

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Chronic kidney disease (CKD) is associated with a mineral and bone disorder (CKD-MBD) which starts early in the course of the disease and worsens with its progression. The main initial serum biochemistry abnormalities are increases in fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH) and decreases in 1,25 dihydroxy vitamin D (calcitriol) and soluble α-Klotho (Klotho), allowing serum calcium and phosphate to stay normal. Subsequently, serum 25 hydroxy vitamin D (calcidiol) decreases and in late CKD stages hyperphosphatemia develops in the majority of patients. Serum calcium may stay normal, decrease, or increase. More recent reports showed that sclerostin, Dickkopf-1, and activin A also play an important role in the pathogenesis of CKD-MBD. Both the synthesis and the secretion of PTH are continuously stimulated in the course of CKD, resulting in secondary hyperparathyroidism. In addition to the above systemic disturbances, downregulation of vitamin D receptor, calcium-sensing receptor, and Klotho expression in parathyroid tissue further enhances PTH overproduction. Last but not least, miRNAs have also been shown to be involved in the hyperparathyroidism of CKD. The chronic stimulation of parathyroid secretory function is not only characterized by a progressive rise in serum PTH but also by parathyroid gland hyperplasia. It results from an increase in parathyroid cell proliferation which is not fully compensated by a concomitant increase in parathyroid cell apoptosis. Parathyroid hyperplasia is initially of the diffuse, polyclonal type. In late CKD stages it often evolves towards a nodular, monoclonal or multiclonal type of growth. Enhanced parathyroid expression of transforming growth factor-α and its receptor, the epidermal growth factor receptor, is involved in polyclonal hyperplasia. Chromosomal changes have been found to be associated with clonal outgrowth in some, but not the majority of benign parathyroid tumors removed from patients with end-stage kidney disease. In initial CKD stages skeletal resistance to the action of PTH may explain why low bone turnover predominates in a significant proportion of patients, together with other conditions inhibiting bone turnover such as reduced calcitriol levels, sex hormone deficiency, diabetes, Wnt inhibitors, and uremic toxins. High turnover bone disease (osteitis fibrosa) occurs only later on, when increased serum PTH levels are able to overcome skeletal PTH resistance. The diagnosis of secondary uremic hyperparathyroidism and osteitis fibrosa relies mainly on serum biochemistry. X-ray and other imaging methods of the skeleton provide diagnostically relevant information only in severe forms. From a therapeutic point of view, it is important to prevent the development of secondary hyperparathyroidism as early as possible in the course of CKD. A variety of prophylactic and therapeutic approaches are available, as outlined in the final part of the chapter. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Chronic kidney disease (CKD) is almost constantly associated with a systemic disorder of mineral and bone metabolism, at present named CKD-MBD (1). According to this definition, the disorder is manifested by either one or a combination of biochemical abnormalities (abnormal calcium, phosphate, PTH, or vitamin D metabolism), bone abnormalities (abnormal bone turnover, mineralization, volume, linear growth, or strength) and vascular or other soft tissue calcification. More recently, the underlying pathophysiology has become more complex, with the progressive awareness that fibroblast growth factor 23 (FGF23), a-Klotho (subsequently called "Klotho") as well as the Wnt-b-catenin signaling pathway also play an important role (see below). CKD-MBD generally becomes apparent in CKD stage G3, i.e., at a glomerular filtration rate between 60 and 30 ml/min x 1.73 m2. Initially, it is characterized by a tendency towards hypocalcemia, fasting normo- or hypophosphatemia, and diminished plasma 25OH vitamin D (calcidiol) and 1,25diOH vitamin D (calcitriol) concentrations, together with a progressive increase in plasma FGF23 and intact parathyroid hormone (iPTH), a decrease in plasma soluble Klotho (2–5) and the development of renal osteodystrophy. Renal osteodystrophy often presents initially as adynamic bone disease and subsequently transforms into osteitis fibrosa or mixed bone disease (6). Pure osteomalacia is seen only infrequently. The low bone turnover observed in a significant proportion of patients in early stages of CKD could be due to the initial predominance of bone turnover inhibitory conditions such as resistance to the action of PTH, reduced serum calcitriol levels, sex hormone deficiency, diabetes, inflammation and malnutrition, and uremic toxins leading to the repression of osteocyte Wnt-β-catenin signaling and increased expression of Wnt antagonists such as sclerostin, Dickkopf-1, and secreted frizzled-related protein 4 (7,8). According to this scenario, high turnover bone disease occurs only later on, when sufficiently elevated serum PTH levels are able to overcome the skeletal resistance to its action. Even at that stage, over suppression of PTH by the administration of excessive calcium and/or vitamin D supplements can again induce adynamic bone disease (9). Nephrologists became progressively aware of the fact that the abnormally high serum phosphorus levels in late CKD stages, associated with either hyperparathyroidism or (mostly iatrogenically induced) hypoparathyroidism, may be detrimental to the patients not only in terms of abnormal bone structure and strength, but also in terms of the relative risk of soft-tissue calcifications and cardiovascular as well as all-cause mortality (10–13). As regards serum PTH levels, observational studies have consistently reported an increased relative risk of death in patients with CKD stage G5 and PTH values at the extremes, that is less than two or greater than nine times the upper normal limit of the assay (14,15). For PTH values within the range of two to nine times the upper normal limit reports of associations with relative risk of cardiovascular events or death in patients with CKD have been inconsistent. Of note, however, a report in elderly men in the community showed a strong association between plasma iPTH in the normal range and cardiovascular mortality (16).
SECONDARY HYPERPARATHYROIDISM IN CKD – SEQUENCE OF PLASMA BIOCHEMISTRY CHANGES IN EARLY CKD STAGES (FIGURE 1)
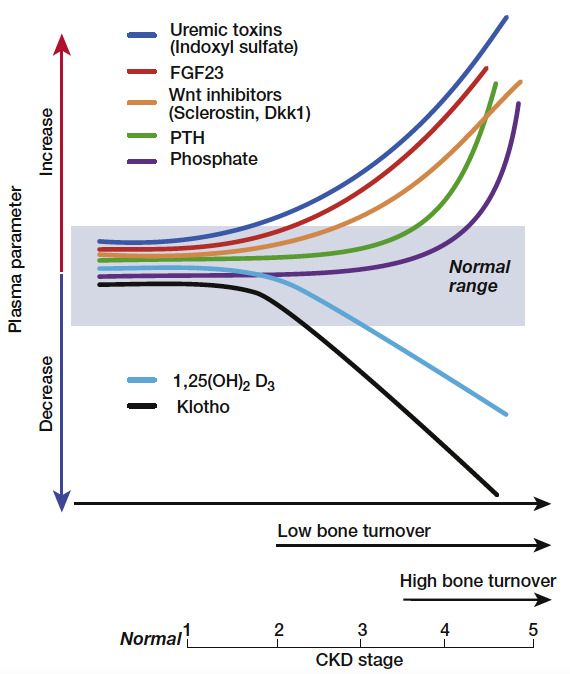
Figure 1.
Schematic view of the time profile of disturbances in mineral hormones and bone turnover with progression of chronic kidney disease (CKD). From Drueke & Massy (6).
Phosphate Retention
The precise sequence of metabolic anomalies in incipient CKD leading to secondary hyperparathyroidism remains a matter of debate. Many years ago, it was postulated that a retention of phosphate in the extracellular space due to the decrease in glomerular filtration rate and the accompanying reduction in plasma ionized calcium concentration was the primary event in the pathogenesis of secondary hyperparathyroidism. These anomalies would only be transient and a new steady state would rapidly be reached, with normalization of plasma calcium and phosphate in response to increased PTH secretion and the well-known inhibitory effect of this hormone on the tubular reabsorption of phosphate (“trade-off hypothesis” of Bricker and Slatopolsky) (17). However, this hypothesis has become less attractive since it was demonstrated that plasma phosphate is only rarely elevated in early CKD, and phosphate balance was found to be not positive but negative, at least in rats with moderate-degree CKD (18). Most often, plasma phosphate remains normal until CKD stages G4-G5 (2,19). It may even be moderately diminished in CKD (20). Oral phosphate absorption remains normal in early stages of experimental CKD (18), and urinary phosphate excretion after an oral overload in patients with mild CKD was actually found to be accelerated (20). Nonetheless, one could argue that in early kidney failure normal or even subnormal concentrations of plasma phosphate might be observed after a slight, initial plasma phosphate increase following phosphate ingestion and stimulation of the secretion of FGF23 and PTH, which in turn could overcorrect plasma phosphate rapidly, due to a more potent inhibition of tubular phosphate reabsorption. However, a more recent study identified slight increases of plasma phosphate in a large US population sample (NHANES III) with CKD stage G3 as compared to a healthy control population without evidence of kidney disease (21). Probably both the time of plasma phosphate determinations during the day as well as subtle changes in circulating and local factors involved in the control of phosphate balance determine the actual plasma level of phosphate in patients with CKD.
Fibroblast Growth Factor 23 (FGF23) and Klotho
FGF23 is recognized at present as a major, if not the most important player in the control of phosphate metabolism. It is mainly produced by osteocytes and osteoblasts. It decreases plasma phosphate by reducing tubular phosphate reabsorption similar to, but independent of PTH. Moreover, in contrast to PTH it decreases the renal synthesis of calcitriol. To activate its receptors FGFR-1 and FGFR-3 on tubular epithelial cells it requires the presence of Klotho (or more precisely α-Klotho), which in its function as a co-receptor confers FGF receptor specificity for FGF23 (22). Although initially Klotho expression was found only in the distal tubule, it has subsequently been demonstrated to occur in the proximal tubule as well. In line with this finding, ablation of Klotho specifically from the distal tubules certainly resulted in a hyperphosphatemic phenotype, but to a lesser degree than in systemic or whole nephron Klotho knockout models (23). The regulation of FGF23 production and its interrelations with PTH, calcitriol, calcium, phosphate, and Klotho are complex, being only progressively unraveled. Isakova et al. provided evidence that serum FGF23 increased earlier than serum iPTH in patients with CKD (4). This observation is also supported by experiments in an animal model of CKD and the use of anti-FGF23 antibodies (24). However, the authors of a subsequent large-scale population study took issue with the claim that the increase in circulating FGF23 preceded that of PTH (25). Klotho expression in kidney, Klotho plasma levels, and Klotho urinary excretion decrease with progressive CKD (26,27). The presence of Klotho is required to allow FGF23 to exert its action in the kidney. In addition, Klotho also exerts FGF23 independent effects. It acts from the tubular luminal side as an autocrine or paracrine enzyme to regulate transporters and ion channels. By modifying the Na-phosphate cotransporter NaPi2a it can enhance phosphaturia directly (28). However, its purported glycosidase activity has been put into question recently (29). The issue then arises which comes first in CKD, an increase in FGF23 or a decrease in Klotho? The answer remains a matter of debate (30). Some studies showed that secreted soluble Klotho levels decrease before FGF23 levels increase (31,32) but the sequence of events may differ depending on experimental models and diverse clinical conditions (33). CKD is probably the most common cause of chronically elevated serum FGF23 levels (34). FGF23 production in bone is increased by phosphate, calcitriol, calcium, PTH, Klotho, and iron. Not all of these effects are necessarily direct. The effect of PTH clearly is both direct, via stimulation of PTH receptor-1 (PTH-R1) (35) and the orphan nuclear receptor Nurr1 (36), and indirect, via an increase in calcitriol synthesis (37). On the other hand, FGF23 inhibits PTH synthesis and secretion although in CKD this effect is mitigated by reduced Klotho and FGFR-1 expression in parathyroid tissue (38–40).
The increase in circulating FGF23 with the progression of CKD is independently associated with serum phosphate, calcium, iPTH, and calcitriol (41,42). Despite its direct inhibitory action on the parathyroid tissue FGF23 contributes to the progression of secondary hyperparathyroidism by reducing renal calcitriol synthesis and subsequently decreasing active intestinal calcium transport. Figure 2 shows the complex interrelations between serum FGF23, Klotho, phosphate, calcium, calcitriol, and parathyroid function in CKD.

Figure 2.
Chronic kidney disease-associated mineral and bone disorder (CKD-MBD). Complex interactions between phosphate, FGF23, FGF receptor-1c (FGFR1c), Klotho, 1,25diOH vitamin D (calcitriol), renal 1α 25OH vitamin D hydroxylase (1α hydroxylase), vitamin D receptor (VDR), calcium, Ca-sensing receptor (CaSR), and parathyroid hormone (PTH). From Komaba & Fukagawa (43), modified.
Calcium Deficiency
In early CKD stages, disturbances of calcium metabolism may already be present. They include a calcium deficiency state due to a negative calcium balance resulting from low oral calcium intakes and impaired active intestinal calcium absorption (although a positive calcium balance can be induced by the ingestion of high amounts of calcium-containing phosphate binders) (44,45), a tendency towards hypocalcemia due to skeletal resistance to the action of PTH (46), and reduced calcium-sensing receptor (CaSR) expression in the parathyroid cell. All these factors contribute to the development of parathyroid over function (46,47). Their relative importance increases with the progression of CKD. It also depends on individual patient characteristics such as the underlying type of nephropathy, comorbidities, dietary habits, and amount of food intake.
Inhibition of Calcitriol Synthesis
The progressive loss of functioning nephrons and increased production of FGF23 are mainly responsible for the reduction in renal calcitriol synthesis, favoring the development of parathyroid over function. Although PTH in turn stimulates renal tubular 1α-OH vitamin D hydroxylase activity resistance to its action probably attenuates this counter-regulatory mechanism. Whether the direct inhibition of 1α-OH vitamin D hydroxylase activity by FGF23 is more powerful than its stimulation by PTH depends on several other additional factors such as the presence of hyperphosphatemia, metabolic acidosis, and uremic toxins. The marked disturbances of the calcitriol synthesis pathway probably explain the long reported direct relation in CKD patients between plasma calcidiol and calcitriol, and between plasma calcitriol and glomerular filtration rate (48). Such relations are not observed in people with normal kidney function.
Yet another hypothesis is based on the observation that calcidiol does not penetrate into proximal tubular epithelium from the basolateral side, but only from the luminal side. The complex formed by calcidiol and its binding protein (DBP) is ultrafiltered by the glomerulus, subsequently enters the tubular epithelium from the apical side via the multifunctional brush border membrane receptor megalin, and then serves as substrate for the renal enzyme, 1α-OH vitamin D hydroxylase for calcitriol synthesis (Figure 3) (49). Reduced glomerular filtration leads to a decrease in calcidiol-DBP complex transfer into the proximal tubular fluid and hence reduced availability of calcidiol substrate for luminal reabsorption and calcitriol formation. However, the validity for the human situation of this mechanism established in the mouse has subsequently been questioned since 1α-OH vitamin D hydroxylase expression was found not only in proximal, but also in distal tubular epithelium of human kidney, that is in tubular areas in which megalin apparently is not expressed (50).

Figure 3.
Schematic representation of the role of megalin in renal tubular 25 OH vitamin D reabsorption. Megalin is a multifunctional brush border membrane receptor expressed in the proximal renal tubule. It enables endocytic reabsorption of 25 OH vitamin D (calcidiol) filtered by the glomerulus and the subsequent synthesis of 1,25 diOH vitamin D (calcitriol) by mitochondrial 1-a 25 OH vitamin D hydroxylase. After Nykjaer et al (49).
Finally, the concentration of plasma calcidiol is diminished in the majority of patients with CKD (51,52). The reasons for this vitamin D deficiency state include insufficient hours of sunshine or sun exposure especially in the elderly, skin pigmentation, intake of antiepileptic drugs (like in general population), and in addition enhanced urinary excretion of calcidiol complexed to vitamin D binding protein (DBP) in the presence of proteinuria, and loss into the peritoneal cavity in those on peritoneal dialysis treatment. All these factors may also contribute to the reduction in calcitriol synthesis (53). However, low plasma calcidiol has also been postulated to be a risk factor per se for secondary hyperparathyroidism, as suggested by an observational study in Algerian hemodialysis patients with insufficient exposure to sunshine (54) and the observation that calcidiol is able to directly suppress PTH synthesis and secretion in bovine parathyroid cells in vitro, although with much less potency than calcitriol (55).
SECONDARY HYPERPARATHYROIDISM IN CKD – PLASMA BIOCHEMISTRY CHANGES IN ADVANCED CKD STAGES (FIGURE 1)
The above-mentioned roles of relative or absolute deficiency states of calcium and vitamin D are steadily gaining importance with the progression of CKD, and phosphate becomes a major player.
Role of Hyperphosphatemia
In CKD stages G4-G5 hyperphosphatemia becomes an increasingly frequent feature (19), due to phosphate retention caused by the progressive loss of functioning nephrons and the increasing difficulty in augmenting glomerular phosphate ultrafiltration and to further reduce its tubular reabsorption when it is already maximally inhibited by high serum FGF23 and PTH levels.
FGF23 Excess and Klotho Deficiency
Circulating FGF23 may reach extremely high, maladaptive concentrations in patients with end-stage kidney disease (ESKD) (56). In parallel, a reduction of Klotho expression is observed in kidney and parathyroid tissue, as well as of soluble Klotho in the plasma and urine of patients and animals with CKD (26,27,30). The reduction is particularly marked in advanced stages of CKD. The resulting resistance to the action of FGF23 in kidney and parathyroid tissue favors hyperparathyroidism (see below).
The uremic syndrome itself could also play a role. In addition to phosphate many other so-called uremic toxins, that is substances which accumulate in the uremic state, are known to interfere with vitamin D metabolism and action (57,58). Indoxyl sulfate has been shown to participate in the pathogenesis of skeletal resistance to the action of PTH (59), in addition to direct inhibitory effects on bone turnover (60).
MECHANISMS INVOLVED IN THE PATHOGENESIS OF SECONDARY HYPERPARATHYROIDISM
Generally speaking, there are at least two major different mechanisms which determine the magnitude of secondary hyperparathyroidism in CKD. The first is an increase in PTH synthesis and secretion, and the second an increase in parathyroid gland mass, mostly due to enhanced cell proliferation (hyperplasia), and to a lesser degree also an increase in cell size (hypertrophy) (see schematic representation in Figure 4). Whereas acute stimulation of PTH synthesis and/or release generally occurs in the absence of cell growth stimulation, these two processes appear to be tightly linked whenever there is chronic stimulation. The main factors involved in the control of the two processes are again calcitriol, calcium, and phosphate whereas the direct effects of FGF23 appear to be essentially limited to the control of PTH synthesis and secretion. In the following, the disturbances of the mechanisms controlling parathyroid function will be discussed subsequently for each of these three factors, although there are numerous interactions between them. Subsequently, the influence of other factors and comorbid conditions related to CKD will be presented.
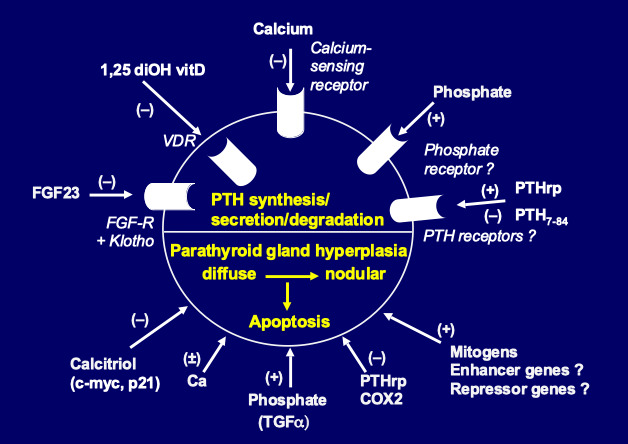
Figure 4.
Pathogenesis of secondary hyperparathyroidism. Schematic representation of parathyroid hormone (PTH) synthesis and secretion (upper part) and parathyroid cell proliferation and apoptosis (lower part), as regulated by a number of hormones and growth factors.
Calcitriol
The above-mentioned decrease in plasma calcitriol aggravates hyperparathyroidism via several mechanisms. The first is direct and results from an insufficient inhibition of PTH synthesis due to low circulating calcitriol levels and a disturbed action of calcitriol at the level of the preproPTH gene. It is well established that calcitriol, after forming a complex with its receptor, vitamin D receptor (VDR) and heterodimerizing with the retinoic acid receptor (RXR), directly inhibits preproPTH gene transcription by binding to a specific DNA response element (VDRE) located in the 5’-flanking region of the gene. In CKD, in addition to low extracellular concentrations of calcitriol, at least two other factors interfere with calcitriol’s action on the preproPTH gene (61). The first factor is a reduced expression of the VDR gene in hyperplastic parathyroid tissue of CKD patients (62). This reduction is particularly marked in nodular, as compared to diffusely hyperplastic parathyroid tissue. The second factor is reduced binding of calcitriol to VDR, slowed nuclear migration of the calcitriol–VDR complex, and less efficient inhibitory action on the preproPTH gene, in association with the uremic state (58,63). Of note, extracellular Ca2+ concentration [Ca2+e] appears to play a role in the regulation of VDR expression. In rat parathyroid glands, low [Ca2+e] reduced VDR expression independently of calcitriol, whereas high [Ca2+e] increased it (64). Hypocalcemia may attenuate by this mechanism the feedback of increased plasma calcitriol concentrations on the parathyroids.
The second level at which calcitriol regulates PTH gene expression involves calreticulin. Calreticulin is a calcium binding protein which is present in the endoplasmic reticulum of the cell, and also may have a nuclear function. It regulates gene transcription via its ability to bind a protein motif in the DNA-binding domain of nuclear hormone receptors of sterol hormones. Sela-Brown et al. proposed that calreticulin might inhibit vitamin D's action on the PTH gene, based on in vitro and in vivo experiments (65). They fed rats either a control diet or a low calcium diet, which led to increased PTH mRNA levels despite high serum calcitriol levels that would be expected to inhibit PTH gene transcription. Their postulate that high calreticulin levels in the nuclear fraction might prevent the effect of calcitriol on the PTH gene was strongly supported by the observation that hypocalcemic rats had increased levels of calreticulin protein in parathyroid nuclear fraction. This could explain why hypocalcemia leads to increased PTH gene expression despite high serum calcitriol levels, and might also be relevant for the refractoriness of secondary hyperparathyroidism to calcitriol treatment observed in many patients with CKD.
The third mechanism of calcitriol’s action could be indirect, via a stimulatory effect on parathyroid CaSR expression, as shown by Brown et al (66) and subsequently confirmed by Mendoza et al (67).
The fourth mechanism is again a direct one. It concerns the well-known inhibitory effect of vitamin D on cell proliferation and the induction of differentiation towards mature, slowly growing cells. A decrease in plasma calcitriol and a perturbed action at molecular targets favors abnormal cell growth. This is the case with parathyroid tissue as well, and parathyroid hyperplasia ensues (68). The importance of vitamin D in the pathogenesis of parathyroid hyperplasia of experimental uremia has first been shown by Szabo et al (69). These authors administered increasing doses of calcitriol to rats either at the time of inducing chronic kidney failure or at a later time point, when uremia was already well established. They were able to prevent parathyroid cell proliferation entirely when calcitriol was given in initial CKD stages, but not when given later on. Fukagawa et al showed that pharmacologic doses of calcitriol repressed c-myc expression in the parathyroid tissue of uremic rats and suggested that the hormone might suppress parathyroid hyperplasia by this pathway (70). In contrast, Naveh-Many et al. (71) failed to observe such an antiproliferative effect of calcitriol in parathyroid cells of uremic rats but they administered the hormone for only three days. Such short-term administration may not have been sufficient for an efficacious suppression of cell turnover.
To answer the question of a possible direct calcitriol action on parathyroid cells, several studies were performed in experimental models in vitro. Nygren et al. (72) showed in primary cultures of bovine parathyroid cells, maintained in short-term culture, that these cells underwent significant increases both in number and size in response to fetal calf serum, and that the addition of 10-100 ng/mL calcitriol almost completely inhibited cell proliferation whereas cell hypertrophy was unaffected. Kremer et al (73) subsequently confirmed in the same parathyroid cell model that calcitriol exerted an anti-proliferative action. They further suggested that this inhibition occurred via a reduction of c-myc mRNA expression. One report showed an inhibitory action under long-term culture conditions (up to 5 passages) of the effect of calcitriol on bovine parathyroid cell proliferation (74). Our group subsequently confirmed such a direct antiproliferative effect of calcitriol in a human parathyroid cell culture system derived from hyperplastic parathyroid tissue of patients with severe secondary uremic hyperparathyroidism (75) (Figure 5).

