By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2001, Garland Science.
Bookshelf ID: NBK27155

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001.

Autoimmune disease occurs when a specific adaptive immune response is mounted against self antigens. The normal consequence of an adaptive immune response against a foreign antigen is the clearance of the antigen from the body. Virus-infected cells, for example, are destroyed by cytotoxic T cells, whereas soluble antigens are cleared by formation of immune complexes of antibody and antigen, which are taken up by cells of the mononuclear phagocytic system such as macrophages. When an adaptive immune response develops against self antigens, however, it is usually impossible for immune effector mechanisms to eliminate the antigen completely, and so a sustained response occurs. The consequence is that the effector pathways of immunity cause chronic inflammatory injury to tissues, which may prove lethal. The mechanisms of tissue damage in autoimmune diseases are essentially the same as those that operate in protective immunity and in hypersensitivity diseases. Some common autoimmune diseases are listed in Fig. 13.1.
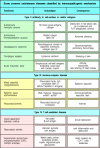
Autoimmune diseases classified by the mechanism of tissue damage. Autoimmune diseases can be grouped in the same way as hypersensitivity reactions, according to the type of immune response and the mechanism by which it damages tissues. The immunopathological (more...)
Adaptive immune responses are initiated by the activation of antigen-specific T cells, and it is believed that autoimmunity is initiated in the same way. T-cell responses to self antigens can inflict tissue damage either directly or indirectly. Cytotoxic T-cell responses and inappropriate activation of macrophages by TH1 cells can cause extensive tissue damage, whereas inappropriate T-cell help to self-reactive B cells can initiate harmful autoantibody responses. Autoimmune responses are a natural consequence of the open repertoires of both B-cell and T-cell receptors, which allow them to recognize any pathogen. Although these repertoires are purged of most receptors that bind with high affinity to self antigens encountered during development, they still include receptors of lower affinity reactive to some self antigens. It is not known what triggers autoimmunity, but both environmental and genetic factors, especially MHC genotype, are clearly important. Transient autoimmune responses are common, but it is only when they are sustained and cause lasting tissue damage that they attract medical attention. In this section, we will examine the nature of autoimmune responses and how autoimmunity leads to tissue damage. In the last section of this chapter, we will examine the mechanisms by which self-tolerance is lost and autoimmune responses are initiated.
Early in the study of immunity it was realized that the powerful effector mechanisms used in host defense could, if turned against the host, cause severe tissue damage; Ehrlich termed this horror autotoxicus. Healthy individuals do not mount sustained adaptive immune responses to their own antigens and, although transient responses to damaged self tissues occur, these rarely cause additional tissue damage. But although self-tolerance is the general rule, sustained immune responses to self tissues occur in some individuals, and these autoimmune responses cause the severe tissue damage that Ehrlich predicted.
In certain genetically susceptible strains of experimental animals, autoimmune disease can be induced artificially by injection of ‘self’ tissues from a genetically identical animal mixed with strong adjuvants containing bacteria (see Appendix I, Section A-4). This shows that autoimmunity can be provoked by inducing a specific, adaptive immune response to self antigens and forms the basis for our understanding of how autoimmune disease arises. In humans, autoimmunity usually arises spontaneously; that is, we do not know what events initiate the immune response to self that leads to the autoimmune disease. There is evidence, as we will learn in the last part of this chapter, that some autoimmune disorders, such as rheumatic fever, may be triggered by infectious agents. There is, however, also evidence, particularly from animal models of autoimmunity, that many autoimmune disorders occur through internal dysregulation of the immune system without the participation of infectious agents.
The classification of disease is an uncertain science, especially in the absence
of a precise understanding of causative mechanisms. This is well illustrated by
the difficulty in classifying the autoimmune diseases. It is useful to
distinguish two major patterns of autoimmune disease, the diseases in which the
expression of autoimmunity is restricted to specific organs of the body, known
as ‘organ-specific’ autoimmune diseases, and those in which many tissues of the
body are affected, the ‘systemic’ autoimmune diseases. Examples of
organ-specific autoimmune diseases are Hashimoto's thyroiditis and Graves'
disease, each predominantly affecting the thyroid gland, and type I
insulin-dependent diabetes mellitus (IDDM), which affects the pancreatic islets.
Examples of systemic autoimmune disease are systemic lupus erythematosus (SLE)
and primary Sjögren's syndrome, in which tissues as diverse as the skin,
kidneys, and brain may all be affected. ( Insulin-Dependent Diabetes Mellitus and Systemic Lupus Erythematosus, in
Case Studies in Immunology, see Preface for details)
Insulin-Dependent Diabetes Mellitus and Systemic Lupus Erythematosus, in
Case Studies in Immunology, see Preface for details)
The autoantigens recognized in these two categories of disease are themselves respectively organ-specific and systemic. Thus, Graves' disease is characterized by the production of antibodies to the thyroid-stimulating hormone (TSH) receptor in the thyroid gland; Hashimoto's thyroiditis by antibodies to thyroid peroxidase; and type I diabetes by anti-insulin antibodies. By contrast, SLE is characterized by the presence of antibodies to antigens that are ubiquitous and abundant in every cell of the body, such as anti-chromatin antibodies and antibodies to proteins of the pre-mRNA splicing machinery—the spliceosome complex—within the cell.
It is likely that the organ-specific and systemic autoimmune diseases have somewhat different etiologies, which provides a biological basis for their division into two broad categories. Evidence for the validity of this classification also comes from observations that different autoimmune diseases cluster within individuals and within families. The organ-specific autoimmune diseases frequently occur together in many combinations; for example, autoimmune thyroid disease and the autoimmune depigmenting disease vitiligo are often found in the same person. Similarly, SLE and primary Sjögren's syndrome can coexist within a single individual or among different members of a family.
