, Donaldson MD. Auxological outcome and time to menarche following long-acting goserelin therapy in girls with central precocious or early puberty. Clin Endocrinol (Oxf). 2004;61(5):626-34. [PubMed: 15521967]
- 195.
- Traggiai C, Perucchin PP, Zerbini K, Gastaldi R, De Biasio P, Lorini R. Outcome after depot gonadotrophin-releasing hormone agonist treatment for central precocious puberty: effects on body mass index and final height. Eur J Endocrinol. 2005;153(3):463-4. [PubMed: 16131610]
- 196.
- Wolters B, Lass N, Reinehr T. Treatment with gonadotropin-releasing hormone analogues: different impact on body weight in normal-weight and overweight children. Horm Res Paediatr. 2012;78(5-6):304-11. [PubMed: 23257624]
- 197.
- Palmert MR, Mansfield MJ, Crowley WF, Crigler JF, Crawford JD, Boepple PA. Is obesity an outcome of gonadotropin-releasing hormone agonist administration? Analysis of growth and body composition in 110 patients with central precocious puberty. J Clin Endocrinol Metab. 1999;84(12):4480-8. [PubMed: 10599706]
- 198.
- Boot AM, De Muinck Keizer-Schrama S, Pols HA, Krenning EP, Drop SL. Bone mineral density and body composition before and during treatment with gonadotropin-releasing hormone agonist in children with central precocious and early puberty. J Clin Endocrinol Metab. 1998;83(2):370-3. [PubMed: 9467543]
- 199.
- Lazar L, Lebenthal Y, Yackobovitch-Gavan M, Shalitin S, de Vries L, Phillip M, et al. Treated and untreated women with idiopathic precocious puberty: BMI evolution, metabolic outcome, and general health between third and fifth decades. J Clin Endocrinol Metab. 2015;100(4):1445-51. [PubMed: 25650898]
- 200.
- Guaraldi F, Beccuti G, Gori D, Ghizzoni L. MANAGEMENT OF ENDOCRINE DISEASE: Long-term outcomes of the treatment of central precocious puberty. Eur J Endocrinol. 2016;174(3):R79-87. [PubMed: 26466612]
- 201.
- Lee JW, Kim HJ, Choe YM, Kang HS, Kim SK, Jun YH, et al. Significant adverse reactions to long-acting gonadotropin-releasing hormone agonists for the treatment of central precocious puberty and early onset puberty. Ann Pediatr Endocrinol Metab. 2014;19(3):135-40. [PMC free article: PMC4208261] [PubMed: 25346917]
- 202.
- Omar AA, Nyaga G, Mungai LNW. Pseudotumor cerebri in patient on leuprolide acetate for central precocious puberty. Int J Pediatr Endocrinol. 2020;2020(1):22. [PMC free article: PMC7712604] [PubMed: 33292495]
- 203.
- Lazar L, Meyerovitch J, de Vries L, Phillip M, Lebenthal Y. Treated and untreated women with idiopathic precocious puberty: long-term follow-up and reproductive outcome between the third and fifth decades. Clin Endocrinol (Oxf). 2014;80(4):570-6. [PubMed: 24033561]
- 204.
- Bertelloni S, Baroncelli GI, Ferdeghini M, Menchini-Fabris F, Saggese G. Final height, gonadal function and bone mineral density of adolescent males with central precocious puberty after therapy with gonadotropin-releasing hormone analogues. Eur J Pediatr. 2000;159(5):369-74. [PubMed: 10834524]
- 205.
- Thornton P, Silverman LA, Geffner ME, Neely EK, Gould E, Danoff TM. Review of outcomes after cessation of gonadotropin-releasing hormone agonist treatment of girls with precocious puberty. Pediatr Endocrinol Rev. 2014;11(3):306-17. [PubMed: 24719967]
- 206.
- Arcari AJ, Freire AV, Ballerini MG, Escobar ME, Diaz Marsiglia YM, Bergada I, et al. Prevalence of Polycystic Ovarian Syndrome in Girls with a History of Idiopathic Central Precocious Puberty. Horm Res Paediatr. 2023:1-6. [PubMed: 37552972]
- 207.
- Eugster EA. Treatment of Central Precocious Puberty. J Endocr Soc. 2019;3(5):965-72. [PMC free article: PMC6486823] [PubMed: 31041427]
- 208.
- Schoelwer MJ, Donahue KL, Didrick P, Eugster EA. One-Year Follow-Up of Girls with Precocious Puberty and Their Mothers: Do Psychological Assessments Change over Time or with Treatment? Horm Res Paediatr. 2017;88(5):347-53. [PMC free article: PMC5808430] [PubMed: 28926827]
- 209.
- Kaplowitz PB, Backeljauw PF, Allen DB. Toward More Targeted and Cost-Effective Gonadotropin-Releasing Hormone Analog Treatment in Girls with Central Precocious Puberty. Horm Res Paediatr. 2018;90(1):1-7. [PubMed: 30048994]
- 210.
- Popovic J, Geffner ME, Rogol AD, Silverman LA, Kaplowitz PB, Mauras N, et al. Gonadotropin-releasing hormone analog therapies for children with central precocious puberty in the United States. Frontiers in Pediatrics. 2022;10. [PMC free article: PMC9577333] [PubMed: 36268040]
- 211.
- Eugster EA. Treatment of Central Precocious Puberty. Journal of the Endocrine Society. 2019;3(5):965-72. [PMC free article: PMC6486823] [PubMed: 31041427]
- 212.
- Pasquino AM, Municchi G, Pucarelli I, Segni M, Mancini MA, Troiani S. Combined treatment with gonadotropin-releasing hormone analog and growth hormone in central precocious puberty. J Clin Endocrinol Metab. 1996;81(3):948-51. [PubMed: 8772556]
- 213.
- Shi Y, Ma Z, Yang X, Ying Y, Luo X, Hou L. Gonadotropin-releasing hormone analogue and recombinant human growth hormone treatment for idiopathic central precocious puberty in girls. Front Endocrinol (Lausanne). 2022;13:1085385. [PMC free article: PMC9794601] [PubMed: 36589818]
- 214.
- Cho AY, Shim YS, Lee HS, Hwang JS. Effect of gonadotropin-releasing hormone agonist monotherapy and combination therapy with growth hormone on final adult height in girls with central precocious puberty. Sci Rep. 2023;13(1):1264. [PMC free article: PMC9870989] [PubMed: 36690835]
- 215.
- Pucarelli I, Segni M, Ortore M, Arcadi E, Pasquino AM. Effects of combined gonadotropin-releasing hormone agonist and growth hormone therapy on adult height in precocious puberty: a further contribution. J Pediatr Endocrinol Metab. 2003;16(7):1005-10. [PubMed: 14513877]
- 216.
- Li YH, Zhu SY, Ma HM, Su Z, Chen HS, Chen QL, et al. [Effect of gonadotropin-releasing hormone analog combined with stanazolol on final height in girls with idiopathic central precocious puberty and apparent decrease of linear growth]. Zhonghua Er Ke Za Zhi. 2013;51(11):807-12. [PubMed: 24484553]
- 217.
- Zhu S, Long L, Hu Y, Tuo Y, Li Y, Yu Z. GnRHa/Stanozolol Combined Therapy Maintains Normal Bone Growth in Central Precocious Puberty. Front Endocrinol (Lausanne). 2021;12:678797. [PMC free article: PMC8221533] [PubMed: 34177807]
- 218.
- Vottero A, Pedori S, Verna M, Pagano B, Cappa M, Loche S, et al. Final height in girls with central idiopathic precocious puberty treated with gonadotropin-releasing hormone analog and oxandrolone. J Clin Endocrinol Metab. 2006;91(4):1284-7. [PubMed: 16449342]
- 219.
- Liu S, Liu Q, Cheng X, Luo Y, Wen Y. Effects and safety of combination therapy with gonadotropin-releasing hormone analogue and growth hormone in girls with idiopathic central precocious puberty: a meta-analysis. J Endocrinol Invest. 2016;39(10):1167-78. [PubMed: 27225286]
- 220.
- Wit JM. Should Skeletal Maturation Be Manipulated for Extra Height Gain? Front Endocrinol (Lausanne). 2021;12:812196. [PMC free article: PMC8716689] [PubMed: 34975773]
- 221.
- Dotremont H, France A, Heinrichs C, Tenoutasse S, Brachet C, Cools M, et al. Efficacy and safety of a 4-year combination therapy of growth hormone and gonadotropin-releasing hormone analogue in pubertal girls with short predicted adult height. Front Endocrinol (Lausanne). 2023;14:1113750. [PMC free article: PMC10064858] [PubMed: 37008942]
- 222.
- Trujillo MV, Lee PA, Reifschneider K, Backeljauw PF, Dragnic S, Van Komen S, et al. Using change in predicted adult height during GnRH agonist treatment for individualized treatment decisions in girls with central precocious puberty. J Pediatr Endocrinol Metab. 2023;36(3):299-308. [PubMed: 36473097]
- 223.
- Vargas Trujillo M, Dragnic S, Aldridge P, Klein KO. Importance of individualizing treatment decisions in girls with central precocious puberty when initiating treatment after age 7 years or continuing beyond a chronological age of 10 years or a bone age of 12 years. J Pediatr Endocrinol Metab. 2021;34(6):733-9. [PubMed: 33856747]
- 224.
- Mendle J, Ryan RM, McKone KMP. Age at Menarche, Depression, and Antisocial Behavior in Adulthood. Pediatrics. 2018;141(1). [PMC free article: PMC5744273] [PubMed: 29279324]
- 225.
- Mendle J, Ryan RM, McKone KMP. Early Menarche and Internalizing and Externalizing in Adulthood: Explaining the Persistence of Effects. J Adolescent Health. 2019;65(5):599-606. [PMC free article: PMC6814541] [PubMed: 31500947]
- 226.
- Schoelwer MJ, Donahue KL, Didrick P, Eugster EA. One-Year Follow-Up of Girls with Precocious Puberty and Their Mothers: Do Psychological Assessments Change over Time or with Treatment? Hormone Research in Paediatrics. 2017;88(5):347-53. [PMC free article: PMC5808430] [PubMed: 28926827]
- 227.
- Wojniusz S, Callens N, Sütterlin S, Andersson S, De Schepper J, Gies I, et al. Cognitive, Emotional, and Psychosocial Functioning of Girls Treated with Pharmacological Puberty Blockage for Idiopathic Central Precocious Puberty. Front Psychol. 2016;7:1053. [PMC free article: PMC4940404] [PubMed: 27462292]
- 228.
- Williams VSL, Soliman AM, Barrett AM, Klein KO. Review and evaluation of patient-centered psychosocial assessments for children with central precocious puberty or early puberty. J Pediatr Endocrinol Metab. 2018;31(5):485-95. [PubMed: 29649000]
- 229.
- Lewis KA, Eugster EA. Random luteinizing hormone often remains pubertal in children treated with the histrelin implant for central precocious puberty. J Pediatr. 2013;162(3):562-5. [PMC free article: PMC4094029] [PubMed: 23040793]
- 230.
- Neely EK, Silverman LA, Geffner ME, Danoff TM, Gould E, Thornton PS. Random unstimulated pediatric luteinizing hormone levels are not reliable in the assessment of pubertal suppression during histrelin implant therapy. Int J Pediatr Endocrinol. 2013;2013(1):20. [PMC free article: PMC4220797] [PubMed: 24295437]
- 231.
- Klein KO, Lee PA. Gonadotropin-Releasing Hormone (GnRHa) Therapy for Central Precocious Puberty (CPP): Review of Nuances in Assessment of Height, Hormonal Suppression, Psychosocial Issues, and Weight Gain, with Patient Examples. Pediatr Endocrinol Rev. 2018;15(4):298-312. [PubMed: 29806750]
- 232.
