Although genetic variation is important for evolution, the survival of the individual demands genetic stability. Maintaining genetic stability requires not only an extremely accurate mechanism for replicating DNA, but also mechanisms for repairing the many accidental lesions that occur continually in DNA. Most such spontaneous changes in DNA are temporary because they are immediately corrected by a set of processes that are collectively called DNA repair. Of the thousands of random changes created every day in the DNA of a human cell by heat, metabolic accidents, radiation of various sorts, and exposure to substances in the environment, only a few accumulate as mutations in the DNA sequence. We now know that fewer than one in 1000 accidental base changes in DNA results in a permanent mutation; the rest are eliminated with remarkable efficiency by DNA repair.
The importance of DNA repair is evident from the large investment that cells make in DNA repair enzymes. For example, analysis of the genomes of bacteria and yeasts has revealed that several percent of the coding capacity of these organisms is devoted solely to DNA repair functions. The importance of DNA repair is also demonstrated by the increased rate of mutation that follows the inactivation of a DNA repair gene. Many DNA repair pathways and the genes that encode them—which we now know operate in a wide variety of organisms, including humans—were originally identified in bacteria by the isolation and characterization of mutants that displayed an increased mutation rate or an increased sensitivity to DNA-damaging agents.
Recent studies of the consequences of a diminished capacity for DNA repair in humans have linked a variety of human diseases with decreased repair (). Thus, we saw previously that defects in a human gene that normally functions to repair the mismatched base pairs in DNA resulting from replication errors can lead to an inherited predisposition to certain cancers, reflecting an increased mutation rate. In another human disease, xeroderma pigmentosum (XP), the afflicted individuals have an extreme sensitivity to ultraviolet radiation because they are unable to repair certain DNA photoproducts. This repair defect results in an increased mutation rate that leads to serious skin lesions and an increased susceptibility to certain cancers.
Inherited Syndromes with Defects in DNA Repair.
Without DNA Repair, Spontaneous DNA Damage Would Rapidly Change DNA Sequences
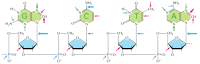
Although DNA is a highly stable material, as required for the storage of genetic information, it is a complex organic molecule that is susceptible, even under normal cellular conditions, to spontaneous changes that would lead to mutations if left unrepaired (). DNA undergoes major changes as a result of thermal fluctuations: for example, about 5000 purine bases (adenine and guanine) are lost every day from the DNA of each human cell because their N-glycosyl linkages to deoxyribose hydrolyze, a spontaneous reaction called depurination. Similarly, a spontaneous deamination of cytosine to uracil in DNA occurs at a rate of about 100 bases per cell per day (). DNA bases are also occasionally damaged by an encounter with reactive metabolites (including reactive forms of oxygen) or environmental chemicals. Likewise, ultraviolet radiation from the sun can produce a covalent linkage between two adjacent pyrimidine bases in DNA to form, for example, thymine dimers (). If left uncorrected when the DNA is replicated, most of these changes would be expected to lead either to the deletion of one or more base pairs or to a base-pair substitution in the daughter DNA chain (). The mutations would then be propagated throughout subsequent cell generations as the DNA is replicated. Such a high rate of random changes in the DNA sequence would have disastrous consequences for an organism.
A summary of spontaneous alterations likely to require DNA repair. The sites on each nucleotide that are known to be modified by spontaneous oxidative damage (red arrows), hydrolytic attack (blue arrows), and uncontrolled methylation by the methyl group (more...)
Depurination and deamination. These two reactions are the most frequent spontaneous chemical reactions known to create serious DNA damage in cells. Depurination can release guanine (shown here), as well as adenine, from DNA. The major type of deamination (more...)
The thymine dimer. This type of damage is introduced into DNA in cells that are exposed to ultraviolet irradiation (as in sunlight). A similar dimer will form between any two neighboring pyrimidine bases (C or T residues) in DNA.
How chemical modifications of nucleotides produce mutations. (A) Deamination of cytosine, if uncorrected, results in the substitution of one base for another when the DNA is replicated. As shown in Figure 5-47, deamination of cytosine produces uracil. (more...)
The DNA Double Helix Is Readily Repaired
The double-helical structure of DNA is ideally suited for repair because it carries two separate copies of all the genetic information—one in each of its two strands. Thus, when one strand is damaged, the complementary strand retains an intact copy of the same information, and this copy is generally used to restore the correct nucleotide sequences to the damaged strand.