These clusters of autoimmune diseases provide the most useful classification into different subtypes, each of which may turn out to have a distinct mechanism. A working classification of autoimmune diseases based on clustering is given in Fig. 13.2. It can be seen that the strict separation of diseases into ‘organ-specific’ and ‘systemic’ categories breaks down to some extent. Not all autoimmune diseases can be usefully classified in this manner. Autoimmune hemolytic anemia, for example, sometimes occurs as a solitary entity and could be classified as an organ-specific disease. In other circumstances it may occur in conjunction with SLE as part of a systemic autoimmune disease.

Some common autoimmune diseases classified according to their ‘organ-specific’ or ‘systemic’ nature. Diseases that tend to occur in clusters are grouped in single boxes. Clustering is defined as more than one disease affecting (more...)
Although anyone can, in principle, develop an autoimmune disease, it seems that some individuals are more at risk than others of developing particular diseases. We will first consider those factors that contribute to susceptibility.
The best evidence in humans for susceptibility genes for autoimmunity comes from family studies, especially studies of twins. A semiquantitative technique for measuring what proportion of the susceptibility to a particular disease arises from genetic factors is to compare the incidence of disease in monozygotic and dizygotic twins. If a disease shows a high concordance in all twins, it could be caused by shared genetic or environmental factors. This is because both monozygotic and dizygotic twins tend to be brought up in shared environmental conditions. If the high concordance is restricted to monozygotic rather than dizygotic twins, however, then genetic factors are likely to be more important than environmental factors.
Studies with twins have been undertaken for several human diseases in which autoimmunity is important, including type I IDDM, rheumatoid arthritis, multiple sclerosis, and SLE. In each case, around 20% of pairs of monozygotic twins show disease concordance, compared with fewer than 5% of dizygotic twins. A similar technique is to compare the frequency of a disease such as diabetes in the siblings of patients who have diabetes with the frequency of that disease in the general population. The ratio of these two frequencies gives a measure of the heritability of the disease, although shared environmental factors within families could also be at least partly responsible for an increased frequency.
Results from both twin and family studies show an important role for both inherited and environmental factors in the induction of autoimmune disease. In addition to this evidence from humans, certain inbred mouse strains have an almost uniform susceptibility to particular spontaneous or experimentally induced autoimmune diseases, whereas other strains do not. These findings have led to an extensive search for genes that determine susceptibility to autoimmune disease.
So far, susceptibility to autoimmune disease has been most consistently associated with MHC genotype. Human autoimmune diseases that show associations with HLA type are shown in Fig. 13.3. For most of these diseases, susceptibility is linked most strongly with MHC class II alleles, but in some cases there are strong associations with particular MHC class I alleles.

Associations of HLA serotype and sex with susceptibility to autoimmune disease. The ‘relative risk’ for an HLA allele in an autoimmune disease is calculated by comparing the observed number of patients carrying the HLA allele with the (more...)
The association of MHC genotype with disease is assessed initially by comparing the frequency of different alleles in patients with their frequency in the normal population. For IDDM, this approach originally demonstrated an association with HLA-DR3 and HLA-DR4 alleles identified by serotyping (Fig. 13.4). Such studies also showed that the MHC class II allele HLA-DR2 has a dominant protective effect; individuals carrying HLA-DR2, even in association with one of the susceptibility alleles, rarely develop diabetes. Another way of determining whether MHC genes are important in autoimmune disease is to study the families of affected patients; it has been shown that two siblings affected with the same autoimmune disease are far more likely than expected to share the same MHC haplotypes (Fig. 13.5).
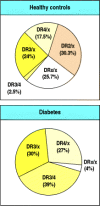
Population studies show association of susceptibility to IDDM with HLA genotype. The HLA genotypes (determined by serotyping) of diabetic patients (bottom panel) are not representative of those found in the population (top panel). Almost all diabetic (more...)

Family studies show strong linkage of susceptibility to IDDM with HLA genotype. In families in which two or more siblings have IDDM, it is possible to compare the HLA genotypes of affected siblings. Affected siblings share two HLA haplotypes much more (more...)
As HLA genotyping has become more exact through the sequencing of HLA alleles, disease associations that were originally discovered through HLA serotyping using antibodies have been defined more precisely. For example, the association between IDDM and the DR3 and DR4 alleles is now known to be due to their tight genetic linkage to DQβ alleles that confer susceptibility to disease. Indeed, disease susceptibility is most closely associated with polymorphisms at a particular position in the DQβ amino acid sequence. The most abundant DQβ amino acid sequence has an aspartic acid at position 57 that is able to form a salt bridge across the end of the peptidebinding cleft of the DQ molecule. By contrast, the diabetic patients in Caucasoid populations mostly have valine, serine, or alanine at that position and thus make DQ molecules that lack this salt bridge (Fig. 13.6). The nonobese diabetic (NOD) strain of mice, which develops spontaneous diabetes, also has a serine at that position in the homologous MHC class II molecule, known as I-Ag7.

Amino acid changes in the sequence of an MHC class II protein correlate with susceptibility to and protection from diabetes. The HLA-DQβ1 chain contains an aspartic acid (Asp) at position 57 in most people; in Caucasoid populations, patients with IDDM (more...)
The association of MHC genotype with autoimmune disease is not surprising, because autoimmune responses involve T cells, and the ability of T cells to respond to a particular antigen depends on MHC genotype. Thus the associations can be explained by a simple model in which susceptibility to an autoimmune disease is determined by differences in the ability of different allelic variants of MHC molecules to present autoantigenic peptides to autoreactive T cells. This would be consistent with what we know of T-cell involvement in particular diseases. In diabetes, for example, there are associations with both MHC class I and MHC class II alleles and this is consistent with the finding that both CD8 and CD4 T cells, which respond to antigens presented by MHC class I and MHC class II molecules, respectively, mediate the autoimmune response.