- Houk CP, Kunselman AR, Lee PA. The diagnostic value of a brief GnRH analogue stimulation test in girls with central precocious puberty: a single 30-minute post-stimulation LH sample is adequate. J Pediatr Endocrinol Metab. 2008;21(12):1113-8. [PubMed: 19189683]
- 233.
- Yazdani P, Lin Y, Raman V, Haymond M. A single sample GnRHa stimulation test in the diagnosis of precocious puberty. Int J Pediatr Endocrinol. 2012;2012(1):23. [PMC free article: PMC3438042] [PubMed: 22809285]
- 234.
- Ohyama K, Tanaka T, Tachibana K, Niimi H, Fujieda K, Matsuo N, et al. Timing for discontinuation of treatment with a long-acting gonadotropin-releasing hormone analog in girls with central precocious puberty. TAP-144SR CPP Study Group. Endocr J. 1998;45(3):351-6. [PubMed: 9790269]
- 235.
- Carel JC, Roger M, Ispas S, Tondu F, Lahlou N, Blumberg J, et al. Final height after long-term treatment with triptorelin slow release for central precocious puberty: importance of statural growth after interruption of treatment. French study group of Decapeptyl in Precocious Puberty. J Clin Endocrinol Metab. 1999;84(6):1973-8. [PubMed: 10372696]
- 236.
- Carel JC, Eugster EA, Rogol A, Ghizzoni L, Palmert MR, Antoniazzi F, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics. 2009;123(4):e752-62. [PubMed: 19332438]
- 237.
- Boyce AM, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of Galpha s Activation. Endocr Rev. 2020;41(2):345-70. [PMC free article: PMC7127130] [PubMed: 31673695]
- 238.
- Tufano M, Ciofi D, Amendolea A, Stagi S. Auxological and Endocrinological Features in Children With McCune Albright Syndrome: A Review. Front Endocrinol (Lausanne). 2020;11:522. [PMC free article: PMC7417367] [PubMed: 32849305]
- 239.
- Spencer T, Pan KS, Collins MT, Boyce AM. The Clinical Spectrum of McCune-Albright Syndrome and Its Management. Horm Res Paediatr. 2019;92(6):347-56. [PMC free article: PMC7302983] [PubMed: 31865341]
- 240.
- Kim IS, Kim ER, Nam HJ, Chin MO, Moon YH, Oh MR, et al. Activating mutation of GS alpha in McCune-Albright syndrome causes skin pigmentation by tyrosinase gene activation on affected melanocytes. Horm Res. 1999;52(5):235-40. [PubMed: 10844413]
- 241.
- Szymczuk V, Taylor J, Michel Z, Sinaii N, Boyce AM. Skeletal Disease Acquisition in Fibrous Dysplasia: Natural History and Indicators of Lesion Progression in Children. J Bone Miner Res. 2022;37(8):1473-8. [PubMed: 35695414]
- 242.
- Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, et al. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res. 2001;16(5):806-13. [PubMed: 11341325]
- 243.
- Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27(5):1132-41. [PMC free article: PMC7448291] [PubMed: 22247037]
- 244.
- Boyce AM, Casey RK, Ovejero Crespo D, Murdock CM, Estrada A, Guthrie LC, et al. Gynecologic and reproductive outcomes in fibrous dysplasia/McCune-Albright syndrome. Orphanet J Rare Dis. 2019;14(1):90. [PMC free article: PMC6489337] [PubMed: 31036049]
- 245.
- Boyce AM, Chong WH, Shawker TH, Pinto PA, Linehan WM, Bhattacharryya N, et al. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab. 2012;97(9):E1782-90. [PMC free article: PMC3431566] [PubMed: 22745241]
- 246.
- Tatsi C, Stratakis CA. Neonatal Cushing Syndrome: A Rare but Potentially Devastating Disease. Clin Perinatol. 2018;45(1):103-18. [PMC free article: PMC5806137] [PubMed: 29406000]
- 247.
- Gaujoux S, Salenave S, Ronot M, Rangheard AS, Cros J, Belghiti J, et al. Hepatobiliary and Pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2014;99(1):E97-101. [PubMed: 24170100]
- 248.
- Zacharin M, Bajpai A, Chow CW, Catto-Smith A, Stratakis C, Wong MW, et al. Gastrointestinal polyps in McCune Albright syndrome. J Med Genet. 2011;48(7):458-61. [PMC free article: PMC4034052] [PubMed: 21357941]
- 249.
- Majoor BC, Boyce AM, Bovee JV, Smit VT, Collins MT, Cleton-Jansen AM, et al. Increased Risk of Breast Cancer at a Young Age in Women with Fibrous Dysplasia. J Bone Miner Res. 2018;33(1):84-90. [PubMed: 28856726]
- 250.
- Lumbroso S, Paris F, Sultan C, European Collaborative S. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study. J Clin Endocrinol Metab. 2004;89(5):2107-13. [PubMed: 15126527]
- 251.
- Roszko KL, Guthrie L, Li X, Collins MT, de Castro LF, Boyce AM. Identification of GNAS Variants in Circulating Cell-Free DNA from Patients with Fibrous Dysplasia/McCune Albright Syndrome. J Bone Miner Res. 2023;38(3):443-50. [PubMed: 36593655]
- 252.
- Couch RM, Muller J, Perry YS, Winter JS. Kinetic analysis of inhibition of human adrenal steroidogenesis by ketoconazole. J Clin Endocrinol Metab. 1987;65(3):551-4. [PubMed: 3497939]
- 253.
- Estrada A, Boyce AM, Brillante BA, Guthrie LC, Gafni RI, Collins MT. Long-term outcomes of letrozole treatment for precocious puberty in girls with McCune-Albright syndrome. Eur J Endocrinol. 2016;175(5):477-83. [PMC free article: PMC5066167] [PubMed: 27562402]
- 254.
- Eugster EA, Rubin SD, Reiter EO, Plourde P, Jou HC, Pescovitz OH, et al. Tamoxifen treatment for precocious puberty in McCune-Albright syndrome: a multicenter trial. J Pediatr. 2003;143(1):60-6. [PubMed: 12915825]
- 255.
- Hu R, Hilakivi-Clarke L, Clarke R. Molecular mechanisms of tamoxifen-associated endometrial cancer (Review). Oncol Lett. 2015;9(4):1495-501. [PMC free article: PMC4356269] [PubMed: 25788989]
- 256.
- Sims EK, Garnett S, Guzman F, Paris F, Sultan C, Eugster EA, et al. Fulvestrant treatment of precocious puberty in girls with McCune-Albright syndrome. Int J Pediatr Endocrinol. 2012;2012(1):26. [PMC free article: PMC3488024] [PubMed: 22999294]
- 257.
- Nabhan ZM, West KW, Eugster EA. Oophorectomy in McCune-Albright syndrome: a case of mistaken identity. J Pediatr Surg. 2007;42(9):1578-83. [PubMed: 17848252]
- 258.
- Corica D, Aversa T, Pepe G, De Luca F, Wasniewska M. Peculiarities of Precocious Puberty in Boys and Girls With McCune-Albright Syndrome. Front Endocrinol (Lausanne). 2018;9:337. [PMC free article: PMC6023984] [PubMed: 29988390]
- 259.
- Tessaris D, Matarazzo P, Mussa A, Tuli G, Verna F, Fiore L, et al. Combined treatment with bicalutamide and anastrozole in a young boy with peripheral precocious puberty due to McCune-Albright Syndrome. Endocr J. 2012;59(2):111-7. [PubMed: 22068112]
- 260.
- Partsch CJ, Kreller-Laugwitz G, Sippell WG. [Transitory precocious puberty caused by autonomous ovarian cysts. Clinical, endocrinologic and sonographic course]. Monatsschr Kinderheilkd. 1989;137(4):235-8. [PubMed: 2499771]
- 261.
- de Sousa G, Wunsch R, Andler W. Precocious pseudopuberty due to autonomous ovarian cysts: a report of ten cases and long-term follow-up. Hormones. 2008;7(2):170-4. [PubMed: 18477555]
- 262.
- Nayak S, Witchel SF, Sanfilippo JS. Vaginal foreign body: a delayed diagnosis. J Pediatr Adolesc Gynecol. 2014;27(6):e127-9. [PubMed: 24656699]
- 263.
- Ganer Herman H, Shalev A, Ginat S, Kerner R, Keidar R, Bar J, et al. Clinical characteristics of adnexal torsion in premenarchal patients. Arch Gynecol Obstet. 2016;293(3):603-8. [PubMed: 26288977]
- 264.
- Adeyemi-Fowode O, Lin EG, Syed F, Sangi-Haghpeykar H, Zhu H, Dietrich JE. Adnexal Torsion in Children and Adolescents: A Retrospective Review of 245 Cases at a Single Institution. J Pediatr Adolesc Gynecol. 2019;32(1):64-9. [PubMed: 30012428]
- 265.
- Spinelli C, Piscioneri J, Strambi S. Adnexal torsion in adolescents: update and review of the literature. Curr Opin Obstet Gynecol. 2015;27(5):320-5. [PubMed: 26204167]
- 266.
- Spinelli C, Buti I, Pucci V, Liserre J, Alberti E, Nencini L, et al. Adnexal torsion in children and adolescents: new trends to conservative surgical approach -- our experience and review of literature. Gynecol Endocrinol. 2013;29(1):54-8. [PubMed: 22817767]
- 267.
- Heo S, Shim YS, Lee HS, Hwang JS. Clinical course of peripheral precocious puberty in girls due to autonomous ovarian cysts. Clin Endocrinol (Oxf). 2023. [PubMed: 37386805]
- 268.
- Poonai N, Poonai C, Lim R, Lynch T. Pediatric ovarian torsion: case series and review of the literature. Can J Surg. 2013;56(2):103-8. [PMC free article: PMC3617114] [PubMed: 23351494]
- 269.
- Gaikwad PM, Goswami S, Sengupta N, Baidya A, Das N. Transformation of Peripheral Sexual Precocity to Central Sexual Precocity Following Treatment of Granulosa Cell Tumor of the Ovary. Cureus. 2022;14(2):e22676. [PMC free article: PMC8966063] [PubMed: 35371651]
- 270.
- Bessiere L, Todeschini AL, Auguste A, Sarnacki S, Flatters D, Legois B, et al. A Hot-spot of In-frame Duplications Activates the Oncoprotein AKT1 in Juvenile Granulosa Cell Tumors. EBioMedicine. 2015;2(5):421-31. [PMC free article: PMC4485906] [PubMed: 26137586]
- 271.
- Fuller PJ, Leung D, Chu S. Genetics and genomics of ovarian sex cord-stromal tumors. Clin Genet. 2017;91(2):285-91. [PubMed: 27813081]
- 272.
- Yamamoto H, Sakamoto H, Kumagai H, Abe T, Ishiguro S, Uchida K, et al. Clinical Guidelines for Diagnosis and Management of Peutz-Jeghers Syndrome in Children and Adults. Digestion. 2023:1-13. [PubMed: 37054692]
- 273.
- De Paolis E, Paragliola RM, Concolino P. Spectrum of DICER1 Germline Pathogenic Variants in Ovarian Sertoli-Leydig Cell Tumor. J Clin Med. 2021;10(9). [PMC free article: PMC8123016] [PubMed: 33922805]
- 274.
- Patel SS, Carrick KS, Carr BR. Virilization persists in a woman with an androgen-secreting granulosa cell tumor. Fertil Steril. 2009;91(3):933 e13-5. [PubMed: 19062005]
- 275.
- Kalfa N, Meduri G, Philibert P, Patte C, Boizet-Bonhoure B, Thibaut E, et al. Unusual virilization in girls with juvenile granulosa cell tumors of the ovary is related to intratumoral aromatase deficiency. Horm Res Paediatr. 2010;74(2):83-91. [PubMed: 20395670]
- 276.
- Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res. 2018;24(10):2251-61. [PMC free article: PMC6260592] [PubMed: 29343557]
- 277.
- Trahmono, Wahyudi I, Rodjani A, Situmorang GR, Marzuki NS. Precocious Pseudo-Puberty with Testicular Enlargement: Two Cases of Leydig Cell Tumor with Different Histopathological Results. Res Rep Urol. 2020;12:577-82. [PMC free article: PMC7695686] [PubMed: 33262958]
- 278.
- Luckie TM, Danzig M, Zhou S, Wu H, Cost NG, Karaviti L, et al. A Multicenter Retrospective Review of Pediatric Leydig Cell Tumor of the Testis. J Pediatr Hematol Oncol. 2019;41(1):74-6. [PubMed: 29554024]
- 279.
- De Felici M, Klinger FG, Campolo F, Balistreri CR, Barchi M, Dolci S. To Be or Not to Be a Germ Cell: The Extragonadal Germ Cell Tumor Paradigm. Int J Mol Sci. 2021;22(11). [PMC free article: PMC8199495] [PubMed: 34205983]
- 280.
- Cattoni A, Albanese A. Case report: Fluctuating tumor markers in a boy with gonadotropin-releasing hormone-independent precocious puberty induced by a pineal germ cell tumor. Front Pediatr. 2022;10:940656. [PMC free article: PMC9445167] [PubMed: 36081625]
- 281.
- . !!! INVALID CITATION !!! (269, 271, 272).
- 282.
- Yhoshu E, Lone YA, Mahajan JK, Singh UB. Hepatoblastoma with Precocious Puberty. J Indian Assoc Pediatr Surg. 2019;24(1):68-71. [PMC free article: PMC6322182] [PubMed: 30686892]
- 283.
- Hersmus R, van Bever Y, Wolffenbuttel KP, Biermann K, Cools M, Looijenga LH. The biology of germ cell tumors in disorders of sex development. Clin Genet. 2017;91(2):292-301. [PubMed: 27716895]
- 284.
- Shenker A, Laue L, Kosugi S, Merendino JJ, Jr., Minegishi T, Cutler GB, Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365(6447):652-4. [PubMed: 7692306]
- 285.
- Themmen APN, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21(5):551-83. [PubMed: 11041448]
- 286.
- Gurnurkar S, DiLillo E, Carakushansky M. A Case of Familial Male-limited Precocious Puberty with a Novel Mutation. J Clin Res Pediatr Endocrinol. 2021;13(2):239-44. [PMC free article: PMC8186329] [PubMed: 32757547]
- 287.
- Haddad NG, Eugster EA. Peripheral precocious puberty including congenital adrenal hyperplasia: causes, consequences, management and outcomes. Best Pract Res Clin Endocrinol Metab. 2019;33(3):101273. [PubMed: 31027974]
- 288.
- Cunha-Silva M, Brito VN, Macedo DB, Bessa DS, Ramos CO, Lima LG, et al. Spontaneous fertility in a male patient with testotoxicosis despite suppression of FSH levels. Hum Reprod. 2018;33(5):914-8. [PubMed: 29538680]
- 289.
- Kooij CD, Mavinkurve-Groothuis AMC, Kremer Hovinga ICL, Looijenga LHJ, Rinne T, Giltay JC, et al. Familial Male-limited Precocious Puberty (FMPP) and Testicular Germ Cell Tumors. J Clin Endocrinol Metab. 2022;107(11):3035-44. [PMC free article: PMC9681611] [PubMed: 36071555]
- 290.
- Reiter EO, Mauras N, McCormick K, Kulshreshtha B, Amrhein J, De Luca F, et al. Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testotoxicosis: a phase II, open-label pilot study (BATT). J Pediatr Endocrinol Metab. 2010;23(10):999-1009. [PubMed: 21158211]
- 291.
- Leschek EW, Flor AC, Bryant JC, Jones JV, Barnes KM, Cutler GB, Jr. Effect of Antiandrogen, Aromatase Inhibitor, and Gonadotropin-releasing Hormone Analog on Adult Height in Familial Male Precocious Puberty. J Pediatr. 2017;190:229-35. [PMC free article: PMC5726420] [PubMed: 29144249]
- 292.
- Flippo C, Kolli V, Andrew M, Berger S, Bhatti T, Boyce AM, et al. Precocious Puberty in a Boy With Bilateral Leydig Cell Tumors due to a Somatic Gain-of-Function LHCGR Variant. J Endocr Soc. 2022;6(10):bvac127. [PMC free article: PMC9469925] [PubMed: 36111273]
- 293.
- Latronico AC, Lins TS, Brito VN, Arnhold IJ, Mendonca BB. The effect of distinct activating mutations of the luteinizing hormone receptor gene on the pituitary-gonadal axis in both sexes. Clin Endocrinol (Oxf). 2000;53(5):609-13. [PubMed: 11106922]
- 294.
- Vanwyk JJ, Grumbach MM. Syndrome of Precocious Menstruation and Galactorrhea in Juvenile Hypothyroidism - an Example of Hormonal Overlap in Pituitary Feedback. J Pediatr-Us. 1960;57(3):416-35.
- 295.
- Kusuma Boddu S, Ayyavoo A, Hebbal Nagarajappa V, Kalenahalli KV, Muruda S, Palany R. Van Wyk Grumbach Syndrome and Ovarian Hyperstimulation in Juvenile Primary Hypothyroidism: Lessons From a 30-Case Cohort. J Endocr Soc. 2023;7(6):bvad042. [PMC free article: PMC10184442] [PubMed: 37197410]
- 296.
- Reddy P, Tiwari K, Kulkarni A, Parikh K, Khubchandani R. Van Wyk Grumbach Syndrome: A Rare Consequence of Hypothyroidism. Indian J Pediatr. 2018;85(11):1028-30. [PubMed: 29777468]
- 297.
- Zhang H, Geng N, Wang Y, Tian W, Xue F. Van Wyk and Grumbach syndrome: two case reports and review of the published work. J Obstet Gynaecol Res. 2014;40(2):607-10. [PubMed: 24118179]
- 298.
- Baranowski E, Hogler W. An unusual presentation of acquired hypothyroidism: the Van Wyk-Grumbach syndrome. Eur J Endocrinol. 2012;166(3):537-42. [PubMed: 22170796]
- 299.
- Anasti JN, Flack MR, Froehlich J, Nelson LM, Nisula BC. A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J Clin Endocrinol Metab. 1995;80(1):276-9. [PubMed: 7829625]
- 300.
- Polat S, Kulle A, Karaca Z, Akkurt I, Kurtoglu S, Kelestimur F, et al. Characterisation of three novel CYP11B1 mutations in classic and non-classic 11beta-hydroxylase deficiency. Eur J Endocrinol. 2014;170(5):697-706. [PubMed: 24536089]
- 301.
- Armengaud JB, Charkaluk ML, Trivin C, Tardy V, Breart G, Brauner R, et al. Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J Clin Endocrinol Metab. 2009;94(8):2835-40. [PubMed: 19454583]
- 302.
- Miller WL. Congenital Adrenal Hyperplasia: Time to Replace 17OHP with 21-Deoxycortisol. Horm Res Paediatr. 2019;91(6):416-20. [PubMed: 31450227]
- 303.
- Sarafoglou K, Merke DP, Reisch N, Claahsen-van der Grinten H, Falhammar H, Auchus RJ. Interpretation of Steroid Biomarkers in 21-Hydroxylase Deficiency and Their Use in Disease Management. J Clin Endocrinol Metab. 2023;108(9):2154-75. [PMC free article: PMC10438890] [PubMed: 36950738]
- 304.
- Mallappa A, Merke DP. Management challenges and therapeutic advances in congenital adrenal hyperplasia. Nat Rev Endocrinol. 2022;18(6):337-52. [PMC free article: PMC8999997] [PubMed: 35411073]
- 305.
- Witchel SF. Congenital Adrenal Hyperplasia. J Pediatr Adolesc Gynecol. 2017;30(5):520-34. [PMC free article: PMC5624825] [PubMed: 28450075]
- 306.
- Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27(7):867-81. [PubMed: 12826878]
- 307.
- . !!! INVALID CITATION !!! (297).
- 308.
- Custodio G, Komechen H, Figueiredo FR, Fachin ND, Pianovski MA, Figueiredo BC. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell Endocrinol. 2012;351(1):44-51. [PubMed: 22056871]
- 309.
- Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet. 2005;137C(1):53-71. [PubMed: 16010678]
- 310.
- Clay MR, Pinto EM, Fishbein L, Else T, Kiseljak-Vassiliades K. Pathological and Genetic Stratification for Management of Adrenocortical Carcinoma. J Clin Endocrinol Metab. 2022;107(4):1159-69. [PMC free article: PMC8947319] [PubMed: 34850906]
- 311.
- Kaplowitz PB. For Premature Thelarche and Premature Adrenarche, the Case for Waiting before Testing. Horm Res Paediatr. 2020;93(9-10):573-6. [PubMed: 33352558]
- 312.
- Freedman SM, Kreitzer PM, Elkowitz SS, Soberman N, Leonidas JC. Ovarian microcysts in girls with isolated premature thelarche. J Pediatr. 1993;122(2):246-9. [PubMed: 8429440]
- 313.
- Witchel SF, Pinto B, Burghard AC, Oberfield SE. Update on adrenarche. Curr Opin Pediatr. 2020;32(4):574-81. [PMC free article: PMC7891873] [PubMed: 32692055]
- 314.
- Kaya G, Abali ZY, Bas F, Poyrazoglu S, Darendeliler F. Body mass index at the presentation of premature adrenarche is associated with components of metabolic syndrome at puberty. European Journal of Pediatrics. 2018;177(11):1593-601. [PubMed: 30056577]
- 315.
- Ibanez L, Ong K, de Zegher F, Marcos MV, del Rio L, Dunger DB. Fat distribution in non-obese girls with and without precocious pubarche: central adiposity related to insulinaemia and androgenaemia from prepuberty to postmenarche. Clin Endocrinol (Oxf). 2003;58(3):372-9. [PubMed: 12608944]
- 316.
- Ibanez L, Potau N, Virdis R, Zampolli M, Terzi C, Gussinye M, et al. Postpubertal outcome in girls diagnosed of premature pubarche during childhood: increased frequency of functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1993;76(6):1599-603. [PubMed: 8501168]
- 317.
- Tennila J, Jaaskelainen J, Utriainen P, Voutilainen R, Hakkinen M, Auriola S, et al. PCOS Features and Steroid Profiles Among Young Adult Women with a History of Premature Adrenarche. J Clin Endocrinol Metab. 2021;106(9):e3335-e45. [PMC free article: PMC8372635] [PubMed: 34060603]
- 318.
- Ramsey JT, Li Y, Arao Y, Naidu A, Coons LA, Diaz A, et al. Lavender Products Associated With Premature Thelarche and Prepubertal Gynecomastia: Case Reports and Endocrine-Disrupting Chemical Activities. J Clin Endocrinol Metab. 2019;104(11):5393-405. [PMC free article: PMC6773459] [PubMed: 31393563]
- 319.
- Kim SH, Park MJ. Effects of phytoestrogen on sexual development. Korean J Pediatr. 2012;55(8):265-71. [PMC free article: PMC3433562] [PubMed: 22977438]
- 320.
- Cleemann L, Holm K. [Precocious pseudopuberty in a seven year-old girl due to estrogen treatment of labial adhesion]. Ugeskr Laeger. 2011;173(20):1435-6. [PubMed: 21586251]
- 321.