Figure 5.
Antiproliferative effect of 1,25 diOH vitamin D on parathyroid cells. Reduction of parathyroid cell proliferation in response to increasing medium 1,25diOH vitamin D (calcitriol) concentrations in the incubation milieu of a human parathyroid cell culture system, with parathyroid cells derived from hyperplastic parathyroid tissue of patients with severe secondary uremic hyperparathyroidism. From Roussanne et al (75).
A fifth mechanism is the potential association between parathyroid function and vitamin D receptor (VDR) polymorphism. Fernandez et al (76) separated hemodialysis patients with same serum calcium and time on dialysis treatment into two groups, according to their serum iPTH levels, namely low PTH (<12 pmol/L) or high PTH (>60 pmol/L). They found that the BB genotype and the B allele were significantly more frequent in the low PTH than in the high PTH group (32.3% vs 12.5%, and 58.8% vs 39.1%, respectively). This information suggests that VDR gene polymorphism influences parathyroid function in CKD. Similar results have been reported by an Italian group (77) and in a large sample of Japanese hemodialysis patients (78). In this latter study, after excluding patients with diabetes and patients with a dialysis vintage of less than ten years, the authors observed lower plasma iPTH levels in ESKD patients with BB than with Bb or bb alleles. A relationship between Apa I polymorphism (A/a alleles) and the severity of hyperparathyroidism has also been sought in Japanese hemodialysis patients (79). Plasma PTH levels in AA and Aa groups were approximately half that of the aa group. However, other groups found no difference in PTH levels for various VDR polymorphisms (80–82). Moreover, although in some clinical conditions VDR polymorphism may be associated with variations of the half-life of the VDR gene transcript (83) or of VDR function (84), there has been no report showing that in uremic patients with secondary hyperparathyroidism the density of parathyroid cell VDR varies with different VDR genotypes. In addition, although VDR genotypes may have some influence on the degree of parathyroid cell proliferation, the mechanism by which this could occur remains unknown.
Finally, Egstrand et al recently provided experimental evidence for the role of a circadian clock operating in parathyroid glands. This clock and downstream cell cycle regulators were shown to be disturbed in uremic rats, potentially contributing to dysregulated parathyroid proliferation in secondary hyperparathyroidism (85).
Calcium
It has long been known that [Ca2+e] is the primary regulator of PTH secretion. Small changes in serum Ca2+ concentration result in immediate changes of PTH release which are short-lived or long-lived, depending on the velocity of the restoration of serum Ca2+ towards normal. Thus, postprandial urinary calcium excretion was increased in patients with CKD as it was in healthy volunteers, but only in the patients was this accompanied by significantly reduced serum Ca2+ and increased PTH levels (86). The inverse relation between Ca2+ and PTH in the circulation obeys a sigmoidal curve (87). While the majority of in vitro studies have reported a decreased responsiveness of hyperplastic parathyroid cells to changes in [Ca2+e] in vivo studies have not always confirmed this. Such discrepant findings are likely due to different methods used to assess the dynamics of PTH secretion (88).
Several in vitro studies have shown that the set point of calcium for PTH secretion (that is the Ca2+ concentration required to produce half maximal PTH secretion) is greater in parathyroid cells from primary adenomas and secondary (uremic) hyperplastic parathyroid glands than in normal parathyroid cells (89). Such a relatively poor response to [Ca2+e] should contribute to the increased PTH levels observed in uremic patients with secondary hyperparathyroidism.
We and others have demonstrated that both primary parathyroid adenoma and secondary uremic, hyperplastic parathyroid gland tissue exhibit a decrease in the expression of CaSR protein (90,91). In secondary uremic hyperparathyroidism, there is a significant decrease of CaSR in diffusely growing hyperplastic tissue, with the decrease being even more marked in nodular areas (characteristic of advanced hyperparathyroidism with autonomously growing cells) (90) (Figure 6). Since changes in intracellular Ca2+ elicited by hyper or hypocalcemia depend on the expression and activity of the CaSR, any decrease explains, at least in part, an impaired intracellular calcium response to [Ca2+e] and hence a reduced inhibitory effect of the cation on PTH secretion. Several factors contribute to the downregulation of CaSR expression and/or activity in CKD including reduced calcitriol levels (66,67), low magnesium levels (92), dietary phosphate (probably indirect action) (93), and metabolic acidosis (94). However, raising extracellular phosphate has been recently shown to also exert a direct inhibitory action on parathyroid cell CaSR activity of isolated human parathyroid cells resulting in an increase in PTH secretion (95). Almaden et al studied calcium-regulated PTH response in vitro, using respectively primary parathyroid adenoma and uremic hyperplastic tissue, the latter either of the nodular or the diffuse type (96). They found that in primary adenoma tissue PTH secretion was less responsive to an increase in [Ca2+e] than in uremic hyperplastic parathyroid tissue; among the latter, nodular tissue was less responsive than diffusely hyperplastic tissue. The decreased secretory response to Ca2+ observed in nodular uremic hyperplasia may be explained by the markedly reduced CaSR expression in CKD, as demonstrated by Gogusev et al (90). This decrease can be overcome, at least partially, by PTHrp, as shown by Lewin et al (97), who observed that the administration of PTHrp significantly stimulated the impaired secretory capacity of the parathyroid glands of uremic rats in response to hypocalcemia. Of note, this observation also implies that the PTH/PTHrp receptor is expressed on the parathyroid cell.

Figure 6.
Calcium-sensing receptor (CaSR) expression in normal and hyperplastic parathyroid glands. Normal parathyroid tissue (in blue), secondary (2°) hyperparathyroidism from dialysis patients (glands with diffuse hyperplasia in yellow; glands with nodular hyperplasia in green), and primary (1°) adenomatous hyperparathyroidism from patients with conserved kidney function (in orange). Decreased expression of both CaSR protein and mRNA in the majority of hyperplastic glands, with a particularly marked decrease in nodular type secondary uremic hyperparathyroidism. After Gogusev et al (90).
The shift of the calcium set point to the right in dialysis patients in vivo has been a much less constant finding than the right shift observed in the above-mentioned studies in uremic parathyroid tissue in vitro. While in CKD patients with a mild to moderate degree of hyperparathyroidism the set point was most often found to be normal, an altered set point was observed in presence of severe parathyroid over function with hypercalcemia (98). This anomaly could at least in part be due to CaSR down-regulation. As regards CKD patients with less severe parathyroid over function, a considerable controversy took place regarding the results of in vivo assessments of parathyroid gland function (99,100). In part, disparities among study results reflected technical differences in experimental methods and/or variations in the mathematical modeling of PTH secretion in vivo (101). Another difficulty in interpreting the results of in vivo dynamic tests of parathyroid gland function relates to the issue of parathyroid gland size. Because there is a basal, or non-suppressible, component of PTH release from the parathyroid cell even at high [Ca2+e], excessive PTH secretion may result solely from increases in parathyroid gland mass (98). This can theoretically occur in the absence of any defect in calcium sensing at the level of the parathyroid cell. Since parathyroid gland hyperplasia is present to some extent in nearly all patients with CKD stages G3-G5, alterations in PTH secretion due to increases in parathyroid gland mass cannot readily be distinguished from those attributable to changes in calcium-sensing by the parathyroid cell using the four-parameter model for in vivo studies (100).
The role of calcium in parathyroid cell proliferation is less clear than is generally assumed. Calcium deficiency, in the presence or absence of hypocalcemia, together with vitamin D deficiency or reduced production of calcitriol, probably is a major stimulus of parathyroid hyperplasia. Naveh-Many et al showed that calcium deprivation, together with vitamin D deficiency, greatly enhanced the rate of parathyroid cell proliferation in normal rats and also in rats with CKD, using the cell cycle-linked antigen, PCNA (71). The concomitant decrease in CaSR expression in CKD, as observed in parathyroid glands of both dialysis patients and uremic rats (90,102), should theoretically enhance parathyroid tissue hyperplasia further. Indirect support for this contention came from the observation that the administration of the calcimimetic compound NPS R-568, a CaSR agonist, led to the suppression of parathyroid cell proliferation in rats with CKD (103). However, in the study by Naveh-Many et al the dietary regimen was poor in both calcium and vitamin D. In contrast, when feeding normal rats on a calcium-deficient diet alone, in the absence of concomitant vitamin D deficiency, Wernerson et al observed parathyroid cell hypertrophy, not hyperplasia (104).
The question whether the effect of calcium is direct or indirect remains therefore unsolved at present. It can only be answered by in vitro studies. For a long time, available culture systems using normal parathyroid cells did not allow the maintenance of functionally active cells for prolonged time periods. They were all characterized by a rapid and significant loss of PTH secretion within 3 to 4 days (105–107). One culture model has been described, using bovine parathyroid cell organoids, which maintained the ability to modulate PTH secretion in response to [Ca2+e] and tissue-like morphology for 2 weeks (108). However, only one long-term study using bovine parathyroid cells demonstrated a release of bioactive bovine PTH but with reduced sensitivity to [Ca2+e] (109). Other reports showed that the rapid decrease in PTH responsiveness of cultured bovine parathyroid cells to changes in [Ca2+e] was associated with a marked reduction in CaSR expression (110,111). Yet other parathyroid cell-derived culture models proposed in the literature were in fact devoid of any PTH secretory capacity (112,113).
To study direct effects of [Ca2+e] on the parathyroid cell in vitro, we developed a functional human parathyroid cell culture system capable of maintaining regulation of its secretory activity and the expression of extracellular CaSR mRNA and protein for several weeks. For this purpose, we used parathyroid cells derived from hyperplastic parathyroid tissue of hemodialysis patients with severe secondary hyperparathyroidism (114). In a subsequent study with this experimental model, we surprisingly obtained evidence that parathyroid cell proliferation index, as estimated by [3H]-thymidine incorporation into an acid-precipitable fraction as a measure of DNA synthesis, could be directly stimulated by high [Ca2+e] in the incubation medium, compared with low [Ca2+e] (75) (Figure 7).

Figure 7.
Effect of medium calcium concentration on parathyroid cell proliferation. Stimulatory effect on parathyroid cell proliferation (measured by KI-67 staining method) of high medium calcium concentrations in the incubation milieu of a human parathyroid cell culture system derived from hyperplastic parathyroid tissue of patients with severe secondary uremic hyperparathyroidism. From Roussanne et al (75).
We confirmed this finding in independent experiments using the cell cycle-linked antigen Ki-67 to determine parathyroid cell proliferation. However, the addition of the calcimimetic NPS R-467 to the incubation medium led to a decrease in cell proliferation (Figure 8).

Figure 8.
Inhibitory effect of calcimimetic on parathyroid cell proliferation. Human parathyroid cells derived from hyperplastic parathyroid tissue of patients with severe secondary uremic hyperparathyroidism were maintained in high medium calcium incubation milieu, and exposed to increasing concentrations of calcimimetic NPS R-467. Determination of cell proliferation by [3H]-thymidine incorporation method. After Roussanne et al (75).
Of interest, calcimimetics have subsequently been shown to upregulate the expression of both CaSR (67,115) and VDR (67) in parathyroid glands of uremic rats. In an attempt to unify our apparently contradictory in-vitro observations with respect to findings made in vivo, we proposed the following hypothesis. The effect of calcium on parathyroid cell proliferation could occur along two different pathways, via two distinct mechanisms. Inhibition of proliferation would occur via the well-known parathyroid CaSR-dependent pathway, whereas stimulation of proliferation would occur via an alternative pathway (Figure 9). Note that the parathyroid tissue samples used in our study stemmed from uremic patients with long-term ESKD and severe secondary hyperparathyroidism. Since such parathyroid tissue generally exhibits decreased CaSR expression, it is possible that the number of CaSR expressed in the parathyroid cell membranes of our culture model was insufficient to inhibit cell proliferation. Of note, the human CaSR gene has two promoters and two 5’ untranslated exons; therefore, the alternative usage of these exons leads to production of multiple CaSR mRNAs in parathyroid cells (116). The expression of CaSR mRNA produced by one of the two promoters of CaSR gene is specifically reduced in parathyroid adenomas, suggesting a role in PTH hypersecretion and proliferation. Moreover, the membrane-bound 550-kD Ca2+-binding glycoprotein megalin, belonging to the low-density lipoprotein receptor superfamily, has been identified in parathyroid chief cells as another putative calcium-sensing molecule which could be involved in calcium-regulated cellular signaling processes as well (117). Based on these observations, one can postulate that parathyroid cells express multiple CaSR-like molecules. Consequently, if the well-known parathyroid CaSR is downregulated, parathyroid cell proliferation induced by increases in [Ca2+e] may occur via a different type of CaSR. Another possibility is an alteration in post-receptor signal transduction that could occur in hyperparathyroid states or under cell culture conditions. Our observations are in line with findings by Ishimi et al. which were incompatible with a direct effect of low [Ca2+e] in the pathogenesis of parathyroid hyperplasia (74). However, any extrapolation from such in vitro observations to the in vivo setting should be done with caution, and further work is needed to define the precise pathway(s) by which calcium regulates parathyroid tissue growth.
![Figure 9. . Hypothesis of the regulation of parathyroid cell proliferation by extracellular [Ca2+].](/books/NBK278975/bin/hyprparathyroid-kdny-Image009.jpg)
Figure 9.
Hypothesis of the regulation of parathyroid cell proliferation by extracellular [Ca2+]. 1) Inhibitory pathway via the calcium-sensing receptor (CaR). 2) Stimulatory pathway via an unknown transmembrane transduction mechanism. Physiologically, pathway 1 predominates over pathway 2. In presence of parathyroid hyperplasia with calcium-sensing receptor down-regulation pathway 2 could become dominant and favor parathyroid cell proliferation over suppression. After Roussanne et al (75).
Phosphate
Hyperphosphatemia is associated with increased PTH secretion. The stimulation of PTH release occurs via direct and indirect mechanisms. The initially proposed indirect mechanism, which remains true according to present knowledge, is via a decrease in plasma Ca2+ concentration (see above). Hyperphosphatemia also leads to an inhibition of the renal synthesis of calcitriol, probably mostly via stimulation of FGF23 production.
A direct action of phosphate on PTH secretion by the parathyroid cell has long been suspected. However, it has been formally demonstrated in vitro only in 1996 (118–120). This demonstration required the use of either intact parathyroid glands (from rats) (Figure 10) or parathyroid tissue slices (from cows) whereas it had not been possible to obtain such direct stimulation using the classic model of isolated bovine parathyroid cells. Elevating plasma phosphate concentration in the incubation milieu of experimental models using intact (or partially intact) parathyroid tissue leads to a stimulation of PTH secretion within some hours, in the absence of any change in [Ca2+e]. It can however be abrogated by an increase in cytosolic Ca2+ concentration (121).

Figure 10.
Direct inhibition of parathyroid hormone (PTH) secretion by phosphate. Intact parathyroid glands obtained from normal rats were maintained in culture and exposed to increasing in phosphate concentrations in the incubation medium. After Almaden et al (121).
Silver’s group reported subsequently that phosphate, like calcium, regulates pre-pro-PTH gene expression post-transcriptionally by changes in protein-PTH mRNA interactions at the 3'-UTR which determine PTH mRNA stability. They identified the minimal sequence required for protein binding in the PTH mRNA 3'-UTR and determined its functionality. They found that the conserved PTH RNA protein-binding region conferred responsiveness to calcium and phosphate and determined PTH mRNA stability and levels (122). Thus, a low calcium diet increased stability, whereas a low phosphate diet decreased stability of PTH mRNA (123) (Figure 11). The PTH mRNA 3’-untranslated region-binding protein was subsequently identified by this research group as adenylate-uridylate-rich element RNA binding protein 1 (AUF1) (124).

Figure 11.
Post-transcriptional regulation of PTH mRNA stability by calcium, phosphate, and kidney failure. Pre-pro-PTH gene expression is modulated via changes in protein-PTH mRNA interactions at the 3'-UTR region which determine PTH mRNA stability. Low calcium diet increases stability, whereas low phosphate diet decreases stability of PTH mRNA. PTH mRNA protective factor AUF1 in yellow, PTH mRNA degrading endonuclease in orange. After Yalcindag et al (123).
In addition to its stimulatory effect on PTH secretion a high phosphate diet also rapidly induces parathyroid over function and hyperplasia, as shown in experimental animal models (125). Subsequent studies showed that hyperphosphatemia induced by phosphate-rich diets in animals with CKD induced parathyroid hyperplasia even when changes in plasma Ca2+ and calcitriol concentration were carefully avoided, pointing to a direct effect of phosphate on cell proliferation (71,120). Conversely, early dietary phosphate restriction in the course of CKD was capable of preventing both PTH over secretion and parathyroid hyperplasia (71,120,126). Interestingly, dietary phosphate restriction following phosphate overload in rats led to an immediate decrease in PTH secretion despite no regression of parathyroid gland size (127).
Our group wished to know whether the stimulatory effect of phosphate on parathyroid cell proliferation was direct or indirect. To answer this question, we used the above described in vitro model of human parathyroid cells maintained in long-term culture (114). We could show that cell proliferation index was directly stimulated by high phosphate concentrations in the incubation medium, compared with low phosphate concentration (75) (Figure 12). These experiments demonstrated that phosphate is capable of stimulating not only PTH secretion, but also of inducing parathyroid tissue hyperplasia by a direct mode of action.

Figure 12.
Direct stimulatory effect of extracellular phosphate on parathyroid cell proliferation. Response of parathyroid cell growth to increasing phosphate concentrations in the incubation milieu of a human parathyroid cell culture system derived from hyperplastic parathyroid tissue of patients with severe secondary uremic hyperparathyroidism. Determination of cell proliferation by [3H]-thymidine incorporation method. After Roussanne et al (128).
FGF23 plays an important role in the control of plasma phosphate. Elevated FGF23 in CKD allows efficient inhibition of proximal tubular phosphate reabsorption and maintenance of plasma phosphorus in the normal range. However, since hyperphosphatemia directly stimulates PTH secretion, its correction by FGF23 indirectly leads to a reduction of PTH release, in addition to the direct inhibitory action of FGF23 on parathyroid secretory activity (see above).
FGF23 and Klotho
As mentioned before FGF23 directly inhibits PTH synthesis and secretion via its action on parathyroid FGFR-1 (129). FGF23 also increases parathyroid CaSR and VDR expression, further contributing to the suppression of PTH by this hormone (Canalejo 2010). In advanced stages of CKD FGF23’s effect is partially or even completely abolished owing to downregulation of the expression of its receptor and co-receptor Klotho (38–40). Of interest, in early stages of CKD there could be an initial upregulation of FGFR-1 and Klotho, with enhanced PTH secretion in response to FGF23 via an Na+/K+ -ATPase driven pathway (130). Subsequent findings suggested a function for Klotho in suppressing PTH biosynthesis and parathyroid gland growth, even in the absence of CaSR (131). Moreover, they pointed to a physical interaction between Klotho and CaSR. Specific deletion of CaSR in parathyroid tissue led to elevated serum PTH levels and parathyroid gland hyperplasia, and additional deletion of Klotho in parathyroid glands exacerbated this condition. However, a recent review concluded that role of parathyroid Klotho remains controversial (132).
MicroRNAs
More recently, Shilo et al provided evidence for the important role of microRNAs (miRNAs) in the physiological regulation of parathyroid function, and its dysregulation in the secondary hyperparathyroidism of CKD (133,134). The authors found an abnormal regulation of many miRNAs in experimental uremic hyperparathyroidism supporting a key role for miRNAs in this condition. Specifically, their studies showed that inhibition of the abundant let-7 family increased PTH secretion in normal and uremic rats, as well as in mouse parathyroid organ cultures. Conversely, inhibition of the upregulated miRNA-148 family prevented the increase in serum PTH of uremic rats, and inhibition of let-7 family also reduced PTH secretion in parathyroid cultures. Thus, miRNA dysregulation represents yet another crucial step in the pathogenesis of secondary hyperparathyroidism.
Other Factors and Conditions
As already pointed out above the uremic state with its accumulation of numerous uremic toxins is another long suspected, albeit yet ill-defined factor in the pathogenesis of secondary hyperparathyroidism. Recently, several pieces of evidence have been provided in favor of a role of the uremic state which interferes with the binding of calcitriol to VDR (58) and with the nuclear uptake of the hormone-receptor complex (63). This should have consequences not only for PTH synthesis and secretion, but also for parathyroid cell proliferation. Another mechanism of excessive proliferation involves the mTOR pathway, which has been shown to be activated in secondary hyperparathyroidism (135). Inhibition of mTOR complex 1 by rapamycin decreased parathyroid cell proliferation in vivo and in vitro.
Patients with diabetes receiving dialysis therapy have relatively low plasma PTH levels, as compared to those without diabetes. The high incidence of low bone turnover in uremic patients with diabetes (136–139) has been attributed to low levels of biologically active PTH, possibly via an inhibition of PTH secretion or a modification of the PTH peptide by the accumulation of advanced glycation end-products such as pentosidine (140) or else oxidation of PTH (141,142). However, experimental studies have demonstrated that the metabolic abnormalities associated with diabetes can also directly decrease bone turnover, independent of PTH (143). In general, patients with low bone turnover tend to develop hypercalcemia when on a normal or high dietary calcium intake, probably due to the diminished skeletal capacity of calcium uptake. This in turn tends to reduce plasma PTH. Thus, not only does hypoparathyroidism promote adynamic bone disease but adynamic bone disease also favors hypoparathyroidism. Another issue is whether in patients with diabetes abnormalities such as hyperglycemia and insulin deficiency or resistance may directly affect parathyroid function. In an in vitro study using dispersed bovine parathyroid cells, high glucose and low insulin concentrations suppressed the PTH response to low Ca2+ concentration (144). These results are compatible with the view that diabetes directly inhibits parathyroid function. However, when uremic rats were fed on a high phosphate diet to induce secondary hyperparathyroidism, the presence of diabetes did not affect the development of parathyroid over function (143).
Aluminum bone disease is generally associated with low serum PTH levels (145,146) and a decreased PTH response to stimulation by hypocalcemia (147,148). In aluminum intoxicated patients, high amounts of aluminum are also found in parathyroid tissue (149). The relatively low PTH levels may reflect either an inhibition of PTH secretion by the hypercalcemia commonly observed in this condition (150) or a direct inhibitory effect of aluminum on parathyroid cell function (151). Direct toxic effects of the trace element have also been demonstrated in studies in vitro (152,153). Observations made in experimental animals and results of clinical studies have been less clear. Whereas some experiments indicated that aluminum overload did not decrease plasma PTH levels in vivo (152,153), other experiments reported a decrease (154,155). Whatever the mechanisms involved, subsequent clinical data clearly showed that the introduction of an aluminum-free dialysis fluid and the discontinuation of aluminum contamination of the dialysate or aluminum removal with deferoxamine resulted in an increase in plasma PTH levels and in PTH response to hypocalcemia (156). Thus, although there appears to be an association between aluminum toxicity and parathyroid gland function, the interaction is complex.
Post-Receptor Mechanisms Involved in Polyclonal Parathyroid Tissue Growth
As pointed out above, calcitriol reduces parathyroid cell proliferation by decreasing the expression of the early gene, c-myc. This gene modulates cell cycle progression from G1 to S phase. A decrease in plasma calcitriol and/or a disturbance of its action at the level of the parathyroid cell, which are both frequently observed in uremic patients, may cause disinhibition of c-myc expression and progression into the cell cycle. Another mode of action involves the cyclin kinase inhibitor p21WAF1. Calcitriol has long been shown to induce the differential expression of p21WAF1 in the myelo-monocytic cell line U937 and to activate the p21 gene transcriptionally in a VDR-dependent, but p53-independent, manner, thereby arresting parathyroid growth (157). Slatopolsky’s group further showed that the administration of calcitriol to moderately uremic rats enhanced parathyroid p21 expression and prevented high phosphate-induced increase in parathyroid TGF-α content (157). In addition, they found that calcitriol altered membrane trafficking of the epithelial growth factor receptor (EGFR), which binds both EGF and TGF-α, and down-regulated EGFR mediated growth signaling (158). Induction of p21 and reduction of TGF-α content in the parathyroid glands also occurred when uremia-induced parathyroid hyperplasia was suppressed by high dietary Ca intake. The mechanisms by which a phosphate-rich diet and hyperphosphatemia induce parathyroid hyperplasia, and conversely a phosphate-poor diet and hypophosphatemia inhibit parathyroid tissue growth have also been examined by this group in a detailed fashion. Thus, Dusso et al showed that feeding a low phosphate diet to uremic rats increased parathyroid p21 gene expression through a vitamin D-independent mechanism (159). When administering a high phosphate diet, p21 expression was not suppressed. In this condition, they observed an increase in parathyroid tissue TGF-α expression and a direct correlation between this expression and parathyroid cell proliferation rate. This finding is in line with the previous observation by our group of de novo TGF-α expression in severely hyperplastic parathyroid tissue of patients with ESKD (160). The inducer of TGF-α gene transcription could be activator protein 2α (AP2), whose expression and transcriptional activity at the TGF-α promoter is increased in the secondary hyperparathyroidism of CKD (161).
Although these findings provide more insight into the pathways by which changes in phosphate intake, and ultimately variations in extracellular phosphate concentration, control parathyroid tissue growth the exciting question of the transmembrane signal transduction mechanism and subsequent nuclear events triggered by phosphate remains yet to be answered.
In addition to p21 and TGF-α, a variety of other growth factors and inhibitors are probably involved in polyclonal parathyroid hyperplasia. Thus, PTHrp has been proposed as a possible growth suppressor in the human parathyroid (162). PTHrp, and probably PTH itself, also exert an inhibitory effect on PTH secretion by acting via a negative feedback loop on PTH-R1 which appears to be expressed in the parathyroid cell membrane as well (97). Table 1 summarizes various changes in gene and growth factor expression, which are potentially involved in the parathyroid tissue hyperplasia of secondary uremic hyperparathyroidism. Gcm2 has been identified as a master regulatory gene of parathyroid gland development, since Gcm2 knockout mice lack parathyroid glands (163). Correa et al. found high Gcm2 mRNA expression in human parathyroid glands in comparison with other non-neural tissues and under expression in parathyroid adenomas but not in lesions of HPT secondary to uremia (164). Gcm2 expression itself is regulated by Gata3, and Gata3, in cooperation with Gcm2 and MafB, stimulates PTH gene expression, by interacting with the ubiquitous transcription factor SP1 (165). MafB probably plays a role in uremic hyperparathyroidism as well. Thus, stimulation of the parathyroid by CKD in MafB+/-mice resulted in an impaired increase in serum PTH, PTH mRNA, and parathyroid cell proliferation (166,167).
Table 1.
Changes in Gene and Growth Factor Expression Potentially Involved in Parathyroid Tissue Hyperplasia of Secondary Uremic Hyperparathyroidism
| Early immediate genes and receptor/coreceptor genes -Enhanced c-myc gene expression (Fukagawa et al, Kidney Int 1991; 39: 874-81) -Decreased calcium-sensing receptor (CaSR) gene expression (Kifor et al, J Clin Endocrinol Metab 1996; 81: 1598-1606. Gogusev et al, Kidney Int 1997; 51: 328-36) -Decreased vitamin D receptor (VDR) gene expression (Fukuda et al, J Clin Invest 1993; 92: 1436-42) -Decrease in parathyroid Klotho and FGFR1c gene expression (Galitzer et al, Kidney Int 2010; 77: 211-8. Canalejo et al, JASN 2010; 21: 1125-35. Komaba et al, Kidney Int 2010; 77: 232-8) |
| Gene polymorphisms -Vitamin-D receptor (VDR) gene polymorphism (Olmos et al, Methods Find Exp Clin Pharmacol 1998; 20: 699-707. Fernandez et al, J Am Soc Nephrol 1997; 8: 1546-52. Tagliabue et al, Am J Clin Pathol 1999; 112: 366-70) |
| Growth factors and cell cycle inhibitors =Increased acidic growth factor (aFGF) gene expression (Sakaguchi, J Biol Chem 1992; 267: 24554-62) -Decreased parathyroid hormone-related peptide (PTHrp) gene expression (Matsushita et al, Kidney Int 1999; 55: 130-8) -De novo transforming growth factor-α (TGF-α) gene expression (Gogusev et al, Nephrol Dial Transplant 1996; 11: 2155-62) -Induction of TGF-α by high phosphate diet (Dusso et al, Kidney Int 2001; 59: 855-865) -Insufficient inhibition of cyclin kinase inhibitor p21WAF1 (Dusso et al, Kidney Int 2001; 59: 855-65); p21WAF1can be induced by calcitriol (Cozzolino et al, Kidney Int 2001; 60: 2109-2117) -mTOR activation and rpS6 phosphorylation (Volovelsky et al, JASN 2016; 27: 1091–1101) |
| Gene mutations: association with monoclonal or multiclonal growth -Mutation of menin gene (Falchetti et al, J Clin Endocrinol Metab 1993; 76: 139-44. Tahara et al, J Clin Endocrinol Metab 2000; 85: 4113-7. Imanishi et al, J Am Soc Nephrol 2002;13:1490-8) -Mutation of Ha-ras gene (Inagaki et al, Nephrol Dial Transplant 1998; 13: 350-7) -No involvement of VDR or CaSR gene mutations (Degenhardt et al, Kidney Int 1998; 53: 556-61. Brown et al, J Clin Endocrinol Metab 2000; 85: 868-72) |
SECONDARY HYPERPARATHYROIDISM IN CKD – MECHANISMS INVOLVED IN THE TRANSFORMATION OF POLYCLONAL TO MONOCLONAL PARATHYROID GROWTH
In severe forms of secondary hyperparathyroidism nodular formations within diffusely hyperplastic tissue are a frequent finding (168). This observation probably corresponds to the occurrence of a monoclonal type of cell proliferation within a given tissue, which initially exhibits polyclonal growth. Clonal, benign tumoral growth was initially shown by Arnold et al using chromosome X-inactivation analysis method (169), and subsequently confirmed by other groups (170,171). After the initially diffuse, polyclonal hyperplasia, with the progression of CKD towards ESKD foci of nodular, monoclonal growth may arise within one or several parathyroid glands which eventually may transform to diffuse monoclonal neoplasia leading to an aspect comparable to that of primary parathyroid adenoma. Several different clones often coexist in same patient, and sometimes even in a single parathyroid gland. Figure 13 shows the progression from polyclonal to monoclonal and/or multiclonal parathyroid hyperplasia (172). It also shows corresponding changes in ultrasonographic features.