An alternative hypothesis for the association between MHC genotype and susceptibility to autoimmune diseases emphasizes the role of MHC alleles in shaping the T-cell receptor repertoire (see Chapter 7). This hypothesis proposes that self peptides associated with certain MHC molecules may drive the positive selection of developing thymocytes that are specific for particular autoantigens. Such autoantigenic peptides might be expressed at too low a level or bind too poorly to self MHC molecules to drive negative selection in the thymus, but be present at a sufficient level or bind strongly enough to drive positive selection. This hypothesis is supported by observations that I-Ag7, the disease-associated MHC class II molecule in the diabetes-prone NOD mice, binds many peptides very poorly and may therefore be less effective in driving intrathymic negative selection of T cells that bind self peptides.
However, MHC genotype alone does not determine genetic susceptibility to disease. Identical twins, sharing all of their genes, are far more likely to develop the same autoimmune disease than MHC-identical siblings, demonstrating that genetic factors other than the MHC also affect whether an individual develops disease. Recent studies of the genetics of autoimmune diabetes in humans and mice have shown that there are several independently segregating disease susceptibility loci in addition to the MHC.
There is also evidence that variation in the level of a potential autoantigen within the thymus can influence disease development. In the case of human insulin, which can act as an autoantigen in type I IDDM, the level of transcription of the insulin gene shows genetic variation between individuals; this is associated with a polymorphic minisatellite sequence located upstream of the gene. Gene variants that are transcribed at a high level in the thymus tend to protect against the development of diabetes, whereas variants transcribed at a lower level are associated with disease susceptibility. This is because the expression of high levels of insulin in the thymus may cause the deletion of T cells specific for the insulin peptides (see Section 7-24).
The major serological abnormality in SLE is the presence of autoantibodies to ubiquitous and abundant intracellular antigens, such as chromatin. How is tolerance broken to such all-pervasive self antigens? A number of genes have been implicated in the etiology of SLE in humans and mice (Fig. 13.7). These can be classified into three categories on the basis of their physiological function. The first comprises genes whose products are active in the body's mechanisms for disposing of dead and dying cells, which could provide a source of autoantigens. Genetic knockout in mice (see Appendix I, Section A-47) of four genes in this category has produced animal models of SLE. One of these genes codes for the complement protein C1q, which, together with other complement proteins, is involved in the effective clearance of immune complexes and apoptotic cells. A second gene in this category encodes serum amyloid P component, which binds chromatin and may mask it from the immune system. Its deletion results in the development of antibodies against chromatin and development of glomerulonephritis caused by deposition of immune complexes of these antibodies in the kidney. Third, deletion of DNase I, an enzyme that digests extracellular chromatin, results in the development of anti-chromatin antibodies and glomerulonephritis. Fourth, a similar phenotype has been seen in mice in which the secretory portion of the immunoglobulin μ chain is deleted, and which thus lack secreted IgM, which may have an important role in the clearance of effete cells. However, the majority of cases of spontaneous SLE are likely to be influenced by far more complex genetic factors than these single-gene defects.

Genes encoding three functional classes of proteins are implicated in the etiology of SLE. The first class comprises genes encoding proteins that may have a role in the clearance of cellular debris and masking it from the immune system. The second comprises (more...)
The second category of disease susceptibility genes for SLE includes those encoding proteins that regulate the thresholds for tolerance and activation of T and B lymphocytes, such as Fas, Fas ligand, the signaling molecule SHP-1, the B-cell inhibitory receptor CD22, FcγRIIB, and the cell-cycle inhibitor p21. The third category of genes encode proteins that could modify the expression of SLE in individual organs by their involvement in immune complex-mediated inflammation. Examples are the polymorphic genes for FcγRIIa and FcγRIII, where the variant proteins are thought to differ in their ability to bind immune complexes and are associated with the presence of nephritis in SLE.
A further very important factor in disease susceptibility to SLE is the hormonal status of the patient. Indeed, many autoimmune diseases show a strong sex bias (see Fig. 13.3). Where a bias towards disease in one sex is observed in experimental animals, castration or the administration of estrogen to males usually normalizes disease incidence between the two sexes. Furthermore, many autoimmune diseases that are more common in females show peak incidence in the years of active child bearing, when production of the female sex hormones estrogen and progesterone is at its greatest. A thorough understanding of how these genetic and hormonal factors contribute to disease susceptibility might allow us to prevent the autoimmune response.
Tissue injury in autoimmune disease results because the self antigen is an intrinsic component of the body and, consequently, the effector mechanisms of the immune system are directed at the body's own tissues. Also, because the adaptive immune response is incapable of removing the offending autoantigen from the body, the immune response persists, and there is a constant supply of new autoantigen, which amplifies the response. An important exception to this rule is type I IDDM, in which the autoimmune response destroys the target organ completely. This leads to a failure to produce insulin—one of the major autoantigens in this disease. Lack of insulin is in turn responsible for the phenotype of diabetes mellitus.
The mechanisms of tissue injury in autoimmunity can be classified according to the scheme adopted for hypersensitivity reactions (see Figs 13.1 and Figs 12.2). As with the hypersensitivity reactions, tissue damage can be mediated by the effector actions of both T cells and B cells. The antigen, or group of antigens, against which the autoimmune response is directed, and the mechanism by which the antigen-bearing tissue is damaged, together determine the pathology and clinical expression of the disease (see Fig. 13.1).
Autoimmune diseases differ from hypersensitivity responses in that type I IgE-mediated responses do not seem to have a major role. IgE autoantibodies have, however, been found in autoimmune disease, and although there is no proof that they mediate any autoimmune disease, there are diseases where this may be so. For example, asthma and eosinophilia (see Chapter 12) are found in a rare autoimmune vasculitis, an inflammatory disease of blood vessels that is known as Churg-Strauss vasculitis.