- Guarneri MP, Brambilla G, Loizzo A, Colombo I, Chiumello G. Estrogen exposure in a child from hair lotion used by her mother: clinical and hair analysis data. Clin Toxicol (Phila). 2008;46(8):762-4. [PubMed: 18763154]
- 322.
- Martinez-Pajares JD, Diaz-Morales O, Ramos-Diaz JC, Gomez-Fernandez E. Peripheral precocious puberty due to inadvertent exposure to testosterone: case report and review of the literature. J Pediatr Endocrinol Metab. 2012;25(9-10):1007-12. [PubMed: 23426834]
- 323.
- Castiello F, Freire C. Exposure to non-persistent pesticides and puberty timing: a systematic review of the epidemiological evidence. Eur J Endocrinol. 2021;184(6):733-49. [PubMed: 33769962]
- 324.
- Watkins DJ, Sanchez BN, Tellez-Rojo MM, Lee JM, Mercado-Garcia A, Blank-Goldenberg C, et al. Phthalate and bisphenol A exposure during in utero windows of susceptibility in relation to reproductive hormones and pubertal development in girls. Environ Res. 2017;159:143-51. [PMC free article: PMC5623649] [PubMed: 28800472]
- 325.
- Lee JE, Jung HW, Lee YJ, Lee YA. Early-life exposure to endocrine-disrupting chemicals and pubertal development in girls. Ann Pediatr Endocrinol Metab. 2019;24(2):78-91. [PMC free article: PMC6603611] [PubMed: 31261471]
- 326.
- Franssen D, Svingen T, Lopez Rodriguez D, Van Duursen M, Boberg J, Parent AS. A Putative Adverse Outcome Pathway Network for Disrupted Female Pubertal Onset to Improve Testing and Regulation of Endocrine Disrupting Chemicals. Neuroendocrinology. 2022;112(2):101-14. [PubMed: 33640887]
- 327.
- Taylor KW, Howdeshell KL, Bommarito PA, Sibrizzi CA, Blain RB, Magnuson K, et al. Systematic evidence mapping informs a class-based approach to assessing personal care products and pubertal timing. Environ Int. 2023;181:108307. [PubMed: 37948866]
- 328.
- Ahn C, Jeung EB. Endocrine-Disrupting Chemicals and Disease Endpoints. Int J Mol Sci. 2023;24(6). [PMC free article: PMC10049097] [PubMed: 36982431]
- 329.
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, et al. EDC-2: The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev. 2015;36(6):E1-E150. [PMC free article: PMC4702494] [PubMed: 26544531]
- 330.
- Uldbjerg CS, Koch T, Lim YH, Gregersen LS, Olesen CS, Andersson AM, et al. Prenatal and postnatal exposures to endocrine disrupting chemicals and timing of pubertal onset in girls and boys: a systematic review and meta-analysis. Hum Reprod Update. 2022;28(5):687-716. [PMC free article: PMC9434240] [PubMed: 35466359]
- 331.
- De Sanctis V, Rigon F, Bernasconi S, Bianchin L, Bona G, Bozzola M, et al. Age at Menarche and Menstrual Abnormalities in Adolescence: Does it Matter? The Evidence from a Large Survey among Italian Secondary Schoolgirls. Indian J Pediatr. 2019;86(Suppl 1):34-41. [PubMed: 30628040]
- 332.
- Reindollar RH, Byrd JR, McDonough PG. Delayed sexual development: a study of 252 patients. Am J Obstet Gynecol. 1981;140(4):371-80. [PubMed: 7246652]
- 333.
- Herman-Giddens ME, Steffes J, Harris D, Slora E, Hussey M, Dowshen SA, et al. Secondary sexual characteristics in boys: data from the Pediatric Research in Office Settings Network. Pediatrics. 2012;130(5):e1058-68. [PubMed: 23085608]
- 334.
- Jonsdottir-Lewis E, Feld A, Ciarlo R, Denhoff E, Feldman HA, Chan YM. Timing of Pubertal Onset in Girls and Boys With Constitutional Delay. J Clin Endocrinol Metab. 2021;106(9):e3693-e703. [PMC free article: PMC8372671] [PubMed: 33890108]
- 335.
- Barroso PS, Jorge AAL, Lerario AM, Montenegro LR, Vasques GA, Lima Amato LG, et al. Clinical and Genetic Characterization of a Constitutional Delay of Growth and Puberty Cohort. Neuroendocrinology. 2020;110(11-12):959-66. [PubMed: 31726455]
- 336.
- Sukumar SP, Bhansali A, Sachdeva N, Ahuja CK, Gorsi U, Jarial KD, et al. Diagnostic utility of testosterone priming prior to dynamic tests to differentiate constitutional delay in puberty from isolated hypogonadotropic hypogonadism. Clin Endocrinol (Oxf). 2017;86(5):717-24. [PubMed: 28261833]
- 337.
- Galazzi E, Improda N, Cerbone M, Soranna D, Moro M, Fatti LM, et al. Clinical benefits of sex steroids given as a priming prior to GH provocative test or as a growth-promoting therapy in peripubertal growth delays: Results of a retrospective study among ENDO-ERN centres. Clin Endocrinol (Oxf). 2021;94(2):219-28. [PubMed: 32969044]
- 338.
- Vezzoli V, Hrvat F, Goggi G, Federici S, Cangiano B, Quinton R, et al. Genetic architecture of self-limited delayed puberty and congenital hypogonadotropic hypogonadism. Front Endocrinol. 2023;13. [PMC free article: PMC9884699] [PubMed: 36726466]
- 339.
- Aung Y, Kokotsis V, Yin KN, Banerjee K, Butler G, Dattani MT, et al. Key features of puberty onset and progression can help distinguish self-limited delayed puberty from congenital hypogonadotrophic hypogonadism. Front Endocrinol (Lausanne). 2023;14:1226839. [PMC free article: PMC10493306] [PubMed: 37701896]
- 340.
- Gohil A, Eugster EA. Delayed and Precocious Puberty: Genetic Underpinnings and Treatments. Endocrin Metab Clin. 2020;49(4):741-+. [PMC free article: PMC7705597] [PubMed: 33153677]
- 341.
- Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. European Journal of Endocrinology. 2018;178(4):377-88. [PMC free article: PMC5863472] [PubMed: 29419413]
- 342.
- Zhu J, Choa RE, Guo MH, Plummer L, Buck C, Palmert MR, et al. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2015;100(4):E646-54. [PMC free article: PMC4399304] [PubMed: 25636053]
- 343.
- Cui X, Cui Y, Shi L, Luan J, Zhou X, Han J. A basic understanding of Turner syndrome: Incidence, complications, diagnosis, and treatment. Intractable Rare Dis Res. 2018;7(4):223-8. [PMC free article: PMC6290843] [PubMed: 30560013]
- 344.
- Cameron-Pimblett A, La Rosa C, King TFJ, Davies MC, Conway GS. The Turner syndrome life course project: Karyotype-phenotype analyses across the lifespan. Clin Endocrinol (Oxf). 2017;87(5):532-8. [PubMed: 28617979]
- 345.
- Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1-G70. [PubMed: 28705803]
- 346.
- Chan YM, Lippincott MF, Sales Barroso P, Alleyn C, Brodsky J, Granados H, et al. Using Kisspeptin to Predict Pubertal Outcomes for Youth With Pubertal Delay. J Clin Endocrinol Metab. 2020;105(8):e2717-25. [PMC free article: PMC7282711] [PubMed: 32232399]
- 347.
- Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab. 2003;88(2):622-6. [PubMed: 12574191]
- 348.
- Tian SY, Li Y, Zhao LM, He HY. [Clinicopathological characteristics of Klinefelter syndrome: a testicular biopsy analysis of 87 cases]. Zhonghua Bing Li Xue Za Zhi. 2023;52(4):341-6. [PubMed: 36973193]
- 349.
- Hamzik MP, Gropman AL, Brooks MR, Powell S, Sadeghin T, Samango-Sprouse CA. The Effect of Hormonal Therapy on the Behavioral Outcomes in 47,XXY (Klinefelter Syndrome) between 7 and 12 Years of Age. Genes (Basel). 2023;14(7). [PMC free article: PMC10379663] [PubMed: 37510306]
- 350.
- Lazos-Ochoa M, Aguirre D, Nieto K, Pena R, Palma I, Kofman S, et al. Extragonadal mediastinal germ cell tumors are often associated with Klinefelter syndrome. Modern Pathol. 2006;19:137-. [PubMed: 16564924]
- 351.
- Lopez Krabbe HV, Holm Petersen J, Asserhoj LL, Johannsen TH, Christiansen P, Jensen RB, et al. Reproductive hormones, bone mineral content, body composition, and testosterone therapy in boys and adolescents with Klinefelter syndrome. Endocr Connect. 2023;12(7). [PMC free article: PMC10305500] [PubMed: 37010084]
- 352.
- Rohayem J, Nieschlag E, Zitzmann M, Kliesch S. Testicular function during puberty and young adulthood in patients with Klinefelter's syndrome with and without spermatozoa in seminal fluid. Andrology. 2016;4(6):1178-86. [PubMed: 27611179]
- 353.
- Masterson TA, 3rd, Nassau DE, Ramasamy R. A clinical algorithm for management of fertility in adolescents with the Klinefelter syndrome. Curr Opin Urol. 2020;30(3):324-7. [PubMed: 32235276]
- 354.
- Vena W, Carrone F, Delbarba A, Akpojiyovbi O, Pezzaioli LC, Facondo P, et al. Body composition, trabecular bone score and vertebral fractures in subjects with Klinefelter syndrome. J Endocrinol Invest. 2023;46(2):297-304. [PubMed: 36030302]
- 355.
- Butler G, Srirangalingam U, Faithfull J, Sangster P, Senniappan S, Mitchell R. Klinefelter syndrome: going beyond the diagnosis. Arch Dis Child. 2023;108(3):166-71. [PMC free article: PMC7614197] [PubMed: 35948402]
- 356.
- Tanner M, Miettinen PJ, Hero M, Toppari J, Raivio T. Onset and progression of puberty in Klinefelter syndrome. Clin Endocrinol (Oxf). 2022;96(3):363-70. [PubMed: 34523156]
- 357.
- Cools M, Nordenstrom A, Robeva R, Hall J, Westerveld P, Fluck C, et al. Caring for individuals with a difference of sex development (DSD): a Consensus Statement. Nat Rev Endocrinol. 2018;14(7):415-29. [PMC free article: PMC7136158] [PubMed: 29769693]
- 358.
- Rosario R, Anderson R. The molecular mechanisms that underlie fragile X-associated premature ovarian insufficiency: is it RNA or protein based? Mol Hum Reprod. 2020;26(10):727-37. [PMC free article: PMC7566375] [PubMed: 32777047]
- 359.
- Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur J Hum Genet. 2015;23(4):417-25. [PMC free article: PMC4666582] [PubMed: 25227148]
- 360.
- Yatsenko SA, Rajkovic A. Genetics of human female infertilitydagger. Biol Reprod. 2019;101(3):549-66. [PMC free article: PMC8127036] [PubMed: 31077289]
- 361.
- Flechtner I, Viaud M, Kariyawasam D, Perrissin-Fabert M, Bidet M, Bachelot A, et al. Puberty and fertility in classic galactosemia. Endocr Connect. 2021;10(2):240-7. [PMC free article: PMC7983486] [PubMed: 33491660]
- 362.
- Gubbels CS, Land JA, Rubio-Gozalbo ME. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstet Gynecol Surv. 2008;63(5):334-43. [PubMed: 18419833]
- 363.
- Frederick AB, Zinsli AM, Carlock G, Conneely K, Fridovich-Keil JL. Presentation, progression, and predictors of ovarian insufficiency in classic galactosemia. J Inherit Metab Dis. 2018;41(5):785-90. [PMC free article: PMC6128750] [PubMed: 29721917]
- 364.