Figure 13.
Schematic representation of the transformation of parathyroid hyperplasia from polyclonal to nodular, monoclonal/multiclonal growth with the progression of CKD towards ESKD. After Tominaga et al (172).
Acquired mutations of tumor enhancer or tumor suppressor genes are almost certainly involved in the development of such cell clones but precise knowledge about acquired genetic abnormalities remains limited (170). To identify new locations of parathyroid oncogenes or tumor suppressor genes important in this disease, Imanishi et al performed both comparative genomic hybridization (CGH) and genome-wide molecular allelotyping on a large number of uremia-associated parathyroid tumors (173). One or more chromosomal changes were present in 24% of tumors, markedly different from the values in common sporadic adenomas (28%), whereas no gains or losses were found in 76% of tumors. Two recurrent abnormalities were found, namely gain of chromosome 7 (9% of tumors) and gain of chromosome 12 (11% of tumors). Losses on chromosome 11, the location of the MEN1 tumor suppressor gene, occurred in only one uremia-associated tumor (2%), as compared to 34% in adenomas. The additional search for allelic losses with polymorphic microsatellite markers led to the observation of recurrent allelic loss on 18q (13% of informative tumors). Lower frequency loss was detected on 7p, 21q, and 22q. Interestingly, the cyclin D1 oncogene, activated and overexpressed by clonal gene rearrangement or other mechanisms in 20-40% of parathyroid adenomas (174,175) has not been found to be overexpressed in uremia-associated tumors (175).
Another interesting question was if somatic genes played a major role in the normal regulation of parathyroid function, such as the CaSR and VDR genes. The expression of these two genes was found to be decreased in the hyperplastic parathyroid tissue of uremic patients (62,90,91). The decrease was particularly marked in nodular areas, as compared to diffuse areas of parathyroid gland hyperplasia. Moreover, in uremic rats the decrease in CaSR expression was inversely related to the degree of parathyroid cell proliferation (93). However, the search for mutations or deletions of the VDR gene or the CaSR gene in uremic hyperparathyroidism has remained unsuccessful (170,176,177). The question remains unsolved whether the downregulation of CaSR and VDR expression is a primary event or whether it is secondary to hyperplasia.
Whether benign parathyroid tumors may evolve towards malignant forms is still subject to debate. Since in patients on dialysis therapy parathyroid carcinoma is a rare event (178–180), malignant transformation of clonal parathyroid neoplasms is probably exceptional.
Genome-wide allelotyping and CGH have directly confirmed the presence of monoclonal parathyroid neoplasms in uremic patients with refractory secondary hyperparathyroidism whereas the candidate gene approach has led to only modest results. Somatic inactivation of the MEN1 gene does contribute to the pathogenesis of uremia-associated parathyroid tumors, but its role in this disease appears to be limited, and there is probably no role for DNA changes of the CaSR and VDR genes. Recurrent DNA abnormalities suggest the existence of new oncogenes on chromosomes 7 and 12, and tumor suppressor genes on 18q and 21q, involved in uremic hyperparathyroidism. Finally, patterns of somatic DNA alterations indicate that markedly different molecular pathogenetic pathways exist for clonal outgrowth in severe uremic hyperparathyroidism, as compared to common sporadic parathyroid adenomas. Our group did not find a correlation between the presence of microscopically evident nodules and the clonal character of resected parathyroid tissue, and appearances of several glands with histologic patterns of diffuse hyperplasia also were unequivocally monoclonal in the absence of detectable nodular formations, suggesting that the current criteria for pathological diagnosis do not reflect the genetic differences among these two histopathological types (169).
Parathyroid Cell Apopotosis
It remains uncertain whether reduced apoptosis rates can also contribute to parathyroid tissue hyperplasia (68,181,182). One research group examined this issue in rats with short-term kidney failure (5 days). They were unable to detect apoptosis in hyperplastic parathyroid glands (183). However, this failure could be due to lack of sensitivity of the employed methods.
Negative findings in rats, with no identifiable apoptotic figures at all in parathyroid glands (68,182,183), contrast with subsequent positive observations in rats by others (184,185) and with personal observations of significant apoptotic figures in hyperplastic parathyroid glands removed from uremic, severely hyperparathyroid patients during surgery (186). In our study of human parathyroid glands from patients with ESKD approximately ten times higher apoptotic cell numbers were observed than in normal parathyroid tissue, using Tunel method (Figure 14) (186).

Figure 14.
Increased proportion of apoptotic (TUNEL positive) cells in parathyroid glands from patients with primary or secondary uremic hyperparathyroidism, as compared to normal parathyroid tissue. After Zhang et al (186).
Of note, the uremic state appears to stimulate apoptosis in other cell types as well such as circulating monocytes (187), possibly via the well-known increase of cytosolic Ca2+ which has been observed in a variety of cell types in kidney failure (188), and also possibly via the noxious effect of bioincompatible dialysis membranes used for renal replacement therapy (189). The observed enhancement of parathyroid tissue apoptosis could compensate, at least in part, for the increase in parathyroid cell proliferation observed in secondary uremic hyperparathyroidism.
SECONDARY HYPERPARATHYROIDISM IN CKD – REGRESSION OF PARATHYROID HYPERPLASIA?
Whether regression of parathyroid hyperplasia occurs in animals or patients with advanced stages of CKD remains a matter of debate. According to some authors regression must be an extremely slow process, if it occurs at all (71,182). This is in sharp contrast to the rapid reversibility of excessive PTH secretion in uremic rats which was observed after normalization of renal function by kidney transplantation (190), although parathyroid mass probably did not rapidly decrease in this acute experimental model.
The issue of regression is of clinical importance. As an example, if a patient on dialysis therapy has a dramatic increase in total parathyroid mass there is practically no chance to obtain gland mass regression after successful kidney transplantation. In this condition it would seem appropriate to perform a surgical parathyroidectomy prior to transplantation. If, however significant regression of hyperplasia can occur as an active or passive process, namely by enhanced apoptosis or reduced proliferation, prophylactic surgery could be avoided. That regression of parathyroid hyperplasia secondary to vitamin D deficiency can occur has been convincingly demonstrated many years ago in experiments done in chicks (191). The administration of cholecalciferol to these birds that had developed an increase in parathyroid gland mass when fed a rachitogenic, vitamin D-free diet for 8-10 weeks led to a significant (50%) reduction in gland weight. Calcitriol failed to achieve same effect at a low, albeit hypercalcemic, dose but was capable of reducing gland mass at a higher dose. However, in an experimental dog model no parathyroid mass regression was found when the animals were first treated with a low-calcium, low-sodium, and vitamin D deficient diet for two years and subsequently a normal diet for another 17 months (192). In uremic animals, evidence for or against the possibility of regression of increased parathyroid tissue mass remains sparse and inconclusive.
The calcimimetic drug NPS R-568 was shown to decrease parathyroid cell proliferation and to prevent parathyroid hyperplasia in 5/6th nephrectomized rats; however, it was unable to entirely revert established hyperplasia (183,193). In apparent contrast, Miller et al showed that in rats with established secondary hyperparathyroidism, cinacalcet administration led to complete regression of parathyroid hyperplasia (194). The cinacalcet-mediated decrease in parathyroid gland size was accompanied by increased expression of the cyclin-dependent kinase inhibitor p21. However, these were short-term experiments over an 11-week time period. Interestingly, the prevention of cellular proliferation with cinacalcet occurred despite increased serum phosphorus and decreased serum calcium levels.
In patients with primary hyperparathyroidism spontaneous remission of parathyroid over function has been observed in rare instances, caused by parathyroid “apoplexy” due to tissue necrosis (195). The diagnosis of parathyroid tissue necrosis is more difficult to ascertain in secondary than in primary forms of hyperparathyroidism because the hyperplasia of the former is not limited to a single gland.
Regression of parathyroid hyperplasia in hemodialysis patients in response to intravenous calcitriol pulse therapy for 12 weeks has been reported by Fukagawa et al using ultrasonography (196). These authors observed a significant decrease in mean gland volume from 0.87 to 0.51 cm3 of this time period, together with a reduction in serum iPTH of more than 50%. In contrast, Quarles et al who also examined parathyroid gland morphology in hemodialysis patients in vivo in response to intermittent intravenous or oral calcitriol treatment for 36 weeks failed to observe a decrease in parathyroid gland size as assessed by high resolution ultrasound and/or magnetic resonance imaging (197). Mean gland size was 1.9 and 2.1 cm3 before and 3.3 and 2.3 cm3 after oral and intravenous calcitriol therapy, respectively. The authors achieved an overall maximum average serum PTH reduction of 43% over this time period. There were marked differences between these two studies which may explain the apparently diverging results. Hyperparathyroidism probably was more severe in the latter than in the former. Although initial mean serum iPTH levels were similar, serum phosphorus was higher and the decrease in serum PTH achieved in response to calcitriol was less marked in the latter. Moreover, parathyroid mass was more than double. In another study, Fukagawa et al examined the possible relation between parathyroid size and the long-term outcome after calcitriol pulse therapy, by subdividing patients into different groups according to initial parathyroid gland volume assessments (198). In two hemodialysis patients with detectable gland(s), in whom the size of all parathyroid glands as well as PTH hypersecretion regressed to normal, secondary hyperparathyroidism remained controllable for at least 12 months after switching to conventional oral active vitamin D therapy. In contrast, in seven hemodialysis patients, in whom the size of all parathyroid glands did not regress to normal by calcitriol pulse therapy, secondary hyperparathyroidism relapsed after switching to conventional therapy although PTH hypersecretion could be controlled temporarily. Similarly, Okuno et al. showed in a study in hemodialysis patients that plasma PTH levels and the number of detectable parathyroid glands decreased in response to the active vitamin D derivative maxacalcitol (22-oxacalcitriol) given for 24 weeks only when the mean value of the maximum diameter of one of the parathyroid glands was less than 11.0 mm, but not when it was above that value (199).
Taken together, these findings suggest that the degree of parathyroid hyperplasia, as detected by ultrasonography, is an important determinant for regression in response to calcitriol therapy. It is probable, although not proven, that the type of hyperplasia, namely monoclonal/multiclonal vs polyclonal growth, is even more important as regards the potential of regression than the mere size of each gland.
Figure 2 (see above) summarizes in a schematic view the main mechanisms involved in the abnormal PTH synthesis and secretion and in parathyroid tissue hyperplasia. Figure 2 further points to the possible counterregulatory role of apoptosis.
ALTERED PTH METABOLISM AND RESISTANCE TO PTH ACTION
PTH metabolism is greatly disturbed in CKD. Normally, most of full-length PTH1-84 is transformed in the liver to the biologically active N-terminal PTH1-34 fragment and several other, inactive C-terminal fragments. The latter are mainly catabolized in the kidney and the degradation process involves solely glomerular filtration and tubular reabsorption, whereas the N-terminal PTH1-34 fragment undergoes both tubular reabsorption and peritubular uptake, as does the full-length PTH1-84 molecule (200). Tubular reabsorption involves the multifunctional receptor megalin (201).
With the progression of CKD, both pathways of renal PTH degradation are progressively impaired. This leads to a marked prolongation of the half-life of C-terminal PTH fragments in the circulation (202–204) and their accumulation in the extracellular space. Moreover, there is no peritubular metabolism of PTH1-84 in uremic non-filtering kidneys, in contrast to peritubular uptake by normal, filtering kidneys (205). Hepatic PTH catabolism appears however to be unchanged in CKD since uremic livers and control livers released equal amounts of immunoreactive C-terminal PTH fragments (205).
A decreased response to the action of PTH may be another factor involved in the stimulation of the parathyroid glands in CKD. A diminished calcemic response to the infusion of PTH has long been reported, suggesting that PTH over secretion was necessary to maintain eucalcemia. The skeletal resistance to PTH has been attributed to various mechanisms, including impaired vitamin D action in association with hyperphosphatemia, overestimation of true PTH(1-84) by assays measuring iPTH (see below), accumulation of inhibitory PTH fragments, oxidative modification of PTH, increase in circulating osteoprotegerin and sclerostin levels, administration of active vitamin D derivatives and calcimimetics, and altered PTH-R1 expression (7,141,206,207). Concerning the latter mechanism, studies have suggested the presence of PTH receptor isoforms in various organs of normal rats. Downregulation of PTH-R1 mRNA has been observed in various tissues in uremic rats (208–211) and also in osteoblasts of patients with end-stage renal disease (212). However, the issue of PTH-R1 expression in bone tissue remains a matter of controversy since one group found it to be upregulated in patients with moderate to severe renal hyperparathyroid bone disease (213). A recent study claimed that inhibition of PTH binding to PTH-R1 by soluble Klotho could represent yet another mechanism of PTH resistance (214). This observation would be compatible with the presence of upregulated, yet biologically inactive PTH-R1.
Other mechanisms involved in the control of the normal balance between bone formation and resorption and their response to PTH are the Wnt-β-catenin signaling pathway and its inhibition by sclerostin and Dickkopf-related protein 1 (Dkk1) (7,56,215), and the activin A pathway with its inhibition by a decoy receptor (216). Wnt-β-catenin inhibitors are expressed predominantly in osteocytes. Whereas reduced activity of sclerostin and Dkk1 leads to increased bone mass and strength, the opposite occurs in animal models with overexpression of both sclerostin and Dkk1. In CKD, circulating levels of both Wnt—β-catenin inhibitors have generally been found to be increased (56), and serum sclerostin was found to correlate negatively with serum PTH (217,218), and PTH has been shown to blunt osteocytic production of this Wnt inhibitor (219). Since high PTH and sclerostin levels coexist in CKD this raises the suspicion that sclerostin contributes to PTH resistance in CKD (7). Calcitonin and bone morphogenetic proteins stimulate, whereas PTH and estrogens suppress the expression of sclerostin and/or Dkk1 (220,221). Bone formation induced by intermittent PTH administration to patients with osteoporosis could be explained, at least in part, by the ability of PTH to downregulate sclerostin expression in osteocytes, permitting the anabolic Wnt signaling pathway to proceed (222). In patients with ESKD sclerostin is a strong predictor of bone turnover and osteoblast number (223). Serum levels of sclerostin correlate negatively with serum iPTH in such patients. Sclerostin was superior to iPTH for the positive prediction of high bone turnover and number of osteoblasts. In contrast, iPTH was superior to sclerostin for the negative prediction of high bone turnover and had similar predictive values as sclerostin for the number of osteoblasts. Serum sclerostin levels increase after parathyroidectomy (7). As regards activin A, a member of the transforming growth factor-b superfamily, Hruska’s group has demonstrated increased serum levels and systemic activation of its receptors in mouse models of CKD (216). In humans, serum activin A levels increase already at early stages of CKD, before elevations in intact PTH and FGF23, equally pointing to a role in CKD-MBD and PTH resistance (224).
Interesting new pathways have recently been identified by Pacifici’s group. First, they used various mouse models to demonstrate a permissive activity of butyrate produced by the gut microbiota, required to allow stimulation of bone formation by PTH (225). Butyrate’s effect was mediated by short-chain fatty acid receptor GPR43 signaling in dendritic cells and by GPR43-independent signaling in T cells. Second, the group showed that intestinal segmented filamentous bacteria (SFB) enabled PTH to expand intestinal TNF+ T and Th17 cells and thereby increase their egress from the intestine and recruitment to the bone marrow to cause bone loss (226). Figure 15 shows these recently detected pathways involving the gut microbiota.
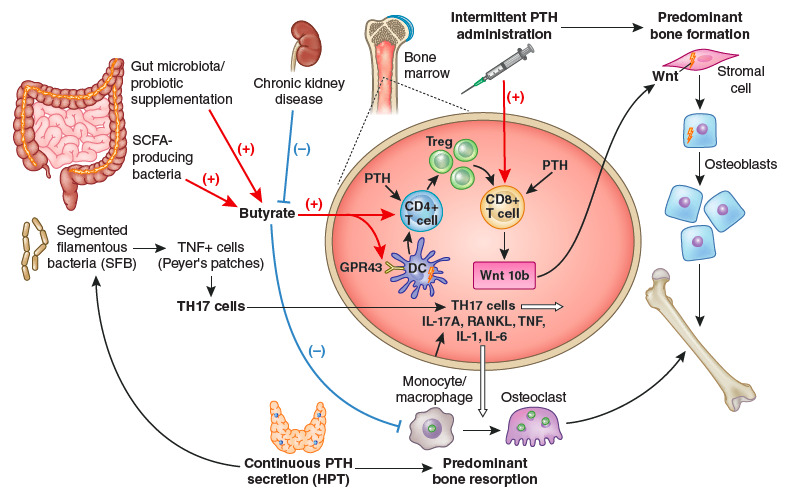
Figure 15.
Importance of intestinal microbiota for PTH action in bone. The stimulation of bone anabolism by PTH requires butyrate formation by short chain fatty acid (SCFA) producing gut bacteria. CKD probably reduces its production. Butyrate increases the frequency of regulatory T (Treg) cells in the intestine and in the bone marrow and potentiates the capacity of intermittently administered PTH to induce the differentiation of naïve CD4+ T cells into Tregs, a population of T cells which induces conventional CD8+ T cells to release Wnt10b. This osteogenic Wnt ligand activates Wnt signaling in osteoblastic cells and stimulates bone formation. Butyrate enables intermittent PTH dosing to expand Tregs via GPR43 signaling in dendritic cells (DCs) and GPR43 independent targeting of T cells. Butyrate may also affect bone remodeling by modulating osteoclast genes. The stimulation of bone resorption by PTH requires the presence of segmented filamentous bacteria (SFB) within gut microbiota for the production of Th17 cells in intestinal Peyers' plaques. Continuously elevated PTH levels lead to TNF+ T cell expansion in the gut and the bone marrow via a microbiota-dependent, but SFB independent mechanism. Furthermore, intestinal TNF producing T cells are required for PTH to increase the number of intestinal Th17 cells, and TNF mediates the migration of intestinal Th17 cells to the bone marrow. This migration depends on upregulation of chemokine receptor CXCR3 and chemokine CCL20. Bone marrow Th17 cells then induce osteoclastogenesis by secreting IL-17A, RANKL, TNF, IL-1, and IL-6. From Massy & Drueke (227).
It will be interesting to examine the hypotheses that the excessive bone resorption associated with secondary hyperparathyroidism in CKD is at least partially due to either insufficient intestinal butyrate availability, excessive intestinal SFB activity, or both (227).
SECONDARY HYPERPARATHYROIDISM IN CKD – CLINICAL FEATURES
In most patients with ESKD, even advanced secondary hyperparathyroidism remains a clinically silent disease. Clinical manifestations are generally related to severe osteitis fibrosa and to the consequences of hypercalcemia and/or hyperphosphatemia.
Osteoarticular pain may be present. When patients become symptomatic, they usually complain of pain on exertion in skeletal sites that are subjected to biomechanical stress. Pain at rest and localized pain are rather unusual and suggest other underlying causes. Severe proximal myopathy is seen in some patients, even in the absence of vitamin D deficiency. These symptoms and signs are more frequent in patients who suffer from mixed renal osteodystrophy, resulting from a combination of parathyroid over function and vitamin D deficiency. Skeletal fractures may occur after only minor injury. They may also develop on the ground of cystic bone lesions, the so-called “brown tumors”, which occur for still unknown reasons in a small number of uremic patients with secondary hyperparathyroidism. Rupture of the patella or avulsion of tendons may be seen in advanced cases.
Uremic pruritus is most often associated with an elevated Ca x P product although other factors may also be involved. Related symptoms and signs are the red eye syndrome due to the deposition of calcium in the conjunctiva, cutaneous calcification, and pseudogout. The latter is a form of painful arthralgia of acute or subacute onset caused by intra-articular deposition of radio-opaque crystals of calcium pyrophosphate dehydrate.
The syndrome of “calciphylaxis” is an infrequent manifestation of cutaneous and vascular calcification in uremic patients which may occur in association with secondary hyperparathyroidism, although this association is by no means constant. It is characterized by a rapidly progressive skin necrosis involving buttocks and the legs, particularly the thighs. It can produce gangrene and may be fatal. It occurs as the result of arteriolar calcification and has also been termed “calcific uremic arteriolopathy” to reflect more accurately the nature of the lesion (228). Of interest, a post-hoc analysis of the EVOLVE trial in chronic hemodialysis patients recently showed that cinacalcet administration, which allows improved PTH control, resulted in a significant decrease in the incidence of calcific uremic arteriolopathy as compared to placebo (229).
SECONDARY HYPERPARATHYROIDISM IN CKD -- DIAGNOSIS
The biochemical diagnosis relies on the determination of plasma iPTH. This is also true for primary hyperparathyroidism. In patients with CKD, it has however become apparent in recent years that there are several limitations to the measurement of iPTH, in addition to the usual day-to-day variations in healthy people (230). Physiological iPTH plasma values are not normal for uremic patients since values in the normal range are often associated with low bone turnover (adynamic bone disease) whereas normal bone turnover may be observed in presence of elevated plasma intact PTH levels (231–234). It is currently unclear to what extent this is due to imperfections in the PTH assays used (see below), PTH receptor status, post-receptor events, non-PTH-mediated changes in bone metabolism (e.g., supply of vitamin D or its metabolites, supply of estrogens or androgens), or a combination of these factors.
The accumulation of a large non (1-84) molecular form of PTH, which is detected by iPTH (so-called "intact" PTH) assays, has been described in patients with CKD (235). The large PTH fragment was tentatively identified as hPTH(7-84) (236). This finding is of importance in the interpretation of PTH values, since true hPTH(1-84) represents only about 50-60% of the levels detected by the currently used intact PTH assays, and since PTH(7-84) antagonizes PTH(1-84) effects on serum calcium and on osteoblasts (237). Moreover, the secretory responses of hPTH(1-84) and non-hPTH(1-84) to changes in [Ca2+e] are not proportional for these two PTH moieties (87). Moreover, a large variability has been found between different assay methods used for plasma PTH measurement in patients with CKD, recognizing PTH(7-84) with various cross-reactivities (238). Varying plasma sampling and storage conditions may further complicate the interpretation of PTH results provided by clinical laboratories (239). The development of assays which detect full-length (whole) human PTH, but not amino-terminally truncated fragments (240), was initially considered as a major progress in this field. To further improve the assessment of uremic hyperparathyroidism and the associated increase in bone turnover Monier-Faugere et al proposed to calculate the ratio of PTH-(1-84) to large C-PTH fragments (241). The usefulness in the clinical setting of the whole PTH assay and of the ratio of whole PTH to PTH fragments has however not been convincingly established for the diagnosis of parathyroid over function in adult (242,243) or pediatric (244) dialysis patients. From a practical point of view, it must be pointed out that at present measurement of PTH with third-generation assays is not widely available. Another potential issue is the presence of oxidized, inactive PTH in the circulation of patients with CKD, with concentrations much higher than those of iPTH (141), although there were large interindividual variations (141). Whereas one study showed a U-shaped association of non-oxidized, but not oxidized, PTH with survival in patients on hemodialysis therapy (245), a subsequent study done in CKD stage 2-4 patients found iPTH, but not non-oxidized PTH, to be associated with all-cause death in multivariable analysis (246). The reasons for these apparently opposite findings are unclear. The assertion that PTH oxidation is a vitro artifact has been disproven recently (247). Based on personal findings, Hocher and Zeng postulated that oxidized and non-oxidized PTH should be measured separately to correctly evaluate the degree of severity and clinical relevance of parathyroid over function in CKD (142). However, in a very recent study Ursem et al observed a strong correlation between serum non-oxidized PTH and total PTH in patients with ESKD (248). Most importantly, they found that both histomorphometric and circulating bone turnover markers exhibited similar correlations with non-oxidized PTH and total PTH. The authors therefore concluded that non-oxidized PTH is not superior to total PTH as a biomarker of bone turnover in ESKD. However, presently available methods do not enable a precise distinction between biologically active and inactive PTH forms, be it through oxidative or other post-translational modifications of the hormone (249). Most importantly, we will hopefully be able in the future to rely not only on serum PTH but also on appropriate direct markers of bone structure and function for the assessment of renal osteodystrophy and on markers of cardiovascular disease related to secondary hyperparathyroidism (250).
Bone x-ray diagnosis is impossible in mild to moderate forms of secondary hyperparathyroidism, but relatively easy in severe forms. Nevertheless, to date x-ray diagnosis is rarely used in routine clinical praxis. Typical lesions include resorptive defects on the external and internal surfaces of cortical bone, with the resorption particularly pronounced on the subperiosteal surface. Resorption within cortical bone enlarges the Haversian channels, resulting in longitudinal striation; resorption at the endosteal surface causes cortical thinning. These lesions can be generally detected first in the hand skeleton, most characteristically at the periosteal surface of the middle phalanges (Figure 16).
Accelerated bone deposition at this site (periosteal neostosis) can also be seen. Another characteristic feature is resorptive loss of acral bone (acro-osteolysis), in particular at the terminal phalanges, at the distal end of the clavicles, and in the skull (‘pepper-pot’ aspect) (Figure 17). Whereas cortical bone is progressively thinning, the mass of spongy bone tends to increase, particularly in the metaphyses. The latter phenomenon results in a characteristic sclerotic aspect of the upper and lower thirds of the vertebrae, contrasting with rarefaction of the center (‘rugger jersey spine’). Osteosclerosis is also commonly seen in radiographs of the metaphyses of the radius and tibia.