An indication of the importance of a double-stranded helix to the safe storage of genetic information is that all cells use it; only a few small viruses use single-stranded DNA or RNA as their genetic material. The types of repair processes described in this section cannot operate on such nucleic acids, and the chance of a permanent nucleotide change occurring in these single-stranded genomes of viruses is thus very high. It seems that only organisms with tiny genomes can afford to encode their genetic information in any molecule other than a DNA double helix.
Each cell contains multiple DNA repair systems, each with its own enzymes and preferences for the type of damage recognized. As we see in the rest of this section, most of these systems use the undamaged strand of the double helix as a template to repair the damaged strand.
DNA Damage Can Be Removed by More Than One Pathway
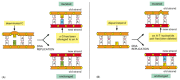
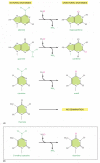
There are multiple pathways for DNA repair, using different enzymes that act upon different kinds of lesions. Two of the most common pathways are shown in . In both, the damage is excised, the original DNA sequence is restored by a DNA polymerase that uses the undamaged strand as its template, and the remaining break in the double helix is sealed by DNA ligase (see ).
A comparison of two major DNA repair pathways. (A) Base excision repair. This pathway starts with a DNA glycosylase. Here the enzyme uracil DNA glycosylase removes an accidentally deaminated cytosine in DNA. After the action of this glycosylase (or another (more...)
The two pathways differ in the way in which the damage is removed from DNA. The first pathway, called base excision repair, involves a battery of enzymes called DNA glycosylases, each of which can recognize a specific type of altered base in DNA and catalyze its hydrolytic removal. There are at least six types of these enzymes, including those that remove deaminated Cs, deaminated As, different types of alkylated or oxidized bases, bases with opened rings, and bases in which a carbon–carbon double bond has been accidentally converted to a carbon–carbon single bond.
As an example of the general mechanism of base excision repair, the removal of a deaminated C by uracil DNA glycosylase is shown in . How is the altered base detected within the context of the double helix? A key step is an enzyme-mediated “flipping-out” of the altered nucleotide from the helix, which allows the enzyme to probe all faces of the base for damage (). It is thought that DNA glycosylases travel along DNA using base-flipping to evaluate the status of each base pair. Once a damaged base is recognized, the DNA glycosylase reaction creates a deoxyribose sugar that lacks its base. This “missing tooth” is recognized by an enzyme called AP endonuclease, which cuts the phosphodiester backbone, and the damage is then removed and repaired (see ). Depurination, which is by far the most frequent type of damage suffered by DNA, also leaves a deoxyribose sugar with a missing base. Depurinations are directly repaired beginning with AP endonuclease, following the bottom half of the pathway in .
The recognition of an unusual nucleotide in DNA by base-flipping. The DNA glycosylase family of enzymes recognizes specific bases in the conformation shown. Each of these enzymes cleaves the glycosyl bond that connects a particular recognized base (yellow) (more...)
The second major repair pathway is called nucleotide excision repair. This mechanism can repair the damage caused by almost any large change in the structure of the DNA double helix. Such “bulky lesions” include those created by the covalent reaction of DNA bases with large hydrocarbons (such as the carcinogen benzopyrene), as well as the various pyrimidine dimers (T-T, T-C, and C-C) caused by sunlight. In this pathway, a large multienzyme complex scans the DNA for a distortion in the double helix, rather than for a specific base change. Once a bulky lesion has been found, the phosphodiester backbone of the abnormal strand is cleaved on both sides of the distortion, and an oligonucleotide containing the lesion is peeled away from the DNA double helix by a DNA helicase enzyme. The large gap produced in the DNA helix is then repaired by DNA polymerase and DNA ligase ().
The Chemistry of the DNA Bases Facilitates Damage Detection
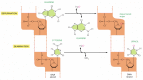
The DNA double helix seems to be optimally constructed for repair. As noted above, it contains a backup copy of the genetic information, so that if one strand is damaged, the other undamaged strand can be used as a template for repair. The nature of the bases also facilitates the distinction between undamaged and damaged bases. Thus, every possible deamination event in DNA yields an unnatural base, which can therefore be directly recognized and removed by a specific DNA glycosylase. Hypoxanthine, for example, is the simplest purine base capable of pairing specifically with C, but hypoxanthine is the direct deamination product of A (). The addition of a second amino group to hypoxanthine produces G, which cannot be formed from A by spontaneous deamination, and whose deamination product is likewise unique.