- Derks B, Rivera-Cruz G, Hagen-Lillevik S, Vos EN, Demirbas D, Lai K, et al. The hypergonadotropic hypogonadism conundrum of classic galactosemia. Hum Reprod Update. 2023;29(2):246-58. [PMC free article: PMC9976963] [PubMed: 36512573]
- 365.
- Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Menheere PP, Wodzig WK, Land JA. Gonadal function in male and female patients with classic galactosemia. Hum Reprod Update. 2010;16(2):177-88. [PubMed: 19793842]
- 366.
- Schweitzer S, Shin Y, Jakobs C, Brodehl J. Long-term outcome in 134 patients with galactosaemia. Eur J Pediatr. 1993;152(1):36-43. [PubMed: 8444204]
- 367.
- Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13(6):802-18. [PubMed: 1706789]
- 368.
- Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, Boepple PA, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81(12):4388-95. [PubMed: 8954047]
- 369.
- Sykiotis GP, Pitteloud N, Seminara SB, Kaiser UB, Crowley WF, Jr. Deciphering genetic disease in the genomic era: the model of GnRH deficiency. Sci Transl Med. 2010;2(32):32rv2. [PMC free article: PMC3936248] [PubMed: 20484732]
- 370.
- Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A. 2010;107(34):15140-4. [PMC free article: PMC2930591] [PubMed: 20696889]
- 371.
- Linscott ML, Chung WCJ. Epigenomic control of gonadotrophin-releasing hormone neurone development and hypogonadotrophic hypogonadism. J Neuroendocrinol. 2020;32(6):e12860. [PubMed: 32452569]
- 372.
- Harrington J, Palmert MR. An Approach to the Patient With Delayed Puberty. J Clin Endocrinol Metab. 2022;107(6):1739-50. [PubMed: 35100608]
- 373.
- Molsted K, Kjaer I, Giwercman A, Vesterhauge S, Skakkebaek NE. Craniofacial morphology in patients with Kallmann's syndrome with and without cleft lip and palate. Cleft Palate Craniofac J. 1997;34(5):417-24. [PubMed: 9345610]
- 374.
- Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol. 2018;178(4):377-88. [PMC free article: PMC5863472] [PubMed: 29419413]
- 375.
- Husebye ES, Anderson MS, Kampe O. Autoimmune Polyendocrine Syndromes. N Engl J Med. 2018;378(12):1132-41. [PMC free article: PMC6007870] [PubMed: 29562162]
- 376.
- McElreavey K, Jorgensen A, Eozenou C, Merel T, Bignon-Topalovic J, Tan DS, et al. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet Med. 2020;22(1):150-9. [PMC free article: PMC6944638] [PubMed: 31337883]
- 377.
- Foster KL, Lee DJ, Witchel SF, Gordon CM. Ovarian Insufficiency and Fertility Preservation During and After Childhood Cancer Treatment. J Adolesc Young Adult Oncol. 2024. [PubMed: 38265460]
- 378.
- Matalliotakis M, Koliarakis I, Matalliotaki C, Trivli A, Hatzidaki E. Clinical manifestations, evaluation and management of hyperprolactinemia in adolescent and young girls: a brief review. Acta Biomed. 2019;90(1):149-57. [PMC free article: PMC6502148] [PubMed: 30889169]
- 379.
- Lee SL, Lim A, Munns C, Simm PJ, Zacharin M. Effect of Testosterone Treatment for Delayed Puberty in Duchenne Muscular Dystrophy. Horm Res Paediatr. 2020;93(2):108-18. [PubMed: 32610327]
- 380.
- Ward LM, Weber DR. Growth, pubertal development, and skeletal health in boys with Duchenne Muscular Dystrophy. Curr Opin Endocrinol Diabetes Obes. 2019;26(1):39-48. [PMC free article: PMC6402320] [PubMed: 30507696]
- 381.
- Esquivel-Zuniga R, Rogol AD. Functional hypogonadism in adolescence: an overlooked cause of secondary hypogonadism. Endocr Connect. 2023;12(11). [PMC free article: PMC10563622] [PubMed: 37615381]
- 382.
- Thavaraputta S, Ungprasert P, Witchel SF, Fazeli PK. Anorexia nervosa and adrenal hormones: a systematic review and meta-analysis. Eur J Endocrinol. 2023;189(3):S64-S73. [PMC free article: PMC10498414] [PubMed: 37669399]
- 383.
- Misra M, Golden NH, Katzman DK. State of the art systematic review of bone disease in anorexia nervosa. Int J Eat Disord. 2016;49(3):276-92. [PMC free article: PMC4769683] [PubMed: 26311400]
- 384.
- Odle AK, Akhter N, Syed MM, Allensworth-James ML, Benes H, Melgar Castillo AI, et al. Leptin Regulation of Gonadotrope Gonadotropin-Releasing Hormone Receptors As a Metabolic Checkpoint and Gateway to Reproductive Competence. Front Endocrinol (Lausanne). 2017;8:367. [PMC free article: PMC5760501] [PubMed: 29354094]
- 385.
- Vazquez MJ, Velasco I, Tena-Sempere M. Novel mechanisms for the metabolic control of puberty: implications for pubertal alterations in early-onset obesity and malnutrition. J Endocrinol. 2019;242(2):R51-R65. [PubMed: 31189134]
- 386.
- Cano Sokoloff N, Misra M, Ackerman KE. Exercise, Training, and the Hypothalamic-Pituitary-Gonadal Axis in Men and Women. Front Horm Res. 2016;47:27-43. [PMC free article: PMC7043068] [PubMed: 27348623]
- 387.
- Gordon CM, Ackerman KE, Berga SL, Kaplan JR, Mastorakos G, Misra M, et al. Functional Hypothalamic Amenorrhea: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2017;102(5):1413-39. [PubMed: 28368518]
- 388.
- De Lorenzo A, Noce A, Moriconi E, Rampello T, Marrone G, Di Daniele N, et al. MOSH Syndrome (Male Obesity Secondary Hypogonadism): Clinical Assessment and Possible Therapeutic Approaches. Nutrients. 2018;10(4). [PMC free article: PMC5946259] [PubMed: 29649106]
- 389.
- Federici S, Goggi G, Quinton R, Giovanelli L, Persani L, Cangiano B, et al. New and Consolidated Therapeutic Options for Pubertal Induction in Hypogonadism: In-depth Review of the Literature. Endocr Rev. 2022;43(5):824-51. [PubMed: 34864951]
- 390.
- Szeliga A, Podfigurna A, Bala G, Meczekalski B. Kisspeptin and neurokinin B analogs use in gynecological endocrinology: where do we stand? J Endocrinol Invest. 2020;43(5):555-61. [PubMed: 31838714]
- 391.
- Alexander EC, Faruqi D, Farquhar R, Unadkat A, Ng Yin K, Hoskyns R, et al. Gonadotropins for pubertal induction in males with hypogonadotropic hypogonadism: systematic review and meta-analysis. Eur J Endocrinol. 2024;190(1):S1-S11. [PMC free article: PMC10773669] [PubMed: 38128110]
- 392.
- Stancampiano MR, Lucas-Herald AK, Russo G, Rogol AD, Ahmed SF. Testosterone Therapy in Adolescent Boys: The Need for a Structured Approach. Horm Res Paediatr. 2019;92(4):215-28. [PubMed: 31851967]
- 393.
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357(9):863-73. [PubMed: 17761590]
- 394.
- Sun T, Xu W, Chen Y, Niu Y, Wang T, Wang S, et al. Reversal of idiopathic hypogonadotropic hypogonadism in a Chinese male cohort. Andrologia. 2022;54(11):e14583. [PubMed: 36123965]
- 395.
- Klein KO, Phillips SA. Review of Hormone Replacement Therapy in Girls and Adolescents with Hypogonadism. J Pediatr Adolesc Gynecol. 2019;32(5):460-8. [PubMed: 31059821]
- 396.
- Cheuiche AV, da Silveira LG, de Paula LCP, Lucena IRS, Silveiro SP. Diagnosis and management of precocious sexual maturation: an updated review. Eur J Pediatr. 2021;180(10):3073-87. [PubMed: 33745030]
- 397.
- Ibanez L, Ferrer A, Marcos MV, Hierro FR, de Zegher F. Early puberty: rapid progression and reduced final height in girls with low birth weight. Pediatrics. 2000;106(5):E72. [PubMed: 11061809]
- 398.
- Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, et al. Clinical Management of Congenital Hypogonadotropic Hypogonadism. Endocr Rev. 2019;40(2):669-710. [PubMed: 30698671]
- 399.
- Varimo T, Hero M, Laitinen EM, Miettinen PJ, Tommiska J, Kansakoski J, et al. Childhood growth in boys with congenital hypogonadotropic hypogonadism. Pediatr Res. 2016;79(5):705-9. [PubMed: 26720605]
- 400.
- Varimo T, Miettinen PJ, Kansakoski J, Raivio T, Hero M. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center. Hum Reprod. 2017;32(1):147-53. [PubMed: 27927844]
- 401.
- Neely EK, Hintz RL, Wilson DM, Lee PA, Gautier T, Argente J, et al. Normal ranges for immunochemiluminometric gonadotropin assays. J Pediatr. 1995;127(1):40-6. [PubMed: 7608809]
- 402.
- Martinez-Aguayo A, Hernández MI, Capurro T, Peña V, Avila A, Salazar T, et al. Leuprolide acetate gonadotrophin response patterns during female puberty. Clin Endocrinol (Oxf). 2010;72(4):489-95. [PubMed: 19863573]
- 403.
- Cao R, Liu J, Fu P, Zhou Y, Li Z, Liu P. The Diagnostic Utility of the Basal Luteinizing Hormone Level and Single 60-Minute Post GnRH Agonist Stimulation Test for Idiopathic Central Precocious Puberty in Girls. Front Endocrinol (Lausanne). 2021;12:713880. [PMC free article: PMC8387794] [PubMed: 34456870]
- 404.
- Howard SR. Interpretation of reproductive hormones before, during and after the pubertal transition-Identifying health and disordered puberty. Clin Endocrinol (Oxf). 2021;95(5):702-15. [PMC free article: PMC9291332] [PubMed: 34368982]
- 405.
- Bizzarri C, Spadoni GL, Bottaro G, Montanari G, Giannone G, Cappa M, et al. The response to gonadotropin releasing hormone (GnRH) stimulation test does not predict the progression to true precocious puberty in girls with onset of premature thelarche in the first three years of life. J Clin Endocrinol Metab. 2014;99(2):433-9. [PubMed: 24297793]
- 406.
- Vestergaard ET, Schjorring ME, Kamperis K, Petersen KK, Rittig S, Juul A, et al. The follicle-stimulating hormone (FSH) and luteinizing hormone (LH) response to a gonadotropin-releasing hormone analogue test in healthy prepubertal girls aged 10 months to 6 years. Eur J Endocrinol. 2017;176(6):747-53. [PubMed: 28348072]
- 407.
- Harrington J, Palmert MR. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab. 2012;97(9):3056-67. [PubMed: 22723321]
- 408.
- Degros V, Cortet-Rudelli C, Soudan B, Dewailly D. The human chorionic gonadotropin test is more powerful than the gonadotropin-releasing hormone agonist test to discriminate male isolated hypogonadotropic hypogonadism from constitutional delayed puberty. European Journal of Endocrinology. 2003;149(1):23-9. [PubMed: 12824862]
- 409.
- Chaudhary S, Walia R, Bhansali A, Dayal D, Sachdeva N, Singh T, et al. FSH-stimulated Inhibin B (FSH-iB): A Novel Marker for the Accurate Prediction of Pubertal Outcome in Delayed Puberty. J Clin Endocrinol Metab. 2021;106(9):e3495-e505. [PubMed: 34010394]
- 410.