By contrast, autoimmunity that damages tissues by mechanisms analogous to type II hypersensitivity reactions is quite common. In this form of autoimmunity, IgG or IgM responses to autoantigens located on cell surfaces or extracellular matrix cause the injury. In other cases of autoimmunity, tissue damage can be due to type III responses, which involve immune complexes containing autoantibodies to soluble autoantigens; these autoimmune diseases are systemic and are characterized by autoimmune vasculitis. Finally, in a number of organ-specific autoimmune diseases, T-cell responses are directly involved in causing the tissue damage. In most autoimmune diseases, several mechanisms of immunopathogenesis operate. Examples include SLE, type I IDDM, and rheumatoid arthritis, in which there is evidence that both T-cell and antibody-mediated pathways cause tissue injury. We will examine how autoantibodies cause tissue damage, before ending with a consideration of self-reactive T-cell responses and their role in autoimmune disease.
IgG or IgM responses to antigens located on the surface of blood cells lead to
the rapid destruction of these cells. An example of this is autoimmune hemolytic anemia (Autoimmune Hemolytic Anemia, in
Case Studies in Immunology, see Preface for details), where
antibodies against self antigens on red blood cells trigger destruction of the
cells, leading to anemia. This can occur in two ways (Fig. 13.8). Red cells with bound IgG or IgM antibody are
rapidly cleared from the circulation by interaction with Fc or complement
receptors, respectively, on cells of the fixed mononuclear phagocytic system;
this occurs particularly in the spleen. Alternatively, the
autoantibody-sensitized red cells are lysed by formation of the membrane-attack complex of complement. In autoimmune thrombocytopenic purpura, autoantibodies against the GpIIb:IIIa
fibrinogen receptor on platelets can cause thrombocytopenia (a depletion of
platelets), which can in turn cause hemorrhage.
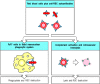
Antibodies specific for cell-surface antigens can destroy cells. In autoimmune hemolytic anemias, red cells coated with IgG autoantibodies against a cell-surface antigen are rapidly cleared from the circulation by uptake by Fc receptor-bearing macrophages (more...)
Lysis of nucleated cells by complement is less common because these cells are better defended by complement regulatory proteins. These proteins protect cells against immune attack by interfering with the activation of complement components and their assembly into a membrane-attack complex (see Section 2-14). Although the activation of complement by the bound autoantibody can proceed to a limited degree, nucleated cells are able to resist lysis by exocytosis or endocytosis of parts of the cell membrane bearing the membrane-attack complex. Nevertheless, nucleated cells targeted by autoantibodies are still destroyed by cells of the mononuclear phagocytic system. Autoantibodies against neutrophils, for example, cause neutropenia, which increases susceptibility to infection with pyogenic bacteria. In all of these cases, accelerated clearance of autoantibody-sensitized cells is the cause of their depletion in the blood. One therapeutic approach to this type of autoimmunity is removal of the spleen, the organ in which the main clearance of red cells, platelets, and leukocytes occurs.
The binding of IgG and IgM antibodies to cells in tissues causes inflammatory injury by a variety of mechanisms. One of these is fixation of complement. Although nucleated cells are relatively resistant to lysis by complement, the assembly of sublytic amounts of the membrane-attack complex on their surface provides a powerful activating stimulus. Depending on the type of cell, the interaction of sublytic doses of the membrane-attack complex with the cell membrane can cause cytokine release, generation of a respiratory burst, or the mobilization of membrane phospholipids to generate arachidonic acid—the precursor of prostaglandins and leukotrienes (lipid mediators of inflammation).
Most cells in tissues are fixed in place and cells of the inflammatory system are attracted to them by chemoattractant molecules. One such is the complement fragment C5a, which is released as a result of complement activation triggered by autoantibody binding. Other chemoattractants, such as leukotriene B4, can be released by the autoantibody-targeted cells. Inflammatory leukocytes are further activated by binding to autoantibody Fc regions and fixed complement C3 fragments on the tissue cells. Tissue injury can then result from the products of the activated leukocytes and by antibody-dependent cellular cytotoxicity mediated by natural killer (NK) cells (see Section 9-21).
A probable example of this type of autoimmunity is Hashimoto's thyroiditis, in which autoantibodies against tissue-specific antigens such as thyroid peroxidase and thyroglobulin are found at extremely high levels for prolonged periods. Direct T cell-mediated cytotoxicity, which we will discuss later, is probably also important in this disease.
A special class of type II hypersensitivity reaction occurs when the autoantibody binds to a cell-surface receptor. Antibody binding to a receptor can either stimulate the receptor or block its stimulation by its natural ligand. In Graves' disease, autoantibody against the thyroid-stimulating hormone receptor on thyroid cells stimulates the excessive production of thyroid hormone. The production of thyroid hormone is normally controlled by feedback regulation; high levels of thyroid hormone inhibit release of thyroid-stimulating hormone (TSH) by the pituitary. In Graves' disease, feedback inhibition fails because the autoantibody continues to stimulate the TSH receptor in the absence of TSH, and the patients become hyperthyroid (Fig. 13.9).

Feedback regulation of thyroid hormone production is disrupted in Graves' disease. Graves' disease is caused by autoantibodies specific for the receptor for thyroidstimulating hormone (TSH). Normally, thyroid hormones are produced in response to TSH and (more...)
In myasthenia gravis (Myasthenia Gravis, in
Case Studies in Immunology, see Preface for details), autoantibodies
against the α chain of the nicotinic acetylcholine receptor, which is present on
skeletal muscle cells at neuromuscular junctions, can block neuromuscular
transmission. The antibodies are believed to drive the internalization and
intracellular degradation of acetylcholine receptors (Fig. 13.10). Patients with myasthenia gravis develop
potentially fatal progressive weakness as a result of their autoimmune disease.