The deamination of DNA nucleotides. In each case the oxygen atom that is added in this reaction with water is colored red. (A) The spontaneous deamination products of A and G are recognizable as unnatural when they occur in DNA and thus are readily recognized (more...)
As discussed in Chapter 6, RNA is thought, on an evolutionary time-scale, to have served as the genetic material before DNA, and it seems likely that the genetic code was initially carried in the four nucleotides A, C, G, and U. This raises the question of why the U in RNA was replaced in DNA by T (which is 5-methyl U). We have seen that the spontaneous deamination of C converts it to U, but that this event is rendered relatively harmless by uracil DNA glycosylase. However, if DNA contained U as a natural base, the repair system would be unable to distinguish a deaminated C from a naturally occuring U.
A special situation occurs in vertebrate DNA, in which selected C nucleotides are methylated at specific C-G sequences that are associated with inactive genes (discussed in Chapter 7). The accidental deamination of these methylated C nucleotides produces the natural nucleotide T () in a mismatched base pair with a G on the opposite DNA strand. To help in repairing deaminated methylated C nucleotides, a special DNA glycosylase recognizes a mismatched base pair involving T in the sequence T-G and removes the T. This DNA repair mechanism must be relatively ineffective, however, because methylated C nucleotides are common sites for mutations in vertebrate DNA. It is striking that, even though only about 3% of the C nucleotides in human DNA are methylated, mutations in these methylated nucleotides account for about one-third of the single-base mutations that have been observed in inherited human diseases.
Double-Strand Breaks are Efficiently Repaired
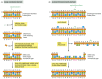
A potentially dangerous type of DNA damage occurs when both strands of the double helix are broken, leaving no intact template strand for repair. Breaks of this type are caused by ionizing radiation, oxidizing agents, replication errors, and certain metabolic products in the cell. If these lesions were left unrepaired, they would quickly lead to the breakdown of chromosomes into smaller fragments. However, two distinct mechanisms have evolved to ameliorate the potential damage. The simplest to understand is nonhomologous end-joining, in which the broken ends are juxtaposed and rejoined by DNA ligation, generally with the loss of one or more nucleotides at the site of joining (). This end-joining mechanism, which can be viewed as an emergency solution to the repair of double-strand breaks, is a common outcome in mammalian cells. Although a change in the DNA sequence (a mutation) results at the site of breakage, so little of the mammalian genome codes for proteins that this mechanism is apparently an acceptable solution to the problem of keeping chromosomes intact. As previously discussed, the specialized structure of telomeres prevents the ends of chromosomes from being mistaken for broken DNA, thereby preserving natural DNA ends.
Two different types of end-joining for repairing double-strand breaks. (A) Nonhomologous end-joining alters the original DNA sequence when repairing broken chromosomes. These alterations can be either deletions (as shown) or short insertions. (B) Homologous (more...)
An even more effective type of double-strand break repair exploits the fact that cells that are diploid contain two copies of each double helix. In this second repair pathway, called homologous end-joining, general recombination mechanisms are called into play that transfer nucleotide sequence information from the intact DNA double helix to the site of the double-strand break in the broken helix. This type of reaction requires special recombination proteins that recognize areas of DNA sequence matching between the two chromosomes and bring them together. A DNA replication process then uses the undamaged chromosome as the template for transferring genetic information to the broken chromosome, repairing it with no change in the DNA sequence (). In cells that have replicated their DNA but not yet divided, this type of DNA repair can readily take place between the two sister DNA molecules in each chromosome; in this case, there is no need for the broken ends to find the matching DNA sequence in the homologous chromosome. The molecular details of the homologous end-joining reaction are considered later in this chapter because they require a general understanding of the way in which cells carry their genetic recombination events. Although present in humans, this type of DNA double-strand break repair predominates in bacteria, yeasts, and Drosophila—all organisms in which little nonhomologous DNA end-joining is observed.
Cells Can Produce DNA Repair Enzymes in Response to DNA Damage
Cells have evolved many mechanisms that help them survive in an unpredictably hazardous world. Often an extreme change in a cell's environment activates the expression of a set of genes whose protein products protect the cell from the deleterious effects of this change. One such mechanism shared by all cells is the heat-shock response, which is evoked by the exposure of cells to unusually high temperatures. The induced “heat-shock proteins” include some that help stabilize and repair partly denatured cell proteins, as discussed in Chapter 6.