- Turcu AF, Mallappa A, Nella AA, Chen X, Zhao L, Nanba AT, et al. 24-Hour Profiles of 11-Oxygenated C(19) Steroids and Delta(5)-Steroid Sulfates during Oral and Continuous Subcutaneous Glucocorticoids in 21-Hydroxylase Deficiency. Front Endocrinol (Lausanne). 2021;12:751191. [PMC free article: PMC8636728] [PubMed: 34867794]
- 411.
- Trost LW, Mulhall JP. Challenges in Testosterone Measurement, Data Interpretation, and Methodological Appraisal of Interventional Trials. J Sex Med. 2016;13(7):1029-46. [PMC free article: PMC5516925] [PubMed: 27209182]
- 412.
- French D. Clinical utility of laboratory developed mass spectrometry assays for steroid hormone testing. J Mass Spectrom Adv Clin Lab. 2023;28:13-9. [PMC free article: PMC9900367] [PubMed: 36756146]
- 413.
- Chin VL, Cai Z, Lam L, Shah B, Zhou P. Evaluation of puberty by verifying spontaneous and stimulated gonadotropin values in girls. J Pediatr Endocrinol Metab. 2015;28(3-4):387-92. [PMC free article: PMC4767152] [PubMed: 25514323]
- 414.
- Abbara A, Eng PC, Phylactou M, Clarke SA, Mills E, Chia G, et al. Kisspeptin-54 Accurately Identifies Hypothalamic Gonadotropin-Releasing Hormone Neuronal Dysfunction in Men with Congenital Hypogonadotropic Hypogonadism. Neuroendocrinology. 2021;111(12):1176-86. [PubMed: 33227799]
- 415.
- Rosenfield RL. Normal and Premature Adrenarche. Endocr Rev. 2021;42(6):783-814. [PMC free article: PMC8599200] [PubMed: 33788946]
- 416.
- Chesover AD, Millar H, Sepiashvili L, Adeli K, Palmert MR, Hamilton J. Screening for Nonclassic Congenital Adrenal Hyperplasia in the Era of Liquid Chromatography-Tandem Mass Spectrometry. J Endocr Soc. 2020;4(2):bvz030. [PMC free article: PMC7041698] [PubMed: 32110745]
- 417.
- Gao Y, Du Q, Liu L, Liao Z. Serum inhibin B for differentiating between congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty: a systematic review and meta-analysis. Endocrine. 2021;72(3):633-43. [PMC free article: PMC8159787] [PubMed: 33464540]
- 418.
- Binder G, Schweizer R, Blumenstock G, Braun R. Inhibin B plus LH vs GnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys. Clin Endocrinol (Oxf). 2015;82(1):100-5. [PubMed: 25207430]
- 419.
- Mosbah H, Bouvattier C, Maione L, Trabado S, De Filippo G, Cartes A, et al. GnRH stimulation testing and serum inhibin B in males: insufficient specificity for discriminating between congenital hypogonadotropic hypogonadism from constitutional delay of growth and puberty. Hum Reprod. 2020;35(10):2312-22. [PubMed: 32862222]
- 420.
- Albrethsen J, Ljubicic ML, Juul A. Longitudinal Increases in Serum Insulin-like Factor 3 and Testosterone Determined by LC-MS/MS in Pubertal Danish Boys. J Clin Endocrinol Metab. 2020;105(10). [PubMed: 32761207]
- 421.
- Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Clin Endocrinol Metab. 2009;94(3):780-5. [PubMed: 19017752]
- 422.
- Greulich WW PS. Radiographic Atlas of Skeletal Development of the Hand and Wrist. 2nd ed: Stanford University Press; 1999.
- 423.
- Tanner JM WR, Cameron N, et al., Assessment of Skeletal Maturity and Prediction of Adult Height (TW2 Method). 2nd ed: Goldstein H (Ed), Academic Press; 1983.
- 424.
- Klein KO, Newfield RS, Hassink SG. Bone maturation along the spectrum from normal weight to obesity: a complex interplay of sex, growth factors and weight gain. J Pediatr Endocrinol Metab. 2016;29(3):311-8. [PubMed: 26565541]
- 425.
- Oh MS, Kim S, Lee J, Lee MS, Kim YJ, Kang KS. Factors associated with Advanced Bone Age in Overweight and Obese Children. Pediatr Gastroentero. 2020;23(1):89-97. [PMC free article: PMC6966218] [PubMed: 31988879]
- 426.
- Bar A, Linder B, Sobel EH, Saenger P, Dimartinonardi J. Bayley-Pinneau Method of Height Prediction in Girls with Central Precocious Puberty - Correlation with Adult Height. J Pediatr-Us. 1995;126(6):955-8. [PubMed: 7776106]
- 427.
- Eitel KB, Eugster EA. Differences in Bone Age Readings between Pediatric Endocrinologists and Radiologists. Endocrine Practice. 2020;26(3):328-31. [PubMed: 31859549]
- 428.
- Wang X, Zhou B, Gong P, Zhang T, Mo Y, Tang J, et al. Artificial Intelligence-Assisted Bone Age Assessment to Improve the Accuracy and Consistency of Physicians With Different Levels of Experience. Frontiers in Pediatrics. 2022;10. [PMC free article: PMC8908427] [PubMed: 35281250]
- 429.
- Thodberg HH, Thodberg B, Ahlkvist J, Offiah AC. Autonomous artificial intelligence in pediatric radiology: the use and perception of BoneXpert for bone age assessment. Pediatr Radiol. 2022;52(7):1338-46. [PMC free article: PMC9192461] [PubMed: 35224658]
- 430.
- de Vries L, Horev G, Schwartz M, Phillip M. Ultrasonographic and clinical parameters for early differentiation between precocious puberty and premature thelarche. Eur J Endocrinol. 2006;154(6):891-8. [PubMed: 16728550]
- 431.
- Sathasivam A, Rosenberg HK, Shapiro S, Wang H, Rapaport R. Pelvic ultrasonography in the evaluation of central precocious puberty: comparison with leuprolide stimulation test. J Pediatr. 2011;159(3):490-5. [PubMed: 21489559]
- 432.
- Lee SH, Joo EY, Lee JE, Jun YH, Kim MY. The Diagnostic Value of Pelvic Ultrasound in Girls with Central Precocious Puberty. Chonnam Med J. 2016;52(1):70-4. [PMC free article: PMC4742613] [PubMed: 26866003]
- 433.
- Battaglia C, Mancini F, Regnani G, Persico N, Iughetti L, De Aloysio D. Pelvic ultrasound and color Doppler findings in different isosexual precocities. Ultrasound Obstet Gynecol. 2003;22(3):277-83. [PubMed: 12942501]
- 434.
- Long MG, Boultbee JE, Hanson ME, Begent RH. Doppler time velocity waveform studies of the uterine artery and uterus. Br J Obstet Gynaecol. 1989;96(5):588-93. [PubMed: 2667631]
- 435.
- Paesano PL, Colantoni C, Mora S, di Lascio A, Ferrario M, Esposito A, et al. Validation of an Accurate and Noninvasive Tool to Exclude Female Precocious Puberty: Pelvic Ultrasound With Uterine Artery Pulsatility Index. Am J Roentgenol. 2019;213(2):451-7. [PubMed: 31039031]
- 436.
- Cheuiche AV, Moro C, Lucena IRS, de Paula LCP, Silveiro SP. Accuracy of doppler assessment of the uterine arteries for the diagnosis of pubertal onset in girls: a scoping review. Sci Rep. 2023;13(1):5791. [PMC free article: PMC10082829] [PubMed: 37031290]
- 437.
- Kafi SE, Alagha E, Shazly MA, Al-Agha A. Pseudo-precocious Puberty Associated with an Adrenocortical Tumor in a Young Child. Cureus. 2019;11(12). [PMC free article: PMC6973539] [PubMed: 31998569]
- 438.
- Alagha E, Kafi SE, Shazly MA, Al-Agha A. Precocious Puberty Associated with Testicular Hormone-secreting Leydig Cell Tumor. Cureus. 2019;11(12):e6441. [PMC free article: PMC6929247] [PubMed: 31893190]
- 439.
- Eyer de Jesus L, Paz de Oliveira AP, Porto LC, Dekermacher S. Testicular adrenal rest tumors - Epidemiology, diagnosis and treatment. J Pediatr Urol. 2023. [PubMed: 37845103]
- 440.
- Cantas-Orsdemir S, Garb JL, Allen HF. Prevalence of cranial MRI findings in girls with central precocious puberty: a systematic review and meta-analysis. J Pediatr Endocrinol Metab. 2018;31(7):701-10. [PubMed: 29902155]
- 441.
- Pedicelli S, Alessio P, Scire G, Cappa M, Cianfarani S. Routine screening by brain magnetic resonance imaging is not indicated in every girl with onset of puberty between the ages of 6 and 8 years. J Clin Endocrinol Metab. 2014;99(12):4455-61. [PubMed: 25238205]
- 442.
- Kaplowitz PB. Do 6-8 year old girls with central precocious puberty need routine brain imaging? Int J Pediatr Endocrinol. 2016;2016:9. [PMC free article: PMC4855709] [PubMed: 27148371]
- 443.
- Mogensen SS, Aksglaede L, Mouritsen A, Sorensen K, Main KM, Gideon P, et al. Pathological and Incidental Findings on Brain MRI in a Single-Center Study of 229 Consecutive Girls with Early or Precocious Puberty. Plos One. 2012;7(1). [PMC free article: PMC3257249] [PubMed: 22253792]
- 444.
- Hansen AB, Renault CH, Wojdemann D, Gideon P, Juul A, Jensen RB. Neuroimaging in 205 consecutive Children Diagnosed with Central Precocious Puberty in Denmark. Pediatr Res. 2023;93(1):125-30. [PubMed: 35365758]
- 445.
- Kim SH, Ahn MB, Cho WK, Cho KS, Jung MH, Suh BK. Findings of Brain Magnetic Resonance Imaging in Girls with Central Precocious Puberty Compared with Girls with Chronic or Recurrent Headache. J Clin Med. 2021;10(10). [PMC free article: PMC8160955] [PubMed: 34069752]
- 446.
- Eugster EA. Update on Precocious Puberty in Girls. J Pediatr Adol Gynec. 2019;32(5):455-9. [PubMed: 31158483]
- 447.
- Yoon JS, So CH, Lee HS, Lim JS, Hwang JS. The prevalence of brain abnormalities in boys with central precocious puberty may be overestimated. Plos One. 2018;13(4). [PMC free article: PMC5882100] [PubMed: 29614125]
- 448.
- Kang JY, Kim SH, Kim H, Ki H, Lee MH. Pituitary magnetic resonance imaging abnormalities in young female patients with hypogonadotropic hypogonadism. Obstet Gynecol Sci. 2019;62(4):249-57. [PMC free article: PMC6629987] [PubMed: 31338342]
- 449.
- Hacquart T, Ltaief-Boudrigua A, Jeannerod C, Hannoun S, Raverot G, Pugeat M, et al. Reconsidering olfactory bulb magnetic resonance patterns in Kallmann syndrome. Ann Endocrinol (Paris). 2017;78(5):455-61. [PubMed: 28807454]
- 450.
- Sarfati J, Saveanu A, Young J. Pituitary stalk interruption and olfactory bulbs aplasia/hypoplasia in a man with Kallmann syndrome and reversible gonadotrope and somatotrope deficiencies. Endocrine. 2015;49(3):865-6. [PubMed: 25381604]
- 451.