Figure 16.
Periosteal resorption and small vessel calcification in severe secondary uremic hyperparathyroidism. (a) X-ray aspect of periosteal resorption within cortical bone of middle phalanges of the hand, indicative of osteitis fibrosa, and extensive finger artery calcification in a CKD stage 5 patient with severe secondary hyperparathyroidism. (b) One year after surgical parathyroidectomy: complete bone lesion healing and disappearance of arterial calcification.

Figure 17.
X-ray pepper-and-salt aspect of the skull in in a chronic hemodialysis patient with severe secondary hyperparathyroidism.
In addition to the skeletal lesions, radiographs often reveal various types of soft tissue calcification. These comprise vascular calcifications, i.e., calcification of intimal plaques (aorta, iliac arteries) (Figure 18a), as well as diffuse calcification (Mönckeberg type) of the media of peripheral muscular arteries (Figure 18b) (251).

Figure 18.
Massive intima (a) and media (b) calcification of hypogastric artery in a chronic hemodialysis patient.
Of interest, media calcification of digital arteries can entirely regress after surgical parathyroidectomy (Figure 16). Calcium deposits may also be seen in periarticular tissue or bursas and may exhibit tumor-like features (Figure 19).

Figure 19.
X-ray feature of a tumor-like periarticular calcification in the shoulder of a chronic hemodialysis patient with adynamic bone disease due to aluminum intoxication.
Since the development of electron-beam computed tomography (EBCT) and multiple slice computed tomography (MSCT) more reliable means have become available to assess quantitatively vascular calcification and its progression in uremic patients (252). However, these techniques are not universally available and they are costly. Moreover, they do not allow a distinction between arterial intima and media calcifications. Such a distinction can be obtained by radiograms of the pelvis and the thigh, combined with ultrasonography of the common carotid artery. Using these simple methods, London et al could show that hemodialysis patients with arterial media calcification had a longer survival than hemodialysis patients with arterial intima calcification, but in turn their survival was significantly shorter than that of hemodialysis patients without calcifications (253). Of note, both severe hyperparathyroidism and marked hypoparathyroidism favor the occurrence of the two types of calcifications in patients with ESKD (254–256). In contrast to permanent elevations in serum PTH, the intermittent administration of PTH1-34 has been shown to decrease arterial calcification in uremic rats (257) and in diabetic mice with LDL receptor deletion (258). This observation tends to demonstrate that normal parathyroid function is required not only for the maintenance of optimal bone structure and function, but also as an efficacious defense against soft tissue calcification, and that intermittent PTH administration may not only improve osteoporosis (259), but also reduce vascular calcification, at least in experimental animals.
SECONDARY HYPERPARATHYROIDISM IN CKD - TREATMENT
Medical Management
Presently available options of medical treatment should take into account the levels of plasma biochemistry and x-ray findings, and as a more recently recognized parameter also the dimensions of the largest parathyroid glands, as assessed by ultrasonography. A gland diameter of 5-10 mm or more is considered as being indicative of autonomous growth, which often is resistant to medical treatment (198).
Schematically, there are five major medical treatment options which can be combined in some cases, but not in others, namely the restriction of phosphate intake and/or the administration of calcium supplements, oral phosphate binders, vitamin D derivatives, and calcimimetics (260,261). In dialysis patients the weekly dose of renal replacement therapy is an additional important factor. An optimal dialysis technique allows controlling hyperphosphatemia, and providing enough calcium to avoid PTH stimulation by hypocalcemia during dialysis sessions.
When trying to control hyperparathyroidism it is important to avoid both hypocalcemia and hypercalcemia and to reduce or correct hyperphosphatemia as well. In patients with controlled plasma phosphate, this can be achieved by giving either calcitriol or one of its synthetic analogs, or by administering oral calcium supplements. For a long time, calcitriol or alfacalcidol was the preferred therapy in uremic patients with high to very high plasma intact PTH values and normal to moderately elevated plasma calcium levels, when plasma phosphate did not exceed recommended levels, namely 1.5 mmol/L for CKD stages 3-4 and 1.8 mmol/L for CKD stage 5, according to the K/DOQI guidelines of 2003 (262). However, the administration of active vitamin D derivatives often induces hypercalcemia and/or hyperphosphatemia. The KDIGO CKD-MBD guideline of 2009 (263) and its -update in 2017/2018 (264,265) suggest "maintaining iPTH levels in CKD stage 5D patients (i.e., patients receiving dialysis therapy) in the range of approximately two to nine times the upper normal limit for the assay, to keep serum calcium normal, and to decrease serum phosphorus towards the normal range." Thus, the recommended iPTH target range has become larger than with the prior K/DOQI guidelines. The KDIGO guideline further suggests that marked changes in iPTH levels in either direction within the newly defined, broadened range should "prompt initiation or change in therapy to avoid progression to levels outside of this range." The updated guideline recommends that patients with CKD stages G3a-G5 not on dialysis whose levels of intact PTH are progressively rising or persistently above the upper normal limit for the assay be evaluated for modifiable factors, including hyperphosphatemia, hypocalcemia, high phosphate intake, and vitamin D deficiency (grade 2C recommendation).
Vitamin D and Active Vitamin D Derivatives
A satisfactory degree of vitamin D repletion should probably be aimed at in case of vitamin D deficiency since the majority of patients with CKD have at least some degree of vitamin D deficiency (51,266). Relative vitamin D depletion has been shown to be an independent risk factor for secondary hyperparathyroidism in hemodialysis patients (54). Repletion with native vitamin D may lead to improved control of secondary hyperparathyroidism in patients with CKD not yet on dialysis (267) and in those treated by dialysis (268) but a beneficial effect has not been observed in a subsequent meta-analysis (269). Vitamin D repletion may allow optimal bone formation, help to avoid osteomalacia, and exert numerous other positive effects due to the pleiotropic actions of vitamin D, but most of these presumably positive actions remain a matter of debate (269,270). Most importantly, randomized controlled trials with native vitamin D or calcidiol have not been performed so far to evaluate hard clinical outcomes of patients with CKD.
As regards the administration of active vitamin D sterols during the course of CKD, the updated KDIGO guideline suggests that calcitriol and vitamin D analogues not be routinely used in patients with CKD G3a-G5 (Grade 2C recommendation). It further states that it is reasonable to reserve the use of these agents for patients with CKD G4-G5 with severe and progressive hyperparathyroidism (264,265).
To correct secondary hyperparathyroidism of moderate to severe degree the oral administration of active vitamin D derivatives is generally more efficient than that of native vitamin D. In hemodialysis patients, calcitriol or its analogs can be given either orally or intravenously. The oral administration can be on a daily basis (for instance 0.125 to 0.5 µg of calcitriol) or as intermittent bolus ingestions (for instance 0.5 to 2.0 µg of calcitriol for each dose) whereas the intravenous administration is always intermittent (also 0.5 to 2.0 µg of calcitriol or more per injection). The route and mode of administration of calcitriol or alfacalcidol probably play only a minor role. Since the highly active 1α-hydroxylated vitamin D derivatives can easily induce hypercalcemia, intensive research has focused on the development of various non-hypercalcemic analogs, including the natural vitamin D compound 24,25(OH)2 vitamin D3, 22-oxa-calcitriol (maxacalcitol), 19-nor-1,25(OH)2 vitamin D3 (paricalcitol), and 1α-(OH) vitamin D2 (hectorol). Despite numerous studies done in many patients, none of them has been shown to be entirely devoid of inducing increases in plasma calcium or phosphate, and none has been demonstrated thus far to be superior to calcitriol or alfacalcidol in the long run in controlling secondary hyperparathyroidism (271,272). An observational study by Teng et al. showed that paricalcitol administration to a large cohort of hemodialysis patients conferred a remarkable (16%) survival advantage over the administration of calcitriol (273). Numerous subsequent observational studies reported a survival benefit, either comparing treatment with active vitamin D derivatives to no treatment, or novel active vitamin D derivatives to calcitriol in CKD patients not yet on dialysis (274) or those receiving dialysis treatment (275–277). Another observational study conducted in hemodialysis patients, however, did not find a survival advantage with paricalcitol, as compared to calcitriol (278). In the absence of randomized controlled trials, it is impossible to conclude that paricalcitol treatment is superior to calcitriol or alfacalcidol in terms of patient survival. Findings of observational studies can only be considered as hypothesis-generating. They need to be confirmed by a properly designed prospective investigation (279).
Calcimimetics
The introduction of the calcimimetic cinacalcet into clinical practice led to a change in the above treatment strategy since it enables parathyroid over function control without increasing plasma calcium or phosphorus. Calcimimetics modify the configuration of the CaSR, a receptor cloned by Brown et al in 1993 (280) They make the CaSR more sensitive to [Ca2+e] in contrast to the so-called calcilytics which decrease its sensitivity, as schematically shown in Figure 20.

Figure 20.
Schematic representation of the modulation of the calcium-sensing receptor (CaSR) by calcimimetics and calcilytics. CaSR is expressed on cell membrane. Calcimimetics increase its sensitivity to calcium ions whereas calcilytics decrease it.
Initial acute studies in chronic hemodialysis patients showed that the calcimimetic cinacalcet was capable of reducing plasma PTH within hours, immediately followed by a rapid decrease in plasma calcium and a minor decrease in plasma phosphate (281–283). In addition, calcimimetics can also reduce parathyroid cell proliferation. Both short-term and long-term studies performed in rats and mice with CKD showed that the administration of the calcimimetic NPS R-568, starting at the time of CKD induction, allowed the prevention of parathyroid hyperplasia (183,193,284). This effect is probably due to a direct inhibitory action on the parathyroid cell, as shown by our group in an experimental study in which we exposed human uremic parathyroid cells to the calcimimetic NPS-R467 (75). An interesting finding of a yet unexplained mechanism and significance is the observation that calcimimetic treatment led to an approximately 5-fold increase in the proportion of oxyphil cells, as compared to chief cells, in parathyroid glands removed from CKD patients with refractory hyperparathyroidism (285–287). Of note, oxyphil cells also exhibited higher CaSR expression than chief cells in such glands (288).
Perhaps more important from a clinical point of view, the administration of calcimimetics enabled an improvement of osteitis fibrosa (103), halted the progression of vascular calcification both in uremic animals (284,289) and probably also in dialysis patients (290), prevented vascular remodeling (291), improved cardiac structure and function (292), and prolonged survival (293) in uremic animals with secondary hyperparathyroidism.
The long-term administration of cinacalcet to chronic hemodialysis patients proved to be superior to optimal standard therapy in controlling secondary uremic hyperparathyroidism, in that it was able to induce not only a decrease in plasma PTH but also in plasma calcium and phosphate (294–297). Figure 21 shows the superior control of severe secondary hyperparathyroidism by cinacalcet as compared to placebo treatment with standard of care (298). The initial daily dose is 30 mg orally, which can be increased up to 180 mg if necessary. Cinacalcet is generally well tolerated, with the exception of gastrointestinal side effects, which however cease in the majority of patients with time. Since its administration generally leads to a decrease in serum calcium, a close follow-up is required, at least initially, to avoid hypocalcemia with possible adverse clinical consequences. Cinacalcet can be associated with calcium-containing and non-calcium containing phosphate binders and also with vitamin D derivatives. For PTH lowering a combination therapy may lead to more complete correction than single drug treatment because of less side-effects and greater efficacy in the control of parathyroid over function (299,300).

Figure 21.
Effect of cinacalcet on need of parathyroidectomy in patients on hemodialysis therapy. In the EVOLVE trial, parathyroidectomy was performed in 140 (7%) cinacalcet-treated and 278 (14%) placebo-treated patients. Key independent predictors of parathyroidectomy included younger age, female sex, geographic region, and absence of history of peripheral vascular disease. One hundred and forty-three (7%) cinacalcet-treated and 304 (16%) placebo-treated patients met the biochemical definition of severe, unremitting (tertiary) hyperparathyroidism. Considering the pre-specified biochemical composite or surgical parathyroidectomy as an endpoint, 240 (12%) cinacalcet-treated and 470 (24%) placebo-treated patients developed severe, unremitting hyperparathyroidism (298).
The subsequent development of an intravenously active calcimetic led to another series of clinical studies aimed at controlling secondary hyperparathyroidism in patients on hemodialysis with an easy access to parenteral drug administration, thereby reducing oral pill overload. Two randomized controlled trials were conducted in such patients with moderate to severe secondary hyperparathyroidism, evaluating the efficacy and safety of the intravenous calcimimetic, etelcalcetide as compared to placebo (301). Thrice weekly administration of active drug after hemodialysis led to a greater than 30% reduction in serum PTH compared with less than 8.9% of patients receiving placebo. The reduction in PTH was rapid and sustained over 26 weeks. Treatment with etelcalcetide lowered serum calcium in the majority of patients, with overt symptomatic hypocalcemia reported in 7%. Adverse events occurred in 92% of etelcalcetide-treated and 80% of placebo-treated patients. Nausea, vomiting, and diarrhea were more common in etelcalcetide-treated patients, as were symptoms potentially related to hypocalcemia. A subsequent double-blind, double-dummy randomized controlled trial compared intravenous etelcalcetide to oral cinacalcet in patients on hemodialysis with moderate to severe secondary hyperparathyroidism (302). It showed that the use of etelcalcetide was not inferior to cinacalcet in reducing serum PTH concentrations over 26 weeks. In addition, etelcalcetide met several superiority criteria, including a greater reduction in serum PTH concentrations from baseline, and more potent reductions in serum concentrations of FGF23 and two markers of high-turnover bone disease.
How about hard patient outcomes? The randomized controlled trial EVOLVE examined the question whether better control of secondary uremic hyperparathyroidism by cinacalcet, as compared to placebo treatment with standard of care, reduced the incidence of cardiovascular events and mortality (298). The study enrolled 3803 patients receiving long-term hemodialysis therapy. Using intention-to-treat analysis the study outcome was negative (Figure 22, upper part). However, after adjustment for age and other confounders, and also when using lag-censoring analysis (Figure 22, lower part), there was a nominally significant reduction in the primary cardiovascular endpoint including mortality in the cinacalcet treatment group in whom serum PTH, calcium, and phosphate were better controlled than in the placebo treatment group. Moreover, a post-hoc lag-censoring analysis of EVOLVE further showed that the incidence of clinically ascertained fractures was lower in the cinacalcet than the placebo arm (303).