Diseases caused by autoantibodies that act as agonists or antagonists for
cell-surface receptors are listed in Fig.
13.11.
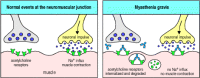
Autoantibodies inhibit receptor function in myasthenia gravis. In normal circumstances, acetylcholine released from stimulated motor neurons at the neuromuscular junction binds to acetylcholine receptors on skeletal muscle cells, triggering muscle contraction (left (more...)

Autoimmune diseases caused by autoantibodies against cell-surface receptors. These antibodies produce different effects depending on whether they are agonists (which stimulate) or antagonists (which inhibit) the receptor. Note that different autoantibodies (more...)
Antibody responses to extracellular matrix molecules are infrequent, but can be very damaging when they occur. In Goodpasture's syndrome, an example of a type II hypersensitivity reaction (see Fig. 12.2), antibodies are formed against the α3 chain of basement membrane collagen (type IV collagen). These antibodies bind to the basement membranes of renal glomeruli (Fig. 13.12a) and, in some cases, to the basement membranes of pulmonary alveoli, causing a rapidly fatal disease if untreated. The autoantibodies bound to basement membrane ligate Fcγ receptors, leading to activation of monocytes, neutrophils, and tissue basophils and mast cells. These release chemokines that attract a further influx of neutrophils into the glomeruli, causing severe tissue injury (Fig. 13.12b). The autoantibodies also cause local activation of complement, which may amplify the tissue injury.
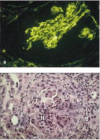
Autoantibodies reacting with glomerular basement membrane cause the inflammatory glomerular disease known as Goodpasture's syndrome. The panels show sections of renal glomeruli in serial biopsies taken from patients with Goodpasture's syndrome. Panel (more...)
Immune complexes are produced whenever there is an antibody response to a soluble antigen (see Appendix I, Section A-8). Normally, they are cleared efficiently by red blood cells bearing complement receptors and by phagocytes of the mononuclear phagocytic system that have both complement and Fc receptors, and such complexes cause little tissue damage. This clearance system can, however, fail in three circumstances. The first follows the injection of large amounts of antigen, leading to the formation of large amounts of immune complexes that overwhelm the normal clearance mechanisms. An example of this is serum sickness (see Section 12-16), which is caused by injection of large amounts of serum proteins. This is a transient disease, lasting only until the immune complexes have been cleared. The second circumstance is seen in chronic infections such as bacterial endocarditis, where the immune response to bacteria lodged on a cardiac valve is incapable of clearing infection. The persistent release of bacterial antigens from the valve infection in the presence of a strong antibacterial antibody response causes widespread immune-complex injury to small blood vessels in organs such as the kidney and the skin.
The third type of failure to clear immune complexes is seen in SLE. This is an immune complex-mediated disease characterized by chronic IgG antibody production directed at ubiquitous self antigens present in all nucleated cells. In SLE, a wide range of autoantibodies are produced to common cellular constituents. The main antigens are three intracellular nucleoprotein particles—the nucleosome, the spliceosome, and a small cytoplasmic ribonucleoprotein complex containing two proteins known as Ro and La (named after the first two letters of the surnames of the two patients in which autoantibodies against these proteins were discovered). In order for these autoantigens to participate in immune-complex formation, they must become extracellular. The autoantigens of SLE are exposed on dead and dying cells and released from injured tissues. In SLE, large quantities of antigen are available, so large numbers of small immune complexes are produced continuously and are deposited in the walls of small blood vessels in the renal glomerulus, in glomerular basement membrane (Fig. 13.13), in joints, and in other organs. This leads to activation of phagocytic cells through their Fc receptors. The consequent tissue damage releases more nucleoprotein complexes, which in turn form more immune complexes. Eventually, the inflammation induced in small blood vessel walls, especially in the kidney, can cause sufficient damage to kill the patient. Mice that lack FcγRIII illustrate the importance of Fc receptors in causing the inflammatory response to immune complexes. Such mice do not develop glomerulonephritis, despite deposition of immune complexes and C3, demonstrating the dominant role of Fc receptors in the autoimmune effector mechanisms of SLE.
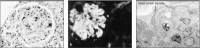
Deposition of immune complexes in the renal glomerulus causes renal failure in systemic lupus erythematosus (SLE). Panel a, a section through a renal glomerulus from a patient with SLE, shows that the deposition of immune complexes has caused thickening (more...)
The presence of an autoantibody by itself is not sufficient to cause autoimmune disease. For disease to occur, the autoantigen must be available for binding by the autoantibody. Two examples illustrate how the availability of autoantigens and the resulting expression of disease can be modulated by environmental cofactors. In untreated Goodpasture's disease, as described in Section 13-9, autoantibodies against type IV collagen typically cause a fatal glomerulonephritis. Type IV collagen is distributed widely in basement membranes throughout the body, including those of the alveoli of the lung, the renal glomeruli, and the cochlea of the inner ear. All patients with Goodpasture's disease develop glomerulonephritis, about 40% develop pulmonary hemorrhage, but none become deaf.
This pattern of disease expression was explained when it was discovered that pulmonary hemorrhage was found almost exclusively in those patients who smoked cigarettes. What differs between basement membrane in glomeruli, alveoli, and the cochlea is the availability of the antigen to antibodies. The major function of glomerular basement membrane is the filtration of plasma, and the endothelium lining glomerular capillaries is fenestrated to allow access of plasma to the basement membrane. Glomerular basement membrane is therefore immediately accessible to circulating autoantibodies. In the alveoli, in contrast, the basement membrane separates the alveolar epithelium from the capillary endothelium, whose cells are joined together by tight junctions. Injury to the endothelial lining of pulmonary capillaries is therefore necessary before antibodies can gain access to the basement membrane. Cigarette smoke stimulates an inflammatory response in the lungs, which damages alveolar capillaries and exposes the autoantigen to antibody. Finally, in the inner ear, the cochlear basement membrane seems to remain inaccessible to autoantibodies at all times.