- Elnaw EAA, Ibrahim AAB, Abdullah MA. Feminizing adrenocortical adenoma in a girl from a resource-limited setting: a case report. J Med Case Rep. 2021;15(1). [PMC free article: PMC8690961] [PubMed: 34930443]
- 452.
- Bulut S, Catli G, Filibeli BE, Manyas H, Ayranci I, Meral R, et al. Virilizing Adrenocortical Carcinoma Oncocytic Variant in a Child with Heterosexual Precocious Puberty and a Literature Review. J Pediatr Res. 2022;9(3):307-13.
- 453.
- Ko JH, Lee HS, Hong J, Hwang JS. Virilizing adrenocortical carcinoma in a child with Turner syndrome and somatic TP53 gene mutation. European Journal of Pediatrics. 2010;169(4):501-4. [PubMed: 19701813]
- 454.
- Elnaw EAA, Ibrahim AAB, Abdullah MA. Feminizing adrenocortical adenoma in a girl from a resource-limited setting: a case report. J Med Case Rep. 2021;15(1):605. [PMC free article: PMC8690961] [PubMed: 34930443]
- 455.
- Gravholt CH, Viuff M, Just J, Sandahl K, Brun S, van der Velden J, et al. The Changing Face of Turner Syndrome. Endocr Rev. 2023;44(1):33-69. [PubMed: 35695701]
- 456.
- Correa Brito L, Rey RA. Taming Idiopathic Central Precocious Puberty: High Frequency of Imprinting Disorders in Familial Forms. J Clin Endocrinol Metab. 2023;108(8):e636-e7. [PMC free article: PMC10348455] [PubMed: 36794430]
- 457.
- Aguirre RS, Eugster EA. Central precocious puberty: From genetics to treatment. Best Pract Res Cl En. 2018;32(4):343-54. [PubMed: 30086862]
- 458.
- Howard SR, Dunkel L. Delayed Puberty-Phenotypic Diversity, Molecular Genetic Mechanisms, and Recent Discoveries. Endocr Rev. 2019;40(5):1285-317. [PMC free article: PMC6736054] [PubMed: 31220230]
- 459.
- Howard SR. Genes underlying delayed puberty. Mol Cell Endocrinol. 2018;476:119-28. [PMC free article: PMC6127442] [PubMed: 29730183]
- 460.
- Villanueva C, Jacobson-Dickman E, Xu C, Manouvrier S, Dwyer AA, Sykiotis GP, et al. Congenital hypogonadotropic hypogonadism with split hand/foot malformation: a clinical entity with a high frequency of FGFR1 mutations. Genet Med. 2015;17(8):651-9. [PMC free article: PMC4430466] [PubMed: 25394172]
- 461.
- LATRONICO A. Deciphering the genetic basis of central precocious puberty. Rev Esp Endocrinol Pediatr. 2023;14 Suppl(2):17-20
- 462.
- Stecchini MF, Macedo DB, Reis AC, Abreu AP, Moreira AC, Castro M, et al. Time Course of Central Precocious Puberty Development Caused by an MKRN3 Gene Mutation: A Prismatic Case. Horm Res Paediatr. 2016;86(2):126-30. [PMC free article: PMC5061599] [PubMed: 27424312]
- 463.
- Teles MG, Bianco SDC, Brito VN, Trarbach EB, Kuohung W, Xu SY, et al. A GPR54-activating mutation in a patient with central precocious puberty. New Engl J Med. 2008;358(7):709-15. [PMC free article: PMC2859966] [PubMed: 18272894]
- 464.
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. New Engl J Med. 2003;349(17):1614-U8. [PubMed: 14573733]
- 465.
- Krstevska-Konstantinova M, Jovanovska J, Tasic VB, Montenegro LR, Beneduzzi D, Silveira LF, et al. Mutational analysis of KISS1 and KISS1R in idiopathic central precocious puberty. J Pediatr Endocrinol Metab. 2014;27(1-2):199-201. [PubMed: 23950571]
- 466.
- Grandone A, Capristo C, Cirillo G, Sasso M, Umano GR, Mariani M, et al. Molecular Screening of MKRN3, DLK1, and KCNK9 Genes in Girls with Idiopathic Central Precocious Puberty. Horm Res Paediatr. 2017;88(3-4):194-200. [PubMed: 28672280]
- 467.
- Macedo DB, Kaiser UB. DLK1, Notch Signaling and the Timing of Puberty. Semin Reprod Med. 2019;37(4):174-81. [PMC free article: PMC8585528] [PubMed: 31972862]
- 468.
- Montenegro L, Labarta JI, Piovesan M, Canton APM, Corripio R, Soriano-Guillen L, et al. Novel Genetic and Biochemical Findings of DLK1 in Children with Central Precocious Puberty: A Brazilian-Spanish Study. J Clin Endocrinol Metab. 2020;105(10). [PubMed: 32676665]
- 469.
- Soares JM, de Holanda FS, Matsuzaki CN, Sorpreso ICE, Veiga ECD, de Abreu LC, et al. Analysis of the PvuII and XbaI polymorphisms in the estrogen receptor alpha gene in girls with central precocious puberty: a pilot study. Bmc Med Genet. 2018;19. [PMC free article: PMC5970514] [PubMed: 29801469]
- 470.
- Lee HS, Park HK, Kim KH, Ko JH, Kim YJ, Yi KH, et al. Estrogen receptor alpha gene analysis in girls with central precocious puberty. J Pediatr Endocr Met. 2013;26(7-8):645-9. [PubMed: 23585209]
- 471.
- Lee HS, Kim KH, Hwang JS. Association of aromatase (TTTA)n repeat polymorphisms with central precocious puberty in girls. Clin Endocrinol (Oxf). 2014;81(3):395-400. [PubMed: 24612204]
- 472.
- Tsuji-Hosokawa A, Matsuda N, Kurosawa K, Kashimada K, Morio T. A Case of MECP2 Duplication Syndrome with Gonadotropin-Dependent Precocious Puberty. Horm Res Paediatr. 2017;87(4):271-6. [PubMed: 27649574]
- 473.
- Canton APM, Krepischi ACV, Montenegro LR, Costa S, Rosenberg C, Steunou V, et al. Insights from the genetic characterization of central precocious puberty associated with multiple anomalies. Hum Reprod. 2021;36(2):506-18. [PubMed: 33313884]
- 474.
- Czako M, Till A, Zima J, Zsigmond A, Szabo A, Maasz A, et al. Xp11.2 Duplication in Females: Unique Features of a Rare Copy Number Variation. Front Genet. 2021;12:635458. [PMC free article: PMC8080037] [PubMed: 33936165]
- 475.
- Ludwig NG, Radaeli RF, da Silva MMX, Romero CM, Carrilho AJF, Bessa D, et al. A boy with Prader-Willi syndrome: unmasking precocious puberty during growth hormone replacement therapy. Arch Endocrin Metab. 2016;60(6):596-600. [PMC free article: PMC10522161] [PubMed: 27982202]
- 476.
- Hoffmann K, Heller R. Uniparental disomies 7 and 14. Best Pract Res Clin Endocrinol Metab. 2011;25(1):77-100. [PubMed: 21396576]
- 477.
- Partsch CJ, Japing I, Siebert R, Gosch A, Wessel A, Sippell WG, et al. Central precocious puberty in girls with Williams syndrome. J Pediatr. 2002;141(3):441-4. [PubMed: 12219071]
- 478.
- Utine GE, Alikasifoglu A, Alikasifoglu M, Tuncbilek E. Central precocious puberty in a girl with Williams syndrome: the result of treatment with GnRH analogue. Eur J Med Genet. 2006;49(1):79-82. [PubMed: 16473313]
- 479.
- Kuroki Y, Katsumata N, Eguchi T, Fukushima Y, Suwa S, Kajii T. Precocious puberty in Kabuki makeup syndrome. J Pediatr. 1987;110(5):750-2. [PubMed: 3572628]
- 480.
- Concolino D, Muzzi G, Pisaturo L, Piccirillo A, Di Natale P, Strisciuglio P. Precocious puberty in Sanfilippo IIIA disease: diagnosis and follow-up of two new cases. Eur J Med Genet. 2008;51(5):466-71. [PubMed: 18586597]
- 481.
- Ray LA, Eckert GJ, Eugster EA. Long-term experience with the use of a single histrelin implant beyond one year in patients with central precocious puberty. J Pediatr Endocrinol Metab. 2023;36(3):309-12. [PubMed: 36625262]
- 482.
- Saengkaew T, Howard SR. Genetics of pubertal delay. Clin Endocrinol (Oxf). 2022;97(4):473-82. [PMC free article: PMC9543006] [PubMed: 34617615]
- 483.
- Akram M, Raza Rizvi SS, Qayyum M, Handelsman DJ. A classification of genes involved in normal and delayed male puberty. Asian J Androl. 2023;25(2):230-9. [PMC free article: PMC10069694] [PubMed: 35532554]
- 484.
- Vilain C, Mortier G, Van Vliet G, Dubourg C, Heinrichs C, de Silva D, et al. Hartsfield Holoprosencephaly-Ectrodactyly Syndrome in Five Male Patients: Further Delineation and Review. American Journal of Medical Genetics Part A. 2009;149a(7):1476-81. [PubMed: 19504604]
- 485.
- Pitteloud N, Meysing A, Quinton R, Acierno J, Dwyer A, Plummer L, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Molecular and Cellular Endocrinology. 2006:60-9. [PubMed: 16764984]
- 486.
- Quennell JH, Mulligan AC, Tups A, Liu X, Phipps SJ, Kemp CJ, et al. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology. 2009;150(6):2805-12. [PMC free article: PMC2732287] [PubMed: 19179437]
- 487.
- Williams KW, Sohn JW, Donato J, Lee CE, Zhao JJ, Elmquist JK, et al. The Acute Effects of Leptin Require PI3K Signaling in the Hypothalamic Ventral Premammillary Nucleus. J Neurosci. 2011;31(37):13147-56. [PMC free article: PMC3319415] [PubMed: 21917798]
- 488.
- Donato J, Cravo RM, Frazao R, Elias CF. Hypothalamic Sites of Leptin Action Linking Metabolism and Reproduction. Neuroendocrinology. 2011;93(1):9-18. [PMC free article: PMC3066240] [PubMed: 21099209]
- 489.
- Farooqi IS, O'Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nat Clin Pract Endoc. 2008;4(10):569-77. [PubMed: 18779842]
- 490.
- Tata B, Huijbregts L, Jacquier S, Csaba Z, Genin E, Meyer V, et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 2014;12(9):e1001952. [PMC free article: PMC4172557] [PubMed: 25248098]
- 491.
- Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368(21):1992-2003. [PMC free article: PMC3738065] [PubMed: 23656588]
- 492.
- Synofzik M, Kernstock C, Haack TB, Schols L. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher-Neuhauser syndrome due to PNPLA6 mutations. J Neurol Neurosurg Psychiatry. 2015;86(5):580-1. [PubMed: 24790214]
- 493.
- Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137(Pt 1):69-77. [PMC free article: PMC3891450] [PubMed: 24355708]
- 494.
- Pingault V, Bodereau V, Baral V, Marcos S, Watanabe Y, Chaoui A, et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet. 2013;92(5):707-24. [PMC free article: PMC3644631] [PubMed: 23643381]
- 495.
- Tziaferi V, Kelberman D, Dattani MT. The role of SOX2 in hypogonadotropic hypogonadism. Sex Dev. 2008;2(4-5):194-9. [PubMed: 18987493]
- 496.
- Alatzoglou KS, Azriyanti A, Rogers N, Ryan F, Curry N, Noakes C, et al. SOX3 deletion in mouse and human is associated with persistence of the craniopharyngeal canal. J Clin Endocrinol Metab. 2014;99(12):E2702-8. [PubMed: 25140394]
- 497.
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547-64. [PubMed: 26194704]
- 498.