Figure 22.
Effect of cinacalcet on cardiovascular outcomes of patients on hemodialysis therapy. The randomized controlled trial EVOLVE examined the question whether a better control of secondary uremic hyperparathyroidism by cinacalcet, as compared to placebo treatment with standard of care, reduced the incidence of cardiovascular events and mortality. The study enrolled 3803 patients receiving long-term hemodialysis therapy. Using intention-to-treat analysis the study outcome was negative (upper part of Figure). However, with lag-censoring analysis there was a nominally significant reduction in the primary composite cardiovascular endpoint in the cinacalcet treatment group in whom serum PTH, calcium, and phosphorus were better controlled than in the placebo treatment group (lower part of Figure). From Chertow et al (298).
Phosphate Binders, Inhibitors of Intestinal Phosphate Absorption, Oral Phosphate Restriction, and Phosphate Removal by Dialysis
Calcium-containing phosphate binders should be given, preferentially during or at the end of phosphate-rich meals, to patients with CKD and uncontrolled hyperphosphatemia who have no hypercalcemia or radiological evidence of marked soft tissue calcifications. In these latter cases non-calcium-containing phosphate binders should be preferred (see below). The administration of calcium salts alone such as calcium carbonate or calcium acetate may be sufficient for the control of hyperphosphatemia in many instances, particularly in patients with CKD stages G3-G5 not yet on dialysis. At the same time these calcium salts will prevent serum iPTH from rising in the majority of patients (304). They may however lead to calcium overload (44,45) and excessive PTH over suppression, resulting eventually in adynamic bone disease (305). In hemodialysis patients, the efficacy and tolerance of this treatment may be enhanced by the concomitant use of low-calcium dialysate, for instance a calcium concentration of 1.25 mmol/L, especially if plasma intact PTH levels are not very high. However, long-term studies have shown that the continuous use of a dialysate calcium of only 1.25 mmol/L requires close monitoring of plasma calcium and PTH because of the risk of inducing excessive PTH secretion (306,307). A dialysate calcium concentration between 1.25 and 1.5 mmol/L is more appropriate in terms of optimal calcium balance and control of secondary hyperparathyroidism (308). The use of a low calcium dialysate also may require higher doses of active vitamin D derivatives (309) or cinacalcet (310) for the control of secondary hyperparathyroidism. Of note, the use of a low calcium bath favors hemodynamic instability during the hemodialysis session (311) and the occurrence of sudden cardiac arrest (312,313). In CAPD patients, the use of calcium carbonate, in the absence of vitamin D, together with a reduction of the dialysate calcium concentration from 1.75 to 1.45 mmol/L prevents the occurrence of hypercalcemia in most patients (314). However, the addition of daily low-dose alfacalcidol may lead to hypercalcemia, despite a further reduction of dialysate calcium to 1.0 mmol/L.
The development of calcium-free, aluminum-free oral phosphate binders such as sevelamer-HCl (315–317), sevelamer carbonate (318,319), lanthanum carbonate (320–322), sucroferric oxyhydroxide (323) and ferric citrate (324) allows controlling hyperphosphatemia without the potential danger of calcium overload. Their phosphate binding capacity is roughly equivalent to that of Ca carbonate or calcium acetate. Sevelamer offers in addition the advantage to lower serum total cholesterol and LDL-cholesterol and to increase serum HDL-cholesterol, to slow the progression of arterial calcification in dialysis patients (316), and possibly to improve survival in such patients (325). The administration of sevelamer is probably more efficient in halting the progression of vascular calcification than calcium carbonate or calcium acetate but this remains a matter of debate (14,326,327). The administration of lanthanum carbonate to uremic animals has been shown to also reduce progression of vascular calcification (328,329), but studies in patients with CKD have led to variable results (330–332). The effects of calcium-free, aluminum-free phosphate binders on serum iPTH are variable, depending on baseline iPTH and concomitant therapies. In general, iPTH levels are higher in response to these binders than to calcium-containing phosphate binders (Figure 23) (333,334).
Figure 23.
Effect of oral calcium vs. sevelamer on serum intact PTH (iPTH) in CKD. In this 54-week, randomized, open-label study the effects of sevelamer hydrochloride on bone structure and various biochemical parameters were compared to that of calcium carbonate in 119 patients on long-term hemodialysis therapy. Serum iPTH was consistently lower with calcium carbonate than with sevelamer treatment. From Ferreira et al (333).
The administration of aluminum-containing phosphate binders should be avoided because of their potential toxicity. They may be given in some treatment resistant cases, but only for short periods of time (263).
Another approach chosen to control hyperphosphatemia and therefore to prevent or delay the development of secondary hyperparathyroidism is pharmacologic interference with active intestinal phosphate transport by oral inhibitors of the phosphate/sodium cotransporter NaPi2b, using either already available drugs such as niacin or nicotinamide (335–337), or recently developed novel inhibitors such as tenapanor (338,339). The rather disappointing results of available studies have not let so far to their introduction into clinical practice (340).
Dietary phosphate intake should be assessed and diminished, if possible. Special attention should be given to the avoidance of foods containing phosphate additives (341). The spontaneous reduction of protein intake with age probably explains the often better control of serum phosphate in elderly as compared to younger patients with ESKD, and this may contribute to the relatively lower PTH levels of the former and their propensity to develop adynamic bone disease (342). However, when reducing dietary phosphate intake and concomitantly protein intake, one has to take care to avoid the induction of a protein malnutrition state. Restricting dietary protein intake excessively may lead to greater mortality (343). In dialysis patients, an attempt should always be made as well to improve the efficiency of the dialysis procedure.
A better correction of metabolic acidosis by bicarbonate-buffered dialysate, as compared to acetate-buffered dialysate, probably helps to delay the progression of osteitis fibrosa in hemodialysis patients (344). One possible mechanism for the beneficial role of acidosis correction is an increase in the sensitivity of the parathyroid gland to plasma ionized calcium (345).
Current recommendations for the medical treatment and prevention of patients with CKD-MBD, including secondary hyperparathyroidism, can be found in the 2009 KDIGO CKD-MBD guideline (263) and its recent update (264,265). It must be pointed out though that there is no definitive proof of a beneficial effect of phosphate lowering on patient-level outcome (247).
Local Injection of Alcohol and Active Vitamin D Derivatives
Since in advanced forms of secondary hyperparathyroidism the hyperplasia of parathyroid glands is asymmetrical, with some glands being grossly enlarged and others remaining relatively small, local injection of ethanol (346,347) or active vitamin D derivatives (348,349) has been proposed as an alternative therapy in patients who become resistant to medical treatment. However, the direct injection technique has not reached widespread use in clinical practice outside of Japan. Other research groups have been unable to obtain convincing results (350,351).
Despite major advances in the medical treatment of CKD-MBD the achievement of the targets for plasma calcium, phosphate, Ca x P product, and PTH, as recommended by the K/DOQI guidelines (262), was found to be far from being optimal in the DOPPS patient population for the years 2002-2004 (352). It was actually rare in the hemodialysis patients of this international cohort to fall within recommended ranges for all four indicators of mineral metabolism, although consistent control of all three main CKD-MBD parameters calcium, phosphate, and PTH was found to be a strong predictor of survival in hemodialysis patients in an observational study (353). A recent report on chronic hemodialysis patients in France confirmed that a satisfactory control of serum calcium, phosphate, or PTH was achieved in less than 20% among them (354).
Surgical Treatment
Surgical correction remains the final, symptomatic therapy of the most severe forms of secondary hyperparathyroidism, which cannot be controlled by medical treatment (355). The most important goal remains to prevent or correct the development of major clinical complications associated with this disease. The presence of severe parathyroid over function must be ascertained by clinical, biochemical and radiological evidence. In general, neck surgery should only be done when plasma iPTH values are greatly elevated (> 600-800 pg/mL), together with an increase in plasma total alkaline phosphatases (or better bone-specific alkaline phosphatase), and only after one or several medical treatment attempts have remained unsuccessful in decreasing plasma iPTH with cinacalcet (in dialysis patients only) and/or active vitamin D derivatives or if their use is relatively or absolutely contraindicated, namely in presence of persistent hypercalcemia, marked hyperphosphatemia, or severe vascular calcifications. Bone histomorphometry examination is rarely needed. Clinical symptoms and signs such as pruritus and osteoarticular pain are non-specific and therefore not good criteria for operation by their own. Similarly, an isolated increase in plasma calcium and/or phosphate, even in case of coexistent soft tissue calcifications, is not a sufficient criterion alone for surgical parathyroidectomy. However, in the presence of a persistently high plasma PTH the latter disturbances may facilitate the decision to proceed to surgery. The results can be spectacular, including in rare instances the complete disappearance of soft tissue calcifications from small peripheral arteries (see Figure 15b). A concomitant aluminum overload should be excluded or treated, if present, before performing surgery.
Two main surgical procedures are generally used, either subtotal parathyroidectomy or total parathyroidectomy with immediate auto-transplantation. There is no substantial difference of operative difficulties and treatment results between the two procedures. We found that the long-term frequency of recurrent hyperparathyroidism was similar (356). One group of authors claimed superiority of total parathyroidectomy without reimplantation of parathyroid tissue in terms of long-term control of parathyroid over function, tolerance, and safety (357), but this claim has been questioned by us and others (358–360). We do not recommend the performance of total parathyroidectomy without auto-transplantation in uremic patients since permanent hypoparathyroidism and adynamic bone disease may ensue, with possible harmful consequences especially for those patients who subsequently undergo kidney transplantation.
As regards the prevalence of parathyroidectomy it was very high before the turn of the century and did not change significantly between 1983 and 1996. According to a survey from Northern Italy in 7371 dialysis patients (361) it was 5.5% in the all patients together but increased with duration of RRT, from 9.2% after 10-15 years to 20.8% after 16-20 years of dialysis therapy. A recent survey from the US showed that parathyroidectomy rates were much lower in the first decade of the 21st century. It decreased from 7.9% in 2003 to a nadir of 3.3% in 2005 - most likely due to the commercial introduction of cinacalcet, then increased again to 5.5% through 2006, and subsequently remained stable until 2011 (362). The authors concluded that despite the use of multiple medical therapies rates of parathyroidectomy in patients with secondary hyperparathyroidism did not decline in recent years. These findings are in contrast with a Canadian study (363) and the international Dialysis Outcomes and Practice Patterns Study (DOPPS) (364). The Canadian study, although restricted to a single province (Quebec), showed a sustained reduction in parathyroidectomy rates after 2006. The DOPPS reported that prescriptions of active vitamin D analogs and cinacalcet increased and that parathyroidectomy rates decreased. Difference in medical treatment modalities between geographic regions and different modes of data analysis may at least partially account for these apparent discrepancies. This is illustrated by the observation that parathyroidectomy rates in Japan fell abruptly after the advent of cinacalcet to approximately 2%, with median serum iPTH around 150 pg/mL between 1996 and 2011 (364).
Parathyroidectomy was associated with higher short-term mortality, but lower long-term mortality among chronic dialysis patients in the US (365). Whether presently available therapeutic and prophylactic measures taken to attenuate secondary hyperparathyroidism play an important role in reducing cardiovascular morbidity and mortality among patients with ESKD remains a matter of debate. The EVOLVE trial points to better clinical outcomes with a more efficient control of parathyroid over function by cinacalcet than by optimal standard treatment but the results, although suggestive, must still be considered as not definitively conclusive (298,303).
ACKNOWLEDGEMENTS
The author wishes to thank Ms Martine Netter, Paris for expert assistance in Figure design.
REFERENCES
- 1.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006;69:1945–1953. [PubMed: 16641930]
- 2.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–38. [PubMed: 17091124]
- 3.
- Fliser D, Kollerits B, Neyer U, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol. 2007;18:2600–2608. [PubMed: 17656479]
- 4.
- Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–1378. [PMC free article: PMC3134393] [PubMed: 21389978]
- 5.
- Hu MC, Kuro-o M, Moe OW. The emerging role of Klotho in clinical nephrology. Nephrol Dial Transplant. 2012;27:2650–2657. [PMC free article: PMC3398064] [PubMed: 22802580]
- 6.
- Drüeke TB, Massy ZA. Changing bone patterns with progression of chronic kidney disease. Kidney International. 2016;89:289–302. [PubMed: 26806832]
- 7.
- Evenepoel P, D’Haese P, Brandenburg V. Sclerostin and DKK1: new players in renal bone and vascular disease. Kidney Int. 2015;88:235–240. [PubMed: 26083653]
- 8.
- Haarhaus M, Evenepoel P. European Renal Osteodystrophy (EUROD) workgroup, Chronic Kidney Disease Mineral and Bone Disorder (CKD-MBD) working group of the European Renal Association–European Dialysis and Transplant Association (ERA-EDTA). Differentiating the causes of adynamic bone in advanced chronic kidney disease informs osteoporosis treatment. Kidney International. 2021;100:546–558. [PubMed: 34102219]
- 9.
- Malluche HH, Mawad HW. MonierFaugere MC. Renal Osteodystrophy in the First Decade of the New Millennium: Analysis of 630 Bone Biopsies in Black and White Patients. J Bone Miner Res. 2011;26:1368–1376. [PMC free article: PMC3312761] [PubMed: 21611975]
- 10.
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Amer Soc Nephrol. 2004;15:2208–2218. [PubMed: 15284307]
- 11.
- Moe SM, Drueke TB. Management of secondary hyperparathyroidism: the importance and the challenge of controlling parathyroid hormone levels without elevating calcium, phosphorus, and calcium-phosphorus product. Am J Nephrol. 2003;23:369–379. [PubMed: 14551461]
- 12.
- Tentori F, Blayney MJ, Albert JM, et al. Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2008;52:519–530. [PubMed: 18514987]
- 13.
- Floege J. Calcium-containing phosphate binders in dialysis patients with cardiovascular calcifications: should we CARE-2 avoid them? Nephrol Dial Transplant. 2008;23:3050–3052. [PubMed: 18625662]
- 14.
- Floege J, Kim J, Ireland E, et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol Dial Transplant. 2011;26:1948–1955. [PMC free article: PMC3107766] [PubMed: 20466670]
- 15.
- Fernandez-Martin JL, Martinez-Camblor P, Dionisi MP, et al. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: the COSMOS study. Nephrol Dial Transplant. 2015;30:1542–1551. [PubMed: 25920921]
- 16.
- Hagstrom E, Hellman P, Larsson TE, et al. Plasma Parathyroid Hormone and the Risk of Cardiovascular Mortality in the Community. Circulation. 2009;119:2765–U34. [PubMed: 19451355]
- 17.
- Slatopolsky E, Caglar S, Pannell J P, et al. On the pathogenesis of hyperparathyroidism in chronic experimental renal insufficiency in the dog. J. Clin. Invest. 1971;50:492–499. [PMC free article: PMC291955] [PubMed: 5545116]
- 18.
- Vorland CJ, Biruete A, Lachcik PJ, et al. Kidney Disease Progression Does Not Decrease Intestinal Phosphorus Absorption in a Rat Model of Chronic Kidney Disease-Mineral Bone Disorder. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research. 2020;35:333–342. [PMC free article: PMC7012714] [PubMed: 31618470]
- 19.
- Moranne O, Froissart M, Rossert J, et al. Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol. 2009;20:164–171. [PMC free article: PMC2615728] [PubMed: 19005010]
- 20.
- Llach F, Massry S G, Koffler A. Secondary hyperparathyroidism in early renal failure: role of phosphate retention. Kidney Int. 1977;12:459–463.
- 21.
- Hsu CY, Chertow GM. Elevations of serum phosphorus and potassium in mild to moderate chronic renal insufficiency. Nephrol Dial Transplant. 2002;17:1419–25. [PubMed: 12147789]
- 22.
- Kurosu H, Kuro OM. The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol Cell Endocrinol. 2009;299:72–78. [PubMed: 19063940]
- 23.
- Olauson, HannesVervloet, Marc GCozzolino, MarioMassy, Ziad AUrena Torres, PabloLarsson, Tobias EengReview2014/12/17 06:0.
- 24.
- Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78:975–980. [PubMed: 20844473]
- 25.
- Dhayat NA, Ackermann D, Pruijm M, et al. Fibroblast growth factor 23 and markers of mineral metabolism in individuals with preserved renal function. Kidney International. 2016;90:648–657. [PubMed: 27370409]
- 26.
- Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. [PMC free article: PMC3014041] [PubMed: 21115613]
- 27.
- Lim K, Lu TS, Molostvov G, et al. Vascular klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125:2243–2255. [PubMed: 22492635]
- 28.
- Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. [PMC free article: PMC2923354] [PubMed: 20466874]
- 29.
- Chen G, Liu Y, Goetz R, et al. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553:461–466. [PMC free article: PMC6007875] [PubMed: 29342138]
- 30.
- Olauson H, Vervloet MG, Cozzolino M, Massy ZA, Urena Torres P, Larsson TE. New insights into the FGF23-Klotho axis. Semin Nephrol. 2014;34:586–597. [PubMed: 25498378]
- 31.
- Pavik I, Jaeger P, Ebner L, et al. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol Dial Transplant. 2013;28:352–359. [PubMed: 23129826]
- 32.
- Shimamura Y, Hamada K, Inoue K, et al. Serum levels of soluble secreted alpha-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. Clin Exp Nephrol. 2012;16:722–729. [PubMed: 22457086]
- 33.
- Smith ER, Holt SG, Hewitson TD. αKlotho-FGF23 interactions and their role in kidney disease: a molecular insight. Cellular and molecular life sciences. CMLS. 2019;76:4705–4724. [PMC free article: PMC11105488] [PubMed: 31350618]
- 34.
- Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012 [PMC free article: PMC3434320] [PubMed: 22622492]
- 35.
- Fan Y, Bi R, Densmore MJ, et al. Parathyroid hormone 1 receptor is essential to induce FGF23 production and maintain systemic mineral ion homeostasis. FASEB J. 2015 [PMC free article: PMC4684518] [PubMed: 26428657]
- 36.
- Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014;86:1106–1115. [PubMed: 24940803]
- 37.
- Lopez I. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 2011;80:475–482. RodriguezOrtiz ME, Almaden Y et al. [PubMed: 21525854]
- 38.
- Galitzer H, BenDov IZ, Silver J. NavehMany T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010;77:211–218. [PubMed: 20016468]
- 39.
- Canalejo R, Canalejo A. FGF23 Fails to Inhibit Uremic Parathyroid Glands. J Amer Soc Nephrol. 2010;21:1125–1135. MartinezMoreno JM et al. [PMC free article: PMC3152229] [PubMed: 20431039]
- 40.
- Komaba H, Goto S, Fujii H, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232–238. [PubMed: 19890272]
- 41.
- Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. [PubMed: 14633152]
- 42.
- Shigematsu T, Kazama JJ, Yamashita T, et al. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44:250–256. [PubMed: 15264182]
- 43.
- Komaba H, Fukagawa M. FGF23-parathyroid interaction: implications in chronic kidney disease. Kidney Int. 2010;77:292–298. [PubMed: 20010546]
- 44.
- Spiegel DM, Brady K. Calcium balance in normal individuals and in patients with chronic kidney disease on low- and high-calcium diets. Kidney Int. 2012;81:1116–1122. [PMC free article: PMC3352985] [PubMed: 22297674]
- 45.
- Hill KM, Martin BR, Wastney ME, et al. Oral calcium carbonate affects calcium but not phosphorus balance in stage 3-4 chronic kidney disease. Kidney Int. 2013;83:959–966. [PMC free article: PMC4292921] [PubMed: 23254903]
- 46.
- Koizumi M, Komaba H, Fukagawa M. Parathyroid function in chronic kidney disease: role of FGF23-Klotho axis. Contrib Nephrol. 2013;180:110–123. [PubMed: 23652554]
- 47.
- Goodman WG, Quarles LD. Development and progression of secondary hyperparathyroidism in chronic kidney disease: lessons from molecular genetics. Kidney Int. 2008;74:276–288. [PubMed: 17568787]
- 48.
- Lucas P A, Brown R C, Woodhead J S, Colesi G A. 1,25 dihydroxycholecalciferol and parathyroid hormone in advanced chronic renal failure : effect of simultaneous protein and phosphorus restriction. Clin. Nephrol. 1986;25:7–10. [PubMed: 3754187]
- 49.
- Nykjaer A, Dragun D, Walther D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D-3. Cell. 1999;96:507–515. [PubMed: 10052453]
- 50.
- Zehnder D, Bland R, Walker EA, et al. Expression of 25-hydroxyvitamin D-3-1 alpha-hydroxylase in the human kidney. J Amer Soc Nephrol. 1999;10:2465–2473. [PubMed: 10589683]
- 51.
- Cuppari L, Garcia-Lopes MG. Hypovitaminosis D in chronic kidney disease patients: prevalence and treatment. J Ren Nutr. 2009;19:38–43. [PubMed: 19121769]
- 52.
- Mehrotra R, Kermah D, Budoff M, et al. Hypovitaminosis D in chronic kidney disease. Clin J Am Soc Nephrol. 2008;3:1144–1151. [PMC free article: PMC2440286] [PubMed: 18417740]
- 53.