A second example of the importance of environmental influences on the expression of autoimmunity is the effect of infection on the vasculitis associated with Wegener's granulomatosis. This disease, which is characterized by a severe necrotizing vasculitis, is strongly associated with the presence of autoantibodies to a granule proteinase of neutrophils (Fig. 13.14); the antibodies are known as anti-neutrophil cytoplasm antibodies (commonly abbreviated as ANCA). The autoantigen is proteinase-3, an abundant serine proteinase of neutrophil granules. Although there is a general correlation between the levels of ANCA and the expression of disease, it is quite common to find patients with high levels of ANCA who remain asymptomatic. If such an individual develops an infection, however, this frequently induces a severe flare-up of the vasculitis.

Serum from patients with Wegener's granulomatosis contains autoantibodies reactive with neutrophil cytoplasmic granules. Normal neutrophils with permeabilized cell membranes have been incubated with serum from a patient with Wegener's granulomatosis. IgG (more...)
It is thought that the reason for this is that resting neutrophils do not express proteinase-3 on the cell surface, and so in the absence of infection the antigen is inaccessible to anti-proteinase-3 autoantibodies. After infection, a variety of cytokines activate neutrophils, with translocation of proteinase-3 to the cell surface. Anti-proteinase-3 antibodies can now bind neutrophils and stimulate degranulation and release of free radicals. In parallel, activation of vascular endothelial cells by the infection causes the expression of vascular adhesion molecules, such as E-selectin, which promote the binding of activated neutrophils to vessel walls with resultant injury. In this way, a variety of nonspecific infections can exacerbate an autoimmune disease.
We have seen that the distribution of organ injury in Goodpasture's syndrome can be explained by the accessibility of basement membrane collagen to autoantibodies and that environmental factors can influence the availability of antigen in different organs. Another example of how the expression of autoimmune inflammation can be modified by anatomical factors is seen in membranous glomerulonephritis. In this disease, patients develop heavy proteinuria (the excretion of protein in the urine), which can cause life-threatening depletion of plasma protein levels. Biopsy of an affected kidney reveals evidence of deposition of antibody and complement beneath the basement membrane of the glomerulus but, in contrast to Goodpasture's syndrome, there is no significant influx of inflammatory cells. The autoantigen in this disease has not been characterized. However, an excellent rodent model of membranous glomerulonephritis is Heymann's nephritis, in which autoantibodies against a glycoprotein on the surface of tubular epithelial cells of the kidney are induced by injection of tubular epithelial tissue. The proteinuria can be abolished by depletion of any of the proteins of the membrane-attack complex of complement but is unaltered by depletion of neutrophils. This shows that the antibodies deposited beneath the glomerular basement membrane in this disease cause tissue injury by activation of complement, but the glomerular basement membrane acts as a complete barrier to inflammatory leukocytes.
In other autoimmune diseases, high levels of autoantibodies against intracellular antigens can be found in the absence of any evidence of antibody-induced inflammation. One such example is a rare myositis (inflammation of muscle) associated with pulmonary fibrosis. Most patients with this disease have high levels of autoantibodies reactive with aminoacyl-tRNA synthetases, the intracellular enzymes responsible for loading tRNAs with amino acids. Addition of these autoantibodies to cell-free extracts in vitro stops translation and protein synthesis completely. There is, however, no evidence that these antibodies cause any injury in vivo, where it is unlikely that they can enter living cells. In this disease, the autoantibody is thought to be a marker of a particular pattern of tissue injury, and does not contribute to the immunopathology of the myositis. Other examples of autoantibodies that are useful diagnostic markers of the presence of disease, but that might play no part in causing organ injury, are antibodies against mitochondrial antigens associated with primary biliary cirrhosis and antibodies against smooth muscle antigens in chronic active hepatitis.
To classify a disease as autoimmune , one must show that an adaptive immune response to a self antigen causes the observed pathology. Initially, the demonstration that antibodies against the affected tissue could be detected in the serum of patients suffering from various diseases was taken as evidence that the diseases had an autoimmune basis. However, such autoantibodies are also found when tissue damage is caused by trauma or infection, though these are typically of much lower affinity than those associated with autoimmune disease. This suggests that autoantibodies can result from, rather than be the cause of, tissue damage. Thus, one must show that the observed autoantibodies are pathogenic before classifying a disease as autoimmune.
It is often possible to transfer disease to experimental animals through the transfer of autoantibodies, causing pathology similar to that seen in the patient from whom the antibodies were obtained (Fig. 13.15). This does not always work, however, presumably because of species differences in autoantigen structure. Some autoimmune diseases can also be transferred from mother to fetus (Fig. 13.16) and are observed in the newborn babies of diseased mothers. When babies are exposed to IgG autoantibodies transferred across the placenta, they will often manifest pathology similar to the mother's (Fig. 13.17). This natural experiment is one of the best proofs that particular autoantibodies exert pathogenic effects. The symptoms of the disease in the newborn typically disappear rapidly as the maternal antibody is catabolized, although they may cause chronic organ injury, such as damage to the heart in babies of mothers with SLE or Sjögren's syndrome. The clearance can be speeded up by a complete exchange of the infant's blood or plasma (plasmapheresis), though this is of no clinical use after permanent injury has occurred, as in congenital heart block.
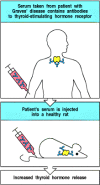
Serum from some patients with autoimmune disease can transfer the same disease to experimental animals. When the autoantigen is very similar in humans and mice or rats, the transfer of antibody from an affected human can cause the same symptoms in an (more...)

Some autoimmune diseases that can be transferred across the placenta by pathogenic IgG autoantibodies. These diseases are caused mostly by autoantibodies to cell-surface or tissue-matrix molecules. This suggests that an important factor determining whether (more...)