- Joustra SD, Wehkalampi K, Oostdijk W, Biermasz NR, Howard S, Silander TL, et al. IGSF1 variants in boys with familial delayed puberty. Eur J Pediatr. 2015;174(5):687-92. [PubMed: 25354429]
- 499.
- Niederberger C. Re: Identification of HESX1 mutations in Kallmann syndrome. J Urol. 2014;191(4):1081. [PubMed: 24703149]
- 500.
- Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83(4):511-9. [PMC free article: PMC2561938] [PubMed: 18834967]
- 501.
- Marcos S, Sarfati J, Leroy C, Fouveaut C, Parent P, Metz C, et al. The Prevalence of CHD7 Missense Versus Truncating Mutations is Higher in Patients With Kallmann Syndrome than in Typical CHARGE Patients (vol 99, pg E2138, 2014). J Clin Endocr Metab. 2015;100(1):317-. [PubMed: 25077900]
- 502.
- Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. P Natl Acad Sci USA. 2014;111(50):17953-8. [PMC free article: PMC4273325] [PubMed: 25472840]
- 503.
- Timmons M, Tsokos M, Asab MA, Seminara SB, Zirzow GC, Kaneski CR, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67(11):2066-9. [PMC free article: PMC1950601] [PubMed: 17159124]
- 504.
- Sato I, Onuma A, Goto N, Sakai F, Fujiwara I, Uematsu M, et al. A case with central and peripheral hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome) plus cataract. J Neurol Sci. 2011;300(1-2):179-81. [PubMed: 20884016]
- 505.
- Pelletier F, Perrier S, Cayami FK, Mirchi A, Saikali S, Tran LT, et al. Endocrine and Growth Abnormalities in 4H Leukodystrophy Caused by Variants in POLR3A, POLR3B, and POLR1C. J Clin Endocrinol Metab. 2021;106(2):e660-e74. [PMC free article: PMC7823228] [PubMed: 33005949]
- 506.
- Liu S, Yan L, Zhou X, Chen C, Wang D, Yuan G. Delayed-onset adrenal hypoplasia congenita and hypogonadotropic hypogonadism caused by a novel mutation in DAX1. J Int Med Res. 2020;48(2):300060519882151. [PMC free article: PMC7605007] [PubMed: 31642359]
- 507.
- Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, et al. Mutations in the Dax-1 Gene Give Rise to Both X-Linked Adrenal Hypoplasia Congenita and Hypogonadotropic Hypogonadism. Nature. 1994;372(6507):672-6. [PubMed: 7990958]
- 508.
- Jennings JE, Costigan C, Reardon W. Moebius sequence and hypogonadotrophic hypogonadism. Am J Med Genet A. 2003;123A(1):107-10. [PubMed: 14556256]
- 509.
- Alves C, Franco RR. Prader-Willi syndrome: endocrine manifestations and management. Arch Endocrinol Metab. 2020;64(3):223-34. [PMC free article: PMC10522225] [PubMed: 32555988]
- 510.
- Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome. Int J Pediatr Endocrinol. 2013;2013(1):14. [PMC free article: PMC3751775] [PubMed: 23962041]
- 511.
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8-13. [PMC free article: PMC3522196] [PubMed: 22713813]
- 512.
- Desai A, Jha O, Iyer V, Dada R, Kumar R, Tandon N. Reversible hypogonadism in Bardet-Biedl syndrome. Fertil Steril. 2009;92(1):391 e13-5. [PubMed: 19327768]
- 513.
- Jain V, Foo SH, Chooi S, Moss C, Goodwin R, Berland S, et al. Borjeson-Forssman-Lehmann syndrome: delineating the clinical and allelic spectrum in 14 new families. Eur J Hum Genet. 2023;31(12):1421-9. [PMC free article: PMC10689765] [PubMed: 37704779]
- 514.
- Wang T, Ren W, Fu F, Wang H, Li Y, Duan J. Digenic CHD7 and SMCHD1 inheritance Unveils phenotypic variability in a family mainly presenting with hypogonadotropic hypogonadism. Heliyon. 2024;10(1):e23272. [PMC free article: PMC10750161] [PubMed: 38148819]
- 515.
- Alavi O, Khamirani HJ, Zoghi S, Feili A, Dastgheib SA, Tabei SMB, et al. Two novel Warburg micro syndrome 1 cases caused by pathogenic variants in RAB3GAP1. Hum Genome Var. 2021;8(1):39. [PMC free article: PMC8548584] [PubMed: 34702808]
- 516.
- Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018;19(10):649-66. [PMC free article: PMC7136165] [PubMed: 29995837]
- 517.
- Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng BH, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 Are Identified in Individuals with Congenital Hypogonadotropic Hypogonadism. American Journal of Human Genetics. 2013;92(5):725-43. [PMC free article: PMC3644636] [PubMed: 23643382]
- 518.
- Bianco SDC, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nature Reviews Endocrinology. 2009;5(10):569-76. [PMC free article: PMC2864719] [PubMed: 19707180]
- 519.
- Gonzalez-Martinez D, Kim SH, Hu YL, Guimond S, Schofield J, Winyard P, et al. Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism. J Neurosci. 2004;24(46):10384-92. [PMC free article: PMC6730313] [PubMed: 15548653]
- 520.
- Maione L, Albarel F, Bouchard P, Gallant M, Flanagan CA, Bobe R, et al. R31C GNRH1 mutation and congenital hypogonadotropic hypogonadism. Plos One. 2013;8(7):e69616. [PMC free article: PMC3723855] [PubMed: 23936060]
- 521.
- Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, et al. Isolated Familial Hypogonadotropic Hypogonadism and a GNRH1 Mutation. New Engl J Med. 2009;360(26):2742-8. [PubMed: 19535795]
- 522.
- Layman LC, Cohen DP, Jin M, Xie J, Li Z, Reindollar RH, et al. Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nature Genetics. 1998;18(1):14-5. [PubMed: 9425890]
- 523.
- Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med. 2012;366(7):629-35. [PubMed: 22335740]
- 524.
- Xu C, Messina A, Somm E, Miraoui H, Kinnunen T, Acierno J, et al. KLB, encoding beta-Klotho, is mutated in patients with congenital hypogonadotropic hypogonadism. Embo Molecular Medicine. 2017;9(10):1379-97. [PMC free article: PMC5623842] [PubMed: 28754744]
- 525.
- Fergani C, Navarro VM. Expanding the Role of Tachykinins in the Neuroendocrine Control of Reproduction. Reproduction. 2016;153(1):R1-R14. [PubMed: 27754872]
- 526.
- Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, et al. SEMA3A, a Gene Involved in Axonal Pathfinding, Is Mutated in Patients with Kallmann Syndrome. Plos Genetics. 2012;8(8). [PMC free article: PMC3426548] [PubMed: 22927827]
- 527.
- Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, et al. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab. 2008;93(10):4113-8. [PubMed: 18682503]
- 528.
- Dode C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2(10):e175. [PMC free article: PMC1617130] [PubMed: 17054399]
- 529.
- Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD Protein that Interacts with Transcription Factor EMX1, Is Mutated in Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. American Journal of Human Genetics. 2010;87(4):465-79. [PMC free article: PMC2948809] [PubMed: 20887964]
- 530.
- Saengkaew T, Ruiz-Babot G, David A, Mancini A, Mariniello K, Cabrera CP, et al. Whole exome sequencing identifies deleterious rare variants in CCDC141 in familial self-limited delayed puberty. NPJ Genom Med. 2021;6(1):107. [PMC free article: PMC8688425] [PubMed: 34930920]
- 531.
- Kotan LD, Hutchins BI, Ozkan Y, Demirel F, Stoner H, Cheng PJ, et al. Mutations in FEZF1 cause Kallmann syndrome. Am J Hum Genet. 2014;95(3):326-31. [PMC free article: PMC4157145] [PubMed: 25192046]
- 532.
- Lofrano-Porto A, Barra GB, Giacomini LA, Nascimento PP, Latronico AC, Casulari LA, et al. Luteinizing hormone beta mutation and hypogonadism in men and women. N Engl J Med. 2007;357(9):897-904. [PubMed: 17761593]
- 533.
- Layman LC, Porto AL, Xie J, da Motta LA, da Motta LD, Weiser W, et al. FSH beta gene mutations in a female with partial breast development and a male sibling with normal puberty and azoospermia. J Clin Endocrinol Metab. 2002;87(8):3702-7. [PubMed: 12161499]
- 534.
- Berger K, Souza H, Brito VN, d'Alva CB, Mendonca BB, Latronico AC. Clinical and hormonal features of selective follicle-stimulating hormone (FSH) deficiency due to FSH beta-subunit gene mutations in both sexes. Fertil Steril. 2005;83(2):466-70. [PubMed: 15705395]
- 535.
- Mancini A, Howard SR, Cabrera CP, Barnes MR, David A, Wehkalampi K, et al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Human Molecular Genetics. 2019;28(8):1357-68. [PMC free article: PMC6452208] [PubMed: 30608578]
- 536.
- Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature. 2013;497(7450):517-20. [PubMed: 23644456]
- 537.
- Zang S, Yin X, Li P. FTO-mediated m(6)A demethylation regulates GnRH expression in the hypothalamus via the PLCbeta3/Ca(2+)/CAMK signalling pathway. Commun Biol. 2023;6(1):1297. [PMC free article: PMC10739951] [PubMed: 38129517]
- ABSTRACT
- INTRODUCTION
- CLINICAL FEATURES OF NORMAL PUBERTAL DEVELOPMENT
- STAGING OF PUBERTY
- DISCOVERY OF THE HYPOTHALAMIC-PITUITARY-GONADAL (HPG) AXIS
- MINI-PUBERTY
- SECULAR TRENDS REGARDING PUBERTY
- VARIATIONS IN PUBERTAL DEVELOPMENT
- PRECOCIOUS PUBERTY
- PREMATURE THELARCHE
- PREMATURE ADRENARCHE
- EXPOSURE TO EXOGENOUS SEX STERIODS
- DELAYED PUBERTY
- EVALUATION OF A CHILD WITH A VARIATION IN PUBERTAL DEVELOPMENT
- SUMMARY
- REFERENCES
- Review Physiology of GnRH and Gonadotrophin Secretion.[Endotext. 2000]Review Physiology of GnRH and Gonadotrophin Secretion.Marques P, De Sousa Lages A, Skorupskaite K, Rozario KS, Anderson RA, George JT. Endotext. 2000
- Review Diagnosis and management of precocious sexual maturation: an updated review.[Eur J Pediatr. 2021]Review Diagnosis and management of precocious sexual maturation: an updated review.Cheuiche AV, da Silveira LG, de Paula LCP, Lucena IRS, Silveiro SP. Eur J Pediatr. 2021 Oct; 180(10):3073-3087. Epub 2021 Mar 21.
- Review Normal Puberty.[Endocrinol Metab Clin North Am...]Review Normal Puberty.Bangalore Krishna K, Witchel SF. Endocrinol Metab Clin North Am. 2024 Jun; 53(2):183-194. Epub 2024 Mar 4.
- Pubertal development: normal, precocious and delayed.[Clin Endocrinol Metab. 1982]Pubertal development: normal, precocious and delayed.Ducharme JR, Collu R. Clin Endocrinol Metab. 1982 Mar; 11(1):57-87.
- The effect of medroxyprogesterone acetate on gonadotropin secretion in girls with precocious puberty.[Am J Med Sci. 1975]The effect of medroxyprogesterone acetate on gonadotropin secretion in girls with precocious puberty.Warren MP, Mathews JH, Morishima A, Vande Wiele R. Am J Med Sci. 1975 May-Jun; 269(3):375-81.
- Normal and Abnormal Puberty - EndotextNormal and Abnormal Puberty - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...