- Cunningham J, Makin H. How important is vitamin D deficiency in uraemia ? (Editorial Comment). Nephrol. Dial. Transplant. 1997;12:16–18. [PubMed: 9027765]
- 54.
- Ghazali A, Fardellone P, Pruna A, et al. Is low plasma 25-(OH)vitamin D a major risk factor for hyperparathyroidism and Looser’s zones independent of calcitriol? Kidney Int. 1999;55:2169–2177. [PubMed: 10354266]
- 55.
- Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006;70:654–659. [PubMed: 16807549]
- 56.
- Vervloet MG, Massy ZA, Brandenburg VM, et al. Bone: a new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014;2:427–436. [PubMed: 24795256]
- 57.
- Glorieux G, Hsu CH, de Smet R, et al. Inhibition of calcitriol-induced monocyte CD14 expression by uremic toxins: role of purines. J Am Soc Nephrol. 1998;9:1826–1831. [PubMed: 9773783]
- 58.
- Patel S R, Ke H Q, Vanholder R, Koenig R J, Hsu C H. Inhibition of calcitriol receptor binding to vitamin D response elements by uremic toxins. J. Clin. Invest. 1995;96:50–59. [PMC free article: PMC185172] [PubMed: 7615822]
- 59.
- Goto S, Fujii H, Hamada Y, Yoshiya K, Fukagawa M. Association between indoxyl sulfate and skeletal resistance in hemodialysis patients. Ther Apher Dial. 2012:417–423. [PubMed: 20649763]
- 60.
- Watanabe K, Tominari T, Hirata M, et al. Indoxyl sulfate, a uremic toxin in chronic kidney disease, suppresses both bone formation and bone resorption. FEBS open bio. 2017;7:1178–1185. [PMC free article: PMC5536993] [PubMed: 28781957]
- 61.
- Dusso AS. Vitamin D receptor: Mechanisms for vitamin D resistance in renal failure. Kidney Int Suppl. 2003:6–9. [PubMed: 12753256]
- 62.
- Fukuda N, Tanaka H, Tominaga Y, Fukagawa M, Kurokawa K, Seino Y. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J. Clin. Invest. 1993;92:1436–1442. [PMC free article: PMC288288] [PubMed: 8397225]
- 63.
- Patel S, Ke H Q, Vanholder R, Hsu C H. Inhibition of nuclear uptake of calcitriol receptor by uremic ultrafiltrate. Kidney Int. 1994;46:129–133. [PubMed: 7933830]
- 64.
- Garfia B, Canadillas S, Canalejo A, et al. Regulation of parathyroid vitamin D receptor expression by extracellular calcium. J Amer Soc Nephrol. 2002;13:2945–2952. [PubMed: 12444213]
- 65.
- SelaBrown A. Russell J, Koszewski NJ, Michalak M, NavehMany T, Silver J. Calreticulin inhibits vitamin D’s action on the PTH gene in vitro and may prevent vitamin D’s effect in vivo in hypocalcemic rats. Mol Endocrinol. 1998;12:1193–1200. [PubMed: 9717845]
- 66.
- Brown A J, Zhong M, Finch J, et al. Rat calcium-sensing receptor is regulated by vitamin D but not by calcium. Am. J. Physiol. 1996;270:F454–F460. [PubMed: 8780248]
- 67.
- Mendoza FJ, Lopez I, Canalejo R, et al. Direct upregulation of parathyroid calcium-sensing receptor and vitamin D receptor by calcimimetics in uremic rats. Amer J Physiol Renal Physiol. 2009;296:F605–F613. [PubMed: 19091789]
- 68.
- Silver J, Sela S B, Naveh-Many T. Regulation of parathyroid cell proliferation. Curr. Opin. Nephrol. Hypertens. 1997;6:321–326. [PubMed: 9263680]
- 69.
- Szabo A, Merke J, Beier E, Mall G, Ritz E. 1,25(OH)2 vitamin D3 inhibits parathyroid cell proliferation in experimental uremia. Kidney Int. 1989;35:1045–1056. [PubMed: 2709685]
- 70.
- Fukagawa M, Kaname S-Y, Igarashi T, Ogata E, Kurokawa K. Regulation of parathyroid hormone synthesis in chronic renal failure in rats. Kidney Int. 1991;39:874–881. [PubMed: 2067203]
- 71.
- Naveh-Many T, Rahamimov R, Livni N, Silver J. Parathyroid cell proliferation in normal and chronic renal failure rats. The effects of calcium, phosphate, and vitamin D. J. Clin. Invest. 1995;96:1786–1793. [PMC free article: PMC185815] [PubMed: 7560070]
- 72.
- Nygren P, Larsson R, Johannson H, Ljunghall S, Rastad J, Akerstrom G. 1,25 (OH)2 D3 inhibits hormone secretion and proliferation but not functional dedifferentiation of cultured bovine parathyroid cells. Calcif. Tissue Int. 1988;43:213–218. [PubMed: 3145126]
- 73.
- Kremer R, Bolivar I, Goltzman D, Hendy G N. Influence of calcium and 1,25-dihydroxycholecalciferol on proliferation and proto-oncogene expression in primary cultures of bovine parathyroid cells. Endocrinology. 1989;125:935–941. [PubMed: 2502380]
- 74.
- Ishimi Y, Russel J, Sherwood L M. Regulation by calcium and 1,25-(OH)2D3 of cell proliferation and function of bovine parathyroid cells in culture. J. Bone Min. Res. 1990;5:755–760. [PubMed: 2396502]
- 75.
- Roussanne MC, Lieberherr M, Souberbielle JC, Sarfati E, Drueke T, Bourdeau A. Human parathyroid cell proliferation in response to calcium, NPS R-467, calcitriol and phosphate. Eur J Clin Invest. 2001;31:610–6. [PubMed: 11454016]
- 76.
- Fernandez A, Fibla J, Betriu A, Piulats J M, Almirall J, Montoliu J. Association between vitamin D receptor gene polymorphism and relative hypoparathyroidism in patients with chronic renal failure. J. Am. Soc. Nephrol. 1997;8:1546–1552. [PubMed: 9335382]
- 77.
- Aterini S, Salvadori M, Ippolito E, et al. The role of vitamin D receptor alleles in the secondary hyperparathyroidism of hemodialysis patients. J Nephrol. 1996;9:201–206.
- 78.
- Nagaba Y, Heishi M, Tazawa H, Tsukamoto Y, Kobayashi Y. Vitamin D receptor gene polymorphisms affect secondary hyperparathyroidism in hemodialyzed patients. Am J Kidney Dis. 1998;32:464–469. [PubMed: 9740163]
- 79.
- Yokoyama K, Shigematsu T, Tsukada T, et al. Apa I polymorphism in the vitamin D receptor gene may affect the parathyroid response in Japanese with end-stage renal disease. Kidney Int. 1998;53:454–458. [PubMed: 9461106]
- 80.
- Carling T, Rastad J, Akerstrom G, Westin G. Vitamin D receptor (VDR) and parathyroid hormone messenger ribonucleic acid levels correspond to polymorphic VDR alleles in human parathyroid tumors. J Clin Endocrinol Metab. 1998;83:2255–2259. [PubMed: 9661591]
- 81.
- Schmidt S, Chudek J, Karkoska H, et al. The BsmI vitamin D-recptor polymorphism and secondary hyperparathyroidism. Nephrol Dial Transplant. 1997;12:1771–1772. (Letter) [PubMed: 9269677]
- 82.
- Torres A, Machado M, Concepcion M T, et al. Influence of vitamin D receptor genotype on bone mass changes after renal transplantation. Kidney Int. 1996;50:1726–1733. [PubMed: 8914043]
- 83.
- Hawa N S, Cockerill F J, Vadher S, et al. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin Endocrinol. 1996;45:85–92. [PubMed: 8796143]
- 84.
- Morrison N A, Qi J C, Tokita A, et al. Prediction of bone density from vitamin D receptor alleles. Nature. 1994;367:284–287. [PubMed: 8161378]
- 85.
- Egstrand S, Nordholm A, Morevati M, et al. A molecular circadian clock operates in the parathyroid gland and is disturbed in chronic kidney disease associated bone and mineral disorder. Kidney International. 2020;98:1461–1475. [PubMed: 32721445]
- 86.
- Isakova T, Gutierrez O, Shah A, et al. Postprandial mineral metabolism and secondary hyperparathyroidism in early CKD. J Am Soc Nephrol. 2008;19:615–623. [PMC free article: PMC2391049] [PubMed: 18216315]
- 87.
- Santamaria R, Almaden Y, Felsenfeld A, et al. Dynamics of PTH secretion in hemodialysis patients as determined by the intact and whole PTH assays. Kidney Int. 2003;64:1867–1873. [PubMed: 14531822]
- 88.
- Moyses RMA, Pereira RC, dosReis LM, Sabbaga E, Jorgetti V. Dynamic tests of parathyroid hormone secretion using hemodialysis and calcium infusion cannot be compared. Kidney Int. 1999;56:659–665. [PubMed: 10432406]
- 89.
- Brown E M. Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. J. Clin. Endocrinol. Metab. 1983;56:572–581. [PubMed: 6822654]
- 90.
- Gogusev J, Duchambon P, Hory B, Giovannini M, Sarfati E, Drüeke T B. Depressed expression of calcium receptor in parathyroid gland tissue of patients with primary or secondary uremic hyperparathyroidism. Kidney Int. 1997;51:328–336. [PubMed: 8995751]
- 91.
- Kifor O, Moore F D, Wang P, et al. Reduced immunostaining for the extracellular Ca2+ - sensing receptor in primary and uremic secondary hyperparathyroidism. J. Clin. Endocrinol. Metab. 1996;81:1598–1606. [PubMed: 8636374]
- 92.
- Rodríguez-Ortiz ME, Canalejo A, Herencia C, et al. Magnesium modulates parathyroid hormone secretion and upregulates parathyroid receptor expression at moderately low calcium concentration. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29:282–289. [PMC free article: PMC3910342] [PubMed: 24103811]
- 93.
- Brown AJ, Ritter CS, Finch JL, Slatopolsky EA. Decreased calcium-sensing receptor expression in hyperplastic parathyroid glands of uremic rats: Role of dietary phosphate. Kidney Int. 1999;55:1284–1292. [PubMed: 10200992]
- 94.
- Campion KL, McCormick WD, Warwicker J, et al. Pathophysiologic Changes in Extracellular pH Modulate Parathyroid Calcium-Sensing Receptor Activity and Secretion via a Histidine-Independent Mechanism. Journal of the American Society of Nephrology: JASN. 2015;26:2163–2171. [PMC free article: PMC4552114] [PubMed: 25556167]
- 95.
- Centeno PP, Herberger A, Mun H-C, et al. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nature Communications. 2019;10:4693. [PMC free article: PMC6795806] [PubMed: 31619668]
- 96.
- Almadén Y, Hernandez A, Torregrosa V, et al. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. J Amer Soc Nephrol. 1998;9:1845–1852. [PubMed: 9773785]
- 97.
- Lewin E, Garfia B, Almaden Y, Rodriguez M, Olgaard K. Autoregulation in the parathyroid glands by PTH/PTHrP receptor ligands in normal and uremic rats. Kidney Int. 2003;64:63–70. [PubMed: 12787396]
- 98.
- Goodman WG, Veldhuis JD, Belin TR, VanHerle AJ, Juppner H, Salusky IB. Calcium-sensing by parathyroid glands in secondary hyperparathyroidism. J Clin Endocrinol Metab. 1998;83:2765–2772. [PubMed: 9709944]
- 99.
- Felsenfeld A J, Rodriguez M. Parathyroid gland function in hemodialysis patients. Semin. Dial. 1996;9:303–309.
- 100.
- Goodman W G, Belin T R, Salusky I B. In vivo assessments of calcium-regulated parathyroid hormone release in secondary hyperparathyroidism. Kidney Int. 1996;50:1834–1844. [PubMed: 8943464]
- 101.
- Ouseph R, Leiser J D, Moe S M. Calcitriol and the parathyroid hormone-ionized calcium curve: a comparison of methodologic approaches. J. Am. Soc. Nephrol. 1996;7:497–505. [PubMed: 8704117]
- 102.
- Mathias RS, Nguyen HT, Zhang MYH, Portale AA. Reduced expression of the renal calcium-sensing receptor in rats with experimental chronic renal insufficiency. J Amer Soc Nephrol. 1998;9:2067–2074. [PubMed: 9808092]
- 103.
- Wada M, Furuya Y, Sakiyama J, et al. NPS R-568 halts or reverses osteitis fibrosa in uremic rats. Kidney Int. 1997;53:448–453. [PubMed: 9461105]
- 104.
- Wernerson A, Windholm S M, Svensson O, Reinholt F P. Parathyroid cell number and size in hypocalcemic young rats. APMIS. 1991;99:1096–1102. [PubMed: 1772646]
- 105.
- LeBoff M S, Rennke H G, Brown E M. Abnormal regulation of parathyroid cell secretion and proliferation in primary cultures of bovine parathyroid cells. Endocrinology. 1983;113:277–284. [PubMed: 6407823]
- 106.
- LeBoff M S, Shoback D, Brown E M, et al. Regulation of parathyroid hormone release and cytosolic calcium by extracellular calcium in dispersed and cultured bovine and pathological human parathyroid cells. J. Clin. Invest. 1985;75:49–57. [PMC free article: PMC423397] [PubMed: 3965511]
- 107.
- McGregor R R, Sarras M P, Houle H, Cohn D V. Primary monolayer culture of bovine parathyroids: effect of calcium, isoproterenol, and growth factors. Mol. Cell. Endocrinol. 1983;30:313–328. [PubMed: 6862097]
- 108.
- R D Ridgeway, J W Hamilton, R R M Gregor. Characteristics of bovine parathyroid cell organoid in culture. In Vitro Cell Dev. 1998; 22: 91–99. [PubMed: 3949675]
- 109.
- Brandi M L, Fitzpatrick L A, Coon H G, Aurbach G D. Bovine parathyroid cell: cultures maintained for more than 140 population doublings. Proc. Natl. Acad. Sci. USA. 1986;83:1709–1713. [PMC free article: PMC323153] [PubMed: 3006065]
- 110.
- Brown A J, Zhong M, Ritter C S, Brown E M, Slatopolsky E. Loss of calcium responsiveness in cultured parathyroid cells is associated with decreased calcium receptor expression. Biochem. Biophys. Res. Commun. 1995;212:861–867. [PubMed: 7626122]
- 111.
- Mithal A, Kifor O, Kifor I, et al. The reduced responsiveness of cultured bovine parathyroid cells to extracellular Ca2+ is associated with marked reduction in the expression of extracellular Ca2+-sensing receptor messenger ribonucleic acid and protein. Endocrinology. 1995;136:3087–3092. [PubMed: 7789335]
- 112.
- Brandi M L, Ornberg R L, Sakaguchi K, et al. Establishment and characterization of a clonal line of parathyroid endothelial cells. FASEB J. 1990;4:3152–3158. [PubMed: 1698682]
- 113.
- Sakaguchi K. Acidic fibroblast growth factor autocrine system as a mediator of calcium-regulated parathyroid cell growth. J. Biol. Chem. 1992;267:24554–24562. [PubMed: 1280262]
- 114.
- Roussanne M-C, Gogusev J, Hory B, et al. Persistence of Ca2+-sensing receptor expression in functionally active, long-term human parathyroid cell cultures. J. Bone Min. Res. 1998;13:1–9. [PubMed: 9525335]
- 115.
- Mizobuchi M, Hatamura I, Ogata H, et al. Calcimimetic compound upregulates decreased calcium-sensing receptor expression level in parathyroid glands of rats with chronic renal insufficiency. J Am Soc Nephrol. 2004;15:2579–2587. [PubMed: 15466262]
- 116.
- Chikatsu N, Fukumoto S, Takeuchi Y, et al. Cloning and characterization of two promoters for the human calcium-sensing receptor (CaSR) and changes of CaSR expression in parathyroid adenomas. J Biol Chem. 2000;275:7553–7557. [PubMed: 10713061]
- 117.
- Lundgren S, Carling T, Hjälm G, et al. Tissue distribution of human gp330/megalin, a putative Ca2+-sensing protein. J. Histochem. Cytochem. 1997;45:383–392. [PubMed: 9071320]
- 118.
- Almadén Y, Canalejo A, Hernandez A, et al. Direct effect of phosphorus on parathyroid hormone secretion from whole rat parathyroid glands in vitro. J Bone Min Res. 1996;11:970–976. [PubMed: 8797118]
- 119.
- Nielsen P K, Feldt-Rasmussen U, Olgaard K. A direct effect of phosphate on PTH release from bovine parathyroid tissue slices but not from dispersed parathyroid cells. Nephrol. Dial. Transplant. 1996;11:1762–1768. [PubMed: 8918619]
- 120.
- Slatopolsky E, Finch J, Denda M, et al. Phosphorus restriction prevents parathyroid gland growth: high phosphorus directly stimulates PTH secretion in vitro. J. Clin. Invest. 1996;97:2534–2540. [PMC free article: PMC507339] [PubMed: 8647946]
- 121.
- Almaden Y, Canalejo A, Ballesteros E, Anon G, Canadillas S, Rodriguez M. Regulation of arachidonic acid production by intracellular calcium in parathyroid cells: Effect of extracellular phosphate. J Amer Soc Nephrol. 2002;13:693–698. [PubMed: 11856773]
- 122.
- Moallem E, Kilav R, Silver J. NavehMany T. RNA-protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–5259. [PubMed: 9478982]
- 123.
- Yalcindag C, Silver J, Naveh-Many T. Mechanism of increased parathyroid hormone mRNA in experimental uremia: roles of protein RNA binding and RNA degradation. Journal of the American Society of Nephrology: JASN. 1999;10:2562–2568. [PubMed: 10589695]
- 124.
- SelaBrown A. Silver J, Brewer G, NavehMany T. Identification of AUF1 as a parathyroid hormone mRNA 3’-untranslated region-binding protein that determines parathyroid hormone mRNA stability. J Biol Chem. 2000;275:7424–7429. [PubMed: 10702317]
- 125.
- Slatopolsky E, Delmez E. Pathogenesis of secondary hyperparathyroidism. Miner. Electrolyte Metab. 1995;21:91–96. [PubMed: 7565472]
- 126.
- Yi H, Fukagawa M, Yamato H, Kumagai M, Watanabe T, Kurokawa K. Prevention of enhanced parathyroid hormone secretion, synthesis and hyperplasia by mild dietary phosphorus restriction in early chronic renal failure in rats: possible direct role of phosphorus. Nephron. 1995;70:242–248. [PubMed: 7566311]
- 127.
- Takahashi F, Denda M, Finch JL, Brown AJ, Slatopolsky E. Hyperplasia of the parathyroid gland without secondary hyperparathyroidism. Kidney Int. 2002;61:1332–1338. [PubMed: 11918740]
- 128.
- M-C Roussanne, J Gogusev, E Sarfati, T Drüeke, A Bourdeau. Effect of phosphate on PTH secretion and proliferation in human parathyroid cells in culture (Abstract). J. Bone Min. Res. (suppl. 1) 1997; 12: S387.
- 129.
- Silver J, Naveh-Many T. FGF23 and the parathyroid. Advances in Experimental Medicine and Biology. 2012;728:92–99. [PubMed: 22396164]
- 130.
- HofmanBang J. Martuseviciene G, Santini MA, Olgaard K, Lewin E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010;78:1119–1127. [PubMed: 20631679]
- 131.
- Fan Y, Liu W, Bi R, et al. Interrelated role of Klotho and calcium-sensing receptor in parathyroid hormone synthesis and parathyroid hyperplasia. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E3749–E3758. [PMC free article: PMC5910831] [PubMed: 29618612]
- 132.
- Mace ML, Olgaard K, Lewin E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. International Journal of Molecular Sciences. 2020;21:E8810. [PMC free article: PMC7699902] [PubMed: 33233840]
- 133.
- Shilo V, Ben-Dov IZ, Nechama M, Silver J, Naveh-Many T. Parathyroid-specific deletion of dicer-dependent microRNAs abrogates the response of the parathyroid to acute and chronic hypocalcemia and uremia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:3964–3976. [PubMed: 26054367]
- 134.
- Shilo V, Mor-Yosef Levi I, Abel R, et al. Let-7 and MicroRNA-148 Regulate Parathyroid Hormone Levels in Secondary Hyperparathyroidism. Journal of the American Society of Nephrology: JASN. 2017;28:2353–2363. [PMC free article: PMC5533223] [PubMed: 28298326]
- 135.
- Volovelsky O, Cohen G, Kenig A, et al. Phosphorylation of Ribosomal Protein S6 Mediates Mammalian Target of Rapamycin Complex 1-Induced Parathyroid Cell Proliferation in Secondary Hyperparathyroidism. Journal of the American Society of Nephrology: JASN. 2016;27:1091–1101. [PMC free article: PMC4814192] [PubMed: 26283674]
- 136.
- Aubia J, Serrano S, Mariosso L, et al. Osteodystrophy of diabetics in chronic dialysis: a histomorphometric study. Calcif Tissue Int. 1988;42:297–301. [PubMed: 3135097]
- 137.
- Hernandez D, Concepcion M T, Lorenzo V, et al. Adynamic bone disease with negative aluminum staining in predialysis patients: prevalence and evolution after maintenance dialysis. Nephrol Dial Transplant. 1994;9:517–523. [PubMed: 7522307]
- 138.
- Pei Y, Hercz G, Greenwood C, et al. Renal osteodystrophy in diabetic patients. Kidney Int. 1993;44:159–164. [PubMed: 8355457]
- 139.
- Vicenti F, Arnaud S B, Recker R, et al. Parathyroid and bone response of the diabetic patient to uremia. Kidney Int. 1984;25:677–682. [PubMed: 6482171]
- 140.
- Panuccio V, Mallamaci F, Tripepi G, et al. Low parathyroid hormone and pentosidine in hemodialysis patients. Am J Kidney Dis. 2002;40:810–5. [PubMed: 12324917]
- 141.
- Hocher B, Armbruster FP, Stoeva S, et al. Measuring parathyroid hormone (PTH) in patients with oxidative stress--do we need a fourth generation parathyroid hormone assay? PLoS One. 2012;7:e40242. [PMC free article: PMC3391306] [PubMed: 22792251]
- 142.
- Hocher B, Zeng S. Clear the Fog around Parathyroid Hormone Assays: What Do iPTH Assays Really Measure? Clinical journal of the American Society of Nephrology: CJASN. 2018;13:524–526. [PMC free article: PMC5969453] [PubMed: 29507007]
- 143.
- Jara A, Gonzalez S, Felsenfeld AJ, et al. Failure of high doses of calcitriol and hypercalcaemia to induce apoptosis in hyperplastic parathyroid glands of azotaemic rats. Nephrol Dial Transplant. 2001;16:506–12. [PubMed: 11239023]
- 144.
- Sugimoto T, Ritter C, Morrisey J, Hayes C, Slatopolsky E. Effects of high concentrations of glucose on PTH secretion in parathyroid cells. Kidney Int. 1990;37:1522–1527. [PubMed: 2194068]
- 145.
- Cannata J B, Briggs J D, Junor B J R, Fell J S. The influence of aluminium on parathyroid hormone levels in hemodialysis patients. Proc Eur Dial Transplant Assoc. 1982;19:244–247. [PubMed: 6878239]
- 146.
- Llach F, Felsenfeld A J, Coleman M D, Keveney J J Jr, Pederson J A, Medlock T R. The natural course of dialysis osteomalacia. Kidney Int. 1986;29 suppl 18:S74–S79. [PubMed: 3458001]
- 147.
- Andress D, Felsenfeld A J, Voigts A, Llach F. Parathyroid hormone response to hypocalcemia in hemodilaysis patients with osteomalacia. Kidney Int. 1983;24:363–370. [PubMed: 6645210]
- 148.
- Kraut J A, Shinaberger J H, Singer F R, et al. Parathyroid hormone response to acute hypocalcemia in dialysis osteomalacia. Kidney Int. 1983;23:725–730. [PubMed: 6876568]
- 149.
- Cann C E, Prussin S G, Gordan G S. Aluminum uptake by the parathyroid glands. J Clin Endocrinol Metab. 1979;49:543–545. [PubMed: 479346]
- 150.
- Rodriguez M, Felsenfeld A J, Llach F. The role of aluminum in the development of hypercalcemia in the rat. Kidney Int. 1987;31:766–771. [PubMed: 3573539]
- 151.
- Morrissey J, Slatopolsky S. Effect of aluminum on parathyroid hormone secretion. Kidney Int. 1986;29 suppl 18:S41–S44. [PubMed: 3457994]
- 152.
- Alfrey A C, Sedman A, Chan Y. The compartmentalization and metabolism of aluminum in uremic rats. J Lab Clin Med. 1985;105:227–233. [PubMed: 3973462]
- 153.
- Henry D A, Goodman W G, Nudelman R K, et al. Parenteral aluminum administration in the dog: I. Plasma kinetics, tissue levels, calcium metabolism, and parathyroid hormone. Kidney Int. 1984;25:362–369. [PubMed: 6427508]
- 154.
- J B DiazLopez. P C D’Haese, E J Noueven, L V Lamberts, J B Cannata, M E De Broe. Estudio del contenido de aluminio en paratiroides de ratas con insuficiencia renal e intoxicacion aluminica cronica. Nefrologia. 1988;8:35–41.
- 155.
- Finch J L, Bergfeld M, Martin K J, Chan Y L, Teitelbaum S, Slatopolsky E. The effects of discontinuation of aluminum exposure on aluminum-induced osteomalacia. Kidney Int. 1986;30:318–324. [PubMed: 3784278]
- 156.
- Felsenfeld A J, Rofdriguez M, Coleman M, Ross D, Llach F. Desferrioxamine therapy in hemodialysis patients with aluminum-associated bone disease. Kidney Int. 1989;35:1371–1378. [PubMed: 2770115]
- 157.
- Cozzolino M, Lu Y, Finch J, Slatopolsky E, Dusso AS. p21(WAF1) and TGF-alpha mediate parathyroid growth arrest by vitamin D and high calcium. Kidney Int. 2001;60:2109–2117. [PubMed: 11737585]
- 158.
- Cordero JB, Cozzolino M, Lu Y, et al. 1,25-Dihydroxyvitamin D down-regulates cell membrane growth- and nuclear growth-promoting signals by the epidermal growth factor receptor. J Biol Chem. 2002;277:38965–38971. [PubMed: 12181310]
- 159.
- Dusso AS, Pavlopoulos T, Naumovich L, et al. P21(WAF1) and transforming growth factor-alpha mediate dietary phosphate regulation of parathyroid cell growth. Kidney Int. 2001;59:855–865. [PubMed: 11231340]
- 160.
- Gogusev J, Duchambon P, Stoermann-Chopard C, Giovannini M, Sarfati E, Drüeke T B. De novo expression of transforming growth factor-alpha in parathyroid gland tissue of patients with primary or secondary uraemic hyperparathyroidism. Nephrol. Dial. Transplant. 1996;11:2155–2162. [PubMed: 8941573]
- 161.
- Arcidiacono MV, Cozzolino M, Spiegel N, et al. Activator protein 2alpha mediates parathyroid TGF-alpha self-induction in secondary hyperparathyroidism. J Am Soc Nephrol. 2008;19:1919–1928. [PMC free article: PMC2551566] [PubMed: 18579641]