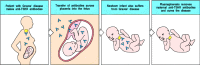
Antibody-mediated autoimmune diseases can appear in the infants of affected mothers as a consequence of transplacental antibody transfer. In pregnant women, IgG antibodies cross the placenta and accumulate in the fetus before birth (see Fig. 9.22). Babies (more...)
Activated effector T cells specific for self peptide:self MHC complexes can cause local inflammation by activating macrophages or can damage tissue cells directly. Diseases in which these actions of T cells are likely to be important include type I IDDM, rheumatoid arthritis, and multiple sclerosis. Affected tissues in patients with these diseases are heavily infiltrated with T lymphocytes and activated macrophages. These autoimmune diseases are mediated by T cells specific for the autoantigen presented by self MHC. T cells are, of course, also required to sustain all autoantibody responses.
It is much more difficult to demonstrate the existence of autoreactive T cells than it is to demonstrate the presence of autoantibodies. First, autoreactive human T cells cannot be used to transfer disease to experimental animals because T-cell recognition is MHC-restricted and animals and humans have different MHC alleles. Second, it is difficult to identify the antigen recognized by a T cell; for example, autoantibodies can be used to stain self tissues to reveal the distribution of the autoantigen, whereas T cells cannot. Nevertheless, there is strong evidence for the involvement of autoreactive T cells in several autoimmune diseases. In type I IDDM, the insulin-producing β cells of the pancreatic islets are selectively destroyed by specific T cells. When such diabetic patients are transplanted with half a pancreas from an identical twin donor, the β cells in the grafted tissue are rapidly and selectively destroyed by CD8 T cells. Recurrence of disease can be prevented by the immunosuppressive drug cyclosporin A (see Chapter 14), which inhibits T-cell activation. Progress towards identifying the targets of such autoreactive T cells and proving that these cells cause disease will be discussed in Section 13-15.
Autoantibodies can be used to purify an autoantigen so that it can be identified. This approach is particularly useful if the autoantibody causes disease in animals, from which large amounts of tissue can be obtained. Autoantibodies can also be used to examine the distribution of the target antigen in cells and tissues by immunohistology, often providing clues to the pathogenesis of the disease.
The identification of a critical autoantigen can also lead to the identification of the CD4 T cells responsible for stimulating autoantibody production. As we learned in Chapter 8, CD4 T cells selectively activate those B cells that bind epitopes that are physically linked to the peptide recognized by the T cell. It follows that the proteins or protein complexes isolated by means of autoantibodies should contain the peptide recognized by the autoreactive CD4 T cell. For example, in myasthenia gravis the autoantibodies that cause disease bind mainly to the α chain of the acetylcholine receptor and can be used to isolate the receptor from lysates of skeletal muscle cells. CD4 T cells that recognize peptide fragments of this receptor subunit can also be found in patients with myasthenia gravis (Fig. 13.18). Thus, both autoreactive B cells and autoreactive T cells are required for this disease.
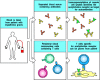
Autoimmune disease caused by antibodies also requires autoreactive T cells. Autoantibodies from the serum of myasthenia gravis patients immunoprecipitate the acetylcholine receptor from lysates of skeletal muscle cells (top panels). To be able to produce antibodies, (more...)
The same phenomenon is seen in SLE. Tissue damage in this disease is caused by immune complexes of autoantibodies directed against a variety of nucleoprotein antigens (see Section 13-9). These autoantibodies show a high degree of somatic hypermutation, which has all the hallmarks of being antigen-driven (see Section 4-9), and the B cells that produce them can be shown to have undergone extensive clonal expansion. Thus, the autoantibodies have the characteristic properties of antibodies formed in response to chronic stimulation of B cells by antigen and specific CD4 T cells, strongly suggesting that they are produced in response to autoantigens containing peptides recognized by specific autoreactive CD4 T cells. Further evidence for this comes from the collective autoantibody specificities observed in individual patients. The autoantibodies in any one individual tend to bind all constituents of a particular nucleoprotein particle; this strongly suggests that there must be CD4 T cells present that are specific for a peptide constituent of this particle. A B cell whose receptor binds a component of this particle will internalize and process the particle, present the peptide to these autoreactive T cells, and receive help from them (Fig. 13.19). Such B-cell-T-cell interactions initiate the antibody response and promote clonal expansion and somatic hypermutation, thus accounting for the observed characteristics of the autoantibody response as well as the clustering of autoantibody specificities in individual patients. This allows the spreading of the autoimmune response to different components of multimolecular complexes, known as antigen spreading or determinant spreading.

Autoreactive helper T cells of one specificity can drive the production of autoantibodies with several different specificities, in a phenomenon known as antigen spreading. In an SLE patient, a B cell specific for the H1 histone protein in nucleosomes, (more...)
Although there is good evidence that T cells are involved in many autoimmune diseases, the T cells that cause particular diseases are hard to isolate, and their targets are difficult to identify. Also, the cells are hyporesponsive and thus difficult to assay. It is also difficult to assay the T cells for their ability to cause disease, because any assay requires target cells of the same MHC genotype as the patient. This problem becomes more tractable in animal models. As many autoimmune diseases in animals are induced by immunization with self tissue, the nature of the autoantigen can be determined by fractionating an extract of the tissue and testing the fractions for their ability to induce disease. It is also possible to clone T-cell lines that will transfer the disease from an affected animal to another animal with the same MHC genotype.