Cells also have mechanisms that elevate the levels of DNA repair enzymes, as an emergency response to severe DNA damage. The best-studied example is the so-called SOS response in E. coli. In this bacterium, any block to DNA replication caused by DNA damage produces a signal that induces an increase in the transcription of more than 15 genes, many of which code for proteins that function in DNA repair. The signal (thought to be an excess of single-stranded DNA) first activates the E. coli RecA protein (see ), so that it destroys a gene regulatory protein that normally represses the transcription of a large set of SOS response genes.
Studies of mutant bacteria deficient in different parts of the SOS response demonstrate that the newly synthesized proteins have two effects. First, as would be expected, the induction of these additional DNA repair enzymes increases cell survival after DNA damage. Second, several of the induced proteins transiently increase the mutation rate by increasing the number of errors made in copying DNA sequences. The errors are caused by the production of low-fidelity DNA polymerases that can efficiently use damaged DNA as a template for DNA synthesis. While this “error-prone” DNA repair can be harmful to individual bacterial cells, it is presumed to be advantageous in the long term because it produces a burst of genetic variability in the bacterial population that increases the likelihood of a mutant cell arising that is better able to survive in the altered environment.
Human cells contain more than ten minor DNA polymerases, many of which are specifically called into play, as a last resort, to copy over unrepaired lesions in the DNA template. These enzymes can recognize a specific type of DNA damage and add the nucleotides that restore the initial sequence. Each such polymerase molecule is given a chance to add only one or a few nucleotides, because these enzymes are extremely error-prone when they copy a normal DNA sequence. Although the details of these fascinating reactions are still being worked out, they provide elegant testimony to the care with which organisms maintain their DNA sequences.
DNA Damage Delays Progression of the Cell Cycle
We have just seen that cells contain multiple enzyme systems that can recognize DNA damage and promote the repair of these lesions. Because of the importance of maintaining intact, undamaged DNA from generation to generation, cells have an additional mechanism that helps them respond to DNA damage: they delay progression of the cell cycle until DNA repair is complete. For example, one of the genes expressed in response to the E. coli SOS signal is sulA, which encodes an inhibitor of cell division. Thus, when the SOS functions are turned on in response to DNA damage, a block to cell division extends the time for repair. When DNA repair is complete, the expression of the SOS genes is repressed, the cell cyle resumes, and the undamaged DNA is segregated to the daughter cells.
Damaged DNA also generates signals that block cell-cycle progression in eucaryotes. As discussed in detail in Chapter 17, the orderly progression of the cell cycle is maintained through the use of checkpoints that ensure the completion of one step before the next step can begin. At several of these cell-cycle checkpoints, the cycle stops if damaged DNA is detected. Thus, in yeast, the presence of DNA damage can block entry into the G1 phase; it can slow DNA replication once begun; and it can block the transition from S phase to M phase. The DNA damage results in an increased synthesis of some DNA repair enzymes, and the delays further facilitate repair by providing the time needed for repair to reach completion.
The importance of the special signaling mechanisms that respond to DNA damage is indicated by the phenotype of humans who are born with defects in the gene that encodes the ATM protein, a large protein kinase. These individuals have the disease ataxia–telangiectasia (AT), whose symptoms include neurodegeneration, a predisposition to cancer, and genome instability. In both humans and yeasts, the ATM protein is needed to generate the initial intracellular signals that produce a response to oxygen-inflicted DNA damage, and individual organisms with defects in this protein are hypersensitive to agents that cause such damage, such as ionizing radiation.
Summary
Genetic information can be stored stably in DNA sequences only because a large set of DNA repair enzymes continuously scan the DNA and replace any damaged nucleotides. Most types of DNA repair depend on the presence of a separate copy of the genetic information in each of the two strands of the DNA double helix. An accidental lesion on one strand can therefore be cut out by a repair enzyme and a corrected strand resynthesized by reference to the information in the undamaged strand.
Most of the damage to DNA bases is excised by one of two major DNA repair pathways. In base excision repair, the altered base is removed by a DNA glycosylase enzyme, followed by excision of the resulting sugar phosphate. In nucleotide excision repair, a small section of the DNA strand surrounding the damage is removed from the DNA double helix as an oligonucleotide. In both cases, the gap left in the DNA helix is filled in by the sequential action of DNA polymerase and DNA ligase, using the undamaged DNA strand as the template.
Other critical repair systems—based on either nonhomologous or homologous end-joining mechanisms—reseal the accidental double-strand breaks that occur in the DNA helix. In most cells, an elevated level of DNA damage causes both an increased synthesis of repair enzymes and a delay in the cell cycle. Both factors help to ensure that DNA damage is repaired before a cell divides.