- 162.
- Matsushita H, Hara M, Endo Y, et al. Proliferation of parathyroid cells negatively correlates with expression of parathyroid hormone-related protein in secondary parathyroid hyperplasia. Kidney Int. 1999;55:130–138. [PubMed: 9893121]
- 163.
- Gunther T, Chen ZF, Kim J, et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. 2000;406:199–203. [PubMed: 10910362]
- 164.
- Correa P, Akerstrom G, Westin G. Underexpression of Gcm2, a master regulatory gene of parathyroid gland development, in adenomas of primary hyperparathyroidism. Clin Endocrinol (Oxf). 2002;57:501–505. [PubMed: 12354132]
- 165.
- Han SI, Tsunekage Y, Kataoka K. Gata3 cooperates with Gcm2 and MafB to activate parathyroid hormone gene expression by interacting with SP1. Mol Cell Endocrinol. 2015;411:113–120. [PubMed: 25917456]
- 166.
- Morito N, Yoh K, Usui T, et al. Transcription factor MafB may play an important role in secondary hyperparathyroidism. Kidney International. 2018;93:54–68. [PubMed: 28964572]
- 167.
- Naveh-Many T, Silver J. Transcription factors that determine parathyroid development power PTH expression. Kidney International. 2018;93:7–9. [PubMed: 29291826]
- 168.
- Mendes V, Jorgetti V, Nemeth J, et al. Secondary hyperparathyroidism in chronic haemodialysis patients: a clinico-pathologic study. Proc. Europ. Dial. Transplant. Assoc. 1983;20:731–738. [PubMed: 6657693]
- 169.
- Arnold A, Brown M F, Ureña P, Gaz R D, Sarfati E, Drüeke T B. Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest. 1995;95:2047–2054. [PMC free article: PMC295791] [PubMed: 7738171]
- 170.
- Chudek J, Ritz E, Kovacs G. Genetic abnormalities in parathyroid nodules of uremic patients. Clin. Cancer Res. 1998;4:211–214. [PubMed: 9516973]
- 171.
- Tominaga Y, Kohara S, Namii Y, et al. Clonal analysis of nodular parathyroid hyperplasia in renal hyperparathyroidism. World J. Surg. 1996;20:744–752. [PubMed: 8678945]
- 172.
- Tominaga Y, Takagi H. Molecular genetics of hyperparathyroid disease. Current Opinion in Nephrology and Hypertension. 1996;5:336–341. [PubMed: 8823531]
- 173.
- Imanishi Y, Tahara H, Palanisamy N, et al. Clonal chromosomal defects in the molecular pathogenesis of refractory hyperparathyroidism of uremia. J Amer Soc Nephrol. 2002;13:1490–1498. [PubMed: 12039978]
- 174.
- Hsi E D, Zukerberg L R, Yang W-I, Arnold A. CyclinD1/PRAD1 expression in parathyroid adenomas: an immunohistochemical study. J. Clin. Endocrinol. Metab. 1996;81:1736–1739. [PubMed: 8626826]
- 175.
- Tominaga Y, Tsuzuki T, Uchida K, et al. Expression of PRAD1 cyclin D1, retinoblastoma gene products, and Ki67 in parathyroid hyperplasia caused by chronic renal failure versus primary adenoma. Kidney Int. 1999;55:1375–1383. [PubMed: 10201002]
- 176.
- Brown SB, Brierley TT, Palanisamy N, et al. Vitamin D receptor as a candidate tumor-suppressor gene in severe hyperparathyroidism of uremia. J Clin Endocrinol Metab. 2000;85:868–872. [PubMed: 10690903]
- 177.
- Degenhardt S, Toell A, Weideman W, Dotzenrath C, Spindler K-D, Grabensee B. Point mutations of the human parathyroid calcium receptor gene are not responsible for non-suppresible renal hyperparathyroidism. Kidney Intern. 1998;53:556–561. [PubMed: 9507199]
- 178.
- Djema AI, Mahmoud MD, Collin P, Heyman MF. Hyperparathyroïdie tertiaire: cancer parathyroïdien avec métastases hépatiques chez un hémodialysé. Néphrologie. 1998;19:121–123. [PubMed: 9633054]
- 179.
- Miki H, Sumitomo M, Inoue H, Kita S, Monden Y. Parathyroid carcinoma in patients with chronic renal failure on maintenance hemodialysis. Surgery. 1996;120:897–901. [Review] [10 refs] [PubMed: 8909528]
- 180.
- Takami H, Kameyama K, Nagakubo I. Parathyroid carcinoma in a patient receiving long-term hemodialysis. Surgery. 1999;125:239–240. [PubMed: 10026762]
- 181.
- Drüeke T B, Zhang P, Gogusev J. Apoptosis: background and possible role in secondary hyperparathyroidism (Invited Comment). Nephrol. Dial. Transplant. 1997;12:2228–2233. [PubMed: 9394304]
- 182.
- Parfitt A M. The hyperparathyroidism of chronic renal failure: a disorder of growth. Kidney Int. 1997;52:3–9. [PubMed: 9211340]
- 183.
- Wada M, Furuya Y, Sakiyama J, et al. The calcimimetic compound NPS R-568 suppresses parathyroid cell proliferation in rats with renal insufficiency. Control of parathyroid cell growth via a calcium receptor. J. Clin. Invest. 1997;100:2977–2983. [PMC free article: PMC508509] [PubMed: 9399943]
- 184.
- Canalejo A, Almaden Y, Torregrosa V, et al. The in vitro effect of calcitriol on parathyroid cell proliferation and apoptosis. J Amer Soc Nephrol. 2000;11:1865–1872. [PubMed: 11004217]
- 185.
- Jara A, Bover J, Felsenfeld A J. Development of secondary hyperparathyroidism and bone disease in diabetic rats with renal failure. Kidney Int. 1995;47:1746–1751. [PubMed: 7643545]
- 186.
- Zhang P, Duchambon P, Gogusev J, et al. Apoptosis in parathyroid hyperplasia of patients with primary or secondary uremic hyperparathyroidism. Kidney Int. 2000;57:437–445. [PubMed: 10652020]
- 187.
- Heidenreich S, Schmidt M, Bachmann J, Harrach B. Apoptosis of monocytes cultured from long-term hemodialysis patients. Kidney Int. 1996;49:792–799. [PubMed: 8648922]
- 188.
- Fadda S G, Massry S G. Chronic renal failure is a state of cellular calcium toxicity. Am. J. Kidney Dis. 1993;21:81–86. (Review) [PubMed: 8418632]
- 189.
- Carracedo J, Ramirez R, Martin-Malo A, Rodriguez M, Aljama P. Nonbiocompatible hemodialysis membranes induce apoptosis in mononuclear cells: the role of G-proteins. J. Am. Soc. Nephrol. 1998;9:46–53. [PubMed: 9440086]
- 190.
- Lewin E, Wang W, Olgaard K. Reversibility of experimental secondary hyperparathyroidism. Kidney Int. 1997;52:1232–1241. [PubMed: 9350646]
- 191.
- Henry H L, Taylor A N, Norman A W. Response of chick parathyroid glands to the vitamin D metabolites, 1,25-dihydroxycholecalciferol and 24,25-dihydroxycholecalciferol. J. Nutr. 1977;107:1918–1926. [PubMed: 903834]
- 192.
- Cloutier M, Brossard JH, Gascon-Barre M, D’Amour P. Lack of involution of hyperplastic parathyroid glands in dogs: adaptation via a decrease in the calcium stimulation set point and a change in secretion profile. J Bone Miner Res. 1994;9:621–629. [PubMed: 8053390]
- 193.
- Colloton M, Shatzen E, Miller G, et al. Cinacalcet HCl attenuates parathyroid hyperplasia in a rat model of secondary hyperparathyroidism. Kidney Int. 2005;67:467–476. [PubMed: 15673294]
- 194.
- Miller G, Davis J, Shatzen E, Colloton M, Martin D, Henley CM. Cinacalcet HCl prevents development of parathyroid gland hyperplasia and reverses established parathyroid gland hyperplasia in a rodent model of CKD. Nephrol Dial Transplant. 2012;27:2198–2205. [PMC free article: PMC3363978] [PubMed: 22036941]
- 195.
- Nylen E, Shah A, Hall J. Spontaneous remission of primary hyperparathroidism from parathroid apoplexy. J. Clin. Endocrinol. Metab. 1996;81:1326–1328. [PubMed: 8636326]
- 196.
- Fukagawa M, Okazaki R, Takano K, et al. Regression of parathyroid hyperplasia by calcitriol-pulse therapy in patients on long-term dialysis. N. Engl. J. Med. 1990;323:421–422. [PubMed: 2370898]
- 197.
- Quarles L D, Yohay D A, Carroll B A, et al. Prospective trial of pulse oral versus intravenous calcitriol treatment of hyperparathyroidism in ESRD. Kidney Int. 1994;45:1710–1721. [PubMed: 7933819]
- 198.
- Fukagawa M, Kitaoka M, Yi H, Fukuda N, Matsumoto T, Ogata E. Serial evaluation of parathyroid size by ultrasonography is another useful marker for the long-term prognosis of calcitriol pulse therapy in chronic dialysis patients. Nephron. 1994;68:221–228. [PubMed: 7830860]
- 199.
- Okuno S, Ishimura E, Kitatani K, et al. Relationship between parathyroid gland size and responsiveness to maxacalcitol therapy in patients with secondary hyperparathyroidism. Nephrol Dial Transplant. 2003;18:2613–2621. [PubMed: 14605286]
- 200.
- Martin K J, Hruska K A, Lewis J, Anderson C, Slatopolsky E. The renal handling of parathyroid hormone: role of peritubular uptake and glomerular filtration. J. Clin. Invest. 1977;60:808–814. [PMC free article: PMC372428] [PubMed: 893678]
- 201.
- Hilpert J, Nykjaer A, Jacobsen C, et al. Megalin antagonizes activation of the parathyroid hormone receptor. J Biol Chem. 1999;274:5620–5625. [PubMed: 10026179]
- 202.
- Freitag JJ, Martin KJ, Hruska KA, Slatopolsky E. Impaired parathyroid hormone metabolism in patients with chronic renal failure. N. Engl. J. Med. 1978;298:29–34. [PubMed: 337145]
- 203.
- Hruska K A, Kopelman R, Rutherford W E, Klahr S, Slatopolsky E. Metabolism of immunoreactive parathyroid hormone in the dog: the role of the kidney and the effects of chronic renal disease. J. Clin. Invest. 1975;56:39–46. [PMC free article: PMC436553] [PubMed: 1141439]
- 204.
- Martin KJ, Hruska KA, Freitag JJ, Klahr S, Slatopolsky E. The peripheral metabolism of parathyroid hormone in patients with chronic renal failure. N. Engl. J. Med. 1979;301:1092–1098. [PubMed: 386126]
- 205.
- Daugaard H, Egfjord M, Lewin E, Olgaard K. Metabolism of intact PTH by isolated perfused kidney and liver from uremic rats. Exp. Nephrol. 1994;2:240–248. [PubMed: 8069660]
- 206.
- Fukagawa M, Kazama JJ, Shigematsu T. Skeletal resistance to PTH as a basic abnormality underlying uremic bone diseases. Am J Kidney Dis. 2001;38:S152–5. [PubMed: 11576943]
- 207.
- Kazama JJ, Shigematsu T, Yano K, et al. Increased circulating levels of osteoclastogenesis inhibitory factor (osteoprotegerin) in patients with chronic renal failure. Am J Kidney Dis. 2002;39:525–532. [PubMed: 11877571]
- 208.
- Smogorzewski M, Tian J, Massry S G. Down-regulation of PTH-PTHrP receptor of heart in CRF: role of [Ca2+]i. Kidney Int. 1995;47:1182–1186. [PubMed: 7783417]
- 209.
- Ureña P, Ferreira A, Morieux C, Drüeke T B, deVernejoul M C. PTH/PTHrP receptor mRNA is down-regulated in epiphyseal cartilage growth plate of uraemic rats. Nephrol. Dial. Transplant. 1996;11:2008–2016. [PubMed: 8918715]
- 210.
- Ureña P, Kubrusly M, Mannstadt M, et al. The renal PTH/PTHrP receptor is down-regulated in rats with chronic renal failure. Kidney Int. 1994;45:605–611. [PubMed: 8164450]
- 211.
- Ureña P, Mannstadt M, Hruby M, et al. Parathyroidectomy does not prevent the renal PTH/PTHrP receptor down-regulation in uremic rats. Kidney Int. 1995;47:1797–1805. [PubMed: 7643551]
- 212.
- Picton ML, Moore PR, Mawer EB, et al. Down-regulation of human osteoblast PTH/PTHrP receptor mRNA in end-stage renal failure. Kidney Int. 2000;58:1440–1449. [PubMed: 11012879]
- 213.
- Langub MC. MonierFaugere MC, Qi QL, Geng Z, Koszewski NJ, Malluche HH. Parathyroid hormone/parathyroid hormone-related peptide type 1 receptor in human bone. J Bone Miner Res. 2001;16:448–456. [PubMed: 11277262]
- 214.
- Takenaka T, Inoue T, Miyazaki T, Hayashi M, Suzuki H. Xeno-Klotho Inhibits Parathyroid Hormone Signaling. J Bone Miner Res. 2015;31:455–462. [PubMed: 26287968]
- 215.
- Drueke TB, Lafage-Proust MH. Sclerostin: just one more player in renal bone disease? Clin J Am Soc Nephrol. 2011;6:700–703. [PubMed: 21441122]
- 216.
- Sugatani T. Systemic Activation of Activin A Signaling Causes Chronic Kidney Disease-Mineral Bone Disorder. International Journal of Molecular Sciences. 2018;19:E2490. [PMC free article: PMC6163495] [PubMed: 30142896]
- 217.
- Viaene L, Behets GJ, Claes K, et al. Sclerostin: another bone-related protein related to all-cause mortality in haemodialysis? Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28:3024–3030. [PubMed: 23605174]
- 218.
- Drechsler C, Evenepoel P, Vervloet MG, et al. High levels of circulating sclerostin are associated with better cardiovascular survival in incident dialysis patients: results from the NECOSAD study. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2015;30:288–293. [PubMed: 25248363]
- 219.
- Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–4583. [PubMed: 16081646]
- 220.
- Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25:178–189. [PMC free article: PMC3153379] [PubMed: 19594304]
- 221.
- Fujita K, Roforth MM, Demaray S, et al. Effects of estrogen on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in postmenopausal women. J Clin Endocrinol Metab. 2014;99:E81–8. [PMC free article: PMC3879677] [PubMed: 24170101]
- 222.
- Silva BC, Bilezikian JP. Parathyroid hormone: anabolic and catabolic actions on the skeleton. Curr Opin Pharmacol. 2015;22:41–50. [PMC free article: PMC5407089] [PubMed: 25854704]
- 223.
- Cejka D, Herberth J, Branscum AJ, et al. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin J Am Soc Nephrol. 2011;6:877–882. [PMC free article: PMC3069382] [PubMed: 21164019]
- 224.
- Lima F, Mawad H, El-Husseini AA, Davenport DL, Malluche HH. Serum bone markers in ROD patients across the spectrum of decreases in GFR: Activin A increases before all other markers. Clinical Nephrology. 2019;91:222–230. [PMC free article: PMC6595397] [PubMed: 30862350]
- 225.
- Li J-Y, Yu M, Pal S, et al. Parathyroid hormone-dependent bone formation requires butyrate production by intestinal microbiota. The Journal of Clinical Investigation. 2020;130:1767–1781. [PMC free article: PMC7108906] [PubMed: 31917685]
- 226.
- Yu M, Malik Tyagi A, Li J-Y, et al. PTH induces bone loss via microbial-dependent expansion of intestinal TNF+ T cells and Th17 cells. Nature Communications. 2020;11:468. [PMC free article: PMC6981196] [PubMed: 31980603]
- 227.
- Massy ZA, Drueke TB. Gut microbiota orchestrates PTH action in bone: role of butyrate and T cells. Kidney International. 2020;98:269–272. [PubMed: 32600825]
- 228.
- Llach F. Calcific uremic arteriolopathy (calciphylaxis): an evolving entity ? Am J Kidney Dis. 1998;32:513–518. [PubMed: 9740172]
- 229.
- Floege J, Kubo Y, Floege A, Chertow GM, Parfrey PS. The Effect of Cinacalcet on Calcific Uremic Arteriolopathy Events in Patients Receiving Hemodialysis: The EVOLVE Trial. Clin J Am Soc Nephrol. 2015;10:800–807. [PMC free article: PMC4422249] [PubMed: 25887067]
- 230.
- Viljoen A, Singh DK, Twomey PJ, Farrington K. Analytical quality goals for parathyroid hormone based on biological variation. Clin Chem Lab Med. 2008;46:1438–1442. [PubMed: 18844499]
- 231.
- Barreto FC, Barreto DV, Moyses RM, et al. K/DOQI-recommended intact PTH levels do not prevent low-turnover bone disease in hemodialysis patients. Kidney Int. 2008;73:771–777. [PubMed: 18185506]
- 232.
- Cohen-Solal M E, Boudailliez B, Sebert J L, Westeel P F, Bouillon R, Fournier A. Comparison of intact, mid region and carboxyterminal assays of parathyroid hormone for the diagnosis of bone disease in hemodialyzed patients. J. Clin. Endocrinol. Metab. 1991;73:516–524. [PubMed: 1874930]
- 233.
- Quarles L D, Lobaugh B, Murphy G. Intact parathyroid hormone overestimates the presence and severity of parathyroid-mediated osseous abnormalities in uremia. J. Clin. Endocrinol. Metab. 1992;75:145–150. [PubMed: 1619003]
- 234.
- Wang M, Hercz G, Sherrard D J, Maloney N A, Segre G V, Pei Y. Relationship between intact 1-84 parathyroid hormone and bone histomorphometric parameters in dialysis patients without aluminium toxicity. Am. J. Kidney Dis. 1995;26:836–844. [PubMed: 7485142]
- 235.
- Brossard J H, Cloutier M, Roy L, Lepage R, Gascon-Barré M, D’Amour P. Accumulation of a non-(1-84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: importance in the interpretation of PTH values. J. Clin. Endocrinol. Metab. 1996;81:3923–3929. [PubMed: 8923839]
- 236.
- Lepage R, Roy L, Brossard J H, et al. A non-(1-84) circulating parathyroid hormone (PTH fragment interferes significantly with intact commercial PTH assay measurements in uremic samples. Clin. Chem. 1998;44:805–809. [PubMed: 9554492]
- 237.
- Langub MC, Monier-Faugere MC, Wang G, Williams JP, Koszewski NJ, Malluche HH. Administration of PTH-(7-84) antagonizes the effects of PTH-(1-84) on bone in rats with moderate renal failure. Endocrinology. 2003;144:1135–1138. [PubMed: 12639892]
- 238.
- Souberbielle JC, Boutten A, Carlier MC, et al. Inter-method variability in PTH measurement: Implication for the care of CKD patients. Kidney Int. 2006;70:345–350. [PubMed: 16788691]
- 239.
- Joly D, Drueke TB, Alberti C, et al. Variation in serum and plasma PTH levels in second-generation assays in hemodialysis patients: a cross-sectional study. Am J Kidney Dis. 2008;51:987–995. [PubMed: 18430500]
- 240.
- John MR, Goodman WG, Gao P, Cantor TL, Salusky IB, Juppner H. A novel immunoradiometric assay detects full-length human PTH but not amino-terminally truncated fragments: Implications for PTH measurements in renal failure. J Clin Endocrinol Metab. 1999;84:4287–4290. [PubMed: 10566687]
- 241.
- MonierFaugere MC. Improved assessment of bone turnover by the PTH-(1-84) large C-PTH fragments ratio in ESRD patients. Kidney Int. 2001;60:1460–1468. Geng ZP, Mawad H et al. [PubMed: 11576360]
- 242.
- Coen G, Bonucci E, Ballanti P, et al. PTH 1-84 and PTH “7-84” in the noninvasive diagnosis of renal bone disease. Am J Kidney Dis. 2002;40:348–354. [PubMed: 12148108]
- 243.
- Reichel H, Esser A, Roth HJ, Schmidt-Gayk H. Influence of PTH assay methodology on differential diagnosis of renal bone disease. Nephrol Dial Transplant. 2003;18:759–768. [PubMed: 12637646]
- 244.
- Salusky IB, Goodman WG, Kuizon BD, et al. Similar predictive value of bone turnover using first- and second-generation immunometric PTH assays in pediatric patients treated with peritoneal dialysis. Kidney Int. 2003;63:1801–1808. [PubMed: 12675856]
- 245.
- Tepel M, Armbruster FP, Grön HJ, et al. Nonoxidized, biologically active parathyroid hormone determines mortality in hemodialysis patients. The Journal of Clinical Endocrinology and Metabolism. 2013;98:4744–4751. [PubMed: 24171919]
- 246.
- Seiler-Mussler S, Limbach AS, Emrich IE, et al. Association of Nonoxidized Parathyroid Hormone with Cardiovascular and Kidney Disease Outcomes in Chronic Kidney Disease. Clinical journal of the American Society of Nephrology: CJASN. 2018;13:569–576. [PMC free article: PMC5968904] [PubMed: 29507005]
- 247.
- Vervloet MG, van Ballegooijen AJ. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney International. 2018;93:1060–1072. [PubMed: 29580635]
- 248.
- Ursem SR, Heijboer AC, D’Haese PC, et al. Non-oxidized parathyroid hormone (PTH) measured by current method is not superior to total PTH in assessing bone turnover in chronic kidney disease. Kidney International. 2021;99:1173–1178. [PubMed: 33422551]
- 249.
- Drüeke TB, Floege J. Parathyroid hormone oxidation in chronic kidney disease: clinical relevance? Kidney International. 2021;99:1070–1072. [PubMed: 33892858]
- 250.
- Drueke TB, Fukagawa M. Whole or fragmentary information on parathyroid hormone? Clin J Am Soc Nephrol. 2007;2:1106–1107. [PubMed: 17928465]
- 251.
- Amann K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin J Am Soc Nephrol. 2008;3:1599–1605. [PubMed: 18815240]
- 252.
- Bellasi A, Raggi P. Techniques and technologies to assess vascular calcification. Semin Dial. 2007;20:129–133. [PubMed: 17374086]
- 253.
- London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740. [PubMed: 12937218]
- 254.
- Rostand SG, Drueke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int. 1999;56:383–392. [PubMed: 10432376]
- 255.
- Tsuchihashi K, Takizawa H, Torii T, et al. Hypoparathyroidism potentiates cardiovascular complications through disturbed calcium metabolism: Possible risk of vitamin D-3 analog administration in dialysis patients with end-stage renal disease. Nephron. 2000;84:13–20. [PubMed: 10644903]
- 256.
- Galassi A, Spiegel DM, Bellasi A, Block GA, Raggi P. Accelerated vascular calcification and relative hypoparathyroidism in incident haemodialysis diabetic patients receiving calcium binders. Nephrol Dial Transplant. 2006;21:3215–3222. [PubMed: 16877490]
- 257.
- Sebastian EM, Suva LJ, Friedman PA. Differential effects of intermittent PTH(1-34) and PTH(7- 34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone. 2008;43:1022–1030. [PMC free article: PMC2644420] [PubMed: 18761112]
- 258.
- Shao JS, Cheng SL. CharltonKachigian N, Loewy AP, Towler DA. Teriparatide (Human parathyroid hormone (1-34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem. 2003;278:50195–50202. [PubMed: 14504275]
- 259.
- Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357:905–916. [PubMed: 17761594]
- 260.
- Drueke TB, Ritz E. Treatment of secondary hyperparathyroidism in CKD patients with cinacalcet and/or vitamin D derivatives. Clin J Am Soc Nephrol. 2009;4:234–241. [PubMed: 19056615]
- 261.
- Malluche HH, Mawad H, Monier-Faugere MC. Effects of treatment of renal osteodystrophy on bone histology. Clin J Am Soc Nephrol. 2008;3 Suppl 3:S157–63. [PMC free article: PMC3152281] [PubMed: 18988701]
- 262.
- Eknoyan G, Levin A, Levin NW. Bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42:1–201.
- 263.
- KidneyDisease-ImprovingGlobalOutcomes(KDIGO)CKD-MBDWorkGroup. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009:S1–130. [PubMed: 19644521]
- 264.
- Ketteler M, Block GA, Evenepoel P, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: what’s changed and why it matters. Kidney International. 2017;92:26–36. [PubMed: 28646995]
- 265.
- Ketteler M, Block GA, Evenepoel P, et al. Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder: Synopsis of the Kidney Disease: Improving Global Outcomes 2017 Clinical Practice Guideline Update. Annals of Internal Medicine. 2018;168:422–430. [PubMed: 29459980]
- 266.
- Bhan I, Thadhani R. Vitamin D therapy for chronic kidney disease. Semin Nephrol. 2009;29:85–93. [PubMed: 19121478]
- 267.
- Kooienga L, Fried L, Scragg R, Kendrick J, Smits G, Chonchol M. The effect of combined calcium and vitamin D3 supplementation on serum intact parathyroid hormone in moderate CKD. Am J Kidney Dis. 2009;53:408–416. [PubMed: 19185400]
- 268.
- Jean G, Souberbielle JC, Chazot C. Monthly cholecalciferol administration in haemodialysis patients: a simple and efficient strategy for vitamin D supplementation. Nephrol Dial Transplant. 2009;24:3799–3805. [PubMed: 19622574]
- 269.
- Agarwal R, Georgianos PI. Con: Nutritional vitamin D replacement in chronic kidney disease and end-stage renal disease. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2016;31:706–713. [PubMed: 27190392]
- 270.
- Goldsmith DJA. Pro: Should we correct vitamin D deficiency/insufficiency in chronic kidney disease patients with inactive forms of vitamin D or just treat them with active vitamin D forms? Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2016;31:698–705. [PubMed: 27190390]
- 271.
- Drueke TB. Calcimimetics versus vitamin D: what are their relative roles? Blood Purif. 2004;22:38–43. [PubMed: 14732810]
- 272.
- Hansen D, Rasmussen K, Danielsen H, et al. No difference between alfacalcidol and paricalcitol in the treatment of secondary hyperparathyroidism in hemodialysis patients: a randomized crossover trial. Kidney Int. 2011;80:841–850. [PubMed: 21832979]