This has made it possible to identify the autoantigens recognized by T cells in
many experimental autoimmune diseases; they are individual peptides that bind to
specific MHC molecules. In some cases, the peptide antigen, when made
immunogenic, is able to elicit disease symptoms in animals of the appropriate
MHC genotype. An example of such an experimental autoimmune disease is experimental allergic
encephalomyelitis (EAE), which can be induced in certain
susceptible strains of mice and rats by injection of central nervous system
tissue together with Freund's complete adjuvant. This disease resembles human
multiple sclerosis (Multiple Sclerosis, in
Case Studies in Immunology, see Preface for details), in which
characteristic plaques of tissue injury are disseminated throughout the central
nervous system. Plaques of active disease show infiltration of nervous tissue by
lymphocytes, plasma cells, and macrophages, which cause destruction of the
myelin sheaths that surround nerve cell axons in the brain and spinal cord.
Further analysis of EAE showed that injection with various purified components of the myelin sheath, notably myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendroglial protein (MOG), can induce EAE. The disease can be transferred to syngeneic animals by using cloned T-cell lines derived from animals with EAE (Fig. 13.20). Many of these cloned T-cell lines are stimulated by peptides of MBP. EAE can be caused by injection of these MBP peptides into animals that possess MHC alleles capable of presenting such peptides to T cells. Activated T cells specific for myelin proteins have also been identified in patients with multiple sclerosis. Although it has not yet been proved that these cells cause the demyelination in multiple sclerosis, this finding suggests that animal models might provide clues to the identity of autoantigenic proteins in human disease.

T cells specific for myelin basic protein mediate inflammation of the brain in experimental autoimmune encephalomyelitis (EAE). This disease is produced in experimental animals by injecting them with isolated spinal cord homogenized in complete Freunds' (more...)
A variety of inflammatory autoimmune diseases can be mediated by TH1 cells responding to self antigens. EAE, for example, can be caused by TH1 cells specific for MBP, as shown by the ability of specific clones of TH1, but not TH2, cells to cause disease on adoptive transfer. Although MBP is an intracellular protein, it is processed for presentation by the vesicular pathway and thus its peptides are presented by MHC class II molecules and recognized by CD4 T cells. Another inflammatory autoimmune disease, rheumatoid arthritis, may be caused by TH1 cells specific for an as yet unidentified antigen present in joints. Engagement with this antigen triggers the T cells to release lymphokines that initiate local inflammation within the joint. This causes swelling, accumulation of polymorphonuclear leukocytes and macrophages, and damage to cartilage, leading to the destruction of the joint. Rheumatoid arthritis is a complex disease and also involves antibodies, often including an IgM anti-IgG autoantibody called rheumatoid factor. Like the SLE autoantibodies described in Section 13-9, the rheumatoid factors isolated from the joints of patients with rheumatoid arthritis show evidence of a T-cell dependent, antigen-driven B-cell response against the Fc portion of IgG. Some of the tissue damage in this disease is caused by the resultant IgM:IgG immune complexes.
Autoantigens recognized by CD4 T cells can be identified by adding cell extracts to cultures of blood mononuclear cells and testing for recognition by CD4 cells derived from an autoimmune patient. If the autoantigen is present in the cell extract, it should be effectively presented, as phagocytes in the blood cultures can take up extracellular protein, degrade it in intracellular vesicles, and present the resulting peptides bound to MHC class II molecules. Identification of autoantigenic peptides is, however, particularly difficult in autoimmune diseases caused by CD8 T cells, as autoantigens recognized by CD8 T cells are not effectively presented in such cultures. Peptides presented by MHC class I molecules must usually be made by the target cells themselves (see Chapter 5); intact cells of target tissue from the patient must therefore be used to study autoreactive CD8 T cells that cause tissue damage. Conversely, the pathogenesis of the disease can itself give clues to the identity of the antigen in some CD8 T cell-mediated diseases. For example, in type I IDDM, the insulin-producing β cells of the pancreatic islets of Langerhans seem to be specifically targeted and destroyed by CD8 T cells (Fig. 13.21). This suggests that a protein unique to β cells is the source of the peptide recognized by the pathogenic CD8 T cells. Studies in the NOD mouse model of type I diabetes have shown that peptides from insulin itself are recognized by pathogenic CD8 cells, confirming the role of insulin as one of the principal autoantigens in type I diabetes.
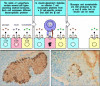
Selective destruction of pancreatic β cells in insulin-dependent diabetes mellitus (IDDM) indicates that the autoantigen is produced in β cells and recognized on their surface. In IDDM, there is highly specific destruction of insulin-producing (more...)
CD4 T cells also seem to be involved in type I IDDM, consistent with the linkage of disease susceptibility to particular MHC class II alleles (see Fig. 13.4). Identifying the autoantigen recognized by CD4 T cells in these diseases is an important goal. Not only might it help us to understand disease pathogenesis but it might also result in several innovative approaches to treatment (see Chapter 14).
For a disease to be defined as autoimmune, the tissue damage must be shown to be caused by an adaptive immune response to self antigens. Autoimmune diseases can be mediated by autoantibodies and/or by auto-reactive T cells, and tissue damage can result from direct attack on the cells bearing the antigen, from immune-complex formation, or from local inflammation. Autoimmune diseases caused by antibodies that bind to cellular receptors, causing either excess activity or inhibition of receptor function, fall into a special class. T cells can be involved directly in inflammation or cellular destruction, and they are also required to sustain autoantibody responses. Similarly, B cells may be important antigen-presenting cells for sustaining autoantigen-specific T-cell responses. The most convincing proof that the immune response is causal in autoimmunity is transfer of disease by transferring the active component of the immune response to an appropriate recipient. The immediate challenge is to identify the autoantigens recognized by T cells in autoimmunity, and to use this information to control the activity of these T cells, or to prevent their activation in the first place. The deeper, more important question is how the autoimmune response is induced. Much has been learned about the induction of immune responses to tissue antigens by examining the response to nonself tissues in transplantation experiments. We will therefore examine the immune response to grafted tissues in the next part of the chapter before turning to the problem of how tolerance is normally maintained, and why immune responses to self antigens occur to cause autoimmune disease.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...