- 273.
- Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–456. [PubMed: 12890843]
- 274.
- Shoben AB, Rudser KD, deBoer IH, Young B, Kestenbaum B. Association of Oral Calcitriol with Improved Survival in Nondialyzed CKD. J Amer Soc Nephrol. 2008;19:1613–1619. [PMC free article: PMC2488261] [PubMed: 18463168]
- 275.
- Shoji T, Shinohara K, Kimoto E, et al. Lower risk for cardiovascular mortality in oral 1alpha-hydroxy vitamin D3 users in a haemodialysis population. Nephrol Dial Transplant. 2004;19:179–184. [PubMed: 14671054]
- 276.
- Teng M, Wolf M, Ofsthun MN, et al. Activated injectable vitamin D and hemodialysis survival: A historical cohort study. J Amer Soc Nephrol. 2005;16:1115–1125. [PubMed: 15728786]
- 277.
- Tentori F, Hunt WC, Stidley CA, et al. Mortality risk among hemodialysis patients receiving different vitamin D analogs. Kidney Int. 2006;70:1858–1865. [PubMed: 17021609]
- 278.
- Tentori F, Albert JM, Young EW, et al. The survival advantage for haemodialysis patients taking vitamin D is questioned: findings from the Dialysis Outcomes and Practice Patterns Study. Nephrol Dial Transplant. 2009;24:963–972. [PubMed: 19028748]
- 279.
- Drueke TB, McCarron DA. Paricalcitol as compared with calcitriol in patients undergoing hemodialysis. N Engl J Med. 2003;349:496–499. [PubMed: 12890849]
- 280.
- Brown E M, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. [PubMed: 8255296]
- 281.
- Antonsen JE, Sherrard DJ, Andress DL. A calcimimetic agent acutely suppresses parathyroid hormone levels in patients with chronic renal failure - rapid communication. Kidney Int. 1998;53:223–227. [PubMed: 9453023]
- 282.
- Goodman WG, Frazao JM, Goodkin DA, Turner SA, Liu W, Coburn JW. A calcimimetic agent lowers plasma parathyroid hormone levels in patients with secondary hyperparathyroidism. Kidney Int. 2000;58:436–45. [PubMed: 10886592]
- 283.
- Goodman WG, Hladik GA, Turner SA, et al. The calcimimetic agent AMG 073 lowers plasma parathyroid hormone levels in hemodialysis patients with secondary hyperparathyroidism. J Am Soc Nephrol. 2002;13:1017–24. [PubMed: 11912261]
- 284.
- Ivanovski O, Nikolov IG, Joki N, et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE-/-) mice. Atherosclerosis. 2009;205:55–62. [PubMed: 19118829]
- 285.
- Basile C, Lomonte C. The function of the parathyroid oxyphil cells in uremia: still a mystery? Kidney International. 2017;92:1046–1048. [PubMed: 29055426]
- 286.
- Lomonte C, Vernaglione L, Chimienti D, et al. Does vitamin D receptor and calcium receptor activation therapy play a role in the histopathologic alterations of parathyroid glands in refractory uremic hyperparathyroidism? Clinical journal of the American Society of Nephrology: CJASN. 2008;3:794–799. [PMC free article: PMC2386693] [PubMed: 18322048]
- 287.
- Ritter C, Miller B, Coyne DW, et al. Paricalcitol and cinacalcet have disparate actions on parathyroid oxyphil cell content in patients with chronic kidney disease. Kidney International. 2017;92:1217–1222. [PubMed: 28750928]
- 288.
- Ritter CS, Haughey BH, Miller B, Brown AJ. Differential gene expression by oxyphil and chief cells of human parathyroid glands. The Journal of Clinical Endocrinology and Metabolism. 2012;97:E1499–1505. [PMC free article: PMC3591682] [PubMed: 22585091]
- 289.
- Lopez I, Aguilera-Tejero E, Mendoza FJ, et al. Calcimimetic R-568 decreases extraosseous calcifications in uremic rats treated with calcitriol. J Am Soc Nephrol. 2006;17:795–804. [PubMed: 16467452]
- 290.
- Raggi P, Chertow GM, Torres PU, et al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant. 2011;26:1327–1339. [PubMed: 21148030]
- 291.
- Koleganova N, Piecha G, Ritz E, Schmitt CP, Gross ML. A calcimimetic (R-568), but not calcitriol, prevents vascular remodeling in uremia. Kidney Int. 2009;75:60–71. [PubMed: 19092814]
- 292.
- Koleganova N, Piecha G, Ritz E, et al. Interstitial fibrosis and microvascular disease of the heart in uremia: amelioration by a calcimimetic. Lab Invest. 2009;89:520–530. [PubMed: 19188910]
- 293.
- Lopez I, Mendoza FJ. The effect of calcitriol, paricalcitol, and a calcimimetic on extraosseous calcifications in uremic rats. Kidney Int. 2008;73:300–307. AguileraTejero E et al. [PubMed: 18004298]
- 294.
- Block GA, Martin KJ, de Francisco AL, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350:1516–1525. [PubMed: 15071126]
- 295.
- Lindberg JS, Moe SM, Goodman WG, et al. The calcimimetic AMG 073 reduces parathyroid hormone and calcium x phosphorus in secondary hyperparathyroidism. Kidney Int. 2003;63:248–254. [PubMed: 12472790]
- 296.
- Quarles LD, Sherrard DJ, Adler S, et al. The calcimimetic AMG 073 as a potential treatment for secondary hyperparathyroidism of end-stage renal disease. J Am Soc Nephrol. 2003;14:575–583. [PubMed: 12595492]
- 297.
- Parfrey PS, Chertow GM, Block GA, et al. The clinical course of treated hyperparathyroidism among patients receiving hemodialysis and the effect of cinacalcet: the EVOLVE trial. J Clin Endocrinol Metab. 2013;98:4834–4844. [PubMed: 24108314]
- 298.
- Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482–2494. [PubMed: 23121374]
- 299.
- Block GA, Zeig S, Sugihara J, et al. Combined therapy with cinacalcet and low doses of vitamin D sterols in patients with moderate to severe secondary hyperparathyroidism. Nephrol Dial Transplant. 2008;23:2311–2318. [PubMed: 18310602]
- 300.
- Wilkie M, Pontoriero G, Macario F, et al. Impact of Vitamin D Dose on Biochemical Parameters in Patients with Secondary Hyperparathyroidism Receiving Cinacalcet. Nephron Clin Pract. 2009;112:c41–c50. [PubMed: 19365139]
- 301.
- Block GA, Bushinsky DA, Cunningham J, et al. Effect of Etelcalcetide vs Placebo on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: Two Randomized Clinical Trials. JAMA. 2017;317:146–155. [PubMed: 28097355]
- 302.
- Block GA, Bushinsky DA, Cheng S, et al. Effect of Etelcalcetide vs Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: A Randomized Clinical Trial. JAMA. 2017;317:156–164. [PubMed: 28097356]
- 303.
- Moe SM, Abdalla S, Chertow GM, et al. Effects of Cinacalcet on Fracture Events in Patients Receiving Hemodialysis: The EVOLVE Trial. J Am Soc Nephrol. 2015;26:1466–1475. [PMC free article: PMC4446874] [PubMed: 25505257]
- 304.
- Shahapuni I, Mansour J, Harbouche L, et al. How do calcimimetics fit into the management of parathyroid hormone, calcium, and phosphate disturbances in dialysis patients? Semin Dial. 2005;18:226–238. [PubMed: 15934970]
- 305.
- Cannata Andia JB. Adynamic bone and chronic renal failure: an overview. Am J Med Sci. 2000;320:81–84. [PubMed: 10981480]
- 306.
- Argilés A, Kerr P G, Canaud B, Flavier J L, Mion C. Calcium kinetics and the long-term effects of lowering dialysate calcium concentration. Kidney Int. 1993;43:630–640. [PubMed: 8455362]
- 307.
- Arenas MD, Alvarez-Ude F, Gil MT, et al. Application of NKF-K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease: changes of clinical practices and their effects on outcomes and quality standards in three haemodialysis units. Nephrol Dial Transplant. 2006;21:1663–1668. [PubMed: 16464885]
- 308.
- Basile C, Libutti P, Di Turo AL, et al. Effect of dialysate calcium concentrations on parathyroid hormone and calcium balance during a single dialysis session using bicarbonate hemodialysis: a crossover clinical trial. Am J Kidney Dis. 2012;59:92–101. [PubMed: 22000728]
- 309.
- Sonikian M, Metaxaki P, Karatzas I, Vlassopoulos D. Paricalcitol treatment of secondary hyperparathyroidism in hemodialysis patients on sevelamer hydrochloride: which dialysate calcium concentration to use? Blood Purif. 2009;27:182–186. [PubMed: 19141997]
- 310.
- Touam M, Menoyo V, Attaf D, Thebaud HE, Drueke TB. High dialysate calcium may improve the efficacy of calcimimetic treatment in hemodialysis patients with severe secondary hyperparathyroidism. Kidney Int. 2005;67:2065–author reply 2065-6. [PubMed: 15840060]
- 311.
- Drüeke TB, Touam M. Calcium balance in haemodialysis--do not lower the dialysate calcium concentration too much (con part). Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24:2990–2993. [PubMed: 19666667]
- 312.
- Pun PH, Horton JR, Middleton JP. Dialysate calcium concentration and the risk of sudden cardiac arrest in hemodialysis patients. Clin J Am Soc Nephrol. 2013;8:797–803. [PMC free article: PMC3641623] [PubMed: 23371957]
- 313.
- Pun PH, Lehrich RW, Honeycutt EF, Herzog CA, Middleton JP. Modifiable risk factors associated with sudden cardiac arrest within hemodialysis clinics. Kidney Int. 2011;79:218–227. [PubMed: 20811332]
- 314.
- Cunningham J, Beer J, Coldwell R D, Noonan K, Sawyer N, Makin H L J. Dialysate calcium reduction in CAPD patients treated with calcium carbonate and alfacalcidol. Nephrol. Dial. Transplant. 1992;7:63–68. [PubMed: 1316583]
- 315.
- Chertow GM, Burke SK, Lazarus JM, et al. Poly[allylamine hydrochloride] (RenaGel): a noncalcemic phosphate binder for the treatment of hyperphosphatemia in chronic renal failure. Am J Kidney Dis. 1997;29:66–71. [PubMed: 9002531]
- 316.
- Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–52. [PubMed: 12081584]
- 317.
- Slatopolsky EA, Burke SK, Dillon MA. RenaGel (R), a nonabsorbed calcium- and aluminum-free phosphate binder, lowers serum phosphorus and parathyroid hormone. Kidney Int. 1999;55:299–307. [PubMed: 9893140]
- 318.
- Delmez J, Block G, Robertson J, et al. A randomized, double-blind, crossover design study of sevelamer hydrochloride and sevelamer carbonate in patients on hemodialysis. Clin Nephrol. 2007;68:386–391. [PubMed: 18184521]
- 319.
- Ketteler M, Rix M, Fan S, et al. Efficacy and tolerability of sevelamer carbonate in hyperphosphatemic patients who have chronic kidney disease and are not on dialysis. Clin J Am Soc Nephrol. 2008;3:1125–1130. [PMC free article: PMC2440270] [PubMed: 18450923]
- 320.
- D’Haese PC, Spasovski GB, Sikole A, et al. A multicenter study on the effects of lanthanum carbonate (Fosrenol(TM)) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int. 2003;63:S73–S78. [PubMed: 12753271]
- 321.
- Hutchison AJ. Oral phosphate binders. Kidney Int. 2009;75:906–914. [PubMed: 19279554]
- 322.
- Hutchison AJ, Barnett ME, Krause R, Kwan JT, Siami GA. Long-term efficacy and safety profile of lanthanum carbonate: results for up to 6 years of treatment. Nephron Clin Pract. 2008;110:c15–c23. [PMC free article: PMC2790759] [PubMed: 18667837]
- 323.
- Sprague SM, Ketteler M. A safety evaluation of sucroferric oxyhydroxide for the treatment of hyperphosphatemia. Expert Opinion on Drug Safety. 2021 [PubMed: 34511018]
- 324.
- Choi YJ, Noh Y, Shin S. Ferric citrate in the management of hyperphosphataemia and iron deficiency anaemia: A meta-analysis in patients with chronic kidney disease. British Journal of Clinical Pharmacology. 2021;87:414–426. [PubMed: 32470149]
- 325.
- Block GA, Raggi P, Bellasi A, Kooienga L, Spiegel DM. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–441. [PubMed: 17200680]
- 326.
- Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23:1407–1415. [PMC free article: PMC3402292] [PubMed: 22822075]
- 327.
- Drueke TB, Massy ZA. Phosphate binders in CKD: bad news or good news? J Am Soc Nephrol. 2012;23:1277–1280. [PubMed: 22797178]
- 328.
- Neven E, Dams G, Postnov A, et al. Adequate phosphate binding with lanthanum carbonate attenuates arterial calcification in chronic renal failure rats. Nephrol Dial Transplant. 2009;24:1790–1799. [PubMed: 19144999]
- 329.
- Nikolov IG, Joki N, Nguyen-Khoa T, et al. Lanthanum carbonate, like sevelamer-HCl, retards the progression of vascular calcification and atherosclerosis in uremic apolipoprotein E-deficient mice. Nephrol Dial Transplant. 2012;27:505–513. [PubMed: 21705467]
- 330.
- Toussaint ND, Lau KK, Polkinghorne KR, Kerr PG. Attenuation of aortic calcification with lanthanum carbonate versus calcium-based phosphate binders in haemodialysis: A pilot randomized controlled trial. Nephrology (Carlton). 2011;16:290–298. [PubMed: 21342323]
- 331.
- Ohtake T, Kobayashi S, Oka M, et al. Lanthanum carbonate delays progression of coronary artery calcification compared with calcium-based phosphate binders in patients on hemodialysis: a pilot study. J Cardiovasc Pharmacol Ther. 2013;18:439–446. [PubMed: 23615577]
- 332.
- Seifert ME, de las Fuentes L, Rothstein M, et al. Effects of phosphate binder therapy on vascular stiffness in early-stage chronic kidney disease. Am J Nephrol. 2013;38:158–167. [PMC free article: PMC3874122] [PubMed: 23941761]
- 333.
- Ferreira A, Frazao JM, Monier-Faugere MC, et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J Am Soc Nephrol. 2008;19:405–412. [PMC free article: PMC2396748] [PubMed: 18199805]
- 334.
- Barreto DV, Barreto FD, deCarvalho AB, et al. Phosphate Binder Impact on Bone Remodeling and Coronary Calcification - Results from the BRiC Study. Nephron Clin Pract. 2008;110:C273–C283. [PubMed: 19001830]
- 335.
- Lenglet A, Liabeuf S, El Esper N, et al. Efficacy and safety of nicotinamide in haemodialysis patients: the NICOREN study. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association. 2017;32:870–879. [PubMed: 27190329]
- 336.
- Lenglet A, Liabeuf S, Guffroy P, Fournier A, Brazier M, Massy ZA. Use of nicotinamide to treat hyperphosphatemia in dialysis patients. Drugs in R&D. 2013;13:165–173. [PMC free article: PMC3784056] [PubMed: 24000048]
- 337.
- Malhotra R, Katz R, Hoofnagle A, et al. The Effect of Extended Release Niacin on Markers of Mineral Metabolism in CKD. Clinical journal of the American Society of Nephrology: CJASN. 2018;13:36–44. [PMC free article: PMC5753310] [PubMed: 29208626]
- 338.
- Pergola PE, Rosenbaum DP, Yang Y, Chertow GM. A Randomized Trial of Tenapanor and Phosphate Binders as a Dual-Mechanism Treatment for Hyperphosphatemia in Patients on Maintenance Dialysis (AMPLIFY). Journal of the American Society of Nephrology: JASN. 2021;32:1465–1473. [PMC free article: PMC8259655] [PubMed: 33766811]
- 339.
- Block GA, Rosenbaum DP, Leonsson-Zachrisson M, et al. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. Journal of the American Society of Nephrology: JASN. 2017;28:1933–1942. [PMC free article: PMC5461797] [PubMed: 28159782]
- 340.
- Drüeke TB, Massy ZA. Lowering Expectations with Niacin Treatment for CKD-MBD. Clinical journal of the American Society of Nephrology: CJASN. 2018;13:6–8. [PMC free article: PMC5753325] [PubMed: 29208625]
- 341.
- D’Alessandro C, Piccoli GB, Cupisti A. The “phosphorus pyramid”: a visual tool for dietary phosphate management in dialysis and CKD patients. BMC Nephrol. 2015;16:9. [PMC free article: PMC4361095] [PubMed: 25603926]
- 342.
- Lorenzo V, Martin M, Rufino M, et al. Protein intake, control of serum phosphorus, and relatively low levels of parathyroid hormone in elderly hemodialysis patients. Am J Kidney Dis. 2001;37:1260–1266. [PubMed: 11382697]
- 343.
- Shinaberger CS, Greenland S, Kopple JD, et al. Is controlling phosphorus by decreasing dietary protein intake beneficial or harmful in persons with chronic kidney disease? Amer J Clin Nutr. 2008;88:1511–1518. [PMC free article: PMC5500249] [PubMed: 19064510]
- 344.
- Lefebvre A, De Vernejoul M C, Gueris J, Goldfarb B, Graulet A M, Morieux C. Optimal correction of acidosis changes progression of dialysis osteodystrophy. Kidney Int. 1989;36:1112–1118. [PubMed: 2557481]
- 345.
- Graham K A, Hoenich N A, Tarbit M, Ward M K, Goodship T H J. Correction of acidosis in hemodialysis patients increases the sensitivity of the parathyroid glands to calcium. J. Am. Soc. Nephrol. 1997;8:627–631. [PubMed: 10495792]
- 346.
- Giangrande A, Castiglioni A, Solbiati L, Allaria P. US-guided percutaneous fine-needle ethanol injection into parathyroid glands in secondary hyperparathyroidism. Nephrol. Dial. Transplant. 1992;7:412–420. [PubMed: 1321377]
- 347.
- Kitakoa M, Fukagawa M, Ogata E, Kurokawa K. Reduction of functioning parathyroid cell mass by ethanol injection in chronic dialysis patients. Kidney Int. 1994;46:1110–1117. [PubMed: 7861705]
- 348.
- Shiizaki K, Hatamura I, Negi S, et al. Percutaneous maxacalcitol injection therapy regresses hyperplasia of parathyroid and induces apoptosis in uremia. Kidney Int. 2003;64:992–1003. [PubMed: 12911549]
- 349.
- Shiizaki K, Negi S, Hatamura I, et al. Biochemical and cellular effects of direct maxacalcitol injection into parathyroid gland in uremic rats. J Am Soc Nephrol. 2005;16:97–108. [PubMed: 15574509]
- 350.
- Fletcher S, Kanagasundaram NS, Rayner HC, et al. Assessment of ultrasound guided percutaneous ethanol injection and parathyroidectomy in patients with tertiary hyperparathyroidism. Nephrol Dial Transplant. 1998;13:3111–3117. [PubMed: 9870475]
- 351.
- de Barros Gueiros JE, Chammas MC, Gerhard R, et al. Percutaneous ethanol (PEIT) and calcitrol (PCIT) injection therapy are ineffective in treating severe secondary hyperparathyroidism. Nephrol Dial Transplant. 2004;19:657–663. [PubMed: 14767023]
- 352.
- Young EW, Akiba T, Albert JM, et al. Magnitude and impact of abnormal mineral metabolism in hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2004;44:34–38. [PubMed: 15486872]
- 353.
- Danese MD, Belozeroff V, Smirnakis K, Rothman KJ. Consistent control of mineral and bone disorder in incident hemodialysis patients. Clin J Am Soc Nephrol. 2008;3:1423–1429. [PMC free article: PMC2518803] [PubMed: 18596117]
- 354.
- Fouque D, Roth H, Darné B, et al. Achievement of Kidney Disease: Improving Global Outcomes mineral and bone targets between 2010 and 2014 in incident dialysis patients in France: the Photo-Graphe3 study. Clinical Kidney Journal. 2018;11:73–79. [PMC free article: PMC5798128] [PubMed: 29423206]
- 355.
- Lau WL, Obi Y, Kalantar-Zadeh K. Parathyroidectomy in the Management of Secondary Hyperparathyroidism. Clinical journal of the American Society of Nephrology: CJASN. 2018;13:952–961. [PMC free article: PMC5989682] [PubMed: 29523679]
- 356.
- Gagné E R, Ureña P, Leite-Silva S, et al. Short and long-term efficacy of total parathyroidectomy with immediate autografting compared with subtotal parathyroidectomy in hemodialysis patients. J. Am. Soc. Nephrol. 1992;3:1008–1017. [PubMed: 1450363]
- 357.
- Stracke S, Keller F, Steinbach G. HenneBruns D, Wuerl P. Long-Term Outcome after Total Parathyroidectomy for the Management of Secondary Hyperparathyroidism. Nephron Clin Pract. 2009;111:C102–C109. [PubMed: 19142022]
- 358.
- Drüeke T, Zingraff J. The dilemma of parathyroidectomy in chronic renal failure. Curr. Opin. Nephrol. Hypertens. 1994;3:386–395. [PubMed: 8076142]
- 359.
- Locatelli F, Cannata-Andia JB, Drueke TB, et al. Management of disturbances of calcium and phosphate metabolism in chronic renal insufficiency, with emphasis on the control of hyperphosphataemia. Nephrol Dial Transplant. 2002;17:723–731. [PubMed: 11981055]
- 360.
- Stratton J, Simcock M, Thompson H, Farrington K. Predictors of recurrent hyperparathyroidism after total parathyroidectomy in chronic renal failure. Nephron Clin Pract. 2003;95:c15–22. [PubMed: 14520017]
- 361.
- Malberti F, Marcelli D, Conte F, Limido A, Spotti D, Locatelli F. Parathyroidectomy in patients on renal replacement therapy: An epidemiologic study. J Amer Soc Nephrol. 2001;12:1242–1248. [PubMed: 11373348]
- 362.
- Kim SM, Long J, Montez-Rath ME, Leonard MB, Norton JA, Chertow GM. Rates and Outcomes of Parathyroidectomy for Secondary Hyperparathyroidism in the United States. Clinical journal of the American Society of Nephrology: CJASN. 2016;11:1260–1267. [PMC free article: PMC4934842] [PubMed: 27269300]
- 363.
- Lafrance J-P, Cardinal H, Leblanc M, et al. Effect of cinacalcet availability and formulary listing on parathyroidectomy rate trends. BMC nephrology. 2013;14:100. [PMC free article: PMC3648401] [PubMed: 23642012]
- 364.
- Tentori F, Wang M, Bieber BA, et al. Recent changes in therapeutic approaches and association with outcomes among patients with secondary hyperparathyroidism on chronic hemodialysis: the DOPPS study. Clinical journal of the American Society of Nephrology: CJASN. 2015;10:98–109. [PMC free article: PMC4284424] [PubMed: 25516917]
- 365.
- Kestenbaum B, Seliger SL, Gillen DL, et al. Parathyroidectomy rates among United States dialysis patients: 1990-1999. Kidney Int. 2004;65:282–288. [PubMed: 14675061]
- ABSTRACT
- INTRODUCTION
- SECONDARY HYPERPARATHYROIDISM IN CKD – SEQUENCE OF PLASMA BIOCHEMISTRY CHANGES IN EARLY CKD STAGES (FIGURE 1)
- SECONDARY HYPERPARATHYROIDISM IN CKD – PLASMA BIOCHEMISTRY CHANGES IN ADVANCED CKD STAGES (FIGURE 1)
- MECHANISMS INVOLVED IN THE PATHOGENESIS OF SECONDARY HYPERPARATHYROIDISM
- SECONDARY HYPERPARATHYROIDISM IN CKD – MECHANISMS INVOLVED IN THE TRANSFORMATION OF POLYCLONAL TO MONOCLONAL PARATHYROID GROWTH
- SECONDARY HYPERPARATHYROIDISM IN CKD – REGRESSION OF PARATHYROID HYPERPLASIA?
- ALTERED PTH METABOLISM AND RESISTANCE TO PTH ACTION
- SECONDARY HYPERPARATHYROIDISM IN CKD – CLINICAL FEATURES
- SECONDARY HYPERPARATHYROIDISM IN CKD -- DIAGNOSIS
- SECONDARY HYPERPARATHYROIDISM IN CKD - TREATMENT
- ACKNOWLEDGEMENTS
- REFERENCES
- [Changes in mineral metabolism in stage 3, 4, and 5 chronic kidney disease (not on dialysis)].[Nefrologia. 2008][Changes in mineral metabolism in stage 3, 4, and 5 chronic kidney disease (not on dialysis)].Lorenzo Sellares V, Torregrosa V. Nefrologia. 2008; 28 Suppl 3:67-78.
- Review Changing bone patterns with progression of chronic kidney disease.[Kidney Int. 2016]Review Changing bone patterns with progression of chronic kidney disease.Drüeke TB, Massy ZA. Kidney Int. 2016 Feb; 89(2):289-302.
- Review 1alpha(OH)D3 One-alpha-hydroxy-cholecalciferol--an active vitamin D analog. Clinical studies on prophylaxis and treatment of secondary hyperparathyroidism in uremic patients on chronic dialysis.[Dan Med Bull. 2008]Review 1alpha(OH)D3 One-alpha-hydroxy-cholecalciferol--an active vitamin D analog. Clinical studies on prophylaxis and treatment of secondary hyperparathyroidism in uremic patients on chronic dialysis.Brandi L. Dan Med Bull. 2008 Nov; 55(4):186-210.
- Review Parathyroid function in chronic kidney disease: role of FGF23-Klotho axis.[Contrib Nephrol. 2013]Review Parathyroid function in chronic kidney disease: role of FGF23-Klotho axis.Koizumi M, Komaba H, Fukagawa M. Contrib Nephrol. 2013; 180:110-23. Epub 2013 May 3.
- Novel insights into parathyroid hormone: report of The Parathyroid Day in Chronic Kidney Disease.[Clin Kidney J. 2019]Novel insights into parathyroid hormone: report of The Parathyroid Day in Chronic Kidney Disease.Ureña-Torres PA, Vervloet M, Mazzaferro S, Oury F, Brandenburg V, Bover J, Cavalier E, Cohen-Solal M, Covic A, Drüeke TB, et al. Clin Kidney J. 2019 Apr; 12(2):269-280. Epub 2018 Jul 20.
- Hyperparathyroidism in Chronic Kidney Disease - EndotextHyperparathyroidism in Chronic Kidney Disease - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...